
Abstract
Canagliflozin is an antidiabetic medicine that inhibits sodium-glucose cotransporter 2 (SGLT2) in proximal tubules. Recently, it was reported to have several noncanonical effects other than SGLT2 inhibiting. However, the effects of canagliflozin on skeletal muscle regeneration remain largely unexplored. Thus, in vivo muscle contractile properties recovery in mice ischemic lower limbs following gliflozins treatment was evaluated. The C2C12 myoblast differentiation after gliflozins treatment was also assessed in vitro. As a result, both in vivo and in vitro data indicate that canagliflozin impairs intrinsic myogenic regeneration, thus hindering ischemic limb muscle contractile properties, fatigue resistance recovery, and tissue regeneration. Mitochondrial structure and activity are both disrupted by canagliflozin in myoblasts. Single-cell RNA sequencing of ischemic tibialis anterior reveals a decrease in leucyl-tRNA synthetase 2 (LARS2) in muscle stem cells attributable to canagliflozin. Further investigation explicates the noncanonical function of LARS2, which plays pivotal roles in regulating myoblast differentiation and muscle regeneration by affecting mitochondrial structure and activity. Enhanced expression of LARS2 restores the differentiation of canagliflozin-treated myoblasts, and accelerates ischemic skeletal muscle regeneration in canagliflozin-treated mice. Our data suggest that canagliflozin directly impairs ischemic skeletal muscle recovery in mice by downregulating LARS2 expression in muscle stem cells, and that LARS2 may be a promising therapeutic target for injured skeletal muscle regeneration.
Similar content being viewed by others


Introduction
Gliflozins are a new class of oral antidiabetic drugs that inhibit sodium-glucose cotransporter 2 (SGLT2), which functions as the main glucose transporter of the kidney and is responsible for the reabsorption of 90% of glucose from primary urine [1]. SGLT2 inhibitor (SGLT2i) reduces the reabsorption of glucose in proximal tubules and therefore enhances urinary glucose excretion, which consequently decreases both fasting and postprandial hyperglycemia. Recently, SGLT2i was reported to have several noncanonical effects other than SGLT2 inhibition [2,3,4]. However, the CANVAS program reported an unexpected increase in the risk of lower extremity amputation in the canagliflozin (CANA)-treated group [5]. In addition, a pharmacovigilance analysis suggested that compared to dapagliflozin (DAPA) and empagliflozin (EMPA), CANA is associated with a higher risk of amputation [6]. Our previous data indeed revealed that glutamate dehydrogenase 1 activity inhibition by canagliflozin interferes with the metabolic activity of mitochondria, which degrades retention and VEGFA secretion of mesenchymal stem cells, hampering angiogenesis and the recovery of blood flow following lower limb ischemia [7]. However, ischemic tissue repair depends not only on blood flow recovery but also on the regeneration of ischemic skeletal muscle.
Skeletal muscle consists of multinucleated myofibers that form through the fusion of mononucleated myoblasts, which is required for skeletal muscle formation during growth and post-injury regeneration [8]. Myogenesis is an important event required for muscle development and regeneration and is a highly coordinated process that includes activation, proliferation, and differentiation of muscle stem cells (MuSC) into fused, multinucleated myotubes. In response to injury, quiescent muscle progenitor cells will be activated and give rise to a transient-amplifying population of myoblasts that migrate to the site of injury and subsequently differentiate and fuse to form newly multinucleated fibers to repair and rebuild the damaged myofiber [9]. Therefore, myogenic regeneration and differentiation is the fundamental step during myogenesis and is regulated by numerous factors, including myogenic regulatory factors (MRFs), noncoding RNAs, micropeptides [10], and mitochondrial maintenance [11, 12]. The question of whether the intrinsic regeneration and differentiation ability of skeletal muscle are directly affected by gliflozins remains completely unexplored.
Therefore, in the present study, we aimed to test the effect of CANA on intrinsic myogenic regeneration and differentiation of ischemic muscle in mice and to uncover the possible underlying mechanism.
Materials and methods
Cell culture and treatments
The C2C12 cell line was purchased from American Type Culture Collection (Manassas, VA, USA). C2C12 cells were cultured in DMEM (Corning, NY, USA) supplemented with 10% (v/v) FBS (Gibco, Carlsbad, CA, USA) (growth medium). Cells were maintained at 37 °C in a 5% CO2 incubator. C2C12 cells were treated with either 10 μM CANA, 1 μM DAPA or 1 μM EMPA (all from Apexbio, Houston, TX, USA). The final concentration for dimethyl sulfoxide was 0.1% (v/v), and 0.1% (v/v) dimethyl sulfoxide was chosen as a negative control. The concentrations chosen for in vitro experiments were on the basis of clinically relevant concentrations of these medicines [13,14,15].
For myoblast differentiation, C2C12 myoblasts at 90% confluency were transferred from growth medium to DMEM containing 2% (v/v) horse serum (differentiation medium, DM). Myotube differentiation was induced for 24, 36, 72, or 120 h.
Western blot analysis
Cell and tissue lysates were prepared using radioimmunoprecipitation assay buffer. The samples were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to a polyvinylidene fluoride membrane, and immunoblotted with the following antibodies: cyclin A2 (CCNA2, 1:1000, ET1612-26, Huabio, Hangzhou, China), cyclin D1 (CCND1, 1:1000, ET1601-31, Huabio), MyoD1 (1:1000, ER1913-45, Huabio), voltage-dependent anion-selective channel (VDAC1, 1:1000, ET1601-20, Huabio), desmin (1:1000, A3736, Abclonal, Wuhan, China), Leucyl-tRNA synthetase 1 (LARS1, 1:1000, A14890, Abclonal), LARS2 (1:1000, DF13122, Affinity, Changzhou, China), embryonic Myosin heavy chain (em-MHC, 1:100, F1.652, DSHB, Lowa, IA, USA), MHC (1:100, MF20, DSHB), myogenin (MyoG, 1:1000, ab1835, Abcam, Cambridge, MA, USA), CHCHD3 (1:1000, MIC19, ab224565, Abcam), OPA1 (1:1000, ab119685, Abcam), Mitofilin (1:1000, ab110329, Abcam), SAM50 (1:1000, ab246987, Abcam), Mitofusin 1 (1:1000, ab221661, Abcam), Mitofusin 2 (1:1000, ab124773, Abcam), DRP1 (1:1000, #8570, Cell Signaling Technology, Danvers, MA, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
QRT-PCR was performed using the ChamQ Universal SYBR qPCR Master Mix kit (Vazyme, Nanjing, China) according to standard protocols. Fold expression relative to the reference gene was calculated using the comparative method 2−ΔΔCt. The primer sequences are indicated in Supplementary Table S1.
Oxygen consumption rate (OCR) analysis
C2C12 cells either treated as previously mentioned or transfected with LARS2 siRNA or control siRNA were subjected to differentiation induction for 36 h. Cells were then detached and collected for OCR analysis. Briefly, a total of 1 × 106 cells were suspended in DMEM (containing 2% FBS), and 2 mL of the cell suspension was added to the Oxygraph-2k chamber (O2k; OROBOROS Instruments, Innsbruck, Austria). The basal OCR of the cells was measured in DMEM with no stimulation, and the maximum OCR of the cells was measured in DMEM after adding 1 μM carbonyl cyanide m-chlorophenylhydrazone.
ATP quantification
Cellular ATP level was detected using an Enhanced ATP Assay Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions and visualized by a luminometer (SpectraMax M5, Molecular Devices, Sunnyvale, CA, USA).
Cell proliferation assay
Cells were treated in a growth medium for 48 h. Cell proliferation was analyzed using a Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) and a 5-ethynyl-2’-deoxyuridine cell proliferation assay kit (RiboBio, Guangzhou, China) according to the manufacturer’s instructions.
Immunocytochemistry study
Cells were fixed with precooled acetone for 15 min, permeabilized with 0.5% (v/v) Triton X-100 and incubated with primary antibodies against Desmin (1:100, Abclonal), MyoG (1:100, Abcam) or MHC (1:20, DSHB) overnight at 4 °C, followed by incubation with FITC dye-conjugated goat anti-rabbit IgG (1:50, Cell Signaling Technology) for 1 h at 37 °C. Nuclei were stained with Hoechst 33258 and viewed using a fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Electron microscopy (TEM)
Cells were collected and immediately fixed at 4 °C overnight in 3% (v/v) glutaraldehyde. Samples were then postfixed with 1% (v/v) osmic acid for 2 h. After being serially dehydrated, the samples were embedded in epoxy resin to form blocks of cells. Ultrathin sections (50 nm) were made using an ultramicrotome (Ultracut UCT, Leica Microsystems, Wetzlar, Germany). The sections were then stained with lead citrate and uranyl acetate and examined under transmission electron microscopy (TEM; T10, FEI, Hillsboro, OR, USA).
RNA interference and plasmid transfection
C2C12 myoblasts at 30%–40% confluency were transfected with 50 nM LARS1 or LARS2 siRNA (Supplementary Table S2, SUNYA, Hangzhou, China) using Lipofectamine RNAiMAX (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol.
For plasmid transfection, C2C12 myoblasts at 60% confluency were transfected with LARS2 plasmid (1 μg/mL) using Lipofectamine 2000 (Life Technologies) according to standard protocols.
Animals
All animal experimental protocols were approved by the Animal Care and Use Committee of Wenzhou Medical University (approval number: wydw2021-0239). Animals were maintained at the animal facility according to protocols of the Review Committee of Animal Care and Use. To establish the diabetic mouse model, male C57BL/6 mice (aged 6 weeks, weighing 18–20 g, Cyagen, Suzhou, China) were fed a high-fat diet (60 kcal%; Biotech Co. Ltd., Beijing, China) for 4 weeks. Experimental diabetes was then induced by daily intraperitoneal injections of streptozotocin (40 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) in citrate buffer for 4 days. Stable blood glucose levels exceeding 13.78 mmol/L (250 mg/dL) were considered to indicate a diabetic mouse.
To mimic the pathological process of muscle injury and regeneration caused by diabetic peripheral artery disease, we induced muscle injury using a femoral artery ligation (FAL) model. Briefly, CANA (10 mg·kg−1·d−1), DAPA (1 mg·kg−1·d−1), EMPA (10 mg·kg−1·d−1) or vehicle control (distilled water containing 0.5% carboxymethyl cellulose and 0.25% Tween-80) was administered to diabetic male C57BL/6 mice via oral gavage daily for 7 days. Ligation and excision of a 3- to 5-mm segment of the femoral artery proximal to the superficial caudal epigastric artery branch was performed. Daily oral gavage of CANA, DAPA, EMPA or vehicle was then continued until 3/7/14/21 days. Laser Doppler perfusion imaging (LDPI, PeriCamPSI, Perimed AB, Sweden) was carried out for limb blood flow evaluation. Mice were killed at 3/7/14/21 days post FAL, and both gastrocnemius and tibialis anterior (TA) were collected for further analysis.
Construction and administration of Lars2 adeno-associated virus vectors
Recombinant adeno-associated virus 9 (AAV9)-expressing mouse Lars2 was constructed and produced by Genechem Co. Ltd. (Shanghai, China). For AAV injection, mice were subjected to right TA and gastrocnemius AAV injection at a concentration of 1 × 1012 μg/mL (2 μL per injection, 5 different sites for each muscle) following anesthesia with isoflurane. Mice that received AAV-vector injection were chosen as a control group (CTRL). Four weeks post injection, mice were further subjected to CANA or vehicle controlled daily gavage for 7 days, followed by right FAL.
Muscle force and fatigue measurements
In situ TA force measurements were performed with the RM6240 signal acquisition system (Chengdu Instrument Factory, Chengdu, China) based on previous protocols [16] with slight modification. In brief, mice were anesthetized using 1% (v/v) pentobarbital on day 14 or 21 post FAL. Right TA was exposed and the distal tendon was freed from connective tissue and attached to the mechanical force transducer using a 6–0 suture. The sciatic nerve was then stimulated with 0.2 s pulses at 5 V, and the muscle was allowed to rest after each stimulation. All isometric measurements were made at an initial muscle length of L0, determined by applying successive single twitch stimulations and adjusting basal muscle tension until maximal isometric twitch force (Pt) was reached. The muscle was then stimulated at different frequencies to obtain absolute maximal tetanic force (P0). Muscle masses (m) were measured to calculate specific tensions (P0/m).
For fatigue resistance, TA was subjected to a series of successive 100 Hz tetanic stimulations delivered every 2 s for 1 min, and the muscle tetanic forces were successively recorded.
Single-cell RNA sequencing (ScRNA-seq) library preparation and sequencing
ScRNA-seq libraries of ischemic TA were prepared using 10× Genomics-Chromium Next GEM Single Cell 3ʹ GEM, Library & Gel Bead Kit v3.1 on a Chromium Controller (10× Genomics, Pleasanton, CA, USA) according to the manufacturer’s recommendation by Beijing Biomarker Technologies Co. Ltd. (Beijing, China). The quality of libraries was assessed using Qubit 4.0 and Agilent 2100. Sequencing was performed on an Illumina NovaSeq with a sequencing depth of at least 50,000 reads per cell and 150 bp (PE150) paired-end reads.
Histological analysis and immunofluorescence study
For succinate dehydrogenase (SDH) staining, frozen sections were incubated in a solution containing 1.5 mM nitroblue tetrazolium, 130 mM sodium succinate, 0.2 mM phenazine methosulfate and 0.1 mM sodium azide for 60 min. Masson staining, Oil-Red O staining, and hematoxylin and eosin (H&E) were conducted according to the standard protocol.
For the immunofluorescence study, frozen sections were fixed with precooled acetone for 15 min, blocked with 5% (v/v) BSA in PBS for 30 min at room temperature, and then incubated with primary antibodies against laminin (1:50, L9393, Sigma-Aldrich, St Louis, MO, USA), em-MHC (1:20, DSHB), MHC (1:20, DSHB) or desmin (1:100, Abclonal) overnight at 4 °C, followed by incubation with FITC dye-conjugated goat anti-rabbit IgG (Cell Signaling Technology) for 1 h at 37 °C. Nuclei were stained with Hoechst and tissue sections were viewed using a fluorescence microscope (Leica) or an A1R-Structured Illumination Superresolution Microscope (A1R-SIM, Nikon, Tokyo, Japan).
Statistical analysis
All the data are presented as mean ± SDs. Statistical analyses were performed using Prism 9.0 software (GraphPad Software Inc., La Jolla, CA, USA). Student’s unpaired t test was performed for comparisons between two groups, and ANOVA followed by Dunnett’s correction was used for comparisons between more than two groups. P < 0.05 was considered statistically significant.
Results
CANA, but not DAPA or EMPA, attenuated ischemic muscle function recovery
We first evaluated the effects of SGLT2 inhibitors on ischemic skeletal muscle function recovery in diabetic mice. The general study design is depicted in Fig. 1a. In situ measurements of TA contraction in response to sciatic nerve stimulation were performed to assess whether the administration of gliflozins had any functional consequences on muscle contractile recovery and fatigue properties after hindlimb ischemia (confirmed using a Laser Doppler, Supplementary Fig. S1a). As shown in Fig. 1b, each group of muscles produced twitches similar to those of their control (CTRL) counterparts, while the body weights and TA mass remained unchanged (Supplementary Fig. S1b, c). In contrast, blood glucose was slightly deceased by CANA and EMPA (Supplementary Fig. S1d). In contrast, CANA treatment significantly decreased tetanic force (Fig. 1c, d) and slightly increased the time-to-peak tension (Supplementary Fig. S1e). After 1 min of repeated stimulation, the contraction force started to drop faster in the CANA group than in the other groups and remained lower until the end of the fatigue protocol (Fig. 1e). Thus, the duration corresponding to a 20% decrease in force (fatigue resistance) was significantly decreased by CANA (Fig. 1f). In addition, we noted that CANA slightly elevated creatine kinase and blood urea nitrogen without affecting the other blood biochemistry parameters (Supplementary Fig. S1f).

a The general in vivo study design using diabetic mice as an experimental model (n = 6 for each group). b In situ TA absolute muscle single twitch force evoked under a 1 Hz stimulation. c TA absolute muscle force–frequency relationship evoked by electrical sciatic nerve stimulation. d TA mass (g)-normalized tetanic forces. e Average curves showing the force decline during a 60-s muscle fatigue protocol. f The duration corresponding to a 20% decrease in force was noted and presented as a bar graph. g Sketchily study design using normoglycemic mice as an in vivo experimental model (n = 6 for each group). h In situ TA absolute muscle single twitch force elicited using a 1 Hz stimulation. i TA absolute muscle force–frequency relationship. j TA mass normalized tetanic forces. k Representative traces of twitch force decline using the method indicated in (e). l Fatigue resistance was determined as indicated in (f). All data are presented as mean ± SD (error bars) and were analyzed by one-way ANOVA with Dunnett’s correction. *P < 0.05, **P < 0.01, and ***P < 0.001.
To exclude the possible interference caused by hyperglycemia, we then performed the same observations in nondiabetic mice (Fig. 1g). Gliflozin-administered mice exhibited similar body weights (Supplementary Fig. S1g), serum glucose levels (Supplementary Fig. S1h) and TA masses (Supplementary Fig. S1i), produced comparable single twitch forces (Fig. 1h) and time-to-peak tension (Supplementary Fig. S1j) compared to the CTRL group. Similarly, only CANA dramatically reduced both tetanic force (Fig. 1i, j) and fatigue resistance (Fig. 1k, l). To further rule out the possible interference caused by a vascular disorder, we also employed a cardiotoxin (CTX)-induced injury model, which directly causes injury to the skeletal muscle without damaging the vasoganglion [17]. In line with our previous findings, CANA treatment did not impair single twitch forces (Supplementary Fig. S1k). However, both specific tetanic force (Supplementary Fig. S1l) and fatigue resistance (Supplementary Fig. S1m) of injured limbs were attenuated by CANA, while blood perfusion of injured limb remained unaffected (Supplementary Fig. S1n). In addition, fatigue resistance of healthy limbs was also unaffected (Supplementary Fig. S1o). Taken together, these findings indicate that CANA delayed the recovery of injured skeletal muscle function in both diabetic and nondiabetic mice, while such effects are blood perfusion independent.
CANA impairs skeletal muscle regeneration post ischemic injury
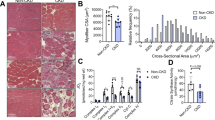
Evans blue staining in the CANA-treated TA was much more profound than that in the other groups on day 14 post FAL (Fig. 2a), suggesting poor regeneration of CANA-treated muscles. We thus assessed whether the functional consequences on muscle contractile and fatigue properties were caused by any morphological changes. H&E staining showed that myofiber nuclei centralization was delayed by CANA and EMPA, and slightly delayed by DAPA (Fig. 2b), and the centrally nucleated myofiber (CNM) ratio in each group on day 14 remained higher than that of the CTRL counterparts (Fig. 2c). Besides, the proportion of CNMs in the CANA group with 1 nucleus was lower, and that with more than 2 nuclei was higher than that in the CTRL and DAPA groups (Fig. 2d) In addition, the cross-sectional area (CSA) of CANA regenerated myofibers was much smaller than that of CTRL on day 7 (Supplementary Fig. S2a) and day 14 (Fig. 2e). Further analysis indicated that the CSA of CANA was mainly within 0–1000 μm2, while that of the remaining counterparts was mainly within 800–2400 μm2 (Fig. 2f). Moreover, the expression of em-MHC and desmin in ischemic myofibers was clearly evident in the CTRL, DAPA and EMPA TA sections 7 days post FAL. In contrast, em-MHC and desmin were nonuniformly expressed in CANA-treated TA (Fig. 2g, h). In line with em-MHC and desmin, CANA significantly disrupted MHC expression on day 14 post FAL (Supplementary Fig. S2b).

a Representative Evans blue staining images of TA from each group of diabetic mice at day 14 post FAL. b H&E staining of cross-sections of diabetic TA from each group at 3/7/14/21 days post FAL (scale bar = 50 μm). c Total and nuclear-centralized myofibers of (b) were counted, and the percentage of centrally nucleated myofibers (CNMs) is presented (n = 5 for each group). d Percentages of CNMs with either 1 nucleus, or 2 or more nuclei among all the CNMs were calculated and presented as a bar graph (n = 5 for each group). e Cryosections of diabetic TA from each group at day 14 post FAL were stained with DAPI for nuclei and laminin for myofiber membrane boundaries. The average CSAs of regenerating myofibers were analyzed, >300 fibers scored per sample, and f the myofiber amount-CSA relationship is presented (n = 5 mice for each group, scale bar = 100 μm). g, h Representative immunofluorescent staining for g em-MHC and laminin, as well as h desmin and laminin of TA 7 days post FAL (scale bar = 20 μm). i Representative Masson staining of cross-sections of diabetic TA from each group at day 14 post FAL. The fibrosis area was calculated and is presented as a bar graph (n = 3 mice for each group, scale bar = 50 μm). All data are presented as mean ± SD (error bars) and were analyzed by one-way ANOVA with Dunnett’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Any disturbances during muscle myolysis or reconstruction may result in unsuccessful regeneration, characterized by the formation of fibrosis [18]. Therefore, Masson staining was performed to delineate the area of fibrosis in skeletal muscle. As expected, the fibrotic area was much greater in the CANA group than in the other three groups (Fig. 2i). Interestingly, we also noticed an accumulation of fatty substances in ischemic muscle (Supplementary Fig. S2c), as well as a lower SDH activity in CANA TA (Supplementary Fig. S2d), indicating muscle fiber transformation in CANA-treated regenerating TA muscles. The qPCR results also showed the same trend of fiber type transformation (Supplementary Fig. S2e). These findings clearly suggest that CANA treatment attenuates skeletal muscle regeneration and promotes a slow-to-fast transition in oxidative muscles.
CANA attenuates myoblast proliferation and hinders the differentiation of myoblasts
Post-injury regeneration of skeletal muscle mainly depends on the proliferation and differentiation of myoblasts. Thus we first evaluated the cell proliferation. As a result, it was remarkably reduced by CANA treatment (Fig. 3a, b). Moreover, the expression levels of CCND1 and CCNA2 were downregulated by CANA (Fig. 3c and Supplementary Fig. S3a).

a C2C12 myoblasts were treated with either 10 μM CANA, 1 μM DAPA, 1 μM EMPA, or vehicle control in growth medium for 48 h, and proliferation was assessed using the CCK-8 method (n = 3). b C2C12 cells were treated as (a) in a growth medium for 48 h and stained with EdU. Representative fluorescent images are shown (scale bar = 100 μm), and the percentage of EdU+ nuclei is presented as a bar graph (n = 3). c Cell lysates were subjected to Western blotting analysis with the indicated antibodies (n = 3). d C2C12 myoblasts were treated as (a) in differentiation medium and subjected to differentiation induction for 72 h. Myotube formation was then assessed by MHC immunofluorescent staining (scale bar = 50 μm). e Myoblast differentiation shown in (d) was quantified by scoring MHC-positive cells with a single nucleus, two to five nuclei, or greater than or equal to six nuclei (n = 3). f C2C12 cells treated as in (a) were induced to differentiate for 24 h, and the cells were then fixed and stained with MyoG antibody and DAPI for immunofluorescence analysis. Representative fluorescent images are shown (scale bar = 50 μm), and the percentage of MyoG+ nuclei is presented as a bar graph (n = 3). g Cell lysates of C2C12 myoblasts that were treated as in (d) were subjected to Western blotting analysis with the indicated antibodies. Relative protein levels are presented as the ratio of the grayscale value against that of beta-actin (n = 3). h C2C12 myoblasts were treated as in (d) for the indicated durations and myogenesis-related mRNA levels were evaluated using qRT-PCR (n = 3). All data are presented as mean ± SD (error bars) and were analyzed by one-way ANOVA with Dunnett’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We next investigated the effects of gliflozins on myoblast differentiation. After 72 h of differentiation induction, CANA-treated myoblasts failed to form syncytial myotubes of comparable size or nucleus number compared to CTRL group (Fig. 3d, e and Supplementary Fig. S3b). Immunocytochemistry revealed that the percentage of MyoG+ nuclei was markedly decreased by CANA (Fig. 3f). Immunoblotting also confirmed the downregulation of MyoD and MyoG by CANA (Fig. 3g). In addition, desmin, which is expressed in newly formed myofibers, was decreased in CANA-treated myotubes that differentiated for indicated time durations (Supplementary Fig. S3c, d). Interestingly, we found that myogenesis-related mRNA levels such as Myog, Myf5, Cadh15, Capn1, and Pgc-1α were all paradoxically upregulated by CANA administration (Fig. 3h and Supplementary Fig. S3e).
Single-cell RNA sequencing reveals that mitochondrial leucyl-tRNA synthetase 2 (LARS2) is downregulated by CANA in muscle stem cells
We then analyzed the ischemic TA 3 days post injury using single-cell RNA sequencing (scRNA-seq, CTRL vs CANA). Unsupervised shared nearest neighbor (SNN) clustering partitioned cells into 12 clusters based on their transcriptomic programs after optimizing the SNN resolution parameter by silhouette analysis (Fig. 4a). In examining the full atlas, we interpreted the clusters as broadly defining 12 different cell populations based on the cluster-average expression of canonical genes (Fig. 4b). Further analysis indicated a notable decrease in MuSCs, as well as a slight increase in fibro-adipogenic progenitors (FAPs) in CANA-treated TA was observed compared to the control counterpart (Fig. 4c, indicated by red boxes).

a Unsupervised SNN clustering resolved at least 12 distinct types of cells (color-coded in legend). b Identification of cell types from SNN clusters based on cluster-average expression of canonical genes. Dot size represents the percentage of cells with a nonzero expression level, and color scale represents the average expression level across all cells within cluster. c Comparison of the cell type composition between CTRL- and CANA-treated ischemic TAs. d All cells within the muscle stem/progenitor clusters were further selected based on e cluster-average expression of canonical genes of MuSCs and myoblasts. f A refined analysis of muscle stem and progenitor subpopulations. These cells were further classified into four subpopulations (clusters 0–4) based on the heterogeneous gene expression listed in (g). g The top 15 heterogeneously expressed genes within the four subpopulations. h Compositional difference of the four subclusters of muscle stem and progenitor subpopulations. i A trajectory inference model was employed to delineate the interrelatedness of the four subpopulations. These four myogenic subclusters were aligned into a one-dimensional “pseudotime” axis, which presented organized, branched progression of cells from relatively early-stage subpopulations (left, clusters 2, 3) to a relatively mature subpopulation (right, cluster 0). j The trajectory inference model in (i) was further analyzed by dividing the MuSC/myoblast population into CTRL- and CANA-treated subgroups. k Top four canonical markers within MuSCs and myoblasts, whose population size were all notably downregulated by CANA compared to the CTRL counterpart.
Myogenesis post-injury requires the activation of MuSCs and thereafter an increase in myoblasts, which subsequently differentiate and fuse to form newly multinucleated fibers. We, therefore, explored the cellular heterogeneity within the myogenic cell population by selecting MuSCs and myoblasts (Fig. 4d) from the unified transcriptomic atlas using the expression of canonical MuSC and myoblast genes (Fig. 4e). The top ten differentially expressed genes in MuSCs compared to myoblasts, and in myoblasts compared to MuSCs are listed in Supplementary Fig. S4a, b. Unbiased SNN clustering revealed four subpopulations within the MuSC and myoblast populations (Fig. 4f) based on a selection of differentially expressed genes (Fig. 4g). Of note, CANA administration drastically decreased the cell number of cell cluster 0 and increased that of cell cluster 2 (Fig. 4h).
Next, given that SNN clustering lacks a hierarchical structure, we sought to organize these subpopulations using a trajectory inference model to delineate their interrelatedness. The monocle analysis focused on differentially expressed genes across these four myogenic clusters and aligned cells into a one-dimensional “pseudotime” axis (Fig. 4i). Pseudotime-ordered single-cell expression trajectories for myogenesis-related genes enriched in the muscle stem and progenitor subpopulations are listed in Supplementary Fig. S4c, d. Interestingly, this refined trajectory model identified CANA-treated MuSCs that were mostly confined to early-stage subpopulations (clusters 2 and 3), while the relatively mature subpopulation was mainly from CTRL MuSCs (Fig. 4j). Furthermore, we identified four canonical markers within the MuSC cluster, nominally, mt-Rnr1, CT010467.1, Lars2 and Gm42418 (Fig. 4k), whose population size was all notably downregulated by CANA.
CANA downregulates LARS2 expression in regenerating skeletal muscle and differentiating myoblasts
The scRNA-seq results indicated that CANA downregulated LARS2+ cells in MuSCs. We then evaluated the effects of gliflozins on LARS2 protein expression in regenerative skeletal muscle, which requires the activation and differentiation of MuSCs. As expected, histological analysis in vivo showed that CANA, instead of DAPA or EMPA, significantly decreased LARS2 protein expression in regenerating myofibers of ischemic TA at all time points post ischemia (Fig. 5a, b). In contrast, LARS2 expression in healthy skeletal muscle was minimally detectable and was not affected by any of the gliflozins (Supplementary Fig. S5a, b). Immunoblotting (Fig. 5c) also indicated that CANA notably downregulated LARS2 expression in differentiating C2C12 myoblasts in vitro.

a Immunohistochemistry (IHC) was performed in ischemic TA from CTRL-, CANA-, DAPA- or EMPA-treated diabetic mice at days 3/7/14 post FAL using an antibody against LARS2. Representative images are shown (scale bar = 50 μm). b Quantification of the density of LARS2 in (a) and the mean density ratio against CTRL (n = 4). c C2C12 myoblasts were treated with either 10 μM CANA, 1 μM DAPA, 1 μM EMPA, or vehicle control and subjected to differentiation induction for 72 h. Cell lysates were then subjected to Western blot analysis with the indicated antibodies. Relative protein levels are presented as a bar graph (n = 3). All data are presented as mean ± SD (error bars) and were analyzed by one-way ANOVA with Dunnett’s correction. **P < 0.01 and ****P < 0.0001.
CANA disrupts mitochondrial activity and mitochondrial structure in vitro
LARS2 is encoded in the nuclear genome, synthesized in the cytoplasm, and transported into mitochondria [19]. Mitochondria are the primary organelles that produce energy, which is critical for myogenic differentiation and muscle contraction, and mitochondrial activity is also an important regulator of myoblast differentiation. We thus questioned whether gliflozins have any effects on myoblast mitochondria. We first evaluated the effects of gliflozins on mitochondrial bioenergetics. Cellular ATP level was reduced by 20% and 8% by CANA administration during C2C12 differentiation induced for 36 and 48 h, respectively. Interestingly, DAPA and EMPA also slightly reduced cellular ATP level (Fig. 6a). However, only CANA reduced both the maximal OCR and basal OCR compared to their CTRL counterparts (Fig. 6b).

a C2C12 cells were treated with 10 μM CANA, 1 μM DAPA, 1 μM EMPA, or vehicle control and subjected to differentiation induction for 36 or 48 h. Cellular ATP level was measured and is presented as bar graphs (n = 3). b C2C12 cells were treated as in (a), and OCR rates were assayed under both basal and maximal conditions (n = 3). c C2C12 cells were treated as in (a) for 36 h, and cellular mitochondrial morphology was then analyzed using MitoTracker staining and observed under an A1R-SIM. d Mitochondrial amount, average area, aspect ratio (AR) and form factor (FF) of (c) were evaluated and are presented as bar graphs (n = 3). e Cells were classified according to their mitochondrial morphology, and the percentage of cells with each mitochondrial morphology was calculated (n = 3). f Representative TEM images of C2C12 cells treated as in (a) for 36 h. The mitochondrial cross-sectional area was measured and is presented as a bar graph (>100 mitochondria for each group). g Cells were treated as in (a) and subjected to differentiation induction for 36 h. Mitochondrial membrane potential was then assessed by JC-1 staining and observed under a fluorescence microscope. Representative images are shown (scale bar = 50 μm), and the fluorescence intensity ratio between red and green fluorescence is presented (n = 3). h Mitochondrial ROS in (g) was assessed by Mitosox red staining and evaluated by flow cytometry. ROS level was presented as a bar graph (n = 3). All data are presented as mean ± SD (error bars) and were analyzed by one-way ANOVA with Dunnett’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We then assessed whether the functional consequences on mitochondrial activities were caused by any morphological changes. Strikingly, a large number of fragmented mitochondria were present in CANA-treated myoblasts (Fig. 6c), while the number of cellular mitochondria was not affected. However, compared to the CTRL and DAPA groups, the average mitochondrial CSA was markedly reduced by CANA. Of note, both aspect ratio (AR, only measures elongation) and form factor (FF, more sensitive to curvature and the irregular shapes of filamentous mitochondria) were employed to depict mitochondrial characteristics. Of note, both AR and FF were downregulated by CANA (Fig. 6d).
We further divided these cells into three different categories based on visual inspection of their mitochondria (Supplementary Fig. S6a). In line with our previous findings, CANA dramatically increased the percentage of cells with fragmented mitochondria (Fig. 6e). TEM also revealed a reduction in mitochondrial CSA by CANA (Fig. 6f). In addition, mitochondrial fusion/fission- and cristae-related proteins (Supplementary Fig. S6b), as well as mitophagy-related proteins (Supplementary Fig. S6c), were not affected by CANA treatment. We finally studied the mitochondrial reactive oxygen species (ROS) level and membrane potential, finding that CANA increased mitochondrial ROS without affecting membrane potential (Fig. 6g, h and Supplementary Fig. S6d). Taken together, these data suggest that CANA disrupted myoblast mitochondrial respiratory activity and mitochondrial structure.
LARS2 is required for skeletal muscle regeneration and is crucial for mitochondrial activity
LARS1 has been proven to be involved in the regulation of myogenesis. Indeed, knockdown of LARS1 enhanced myotube formation (Supplementary Fig. S7a). Based on our preliminary findings, we speculate that LARS2 is also a key regulator of skeletal muscle regeneration by maintaining mitochondrial activity. Therefore, we first evaluated LARS2 expression during myoblast differentiation and skeletal muscle regeneration. LARS2 showed a definite increase along with myoblast differentiation progressed (Fig. 7a–c). However, CANA dramatically delayed the upregulation of LARS2 at all time points during differentiation (Fig. 7b). Likewise, an increase in LARS2 expression in regenerative myofibers was clearly indicated (Figs. 5b and 7d CTRL group). Within the same ischemic skeletal muscle, LARS2 was highly expressed in regenerating myofibers (Supplementary Fig. S7b, black arrow) compared to mature myofibers (Supplementary Fig. S7b, red arrow). These results suggest that LARS2 may be intimately involved in skeletal muscle regeneration.

a C2C12 cells were treated with 10 μM CANA, 1 μM DAPA, 1 μM EMPA, or vehicle control and subjected to differentiation induction for 24/72/120 h. Cell lysates were then subjected to Western blot analysis with the indicated antibodies. b The relative LARS2 protein level in (a) is presented as the ratio of the grayscale value against that of beta-actin (n = 3). c Relative LARS2 protein levels at different time points within the CTRL group in (a) are presented (n = 3). d IHC was performed in ischemic TA from CTRL diabetic mice at days 3/7/14 post FAL using an antibody against LARS2. Representative images are shown (scale bar = 50 μm). e C2C12 myoblasts were transfected with LARS2 siRNA or control siRNA (C2C12LARS2 siRNA or C2C12control siRNA). PBS was served as a blank control. Cells were then subjected to differentiation induction for 72 h. Myotube formation was assessed by immunofluorescent staining (scale bar = 50 μm). Myoblast differentiation was quantified as previously described (n = 3). f C2C12LARS2 siRNA and C2C12control siRNA were induced to differentiate for 72 h and subjected to Western blotting with the indicated antibodies. Relative protein levels are presented as a bar graph (n = 3). g C2C12LARS2 siRNA and C2C12control siRNA were induced to differentiate for 36 h, followed by MyoG and DAPI immunofluorescence staining (scale bar = 50 μm). The percentage of MyoG+ nuclei was quantified and is presented as a bar graph (n = 3). h C2C12LARS2 siRNA and C2C12control siRNA were cultured in a growth medium for 48 h and stained with EdU. Representative fluorescent images are shown (scale bar = 100 μm), and the percentage of EdU+ nuclei is presented (n = 3). i C2C12LARS2 siRNA and C2C12control siRNA were subjected to differentiation induction for 36 h. Cellular ATP level was measured (n = 3). j Cellular OCR in (i) was analyzed (n = 3). k Cells treated as in (i) were then fixed and stained using MitoTracker, and mitochondrial morphology was observed using an A1R-SIM (scale bar = 5 μm). All data are presented as mean ± SD (error bars) and were analyzed by one-way ANOVA with Dunnett’s correction, or by Student’s unpaired t test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
In order to address this hypothesis, LARS2 knockdown C2C12 myoblasts were generated using siRNA transfection (C2C12LARS2 siRNA vs C2C12control siRNA, Supplementary Fig. S7c), followed by differentiation induction for 72 h. As a result, MHC+ cells were notably reduced by LARS2 knockdown. The percentage of nuclei in large myotubes (with ≥6 nuclei) was significantly lower in C2C12LARS2 siRNA than in C2C12control siRNA (Fig. 7e). The protein levels of MyoD, MyoG and MHC (Fig. 7f), as well as the percentage of MyoG+ nuclei (Fig. 7g), were both markedly decreased in C2C12LARS2 siRNA.
We further evaluated mitochondrial activity in C2C12LARS2 siRNA. Knockdown of LARS2 attenuated the proliferation (Fig. 7h) and cellular ATP level (Fig. 7i) of C2C12 myoblasts. Furthermore, both basal and maximum OCR were decreased by LARS2 knockdown (Fig. 7j). We, therefore, wondered whether the mitochondrial structure was affected by LARS2 knockdown. Of note, a large number of fragmented mitochondria appeared in LARS2 knockdown myoblasts (Fig. 7k), and the number of myoblasts with fragmented mitochondria was significantly higher than that of C2C12control siRNA (Supplementary Fig. S7d). Further analysis revealed that AR, FF and average CSA were all decreased except for the cellular mitochondrial amount (Supplementary Fig. S7e), whereas mitochondrial fusion/fission- and cristae-related proteins were not affected (Supplementary Fig. S7f). In addition, mitochondrial ROS levels were dramatically elevated (Supplementary Fig. S7g), whereas the membrane potential remained unchanged (Supplementary Fig. S7h). These results suggest that LARS2 is essential for the maintenance of mitochondrial structure and activity and thus is crucial for skeletal muscle regeneration.
Forced expression of LARS2 restores the differentiation and regeneration of CANA-treated myoblasts and skeletal muscle
The effects of myoblast LARS2 deletion on myofiber differentiation and mitochondrial activity prompted us to investigate the consequences of enhanced LARS2 expression in myoblasts. To this aim, we first generated LARS2-overexpressing myoblasts (C2C12Flag LARS2) using a plasmid-based gene delivery method. Forced expression of LARS2 in C2C12 (C2C12Flag LARS2) promoted MyoG and MHC expression in myoblasts (Supplementary Fig. S8a) and multinucleated syncytial myotube formation (Supplementary Fig. S8b) compared with C2C12Flag vector. In addition, LARS2 overexpression reversed MyoG, MyoD, and MHC expression (Fig. 8a–c), promoted the proliferation (Supplementary Fig. S8c), as well as the multinucleated syncytial myotube formation of CANA-treated myoblasts (Fig. 8d, e). However, we did not observe any reversion in mitochondrial ROS production (Supplementary Fig. S8d).

a C2C12 myoblasts were first transfected either with a Flag-tagged vector plasmid (C2C12Flag vector) or a Flag-tagged LARS2 plasmid (C2C12Flag LARS2), followed by 10 μM CANA treatment and induced to differentiate for 36 h. Representative MyoG immunofluorescent staining images are shown, and the percentage of MyoG+ nuclei was quantified (n = 3, scale bar = 100 μm). b CANA-treated C2C12Flag vector and C2C12Flag LARS2 were induced for 36 and 72 h. Cell lysates were subjected to Western blotting with the indicated antibodies, and c the relative protein levels were calculated as mentioned before (n = 3). d C2C12Flag vector and C2C12Flag LARS2 treated with CANA, or C2C12 treated with PBS (as blank control) were subjected to differentiation induction for 72 h. Myotube formation was assessed by MHC immunofluorescent staining (scale bar = 50 μm), and e myoblast differentiation was quantified as previously mentioned (n = 3). f Sketchily study design using normoglycemic mice as an in vivo experimental model. g In situ TA absolute muscle single twitch force. h The duration corresponding to a 20% decrease in force (fatigue resistance), i TA mass (g)-normalized tetanic forces, and j TA absolute muscle force–frequency relationship were measured and presented as bar graphs (n = 4). k Representative images of H&E staining of TA cross-sections at 14 days post FAL (scale bar = 50 μm). l CSAs of regenerating myofibers in (k) were analyzed (>300 fibers scored per sample), and the myofiber amount-CSA relationship is presented as a bar graph (n = 4). m Representative images of immunofluorescent staining for MHC and WGA (scale bar = 20 μm), as well as n Masson staining of TA cross-sections (scale bar = 50 μm). The fibrosis area was calculated and is presented as a bar graph (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, one-way ANOVA. All data are presented as mean ± SD (error bars).
We, therefore, hypothesized that LARS2 overexpression in vivo could improve ischemic muscle regeneration in CANA-treated mice. An AAV conducted in vivo LARS2 overexpression model was employed to answer this question (Fig. 8f). LARS2 overexpression in TA per se did not affect the blood flow as assessed by LDPI (Supplementary Fig. S8e). However, enhanced LARS2 expression in TA notably improved muscle contractile recovery on D14 post ischemia, as well as fatigue resistance (Fig. 8g–j and Supplementary Fig. S8f), without affecting muscle mass (Supplementary Fig. S8g). Histological analysis indicated that ischemic TA and gastrocnemius myofiber regeneration, including the proportion of CNM with more than 2 nuclei (Supplementary Fig. S8h), CNM ratio (Supplementary Fig. S8i), myofiber morphology assessed by H&E staining (Fig. 8k and Supplementary Fig. S8j), and CSA of the myofiber (Fig. 8l and Supplementary Fig. S8k), were all improved by enhanced LARS2 expression. Compared to CANA-treated miceAAV-vector, CANA-treated miceAAV-LARS2 exhibited more uniform and abundant expression of MHC (Fig. 8m). In addition, LARS2 overexpression dramatically decreased the fibrotic area (Fig. 8n) and reversed the muscle fiber transformation induced by CANA (Supplementary Fig. S8l). Taken together, these data suggest that forced expression of LARS2 may restore the inhibitory effects of CANA on myoblasts and skeletal muscle differentiation and regeneration.
Discussion
Recent clinical findings have indicated that CANA treatment is associated with an increase in amputation risk [5]. It has been speculated that the unexpected side effects might be attributed to changes in limb blood flow that deteriorate with peripheral artery disease. However, our present study revealed that a therapeutic dose of CANA, instead of DAPA or EMPA, downregulated LARS2 in MuSCs, impairing mitochondrial maintenance and further attenuating intrinsic myogenic regeneration and differentiation. As a consequence, ischemic limb muscle contractile properties and fatigue resistance recovery as well as tissue regeneration were delayed by CANA administration.
Accumulating evidence implies that gliflozins exert substance-specific effects independent of SGLT2 inhibition [2, 20,21,22]. In line with these findings, our current study confirms that clinically relevant concentrations of CANA, instead of DAPA or EMPA, impair ischemic limb recovery, implying that SGLT2 is not involved in the off-target effect of CANA. Indeed, Secker et al. [23] emphasized that among gliflozins, only CANA exhibited an SGLT2-independent, off-target effect, characterized by the dual inhibition of glutamate dehydrogenase and complex I of the mitochondrial electron transport chain at pharmacologically relevant concentrations. Hawlay et al. [24] demonstrated that CANA, instead of DAPA, activated the AMP-activated protein kinase pathway in vivo via mitochondrial dysfunction, independent of its effect on glucose uptake, also supporting our interpretation that the CANA findings we report here are SGLT independent.
Several lines of evidence indicate that CANA attenuates cellular mitochondrial activity [7, 23,24,25]. A growing bulk of data suggests that the mitochondrial network plays a pivotal role in the regeneration of skeletal muscle and satellite cells [11, 26]. In addition, several lines of evidence suggest that mitochondrial function and activity are linked to cell differentiation, as has been shown in a wide variety of cell types including myoblasts [12, 27,28,29]. Mitochondrial biogenesis and enzyme activity are notably increased during myoblast differentiation and muscle regeneration [30]. Therefore, attenuated mitochondrial activity is related to an impairment of injured skeletal muscle regeneration [31], which may explain the inhibition of myogenesis caused by CANA. We also noticed a marked fusion/fission-independent transformation of mitochondria in CANA-treated C2C12 myoblasts, evidenced by fragmented mitochondria and unaltered fusion/fission-related proteins. Such remodeling was accompanied by an increase in ROS production in mitochondria, indicating that the precise balance of mitochondrial dynamics is among the most critical features in the control of mitochondrial function [32].
Based on the scRNA-seq findings, we further focused on LARS2, also known as mitochondrial LARS. It is well recognized that the canonical LARS function is to ensure that the genetic code is accurately deciphered by attaching the correct amino acid to the equivalent tRNA. Growing evidence suggests that LARS has broader roles than its translational function. Yoon et al. [33] reported that LARS1 plays a pivotal role in the control of leucine metabolism. The enzyme also serves as a leucine sensor for mTORC1, which regulates protein synthesis, metabolism, autophagy, and cell growth [34, 35]. Recently, LARS1 was found to be positively involved in skeletal muscle differentiation through the activation of mTOR signaling [36]. In contrast, Son et al. reported LARS1 as a negative regulator of myogenic differentiation and injury-induced skeletal muscle regeneration [37]. As indicated by scRNA-seq in our present study, CANA treatment notably decreased LARS2 in MuSC. We thus explored the roles of LARS2 in myoblast differentiation and muscle regeneration. LARS2 expression progressively increased in newly formed myofibers during skeletal muscle regeneration post ischemia, while being minimally detectable in mature myofibers. Knocking down LARS2 in C2C12 myoblasts significantly downregulated the expression of MRFs and hindered myotube formation. In contrast, forced expression of LARS2 markedly enhanced C2C12 differentiation. These data suggest that LARS2, as a conventional aminoacylation enzyme, plays a nonnegligible role in myoblast differentiation and muscle regeneration.
Knocking down of LARS2 significantly caused a shift from tubular-structure mitochondria to fragmented mitochondria, which was accompanied by impairment of mitochondrial activity and an increase in mitochondrial ROS production. These data suggest that LARS2 may be an important regulator of mitochondrial structure. To date, several studies have suggested that mitochondrial structural modifications can regulate bioenergetics [32, 38, 39]. The dynamic nature of mitochondria is controlled by a series of dynamin-like GTPase proteins that mediate fission and fusion events [40]. The fission process involves the division of mitochondria at the inner and outer membranes through the action of the large GTPase dynamin-related protein 1 (DRP1) as well as Fis1, MFF, MiD49 and MiD51 [41,42,43]. In contrast, mitochondrial fusion is mediated by three dynamin-related GTPases: mitofusin 1 (MFN1), mitofusin 2 (MFN2) and optic atrophy protein 1 (OPA1) [44, 45]. However, we did not notice any expression alteration in DRP1, OPA1 or MFN1/2, suggesting that there might be fusion/fission-independent remodeling of mitochondria.
Interestingly, our scRNA-seq data indicated that CANA treatment also affected several other cell populations, especially, the FAPs. While skeletal MuSCs are generally accepted as the direct cellular effectors of muscle regeneration [46], numbers of cell types are emerging as critical regulators of MuSCs [47]. Among them, muscle interstitial FAPs have been proposed to convert environmental perturbations into cues that coordinate MuSCs activity upon acute injury [48]. Restriction of FAPs activity within a specific timeframe during the regeneration process is of vital importance, as an abnormal persistence of FAPs is associated with persistent inflammation, formation of fibrotic scars, fat deposition, and impaired muscle regeneration [49]. As shown by our current research, a population size of FAPs is slightly increased by CANA on day 3 post FAL, which may attribute to the histological changes in CANA-treated TA. However, the effects of CANA on FAPs, as well as the possible crosstalk between MuSCs and FAPs need to be further elucidated.
Taken together, our study suggests that CANA treatment delays ischemic limb function recovery and tissue regeneration in mice. Such impairment is due to the downregulation of LARS2 in MuSCs, which attenuates intrinsic myogenic regeneration and differentiation. Further investigation revealed that LARS2, as an essential factor for mitochondrial maintenance, plays pivotal roles in myoblast differentiation regulation, and may become an ideal therapeutic target for ischemic skeletal muscle regeneration.
References
Birkeland KI, Jorgensen ME, Carstensen B, Persson F, Gulseth HL, Thuresson M, et al. Cardiovascular mortality and morbidity in patients with type 2 diabetes following initiation of sodium-glucose co-transporter-2 inhibitors versus other glucose-lowering drugs (CVD-REAL Nordic): a multinational observational analysis. Lancet Diabetes Endocrinol. 2017;5:709–17.
Xu C, Wang W, Zhong J, Lei F, Xu N, Zhang Y, et al. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem Pharmacol. 2018;152:45–59.
Sun X, Han F, Lu Q, Li X, Ren D, Zhang J, et al. Empagliflozin ameliorates obesity-related cardiac dysfunction by regulating sestrin2-mediated AMPK-mTOR signaling and redox homeostasis in high-fat diet-induced obese mice. Diabetes. 2020;69:1292–305.
Nugrahaningrum D, Marcelina O, Liu C, Wu S, Kasim V. Dapagliflozin promotes neovascularization by improving paracrine function of skeletal muscle cells in diabetic hindlimb ischemia mice through PHD2/HIF-1α axis. Front Pharmacol. 2020;11:1104.
Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644–57.
Fadini GP, Avogaro A. SGLT2 inhibitors and amputations in the US FDA Adverse Event Reporting System. Lancet Diabetes Endocrinol. 2017;5:680–1.
Lin Y, Nan J, Shen J, Lv X, Chen X, Lu X, et al. Canagliflozin impairs blood reperfusion of ischaemic lower limb partially by inhibiting the retention and paracrine function of bone marrow derived mesenchymal stem cells. EBioMedicine. 2020;52:102637.
Park SY, Yun Y, Lim JS, Kim MJ, Kim SY, Kim JE, et al. Stabilin-2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration. Nat Commun. 2016;7:10871.
Brack AS, Rando TA. Tissue-specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell. 2012;10:504–14.
Lin YF, Xiao MH, Chen HX, Meng Y, Zhao N, Yang L, et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death Dis. 2019;10:528.
Nichenko AS, Southern WM, Atuan M, Luan J, Peissig KB, Foltz SJ, et al. Mitochondrial maintenance via autophagy contributes to functional skeletal muscle regeneration and remodeling. Am J Physiol Cell Physiol. 2016;311:C190–200.
Sin J, Andres AM, Taylor DJ, Weston T, Hiraumi Y, Stotland A, et al. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy. 2016;12:369–80.
Kasichayanula S, Chang M, Hasegawa M, Liu X, Yamahira N, LaCreta FP, et al. Pharmacokinetics and pharmacodynamics of dapagliflozin, a novel selective inhibitor of sodium-glucose co-transporter type 2, in Japanese subjects without and with type 2 diabetes mellitus. Diabetes Obes Metab. 2011;13:357–65.
Brand T, Macha S, Mattheus M, Pinnetti S, Woerle HJ. Pharmacokinetics of empagliflozin, a sodium glucose cotransporter-2 (SGLT-2) inhibitor, coadministered with sitagliptin in healthy volunteers. Adv Ther. 2012;29:889–99.
Devineni D, Polidori D, Curtin CR, Murphy J, Wang SS, Stieltjes H, et al. Pharmacokinetics and pharmacodynamics of once- and twice-daily multiple-doses of canagliflozin, a selective inhibitor of sodium glucose co-transporter 2, in healthy participants. Int J Clin Pharmacol Ther. 2015;53:438–46.
Delezie J, Weihrauch M, Maier G, Tejero R, Ham DJ, Gill JF, et al. BDNF is a mediator of glycolytic fiber-type specification in mouse skeletal muscle. Proc Natl Acad Sci USA. 2019;116:16111–20.
Ownby CL, Fletcher JE, Colberg TR. Cardiotoxin 1 from cobra (Naja naja atra) venom causes necrosis of skeletal muscle in vivo. Toxicon. 1993;31:697–709.
Delaney K, Kasprzycka P, Ciemerych MA, Zimowska M. The role of TGF-β1 during skeletal muscle regeneration. Cell Biol Int. 2017;41:706–15.
Bullard JM, Cai YC, Spremulli LL. Expression and characterization of the human mitochondrial leucyl-tRNA synthetase. Biochim Biophys Acta. 2000;1490:245–58.
Nasiri-Ansari Ν, Dimitriadis G, Agrogiannis G, Perrea D, Kostakis I, Kaltsas G, et al. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc Diabetol. 2018;17:106.
Shiba K, Tsuchiya K, Komiya C, Miyachi Y, Mori K, Shimazu N, et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci Rep. 2018;8:2362.
Kuang H, Liao L, Chen H, Kang Q, Shu X, Wang Y. Therapeutic effect of sodium glucose co-transporter 2 inhibitor dapagliflozin on renal cell carcinoma. Med Sci Monit. 2017;23:3737–45.
Secker P, Beneke S, Schlichenmaier N, Delp J, Gutbier S, Leist M, et al. Canagliflozin mediated dual inhibition of mitochondrial glutamate dehydrogenase and complex I: an off-target adverse effect. Cell Death Dis. 2018;9:226.
Hawley SA, Ford RJ, Smith BK, Gowans GJ, Mancini SJ, Pitt RD, et al. The Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes. 2016;65:2784–94.
Villani LA, Smith BK, Marcinko K, Ford RJ, Broadfield LA, Green AE, et al. The diabetes medication Canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration. Mol Metab. 2016;5:1048–56.
Wagatsuma A, Sakuma K. Mitochondria as a potential regulator of myogenesis. ScientificWorldJournal. 2013;2013:593267.
Rochard P, Rodier A, Casas F, Cassar-Malek I, Marchal-Victorion S, Daury L, et al. Mitochondrial activity is involved in the regulation of myoblast differentiation through myogenin expression and activity of myogenic factors. J Biol Chem. 2000;275:2733–44.
Seyer P, Grandemange S, Busson M, Carazo A, Gamaléri F, Pessemesse L, et al. Mitochondrial activity regulates myoblast differentiation by control of c-Myc expression. J Cell Physiol. 2006;207:75–86.
Seyer P, Grandemange S, Rochard P, Busson M, Pessemesse L, Casas F, et al. P43-dependent mitochondrial activity regulates myoblast differentiation and slow myosin isoform expression by control of calcineurin expression. Exp Cell Res. 2011;317:2059–71.
Barbieri E, Battistelli M, Casadei L, Vallorani L, Piccoli G, Guescini M, et al. Morphofunctional and biochemical approaches for studying mitochondrial changes during myoblasts differentiation. J Aging Res. 2011;2011:845379.
Pessemesse L, Tintignac L, Blanchet E, Cortade F, Jublanc E, Demangel R, et al. Regulation of mitochondrial activity controls the duration of skeletal muscle regeneration in response to injury. Sci Rep. 2019;9:12249.
Baker N, Patel J, Khacho M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: how mitochondrial structure can regulate bioenergetics. Mitochondrion. 2019;49:259–68.
Yoon I, Nam M, Kim H, Moon H, Kim S, Jang J, et al. Glucose-dependent control of leucine metabolism by leucyl-tRNA synthetase 1. Science. 2020;367:205–10.
Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149:410–24.
Kim JH, Lee C, Lee M, Wang H, Kim K, Park SJ, et al. Control of leucine-dependent mTORC1 pathway through chemical intervention of leucyl-tRNA synthetase and RagD interaction. Nat Commun. 2017;8:732.
Sato Y, Sato Y, Suzuki R, Obeng K, Yoshizawa F. Leucyl-tRNA synthetase is required for the myogenic differentiation of C2C12 myoblasts, but not for hypertrophy or metabolic alteration of myotubes. Exp Cell Res. 2018;364:184–90.
Son K, You JS, Yoon MS, Dai C, Kim JH, Khanna N, et al. Nontranslational function of leucyl-tRNA synthetase regulates myogenic differentiation and skeletal muscle regeneration. J Clin Invest. 2019;129:2088–93.
Mannella CA. The relevance of mitochondrial membrane topology to mitochondrial function. Biochim Biophys Acta. 2006;1762:140–7.
Zick M, Rabl R, Reichert AS. Cristae formation-linking ultrastructure and function of mitochondria. Biochim Biophys Acta. 2009;1793:5–19.
Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2016;78:505–31.
Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–67.
Otera H, Miyata N, Kuge O, Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol. 2016;212:531–44.
Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–56.
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200.
Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, et al. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–95.
Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, et al. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–56.
Farup J, Madaro L, Puri PL, Mikkelsen UR. Interactions between muscle stem cells, mesenchymal-derived cells and immune cells in muscle homeostasis, regeneration and disease. Cell Death Dis. 2015;6:e1830.
Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153:376–88.
Uezumi A, Fukada S, Yamamoto N, Ikemoto-Uezumi M, Nakatani M, Morita M, et al. Identification and characterization of PDGFRα+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 2014;5:e1186.
Acknowledgements
We gratefully acknowledge the team of the Scientific Research Center of Wenzhou Medical University for their technical and scientific expertise in the immunocytochemistry study. This study was financially supported by the National Natural Science Foundation of China (82070493, 82102564, 81800314), Natural Science Foundation of Zhejiang Province (LY21H020008), and Wenzhou Municipal Science and Technology Bureau Foundation (ZY2020016, Y20210161).
Author information
Authors and Affiliations
Contributions
YNL, ZZL, and XC conceived and designed the project. XHL, XXC, JLN, XML performed the experiments. BBW performed the transmission electron microscopy analysis. RYZ performed animal anesthesia. JS and QLZ performed the animal models. WAC provided supervision of the study. YNL, LS, and ZTW wrote the manuscript, with contributions from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Lv, Xh., Cong, Xx., Nan, Jl. et al. Anti-diabetic drug canagliflozin hinders skeletal muscle regeneration in mice. Acta Pharmacol Sin 43, 2651–2665 (2022). https://doi.org/10.1038/s41401-022-00878-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00878-7
