
Abstract
The phenotypic transformation of microglia in the ischemic penumbra determines the outcomes of ischemic stroke. Our previous study has shown that chemokine-like-factor 1 (CKLF1) promotes M1-type polarization of microglia. In this study, we investigated the cellular source and transcriptional regulation of CKLF1, as well as the biological function of CKLF1 in ischemic penumbra of rat brain. We showed that CKLF1 was significantly up-regulated in cultured rat cortical neurons subjected to oxygen-glucose deprivation/reoxygenation (ODG/R) injury, but not in cultured rat microglia, astrocytes and oligodendrocytes. In a rat model of middle cerebral artery occlusion, we found that CKLF1 was up-regulated and co-localized with neurons in ischemic penumbra. Furthermore, the up-regulated CKLF1 was accompanied by the enhanced nuclear accumulation of NF-κB. The transcriptional activity of CKLF1 was improved by overexpression of NF-κB in HEK293T cells, whereas application of NF-κB inhibitor Bay 11-7082 (1 μM) abolished it, caused by OGD/R. By using chromatin-immunoprecipitation (ChIP) assay we demonstrated that NF-κB directly bound to the promoter of CKLF1 (at a binding site located at −249 bp to −239 bp of CKLF1 promoter region), and regulated the transcription of human CKLF1. Moreover, neuronal conditional medium collected after OGD/R injury or CKLF1-C27 (a peptide obtained from secreted CKLF1) induced the M1-type polarization of microglia, whereas the CKLF1-neutralizing antibody (αCKLF1) or NF-κB inhibitor Bay 11-7082 abolished the M1-type polarization of microglia. Specific knockout of neuronal CKLF1 in ischemic penumbra attenuated neuronal impairments and M1-type polarization of microglia caused by ischemic/reperfusion injury, evidenced by inhibited levels of M1 marker CD16/32 and increased expression of M2 marker CD206. Application of CKLF1-C27 (200 nM) promoted the phosphorylation of p38 and JNK in microglia, whereas specific depletion of neuronal CKLF1 in ischemic penumbra abolished ischemic/reperfusion-induced p38 and JNK phosphorylation. In summary, CKLF1 up-regulation in neurons regulated by NF-κB is one of the crucial mechanisms to promote M1-type polarization of microglia in ischemic penumbra.
Similar content being viewed by others
Introduction
Stroke is the leading cause of mortality and disability as lacking available therapeutic options [1]. An effective strategy to limit brain injury caused by ischemic stroke is to rescue “ischemic penumbra,” which was located around the border of ischemic lesion [2,3,4]. As the innate immune cell, microglia first respond to ischemic injury [5]. The activation of microglia in the peri-infarct area decides the levels of ischemic injury [6,7,8]. Activated microglia are classified into two opposite types: M1-type and M2-type. M1 microglia release various pro-inflammatory cytokines to aggravate the ischemic injury, while M2 microglia produce anti-inflammatory mediators to promote recovery [9]. The activated microglia in the peri-infarct area undergoes transition from M2 to M1 when the lesions expand, which in turn aggravate brain injury [10]. Regulating the shift of M1–M2 in penumbra is considered as a potential strategy to rescue ischemic penumbra [11].
The polarization of microglia seems to be controlled by neurons after stroke. Microglial activation frequently occurs in which the damaged neurons project to [12, 13]. Therefore, it has been suggested that putative fast and direct interactions between endangered neurons and microglia in the brain exist [14]. However, the molecules that participate in this neuron–microglia communication is not yet known. Chemokines and their receptors specialized constitute an elaborate signaling system that mediates cell–cell communication in CNS [13]. Chemokines are secreted from neurons in a temporarily and spatially manner [15]. Microglia respond to chemokine as a message via the appropriate receptors [16, 17]. Chemokine may be an ideal messenger for the communication between neurons and microglia due to such features [18, 19].
CKLF1 (Chemokine-like-factor 1, CKLF1) is one of the member of CC-type chemokines, which plays a vital role in ischemic stroke with broad-spectrum biological functions in inflammation. It has been found that the content of CKLF1 experienced an inverted “U” manner in the ischemic stroke [20, 21]. Inhibiting or knocking out CKLF1 can alleviate cerebral ischemic injury. Our previous study also found that administration of C27 (active peptide of CKLF1) promotes polarization of microglia to M1-type and inhibits M2-type [20]. But the cellular source of CKLF1 and the biological function of CKLF1 in penumbra remained elusive. Considering the influence of CKLF1 on microglia, we hypothesized that CKLF1 might act as a messenger for neuron-microglia communication. By verifying the above hypothesis, we would preliminarily analyze the cellular source and transcriptional regulation mechanism of CKLF1, as well as the biological function of CKLF1 in penumbra after stroke.
Materials and methods
Ethics statement
Our study was approved by the Ethics Committee of the Chinese Academy of Medical Sciences and the Peking Union Medical College (CAMS&PUMC). All animal procedures were performed under the Guide for the Care and Use of Laboratory Animals with the approval of the Chinese Academy of Medical Sciences and the Peking Union Medical College (CAMS&PUMC), Beijing, China.
Rat model of middle cerebral artery occlusion (MCAO)
All Sprague-Dawley rats were purchased from Charles River Laboratories (Beijing, China). Rats underwent MCAO for 1 h, as described previously [22]. Briefly, anesthesia was maintained by 3% isoflurane, mixed with 5% CO2/95% O2 via face mask. The right common carotid artery, internal carotid artery, and external carotid artery were carefully separated. The distal end of the external carotid artery was ligated and then cauterized. A small opening at the stump of the external carotid artery was cut by the ophthalmic forceps. The thread was inserted into the internal carotid artery until there was slight resistance. The thread was pulled out followed by 60 min of occlusion. The thread was purchased from CinonTech Co. LTD (Beijing, China).
Generation of CKLF1fl/fl rats
The CKLF1fl/fl transgenic rat was generated in the Institute of Laboratory Animals Science, the Chinese Academy of Medical Sciences, and the Peking Union Medical College (CAMS&PUMC) (Beijing, China). Two separate single-guide RNAs (sgRNAs) and two single-stranded oligonucleotide (ssODNs) donors with Loxp sequences were used to insert single LoxP site 5′ and 3′ of exon 1 of the rat CKLF1 gene. The removal of exon 1 will result in frameshift mutations and premature translational termination. The validated sgRNA, Cas9 mRNA, and ssODNs were co-microinjected into the cytoplasm of Sprague-Dawley rats. Following microinjection, zygotes were transferred into pseudopregnant ICR-recipient females. Standard methods of genotyping were used to characterize the genotype of founder rats using the following primers listed in Supplementary Table S1.
Virus and infection
To specifically knockdown CKLF1 in the neuron of perspective penumbra, the pAAV-Syn-Cre was injected to the perspective penumbra site of CKLF1fl/fl rat as reported previously [23]. The AAV-Syn-Cre-GFP (AAV-Cre) and AAV-hSyn-GFP (AAV-Con) were provided by Obio Technology Corp (Shanghai, China). Prior to AAV injection, rats were anesthetized with 3% isoflurane mixed with 5% CO2/95% O2. AAV-Syn-Cre (1 μL) or AAV-hSyn-GFP (1 μL) was injected into the middle cortex as follows: AP, −2.76 mm; ML, +2.00 mm; DV, −1.2 mm.
Primary culture of cortical neurons
Primary-cultured rat cortical neurons were obtained from the Sprague-Dawley rats that were born within 24 h, purchased from SPF Biotechnology Co. Ltd. (Beijing, China). The isolated cortex was digested with 0.25% trypsin that contained 0.04% EDTA and DNase (1 mg/mL) at 37 °C, and then suspended in Neurobasal medium (2048567, Gibco) containing 2% B27 supplement (17504044, Gibco). The dissociated cortical neurons were seeded in poly-L-lysine (PLL)-coated plates and incubated in 5% CO2-humidified air at 37 °C incubator.
Microglia/oligodendrocytes/astrocytes cultures
The individual primary glial cell culture was prepared from the cortex of 24 h Sprague–Dawley rats as described previously [24, 25]. The cortices of the rat were dissected and dissociated by a pair of fine scissors. The dissociated cells were digested with 0.25% trypsin that contained 0.04% EDTA and DNase (1 mg/mL) at 37 °C and then seeded in PLL-coated culture flasks in DMEM (2043218, Gibco) containing 10% FBS (1652793, Gibco) and 1% streptomycin/penicillin. The cultures were incubated in a 5% CO2 incubator at 37 °C. After a total of 12–15 days in culture, the loosely adherent microglia were detached by shaking at 200 r/min for 30 min. The detached microglia were suspended in DMEM plus 10% FBS and re-plated onto PLL-coated 6-well at a density of 105 cells/mL. After removing the microglia, the coculture flasks were replaced for fresh medium and shaken at 220 r/min for 18–22 h to obtain the oligodendrocytes. After completing the above steps, the tightly attached cell layer on the culture flasks were astrocytes. The primal cultures of astrocytes were digested with 0.25% trypsin containing 0.04% EDTA and re-plated onto PLL-coated six-well at a density of 105 cells/mL.
Oxygen-glucose deprivation/reoxygenation (ODG/R) injury model
The oxygen-glucose deprivation/reoxygenation (OGD/R) model for the primary neurons was established as reported previously [20]. The culture medium was replaced with Earl’s solution and then put in a hypoxia chamber and filled with 5% CO2/95% N2. The cultures were kept in a chamber for 1 h. After OGD, the primary neurons were returned to their original culture condition and incubated for 24, 48, and 72 h for reperfusion, respectively.
RNA isolation and quantitative reverse transcription
Total RNA was extracted with Trizol reagents. After extraction, complementary DNA (cDNA) was synthesized using cDNA Synthesis SuperMix (#AT311, TransGen). For the gene-expression profile, TransStat Tip Green qPCR SuperMix (#AQ142, TransGen) was used to amplify cDNA level. The mRNA levels were analyzed by real-time PCR using LineGene 9600 Quantitative PCR System (Bioer, China). GAPDH was employed as an internal control. The ∆∆Ct method was used to calculate the expression level. The primers used are listed in Supplementary Table S2.
ELISA
CKLF1 levels in the supernatants of the primary cultures were measured by enzyme-linked immunosorbent assay (ELSIA) kit (NBP2-75291, NOVUS) following the manufacturer’s instructions.
Immunofluorescence
Brain sections were heated in citrate buffer 0.01 mol/L solution antigen retrieval. Then, the sections were immersed in 0.5% TritonX-100 in PBS for permeabilization. After treatment, the sections were washed and blocked in 5% BSA for 1 h at room temperature (RT). Then, the sections were incubated with primary antibodies in a humidified chamber overnight at 4 °C. On the next day, the sections were washed and incubated with secondary antibodies and Hoechst in the dark at RT for 2 h. This section was visualized and captured by the confocal microscope using a Leica TCS SP8 (Lecia Microsystems, Buffalo Grove, IL, USA). Primary antibodies were used as follows: CKLF1 (Abcam, ab180512, rabbit, 1:200), NeuN (Millipore, 2967854, mouse, 1:100), Iba-1 (Wako, 019-19741, rabbit, 1:500), CD16/32 (BD Biosciences, 553142, mouse, 1:200), CD206 (R&D System, AF2535, mouse, 1:200), IL-10 (Santa Cruz, sc-365858, mouse, 1:200), iNOS (Abcam, ab13120, rabbit, 1:200), Iba-1 (Wako, wdl1621, mouse, 1:200), p-p38 (Santa Cruz, sc-7973, mouse, 1:200), p-JNK (Santa Cruz, sc-6254, mouse, 1:200), and Cre (Cell Signal Technology, #15036, rabbit, 1:200). All of the antibodies were diluted in the 3% BSA solution.
Chromatin-immunoprecipitation (ChIP)
Chromatin-immunoprecipitation assay was performed using the ChIP assay kit (P2078, Beyotime), as described previously [26]. The animal’s cortex was quickly removed and triturated into single-cell suspension in PBS. The 1% formaldehyde was added into the obtained single-cell suspension. The samples were incubated at 37 °C for 10 min to cross-link the target protein and the corresponding genomic DNA. Then, the glycine solution was added to stop cross-linking. The obtained sample was cracked in the SDS-lysis buffer containing PMSF solution and then sonicated at 15 W to make DNA break between 200 and 1000 bp. The sonicated sample was centrifuged at speed of 12,000 × g for 2 min and the supernatant was transformed into another tube. The ChIP Dilution Buffer containing PMSF was added to the supernatant to make a final concertation to 2 mL. Protein A+G Agarose/Salmon Sperm DNA was added to the sample and then mixed for 30 min at 4 °C to avoid non-specific binding. The supernatant was transferred to a new tube after centrifugation at 1000 × g for 1 min. Sonicated samples were immunoprecipitated by employing 1 μg of NF-κB antibody or rabbit IgG antibody overnight at 4 °C. The next day, protein A+G Agarose/Salmon Sperm DNA was added to precipitate the primary antibody recognition protein-containing DNA fragments shaking at 4 °C for 2 h. Then, the DNA was eluted. DNA bound to the immunoprecipitated histones was amplified by PCR. The primers used for ChIP were listed in Supplementary Table S3.
Cell culture
HEK293T and BV2 cells were purchased from the Chinese Academy of Medical Sciences and the Peking Union Medical College (CAMS&PUMC) (Beijing, China). Both BV2 and HEK293T cells were cultured with DMEM medium containing 10% FBS in 75-cm2 culture flasks and incubated in 5% CO2 humidified air in 37 °C incubators.
DNA constructs
The 3000 bp fragment of the CKLF1 promoter was cloned into the pGL3.0 luciferase reporter vector (Promega) provided by Hanbio Co. (Shanghai, China). The cDNA construct of NF-κB was purchased from Hanbio Co. (Shanghai, China). The expression efficiency of NF-κB vector was tested by Western blotting (Supplementary Fig. S1). To confirm the key element for CKLF1 promoter activation, we constructed continuous deletion variation of CKLF1-reported plasmids: CKFL1Δ1 (−1139–+1), CKLF1Δ2 (−790–+1), CKLF1Δ3 (−482–+1), and CKLF1Δ4 (−163–+1). The fragments of deletion variation of CKLF1 promoter were cloned into pGL3.0 luciferase reporter vector. Primers used for plasmid vector construction are listed in Supplementary Table S3. The mutant site of the CKLF1 promoter was provided by Hanbio Co. (Shanghai, China). The pRL-TK (Promega) was used as internal control.
Plasmid transfection and dual-luciferase reporter assay
The luciferase reporter plasmids were co transfected into cells with NF-κB or pcDNA-3.1 vector and pRL-TK plasmids by using Lipofectamine 2000 (Invitrogen) solution. Cells were seeded into a 24-well plate at the intensity of 5000 cells/mL the day before transfection. Cells were incubated with Opti-MEM medium for 30–60 min before transfection. Lipofectamine 2000 was dissolved into Opti-MEM. Meanwhile, the plasmid was dissolved to Opti-MEM. The Lipofectamine 2000 Opti-MEM and plasmid Opti-MEM were mixed at 1:1 ratio gently and incubated for 20 min. The mixtures were added into the cell plates by drop and incubated in an incubator for 6 h. The cultures were transferred to the original cultural conditions. Cells were collected 24 h after transfection. The luciferase activity was measured by Dual-Luciferase Reporter Assay Kit (E10191, Promega).
Western blotting
Tissues and cells were lysed in RIPA buffer (C1053, Applygen) containing cocktail protease inhibitors and phosphatase inhibitors. Proteins concentration was detected by BCA assay kits. The protein sample was segregated by 10% SDS-PAGE gel and then transferred to polyvinylidene fluoride membranes in the method of wet electric transfection. The membranes were blocked with 5% BSA diluted in TBST and hybridized to the primary antibody overnight at 4 °C. The secondary antibodies were hybridized at room temperature for 2 h. Protein bands were detected using Image Quant LAS 4000 mini. The band intensities were analyzed by ImageJ. Primary antibodies were used as follows: NF-κB (Santa Cruz, sc-8008, rabbit, 1:500), p38 (Santa Cruz, sc-535, rabbit, 1:500), p-p38 (Santa Cruz, sc-7973, mouse, 1:500), JNK (Santa Cruz, sc-137018, rabbit, 1:500), p-JNK (Santa Cruz, sc-6254, mouse, 1:500), ERK1/2 (Cell Signal Technology, #4695 S, rabbit, 1:500), p-ERK1/2 (Cell Signal Technology, #4377 S, rabbit, 1:500), β-actin (Sigma, A5441, mouse, 1:5000), and α-Tubulin (Cell Signal Technology, #2125, rabbit, 1:1000). All of the antibodies were diluted in the 3% BSA solution.
Statistical analysis
The results are presented as mean±SEM. Differences between two groups were analyzed with the Student t-test (two-tailed), and differences between multiple groups were analyzed with one-way ANOVA and two-way ANOVA. The correlation coefficient was analyzed by Pearson Chi-squared test. P < 0.05 was considered to be significant. Statistical analysis was done by GraphPad Prism 8.0.
Results
Ischemic/reperfusion causes the production of CKLF1 in neurons specifically
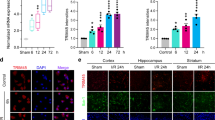
To investigate the cellular source of CKLF1 in the acute phase of ischemic stroke, primary cultures of neurons, microglia, astrocytes, and oligodendrocytes were established, respectively. Twenty-four hours after reoxygenation followed by OGD, the levels of CKLF1 mRNA and its contents in the medium were measured by qPCR and ELISA. Neither neurons nor glial cells have CKLF1 mRNA expression detected under normal conditions (Fig. 1a). Twenty-four hours after OGD, the levels of CKLF1 mRNA only increased significantly in neurons, but not in glial cells, including astrocyte, microglia, and oligodendrocyte. Similar changes were also found in protein expression (Fig. 1b). Our pervious study has confirmed that CKLF1 is upregulated after stroke [27]. Additional experiments were performed to investigate the co-localization of CKLF1 and neurons in vivo. CKLF1 was co-localized with neurons in the 24-h MCAO rat model as determined by double immunofluorescence (Fig. 1c). A positive linear correlation was measured between CKLF1+ cells and NeuN+ cells (Fig. 1d). All of these results suggested that neuron is the main cellular source of CKLF1 in the acute phase of ischemic stroke.

Cell specificity of CKLF1 expression in rat primary astroglia, oligodendroglia, microglia, and neuron cultures exposed to OGD/R. a, b Reoxygenation for 24 h after exposure to OGD for 1 h, only the neuron showed that the mRNA (a) and protein (b) of CKLF1 were significantly increased. Data are expressed as mean± SEM (n = 3). *P < 0.05, ***P < 0.001 by t-test. c Double staining of CKLF1 (green) is co-localized with neurons (red) in the early stage of cerebral ischemia as determined by immunofluorescence after 1 h of MCAO followed by 24 h of reperfusion. The region below the dashed line represents the injured area, while the region above the dashed line indicates the normal area adjacent to the injured area. Scale bar = 50 μm. d Pearson correlation between the number of CKLF1+ and NeuN+ cells was analyzed. The data are expressed as mean±SEM (n = 13). **P < 0.01, ***P < 0.001. One-way ANOVA followed by Tukey test. e LDH was employed to evaluate the level of cell injury at reoxygenation for 24, 48, and 72 h after exposure to OGD for 1 h. CKLF1 mRNA (f) and CKLF1 protein (g) produced by rat cortical neurons were detected after exposure to 24, 48, and 72 h of reoxygenation, following 1 h of OGD. The data are expressed as mean±SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA.
To further investigate the relationship between the neuronal damage and CKLF1 production, neuronal viability and CKLF1 contents were detected at 24, 48, and 72 h of OGD/R, respectively. Extracellular LDH contents were employed to reveal the level of neuronal injury. The contents of LDH were significantly increased from 24 h and reached their peak at 48 h (Fig. 1e). It declined at 72 h, which is still significantly higher than that in the control group (Fig. 1e). Then we explored the mRNA levels and protein contents of CKLF1 at different time points. A significantly increased level of CKLF1 was observed, which remained to be evaluated up to 48 h (Fig. 1f, g). Both mRNA and protein levels of CKLF1 were significantly increased under exposure to ischemic damage, which also showed consistent trends with neuron-injury levels after OGD/R.
NF-κB regulates neuronal CKLF1 production in response to OGD/R
After clarifying its cellular source, we next explored its transcriptional mechanism of CKLF1. According to the predicted results from Jaspar and UCSC, we assumed that NF-κB may be involved in the transcription of CKLF1. We first investigated the nuclear accumulation of NF-κB in primary neurons. After OGD/R, nuclear content of NF-κB was pronouncedly increased (Fig. 2a–c). Moreover, the ratio between nuclear p65 and cytoplasmic p65 also increased in the OGD/R group. The elevated p65 content in the nucleus provides a prerequisite for its regulation of CKLF1 transcription. Next, we tested whether Bay 11-7082 (NF-κB inhibitor) could suppress the expression of CKLF1 under hypoxia. The culture of primary neurons was incubated with Bay 11-7082 and then exposed to 1 h of OGD, followed by 24 h of reoxygenation. qPCR and ELISA showed that CKLF1 mRNA and protein were significantly decreased compared with the OGD group after treatment with Bay 11-7082 (Fig. 2d, e). The results suggest that NF-κB was involved in regulating CKLF1 production.

a The contents of NF-κB were measured in the nucleus and cytoplasma of cortex neuron after OGD/R. b, c Nuclear translocation of NF-κB was significantly increased in the cortex neuron after exposure to OGD/R. The data are expressed as mean± SEM (n = 3), **P < 0.01, ***P < 0.001 by t-test. Cortex neurons were subjected to 1 h OGD and then reoxygenation for 24 h with the treatment of Bay 11-7082 (1 μM) or vehicle (0.1% DMSO). Cells were harvested for RT-PCR and medium were collected for ELISA analysis. d CKLF1 mRNA was detected by RT-PCR. e The contents of CKLF1 protein were detected by ELISA. The data are expressed as mean± SEM (n = 3). ***P < 0.001 by one-way ANOVA.
NF-κB enhances the transcriptional activity of CKLF1
In order to observe whether there is a direct interaction between CKLF1 and NF-κB, chromatin-immunoprecipitation (ChIP) assay was used to investigate whether NF-κB could directly bind to the promoter of CKLF1. As shown in Fig. 3a and b, there is a significant increase of CKLF1 contents in the MCAO group as compared to that in the control group, suggesting that ischemia/reperfusion promoted the binding between p65 and CKLF1 promoter. To find out the drive effect of p65 on the transcriptional level of CKLF1, we cloned a DNA fragment from −2000 to +1 bp of the human CKLF1 5′-flanking region to the pGL3.0 luciferase reporter vector to generate CKLF1-promoter-luciferase constructs (CKLF1-Luc). The overexpression vector for NF-κB was also constructed based on the pcDNA-3.1 vector (pcDNA-3.1-NF-κB). CKLF1-Luc reporter constructs were transfected to 293 T cells with or without NF-κB plasmid as well as the pRL-TK vector as an internal control for normalizing transfection efficiency. Twenty-four hours after transfection, cells were harvested for luciferase assay. CKLF1-Luc and pGL3.0-basic vector were co-transfected to 293 T cells severed as the control group. The luciferase activity in 293 T cells co-transfected by CKLF1-Luc and pcDNA-3.1-NF-κB was significantly higher than that in the control group, which was also positively correlated with the amount of NF-κB plasmid transfection (Fig. 3c). Moreover, CKLF1-Luc showed robust responses to OGD/R; however, Bay 11-7082 impaired this responsitivity (Fig. 3d).

a, b ChIP assay shows that NF-κB interacts with CKLF1 promoter in the cortex of rat after 1 h of MCAO followed by reperfusion for 24 h. The data are expressed as mean± SEM (n = 3), **P < 0.01 by t-test. c Luciferase activity was measured in 293 T cells after co-transfection with human CKLF1 (−2000/+1) promoter reporter and different concentrations of NF-κB vector. Data are expressed as mean± SEM (n = 3), *P < 0.05, ***P < 0.001 by one-way ANOVA. d Luciferase assay shows that inhibiting NF-κB activity decreases human CKLF1 promoter activity in 293 T cells exposed to 1 h of OGD followed 24 h of reoxygenation compared with OGD/R group. Data are expressed as mean ± SEM (n = 3), *P < 0.05, ***P < 0.001 by one-way ANOVA. e The binding site of NF-κB on CKLF1 promoter was predicted in Jaspar. f Scheme of serially truncated human CKLF1 promoter constructs along with co-transfection of the pRL-TK and NF-κB overexpression plasmid in 293 T cells. Data are expressed as mean± SEM (n = 3), **P < 0.01, ***P < 0.001 by t-test; g Scheme of mutation of the binding site of human CKLF1 promoter. h Relative luciferase activity was measured in 293 T cells after co-transfection of CKLF1 mutant promoter or CKLF1 promoter with NF-κB overexpression plasmid. Data are expressed as mean± SEM (n = 3), ***P < 0.001 by one-way ANOVA.
To further identify the NF-κB-binding site in CKLF1 promoter region, a series of continuous deletion variations of 5′-end-truncated CKLF1 promoter constructs were cloned based on the predicted binding sites (Fig. 3e, f). These constructs were transfected to 293 T cells with pcDNA-3.1-NF-κB or pcDNA-3.1 vector. Deletion of the region −482 to −163 bp resulted in the loss of luciferase activity in 293 T cells (Fig. 3f). By computer-based sequence analysis (Fig. 3e), a single-consensus-binding site (5′-GAGGATTTCCC-3′ at −248 to −238 bp) for transcription factor NF-κB was identified in the region from −482 to −163 bp (Fig. 3f). Site-directed mutagenesis was used to mutate the putative site and then its effects on luciferase activity in HEK293T cells were assayed (Fig. 3g). Mutation in the putative binding motif could significantly abolish the increase of luciferase activity by overexpression of NF-κB (Fig. 3h). These results showed that NF-κB could directly participate in the activation of CKLF1 transcription with the binding site located at the CKLF1 promoter region.
Neuronal CKLF1 drives microglia toward M1 phenotype in vitro
We next attempted to clarify the potential role of this neuron-secreted CKLF1 under hypoxia, especially as it was relevant to the neuron–microglia communication. The conditioned medium-transfer experiments were performed to explore whether microglial phenotype could be regulated by neuronal CKLF1. The neuron was subjected to OGD for 1 h to provoke CKLF1 production. The conditioned medium or medium of untreated neurons was collected to stimulate primary microglia after 24 h. (Fig. 4a). Then, the expression of selected genes to characterize microglia polarization was tested. Fresh Neurobasal medium containing CKLF1-C27 served as the positive group. The CKLF1-neutralizing antibody (αCKLF1) was added to neuron-conditioned medium to block the biological function of CKLF1.

a Schematic of the conditioned medium-transfer experiment. The cultures of neuron were subjected to 1 h of OGD followed by 24 h of reoxygenation, with or without 1 μM Bay 11-7082. Conditioned medium was harvested from each experimental condition and transferred individually to the primary microglia in culture. Fresh neuronal medium with or without C27 and α-CKLF1 was used as an additional control. Primary microglia were harvested to RT-PCR after 24 h of incubation with the conditional medium. The gene profiling of M1 polarization marker CD16 (b) and CD86 (c) was assessed. Besides, the mRNA expression of M1 polarization products iNOS (d) and TNF-α (e) was measured. The mRNA expression of M2 polarization marker CD206 (f) and M2 polarization production of IL-10 (g) were assessed. The data are expressed as mean± SEM (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA.
OGD conditioned medium and C27 robustly up regulated M1 genes, including CD16 and CD86 (Fig. 4b, c), and inflammatory production genes, including TNF-α and iNOS (Fig. 4d, e). Such up-regulation of M1 marker could be abolished by αCKLF1 or Bay 11-7082, suggesting that blockade of CKLF1 activity or inhibition of the transcription of CKLF1 could reverse M1 polarization of microglia caused by the neuronal OGD conditioned medium. The expression of M2 gene CD206 and anti-inflammatory gene IL-10 was not affected by any experimental interventions with the unchanged level of the internal control GAPDH (Fig. 4f, g). All of these results suggested that CKLF1 up-regulated the M1 polarization of microglia and had no significant effect on its M2-type polarization. In addition, we also used co-culture system of microglia-neuron to study the effect of inhibiting microglia activation on neuronal CKLF1 expression. It was found that the expression of CKLF1 was partly decreased and the damage of neurons was improved when the microglial activation was inhibited (Supplementary Fig. S2).
Neuronal-specific CKLF1 deficiency protects neuron against ischemia/reperfusion in penumbra area
To further validate the function of neuronal CKLF1 in vivo, a neuronal-specific CKLF1 knockout rat was generated by Cre/loxp system. We constructed rats with two loxp sequences on both sides of the exon 1 of CKLF1 in the same orient (CKLF1fl/fl rat) (Fig. 5b). AAV-Syn-Cre-GFP (neuron-specific adeno-associated virus) or AAV-Syn-GFP (control virus) was delivered into the perspective penumbra of CKLF1fl/fl rat by microinjection (Fig. 5a, b). To assess neuronal CKLF1 deficiency on the neural impairment induced by ischemia/reperfusion, MCAO surgery was carried out after 21 days of virus infection. We measured CKLF1 immunofluorescence of neurons in the injected area followed by MCAO (Fig. 5c). Although CKLF1 was robustly increased in neurons under ischemic injury, the expression of CKLF1 in neurons was significantly reduced in CKLF1fl/fl rats receiving the pAAV-Syn-Cre injection after MCAO (Fig. 5c, e). Meanwhile, the number of injured neurons was pronouncedly decreased in CKLF1fl/fl rat receiving the pAAV-Syn-Cre injection compared with CKLF1fl/fl after MCAO (Fig. 5d, f), which suggested that deficiency of neuronal CKLF1 in penumbra area protects the neuron against ischemic injury.

a Schematic representation of the injection site and the expression of GFP. b A diagram of neuronal CKLF1 depletion in the theoretical penumbra. AAV-Syn-Cre-GFP or AAV-hSyn-GFP was injected anterior to the prospective penumbra of CKLF1fl/fl rat at 21 days prior to stroke. c Immunofluorescence staining for CKLF1 (magenta) and NeuN (red) was performed to assess the knockout efficiency after MCAO. c, e The expression of neuronal CKLF1 was assessed by immunofluorescence staining after MCAO. Scale bar = 50 μm. The data are expressed as mean ± SEM (n = 6), ***P < 0.001 by t-test. d Injured neurons were identified by Nissl staining. Black arrows represent the intact neurons with flush-cell bodies. Red arrows represent apoptotic nerve cells with shrunken and pyknotic nuclei. f The number of injured neurons in the cortex was counted after neuronal-specific CKLF1 depletion in the penumbra. Scale bar = 50 μm. The data are expressed as mean± SEM (n = 9–14), ***P < 0.001 by one-way ANOVA.
CKLF1 deficiency in neurons inhibits M1 polarization of microglia in penumbra after MCAO
We then investigated whether CKLF1 deficiency in neurons affects microglia in the penumbra after MCAO. After MCAO, M1 marker CD16/32 in Iba-1+ microglia were dramatically decreased in CKLF1fl/fl rat receiving the AAV-Syn-Cre injection compared with CKLF1fl/fl rat (Fig. 6a–c). Interestingly, the amount of activated microglia (Iba-1) was significantly decreased in CKLF1fl/fl rat receiving the AAV-Syn-Cre injection under ischemic injury (Fig. 7g). In contrast, neuronal CKLF1 deficiency significantly promoted M2 marker in Iba-1+ microglia compared with CKLF1fl/fl rat receiving the control virus after MCAO (Fig. 6d, e). M1 microglia product iNOS and M2 microglia products were also detected. Neuronal CKLF1 deficiency inhibited the expression of iNOS in Iba-1+ microglia while up-regulated the expression of IL-10 Iba-1+ microglia (Fig. 7a–c). As expected, the number of CD16+Iba-1+ microglia displayed a positive correlation with iNOS+Iba-1+ microglia (Fig. 7h). At the same time, IL-10+Iba-1+ positive microglia and IL-10 intensity in Iba-1+ cells were significantly increased in CKLF1fl/fl rat receiving the pAAV-Syn-Cre injection (Fig. 7d–f). A positive linear correlation was observed between the numbers of CD206+Iba-1+ microglia (Fig. 7i). Based on the above results, we conclude that neuronal CKLF1 regulated the shift of microglial phenotype under ischemia.

a Representative immunofluorescence staining for CD16/32 (pseudocolor green) and Iba-1(red) 1 day after 1 h ischemia, followed by 24 h of reperfusion in the perspective enumbra. Numbers of CD16/32+/Iba-1+ cells (b) and CD16/32 intensity in Iba-1+ cells (c) were quantified (n = 14). d Representative immunofluorescence staining for CD206 (pseudocolor green) and Iba-1 (red) in the perspective penumbra. Numbers of CD206+/Iba-1+ cells (e), and CD206 intensity in Iba-1+ cells (f) were quantified (n = 10). Scale bar = 50 μm. The data are expressed as mean ± SEM (n = 10–14), ***P < 0.001 by t-test.

a Representative double-immunofluorescence staining for iNOS (pseudocolor green) and Iba-1 (red) in the perspective penumbra. Numbers of iNOS+/Iba-1+ (b), and iNOS intensity in Iba-1+ cells (c) were quantified (n = 7). The expression of M2 product IL-10 in microglia was evaluated by immunofluorescence. d Representative double-immunofluorescence staining for IL-10 (pseudocolor green) and Iba-1 (red) in the injected area. Numbers of IL-10+/Iba-1+ (e) and IL-10 intensity in Iba-1+ cells (f) were quantified (n = 8–12). Scale bar = 50 μm. The data are expressed as mean± SEM (n = 7–12), ***P < 0.001 by t-test. g Numbers of Iba-1+ were also measured. The data are expressed as mean ± SEM (n = 12), ***P < 0.001 by t-test. h Correlations between the number of iNOS+/Iba-1+ and CD16/32+/Iba-1+ M1-like microglia were analyzed by Pearson correlation coefficients. i Pearson correlation between the number of IL-10+/Iba-1+ and CD206+/Iba-1+ M2-like microglia was analyzed.
CKLF1 promotes the activation of MAPK-p38 and MAPK-JNK in microglia
Our previous study has reported that administration of αCKLF1 protects the brain against ischemia injury by inhibiting the activation of MAPK signal pathways [21]. Thus, we detected the change of MAPK signal transduction pathways in the BV2 microglia after treated with C27 (CKLF1). BV2 microglia were incubated with 100 nM C27 for 12 h. Then the expression of MAPKs was detected by Western blot. To our surprise, both phosphorylation of p38 and JNK was significantly increased, while administration of C27 showed no effect on ERK (Fig. 8a–d). The activation of P38 and JNK was also validated in vivo. Double immunolabeling for Iba-1 and p-JNK or p-P38 was performed on CKLF1fl/fl rat receiving the AAV-Syn-Cre injection (Fig. 8d, f). Consistent with what we found in BV2 microglia, the expression of pJNK (Fig. 8d, e) and pP38 in Iba-1+ microglia (Fig. 8f, g) was significantly decreased in CKLF1fl/fl rat infected by AAV-Syn-Cre after MCAO.

The phosphorylation of MAPK-ERK, P38, and JNK was measured by western blot after treatment with CKLF1 (200 nM) for 12 h in BV2 microglia. a MAPK-ERK shows no difference after treatment with CKLF1. The phosphorylation of MAPK-p38 (b) and MAPK-JNK (c) was significantly increased. The data are expressed as mean ± SEM (n = 3), NS: not significant, *P < 0.05 by t-test. d Representative double-immunofluorescence staining for p-JNK(magenta) and Iba-1 (red) 24 h after MCAO for 1 h in the perspective penumbra. e Numbers of pJNK+/Iba-1+ were quantified. f Representative immunofluorescence staining for p-P38 (magenta) and Iba-1 (red) 1 day after cerebral ischemia in the injected area. g Numbers of p-P38+/Iba-1+ were quantified. Scale bar = 50 μm. The data are expressed as mean ± SEM (n = 9–12), *P < 0.05, ***P < 0.001 by t-test.
Discussion
This study elucidated the potential effect of CKLF1 in the crosstalk between neurons and microglia in the pathogenesis of ischemic stroke. The production of CKLF1 was initiated by ischemia/reperfusion injury in neurons, which promoted the M1 polarization of microglia (Fig. 9). Conditional knockout of neuronal CKLF1 attenuated the level of M1 polarization of microglia and alleviated the inflammation significantly. By performing the neuron-conditioned medium transfer experiments to primary microglia, we demonstrated that injured neurons produce sufficient amounts of CKLF1 to modulate the microglia polarization. Neutralized CKLF1 activity also blocked the expression of biomarkers of M1 polarization significantly. Mechanism study found that the expression of CKLF1 was regulated by NF-κB directly. The ChIP and luciferase assay revealed that NF-κB directly binds to the promoter of CKLF1. All of these results suggested a new molecule that participates in the interactions between damaged neurons and microglia.

CKLF1 is up-regulated by NF-κB in neuron under cerebral ischemia. CKLF1 secreted from neuron promotes M1 polarization of microglia while inhibits M2 polarization of microglia after stroke.
CKLF1 plays a vital role in ischemic stroke with broad-spectrum biological functions in inflammation [28, 29]. Our group has reported that knocking out CKLF1 or administration of αCKLF1 can significantly improve ischemic injury through reducing cerebral infarct and promoting recovery [21]. IMM-H004, a CKLF1 antagonist developed by our group, shows the ability to improve brain lesions by inhibiting inflammation [30]. Related studies have found that CKLF1 is co-localized with neurons in the early stage of cerebral ischemia in hypertensive rats, but not with other neural cell types as determined by immunofluorescence [31]. Such study enlightened us that the neurons may be a cellular source of CKLF1 after stroke. We measured CKLF1 in various kinds of resident cells of the brain under ischemia to investigate the cellular source of CKLF1. Our results showed that CKLF1 is produced in neurons after cerebral ischemia.
Identifying the cellular source of CKLF1 helps us to further investigate the transcriptional regulation of CKLF1. One of the main purposes of our study was to investigate the regulation mechanism of CKLF1 expression under hypoxia. NF-κB is a ubiquitous transcription factor that regulates immune signaling in ischemic stroke [32, 33]. Compelling evidence shows that NF-κB activation in neurons rather than in glia plays a vital role in the ischemic damage [34]. NF-κB in neurons can be activated by various factors after cerebral ischemia, such as hypoxia, TNF-α, and IL-1β, especially glutamate, while LPS and ROS cannot activate NF-κB in neurons [34,35,36]. As the literature indicates that hypoxia–ischemia can induce 2 peaks of NF-κB at 3–6 h and 24 h, that p65, rather than c-Rel, is rapidly activated in neurons after MCAO and OGD [37,38,39]. The chemokine gene transcripts were strongly induced after NF-κB activation [36]. We hypothesized that the expression of CKLF1 in neurons was regulated by NF-κB based on the predicted results at UCSC and Jaspar. Pretreatment with NF-κB inhibitor would suppress the expression of CKLF1 suggested that CKLF1 could be regulated by NF-κB. To further investigate whether NF-κB was involved in the expression of CKLF1 at transcriptional level, ChIP assay and luciferase assay were performed. ChIP assay showed that NF-κB binds to the promoter region of CKLF1 under ischemia. There are three putative binding sites to NF-κB through analyzing the promoter gene of CKLF1. We finally confirm that the core site of CKLF1 promoter is located at -238 to -249 bp by truncation strategies and directly site mutagenesis.
Timely intervention in the development of the ischemic penumbra to the infarct is an effective strategy for the treatment of stroke [2, 40]. Regulating the polarization of microglia shift from M1 to M2 seems to be an effective means to prevent the deterioration of the penumbra [41]. Microglia suddenly become activated in which neurons are injurious, while microglial activity is likely to be controlled by endangered neurons. It has been suggested that putative fast and direct interactions between neurons and microglia in the brain exist. However, the identity of the molecules involved in this neuron–microglia signaling is not yet known. CKLF1, released by neurons under hypoxia, might contribute to an intercellular signal establishing direct neuron–microglia interactions during ischemic events in the CNS. Therefore, another objective of our study is to answer the question of whether CKLF1 is a neuronal “on” or “off” signal to microglia. Here we showed that neuronal cultures exposed to OGD/R promote M1 polarization of microglia, which was abolished by pretreatment with αCKLF1, indicating that neuronal CKLF1 participated in modulating the M1 phenotype of activated microglia. In vitro studies showed that blockade of CKLF1 activity or inhibiting the expression of CKLF1 in neuron would reverse the effect of neuronal cultures exposed to OGD/R on microglia.
To further investigate whether CKLF1 is involved in neuron–microglia communication, we established neuron-specific CKLF1-depletion rat to examine the role of CKLF1 between neurons and microglia communication in vivo. We employed CD16/32 as a marker for M1, while CD206 for M2 [7, 42]. In rat MCAO model, we found that the CKLF1 deficiency led to M1 microglia shift to M2 microglia. Moreover, the M1 microglia product iNOS was significantly decreased, while the M2 microglia product IL-10 was dramatically increased in neuronal CKLF1 deficiency rat MCAO model. It is worth noting that the number of activated microglia was reduced considerably in CKLF1fl/fl rat that received AAV-Syn-Cre. According to the presence of cysteine motifs near their amino termini, CKLF1 can be regarded as CC chemokines. Based on the known role of CKLF1 in microglia modulation, we suggest that neuronal CKLF1, similar to what has been indicated for CCL21 (a neuronal chemokine) [13], could support neuron-microglia communications, allowing neurons to activate microglia polarization to M1-type directly.
Mitogen-activated protein kinases (MAPKs), which contain three major members (ERK1/2, p38, and JNK), play a vital role in regulating the microglial polarization [43]. The MAPK is central to linking the inflammatory stimuli to the cellular response [44]. M1 polarization of microglia can be caused by p38 and JNK phosphorylation, followed by the activator protein (AP-1) and NF-κB-mediated transcription of inflammatory mediators, such as NO, iNOS, and the proinflammatory cytokines TNF-α, interleukin (IL)-6, and IL-1β [45,46,47,48]. In traumatic brain injury models, knocking out p38 can significantly reduce the expression of M1-type microglia marker CD68, and the expression of M1-type microglia production, such as TNF-α, IL-1β, and IL-6 [49]. The use of JNK inhibitor can also inhibit the microglial activation [46]. Our pervious study found that CKLF1 could activate MAPK pathway in the cerebral cortex after stroke [21]. In this study, we detected the activation of MAPK after treatment with CKLF1. Phosphorylation of both JNK and p38 is significantly increased with no difference in the ERK1/2 after treatment with CKLF1. Additional experiments show that the phosphorylation of p38 and JNK was significantly inhibited after neuronal CKLF1 depletion in the penumbra following ischemic stroke. Studies have demonstrated that the MAPKs were involved in neuronal response to glia-derived neurotoxic molecules. Pharmacological inhibition studies further demonstrated that p38 and JNK, but not ERK1/2, are important signal transduction pathway contributing to glia-induced neuron death [46, 50]. Previous studies have found that CKLF1 can bind to the CCR4 receptor on microglia [51]. However, whether CKLF1 promotes the phosphorylation of p38 and JNK through the CCR4 receptor needs to be further verified.
Collectively, we have now demonstrated that damaged neurons under OGD express CKLF1 through NF-κB. This neuron-secreted CKLF1 then effectively participates in the modulation of the microglial phenotype by activating p38 and JNK pathway.
References
Patel P, Yavagal D, Khandelwal P. Hyperacute management of ischemic strokes: JACC focus Seminar. J Am Coll Cardiol. 2020;75:1844–56.
Baron JC. Protecting the ischaemic penumbra as an adjunct to thrombectomy for acute stroke. Nat Rev Neurol. 2018;14:325–37.
Schiemanck SK, Post MW, Witkamp TD, Kappelle LJ, Prevo AJ. Relationship between ischemic lesion volume and functional status in the 2nd week after middle cerebral artery stroke. Neurorehabil Neural Repair. 2005;19:133–8.
Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia—the ischemic penumbra. Stroke. 1981;12:723–5.
Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol. 2013;5:73–90.
Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–89.
Hu X, Li P, Guo Y, Wang H, Leak Rehana K, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–70.
Rodhe J, Burguillos MA, de Pablos RM, Kavanagh E, Persson A, Englund E, et al. Spatio-temporal activation of caspase-8 in myeloid cells upon ischemic stroke. Acta Neuropathol Commun. 2016;4:92.
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11:56–64.
Lourbopoulos A, Ertürk A, Hellal F. Microglia in action: how aging and injury can change the brain’s guardians. Front Cell Neurosci. 2015;9:54.
Simon DW, McGeachy MJ, Bayır H, Clark RSB, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13:572.
Van Steenwinckel J, Reaux-Le Goazigo A, Pommier B, Mauborgne A, Dansereau MA, Kitabgi P, et al. CCL2 released from neuronal synaptic vesicles in the spinal cord is a major mediator of local inflammation and pain after peripheral nerve injury. J Neurosci. 2011;31:5865–75.
de Jong EK, Dijkstra IM, Hensens M, Brouwer N, van Amerongen M, Liem RS, et al. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci. 2005;25:7548–57.
Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602.
Kraaijeveld AO, de Jager SC, de Jager WJ, Prakken BJ, McColl SR, Haspels I, et al. CC chemokine ligand-5 (CCL5/RANTES) and CC chemokine ligand-18 (CCL18/PARC) are specific markers of refractory unstable angina pectoris and are transiently raised during severe ischemic symptoms. Circulation. 2007;116:1931–41.
Werner Y, Mass E, Ashok Kumar P, Ulas T, Händler K, Horne A, et al. Cxcr4 distinguishes HSC-derived monocytes from microglia and reveals monocyte immune responses to experimental stroke. Nat Neurosci. 2020;23:351–62.
Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. 2017;13:420–33.
Biber K, Vinet J, Boddeke HW. Neuron-microglia signaling: chemokines as versatile messengers. J Neuroimmunol. 2008;198:69–74.
Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;13:411–3.
Chen C, Chu SF, Ai QD, Zhang Z, Guan FF, Wang SS, et al. CKLF1 aggravates focal cerebral ischemia injury at early stage partly by modulating microglia/macrophage toward M1 polarization through CCR4. Cell Mol Neurobiol. 2019;39:651–69.
Kong LL, Wang ZY, Han N, Zhuang XM, Wang ZZ, Li H, et al. Neutralization of chemokine-like factor 1, a novel C-C chemokine, protects against focal cerebral ischemia by inhibiting neutrophil infiltration via MAPK pathways in rats. J Neuroinflammation. 2014;11:112.
Kong LL, Hu JF, Zhang W, Yuan YH, Han N, Chen NH. C19, a C-terminal peptide of chemokine-like factor 1, protects the brain against focal brain ischemia in rats. Neurosci Lett. 2012;508:13–6.
Ashwal S, Tone B, Tian HR, Cole DJ, Liwnicz BH, Pearce WJ. Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion in the rat pup. Pediatr Res. 1999;46:390–400.
Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, et al. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc. 2007;2:1044–51.
Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013;4:e716.
Dou C, Sun L, Wang L, Cheng J, Wu W, Zhang C, et al. Bromodomain-containing protein 9 promotes the growth and metastasis of human hepatocellular carcinoma by activating the TUFT1/AKT pathway. Cell Death Dis. 2020;11:730.
Kong LL, Hu JF, Zhang W, Yuan YH, Ma KL, Han N, et al. Expression of chemokine-like factor 1 after focal cerebral ischemia in the rat. Neurosci Lett. 2011;505:14–8.
Han W, Lou Y, Tang J, Zhang Y, Chen Y, Li Y, et al. Molecular cloning and characterization of chemokine-like factor 1 (CKLF1), a novel human cytokine with unique structure and potential chemotactic activity. Biochem J. 2001;357:127–35.
Li G, Li GY, Wang ZZ, Ji HJ, Wang DM, Hu JF, et al. The chemokine-like factor 1 induces asthmatic pathological change by activating nuclear factor-κB signaling pathway. Int Immunopharmacol. 2014;20:81–8.
Ai QD, Chen C, Chu S, Zhang Z, Luo Y, Guan F, et al. IMM-H004 therapy for permanent focal ischemic cerebral injury via CKLF1/CCR4-mediated NLRP3 inflammasome activation. Transl Res. 2019;212:36–53.
Yang PF, Song XY, Zeng T, Ai QD, Liu DD, Zuo W, et al. IMM-H004, a coumarin derivative, attenuated brain ischemia/reperfusion injuries and subsequent inflammation in spontaneously hypertensive rats through inhibition of VCAM-1. RSC Adv. 2017;7:27480–95.
Seo HH, Lee S, Lee CY, Lee J, Shin S, Song BW, et al. Multipoint targeting of TGF-β/Wnt transactivation circuit with microRNA 384-5p for cardiac fibrosis. Cell Death Differ. 2019;26:1107–23.
Roth Flach RJ, Skoura A, Matevossian A, Danai LV, Zheng W, Cortes C, et al. Endothelial protein kinase MAP4K4 promotes vascular inflammation and atherosclerosis. Nat Commun. 2015;6:8995.
Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, et al. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–9.
Harari OA, Liao JK. NF-kappaB and innate immunity in ischemic stroke. Ann NY Acad Sci. 2010;1207:32–40.
Listwak SJ, Rathore P, Herkenham M. Minimal NF-kappaB activity in neurons. Neuroscience. 2013;250:282–99.
Nijboer CH, Heijnen CJ, Groenendaal F, May MJ, van Bel F, Kavelaars A. A dual role of the NF-kappaB pathway in neonatal hypoxic-ischemic brain damage. Stroke. 2008;39:2578–86.
Sarnico I, Lanzillotta A, Boroni F, Benarese M, Alghisi M, Schwaninger M, et al. NF-kappaB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. J Neurochem. 2009;108:475–85.
Chen H, Lin W, Lin P, Zheng M, Lai Y, Chen M, et al. IL-10 produces a dual effect on OGD-induced neuronal apoptosis of cultured cortical neurons via the NF-κB pathway. Aging (Albany NY). 2019;11:10796–813.
Esposito E, Li W, T. Mandeville E, Park JH, Şencan I, Guo S, et al. Potential circadian effects on translational failure for neuroprotection. Nature. 2020;582:395–8.
Chen AQ, Fang Z, Chen XL, Yang S, Zhou YF, Mao L. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. 2019;10:487.
Ma S, Wang J, Wang Y, Dai X, Xu F, Gao X, et al. Diabetes mellitus impairs white matter repair and long-term functional feficits after cerebral ischemia. Stroke. 2018;49:2453–63.
Bodles AM, Barger SW. Secreted beta-amyloid precursor protein activates microglia via JNK and p38-MAPK. Neurobiol Aging. 2005;26:9–16.
Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405.
Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ. Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J Neuroinflammation. 2011;8:79.
Xing B, Bachstetter AD, Van Eldik LJ. Microglial p38α MAPK is critical for LPS-induced neuron degeneration, through a mechanism involving TNFα. Mol Neurodegener 2011;6:84.
Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554–65.
Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2.
Bachstetter AD, Rowe RK, Kaneko M, Goulding D, Lifshitz J, Van Eldik LJ. The p38α MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci. 2013;33:6143–53.
Xie Z, Smith CJ, Van Eldik LJ. Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia. 2004;45:170–9.
Chen C, Ai Q, Chu S, Zhang Z, Zhou X, Luo P, et al. IMM-H004 protects against oxygen-glucose deprivation/reperfusion injury to BV2 microglia partly by modulating CKLF1 involved in microglia polarization. Int Immunopharmacol. 2019;70:69–79.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81730096, 82074044, 81873026, 81773924), the Natural Science Foundation of Beijing (7192135), the Drug Innovation Major Project (2018ZX09711001-002-007, 2018ZX09711001-003-005, 2018ZX09711001-009-013), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-1-004), and the High-End Foreign Experts Introduction Program (G20200001485).
Author information
Authors and Affiliations
Contributions
XZ, SFC, and NHC designed research. XZ, YNZ, FFL, ZZ, LYC, HYH, and XY performed the experiments and drafted the paper. WBH, SFC, and NHC participated in data analysis. HSS and ZPF were involved in discussion of experiments. SFC and NHC revised the paper. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhou, X., Zhang, Yn., Li, Ff. et al. Neuronal chemokine-like-factor 1 (CKLF1) up-regulation promotes M1 polarization of microglia in rat brain after stroke. Acta Pharmacol Sin 43, 1217–1230 (2022). https://doi.org/10.1038/s41401-021-00746-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00746-w
Keywords
This article is cited by
-
A breakdown of metabolic reprogramming in microglia induced by CKLF1 exacerbates immune tolerance in ischemic stroke
Journal of Neuroinflammation (2023)
-
CZK, a novel alkaloid derivative from Clausena lansium, alleviates ischemic stroke injury through Nrf2-mediated antioxidant effects
Scientific Reports (2023)
-
A novel small-molecular CCR5 antagonist promotes neural repair after stroke
Acta Pharmacologica Sinica (2023)
-
Regulation of oxygen–glucose deprivation/reperfusion-induced inflammatory responses and M1-M2 phenotype switch of BV2 microglia by lobetyolin
Metabolic Brain Disease (2023)
