
Abstract
Heart failure (HF) represents one of the leading causes of cardiovascular diseases with high rates of hospitalization, morbidity and mortality worldwide. Ample evidence has consolidated a crucial role for mitochondrial injury in the progression of HF. It is well established that mitochondrial Ca2+ participates in the regulation of a wide variety of biological processes, including oxidative phosphorylation, ATP synthesis, reactive oxygen species (ROS) generation, mitochondrial dynamics and mitophagy. Nonetheless, mitochondrial Ca2+ overload stimulates mitochondrial permeability transition pore (mPTP) opening and mitochondrial swelling, resulting in mitochondrial injury, apoptosis, cardiac remodeling, and ultimately development of HF. Moreover, mitochondria possess a series of Ca2+ transport influx and efflux channels, to buffer Ca2+ in the cytoplasm. Interaction at mitochondria-associated endoplasmic reticulum membranes (MAMs) may also participate in the regulation of mitochondrial Ca2+ homeostasis and plays an essential role in the progression of HF. Here, we provide an overview of regulation of mitochondrial Ca2+ homeostasis in maintenance of cardiac function, in an effort to identify novel therapeutic strategies for the management of HF.
Similar content being viewed by others
Introduction
Heart failure (HF) is common in many end-stage cardiovascular diseases with high morbidity and mortality [1,2,3]. HF is believed to be responsible for most hospitalizations throughout the world, and its prevalence continues to rise with the aging population [4,5,6,7]. Despite substantial advances in the recent clinical management of HF, the survival rate of patients afflicted with HF remains dismally low. The ESC-HF pilot study revealed that the one-year hospitalization rates for patients with acute and chronic HF were 43.9% and 31.9%, respectively [8]. The 5-year survival rate of patients with HF is only 50% [4]. In this context, it is pertinent to elaborate the precise mechanisms underlying HF in an effort to identify novel therapeutic regimens to retard or reverse the pathological progression of HF, ultimately improving quality of life and reducing mortality [3, 9,10,11,12,13].
The rhythmicity, automaticity and contraction of the heart demand profound energy, which is driven by oxidative phosphorylation within mitochondria [14,15,16,17,18]. Not surprisingly, mitochondria play a crucial role in the etiology of HF [19,20,21]. Mitochondrial dysfunction has been shown to contribute to defects in bioenergetics, Ca2+ transport, redox balance, mitochondrial membrane potential (ΔΨm), metabolic signaling, and apoptosis and other forms of cell death [10, 14, 22,23,24]. Numerous findings have shown that mitochondrial disorders are detrimental to cardiac function, resulting in cardiac hypertrophy and ultimately HF [1, 14, 19, 25,26,27,28]. In the early phase of cardiac hypertrophy, the increased energy demand triggers complementary regulation of mitochondrial oxidative phosphorylation through Ca2+ and ADP. These maladaptive processes are closely associated with disrupted mitochondrial Ca2+ homeostasis in the myocardium.
Ca2+ acts as a ubiquitous second messenger that translates information delivered by an array of cell signals [29, 30]. Ca2+ dynamics play critical roles in the excitation-contraction coupling (ECC) of cardiomyocytes [18, 31, 32]. Transient increases in cytoplasmic Ca2+ levels activate the formation of cross bridges to engage myocardial contraction [32]. Mitochondria accumulate Ca2+ during systole and participate in the process of mitochondrial oxidative phosphorylation and the ATP synthesis electron transport chain (ETC) function [33]. On the other hand, excessive mitochondrial Ca2+ uptake stimulates the production of reactive oxygen species (ROS), opening of the mitochondrial permeability transition pore (mPTP) and initiation of apoptosis, resulting in mitochondrial injury, cardiac remodeling and ultimately HF [34]. In this review, we present a brief update on recent progress in understanding the vital role of mitochondrial Ca2+ homeostasis in the maintenance of cardiac function and how mitochondrial Ca2+ perturbation may contribute to the onset and development of HF.
Maintenance of mitochondrial Ca2+ homeostasis
Cytoplasmic Ca2+ and ER Ca2+
Intracellular Ca2+ controls nearly all cellular biological processes encompassing muscle contraction, secretion and gene transcription [35]. The level of intracellular Ca2+ needs to be maintained at a range of ~100–1000 nmol/L, an ~10,000-fold difference than the extracellular Ca2+ concentration (1.5 mmol/L) maintained through consumption of considerable amounts of energy [35, 36]. Due to the significant electrochemical gradient between the extracellular space and cytoplasm, increases in intracellular Ca2+ can be elicited by both Ca2+ entry from the extracellular pool through ion channels in plasma membranes and Ca2+ release from intracellular stores, including the ER. In addition, Ca2+ may enter the intracellular domains from the extracellular space through voltage-gated Ca2+ channels (VGCCs) and transient receptor potential channels (TRPCs) located in the plasma membrane [37, 38].
The sarcoplasmic reticulum (SR) represents a specialized form of the endoplasmic reticulum (ER) in cardiomyocytes [39]. Both SR and ER are enriched with Ca2+ channels – inositol 1,4,5-triphosphate receptors (InP3Rs) and ryanodine receptors (RyRs) – in the ER and SR, respectively [40]. Both channels mainly release Ca2+ from the ER or SR to the cytoplasm when intracellular Ca2+ levels are low. There are three isoforms of RyRs, of which, RyR2,is mainly localized in the myocardium, although the RyR1 isoform may also be found in cardiac mitochondria [40,41,42].
In addition, relaxation of cardiomyocytes depends on a decrease in intracellular Ca2+ levels that allows myofilament cross-bridge uncoupling [15, 36]. The cyclical decrease in cytosolic Ca2+ occurs mainly through Ca2+ re-uptake by the SR, which is mediated by Ca2+ ATPase 2 (SERCA2) in the SR and, to some extent, sarcolemma Ca2+ ATPase, an Na+/Ca2+ exchanger, and mitochondrial uptake machinery [43].
Mitochondrial Ca2+ influx
Mitochondria are important intracellular organelles in regulating Ca2+ dynamic oscillation due to their high Ca2+-buffering capacity [44]. Mitochondria possess a series of Ca2+ transport channels to maintain tight regulation of Ca2+ influx and efflux, which prevents drastic intracellular Ca2+ oscillations [31]. Mitochondria are structurally segregated into 4 distinct compartments by two membranes, the outer mitochondrial membrane (OMM), the intermembrane space, the inner mitochondrial membrane (IMM), and the mitochondrial matrix. Because of the high permeability of Ca2+ in the OMM, the Ca2+ levels in the intermembrane space are comparable to the Ca2+ levels in the cytoplasm [45]. In cells at rest, the Ca2+ levels in the mitochondrial matrix are comparable to those found in the cytoplasm (~100 nmol/L). When cells are excited, cytoplasmic Ca2+ levels can be in concentrations as to 2–3 μmol/L, and Ca2+ levels can rise to 10 μmol/L or even higher (500 μmol/L) in the mitochondrial matrix [44].
Mitochondrial voltage-dependent anion channels (VDACs)
The accumulation of mitochondrial Ca2+ requires Ca2+ to pass through the OMM and IMM. In living cells, the high conductance protein VDAC, located on the OMM, governs the transport of Ca2+ into the intermembrane space of a mitochondrion. There are three identified isoforms of the VDAC protein: VDAC1, VDAC2, and VDAC3 [46]. The most abundant isoform, VDAC1, is deemed the main Ca2+ transport channel [47]. Ca2+ ions move across the IMM driven by an electrical gradient controlled through the mitochondrial Ca2+ uniporter (MCU) complex and reach the mitochondrial matrix. The Ca2+ levels in the mitochondrial matrix can reach a level of approximately one or two orders of magnitude greater than that in the cytoplasm. VDAC1 levels were reported to be increased in the left ventricular tissues of patients suffering from hypertrophic cardiomyopathy [48]. During hypoxia-reoxygenation of cardiomyocytes, the VDAC1-mediated Ca2+ channeling complex is increased concomitantly with mitochondrial Ca2+ content. The inhibition of VDAC1 channels dramatically suppressed mitochondrial Ca2+ overload and thus protected cells from hypoxia-reoxygenation stress [49].
The mitochondrial Ca2+ uniporter (MCU) complex
The MCU complex is a highly selective ion channel located on the IMM and transports Ca2+ into the mitochondrial matrix [50, 51]. MCU is critical for the mitochondrial energetic adaptation initiated by mitochondrial Ca2+ uptake. A fundamental property of MCU is the low activity of resting cytosolic Ca2+ levels, which prevent mitochondrial Ca2+ overload.
Several proteins were recently indicated to participate in mitochondrial Ca2+ uptake, including MCUa, MCUb and its regulators MICU1/2, mitochondrial Ca2+ uniporter regulator 1 (MCUR1) and the essential MCU regulator (EMRE). The pore-forming subunit molecule MCUa is a 35 kDa protein with a highly conserved DIME motif that is composed of two coiled-coil domains and two transmembrane helices. The DIME motif of MCU are sides at the pore entrance and accounts for selective Ca2+ uptake, while the other transmembrane domain constitutes a hydrophilic pore, with the coiled-coil domains stabilizing the overall architecture [52]. MCUb shares 50% homology with MCUa but the DIME motifs are considerably different, which inhibits mitochondrial Ca2+ uptake. MCUb is considered a dominant-negative form of MCUa, physically binding to MCUa and modulating Ca2+ permeation.
MICU1 binds to the D-ring domain of MCU through the DIME motif to regulate both the Ca2+ transport current and ruthenium red sensitivity of the MCU complex [53]. The high-affinity interaction of the MICU1/2 complex appears to serve as an on-off switch in direct response to cytosolic Ca2+ signals [54,55,56]. At low Ca2+ levels, MICU2 shuts down MCU function, whereas MICU1 allows mitochondria to promptly respond to elevated cytoplasmic Ca2+ [57]. Moreover, EMRE is reported to act as a scaffold protein to mediate the interaction between MCU and the MICU1/2 complex. In the absence of EMRE, MCU complex-mediated mitochondrial Ca2+ conductance is abolished in a manner similar to MCU silencing [58].
The precise pathophysiological effect of the MCU complex in HF needs further exploration. Regarding the role of MCU-mediated mitochondrial Ca2+ uptake in HF, conflicting results from previous studies have led to an ongoing debate [59]. It has been suggested that the level of mitochondrial MCU protein was significantly increased in mouse hearts in cases of pressure overload-induced cardiac hypertrophy, while this level was markedly downregulated in type 1 diabetic mouse hearts [50, 60, 61]. However, the probability of MCU being open was significantly decreased in failing human hearts, as determined by patch clamping of the inner membrane of mitochondria [62]. During acute ischemic injuries, MCU may mediate acute mitochondrial Ca2+ overload, trigger the permeability transition and ultimately induce cardiomyocyte death [63]. In addition, the MCU complex is necessary for increases in heart rate under stress challenge, while MCU inhibitors can lower the inappropriate tachycardia without affecting resting heart rhythm [64]. A compensatory rise in MCU was noted in cardiomyocytes during an energy shortage during the initial phase of transverse aortic constriction (TAC)-induced cardiac hypertrophy. In cardiac-selective MCU-knockout mice, cytosolic Ca2+ elevation was mainly the result of impaired mitochondrial Ca2+-dependent oxidative phosphorylation and ATP generation in the mitochondrial matrix [65]. In the context of elevated MCU levels, mitochondrial Ca2+ uptake is elevated to best facilitate mitochondrial ATP synthesis [50]. These findings have prompted the speculation of a potential role for MCU complex-mediated mitochondrial Ca2+ uptake in therapeutic targeting against HF.
Other channels of mitochondrial Ca2+ influx
More channels have been identified to shuffle Ca2+ into mitochondria. “Rapid-mode” uptake(RaM) allows fast intramitochondrial Ca2+ changes to mirror cytosolic Ca2+ changes in the time frame of milliseconds to turn on the mitochondrial metabolic reaction [66, 67]. Ryanodine receptor isoform (RyR)1 is another channel located in the IMM and plays an important role in the dynamic uptake of Ca2+ into mitochondria during Ca2+ oscillations [68]. Further exploration is required to investigate the mechanism of RyR1 in the progression of HF.
Mitochondrial Ca2+ efflux
To accommodate mitochondrial Ca2+ in response to changes in cytosolic Ca2+ levels, an Ca2+ ions are extruded from the mitochondrial matrix. Two major pathways are identified to counter the MCU complex and trigger Ca2+ efflux from mitochondria, namely, the Na+/Ca2+/Li+ permeable exchanger (NCLX) [69, 70] and the H+/Ca2+ exchanger (HCX) [71].
Mitochondrial Na+/Ca2+/Li+ permeable exchanger (NCLX)
NCLX is the primary mechanism of Ca2+ extrusion from the mitochondrial matrix [72]. NCLX is located on the IMM and appears to be the predominant antiporter in the heart and brain. NCLX exchanges 3 or 4 Na+ for 1 Ca2+. The kinetics of mitochondrial Ca2+ efflux are much slower than those of Ca2+ influx, which accounts for mitochondrial Ca2+ accumulation and sequentially contributes to the activation of the Krebs cycle following β-adrenergic stimulation [73]. The electrochemical gradient for Na+ influx into the mitochondrial matrix is the main driving force for NCLX. Given the intracellular Na+ levels of 4–8 mmol/L in cardiomyocytes, elevated intracellular Na+ in HF accelerates NCLX-mediated Ca2+ efflux and thereby reduce intramitochondrial Ca2+ levels [74].
It was noted that both the mRNA and protein levels of NCLX were decreased in myocardial infarction-induced HF [75]. NCLX overexpression contributes to contractile dysfunction and arrhythmogenesis, probably through compensatory Ca2+ transport [76, 77]. Moreover, tamoxifen-induced cardiac-specific NCLX-knockout mice displayed severe myocardial dysfunction, and less than 13% of these mice survived beyond two weeks. This impressive lethality of the mouse models underscores a critical role of NCLX as the primary mitochondrial Ca2+ efflux mechanism. In addition, ROS production and mPTP opening in cardiomyocytes were significantly increased in the NCLX-knockout mice compared with the control mice [78]. However, cardiac dysfunction from acute myocardial infarction (MI) was rescued in NCLX-knockout mice crossed with cyclophilin D-null mice, indicating that cardiomyocyte apoptosis and cardiac fibrosis may be attributed to Ca2+-induced mPTP opening in the NCLX-knockout mice [78]. Together, these findings suggest that NCLX may have potent therapeutic potential in the management of heart diseases.
Mitochondrial H+/Ca2+ exchanger (HCX)
HCX, another protein participating in mitochondrial Ca2+ extrusion, is also known as leucine zipper EF-hand-containing transmembrane protein 1 (LETM1) [79]. This protein mainly mediates Ca2+ release from mitochondria in the liver and kidney with the exchange of 2 H+ for 1 Ca2+. The LETM1 motif contains only a single transmembrane helix and might oligomerize into a hexameric structure to function as a transport route [80]. However, electroneutral transport of Ca2+ and insensitivity to ruthenium red and CGP-37157 were noted in highly purified LETM1-containing liposomes, confirming a role for LETM1 as a candidate for HCX function [81]. However, conflicting findings were also found for mitochondrial Ca2+ in LETM1-null cells; more supporting evidence is required to clarify the vital role of LETM1 in mitochondrial Ca2+ efflux [80]. LETM1 was reported to be closely associated with Wolf-Hirshhorn syndrome characterized by neurological disorders, although the possible underlying mechanism needs further scrutiny [82].
Finally, the Na+/H+ exchanger (NHE) serves as another critical exchanger for proper mitochondrial Ca2+ influx [83]. NHE is mainly responsible for Na+ flow into the cytoplasm, resulting in the maintenance of Na+ gradients across mitochondrial membranes and mitochondrial Ca2+ homeostasis [84]. The increased activity of NHE-1 was identified in HF, and the resultant increase in intracellular Na+ levels may disturb mitochondrial Ca2+-related processes. Inhibition of NHE-1 was reported to retard cardiac hypertrophy and HF in response to pressure overload [85, 86].
Mitochondria-associated ER membranes (MAMs)
Mitochondrial Ca2+ homeostasis is closely associated with the communication between mitochondria and the ER, which mainly assists in Ca2+ transfer, signal transduction, mitochondrial metabolism, and mitochondrial dynamics [87, 88]. Mitochondria and ER possess contact sites, which account for ~20% of the total mitochondrial network with the distance between two organelles ranging from 10 nm–60 nm, structures commonly termed mitochondria-associated endoplasmic reticulum membranes (MAMs) [89]. At MAMs, Ca2+ directly transfers from the ER to mitochondria and promotes the Krebs cycle to synthesize ATP in the mitochondrial matrix [90]. In cardiomyocytes, SR-mitochondria coupling also serves as an important regulator of biological events, including Ca2+ signaling, lipid metabolism, inflammation, autophagy, and apoptosis [88, 91, 92].
MAMs maintain mitochondrial Ca2+ homeostasis through MAM-related protein bridges. MAMs provide a microenvironment with a high level of Ca2+, which is necessary for mitochondria internalization of Ca2+ [88]. How do MAM-related proteins affect mitochondrial Ca2+ ions? Recent evidence has confirmed that several molecules work as Ca2+ transport complexes to regulate Ca2+ transfer at MAMs (Fig. 1).

ER endoplasmic reticulum, IMM inner mitochondrial membrane, OMM outer mitochondrial membrane, MFN1/2 mitofusin 1/2, MCU mitochondrial calcium uniporter, NCLX Na+/Ca2+/Li+ permeable exchanger, VDAC1 voltage-dependent anion channel, IP3R inositol 1,4,5-triphosphate receptor, GRP75 chaperone 75 kDa glucose-regulated protein, FUNDC1 FUN14 domain containing 1, Sig-1R the sigma-1 receptor, PTPIP51 protein tyrosine phosphatase-interacting protein 51, and VAPB vesicle-associated membrane protein-associated protein B.
IP3R1-GRP75-VDAC1
Multiprotein complexes participate in regulating Ca2+ transfer from the ER to mitochondria. Inositol 1,4,5-trisphosphate receptor type 1 (IP3R1), the major channel in the ER that releases Ca2+, allows the formation of microdomains highly enriched with Ca2+ in the vicinity of the ER [87]. Conversely, VDAC1 in the OMM acts as a Ca2+-uptake channel with high conductance. As the third element of this complex, a 75 kDa chaperone, glucose-regulated protein (GRP75), a member of the Hsp70 family, tethers cytosolic portions of IP3R1 and VDAC1 [93]. Thus, Ca2+ is directly transferred from the ER to the cytosol and then moves across the OMM into the intermembrane space of a mitochondrion. Finally, Ca2+ ions enter the mitochondrial matrix through the MCU complex.
The increased expression of IP3R1 has been demonstrated in heart tissues from HF patients and experimental animals [94]. In addition, genetic or pharmacological inhibition of cyclophilin D, a chaperone of the IP3R1-GRP75-VDAC1 complex, may mitigate Ca2+ transfer from the ER to mitochondria through IP3R1 in adult cardiomyocytes and thus attenuate mitochondrial Ca2+ overload, ultimately protecting cells from hypoxia-reoxygenation [49, 95].
MFN1–MFN2
In addition to the IP3R1-GRP75-VDAC1 complex, mitochondrial fusion also forms bridges that enjoin the ER and mitochondria. Mitochondrial fusion is traditionally realized through the function of mitofusin (MFN)2, a GTPase that resides at the OMM as tethers to juxtaposed mitochondria. MFN2 resides on ER membranes and forms dimers with MFN1 or MFN2 located on the OMM [96]. Mitochondrial Ca2+ uptake is inhibited due to the disassociation of the ER and mitochondria in MFN2-knockout cells, which does not affect the Ca2+ release capacity of the ER [97].
Furthermore, these results not only reflect the distance between these subcellular organelles but also show the coordinated regulation of the mitochondrial network and ER dynamics. It was reported that the levels of MFN1 were increased in mitochondria during HF, yet its GTPase activity was significantly compromised, revealed by an accumulation of defective MFN1 in HF [98]. Several reports have suggested that MFN2 is downregulated in myocardial hypertrophy and doxorubicin-induced cardiotoxicity [99, 100], while MFN2-knockout cells displayed overtly reduced junctional SR–mitochondria contact sites and developed dilated cardiomyopathy in mice. In addition, MFN1/MFN2 cardiac-knockout induces resistance to mPTP opening due to a reduction in SR–mitochondria contacts, mitochondrial Ca2+ overload, and ROS generation during cardiac ischemia/reperfusion(I/R) injury [101, 102].
FUNDC1-IP3R2
FUN14 domain containing 1 (FUNDC1) is a highly conserved OMM molecule that mediates independent mitophagy [103]. A recent study suggested that FUNDC1 localized at MAMs and interacted with IP3R2 to mediate Ca2+ release from SR into both mitochondrial and cytoplasmic cardiomyocytes. Although deletion of IP3R2 did not influence the progression of HF in diabetes-related cardiac disease or pressure overload-induced HF [104], FUNDC1 deletion led to cardiac dysfunction characterized by disrupted MAMs and impaired Ca2+ transfer from the SR to mitochondria [105]. In FUNDC1-knockout cells, both intracellular and mitochondrial Ca2+ levels were reduced, and Ca2+-sensitive CREB-mediated Fis1 expression was suppressed, causing mitochondrial dysregulation, cardiac dysfunction and HF [105].
VAPB-PTPIP51
In addition to the aforementioned molecules, vesicle-associated membrane protein-associated protein B (VAPB), located on the ER surface, is coupled to an OMM called protein tyrosine phosphatase interacting protein 51 (PTPIP51) [106]. Previous work has revealed that modifying the expression of VAPB and PTPIP51 affects the transfer of Ca2+ between the two subcellular organelles. Recently, studies suggested that VAPB-PTPIP51 regulates Ca2+ transfer by influencing the function of IP3R-VDAC1 to regulate autophagy [107]. Further studies are required to elucidate the likely role and mechanism of action of VAPB-PTPIP51 in HF.
Other
Recent findings have also identified several molecular chaperones, including calnexin [108], syntaxin-17 and sigma-1 receptor (Sig-1R) at MAMs, which are involved in the regulation of information exchange, such as Ca2+ ions between the ER and mitochondria. Sig-1R is a chaperone located in MAMs that regulates Ca2+ transfer between mitochondria and ER by binding with IP3R3 [109]. Upon the induction of ER stress, Sig-1R is separated from BIP, and IP3R3 is stabilized to accelerate Ca2+ influx from the ER into mitochondria to increase the production of ATP. It has been suggested that Sig-1R knockout leads to mitochondrial dysfunction and HF [109]. In addition, it has been suggested that syntaxin-17 interacts with Drp1 and regulates the calcium concentration in the ER and cytoplasm [110]. Under various stress conditions, mitochondrial Ca2+ homeostasis is affected by the influence of MAM-related proteins. RyR2 was found to interact with VDAC2 in HL-1 cells and mediate calcium transfer between the SR and mitochondria [111].
Disruption of mitochondrial Ca2+ homeostasis in HF
Mitochondrial Ca2+ and ATP generation
Mitochondria constitute ~30% of the cellular volume in cardiomyocytes, where they supply ATP to meet the heightened energy demand for cardiac function. Ca2+ plays a crucial role in ATP production for communicating cellular energy demands to mitochondria [112, 113]. Oxidative phosphorylation is tightly regulated by mitochondrial Ca2+ as Ca2+ activates tricarboxylic acid dehydrogenases to produce equivalents (NADH/NADPH) for electron transport [114]. Mitochondria constitute a vital buffer function for cytosolic Ca2+, although excess mitochondrial Ca2+ leads to mitochondrial injury and myocardial death.
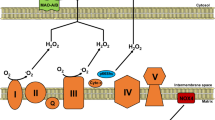
The HF process is closely linked to abnormal mitochondrial energy metabolism, including reduced energy metabolism and impaired energy utilization, which adversely disturb Ca2+ homeostasis [14, 114]. In HF, there is an imbalance between Ca2+ and ATP, which results in the oxidation of pyridine nucleotides in the mitochondrial matrix, where NADH oxidation may reduce ATP synthesis and cardiomyocyte contraction, while NADPH oxidation may provoke oxidative damage with consequent maladaptive cardiac remodeling [115]. Understanding these complicated mechanisms that affect this fine-tuned equilibrium may contribute to the exploration of therapeutic regimens that attenuate cardiac remodeling through a process that differs from classical neuroendocrine inhibition (Fig. 2).

mPTP mitochondrial permeability transition pore, ROS reactive oxygen species.
Mitochondrial Ca2+ and ROS production
Mitochondria are major sources of ROS due to their oxidative reactions. Thus, mitochondria are more prone to oxidative injury than other organelles [116]. Oxidative stress may damage mitochondrial DNA, leading to protein oxidative damage and impaired mitochondria and myocardial energetics in HF. In animal models, ROS generation in mitochondria is causally linked to the onset and progression of cardiac dysfunction triggered by various pathological stimuli, including angiotensin II, pressure overload, and cardiac ischemia, and lead to an increased risk of arrhythmia [14, 117].
In failing hearts, O2-anions form at mitochondrial complex I and are then converted to hydroxyl radicals, the major mitochondrial ROS in the heart. ROS induce injuries to the mitochondrial respiratory chain and slow electron transfer through complex I, thereby causing a reduction in NADH [118]. Furthermore, mitochondria may potentiate ROS through ROS-induced ROS release, which involves ROS-induced activation of anion channels in the IMM, mPTP, and ATP-sensitive K+-channels [20, 119]. Activation of these channels dissipates the ΔΨm and consumes NADH and NADPH, resulting in the emission of H2O2. Finally, hyper acetylation of mitochondrial proteins was found to be significantly increased in the hearts of patients with HF, which may be attributed to increased myocardial levels of acetyl-CoA and acetylation of various proteins [120, 121].
One of the common triggers for ROS-induced ROS release is angiotensin II stimulation or pressure overload, resulting in damage to the ECC through the negative effects of RyR2s, SERCA2, and NCLX [20]. Mitochondrial Ca2+ overload is induced by mitochondrial ROS, which favor Ca2+ efflux via NCLX, thereby eliciting nonspecific Ca2+ flux that reverses the polarization of ΔΨm. In addition, ROS activate Ca2+/calmodulin-dependent protein kinase II (CaMKII), an essential regulator involved in both Ca2+ and Na+ processes, including those involved with RyR2s, phospholamban, L-type Ca2+ channels, and late Na+ current, ultimately contributing to cardiac dysfunction and arrhythmias in HF. CaMKII also induces cardiac remodeling and cardiac hypertrophy through the phosphorylation of histone deacetylase (HDAC), potentially leading to elevated ROS generation. Therefore, ROS and mitochondrial Ca2+ overload contribute to HF through several mechanisms, prompting a vicious cycle resulting in cardiac hypertrophy, interstitial fibrosis, contractile dysfunction and sudden death [20].
Mitochondrial Ca2+ and mitochondrial dynamics in HF
Ample evidence has suggested that mitochondrial architecture is constantly modified by highly dynamic organelle transformations [30, 122,123,124]. Mitochondria undergo continuous fission and fusion to maintain mitochondrial integrity and remove damaged mitochondria [125, 126]. Dynamin-related protein 1 (DRP1) is recruited to mitochondria and forms a constricted ring that drives mitochondrial fragmentation. In contrast, mitochondrial fusion is regulated by MFN1, MFN2, and optic atrophy protein 1 (OPA1) [125,126,127]. MFN1/MFN2 are located on the membrane of two adjacent mitochondria that interact with each other to mediate OMM fusion. OPA1, on the other hand, resides on the IMM and is critical for the for maintenance of crista structures and IMM fusion.
The precise interplay between Ca2+ and mitochondrial dynamics is poorly understood in the context of cardiac hypertrophy. Recent studies indicated that Ca2+ inhibits mitochondrial motility to retain mitochondrial Ca2+ signaling and energy supply levels [128]. It was also indicated that calcineurin, as a Ca2+-dependent phosphatase, is overtly activated by elevated Ca2+ levels to phosphorylate Drp1 at Ser637 and promote Drp1 recruitment to mitochondria, resulting in mitochondrial fission [129, 130]. In addition, it has been noted that the treatment of cardiomyocytes with norepinephrine promotes extensive mitochondrial fission and mitochondrial injury through the activation of the α1-adrenergic-receptor-Ca2+-calcineurin-Drp1 signaling axis [131]. These findings suggest a key role for mitochondrial Ca2+ in mitochondrial dynamics and pathological cardiac remodeling [132].
Mitochondrial Ca2+and mitophagy in HF
Mitochondria account for ~20%–30% of cardiac mass, and their degradation is crucial for mitochondrial quality control. Interfering with mitochondrial quality control results in the accumulation of dysfunctional mitochondria and, ultimately, cardiac dysfunction [26]. Mitophagy is an essential process of selective autophagy, which results in the removal of irreversibly damaged mitochondria [125, 126]. Emerging evidence supports the protective role of mitophagy in the progression of HF [125, 126, 133]. Mitophagy is mainly mediated by the PINK1/Parkin pathway. PINK1-deficient hearts are more susceptible to I/R injury and transition more rapidly to failure in response to pressure overload [134]. Moreover, PRKN-knockout mice showed dysfunctional mitochondrial accumulation following MI and were susceptible to doxorubicin-induced cardiotoxicity and cardiac dysfunction [135,136,137]. These observations revealed a cardioprotective role for mitophagy in HF progression and highlight the utility of promoting mitophagy in therapeutics targeted to HF [138,139,140].
Recent studies revealed an important role for mitochondrial Ca2+ content in mitochondrial autophagy (mitophagy) regulation. In particular, mitochondrial Ca2+ uptake regulates AMP kinase (AMPK) activity and activates autophagy via the AMPK-mammalian target of rapamycin-UNC-51-like kinase 1 (AMPK/mTOR/ULK1) signaling pathway [141]. However, mitochondrial Ca2+ overload perturbs mitochondrial physiology, leading to ROS production, mPTP opening and mitophagy initiation [142]. These findings suggested that aberrant levels of Ca2+ may have opposite effects on autophagy. Moreover, mitochondrial Rho-GTPase (RHOT1) is an OMM protein and a Ca2+-sensitive regulator because of its two EF-hand Ca2+ binding domains [143]. PINK1 may phosphorylate RHOT1 and trigger Parkin-mediated mitophagy [144]. In addition, the accumulation of α-synuclein (SNCA) was found to increase mitochondrial Ca2+ levels and promote oxidative damage, whereas SNCA loss impaired mitochondrial Ca2+ and enhanced autophagy [145, 146]. Future work is warranted to elucidate how mitochondrial Ca2+ participates in the modulation of mitophagy in cardiomyocytes.
Mitochondrial Ca2+ overload, mPTP opening, mitochondrial apoptosis and necrosis
Excess uptake of mitochondrial Ca2+ and mitochondrial ROS production may both result in the depolarization of the ΔΨm, the opening of the mPTP, and a decreased proton gradient. Ca2+ and cytochrome c efflux from mitochondria results in mitochondrial swelling, loss of ΔΨm and apoptosis [147, 148]. The mPTP appears to be a crucial, but still incompletely understood, target of Ca2+-dependent CaMKII. Cardiomyocytes from transgenic mice overexpressing CaMKII inhibitory protein sustained higher Ca2+ levels in mitochondria prior to mPTP opening and were more resistant to apoptosis induced by I/R or MI injury, suggesting that Ca2+ levels promote mPTP opening and myocardial apoptosis partly through the activation of CaMKII [149].
Apoptosis refers to programmed cell death to maintain the stability of the internal environment of cells. Three main branches of signaling pathways are currently proposed, including the death receptor, endoplasmic reticulum (ER) and mitochondrial pathways [150]. The mitochondrial pathway is the most common and core mechanism regulated by mitochondrial Ca2+ [151]. Under apoptosis-inducing stimuli, such as toxins, hypoxia, viral infections, radiation, high cytosolic Ca2+ levels, and oxidative damage by ROS, mitochondria show increased permeability and Ca2+ overload, depolarized transmembrane potential, and cytochrome c and pro-apoptotic mediator release [152].
Mitochondria Ca2+ overload is a key element in the processes that control cellular death and survival through convergence at MAMs [153]. Bcl-2-like protein 1, an antiapoptotic protein, is localized to MAMs and promotes Ca2+ transfer from the ER to mitochondria as an adaptive response to promote mitochondrial bioenergetics and prevent cytosolic Ca2+ overload under stress conditions [154]. The phosphatase PTEN has been identified as another protective component of MAMs. PTEN is commonly referred to as a canonical tumor suppressor due to its inhibition of Akt on plasma membranes. PTEN also resides at MAMs, where it suppresses Akt signaling, interacts with IP3R1 protein, and increases Ca2+ transfer to mitochondria, thus inducing cells to undergo apoptosis [155].
In cardiomyocytes, aldosterone accelerates the mitochondrial apoptotic pathway through the dephosphorylation of the pro-apoptotic molecule Bad, enhancement of mitochondrial permeability, release of cytochrome c and concomitant activation of caspase 3, directly contributing to the progression of HF [156]. However, the effects of aldosterone were suppressed by inhibitors of the Ca2+-dependent phosphatase calcineurin and FK506 [157]. Furthermore, cytochrome c is released from mitochondria and is translocated to the ER, where it selectively binds to InP3Rs, resulting in mitochondrial Ca2+ overload and the induction of additional cytochrome c release, which amplifies the apoptotic signaling response to ischemia stress [158]. These findings revealed a feed-forward modality in which early cytochrome c release facilitates IP3R function, leading to mitochondria-induced apoptosis in ischemic heart diseases.
Notably, the dysregulation of mitochondrial Ca2+ is also a potent trigger of cellular necrosis [156, 159, 160]. With the simultaneous opening of the mPTP pores in mitochondria, ATP is depleted, with the level becoming insufficient to aggregate apoptosomes. The obviously increased cytosolic Ca2+ may activate a number of hydrolytic enzymes, resulting in organelle swelling, structural degeneration and necrotic death. This sequence of events leads to cardiomyocyte necrosis with the leakage of intracellular contents, including troponins, which serve as danger signals to the innate immune system. Later, inflammatory cells and myofibroblasts are recruited to the site of necrosis, resulting in reparative fibrosis, cardiac remodeling, and, not surprisingly, HF [159].
Perspectives and conclusion
The mitochondrion is an intracellular organelle governing energy supply, cell signaling and cell survival. Much attention has been paid to understanding the role of mitochondrial Ca2+ in the etiology of HF. Mitochondrial function is determined by the amplitude and kinetics of Ca2+ cycling and a number of key molecules governing various mitochondrial Ca2+ transport machineries. Much progress has been made in understanding these mitochondrial Ca2+ transport processes, including ion specificity, activation, inhibition, kinetics, electrical conductivity and other characteristics. With the development of recent cutting-edge technical methodologies, such as molecular biology and bioinformatics, more molecular mechanisms of these Ca2+ transport pathways are being revealed. In addition, due to the identification of mitochondrial Ca2+ transport machineries, including MCU, MICU1, and NCLX, the study of mitochondrial Ca2+ transport will shift from the classical cellular level to the molecular level to generate a better understanding of the physiological functions of mitochondrial Ca2+ transport. In animal models, mitochondrial Ca2+ transport inhibitors have been used to discern the physiological function of mitochondrial Ca2+ transport. However, the use of inhibitors such as ruthenium red, ruthenium 360, diltiazem, clonazepam, and CGP37157 often yield disparate and inconsistent results [161]. Therefore, drug development targeting mitochondrial Ca2+ transfer is pertinent for the advance of the field.
Several studies have suggested alterations in mitochondrial function and ER disorders are related to cardiomyopathies, cardiac hypertrophy and HF progression [21, 105]. However, the field of ER–mitochondria communication has received much less attention. Several pieces of evidence have indicated that interactions at MAMs may serve as critical factors in the pathophysiology of HF. The maintenance of mitochondrial Ca2+ homeostasis and mitochondrial function is essential for cardiomyocyte survival and cardiac function. The acquisition of novel technical tools necessary to regulate MAMs is an important objective for future research and therapeutic approaches. The in-depth understanding of mechanisms involved in mitochondrial Ca2+ regulation will contribute to the development of therapeutic strategies towards improving mitochondrial dysfunction in the heart and ultimately HF.
References
Smith JG. Molecular epidemiology of heart failure: translational challenges and opportunities. JACC Basic Transl Sci. 2017;2:757–69.
Wang S, Ren J. Obesity paradox in aging: from prevalence to pathophysiology. Prog Cardiovasc Dis. 2018;61:182–9.
Ren J, Zhang Y. Emerging therapeutic potential targeting genetics and epigentics in heart failure. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1867–9.
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Rev Esp Cardiol (Engl Ed). 2016;69:1167.
Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2015;385:812–24.
Yang M, Li C, Zhang Y, Ren J. Interrelationship between Alzheimer's disease and cardiac dysfunction: the brain-heart continuum? Acta Biochim Biophys Sin (Shanghai). 2020;52:1–8.
Ren J, Zhang Y. Targeting autophagy in aging and aging-related cardiovascular diseases. Trends Pharmacol Sci. 2018;39:1064–76.
Maggioni AP, Dahlstrom U, Filippatos G, Chioncel O, Crespo Leiro M, Drozdz J, et al. EURObservational Research Programme: regional differences and 1-year follow-up results of the Heart Failure Pilot Survey (ESC-HF Pilot). Eur J Heart Fail. 2013;15:808–17.
Yin Z, Ren J, Guo W. Sarcomeric protein isoform transitions in cardiac muscle: a journey to heart failure. Biochim Biophys Acta. 2015;1852:47–52.
Zhang Y, Whaley-Connell AT, Sowers JR, Ren J. Autophagy as an emerging target in cardiorenal metabolic disease: From pathophysiology to management. Pharmacol Ther. 2018;191:1–22.
Zhang Y, Ren J. Metabolic syndrome and cardiovascular health: a look beyond the horizon. Curr Pharmacol Des. 2013;19:4797–8.
Zhang Y, Sowers JR, Ren J. Pathophysiological insights into cardiovascular health in metabolic syndrome. Exp Diabetes Res. 2012;2012:320534.
Zhang Y, Ren J. ALDH2 in alcoholic heart diseases: molecular mechanism and clinical implications. Pharmacol Ther. 2011;132:86–95.
Kimball TH, Vondriska TM. Metabolism, epigenetics, and causal inference in heart failure. Trends Endocrinol Metab. 2020;31:181–91.
Wold LE, Ren J. Mechanical measurement of contractile function of isolated ventricular myocytes. Methods Mol Med. 2007;139:263–70.
Li SY, Golden KL, Jiang Y, Wang GJ, Privratsky JR, Zhang X, et al. Inhibition of sarco(endo)plasmic reticulum Ca2+-ATPase differentially regulates contractile function in cardiac myocytes from normotensive and spontaneously hypertensive rats: role of Ca2+ regulatory proteins. Cell Biochem Biophys. 2005;42:1–12.
Ren J, Zhang X, Scott GI, Esberg LB, Ren BH, Culver B, et al. Adenovirus gene transfer of recombinant endothelial nitric oxide synthase enhances contractile function in ventricular myocytes. J Cardiovasc Pharmacol. 2004;43:171–7.
Ceylan-Isik AF, Ren J. Gender difference in cardiac excitation-contraction coupling: role of estrogen. Cardiology. 2005;1:1–7.
Qiu Z, Wei Y, Song Q, Du B, Wang H, Chu Y, et al. The role of myocardial mitochondrial quality control in heart failure. Front Pharmacol. 2019;10:1404.
Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res. 2018;122:1460–78.
Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128:3716–26.
Aimo A, Borrelli C, Vergaro G, Piepoli MF, Caterina AR, Mirizzi G, et al. Targeting mitochondrial dysfunction in chronic heart failure: current evidence and potential approaches. Curr Pharmacol Des. 2016;22:4807–22.
Liang X, Wang S, Wang L, Ceylan AF, Ren J, Zhang Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol Res. 2020;157:104846.
Pang JJ, Barton LA, Chen YG, Ren J. Mitochondrial aldehyde dehydrogenase in myocardial ischemia-reperfusion injury: from bench to bedside. Sheng Li Xue Bao. 2015;67:535–44.
Dey S, DeMazumder D, Sidor A, Foster DB, O'Rourke B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ Res. 2018;123:356–71.
Chaanine AH, Sreekumaran Nair K, Bergen RH, 3rd, Klaus K, Guenzel AJ, Hajjar RJ, et al. Mitochondrial integrity and function in the progression of early pressure overload-induced left ventricular remodeling. J Am Heart Assoc. 2017;6:e005869.
Wang S, Ren J. Refining the role of autophagy in hypertrophic cardiomyopathy. Int J Cell Sci Mol Biol. 2018;4:555637.
Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–46.
Luo M, Anderson ME. Mechanisms of altered Ca2+ handling in heart failure. Circ Res. 2013;113:690–708.
Tan Y, Chen S, Zhong J, Ren J, Dong M. Mitochondrial injury and targeted intervention in septic cardiomyopathy. Curr Pharmacol Des. 2019;25:2060–70.
Cao JL, Adaniya SM, Cypress MW, Suzuki Y, Kusakari Y, Jhun BS, et al. Role of mitochondrial Ca2+ homeostasis in cardiac muscles. Arch Biochem Biophys. 2019;663:276–87.
Smith GL, Eisner DA. Calcium buffering in the heart in health and disease. Circulation. 2019;139:2358–71.
Kohlhaas M, Maack C. Calcium release microdomains and mitochondria. Cardiovasc Res. 2013;98:259–68.
Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW 2nd, Kitsis RN, et al. Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ Res. 2016;118:1960–91.
Clapham DE. Calcium signaling. Cell. 2007;131:1047–58.
Dong F, Taylor MM, Samson WK, Ren J. Intermedin (adrenomedullin-2) enhances cardiac contractile function via a protein kinase C- and protein kinase A-dependent pathway in murine ventricular myocytes. J Appl Physiol (1985). 2006;101:778–84.
Mulier M, Vriens J, Voets T. TRP channel pores and local calcium signals. Cell Calcium. 2017;66:19–24.
Bano D, Ankarcrona M. Beyond the critical point: an overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci Lett. 2018;663:79–85.
Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest. 2013;123:46–52.
Giorgi C, Danese A, Missiroli S, Patergnani S, Pinton P. Calcium dynamics as a machine for decoding signals. Trends Cell Biol. 2018;28:258–73.
Beutner G, Sharma VK, Lin L, Ryu SY, Dirksen RT, Sheu SS. Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochim Biophys Acta. 2005;1717:1–10.
Altschafl BA, Beutner G, Sharma VK, Sheu SS, Valdivia HH. The mitochondrial ryanodine receptor in rat heart: a pharmaco-kinetic profile. Biochim Biophys Acta. 2007;1768:1784–95.
Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–60.
Dedkova EN, Blatter LA. Mitochondrial Ca2+ and the heart. Cell Calcium. 2008;44:77–91.
Hajnoczky G, Csordas G. Calcium signalling: fishing out molecules of mitochondrial calcium transport. Curr Biol. 2010;20:R888–91.
Messina A, Reina S, Guarino F, De Pinto V. VDAC isoforms in mammals. Biochim Biophys Acta. 2012;1818:1466–76.
Camara AKS, Zhou Y, Wen PC, Tajkhorshid E, Kwok WM. Mitochondrial VDAC1: a key gatekeeper as potential therapeutic target. Front Physiol. 2017;8:460.
Mitra A, Basak T, Datta K, Naskar S, Sengupta S, Sarkar S. Role of alpha-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 2013;4:e582.
Paillard M, Tubbs E, Thiebaut PA, Gomez L, Fauconnier J, Da Silva CC, et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation. 2013;128:1555–65.
Yu Z, Chen R, Li M, Yu Y, Liang Y, Han F, et al. Mitochondrial calcium uniporter inhibition provides cardioprotection in pressure overload-induced heart failure through autophagy enhancement. Int J Cardiol. 2018;271:161–8.
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–5.
Yamamoto T, Ozono M, Watanabe A, Maeda K, Nara A, Hashida M, et al. Functional analysis of coiled-coil domains of MCU in mitochondrial calcium uptake. Biochim Biophys Acta Bioenerg. 2019;1860:148061.
Paillard M, Csordas G, Huang KT, Varnai P, Joseph SK, Hajnoczky G. MICU1 interacts with the D-Ring of the MCU pore to control its Ca2+ flux and sensitivity to Ru360. Mol Cell. 2018;72:778–85 e3.
Bhosale G, Sharpe JA, Koh A, Kouli A, Szabadkai G, Duchen MR. Pathological consequences of MICU1 mutations on mitochondrial calcium signalling and bioenergetics. Biochim Biophys Acta Mol Cell Res. 2017;1864:1009–17.
Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, et al. MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 2016;16:1561–73.
Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, et al. MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 2016;15:1673–85.
Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell. 2014;53:726–37.
De La Fuente S, Lambert JP, Nichtova Z, Fernandez Sanz C, Elrod JW, Sheu SS, et al. Spatial separation of mitochondrial calcium uptake and extrusion for energy-efficient mitochondrial calcium signaling in the heart. Cell Rep. 2018;24:3099–107 e4.
Garbincius JF, Luongo TS, Elrod JW. The debate continues - What is the role of MCU and mitochondrial calcium uptake in the heart? J Mol Cell Cardiol. 2020;143:163–74.
Zaglia T, Ceriotti P, Campo A, Borile G, Armani A, Carullo P, et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc Natl Acad Sci USA. 2017;114:E9006–15.
Suarez J, Cividini F, Scott BT, Lehmann K, Diaz-Juarez J, Diemer T, et al. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J Biol Chem. 2018;293:8182–95.
Michels G, Khan IF, Endres-Becker J, Rottlaender D, Herzig S, Ruhparwar A, et al. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation. 2009;119:2435–43.
Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22.
Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci USA. 2015;112:9129–34.
Holmstrom KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol. 2015;85:178–82.
Buntinas L, Gunter KK, Sparagna GC, Gunter TE. The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim Biophys Acta. 2001;1504:248–61.
Gunter TE, Buntinas L, Sparagna GC, Gunter KK. The Ca2+ transport mechanisms of mitochondria and Ca2+ uptake from physiological-type Ca2+ transients. Biochim Biophys Acta. 1998;1366:5–15.
Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem. 2001;276:21482–8.
Kohlhaas M, Nickel AG, Maack C. Mitochondrial energetics and calcium coupling in the heart. J Physiol. 2017;595:3753–63.
Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA. 2010;107:436–41.
Rottenberg H, Marbach M. The Na+-independent Ca2+ efflux system in mitochondria is a Ca2+/2H+ exchange system. FEBS Lett. 1990;274:65–8.
Boyman L, Williams GS, Khananshvili D, Sekler I, Lederer WJ. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol. 2013;59:205–13.
Parnis J, Montana V, Delgado-Martinez I, Matyash V, Parpura V, Kettenmann H, et al. Mitochondrial exchanger NCLX plays a major role in the intracellular Ca2+ signaling, gliotransmission, and proliferation of astrocytes. J Neurosci. 2013;33:7206–19.
Palty R, Sekler I. The mitochondrial Na+/Ca2+ exchanger. Cell Calcium. 2012;52:9–15.
Shao Q, Ren B, Elimban V, Tappia PS, Takeda N, Dhalla NS. Modification of sarcolemmal Na+-K+-ATPase and Na+/Ca2+ exchanger expression in heart failure by blockade of renin-angiotensin system. Am J Physiol Heart Circ Physiol. 2005;288:H2637–46.
Kostic M, Sekler I. Functional properties and mode of regulation of the mitochondrial Na+/Ca2+ exchanger, NCLX. Semin Cell Dev Biol. 2019;94:59–65.
Sipido KR, Volders PG, Vos MA, Verdonck F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc Res. 2002;53:782–805.
Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, et al. The mitochondrial Na+/Ca2+exchanger is essential for Ca2+ homeostasis and viability. Nature. 2017;545:93–7.
Lin QT, Stathopulos PB. Molecular mechanisms of leucine zipper EF-hand containing transmembrane protein-1 function in health and disease. Int J Mol Sci 2019;20:286.
Shao J, Fu Z, Ji Y, Guan X, Guo S, Ding Z, et al. Leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) forms a Ca2+/H+ antiporter. Sci Rep. 2016;6:34174.
Froschauer E, Nowikovsky K, Schweyen RJ. Electroneutral K+/H+ exchange in mitochondrial membrane vesicles involves Yol027/Letm1 proteins. Biochim Biophys Acta. 2005;1711:41–8.
Li Y, Tran Q, Shrestha R, Piao L, Park S, Park J, et al. LETM1 is required for mitochondrial homeostasis and cellular viability (Review). Mol Med Rep. 2019;19:3367–75.
Romero-Garcia S, Prado-Garcia H. Mitochondrial calcium: transport and modulation of cellular processes in homeostasis and cancer (Review). Int J Oncol. 2019;54:1155–67.
Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Fiolet JW. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–24.
Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Opthof T, et al. Chronic inhibition of Na+/H+-exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc Res. 2005;65:83–92.
Baartscheer A, Hardziyenka M, Schumacher CA, Belterman CN, van Borren MM, Verkerk AO, et al. Chronic inhibition of the Na+/H+ - exchanger causes regression of hypertrophy, heart failure, and ionic and electrophysiological remodelling. Br J Pharmacol. 2008;154:1266–75.
Lopez-Crisosto C, Pennanen C, Vasquez-Trincado C, Morales PE, Bravo-Sagua R, Quest AFG, et al. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat Rev Cardiol. 2017;14:342–60.
Zhou H, Wang S, Hu S, Chen Y, Ren J. ER-mitochondria microdomains in cardiac ischemia-reperfusion injury: a fresh perspective. Front Physiol. 2018;9:755. https://doi.org/10.3389/fphys.2018.00755.
Silva-Palacios A, Zazueta C, Pedraza-Chaverri J. ER membranes associated with mitochondria: possible therapeutic targets in heart-associated diseases. Pharmacol Res. 2020;156:104758.
Dorn GW 2nd, Maack C. SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol. 2013;55:42–9.
De la Fuente S, Sheu SS. SR-mitochondria communication in adult cardiomyocytes: a close relationship where the Ca2+ has a lot to say. Arch Biochem Biophys. 2019;663:259–68.
Ruiz-Meana M, Fernandez-Sanz C, Garcia-Dorado D. The SR-mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc Res. 2010;88:30–9.
Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–11.
Garcia-Rua V, Otero MF, Lear PV, Rodriguez-Penas D, Feijoo-Bandin S, Noguera-Moreno T, et al. Increased expression of fatty-acid and calcium metabolism genes in failing human heart. PLoS ONE. 2012;7:e37505.
Seidlmayer LK, Kuhn J, Berbner A, Arias-Loza PA, Williams T, Kaspar M, et al. Inositol 1,4,5-trisphosphate-mediated sarcoplasmic reticulum-mitochondrial crosstalk influences adenosine triphosphate production via mitochondrial Ca2+ uptake through the mitochondrial ryanodine receptor in cardiac myocytes. Cardiovasc Res. 2016;112:491–501.
Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, et al. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res. 2012;111:1012–26.
Eisner V, Cupo RR, Gao E, Csordas G, Slovinsky WS, Paillard M, et al. Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc Natl Acad Sci USA. 2017;114:E859–E68.
Ferreira JCB, Campos JC, Qvit N, Qi X, Bozi LHM, Bechara LRG, et al. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat Commun. 2019;10:329. https://doi.org/10.1038/s41467-018-08276-6.
Tang H, Tao A, Song J, Liu Q, Wang H, Rui T. Doxorubicin-induced cardiomyocyte apoptosis: role of mitofusin 2. Int J Biochem Cell Biol. 2017;88:55–9.
Sun D, Li C, Liu J, Wang Z, Liu Y, Luo C, et al. Expression profile of microRNAs in hypertrophic cardiomyopathy and effects of microRNA-20 in inducing cardiomyocyte hypertrophy through regulating gene MFN2. DNA Cell Biol. 2019;38:796–807.
Song M, Franco A, Fleischer JA, Zhang L, Dorn GW 2nd. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 2017;26:872–83 e5.
Seidlmayer LK, Mages C, Berbner A, Eder-Negrin P, Arias-Loza PA, Kaspar M, et al. Mitofusin 2 is essential for IP3-mediated SR/mitochondria metabolic feedback in ventricular myocytes. Front Physiol. 2019;10:733.
Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12:689–702.
Cooley N, Ouyang K, McMullen JR, Kiriazis H, Sheikh F, Wu W, et al. No contribution of IP3-R(2) to disease phenotype in models of dilated cardiomyopathy or pressure overload hypertrophy. Circ Heart Fail. 2013;6:318–25.
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, et al. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. 2017;136:2248–66.
Qiao X, Jia S, Ye J, Fang X, Zhang C, Cao Y, et al. PTPIP51 regulates mouse cardiac ischemia/reperfusion through mediating the mitochondria-SR junction. Sci Rep. 2017;7:45379.
Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF, et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun. 2014;5:3996.
Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, et al. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 2012;31:457–70.
Abdullah CS, Alam S, Aishwarya R, Miriyala S, Panchatcharam M, Bhuiyan MAN, et al. Cardiac dysfunction in the sigma 1 receptor knockout mouse associated with impaired mitochondrial dynamics and bioenergetics. J Am Heart Assoc. 2018;7:e009775.
Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell. 2015;32:304–17.
Min CK, Yeom DR, Lee KE, Kwon HK, Kang M, Kim YS, et al. Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem J. 2012;447:371–9.
Lombardi AA, Gibb AA, Arif E, Kolmetzky DW, Tomar D, Luongo TS, et al. Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat Commun. 2019;10:4509.
Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med (Berl). 2010;88:993–1001.
Boyman L, Karbowski M, Lederer WJ. Regulation of mitochondrial ATP production: Ca2+signaling and quality control. Trends Mol Med. 2020;26:21–39.
Griffiths EJ, Balaska D, Cheng WH. The ups and downs of mitochondrial calcium signalling in the heart. Biochim Biophys Acta. 2010;1797:856–64.
Hempel N, Trebak M. Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium. 2017;63:70–96.
Wang S, Guo W, Ren J. Stress signaling in paraquat-induced target organ toxicity. React Oxyg Species. 2016;1:131–40.
Wold LE, Ceylan-Isik AF, Ren J. Oxidative stress and stress signaling: menace of diabetic cardiomyopathy. Acta Pharmacol Sin. 2005;26:908–17.
Xu H, Linn BS, Zhang Y, Ren J. A review on the antioxidative and prooxidative properties of luteolin. React Oxyg Species. 2019;7:136–47.
Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016;2:e84897. https://doi.org/10.1172/jci.insight.84897.
Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. 2016;134:883–94.
Yi M, Weaver D, Hajnoczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol. 2004;167:661–72.
He Q, Harris N, Ren J, Han X. Mitochondria-targeted antioxidant prevents cardiac dysfunction induced by tafazzin gene knockdown in cardiac myocytes. Oxid Med Cell Longev. 2014;2014:654198.
Aroor AR, Mandavia C, Ren J, Sowers JR, Pulakat L. Mitochondria and oxidative stress in the cardiorenal metabolic syndrome. Cardiorenal Med. 2012;2:87–109.
Wu NN, Tian H, Chen P, Wang D, Ren J, Zhang Y. Physical exercise and selective autophagy: benefit and risk on cardiovascular health. Cells. 2019;8:1436. https://doi.org/10.3390/cells8111436.
Wu NN, Zhang Y, Ren J. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid Med Cell Longev. 2019;2019:9825061.
Zhou H, Toan S, Zhu P, Wang J, Ren J, Zhang Y. DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Res Cardiol. 2020;115:11.
Hajnoczky G, Saotome M, Csordas G, Weaver D, Yi M. Calcium signalling and mitochondrial motility. Novartis Found Symp. 2007;287:105–17. discussion17-21
Tong M, Zablocki D, Sadoshima J. The role of Drp1 in mitophagy and cell death in the heart. J Mol Cell Cardiol. 2020;142:138–45.
Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–44.
Fedida D. Modulation of cardiac contractility by alpha 1 adrenoceptors. Cardiovasc Res. 1993;27:1735–42.
Pennanen C, Parra V, Lopez-Crisosto C, Morales PE, Del Campo A, Gutierrez T, et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J Cell Sci. 2014;127:2659–71.
Moyzis AG, Sadoshima J, Gustafsson AB. Mending a broken heart: the role of mitophagy in cardioprotection. Am J Physiol Heart Circ Physiol. 2015;308:H183–92.
Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA. 2011;108:9572–7.
Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308.
Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–26.
Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;301:H1924–31.
Nah J, Miyamoto S, Sadoshima J. Mitophagy as a protective mechanism against myocardial stress. Compr Physiol. 2017;7:1407–24.
Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–63.
Xu H, Yu W, Sun S, Li C, Zhang Y, Ren J. Luteolin attenuates doxorubicin-induced cardiotoxicity through promoting mitochondrial autophagy. Front Physiol. 2020;11:113.
Missiroli S, Bonora M, Patergnani S, Poletti F, Perrone M, Gafa R, et al. PML at mitochondria-associated membranes is critical for the repression of autophagy and cancer development. Cell Rep. 2016;16:2415–27.
Rimessi A, Bonora M, Marchi S, Patergnani S, Marobbio CM, Lasorsa FM, et al. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy. 2013;9:1677–86.
Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA. 2008;105:20728–33.
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906.
Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J Biochem Cell Biol. 2009;41:2015–24.
Cali T, Ottolini D, Negro A, Brini M. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012;287:17914–29.
Vercesi AE, Castilho RF, Kowaltowski AJ, de Oliveira HCF, de Souza-Pinto NC, Figueira TR, et al. Mitochondrial calcium transport and the redox nature of the calcium-induced membrane permeability transition. Free Radic Biol Med. 2018;129:1–24.
Zhang Y, Xia Z, La Cour KH, Ren J. Activation of Akt rescues endoplasmic reticulum stress-impaired murine cardiac contractile function via glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeation pore opening. Antioxid Redox Signal. 2011;15:2407–24.
Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–73.
Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–70.
Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, et al. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006;40:553–60.
Leo S, Bianchi K, Brini M, Rizzuto R. Mitochondrial calcium signalling in cell death. FEBS J. 2005;272:4013–22.
Zecchini E, Siviero R, Giorgi C, Rizzuto R, Pinton P. Mitochondrial calcium signalling: message of life and death. Ital J Biochem. 2007;56:235–42.
Volkmann N, Marassi FM, Newmeyer DD, Hanein D. The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ. 2014;21:206–15.
Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, et al. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013;20:1631–43.
Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–44.
Mano A, Tatsumi T, Shiraishi J, Keira N, Nomura T, Takeda M, et al. Aldosterone directly induces myocyte apoptosis through calcineurin-dependent pathways. Circulation. 2004;110:317–23.
Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–61.
Khan MU, Cheema Y, Shahbaz AU, Ahokas RA, Sun Y, Gerling IC, et al. Mitochondria play a central role in nonischemic cardiomyocyte necrosis: common to acute and chronic stressor states. Pflug Arch. 2012;464:123–31.
Rasola A, Bernardi P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium. 2011;50:222–33.
Gabisonia K, Recchia FA. Gene therapy for heart failure: new perspectives. Curr Heart Fail Rep. 2018;15:340–9.
Acknowledgements
This work is supported in part by National Natural Science Foundation of China (grants 81770261 and 91749128).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Xu, Hx., Cui, Sm., Zhang, Ym. et al. Mitochondrial Ca2+ regulation in the etiology of heart failure: physiological and pathophysiological implications. Acta Pharmacol Sin 41, 1301–1309 (2020). https://doi.org/10.1038/s41401-020-0476-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0476-5
Keywords
This article is cited by
-
SGLT2 inhibitor empagliflozin alleviates cardiac remodeling and contractile anomalies in a FUNDC1-dependent manner in experimental Parkinson’s disease
Acta Pharmacologica Sinica (2024)
-
SZC-6, a small-molecule activator of SIRT3, attenuates cardiac hypertrophy in mice
Acta Pharmacologica Sinica (2023)
-
The BCL-2 family protein BCL-RAMBO interacts and cooperates with GRP75 to promote its apoptosis signaling pathway
Scientific Reports (2023)
-
Changes of energy metabolism in failing heart and its regulation by SIRT3
Heart Failure Reviews (2023)
-
Double-activation of mitochondrial permeability transition pore opening via calcium overload and reactive oxygen species for cancer therapy
Journal of Nanobiotechnology (2022)
