
Abstract
PARP inhibitors are a group of inhibitors targeting poly(ADP-ribose) polymerases (PARP1 or PARP2) involved in DNA repair and transcriptional regulation, which may induce synthetic lethality in BRCAness tumors. Systematic analyzes of genomic sequencing in prostate cancer show that ~10%–19% of patients with primary prostate cancer have inactivated DNA repair genes, with a notably higher proportion of 23%–27% in patients with metastatic castration-resistant prostate cancer (mCRPC). These characteristic genomic alterations confer possible vulnerability to PARP inhibitors in patients with mCRPC who benefit only modestly from other therapies. However, only a small proportion of patients with mCRPC shows sensitivity to PARP inhibitors, and these sensitive patients cannot be fully identified by existing response prediction biomarkers. In this review, we provide an overview of the potential response prediction biomarkers and synergistic combinations studied in the preclinical and clinical stages, which may expand the population of patients with prostate cancer who may benefit from PARP inhibitors.
Similar content being viewed by others
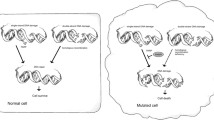

Introduction
Prostate cancer is the most common malignancy and the second most common cause of death among so-called “male cancers.” According to the most recent estimates from the American Cancer Society, there will be ~191,930 new cases of prostate cancer (21% of new cases of male cancers) and 33,330 associated deaths (10% of deaths from male cancers) in the United States in 2020 [1]. Although it is possible to cure prostate cancer in patients with early-stage disease, patients with metastatic disease have poor outcomes. The overall survival of patients with metastatic prostate cancer has not improved over the past 20 years [2].
Since its efficacy was first reported in 1941, androgen deprivation therapy (ADT) has remained the initial treatment for metastatic prostate cancer [3]. However, patients will inevitably progress to a condition that adapts to castration with continuous androgen deprivation, which is termed castration-resistant prostate cancer (CRPC). Although some new therapies have emerged, such as sipuleucel-T, cabazitaxel, abiraterone acetate, enzalutamide, and radium-223, they have conferred only modest benefits to the overall survival [4,5,6,7]. Meanwhile, PARP inhibitors have shown significant benefits in patients with ovarian cancer with BRCA1/2 mutations [8, 9]. In addition, systematic genomic analyzes revealed the enrichment of mutations in the DNA repair pathway in metastatic castration-resistant prostate cancer (mCRPC), implying that patients with mCRPC were vulnerable to PARP inhibitors [10].
PARPs are involved in DNA repair and transcriptional regulation by controlling the pathways needed for prostate malignancy [11]. Moreover, PARP-1 enzymatic activity is elevated during prostate cancer progression and is associated with poor outcomes [12]. Therefore, PARPs are potential targets to treat prostate cancer. PARP inhibitors are a group of inhibitors targeting poly(ADP-ribose) polymerases (PARP1 and PARP2). These drugs reduce the enzymatic activity of PARP and induce double-strand breaks (DSBs) by DNA trapping. The action of PARP inhibitors could induce synthetic lethality in patients with homologous recombination deficiency (HRD). The first PARP inhibitor to be granted a breakthrough therapy designation by the food and drug administration (FDA) for patients with mCRPC was olaparib [10]. Subsequently, other PARP inhibitors—rucaparib and niraparib—were granted breakthrough therapy designation by the FDA for patients with mCRPC with BRCA1/2 mutations in 2018 and 2019, respectively [13, 14]. In addition, to overcome the limitations of the application of PARP inhibitors in patients with HRD, the combination of specific agents offers a new therapeutic strategy. Notably, the combination of abiraterone and olaparib was reported to prolong progression-free survival (PFS), regardless of the mutation status of homologous recombination (HR) repair, in patients with mCRPC [15]. Abiraterone and olaparib mutually increase the efficacy of each other. Although the efficacy of PARP inhibitors in patients with mCRPC has been reported in various studies, the population of patients with prostate cancer who benefit from PARP inhibitors is limited by the lack of biomarkers for the identification of sensitive patients and the intrinsic resistance of the remaining patients.
The main focus of this review is to summarize two options used to widen the applicability of PARP inhibitors: (1) biomarkers, other than BRCA1/2 mutations, to predict response; and (2) synergistic combinations of drugs to induce HRD. The studies discussed are still in the preclinical and clinical stages; hence, they have the potential to facilitate the wider use of PARP inhibitors.
Mechanism of PARP inhibition
PARPs are a family of proteins that participate in the DNA damage response (DDR), genomic stability, and programmed cell death and include PARP1 (the more important protein) and PARP2 [16]. PARP1 and PARP2 are both molecular sensors and signal transducers of DNA breaks [17]. When damage occurs in a DNA strand, it is detected by PARP1 binding. After binding to single-strand breaks (SSBs), PARP1 exerts catalytic activity with the cofactor β-NAD+ and induces PARylation of substrate proteins, which promotes the recruitment of DNA repair effectors. Subsequently, DNA repair effectors mediate DNA repair after PARP1 is released from DNA by autoPARylation. Meanwhile, PARP1 returns to a catalytically inactivated state [18,19,20].
The main function of PARP inhibitors is to bind the catalytic domain of PARP, which blocks a PARP protein from the SSB site and reduces its catalytic activity. In addition, PARP inhibitors hinder the process of DNA repair and finally induce toxic DSBs [21]. Although the DSBs induced by PARP inhibitors could be repaired by competent DSB repair capacity in HR-proficient cells, in HR-deficient cells, DSBs induced by PARP inhibitors remain unrepaired and eventually cause programmed cell death. This process, a typical example of synthetic lethality, is the rationale for the antitumor effect of PARP inhibitors [22, 23].
In addition, as a ubiquitously expressed nuclear enzyme, PARP1 is involved in many biological functions, such as genomic stability, programmed cell death, transcriptional regulation, and chromatin structure modulation [17, 24,25,26]. The role of PARP1 in genomic stability is summarized in Fig. 1a. The increased genomic instability caused by the inhibition of PARP1 promotes the cytotoxicity of PARP inhibitors. Furthermore, there is an increasing body of evidence to support the role of PARP1 in the function of the androgen receptor (AR) and erythroblast transformation-specific (ETS) proteins [24, 27]. Therefore, the anticancer effects of PARP inhibitors are also mediated by impairing the functions of AR and ETS. A schematic overview of the antitumor effect induced by PARP inhibitors is shown in Fig. 1.

a PARP1 participates in multiple cellular processes, such as DNA repair, transcription, chromatin modification, and DNA replication. These roles facilitate genomic stability. b The interaction of PARP inhibitors and genetic aberrations in double-strand breaks (DSBs). PARP inhibitors induce DSBs by trapping PARP1 on DNA. The induced DSBs can be repaired via the HR or NHEJ pathway. Alternatively, impaired DNA repair function, especially impaired HR function, promotes the accumulation of DNA lesions and ultimately leads to cell death. Impaired DNA repair function is due to gene alterations such as BRCA1/2 mutations, ATM alterations, and CHD1 deletion. Androgen receptor inhibition (ADi) also exerts a synergistic effect with PARP inhibitors by impairing DNA repair function. c Synthetic lethality can be induced by PARP inhibitors under specific conditions. In HR-proficient cells, the DSBs induced by PARP inhibitors can be repaired by the proficient HR function. These types of cells are resistant to PARP inhibitors. However, alterations in genes associated with the HR function or the addition of a specific combination agent may induce cells toward the HRD phenotype. Under these conditions, PARP inhibitors can induce lethality. SSB single-strand break, SSBR/BER single-strand break repair/base excision repair, MMEJ microhomology-mediated end joining, NHEJ non-homologous end-joining, HR homologous recombination, HRD homologous recombination.
Clinical trials of PARP inhibitors in prostate cancer
The efficacy of PARP inhibitors in prostate cancer was first reported in the phase I clinical trial of olaparib [9]. This trial included three patients with CRPC and the patient with a BRCA2 mutation showed a 50% decline in the level of prostate-specific antigen (PSA) and a decrease in bone metastases. Furthermore, the efficacy of PARP inhibitors in prostate cancer was assessed in a landmark trial named TOPARP-A (trial of olaparib in patients with advanced castration-resistant prostate cancer), which resulted in the breakthrough therapy designation by the FDA for the treatment of patients with mCRPC with BRCA1/2 or ATM mutations [10]. TOPARP-A was a phase II trial that included 49 evaluable patients with CRPC. In the results of this trial, a 33% (16/49) overall response rate (RR) in unselected patients with CRPC was reported; remarkably, 88% (14/16) of responders were biomarker-positive. In contrast, only 6% (2/33) of biomarker-negative patients responded to olaparib. Among the 16 biomarker-positive patients, all patients with BRCA1/2 germline or somatic alterations responded to olaparib; this accounted for the major proportion of biomarker-positive patients who exhibited a response (8 of 14). This suggested that BRCA1/2 mutants were stronger biomarkers for patient stratification. In addition, 4 of 5 patients with ATM alterations were sensitive to olaparib treatment, and patients with alterations in FANCA, CHEK2, PALB2, HDAC2, and other HR-related genes also responded to olaparib. To validate the biomarkers identified in TOPARP-A, the TOPARP-B study enrolled 98 patients with DDR gene alterations to receive treatment with olaparib [28]. Approximately 83.3% (25/30) of the patients with BRCA1/2 mutations showed a composite overall response, which confirmed the superior predictive power of BRCA1/2 mutations. The response was also identified in other subpopulations, such as patients with PALB2 alterations. However, in contrast to the results of TOPARP-A, only 10.5% (2/19) of the patients with ATM aberrations achieved a RECIST or PSA response in TOPARP-B (enrolled 98 patients), and none of the five patients with ATM aberrations in TRITON2 (enrolled 85 patients) achieved a clinical response [13, 28]. This suggested that the prediction power of ATM alterations alone was not sufficient to identify a population of patients who would benefit from this treatment. Furthermore, PARP inhibitors have been studied in phase III trials, including PROfound and TRITON3, as first-line choices in comparison with AR inhibitors such as abiraterone acetate or enzalutamide. Recent results from PROfound showed significantly better outcomes in patients treated with olaparib than in patients treated with either enzalutamide or abiraterone [29].
In addition to monotherapy with PARP inhibitors, they have also been evaluated in combination with other agents or therapies, such as chemotherapy, radiation therapy, and immunotherapy. As shown in previous preclinical studies, PARP inhibitors could enhance the efficacy of DNA-damaging therapies, such as chemotherapy and radiation therapy, by inhibiting the repair of DNA damage induced by chemotherapy and the growth of tumor cells or by increasing the number of trapped DNA-PARP complexes (exclusive to temozolomide) [30]. However, a pilot study to assess the safety and efficacy of a combination of veliparib with temozolomide showed that the combination was well tolerated in patients with prostate cancer but with less benefit [31]. The low dose and low trapping activity of veliparib may limit the efficacy of the combination. Studies of other combinations of PARP inhibitors and chemotherapy were established as maintenance after several cycles of chemotherapy to avoid major hematologic toxicity and are still ongoing, as are trials of combination therapy with radiation [32].
In contrast, the combination of AR-targeting drugs and PARP inhibitors showed outstanding synergistic efficacy. A phase II trial showed that the combination of abiraterone and olaparib prolonged median PFS from 8.2 months to 13.8 months, regardless of mutation status in the HR pathway, in patients with mCRPC [15]. Further trials, including PROpel, are ongoing to validate the efficacy of the combination of abiraterone and olaparib as first-line therapy. In addition, the combination of PARP inhibitors with emerging immunotherapy has opened up new avenues to explore. Cohort A of a phase I/II study (NCT02484404) preliminarily enrolled 17 patients with mCRPC who were treated with durvalumab and olaparib [33]. This study demonstrated the efficacy of this combination therapy, particularly in patients with DDR aberrations. A further study, KEYNOTE-365, enrolled 41 patients with no DDR aberrations to study the efficacy of the combination of olaparib and pembrolizumab [34]. The results of the trial showed a composite RR of ~15%. Based on this, an ongoing phase III trial (KEYLYNK-010) is comparing the efficacy of pembrolizumab plus olaparib to abiraterone acetate or enzalutamide. In addition, a phase II trial is examining whether olaparib combined with durvalumab could replace the standard treatment for prostate cancer. Another trial focused on whether cetrelimab or abiraterone is the better choice for combination therapy with niraparib (NCT03431350). A summary of ongoing phase II and/or III clinical trials on PARP inhibitors is presented in Table 1.
As reported in the TOPARP-A trial, 2 of the 16 responders could not be recognized using the biomarkers included in this trial. To expand the population of patients with prostate cancer who will benefit from PARP inhibitors, the current target is to discover sensitive response prediction biomarkers. Moreover, patients with intrinsic resistance to PARP inhibitors may also benefit from the combination of PARP inhibitors with synergistic drugs that induce the HRD phenotype.
Response prediction biomarkers
In the past decade, several genomic studies have been conducted on prostate cancer. Unlike other common cancers, prostate cancer was found to have few mutations (0.7 per Mb) [35]. However, mutations occurred more frequently in mCRPC (4.4 per Mb) [36]. Furthermore, it was found that recurrent mutations of mCRPC belong to the DNA repair pathway, and the frequency of somatic mutations in the DNA repair pathway in mCRPC is much higher than that in primary prostate cancer (23%–27% vs. 10%–19%) [36, 37]. These characteristics of genomic alterations in mCRPC may enhance the antitumor effect of PARP inhibitors. Alterations in the DNA repair pathway or those affecting the function of DNA repair, which may be response prediction biomarkers for treatment with PARP inhibitors, are summarized in Table 2. The interactions of PARP inhibitors and genetic aberrations in DSBs are summarized in Fig. 1b.
BRCA1/2 mutations
Both BRCA1 and BRCA2 play key roles in different processes in HR. BRCA1 is involved in ssDNA formation at the DSB site and then joins the RAD51 recombinase onto the forming ssDNA [38]. BRCA2 participates in joining RAD51 onto ssDNA [39]. BRCA1/2 mutations cause a deficiency in HR and lead to synthetic lethality with PARP inhibitors. They are the most explicit response prediction biomarkers, and their detection is necessary not only in patients with mCRPC but also in patients with primary prostate cancer. For example, a patient with mCRPC who exhibited remarkable radiological and biochemical responses to the PARP inhibitor veliparib was retrospectively detected with somatic BRCA2 biallelic (homozygous) loss in the primary tumor, which was collected 18 months before progression to the CRPC stage [40]. Progression to mCRPC may be delayed if a BRCA1/2 mutation assay is performed on the primary tumor and earlier treatment with a PARP inhibitor is applied. The first BRCA1/2 mutation assay, known as the Myriad BRACAnalysis CDx® assay, was approved for detecting a germline BRCA1/2 mutation for the indication of olaparib by the FDA. Another assay, named FOUNDATIONFOCUS™ CDxBRCA, was approved by the FDA as a companion diagnostic test for rucaparib. This is the first assay based on next-generation sequencing (NGS) and was able to detect both germline and somatic BRCA1/2 mutations for the precise administration of a PARP inhibitor.
Ataxia-telangiectasia mutated (ATM) alterations
ATM is a DNA damage sensor that is recruited to DSB lesions and promotes G1/S cell cycle arrest by the activation of CHK2 and the stabilization of p53 [41]. It was reported that ATM inactivation increased sensitivity to PARP inhibitors, as mediated by mitotic catastrophe, independent of apoptosis in lymphoid tumor cells [42]. In Mateo’s study, 4 of 5 patients with CRPC with ATM alterations presented sensitivity to PARP inhibitors [10]. However, recent preclinical and clinical studies found that ATM-deficient tumors did not truly benefit from PARP inhibitors and were much more sensitive to the combination of ATR inhibitors and PARP inhibitors [28, 43,44,45]. This may be because ATM functions as a DNA damage sensor that does not directly execute DNA repair.
Speckle-type POZ (SPOP) mutations
The SPOP protein is a component of the cullin-3-based ubiquitin ligase complex that ubiquitinates target proteins and was demonstrated to be involved in the DDR [46]. Recent genomic analyzes have shown that SPOP is the most frequently mutated gene and that the frequency of SPOP mutations is 11%–13% in patients with prostate cancer [47, 48]. SPOP mutations are exclusive to ETS family gene rearrangements and are enriched with rearrangement-associated deletions such as CHD1 deletion [47, 48]. Moreover, a study demonstrated that SPOP mutations were associated with increased sensitivity to PARP inhibitors by impairing HR and facilitating nonhomologous end-joining (NHEJ) [49]. Although the underlying mechanism is unclear, SPOP mutations are potential biomarkers for PARP inhibitor therapy.
CHD1 deletion
CHD1 deletion, accompanied by SPOP mutations, occurs frequently in prostate cancer [36, 37, 47]. CHD1 equilibrates the selection of HR and NHEJ. The loss of CHD1 stabilizes 53BP1, which induces an increase in error-prone NHEJ. Meanwhile, it impedes the recruitment of HR proteins, which induces impaired error-free HR and genomic instability [50,51,52]. Consequently, a dramatic increase in genomic instability facilitates the effect of PARP inhibitors in prostate cancer. CHD1 deletion may serve as a biomarker for the treatment of PARP inhibitors.
Ribonuclease H2B (RNASEH2B) deletion
A recent CRISPR screening study found that mutations in three genes encoding ribonuclease H2 (RNASEH2A, RNASEH2B, and RNASEH2C) were associated with increased sensitivity to PARP inhibitor therapy [53]. The deletion of ribonuclease H2 leads to impaired ribonucleotide excision repair and induces another ribonucleotide excision pathway mediated by topoisomerase 1, which eventually causes genomic instability and PARP-trapping DNA lesions [54]. Significantly, the loss of RNASEH2B was found in 35.4% of patients with mCRPC [53]. The relationship between RNASEH2B deletion and PARP inhibitor sensitivity needs to be further assessed.
TMPRSS2–ERG fusion and PTEN deletion
Both TMPRSS2–ERG fusion and PTEN deletion are recurrent gene alterations that play important roles in the progression of prostate cancer [55, 56]. PTEN deletion occurs with ERG rearrangement, as the second event following ERG rearrangement [57], and was demonstrated to be sensitive to PARP inhibitors by reducing RAD51 in some sporadic tumors [58, 59]. However, PTEN deletion did not affect HR function or RAD51 recruitment and resulted in only mild sensitivity to PARP inhibitors in prostate cancer [60].
TMPRSS2–ERG fusion consists of an upstream TMPRSS2 gene promoter and a downstream ERG coding region. Among these, the TMPRSS2 gene promoter is driven by AR, and ERG is an oncogenic transcription factor. The fusion product physically interacts with PARP1 and DNA-PKcs, and the overexpressed fusion product induces DNA DSB formation, enhancing the effect of PARP inhibitors [27]. Furthermore, it was found that prostate tumors with ETS gene fusion were sensitive to PARP1 inhibition in a mouse xenograft model [27]. The combination of irradiation and PARP inhibitors appears more effective in patients with TMPRSS2-ERG fusion [61, 62]. A phase I clinical trial found that neither ETS fusion nor PTEN deletion was associated with PARP inhibition in prostate cancer [63]. Consistent with this, a phase II study reported that ETS fusions could not predict the response to PARP inhibitors [64].
Non-BRCA DDR gene alterations
Other non-BRCA DDR gene alterations may cause sensitivity to PARP inhibition by impairing HR function. For example, alterations in PALB2, RAD51B, FANCA, and BRIP1 were also potential predictive biomarkers [13, 28, 65]. However, not all DDR gene alterations, such as gene alterations in ATM, CDK12, and CHEK2, confer clear benefits from PARP inhibitors [13, 28, 65]. In addition, alterations in genes other than DDR, which may also result in increased DNA damage, lead to sensitivity to PARP inhibition. KMT2D is an epigenetic modifier, and the loss of KMT2D causes DNA damage through the accumulation of ROS and increases the sensitivity to PARP inhibitors [66].
The predictive biomarkers for gene alterations are not fully understood. To avoid ignoring alterations in other pathways that may result in HRD, biomarkers in the genomic footprint (HRD mutational signature, genomic instability) or functional assays (γH2AX-RAD51 nuclear foci formation), which directly detect the capacity of DNA repair, should be considered.
Synergistic combination strategies
In addition to determining all beneficiaries of PARP inhibitor therapy by more precise assessment, another way to increase the beneficiaries is to find synergistic combinations with PARP inhibitors. The combination of PARP inhibitors and immunotherapy or AR-targeting drugs showed great potential from clinical data, and targeted drugs such as ATR inhibitors are under clinical investigation. Meanwhile, some drugs that impair the HR function, which may induce synthetic lethality in combination with PARP inhibitors, are still in preclinical testing. Recent studies of potential synergistic combinations in the preclinical and clinical stages are summarized in Table 3.
AR inhibition
A previous study found that AR amplification occurred in 30% of recurrent tumors, whereas no amplification was found in ADT-naive primary tumors [67]. In addition to the increase in AR expression, CRPC tumors were also found to be enriched in alterations in the DNA damage repair pathway. Furthermore, crosstalk between AR and DNA damage repair was discovered recently. There are three major aspects of this process: (1) AR positively regulates the transcription of genes in different DNA repair pathways, such as HR, NHEJ, and MMR [68,69,70]; (2) there is a feedback loop between PARP1 and AR, which means that PARP1 is transcriptionally regulated by AR and is a cofactor of AR transcriptional activity [11, 69]; and (3) AR increases NHEJ capacity by inducing DNA-PKcs expression, and in turn, DNA-PKcs is required for AR transcription activity [68]. AR inhibitors induce synthetic lethality with PARP inhibitors by impairing the DNA repair function. Meanwhile, PARP inhibitors could hinder the transcriptional activity of AR, which facilitates the antitumor effect of AR inhibitors. These interactions between AR and DNA damage repair induce synthetic lethality between AR inhibitors and PARP inhibitors [71, 72]. In a phase II trial, researchers observed a significant increase in radiographic progression-free survival (rPFS) in patients with mCRPC for the combination of olaparib and AR inhibitor compared with the AR inhibitor alone [15]. However, the combination of veliparib and an AR inhibitor did not affect the treatment of abiraterone plus prednisone in PSA RR, measurable disease RR (mRR), or median PFS [64]. It was difficult to determine the cause of the discrepancies between these two trials. Part of the reason for the discrepancies may be the weaker PARP-trapping activity of veliparib. Given the limitation of the small sample size of the two trials, larger trials are needed to clarify the benefit of the combination of an AR inhibitor and a PARP inhibitor. In addition, the frequencies of serious adverse events were reported to be ~34% with the combination of olaparib and abiraterone and 18% with abiraterone alone. The increased serious adverse effects caused by the combination should be considered.
Immune checkpoint blockade
When focusing on programmed death 1/programmed death-ligand 1 (PD-1/PD-L1)-targeting antibodies in prostate cancer, researchers found that a PARP inhibitor could increase the expression of PD-L1 in tumors with wild-type or BRCA mutants and that DNA damage induced by PARP inhibitors could increase neoantigens [73,74,75,76]. This effect of the PARP inhibitor increased the therapeutic targets of immune checkpoint blockade. Based on the rationale of synergism, the combination of durvalumab and olaparib was demonstrated to be efficacious, particularly in men with DDR abnormalities [33]. In addition, the treatment of patients with no DDR aberrations with a combination of olaparib and pembrolizumab resulted in a composite RR of ~15% [34]. The combination of a PARP inhibitor and immune checkpoint blockade increased the population sensitivity to immune checkpoint inhibitors. Moreover, it may be a better choice than immune checkpoint blockade monotherapy for patients with and without DDR abnormalities.
Targeted agents
The combination of PARP inhibitors with some targeted agents is also a potential strategy. In research on ATM alterations, preclinical data showed that olaparib induced reversible G2 arrest but was not cytotoxic in ATM-deficient cell lines [77, 78]. Clinical data also showed that few patients with ATM alterations were sensitive to PARP inhibitors [13, 28]. It is necessary to explore other therapies in patients with ATM alterations [79]. Preclinical studies found that the combination of olaparib and an ATR inhibitor could induce cell death in ATM-deficient cells [77, 78]. This was considered to be mediated by the ablation of G2 arrest induced by ATR inhibitors. Furthermore, the combination was cytotoxic in BRCA-deficient cells [80]. However, the rationale behind the synergism was not clear. Therefore, ATR inhibitors may be a new combination option with PARP inhibitors for patients with ATM alterations or even HR-deficient patients [43]. The TRAP clinical trial is studying the combination of olaparib and the ATR inhibitor AZD6738, with the enrolled patients being grouped into a DNA repair-proficient cohort and a deficient cohort. Similarly, the anti-angiogenic agent cediranib could suppress the expression of BRCA1/2 and RAD51 in addition to having an anti-angiogenic effect [81]. This unexpected function in DDR proteins may induce synthetic lethality with the combination of PARP inhibitors by impairing HR function. It was reported that this combination could significantly prolong rPFS in unselected patients with mCRPC [82]. In addition, as described above, PARP inhibitors show modest efficacy in PTEN-deficient prostate cancer. It was found that olaparib exposure may superactivate the PI3K-Akt signaling pathway [83]. Consequently, the addition of a PI3K inhibitor could greatly increase the efficacy of PARP inhibitors by blocking Akt activation [83]. The efficacy of this combination has been studied in clinical trials (NCT03586661, NCT03586661).
Combination drugs in preclinical testing
NAD(P)H:quinone oxidoreductase 1 (NQO1) catalyzes the two-electron reduction in various quinones, including β-lapachone, by using NADH or NAD(P)H as electron donors. NQO1 is overexpressed in many solid tumors, including non-small cell lung cancer and breast cancer [84]. In NQO1-overexpressing cancer cells, the administration of β-lapachone was found to produce hydrogen peroxide (H2O2) via NQO1-dependent futile redox cycling. H2O2 induced an increase in DNA base and SSB lesions, which caused PARP1 hyperactivation. The hyperactivation of PARP1 resulted in dramatic NAD+/ATP depletion and ultimately led to caspase-independent programmed necrosis [85]. This anticancer effect of β-lapachone is dependent on the high expression of NQO1. Furthermore, it was reported that the addition of PARP inhibitors blocked the function of PARP1 and maintained the level of the NAD(P)H pool, which was required for NQO1-dependent futile redox cycling, and subsequently induced an increase in H2O2 [86]. The significant increase in H2O2, which could not be repaired due to the inhibition of PARP, ultimately induced tumor-selective apoptosis [86]. This mechanism of the combination of β-lapachone and a PARP inhibitor led to synthetic lethality in NQO1-overexpressing cancer cells [86]. Immunohistochemical analysis by Ying Dong et al. found that ~60% of prostate tumors had elevated NQO1 levels compared with paired normal tissue [87]. This suggested that β-lapachone is a potential drug for use in combination with PARP inhibitors for prostate tumors with a high expression of NQO1. More preclinical and clinical studies need to be performed to examine this synergistic combination.
Polo-like kinase 1 (PlK1) is overexpressed in prostate cancer and associated with tumorigenesis and the progression of prostate cancer [88]. As reported by Li et al., the use of a PARP inhibitor caused the accumulation of the cell cycle in the G2/M phase and increased the expression of PlK1 in p53-mutant prostate cancer cells [89]. In these circumstances, the addition of a PlK1 inhibitor dramatically decreased the expression of PARP1 and inhibited tumor growth together with a PARP inhibitor. However, in wild-type p53 cells, the use of a PARP inhibitor did not upregulate the expression of PlK1, and no synergistic effect was found for the combination of a PARP inhibitor and a PlK1 inhibitor. Owing to p53-mediated cell cycle arrest, the DNA damage induced by PARP inhibitors could not induce progression into the G2/M phase and resulted in failure to upregulate PlK1. As expected from this observation, the addition of a p53 inhibitor to the combination of a PARP inhibitor and a PlK1 inhibitor again showed synergism in wild-type p53 cells. It should be noted that the synergism described above was independent of BRCA status. This finding may broaden the population of patients who could benefit from this treatment, regardless of their p53 and BRCA1 status, by the combined inhibition of the PlK1, p53, and PARP proteins.
HDAC inhibitors can decrease the expression of proteins involved in HR and increase the sensitivity to PARP inhibitors in many cancers, including prostate cancer [90,91,92]. Recently, the mechanism behind the synergistic effect of an HDAC inhibitor and a PARP inhibitor was reported to be that the inhibition of HDAC decreased the protein stability of BRCA1 by reducing the protein expression of UHRF1 [93]. In addition, the transcription of UHRF1 could be upregulated by the binding of FOXM1 to the promoter of UHRF1 [94]; simultaneously, FOXM1 inhibition could inhibit the progression of prostate cancer cells by impairing AR function [95, 96]. FOXM1 overexpression is associated with taxane and paclitaxel resistance by regulating the transcription of UHRF1 and ABCC5 in prostate cancer [94, 97]. Moreover, FOXM1 inhibition increased the sensitivity of PARP inhibitors by inducing BRCAness in high-grade serous ovarian cancer or non-serous epithelial ovarian cancer [98, 99]. Nevertheless, it remains unknown whether FOXM1 inhibition can enhance the sensitivity of prostate cancer to PARP inhibitors.
Conclusions
With the rapid development of sequencing technologies, systematic genomic sequencing analyzes have found that recurrent mutations in mCRPC are focused on the DNA repair pathway in both germline and somatic specimens. This suggests the potential efficacy of PARP inhibitors in patients with mCRPC. The results of the TOPARP-A study supported a breakthrough therapy designation granted by the FDA for the treatment of patients with mCRPC and BRCA1/2 or ATM mutations. Moreover, patients with germline DNA damage repair gene mutations were more likely to progress to castration-resistant status. Thus, the early use of PARP inhibitors in patients with prostate cancer or mCRPC and DNA damage repair mutations may slow the progression to castration resistance [40, 100]. Although biomarkers to predict the response of PARP inhibitors extend from germline BRCA1/2 mutations to germline and somatic alterations in the DNA repair pathway, there are still some responders to PARP inhibitors that cannot be recognized by current biomarkers.
Genomic signatures and functional assays are not restricted to the function of a single gene; they are representative of the effect of HRD on the genome and the DNA repair capacity of tumor cells, respectively. Thus, these may serve as supplementary techniques for use with current biomarkers to identify sensitive patients. In addition, the development of new techniques has afforded the possibility of monitoring mutations during disease progression. For example, even if patients were detected to have positive biomarkers at the beginning of treatment, there is still the potential that resistance to PARP inhibitors will be acquired [101]. Conversely, the identification of positive biomarkers, which were not found during the initial treatment but were found by rebiopsy along with disease progression, provides a chance to choose PARP inhibitors [102]. This evidence indicates the necessity of rebiopsy to detect gene alterations related to acquired resistance and terminate ineffective treatment with a PARP inhibitor or to find newly emerged positive biomarkers that warrant the addition of a PARP inhibitor to the treatment plan. Given the rapid development of liquid biopsy, cell-free DNA extracted from plasma has become a priority choice for rebiopsy [103, 104].
However, further research is still required to determine whether the combination of drugs could cause HRD in HR-proficient tumors and ultimately induce synthetic lethality [105]. A phase II clinical trial (NCT01972217) reported the synergistic efficacy of the combination of olaparib and abiraterone in unselected patients with mCRPC. A larger number of patients with or without HRD is required to analyze whether this efficacy is independent of HR repair mutation status. Moreover, the risk of toxicity increases with an increase in effectiveness, and as safety is always the primary concern, the assessment of the risk and effectiveness before administration, dose intensity during administration, and long-term effects after administration must be considered for every patient.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Wu JN, Fish KM, Evans CP, Devere White RW, Dall'Era MA. No improvement noted in overall or cause-specific survival for men presenting with metastatic prostate cancer over a 20-year period. Cancer. 2014;120:818–23.
Huggins C, Stevens RE, Hodges CV. Studies on prostatic cancer: II. The effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209–23.
Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T, et al. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2014;65:467–79.
Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20.
Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12.
Francini E, Gray KP, Shaw GK, Evan CP, Hamid AA, Perry CE, et al. Impact of new systemic therapies on overall survival of patients with metastatic castration-resistant prostate cancer in a hospital-based registry. Prostate Cancer Prostatic Dis. 2019;22:420–7.
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–51.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–708.
Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–49.
Schiewer MJ, Mandigo AC, Gordon N, Huang F, Gaur S, de Leeuw R, et al. PARP-1 regulates DNA repair factor availability. EMBO Mol Med. 2018;10:e8816.
Abida W, Bryce AH, Vogelzang NJ, Amato RJ, Percent I, Shapiro JD, et al. Preliminary results from TRITON2: a phase II study of rucaparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) associated with homologous recombination repair (HRR) gene alterations. Ann Oncol. 2018;29:viii272.
Smith MR, Fizazi K, Sandhu SK, Kelly WK, Efstathiou E, Lara P, et al. Niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD): correlative measures of tumor response in phase II GALAHAD study. J Clin Oncol. 2020;38:118.
Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018;19:975–86.
Herceg Z, Wang ZQ. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res. 2001;477:97–110.
Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–28.
Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–8.
Eustermann S, Wu WF, Langelier MF, Yang JC, Easton LE, Riccio AA, et al. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol Cell. 2015;60:742–54.
Dawicki-McKenna JM, Langelier MF, DeNizio JE, Riccio AA, Cao CD, Karch KR, et al. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol Cell. 2015;60:755–68.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–99.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7.
Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–70.
Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–56.
Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39:8–24.
Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science. 2008;319:819–21.
Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–78.
Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21:162–74.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382:2091–102.
Park HJ, Bae JS, Kim KM, Moon YJ, Park SH, Ha SH, et al. The PARP inhibitor olaparib potentiates the effect of the DNA damaging agent doxorubicin in osteosarcoma. J Exp Clin Cancer Res. 2018;37:107.
Hussain M, Carducci MA, Slovin S, Cetnar J, Qian J, McKeegan EM, et al. Targeting DNA repair with combination veliparib (ABT-888) and temozolomide in patients with metastatic castration-resistant prostate cancer. Invest New Drugs. 2014;32:904–12.
Juan Fita MJ, Heras Lopez L, Mellado B, Mendez Vidal MJ, Anido U, Lorente D, et al. Phase II trial evaluating olaparib maintenance in patients with MCRPC after docetaxel treatment reaching partial or stable response. Ann Oncol. 2018;29:viii301.
Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. 2018;6:141.
Yu EY, Massard C, Retz M, Tafreshi A, Carles J, Hammerer P, et al. Pembrolizumab (pembro) plus olaparib in docetaxel-pretreated patients (pts) with metastatic castrate resistant prostate cancer (mCRPC): cohort A of the phase 1b/2 KEYNOTE-365 study. J Clin Oncol. 2019;37:5027.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501.
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28.
Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018;50:645–51.
Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7:a016600.
Saeki H, Siaud N, Christ N, Wiegant WW, van Buul PP, Han M, et al. Suppression of the DNA repair defects of BRCA2-deficient cells with heterologous protein fusions. Proc Natl Acad Sci USA. 2006;103:8768–73.
Romero-Laorden N, Pineiro-Yanez E, Gutierrez-Pecharroman A, Pacheco MI, Calvo E, Al-Shahrour F, et al. Somatic BRCA2 bi-allelic loss in the primary prostate cancer was associated to objective response to PARPi in a sporadic CRPC patient. Ann Oncol. 2017;28:1158–9.
Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112.
Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–87.
Jette NR, Kumar M, Radhamani S, Arthur G, Goutam S, Yip S, et al. ATM-deficient cancers provide new opportunities for precision oncology. Cancers. 2020;12:687.
Rafiei S, Fitzpatrick K, Liu D, Cai MY, Elmarakeby HA, Park J, et al. ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Res. 2020;80:2094–100.
Wengner AM, Siemeister G, Lucking U, Lefranc J, Wortmann L, Lienau P, et al. The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage-inducing or repair-compromising therapies in preclinical cancer models. Mol Cancer Ther. 2020;19:26–38.
Zhang D, Wang H, Sun M, Yang J, Zhang W, Han S, et al. Speckle-type POZ protein, SPOP, is involved in the DNA damage response. Carcinogenesis. 2014;35:1691–7.
Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–25.
Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9.
Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, Dahija A, et al. SPOP mutation leads to genomic instability in prostate cancer. Elife. 2015;4:e09207.
Kari V, Mansour WY, Raul SK, Baumgart SJ, Mund A, Grade M, et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016;17:1609–23.
Shenoy TR, Boysen G, Wang MY, Xu QZ, Guo W, Koh FM, et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann Oncol. 2017;28:1495–507.
Zhou J, Li J, Serafim RB, Ketchum S, Ferreira CG, Liu JC, et al. Human CHD1 is required for early DNA-damage signaling and is uniquely regulated by its N terminus. Nucleic Acids Res. 2018;46:3891–905.
Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Z, et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature. 2018;559:285–9.
Cerritelli SM, Crouch RJ. The balancing act of ribonucleotides in DNA. Trends Biochem Sci. 2016;41:434–45.
Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22.
Han B, Mehra R, Lonigro RJ, Wang L, Suleman K, Menon A, et al. Fluorescence in situ hybridization study shows association of PTEN deletion with ERG rearrangement during prostate cancer progression. Mod Pathol. 2009;22:1083–93.
Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22.
Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2:53ra75.
Fraser M, Zhao H, Luoto KR, Lundin C, Coackley C, Chan N, et al. PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin Cancer Res. 2012;18:1015–27.
Chatterjee P, Choudhary GS, Alswillah T, Xiong X, Heston WD, Magi-Galluzzi C, et al. The TMPRSS2-ERG gene fusion blocks XRCC4-mediated nonhomologous end-joining repair and radiosensitizes prostate cancer cells to PARP inhibition. Mol Cancer Ther. 2015;14:1896–906.
Chatterjee P, Choudhary GS, Sharma A, Singh K, Heston WD, Ciezki J, et al. PARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cells. PLoS ONE. 2013;8:e60408.
Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–92.
Hussain M, Daignault-Newton S, Twardowski PW, Albany C, Stein MN, Kunju LP, et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol. 2018;36:991–9.
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 study. Clin Cancer Res. 2020;26:2487–96.
Lv S, Wen H, Shan X, Li J, Wu Y, Yu X, et al. Loss of KMT2D induces prostate cancer ROS-mediated DNA damage by suppressing the enhancer activity and DNA binding of antioxidant transcription factor FOXO3. Epigenetics. 2019;14:1194–208.
Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6.
Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–71.
Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3:1245–53.
Schiewer MJ, Knudsen KE. DNA damage response in prostate cancer. Cold Spring Harb Perspect Med. 2019;9:a030486.
Asim M, Tarish F, Zecchini HI, Sanjiv K, Gelali E, Massie CE, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun. 2017;8:374.
Li L, Karanika S, Yang G, Wang J, Park S, Broom BM, et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal. 2017;10:eaam7479.
Teo MY, Seier K, Ostrovnaya I, Regazzi AM, Kania BE, Moran MM, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36:1685–94.
De Bono JS, Goh JCH, Ojamaa K, Piulats Rodriguez JM, Drake CG, Hoimes CJ, et al. KEYNOTE-199: Pembrolizumab (pembro) for docetaxel-refractory metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2018;36:5007.
Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi triggers the STING-dependent Immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79:311–9.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711–20.
Jette NR, Radhamani S, Arthur G, Ye R, Goutam S, Bolyos A, et al. Combined poly-ADP ribose polymerase and ataxia-telangiectasia mutated/Rad3-related inhibition targets ataxia-telangiectasia mutated-deficient lung cancer cells. Br J Cancer. 2019;121:600–10.
Jette NR, Radhamani S, Ye R, Yu Y, Arthur G, Goutam S, et al. ATM-deficient lung, prostate and pancreatic cancer cells are acutely sensitive to the combination of olaparib and the ATR inhibitor AZD6738. Genome Instab Dis. 2020;1:197–205.
Marshall CH, Sokolova AO, McNatty AL, Cheng HH, Eisenberger MA, Bryce AH, et al. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur Urol. 2019;76:452–8.
Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C, et al. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin Cancer Res. 2017;23:3097–108.
Kaplan AR, Gueble SE, Liu Y, Oeck S, Kim H, Yun Z, et al. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci Transl Med. 2019;11:eaav4508.
Kim JW, McKay RR, Taplin ME, Davis NB, Monk P, Appleman LJ, et al. Randomized phase II study of olaparib with or without cediranib in men with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2020;38:111
Gonzalez-Billalabeitia E, Seitzer N, Song SJ, Song MS, Patnaik A, Liu XS, et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Discov. 2014;4:896–904.
Siegel D, Ross D. Immunodetection of NAD(P)H:quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic Biol Med. 2000;29:246–53.
Huang X, Dong Y, Bey EA, Kilgore JA, Bair JS, Li LS, et al. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis. Cancer Res. 2012;72:3038–47.
Huang X, Motea EA, Moore ZR, Yao J, Dong Y, Chakrabarti G, et al. Leveraging an NQO1 bioactivatable drug for tumor-selective use of poly(ADP-ribose) polymerase inhibitors. Cancer Cell. 2016;30:940–52.
Dong Y, Bey EA, Li LS, Kabbani W, Yan J, Xie XJ, et al. Prostate cancer radiosensitization through poly(ADP-Ribose) polymerase-1 hyperactivation. Cancer Res. 2010;70:8088–96.
Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, et al. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate. 2004;60:240–5.
Li J, Wang R, Kong Y, Broman MM, Carlock C, Chen L, et al. Targeting Plk1 to enhance efficacy of olaparib in castration-resistant prostate cancer. Mol Cancer Ther. 2017;16:469–79.
Chao OS, Goodman OB. Synergistic loss of prostate cancer cell viability by coinhibition of HDAC and PARP. Mol Cancer Res. 2014;12:1755–66.
Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, et al. Histone deacetylase inhibitor treatment induces ‘BRCAness' and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget. 2014;5:5637–50.
Weberpals JI, O'Brien AM, Niknejad N, Garbuio KD, Clark-Knowles KV, Dimitroulakos J. The effect of the histone deacetylase inhibitor M344 on BRCA1 expression in breast and ovarian cancer cells. Cancer Cell Int. 2011;11:29
Yin L, Liu Y, Peng Y, Peng Y, Yu X, Gao Y, et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J Exp Clin Cancer Res. 2018;37:153.
Yuan B, Liu Y, Yu X, Yin L, Peng Y, Gao Y, et al. FOXM1 contributes to taxane resistance by regulating UHRF1-controlled cancer cell stemness. Cell Death Dis. 2018;9:562.
Liu Y, Gong Z, Sun L, Li X. FOXM1 and androgen receptor co-regulate CDC6 gene transcription and DNA replication in prostate cancer cells. Biochim Biophys Acta. 2014;1839:297–305.
Liu Y, Liu Y, Yuan B, Yin L, Peng Y, Yu X, et al. FOXM1 promotes the progression of prostate cancer by regulating PSA gene transcription. Oncotarget. 2017;8:17027–37.
Hou Y, Zhu Q, Li Z, Peng Y, Yu X, Yuan B, et al. The FOXM1-ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis. 2017;8:e2659.
Fang P, Madden JA, Neums L, Moulder RK, Forrest ML, Chien J. Olaparib-induced adaptive response Is disrupted by FOXM1 targeting that enhances sensitivity to PARP inhibition. Mol Cancer Res. 2018;16:961–73.
Tassi RA, Todeschini P, Siegel ER, Calza S, Cappella P, Ardighieri L, et al. FOXM1 expression is significantly associated with chemotherapy resistance and adverse prognosis in non-serous epithelial ovarian cancer patients. J Exp Clin Cancer Res. 2017;36:63.
Wei Y, Wu J, Gu W, Wang J, Lin G, Qin X, et al. Prognostic value of germline DNA repair gene mutations in de novo metastatic and castration-sensitive prostate cancer. Oncologist. 2020;25:e1042–e50.
Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci USA. 2013;110:17041–6.
Purshouse K, Schuh A, Fairfax BP, Knight S, Antoniou P, Dreau H, et al. Whole-genome sequencing identifies homozygous BRCA2 deletion guiding treatment in dedifferentiated prostate cancer. Cold Spring Harb Mol Case Stud. 2017;3:a001362.
Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov. 2017;7:999–1005.
Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017;7:1006–17.
Lim E, Johnson SF, Geyer M, Serra V, Shapiro GI. Sensitizing HR-proficient cancers to PARP inhibitors. Mol Cell Oncol. 2017;4:e1299272.
Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15.
Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010;17:1247–54.
Min A, Im SA, Yoon YK, Song SH, Nam HJ, Hur HS, et al. RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol Cancer Ther. 2013;12:865–77.
Wilkes DC, Sailer V, Xue H, Cheng H, Collins CC, Gleave M, et al. A germline FANCA alteration that is associated with increased sensitivity to DNA damaging agents. Cold Spring Harb Mol Case Stud. 2017;3:a001487.
Ciccone MA, Ricker C, Culver J, Maoz A, Melas M, Idos GE, et al. Inactivation of the tumor suppressor BRCA1 interacting protein C-terminal helicase 1 (BRIP1) gene confers increased susceptibility to platinum antineoplastic agents and augments the synergistic response to poly (ADP-ribose) polymerase (PARP) inhibition in ovarian epithelial cells. Gynecol Oncol. 2016;143:196.
Reichert ZR, Daignault S, Teply BA, Devitt ME, Heath EI. Targeting resistant prostate cancer with ATR and PARP inhibition (TRAP trial): a phase II study. J Clin Oncol. 2020;38:TPS254.
Oing C, Tennstedt P, Simon R, Volquardsen J, Borgmann K, Bokemeyer C, et al. BCL2-overexpressing prostate cancer cells rely on PARP1-dependent end-joining and are sensitive to combined PARP inhibitor and radiation therapy. Cancer Lett. 2018;423:60–70.
Morra F, Merolla F, Napolitano V, Ilardi G, Miro C, Paladino S, et al. The combined effect of USP7 inhibitors and PARP inhibitors in hormone-sensitive and castration-resistant prostate cancer cells. Oncotarget. 2017;8:31815–29.
Chen ST, Okada M, Nakato R, Izumi K, Bando M, Shirahige K. The deubiquitinating enzyme USP7 regulates androgen receptor activity by modulating its binding to chromatin. J Biol Chem. 2015;290:21713–23.
Morra F, Merolla F, Criscuolo D, Insabato L, Giannella R, Ilardi G, et al. CCDC6 and USP7 expression levels suggest novel treatment options in high-grade urothelial bladder cancer. J Exp Clin Cancer Res. 2019;38:90.
Ip LR, Poulogiannis G, Viciano FC, Sasaki J, Kofuji S, Spanswick VJ, et al. Loss of INPP4B causes a DNA repair defect through loss of BRCA1, ATM and ATR and can be targeted with PARP inhibitor treatment. Oncotarget. 2015;6:10548–62.
Hodgson MC, Shao LJ, Frolov A, Li R, Peterson LE, Ayala G, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011;71:572–82.
Li L, Chang W, Yang G, Ren C, Park S, Karantanos T, et al. Targeting poly(ADP-ribose) polymerase and the c-Myb-regulated DNA damage response pathway in castration-resistant prostate cancer. Sci Signal. 2014;7:ra47.
Srivastava SK, Bhardwaj A, Singh S, Arora S, McClellan S, Grizzle WE, et al. Myb overexpression overrides androgen depletion-induced cell cycle arrest and apoptosis in prostate cancer cells, and confers aggressive malignant traits: potential role in castration resistance. Carcinogenesis. 2012;33:1149–57.
Booth L, Cruickshanks N, Ridder T, Dai Y, Grant S, Dent P. PARP and CHK inhibitors interact to cause DNA damage and cell death in mammary carcinoma cells. Cancer Biol Ther. 2013;14:458–65.
Morra F, Luise C, Visconti R, Staibano S, Merolla F, Ilardi G, et al. New therapeutic perspectives in CCDC6 deficient lung cancer cells. Int J Cancer. 2015;136:2146–57.
Luo ML, Zheng F, Chen W, Liang ZM, Chandramouly G, Tan J, et al. Inactivation of the prolyl isomerase Pin1 sensitizes BRCA1-proficient breast cancer to PARP inhibition. Cancer Res. 2020;80:3033–45.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFC1306900), National Natural Science Foundation of China (81874327), Key Research and Development Program of Hunan Province (2019SK2251), and Innovation and Research Project of Development and Reform Committee of Hunan Province (2019-875).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Chen, Yx., Tan, Lm., Gong, Jp. et al. Response prediction biomarkers and drug combinations of PARP inhibitors in prostate cancer. Acta Pharmacol Sin 42, 1970–1980 (2021). https://doi.org/10.1038/s41401-020-00604-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00604-1
