
Abstract
Huntington’s disease (HD) is one of main neurodegenerative diseases, characterized by striatal atrophy, involuntary movements, and motor incoordination. Ginsenoside Rg1 (Rg1), an active ingredient in ginseng, possesses a variety of neuroprotective effects with low toxicity and side effects. In this study, we investigated the potential therapeutic effects of Rg1 in a mouse model of HD and explored the underlying mechanisms. HD was induced in mice by injection of 3-nitropropionic acid (3-NP, i.p.) for 4 days. From the first day of 3-NP injection, the mice were administered Rg1 (10, 20, 40 mg·kg−1, p.o.) for 5 days. We showed that oral pretreatment with Rg1 alleviated 3-NP-induced body weight loss and behavioral defects. Furthermore, pretreatment with Rg1 ameliorated 3-NP-induced neuronal loss and ultrastructural morphological damage in the striatum. Moreover, pretreatment with Rg1 reduced 3-NP-induced apoptosis and inhibited the activation of microglia, inflammatory mediators in the striatum. We revealed that Rg1 exerted neuroprotective effects by suppressing 3-NP-induced activation of the MAPKs and NF-κΒ signaling pathways in the striatum. Thus, our results suggest that Rg1 exerts therapeutic effects on 3-NP-induced HD mouse model via suppressing MAPKs and NF-κΒ signaling pathways. Rg1 may be served as a novel therapeutic option for HD.
Similar content being viewed by others

Introduction
Huntington’s disease (HD) is an inherited neurodegenerative disease caused by a CAG expansion mutation in the huntingtin gene [1, 2]. HD patients exhibit a triad of clinical features, i.e., abnormal motor function, cognitive and neuropsychiatric symptoms, and the hallmark of the disease is motor dysfunction [3]. The loss of striatal neurons is a marked feature of HD pathology that is characterized by progressive striatal atrophy followed by involuntary movements and motor incoordination [4, 5]. There are many theories and hypotheses related to the mechanism of this disease, including oxidative stress, mitochondrial impairment, synaptic dysfunction, defects in energy metabolism and apoptosis of striatal neurons caused by the toxicity of the mHtt protein [6, 7]. However, the exact mechanism underlying the death of striatal neurons is not clear. In addition, there are no ideal therapeutic strategies to prevent the progression of HD.
3-Nitropropionic acid (3-NP) is a natural neurotoxin that is produced by the plant Indigofera [8]. By inhibiting the activity of the mitochondrial complex II enzyme succinate dehydrogenase (SDH), 3-NP mainly disturbs energy metabolism, leading to the degeneration of neurons in the striatum. The 3-NP-administrated animal models exhibit HD-like symptoms, including striatal impairment, motor dysfunction, and memory loss [9,10,11]. As 3-NP-induced effects are similar to the symptoms of HD, 3-NP-treated mice are good research models for investigating the effects of drug candidates for HD.
The mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-κB) signaling pathways are crucial modulators of inflammatory responses [12]. When activated, the MAPK and NF-κB pathways mediate inflammation by promoting the expression of multiple cytokines, such as TNF-α, IL-1β, and IL-6 [13]. The MAPK cascade is composed of numerous subgroups, such as extracellular regulated kinases (ERKs), c-Jun NH2-terminal kinases (JNKs), and p38 MAPKs, which play important roles in regulating multiple signal transduction pathways caused by external stimuli and various cellular physiological functions, such as proliferation, apoptosis and synaptic plasticity [14, 15]. Among MAPK pathways, activation of the JNK and p38 MAPK pathways mainly induces neurotoxicity in numerous HD models [12, 16]. Moreover, ERK’s conditions are more complex in the HD models. Activation of ERKs is largely neuroprotective in some HD models, whereas it can induce multifarious neuronal death-related signaling pathways in other HD models [17,18,19,20]. As an important regulator of inflammatory reactions, the NF-κB family includes five members: NF-κB1 (p105), NF-κB2 (p100), RelA (p65), RelB, and c-Rel. Transactivation of NF-κB takes part in many physiological processes, including inflammation, cellular proliferation, and death [15, 21, 22]. In various HD models, the NF-κB cascade is activated in the striatum.
Ginseng is a highly valued and popular medicinal plant [23]. As one of the active ingredient of ginseng, ginsenoside Rg1 (Rg1) possesses a variety of neuroprotective effects and is considered to have low toxicity and few side effects [24,25,26]. Rg1 can cross the blood-brain barrier (BBB) and exert beneficial effects in many neural diseases, especially neurodegenerative diseases. A large number of experiments suggest that Rg1 can exert neuroprotective effects against Alzheimer’s disease (AD) and Parkinson’s disease (PD) through antioxidative, anti-inflammatory and antiapoptotic effects in both cell and animal models [25,26,27,28,29,30]. However, the neuroprotective effects and related mechanisms of Rg1 in an HD mouse model have not yet been elucidated.
In our study, we established a mouse model of 3-NP-induced HD to investigate whether Rg1 can exert neuroprotective effects to ameliorate symptoms related to HD. Our results showed that Rg1 alleviated body weight and behavioral defects. Additionally, Rg1 ameliorated neuronal loss and ultrastructural morphological damage in the striatum. Furthermore, Rg1 reduced apoptosis and inhibited the activation of microglia and inflammatory mediators, such as TNF-α and IL-1β, in the striatum. Moreover, Rg1 exerted neuroprotective effects by suppressing the activation of the MAPK and NF-κB pathways in the striatum.
Materials and methods
Drugs
Rg1 (HPLC > 98%) was obtained from Jilin University (Changchun, China). 3-NP was purchased from Sigma-Aldrich (St. Louis, MO, USA). Nimodipine was purchased from Bayer AG (Leverkusen, Germany). All other reagents used in the experiment were of analytical grade.
Animals
Male C57BL/6 mice (23–25 g) were supplied by SPF Laboratories (Beijing, China) and were housed at a temperature of 25 °C on a 12 h light-dark cycle. Food and water were provided. All procedures were conducted in strict accordance with the guidelines established by the National Institutes of Health for the Care and Use of Laboratory Animals and approved by the Αnimal Care Committee of the Peking Union Medical College and Chinese Academy of Medical Sciences.
Experimental procedures and methods
After one week of acclimatization, the mice were randomly subdivided into seven groups, including (i) the control group (saline, intraperitoneal [i.p.]), (ii) the model group (60 mg/kg 3-NP [the 1st day] and 80 mg/kg 3-NP [the 2rd-4th day], twice daily, 3-NP dissolved in saline, pH = 7.4, i.p.), (iii) the Rg1-10 mg/kg+3-NP group (10 mg/kg Rg1, peros [p.o.]+3-NP, i.p.), (iv) the Rg1-20 mg/kg+3-NP group (20 mg/kg Rg1, p.o.+3-NP, i.p.), (v) the Rg1-40 mg/kg+3-NP group (40 mg/kg Rg1, p.o.+3-NP, i.p.), (vi) the Rg1-40 mg/kg+saline group (40 mg/kg Rg1, p.o.+saline, i.p.), and (vii) the nimodipine+3-NP group (10 mg/kg nimodipine, p.o.+3-NP, i.p.). The dose of 3-NP administered to the (iii), (iv), (v), and (vii) groups was the same as that administered to the model group. Each group contained 12 mice, and a total of 84 mice were used. Drugs or saline was administered 30 min prior to 3-NP injection. 3-NP was administered intraperitoneally twice daily for 4 days (9:00 and 21:00). On the first day, the dose of 3-NP was 60 mg/kg, and 80 mg/kg 3-NP was administered on the following days. The experimental procedure is shown in Fig. 1.

After a week of acclimatization, HD was induced in mice by injection of 3-NP twice daily for four days. The dose of 3-NP was 60 mg/kg on the first day, and 80 mg/kg 3-NP was administered from the second day to the fourth day. Half an hour before 3-NP injection, Rg1 was administered at doses of 10, 20, 40 mg/kg. The open field test and neurological scoring were carried out on the fifth day. All mice were sacrificed for further study.
Body weight measurement
Prior to 3-NP administration, body weights of the only the surviving experimental mice were obtained.
Behavioral assessments
To test the degree of motor impairment induced by 3-NP in the mice from each group, we used a previously reported behavioral scale, with scores ranging from 0 to 5. We evaluated neurological behavior on the fifth day according to the following criteria: 0, normal behavior; 1, attenuated general activity; 2, abnormal gait with discoordination; 3, hindlimb dystonia; 4, inability to move; and 5, death [31, 32]. The test was performed by the experimenter who was well trained in a double-blinded manner.
Open field test
The open field test (OFT) was used to evaluate the locomotor and exploratory behaviors of rodents. We monitored the motor functions of all experimental mice by performing this test. A wooden square box apparatus measuring 50 cm× 50 cm× 50 cm with a white floor was used for the OFT. Mice from all groups were placed in the testing room 1 h prior to the test to allow them to adapt to the environment before the experiment began. Each mouse was allowed to explore the box for 5 min. The trajectory of each mouse was recorded with a Sony camera and analyzed by SMART v2.5.3 system software (Panlab SL, Barcelona, Spain) [33,34,35]. The total traveled distance was analyzed after the test was complete.
Tissue preparation
Chloral hydrate (6%) was used to anesthetize the mice (0.1 mL/10 g, i.p.). For biochemical analysis, the striatum was separated and stored at −80 °C for subsequent experiments. For histological analysis, the mice were anesthetized and then perfused with 0.1 mol/L phosphate-buffered saline (PBS), followed by 4% paraformaldehyde (PFA) (pH = 7.4). Brain tissues were quickly isolated and put in 4% PFA solution at 4 °C for 12 h [25, 36]. To acquire frozen sections, the brain tissues were dehydrated in different concentrations of sucrose (10%, 20%, 30%, and 30%) dissolved in 0.1 mol/L phosphate-buffers (PB). To obtain paraffin sections, the brain tissues were dehydrated and embedded sequentially in the following reagents: 30%, 50%, 70%, 80%, 95%, 100%, and 100% ethanol, xylene and liquid paraffin. The thickness of the paraffin sections was 4 μm.
Nissl staining
Nissl staining is an effective method for evaluating neuronal damage in the mouse brain. We used xylene to dewax the paraffin sections, and then rinsed them with 100%, 95%, and 70% ethanol. The slides were stained with 0.1% cresyl violet solution for 25 min. Then, the sections were washed quickly in water and differentiated in 95% ethanol. We used 100% ethanol to dehydrate the slides and then rinsed them with xylene solution. Finally, the slides were mounted in the fume hood.
Ultrastructural transmission electron microscopy (TEM)
The ultrastructure of the striatum was imaged by TEM. After the mice were perfused, their brains were rapidly isolated. Then, the striatum was cut into ~1 mm3 cubes soon after it was separated from the brain. The cubes were fixed in 2.5% glutaraldehyde for 2 h and then rinsed with 0.1 M PBS. The tissue cubes were dehydrated in an alcohol gradient and then embedded in Epon resin. The embedded tissues were sectioned into ultrathin slices, which were stained with lead citrate and uranyl acetate. The striatal ultrathin sections were observed and imaged by TEM (H-7650, Hitachi, Tokyo, Japan) [25].
Immunohistochemical staining (IHC)
The paraffin sections were dewaxed and rehydrated as described above for Nissl staining. The slides were boiled in 1× antigen retrieval solution (Vector Laboratories, Burlingame, CA, USA) in a microwave for 15 min. After they were cooled to room temperature (RT), the slides were incubated with 3% H2O2 for 30 min. Then, the slides were treated with blocking buffer for 60 min at RT and incubated with the primary antibody in hybridization buffer (10× diluted blocking buffer) overnight (O/N) at 4 °C. The next day, the slides were treated with secondary antibody for 1 h at RT and then counterstained with DAPI for 10 min. Finally, the sections were mounted with antifade mounting medium (Southern Biotech, Βirmingham, AL, USA). At the same time, the Tyramide Signal Amplification kit (TSA) (Invitrogen, San Diego, CA, USA) was used to increase the labeling density of the target protein by utilizing the catalytic activity of HRP. The TSA protocol is the same as the protocol for normal staining until after primary antibody incubation. The sections were incubated with a biotinylated secondary antibody. After being washed with 1× PBS, the tissue sections were incubated with HRP-conjugated streptavidin for 60 min at RT. Crucially, we prepared the tyramide working solution, which contained 100× diluted tyramide substrate solution (488) in amplification buffer and 0.0015% H2O2. Then, the slides were incubated with the working solution for 10 min at RT. After being washed with 0.1 M PBS, the slides were counterstained with DAPI and then mounted with mounting medium [37, 38]. Finally, the sections were observed and imaged with a Leica laser scanning confocal microscope (TCS SP2, Leica, Heidelberg, Germany).
The following primary antibodies were used in the experiment: mouse anti-Iba-1 (1:500, Wako, Tokyo, Japan), rabbit anti-TNF-α (1:500, Abclonal, Wuhan, China), and rabbit anti-IL-1β (1:500, Abclonal, Wuhan, China). The following secondary antibodies were used in the experiment: biotin-conjugated goat anti-mouse IgG (1:400, KPL, Gaithersburg, MD, USA) and biotin-conjugated goat anti-rabbit IgG (1:400, KPL, Gaithersburg, MD, USA).
Western blotting (WB)
We homogenized the isolated striatum tissues in RIPA buffer (Beyotime, Shanghai, China) with protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) and phosphatase inhibitors (Invitrogen, San Diego, CA, USA) and then centrifuged them at 12,000 rpm for 30 min. We collected the supernatants and analyzed the total concentrations with a BCA kit (Applygen, Beijing, China). Gradient SDS-PAGE gels were used to separate an equal amount of each protein sample (40 μg/lane), and then the proteins were transferred to PVDF membranes (Millipore, Temecula, CA, USA). After being blocked with 3% BSA (Sigma-Aldrich, St. Louis, MO, USA) for 2 h, the membranes that contained the proteins were incubated with primary antibodies at 4 °C O/N. The membranes were rinsed with 1× PBST three times for 10 min and then treated with biotinylated secondary antibodies for 2 h at RT. After being washed with 1× PBST, the membranes were treated with HRP-conjugated streptavidin for 1 h at RT. Finally, we used the ImageQuant LAS 4000 Mini (Ge Healthcare, Diegem, Belgium) to detect the bands. The density of the protein bands was analyzed by ImageJ software (NIH, Bethesda, MD, USA) [39, 40].
The following primary antibodies were used in WB: rabbit anti-DARPP-32 (1:1000, ProteinTech Group, Chicago, IL, USA), rabbit anti-cleaved-Caspase3 (1:500, CST, Danvers, MA, USA), mouse anti-β-actin (1:5000, Sigma-Aldrich, St. Louis, MO, USA), goat anti-Iba-1 (1:500, Santa Cruz Biotech, Santa Cruz, CA, USA), mouse anti-phospho-p38 (p-p38) (1:500, Santa Cruz Biotech, Santa Cruz, CA, USA), rabbit anti-p38 (1:500, Santa Cruz Biotech, Santa Cruz, CA, USA), rabbit anti-ERK1/2 (1:1000, CST, Danvers, MA, USA), rabbit anti-phospho-ERK1/2 (p-ERK1/2) (1:1000, CST, Danvers, MA, USA), mouse anti-phospho-JNK (p-JNK) (1:500, Santa Cruz Biotech, Santa Cruz, CA, USA), mouse anti-JNK1 (1:500, Santa Cruz Biotech, Santa Cruz, CA, USA), mouse anti-NF-κB p65 (1:500, Santa Cruz Biotech, Santa Cruz, CA, USA), and mouse anti-phospho-NF-κB p65 Ser536 (p-NF-κB p65) (1:1000, CST, Danvers, MA, USA). The following secondary antibodies were used in the study: biotin-conjugated goat anti-mouse IgG (1:1000, KPL, Gaithersburg, MD, USA), biotin-conjugated goat anti-rabbit IgG (1:1000, KPL, Gaithersburg, MD, USA), and biotin-conjugated rabbit anti-goat IgG (1:2000, KPL, Gaithersburg, MD, USA).
Standard enzymatic analysis with kits
Striatal tissues were prepared as mentioned above by homogenization and centrifugation in saline. Then, we collected the supernatants, which were used to test the activity of SDH in the striatum. SDH kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The SDH kits were used according to the manufacturer’s protocol.
Image analysis
With the aid of ImageJ software, the number of Nissl-stained cells and cells immunopositive for specific markers was counted by an investigator blinded to the experimental groups. Cells in full-image fields of the striatum from three coronal brain sections per mouse were counted for each experimental index. Three different mice were used from each group [41, 42].
Statistical analysis
We used GraphPad Prism 7.0 (GraphPad Software, San Diego, CA, USA) to perform statistical analysis. One-way analysis of variance (ANOVA) followed by Dunnett’s or Tukey’s post hoc test and Student’s unpaired t test were used for data analysis. The data are expressed as the means ± SEM. “n” represents the number of tissues from different mice or mice used. P < 0.05 was considered statistically significant.
Results
Rg1 alleviated body weight loss and behavioral defects induced by 3-NP
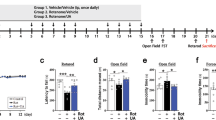
The chemical structure of Rg1 is present in Fig. 2a. In order to investigate the effects of Rg1 on body weight loss and behavioral defects induced by 3-NP, mice were pretreated with Rg1 before being injected with 3-NP. Compared with those of the mice in the control group, the body weights of the mice in the model group were markedly reduced. Furthermore, oral pretreatment with Rg1 alleviated body weight loss in mice in the Rg1 + 3-NP group and the nimodipine+3-NP group compared with mice in the model group (Rg1-40 mg/kg+3-NP group and nimodipine+3-NP group: P < 0.05). Moreover, the mean body weights of mice in the Rg1-40 mg/kg+saline group and those in the control group were not different (Fig. 2b). To evaluate the motor behaviors of mice, each experimental group was subjected to behavioral assessments and the open field test. Based on the behavioral assessment, mice in the model group exhibited serious neurological dysfunction, including attenuated general activity, abnormal gait with discoordination, hindlimb dystonia, and an inability to move. The Rg1 + 3-NP groups and the nimodipine+3-NP group (Rg1-10, 20 and 40 mg/kg+3-NP group; nimodipine+3-NP group: P < 0.01) exhibited significantly lower neurological scores than the model group. Furthermore, the neurological scores of mice from the Rg1-40 mg/kg+saline group were comparable with those of the control mice (Fig. 2c). In the open field test, we found that the total distance traveled by the model group mice was significantly shorter than that traveled by the mice in the control group. The total distance traveled by mice in the Rg1-40 mg/kg+3-NP group and the nimodipine+3-NP group was significantly increased (Rg1-40 mg/kg+3-NP group and nimodipine+3-NP group: P < 0.05) compared with that traveled by the model group. In addition, there were no significant differences in the total distance traveled between the Rg1-40 mg/kg+saline group and the control group (Fig. 2d). These results suggested that pretreatment with Rg1 alleviated body weight loss and motor behavior defects induced by 3-NP.

a The chemical structure of Rg1. b Changes of body weight across groups over 4 experimental days. c Neurological scores across groups on day 5. d Total distance traveled in the open field test across groups. Data are expressed as the mean ± SEM, n = 8. ###P < 0.001 vs control group; *P < 0.05, **P < 0.01 vs model group.
Rg1 ameliorated striatal neuronal loss induced by 3-NP
The striatum is a part of the basal ganglia, which is primarily involved in motor control and action selection. As the principal neurons of the striatum, medium spiny neurons (MSNs) are particularly vulnerable to 3-NP-induced damage in many models [6, 43, 44]. To assess striatal neuron damage, Nissl staining and Western blot analysis were performed. In Nissl-stained sections, the Nissl bodies of neurons were stained pink violet. Images of the striatum for each experimental group are shown in Fig. 3a–g, 3a’–g’. Compared with that in the control group, the number of remaining striatal neurons in the model group was significantly decreased. In addition, the number of striatal neurons was significantly higher in the Rg1 + 3-NP groups and the nimodipine+3-NP group than in the model group (Rg1-10, 20, 40 mg/kg+3-NP groups and nimodipine+3-NP group: P < 0.01). Moreover, there was no significant difference in the number of striatal neurons between the Rg1-40 mg/kg+saline group and the control group (Fig. 3h). Furthermore, the expression of DARPP-32, an established marker of MSNs, was confirmed by Western blot analysis in each experimental group. The expression of DARPP-32 was significantly decreased in the model group compared with the control group. Moreover, compared with that in the model group, the expression of DARPP-32 was significantly increased in the Rg1 + 3-NP groups and the nimodipine+3-NP group (Rg1-20 mg/kg+3-NP group: P < 0.05; Rg1-40 mg/kg+3-NP group and nimodipine+3-NP group: P < 0.01). There was no significant variation in the expression level of DARPP-32 between the Rg1-40 mg/kg+saline group and the control group (Fig. 3i, j). The data indicated that Rg1 ameliorated the loss of striatal neurons induced by 3-NP.

a–g, a’-g’ Nissl staining of striatal neurons across groups. h The counting results of Nissl staining-positive cells in the striatum. i, j The representative blots and densitometry data for DARPP-32 in the striatum across groups. Data are expressed as the mean ± SEM, n = 3. ###P < 0.001 vs control group; *P < 0.05, **P < 0.01 vs model group. Scale bars, (a–g) 500 μm; (a’-g’) 50 μm.
Rg1 alleviated the striatal ultrastructural morphological damage induced by 3-NP
Furthermore, to investigate striatal ultrastructural morphological changes, TEM images were taken for each group. In the control group, the TEM images showed relatively normal ultrastructural morphology in the striatum (Fig. 4a, e, i, m). However, disruption of cytomembranes and condensed chromatin were observed in the model group (Fig. 4b). In subcellular structures, incomplete mitochondrial membranes with few cristae and demyelination of neurites were found in the model group (Fig. 4f, j). In the 3-NP-treated model group, astrocyte end-feet (AEF) surrounding blood vessels were swollen, which was probably caused by impairment of the blood-brain barrier (BBB) [45] (Fig. 4n). The Rg1 and nimodipine groups exhibited more normal cytomembranes, chromatin, mitochondria, neurites, and AEF than the model group. That is, both Rg1 and nimodipine improved the disruption of the cytomembranes, condensation of chromatin, decrease in mitochondrial cristae, demyelination of neurites, and swelling of the AEF (Fig. 4c, d, g, h, k, l, o, p). The results revealed that Rg1 alleviated the striatal ultrastructural morphological damage induced by 3-NP.

a–d The representative photomicrographs of striatal neurons across the groups. e–h The representative TEM images of subcellular structures in the striatum across groups. i–l The representative photomicrographs of myelination in the striatum. m–p The ultrastructural morphology of the BBB across groups. Scale bars, (a–d) 5 μm; (e–p) 2 μm.
Rg1 reduced the striatal apoptosis induced by 3-NP
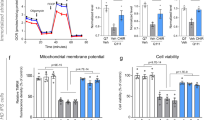
Apoptosis was induced in the striatum of the mouse model of 3-NP-induced HD. As 3-NP induced apoptosis in the striatum by inhibiting SDH activity, we first explored the effects of Rg1 on the activity of SDH by standard enzymatic analysis and then investigated the antiapoptotic effects of Rg1 by WB analysis in each experimental group. Striatal SDH activity was significantly decreased in the model group compared with the control group (Fig. 5b). Moreover, there was a significant increase in striatal SDH activity in the Rg1 + 3-NP groups and the nimodipine+3-NP group mice compared to the model group mice (Rg1-10 mg/kg+3-NP group: P < 0.05; Rg1-20, 40 mg/kg+3-NP group and nimodipine+3-NP group: P < 0.01). There was no difference in striatal activity between the Rg1-40 mg/kg+saline group and the control group. Furthermore, the antiapoptotic effect of Rg1 was evaluated by measuring the expression of the cleaved caspase-3 protein by WB analysis. We found that the protein expression of cleaved caspase-3 was significantly increased in the model group compared with the control group. Compared with that in the model group mice, the expression of cleaved caspase-3 was significantly decreased in the Rg1 + 3-NP groups and the nimodipine+3-NP group (Rg1-20, 40 mg/kg+3-NP groups and nimodipine+3-NP group: P < 0.01). In contrast, there was no difference in the protein expression of cleaved caspase-3 between the Rg1-40 mg/kg+saline group and the control group (Fig. 5a and c). The data suggested that Rg1 reduced 3-NP-induced apoptosis in the striatum.

a and c The representative blots and densitometry data for cleaved caspase-3 protein in the striatum across groups. b The activity of SDH in the striatum across groups. Data are expressed as the mean ± SEM, n = 3. ##P < 0.01 vs control group; *P < 0.05, **P < 0.01 vs model group.
Rg1 inhibited striatal microglial activation induced by 3-NP
3-NP induced striatal microglial activation, which triggers the inflammatory response. To assess the effects of Rg1 on microglial activation, we used both IHC and WB analysis to evaluate the protein expression of Iba-1, a marker of microglia. IHC images of the striatum for each experimental group are presented in Fig. 6a-g, 6a’-g’. In terms of cell morphology, we found that in the model group but not in the control group, Iba-1-positive cells in the striatum exhibited a morphology indicative of activated microglia, with enlarged cell bodies (Fig. 6a, b, 6a’, b’). IHC analysis revealed that the number of Iba-1-positive cells in the striatum in the model group was significantly increased compared with that in the control group (Fig. 6i). Moreover, compared with that in the model group, the number of Iba-1-positive cells in the striatum in the Rg1 + 3-NP groups and the nimodipine+3-NP group was significantly reduced (Rg1-20 mg/kg+3-NP group: P < 0.05; Rg1-40 mg/kg+3-NP group and nimodipine+3-NP group: P < 0.01). WB analysis showed that the protein expression of striatal Iba-1 was markedly increased in the model group compared with the control group (Fig. 6h, j). However, the protein expression of striatal Iba-1 was significantly decreased in the Rg1 + 3-NP groups and the nimodipine+3-NP group compared with the model group (Rg1-20 mg/kg+3-NP group: P < 0.05; Rg1-40 mg/kg+3-NP group and nimodipine+3-NP group: P < 0.01). In contrast, there was no significant difference in the protein expression of striatal Iba-1 between the Rg1-40 mg/kg+saline group and the control group. These results indicated that Rg1 inhibited the striatal microglial activation induced by 3-NP.

a–g, a’–g’ The representative photomicrographs of immunofluorescence histochemical staining of Iba-1 in the striatum. i Quantitative analysis of Iba-1-positive cells in the striatum. h, j The representative blots and densitometry data for Iba-1 in the striatum across groups. Data are expressed as the mean ± SEM, n = 3. ##P < 0.01 vs control group; *P < 0.05, **P < 0.01 vs model group. Scale bars, (a–g, a’–g’) 25 μm.
Rg1 decreased the production of proinflammatory cytokines induced by 3-NP
When inflammation, infection, and alterations in the immune system occur, immune cells are activated through the production of cytokines. These cytokines can be divided into two main types, namely, anti-inflammatory and proinflammatory cytokines; these different types of cytokines make opposing contributions to inflammatory reactions [46, 47]. The production of proinflammatory cytokines was increased in the striatum in the animal model of 3-NP-induced HD, and this increase in proinflammatory cytokine production exacerbated damage to striatal neurons [9, 48]. To investigate whether Rg1 can block the activation of proinflammatory cytokines induced by 3-NP, IHC analysis was performed in the experimental groups (Fig. 7a-g, a’-g’). There was a marked increase in the production of striatal proinflammatory cytokines (TNF-α, IL-1β) in the model group compared to the control group (Fig. 7h, h’). However, the number of striatal TNF-α- and IL-1β-positive neurons was significantly decreased in the Rg1 + 3-NP group and the nimodipine+3-NP group compared with the model group (Rg1-40 mg/kg+3-NP group and nimodipine+3-NP group: P < 0.01). In contrast, the number of striatal TNF-α- and IL-1β-positive neurons in the Rg1-40 mg/kg+saline group was similar to that in the control group. Our results suggested that Rg1 decreased the production of proinflammatory cytokines induced by 3-NP.

a–g, a’–g’ The representative photomicrographs of IHC staining for TNF-α and IL-1β in the striatum across groups. h, h’ Quantitative analysis of TNF-α- and IL-1β-positive cells in the striatum across groups. Data are expressed as the mean ± SEM, n = 3. ###P < 0.001 vs control group; **P < 0.01 vs model group. Scale bars, (a–g, a’–g’) 100 μm.
Rg1 suppressed the activation of the MAPK and NF-κB pathways induced by 3-NP
The MAPK and NF-κB pathways mainly participate in the regulation of inflammatory responses and were broadly activated in the striatum in 3-NP-treated animals. To explore the effects of Rg1 on the MAPK and NF-κB signaling pathways, we evaluated the expression of p38, p-p38, JNK1, p-JNK, ERK1/2, p-ERK1/2, NF-κB p65, and p-NF-κB p65 by WB analysis (Fig. 8a). Activation of p38, ERK1/2, JNK1, and NF-κB p65 was significant in the striatum in the model group compared with the control group. However, there was a significant reduction in the expression of p-p38, p-JNK, p-ERK1/2, and p-NF-κB p65 in the striatum in the Rg1 + 3-NP groups and the nimodipine+3-NP group compared with the model group (Fig. 8b–e). Moreover, there was no significant difference in the expression of p-p38, p-JNK, p-ERK1/2, and p-NF-κB p65 in the striatum between the Rg1-40 mg/kg+saline group and the control group. Our results revealed that Rg1 suppressed the activation of the MAPK and NF-κB pathways induced by 3-NP.

a–e The representative blots and densitometry data for p-p38, p-ERK, p-JNK, p-NF-κB p65 in the striatum across groups. Data are expressed as the mean ± SEM, n = 3. ###P < 0.001 vs control group; *P < 0.05, **P < 0.01 vs model group.
Discussion
In our study, we explored the neuroprotective effects of Rg1 in a mouse model of 3-NP-induced HD. Pretreatment with Rg1 significantly alleviated the loss of body weight and the behavioral defects induced by 3-NP. Moreover, Rg1 ameliorated the striatal neuronal loss and ultrastructural morphological damage induced by 3-NP. Furthermore, we found that Rg1 inhibited SDH inactivity and reduced apoptosis induced by 3-NP in the striatum. Rg1 also inhibited striatal microglial activation and reduced the production of striatal proinflammatory cytokines (TNF-α and IL-1β) induced by 3-NP. Finally, our data showed that Rg1 suppressed the activation of the MAPK and NF-κB signaling pathways induced by 3-NP in the striatum.
The 3-NP-treated animal model is widely used in HD-related research, as the phenotype of 3-NP treatment mimics HD symptoms and neuropathology, including locomotor hypoactivity, discoordination of gait, dystonia, and striatal lesions [49,50,51,52,53]. There is a large body of research indicating that the behavioral defects observed in HD patients are closely associated with striatal lesions. Striatal neurons are mostly made up of MSNs and interneurons. MSNs account for ~95% of all striatal neurons. As a marker of MSNs, DARPP-32 plays vital roles in the integration of the numerous neurotransmitter and neuromodulator signals that arrive at dopaminergic neurons [54, 55]. In the present study, behavioral defects and striatal lesions were induced by 3-NP as previously reported [32, 56, 57]. Compared to control mice, Rg1-treated mice exhibited a marked increase in neurological scores and total distance traveled. Moreover, we found that Rg1 alleviated striatal damage in the Rg1 + 3-NP groups compared with the model group, not only significantly elevating the number of striatal cells, as determined by Nissl staining, but also increasing the protein expression of striatal DARPP-32, as determined by WB analysis (Fig. 3).
Furthermore, to observe striatal damage in the different groups, TEM was used (Fig. 4). 3-NP selectively causes striatal lesions by inhibiting striatal SDH activity. The toxic effects of 3-NP in the striatum are as follows: disturbances of energy metabolism, depolarization of the mitochondrial membrane, dysregulation of intracellular calcium homeostasis, and release of pro-apoptotic proteins from mitochondria [44]. At the ultrastructural level, Rg1 dramatically alleviated disruption of the cytomembrane, condensation of chromatin, and mitochondrial dysfunction in the striatum of the 3-NP-treated mice (Fig. 4). Furthermore, Rg1 prevented a decrease in SDH activity and exerted antiapoptotic effects in the striatum of 3-NP-treated mice (Fig. 5).
A large number of studies have demonstrated that inflammation plays crucial roles in the occurrence and development of neurodegenerative diseases, including HD [58,59,60,61,62,63]. Neuroinflammation is mainly characterized by the following processes: BBB disruption, microglial activation, and increased expression of cytokines, which can initiate multiple neurodegenerative pathways. BBB disruption is associated with inflammatory and immune responses. As major resident immune cells, microglia play crucial roles in the CNS [64]. In the presence of adverse stimuli, resting microglia are activated. Activated microglia can produce a large number of neurotoxic cytokines, such as TNF-α and IL-1β [12, 65]. These results have been reported in many HD models [66,67,68]. Moreover, the BBB can protect neurons from factors in the systemic circulation and regulate the internal environment of the brain. As components of the BBB, astrocyte end-feet play vital roles in the regulation of the brain-to-blood and the blood-to-brain transport of solutes [69]. There have been some reports that administration of 3-NP induces degradation of the BBB in the striatal area in HD animal models [70, 71]. In other words, BBB breakdown may exacerbate the progression of diseases. In this study, we found that the protein expression of Iba-1 was elevated in 3-NP-treated mice compared to control mice by IHC and WB analysis (Fig. 6). In addition, the levels of proinflammatory cytokines, which are produced by activated microglia, were significantly increased in 3-NP-treated mice compared to control mice (Fig. 7). The TEM results confirmed that inflammation occurred, as indicated by demyelination and swelling of astrocyte end-feet in the striatum (Fig. 4j, n). As previously reported, Rg1 can exert neuroprotective effects associated with anti-inflammation in numerous models [72,73,74,75]. In fact, we found that Rg1 relieved 3-NP-induced neuroinflammation in the striatum. Additionally, treatment with Rg1 suppressed 3-NP-induced activation of microglial and proinflammatory cytokines (TNF-α and IL-1β) in the striatum (Fig. 6, 7).
By regulating the activation of the MAPK and NF-κB pathways, 3-NP causes severe impairments in the striatum. Some studies have shown that the symptoms of HD are ameliorated by compounds targeting the MAPK and NF-κB pathways. HD-like syndromes can be alleviated to varying degrees by inhibiting the activation of proteins associated with the MAPK and NF-κB signaling pathways, including p38, JNK, ERK1/2, and NF-κB p65 [7, 12, 76,77,78]. Moreover, there have been some reports on compounds that improve HD-related syndromes by suppressing the Nrf2 signaling pathway [32, 79]. It has been reported that Rg1 exerts neuroprotective activities in many neurological disease models, such as AD, PD, and stroke. In AD, Rg1 alleviates memory impairment by stabilizing the GSK3β/tau pathway and/or attenuating Aβ formation in okadaic acid (OKA)-treated rats [39]. On the other hand, Rg1 can also regulate amyloid precursor protein processing and activate PKA/CREB signaling in transgenic AD mice [80]. In PD, Rg1 can protect dopaminergic neurons from apoptosis by inhibiting oxidative stress and the JNK signaling cascade in a mouse model of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD [81]. Moreover, Rg1 can also protect dopaminergic neurons in the substantia nigra by regulating the activation of the IGF-IR signaling pathway in a rat model of 6-hydroxydopamine (6-OHDA)-induced PD [27] and prevent the degeneration of dopaminergic neurons by modulating the polarization dynamics of microglia and suppressing the NF-κB signaling pathway in a mouse model of lipopolysaccharide (LPS)-induced PD [82]. In stroke, Rg1 attenuates ischemia/reperfusion-induced neuronal damage by mediating the miR-144/Nrf2/ARE signaling pathway in transient middle cerebral artery occlusion (tMCAO) rats [83]. Therefore, we speculate that Rg1 exerts its neuroprotective effects through regulating signaling pathways involved in antioxidative, antiapoptotic, and anti-inflammatory effects. As an anti-inflammatory agent, Rg1 exerts anti-inflammatory effects by regulating the activation of the MAPK and NF-κB pathways in many models [84,85,86,87,88,89,90,91,92,93]. However, there have not yet been any reports that Rg1 can exert neuroprotective effects in HD animal models through the MAPK and NF-κB pathways. The results of the present study showed that Rg1 reduced the activation of p38, JNK, ERK1/2, and NF-κB p65 induced by 3-NP in the damaged striatum (Fig. 8). Our findings indicated that Rg1 can contribute to the survival of striatal neurons by suppressing the activation of the MAPK and NF-κB pathways.
In summary, our data demonstrated that Rg1 can exert neuroprotective effects in a mouse model of 3-NP-induced HD via the MAPK and NF-κB pathways. The possible underlying mechanisms are shown in Fig. 9. After crossing the BBB, 3-NP selectively enters striatal cells, in which the activity of SDH is inhibited, leading to mitochondrial dysfunction and the death of neurons in the striatum [79, 94]. BBB breakdown leads to the leakage of more peripheral proinflammatory cells and microglia into the CNS [95]. In microglia, 3-NP induces the activation of the MAPK and NF-κB signaling pathways. In our study, Rg1 alleviated 3-NP-induced mitochondrial dysfunction and BBB disruption, and it also contributed to the survival of striatal neurons by inhibiting striatal microglial activation, apoptosis, and the expression of proinflammatory mediators induced by 3-NP. Moreover, we found that Rg1 protected against striatal neuronal death by suppressing the activation of proteins in the MAPK and NF-κB pathways, such as JNK, p38, NF-κB p65, and ERK1/2. Thus, Rg1 may exert neuroprotective effects on striatal neurons in a mouse model of 3-NP-induced HD. Nevertheless, further studies are needed to investigate the other exact mechanisms underlying the neuroprotective effect of Rg1 in HD.

3-NP inhibits the activity of SDH in striatal cells, leading to mitochondrial dysfunction and the death of neurons in the striatum, and it also induces BBB disruption, which leads to the leakage of more peripheral proinflammatory cells and microglia into the CNS. In microglia, 3-NP induces the activation of the MAPK and NF-κB signaling pathways. Rg1 alleviates 3-NP-induced mitochondrial dysfunction and BBB breakdown, and it also contributes to the survival of striatal neurons by inhibiting striatal microglial activation, apoptosis, and the expression of proinflammatory cytokines induced by 3-NP. Moreover, Rg1 protects against striatal neuronal death by suppressing the activation of proteins in the MAPK and NF-κB signaling pathways.
Our research together with those of related studies suggest that Rg1 exerts neuroprotective effects in a mouse model of 3-NP-induced HD. Furthermore, Rg1 prevents striatal degeneration and protects striatal neurons through its anti-inflammatory and antiapoptotic effects, inhibiting the activation of the MAPK and NF-κB signaling pathways. Our findings demonstrate that Rg1 can exert neuroprotective effects in a mouse model of 3-NP-induced HD via the MAPK and NF-κB pathways. The study suggests that Rg1 might have potential as a drug candidate for the treatment of HD.
References
Walter C, Clemens LE, Muller AJ, Fallier-Becker P, Proikas-Cezanne T, Riess O, et al. Activation of AMPK-induced autophagy ameliorates Huntington disease pathology in vitro. Neuropharmacology. 2016;108:24–38.
Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. 2010;90:905–81.
Snowden JS. The neuropsychology of Huntington’s disease. Arch Clin Neuropsychol. 2017;32:876–87.
Dargaei Z, Bang JY, Mahadevan V, Khademullah CS, Bedard S, Parfitt GM, et al. Restoring GABAergic inhibition rescues memory deficits in a Huntington’s disease mouse model. Proc Natl Acad Sci USA. 2018;115:E1618–26.
Sepers MD, Smith-Dijak A, LeDue J, Kolodziejczyk K, Mackie K, Raymond LA. Endocannabinoid-specific impairment in synaptic plasticity in striatum of Huntington’s disease mouse model. J Neurosci. 2018;38:544–54.
Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Prim. 2015;1:15005.
Jang M, Cho IH. Sulforaphane ameliorates 3-nitropropionic acid-induced striatal toxicity by activating the Keap1-Nrf2-ARE pathway and inhibiting the MAPKs and NF-κB pathways. Mol Neurobiol. 2015;53:2619–35.
Brouillet E. The 3‐NP model of striatal neurodegeneration. Curr Protoc Neurosci. 2014;67:9.48.
Jamwal S, Kumar P. Spermidine ameliorates 3-nitropropionic acid (3-NP)-induced striatal toxicity: Possible role of oxidative stress, neuroinflammation, and neurotransmitters. Physiol Behav. 2016;155:180–7.
Wang L, Wang J, Yang L, Zhou SM, Guan SY, Yang LK, et al. Effect of Praeruptorin C on 3-nitropropionic acid induced Huntington's disease-like symptoms in mice. Biomed Pharmacother. 2017;86:81–7.
Dhadde SB, Nagakannan P, Roopesh M, Anand Kumar SR, Thippeswamy BS, Veerapur VP, et al. Effect of embelin against 3-nitropropionic acid-induced Huntington’s disease in rats. Biomed Pharmacother. 2016;77:52–8.
Jang M, Lee MJ, Cho IH. Ethyl pyruvate ameliorates 3-nitropropionic acid-induced striatal toxicity through anti-neuronal cell death and anti-inflammatory mechanisms. Brain Behav Immun. 2014;38:151–65.
Lai JL, Liu YH, Liu C, Qi MP, Liu RN, Zhu XF, et al. Indirubin inhibits LPS-induced inflammation via TLR4 abrogation mediated by the NF-κB and MAPK signaling pathways. Inflammation 2017;40:1–12.
Haddad JJ. N-methyl-D-aspartate (NMDA) and the regulation of mitogen-activated protein kinase (MAPK) signaling pathways: a revolving neurochemical axis for therapeutic intervention? Prog Neurobiol. 2005;77:252–82.
Jiao D, Jiang Q, Liu Y, Ji L. Nephroprotective effect of wogonin against cadmium-induced nephrotoxicity via inhibition of oxidative stress-induced MAPK and NF-kB pathway in Sprague Dawley rats. Hum Exp Toxicol. 2019;38:1082–91.
Santana-Martinez RA, Leon-Contreras JC, Barrera-Oviedo D, Pedraza-Chaverri J, Hernandez-Pando R, Maldonado PD. Sustained activation of JNK induced by quinolinic acid alters the BDNF/TrkB axis in the rat striatum. Neuroscience 2018;383:22–32.
Subramaniam S, Unsicker K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010;277:22–9.
Bodai L, Marsh JL. A novel target for Huntington’s disease: ERK at the crossroads of signaling. The ERK signaling pathway is implicated in Huntington’s disease and its upregulation ameliorates pathology. Bioessays. 2012;34:142–8.
Fusco FR, Anzilotti S, Giampa C, Dato C, Laurenti D, Leuti A, et al. Changes in the expression of extracellular regulated kinase (ERK1/2) in the R6/2 mouse model of Huntington’s disease after phosphodiesterase IV inhibition. Neurobiol Dis. 2012;46:225–33.
Yusuf IO, Cheng PH, Chen HM, Chang YF, Chang CY, Yang HI, et al. Fibroblast growth factor 9 suppresses striatal cell death dominantly through ERK signaling in Huntington’s disease. Cell Physiol Biochem. 2018;48:605–17.
Napolitano M, Zei D, Centonze D, Palermo R, Bernardi G, Vacca A, et al. NF-κB/NOS cross-talk induced by mitochondrial complex II inhibition: implications for Huntington’s disease. Neurosci Lett. 2008;434:241–6.
Wang KS, Li J, Wang Z, Mi C, Ma J, Piao LX, et al. Artemisinin inhibits inflammatory response via regulating NF-kappaB and MAPK signaling pathways. Immunopharmacol Immunotoxicol. 2017;39:28–36.
Kim KH, Lee D, Lee HL, Kim C-E, Jung K, Kang KS. Beneficial effects of Panax ginseng for the treatment and prevention of neurodegenerative diseases: past findings and future directions. J Ginseng Res. 2018;42:239–47.
Zhou T, Zu G, Zhang X, Wang X, Li S, Gong X, et al. Neuroprotective effects of ginsenoside Rg1 through the Wnt/β-catenin signaling pathway in both in vivo and in vitro models of Parkinson’s disease. Neuropharmacology. 2016;101:480–9.
Heng Y, Zhang QS, Mu Z, Hu JF, Yuan YH, Chen NH. Ginsenoside Rg1 attenuates motor impairment and neuroinflammation in the MPTP-probenecid-induced parkinsonism mouse model by targeting alpha-synuclein abnormalities in the substantia nigra. Toxicol Lett. 2016;243:7–21.
Jiang W, Wang Z, Jiang Y, Lu M, Li X. Ginsenoside Rg1 ameliorates motor function in an animal model of Parkinson’s disease. Pharmacology. 2015;96:25–31.
Xu L, Chen WF, Wong MS. Ginsenoside Rg1 protects dopaminergic neurons in a rat model of Parkinson’s disease through the IGF-I receptor signalling pathway. Br J Pharmacol. 2009;158:738–48.
Liu Q, Kou JP, Yu BY. Ginsenoside Rg1 protects against hydrogen peroxide-induced cell death in PC12 cells via inhibiting NF-kappaB activation. Neurochem Int. 2011;58:119–25.
Zhu J, Mu X, Zeng J, Xu C, Liu J, Zhang M, et al. Ginsenoside Rg1 prevents cognitive impairment and hippocampus senescence in a rat model of D-galactose-induced aging. PLoS One. 2014;9:e101291.
Nie L, Xia J, Li H, Zhang Z, Yang Y, Huang X, et al. Ginsenoside Rg1 ameliorates behavioral abnormalities and modulates the hippocampal proteomic change in triple transgenic mice of Alzheimer’s disease. Oxid Med Cell Longev. 2017;2017:6473506.
Jang M, Lee MJ, Kim CS, Cho IH. Korean red ginseng extract attenuates 3-nitropropionic acid-induced Huntington’s-like symptoms. Evid Based Complement Altern Med. 2013;2013:237207.
Gao Y, Chu SF, Li JP, Zhang Z, Yan JQ, Wen ZL, et al. Protopanaxtriol protects against 3-nitropropionic acid-induced oxidative stress in a rat model of Huntington's disease. Acta Pharmacol Sin. 2015;36:311–22.
Jamwal S, Kumar P. L-theanine, a component of green tea prevents 3-nitropropionic acid (3-NP)-induced striatal toxicity by modulating nitric oxide pathway. Mol Neurobiol. 2016;54:2327–37.
Navarrete F, Garcia-Gutierrez MS, Laborda J, Manzanares J. Deletion of Dlk2 increases the vulnerability to anxiety-like behaviors and impairs the anxiolytic action of alprazolam. Psychoneuroendocrinology. 2017;85:134–41.
Zhang Y, Jiang YY, Shao S, Zhang C, Liu FY, Wan Y, et al. Inhibiting medial septal cholinergic neurons with DREADD alleviated anxiety-like behaviors in mice. Neurosci Lett. 2017;638:139–44.
Wen L, Zhang QS, Heng Y, Chen Y, Wang S, Yuan YH, et al. NLRP3 inflammasome activation in the thymus of MPTP-induced Parkinsonian mouse model. Toxicol Lett. 2018;288:1–8.
Kroymann J, Mitchell-Olds T. Epistasis and balanced polymorphism influencing complex trait variation. Nature. 2005;435:95–8.
Feng S, Xing C, Shen T, Qiao Y, Wang R, Chen J, et al. Abnormal paraventricular nucleus of hypothalamus and growth retardation associated with loss of nuclear receptor Gene COUP-TFII. Sci Rep. 2017;7:5282.
Song XY, Hu JF, Chu SF, Zhang Z, Xu S, Yuan YH, et al. Ginsenoside Rg1 attenuates okadaic acid induced spatial memory impairment by the GSK3β/tau signaling pathway and the Aβ formation prevention in rats. Eur J Pharmacol. 2013;710:29–38.
Chen C, Chu SF, Ai QD, Zhang Z, Guan FF, Wang SS, et al. CKLF1 aggravates focal cerebral ischemia injury at early stage partly by modulating microglia/macrophage toward M1 polarization through CCR4. Cell Mol Neurobiol. 2019;39:651–69.
Sun H, Tang Y, Guan X, Li L, Wang D. Effects of selective hypothermia on blood-brain barrier integrity and tight junction protein expression levels after intracerebral hemorrhage in rats. Biol Chem. 2013;394:1317–24.
Zuo Y, Huang L, Enkhjargal B, Xu W, Umut O, Travis ZD, et al. Activation of retinoid X receptor by bexarotene attenuates neuroinflammation via PPARγ/SIRT6/FoxO3a pathway after subarachnoid hemorrhage in rats. J Neuroinflammation. 2019;16:47.
Sebastianutto I, Cenci MA, Fieblinger T. Alterations of striatal indirect pathway neurons precede motor deficits in two mouse models of Huntington’s disease. Neurobiol Dis. 2017;105:117–31.
Cho KJ, Cheon SY, Kim GW. Apoptosis signal-regulating kinase 1 mediates striatal degeneration via the regulation of C1q. Sci Rep. 2016;6:18840.
Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood–brain barrier. Nature. 2010;468:557–61.
Andre R, Carty L, Tabrizi SJ. Disruption of immune cell function by mutant huntingtin in Huntington’s disease pathogenesis. Curr Opin Pharmacol. 2016;26:33–8.
Kim YK, Na KS, Myint AM, Leonard BE. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:277–84.
Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87:10–20.
Hanna DM, Tadros MG, Khalifa AE. ADIOL protects against 3-NP-induced neurotoxicity in rats: Possible impact of its anti-oxidant, anti-inflammatory and anti-apoptotic actions. Prog Neuropsychopharmacol Biol Psychiatry. 2015;60:36–51.
Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC. Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med. 2017;7:a024240.
Silva-Palacios A, Ostolga-Chavarría M, Buelna-Chontal M, Garibay C, Hernández-Reséndiz S, Roldán FJ, et al. 3-NP-induced Huntington’s-like disease impairs Nrf2 activation without loss of cardiac function in aged rats. Exp Gerontol. 2017;96:89–98.
Elbaz EM, Helmy HS, El-Sahar AE, Saad MA, Sayed RH. Lercanidipine boosts the efficacy of mesenchymal stem cell therapy in 3-NP-induced Huntington’s disease model rats via modulation of the calcium/calcineurin/NFATc4 and Wnt/beta-catenin signalling pathways. Neurochem Int. 2019;131:104548.
Torres‐Cruz FM, Mendoza E, Vivar‐Cortés IC, García‐Sierra F, Hernández‐Echeagaray E. Do BDNF and NT‐4/5 exert synergistic or occlusive effects on corticostriatal transmission in a male mouse model of Huntington’s disease? J Neurosci Res. 2019;97:1665–77.
Sgambato-Faure V, Qian Y, Forssberg H, Diaz Heijtz R. Motor skill learning is associated with phase-dependent modifications in the striatal cAMP/PKA/DARPP-32 signaling pathway in rodents. PLoS One. 2015;10:e0140974.
Xenias HS, Ibanez-Sandoval O, Koos T, Tepper JM. Are striatal tyrosine hydroxylase interneurons dopaminergic? J Neurosci. 2015;35:6584–99.
Domenici MR, Chiodi V, Averna M, Armida M, Pezzola A, Pepponi R, et al. Neuronal adenosine A2A receptor overexpression is neuroprotective towards 3-nitropropionic acid-induced striatal toxicity: a rat model of Huntington’s disease. Purinergic Signal. 2018;14:235–43.
Ramachandran S, Thangarajan S. Thymoquinone loaded solid lipid nanoparticles counteracts 3-nitropropionic acid induced motor impairments and neuroinflammation in rat model of Huntington’s disease. Metab Brain Dis. 2018;33:1459–70.
Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–69.
Xu L, He D, Bai Y. Microglia-mediated inflammation and neurodegenerative disease. Mol Neurobiol. 2015;53:6709–15.
Thome AD, Harms AS, Volpicelli-Daley LA, Standaert DG. microRNA-155 Regulates alpha-synuclein-induced inflammatory responses in models of Parkinson disease. J Neurosci. 2016;36:2383–90.
Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N. Y.). 2018;4:575–90.
Pido-Lopez J, Andre R, Benjamin AC, Ali N, Farag S, Tabrizi SJ, et al. In vivo neutralization of the protagonist role of macrophages during the chronic inflammatory stage of Huntington’s disease. Sci Rep. 2018;8:11447.
Siew JJ, Chen HM, Chen HY, Chen HL, Chen CM, Soong BW, et al. Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat Commun. 2019;10:3473.
Liu CY, Wang X, Liu C, Zhang HL. Pharmacological targeting of microglial activation: New therapeutic approach. Front Cell Neurosci. 2019;13:514.
Paldino E, Balducci C, La Vitola P, Artioli L, D’Angelo V, Giampà C, et al. Neuroprotective effects of doxycycline in the R6/2 mouse model of Huntington’s disease. Mol Neurobiol. 2020;57:1889–903.
Chang KH, Wu YR, Chen YC, Chen CM. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav Immun. 2015;44:121–7.
Kalonia H, Kumar A. Suppressing inflammatory cascade by cyclo-oxygenase inhibitors attenuates quinolinic acid induced Huntington’s disease-like alterations in rats. Life Sci. 2011;88:784–91.
Valadao PAC, Oliveira BDS, Joviano-Santos JV, Vieira ELM, Rocha NP, Teixeira AL, et al. Inflammatory changes in peripheral organs in the BACHD murine model of Huntington’s disease. Life Sci. 2019;232:116653.
Sweeney MD, Sagare AP, Zlokovic BV. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–50.
Duran-Vilaregut J, del Valle J, Manich G, Camins A, Pallàs M, Vilaplana J, et al. Role of matrix metalloproteinase-9 (MMP-9) in striatal blood-brain barrier disruption in a 3-nitropropionic acid model of Huntington’s disease. Neuropathol Appl Neurobiol. 2011;37:525–37.
Lin CY, Hsu YH, Lin MH, Yang TH, Chen HM, Chen YC, et al. Neurovascular abnormalities in humans and mice with Huntington’s disease. Exp Neurol. 2013;250:20–30.
Gao XQ, Du ZR, Yuan LJ, Zhang WD, Chen L, Teng JJ, et al. Ginsenoside Rg1 exerts anti-inflammatory effects via G protein-coupled estrogen receptor in lipopolysaccharide-induced microglia activation. Front Neurosci. 2019;13:1168.
Sun XC, Ren XF, Chen L, Gao XQ, Xie JX, Chen WF. Glucocorticoid receptor is involved in the neuroprotective effect of ginsenoside Rg1 against inflammation-induced dopaminergic neuronal degeneration in substantia nigra. J Steroid Biochem Mol Biol. 2016;155:94–103.
Li Y, Guan Y, Wang Y, Yu CL, Zhai FG, Guan LX. Neuroprotective effect of the ginsenoside Rg1 on cerebral ischemic injury in vivo and in vitro is mediated by PPARγ-regulated antioxidative and anti-inflammatory pathways. Evid Based Complement Altern Med. 2017;2017:7842082.
Hu JF, Song XY, Chu SF, Chen J, Ji HJ, Chen XY, et al. Inhibitory effect of ginsenoside Rg1 on lipopolysaccharide-induced microglial activation in mice. Brain Res. 2011;1374:8–14.
Laprairie RB, Warford JR, Hutchings S, Robertson GS, Kelly MEM, Denovan-Wright EM. The cytokine and endocannabinoid systems are co-regulated by NF-κB p65/RelA in cell culture and transgenic mouse models of Huntington’s disease and in striatal tissue from Huntington’s disease patients. J Neuroimmunol. 2014;267:61–72.
Mahdy HM, Mohamed MR, Emam MA, Karim AM, Abdel-Naim AB, Khalifa AE. The anti-apoptotic and anti-inflammatory properties of puerarin attenuate 3-nitropropionic-acid induced neurotoxicity in rats. Can J Physiol Pharm. 2014;92:252–8.
Liu SY, Yu XL, Zhu J, Liu XM, Zhang Y, Dong QX, et al. Intravenous immunoglobulin ameliorates motor and cognitive deficits and neuropathology in R6/2 mouse model of Huntington’s disease by decreasing mutant huntingtin protein level and normalizing NF-kappaB signaling pathway. Brain Res. 2018;1697:21–33.
Kulasekaran G, Ganapasam S. Neuroprotective efficacy of naringin on 3-nitropropionic acid-induced mitochondrial dysfunction through the modulation of Nrf2 signaling pathway in PC12 cells. Mol Cell Biochem. 2015;409:199–211.
Fang F, Chen X, Huang T, Lue LF, Luddy JS, Yan SS. Multi-faced neuroprotective effects of ginsenoside Rg1 in an Alzheimer mouse model. Biochim Biophys Acta. 2012;1822:286–92.
Chen XC, Zhou YC, Chen Y, Zhu YG, Fang F, Chen LM. Ginsenoside Rg1 reduces MPTP-induced substantia nigra neuron loss by suppressing oxidative stress. Acta Pharmacol Sin. 2005;26:56–62.
Liu JQ, Zhao M, Zhang Z, Cui LY, Zhou X, Zhang W, et al. Rg1 improves LPS-induced Parkinsonian symptoms in mice via inhibition of NF-kappaB signaling and modulation of M1/M2 polarization. Acta Pharmacol Sin. 2020;41:523–34.
Chu SF, Zhang Z, Zhou X, He WB, Chen C, Luo P, et al. Ginsenoside Rg1 protects against ischemic/reperfusion-induced neuronal injury through miR-144/Nrf2/ARE pathway. Acta Pharmacol Sin. 2019;40:13–25.
Shi Q, He Q, Chen W, Long J, Zhang B. Ginsenoside Rg1 abolish imiquimod‐induced psoriasis‐like dermatitis in BALB/c mice via downregulating NF‐κB signaling pathway. J Food Biochem. 2019;43:e13032.
Mo J, Zhou Y, Yang R, Zhang P, He B, Yang J, et al. Ginsenoside Rg1 ameliorates palmitic acid-induced insulin resistance in HepG2 cells in association with modulating Akt and JNK activity. Pharmacol Rep. 2019;71:1160–7.
Luo M, Yan D, Sun Q, Tao J, Xu L, Sun H, et al. Ginsenoside Rg1 attenuates cardiomyocyte apoptosis and inflammation via the TLR4/NF‐kB/NLRP3 pathway. J Cell Biochem. 2020;121:2994–3004.
Zheng T, Jiang H, Jin R, Zhao Y, Bai Y, Xu H, et al. Ginsenoside Rg1 attenuates protein aggregation and inflammatory response following cerebral ischemia and reperfusion injury. Eur J Pharmacol. 2019;853:65–73.
Li W, Chu Y, Zhang L, Yin L, Li L. Ginsenoside Rg1 attenuates tau phosphorylation in SK-N-SH induced by Aβ‐stimulated THP-1 supernatant and the involvement of p38 pathway activation. Life Sci. 2012;91:809–15.
Shi C, Zheng DD, Fang L, Wu F, Kwong WH, Xu J. Ginsenoside Rg1 promotes nonamyloidgenic cleavage of APP via estrogen receptor signaling to MAPK/ERK and PI3K/Akt. Biochim Biophys Acta. 2012;1820:453–60.
Ge KL, Chen WF, Xie JX, Wong MS. Ginsenoside Rg1 protects against 6-OHDA-induced toxicity in MES23.5 cells via Akt and ERK signaling pathways. J Ethnopharmacol. 2010;127:118–23.
Ye Y, Shan Y, Bao C, Hu Y, Wang L. Ginsenoside Rg1 protects against hind-limb ischemia reperfusion induced lung injury via NF-κB/COX-2 signaling pathway. Int Immunopharmacol. 2018;60:96–103.
Zhang XJ, He C, Li P, Su H, Wan JB. Ginsenoside Rg1, a potential JNK inhibitor, protects against ischemia/reperfusion-induced liver damage. J Funct Foods. 2015;15:580–92.
Fan C, Song Q, Wang P, Li Y, Yang M, Yu SY. Neuroprotective effects of ginsenoside-Rg1 against depression-like behaviors via suppressing glial activation, synaptic deficits, and neuronal apoptosis in rats. Front Immunol. 2018;9:2889.
Liu P, Li Y, Liu D, Ji X, Chi T, Li L, et al. Tolfenamic acid attenuates 3-nitropropionic acid-induced biochemical alteration in mice. Neurochem Res. 2018;43:1938–46.
Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: From physiology to disease and back. Physiol Rev. 2019;99:21–78.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81773924, 81873026, and 81973499), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-1-004), the Drug Innovation Major Project (2018ZX09711001-003-005 and 2018ZX09711001-009-013), and the Beijing Key Laboratory of New Drug Mechanisms and Pharmacological Evaluation Study (BZ0150).
Author information
Authors and Affiliations
Contributions
XY, SFC, and NHC designed the study; XY, ZZW, and FFL performed the experiments; ZZW, FFL, and YHY contributed analytic tools; XY and SFC analyzed the results; and XY wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Yang, X., Chu, Sf., Wang, Zz. et al. Ginsenoside Rg1 exerts neuroprotective effects in 3-nitropronpionic acid-induced mouse model of Huntington’s disease via suppressing MAPKs and NF-κB pathways in the striatum. Acta Pharmacol Sin 42, 1409–1421 (2021). https://doi.org/10.1038/s41401-020-00558-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00558-4
Keywords
This article is cited by
-
Medicinal herbal remedies in neurodegenerative diseases: an update on antioxidant potential
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Ginsenoside Rg1 in neurological diseases: From bench to bedside
Acta Pharmacologica Sinica (2023)
-
Exploring the Potential Therapeutic Approach Using Ginsenosides for the Management of Neurodegenerative Disorders
Molecular Biotechnology (2023)
-
An Overview of the Pathophysiological Mechanisms of 3-Nitropropionic Acid (3-NPA) as a Neurotoxin in a Huntington's Disease Model and Its Relevance to Drug Discovery and Development
Neurochemical Research (2023)
-
Protective role of trametenolic acid B against sevoflurane-induced cognitive impairments by its different regulatory modalities of mir-329-3p in neurons and microglia
Molecular Medicine (2022)
