
Abstract
The novel severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) causes coronavirus disease 2019 (COVID-19) and an ongoing severe pandemic. Curative drugs specific for COVID-19 are currently lacking. Chloroquine phosphate and its derivative hydroxychloroquine, which have been used in the treatment and prevention of malaria and autoimmune diseases for decades, were found to inhibit SARS-CoV-2 infection with high potency in vitro and have shown clinical and virologic benefits in COVID-19 patients. Therefore, chloroquine phosphate was first used in the treatment of COVID-19 in China. Later, under a limited emergency-use authorization from the FDA, hydroxychloroquine in combination with azithromycin was used to treat COVID-19 patients in the USA, although the mechanisms of the anti-COVID-19 effects remain unclear. Preliminary outcomes from clinical trials in several countries have generated controversial results. The desperation to control the pandemic overrode the concerns regarding the serious adverse effects of chloroquine derivatives and combination drugs, including lethal arrhythmias and cardiomyopathy. The risks of these treatments have become more complex as a result of findings that COVID-19 is actually a multisystem disease. While respiratory symptoms are the major clinical manifestations, cardiovascular abnormalities, including arrhythmias, myocarditis, heart failure, and ischemic stroke, have been reported in a significant number of COVID-19 patients. Patients with preexisting cardiovascular conditions (hypertension, arrhythmias, etc.) are at increased risk of severe COVID-19 and death. From pharmacological and cardiovascular perspectives, therefore, the treatment of COVID-19 with chloroquine and its derivatives should be systematically evaluated, and patients should be routinely monitored for cardiovascular conditions to prevent lethal adverse events.
Similar content being viewed by others
Introduction
Coronavirus disease 2019 (COVID-19) is caused by infection with severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), which has spread around the globe and is causing an ongoing severe pandemic with 11,874,226 confirmed cases and 545,481 deaths worldwide that have been reported to the World Health Organization (WHO) as of July 9, 2020 (https://www.who.int/emergencies/diseases/novel-coronavirus-2019). To date, there is still no known approved curative drug therapy specific for COVID-19, especially for patients with the severe and critical forms [1]. However, chloroquine phosphate and its derivative hydroxychloroquine, which have been used for decades in the treatment and prevention of malaria and chronic inflammatory diseases such as rheumatoid arthritis and systemic lupus erythematosus, were discovered to have a high inhibitory potency against SARS-CoV-2 infection in vitro [2,3,4,5] and favorable clinical and virologic benefits in COVID-19 patients [6,7,8,9,10], and they have emerged as important therapies for COVID-19 in several countries, including China, France, USA, and India, although the mechanisms of their anti-COVID-19 effects remain unclear. The desperation to control the pandemic has overridden the concerns regarding the serious adverse effects of chloroquine derivatives and azithromycin, including lethal arrhythmias and cardiomyopathy. Clinical trials in several countries have provided controversial results. The findings that COVID-19 is a multisystem disease involving injuries to many organs, including the lung, heart, and vasculature, have made the risks of using these drugs even more complex. COVID-19 patients with preexisting cardiovascular conditions (hypertension, arrhythmias, heart failure, etc.) are at higher risk of severe COVID-19 and death (Table 1). Here, we provide pharmacological and cardiovascular perspectives on the application of chloroquine derivatives in the treatment of COVID-19. Systematic evaluations of their efficacy and safety, especially of the potential cardiovascular toxicity of chloroquine and hydroxychloroquine and combination therapies with other drugs in the treatment of COVID-19, and genetic variability in the metabolism of these drugs in patients are required to prevent lethal cardiovascular adverse events.
Pharmacology of chloroquine/hydroxychloroquine for COVID-19 therapy
Antiviral effects and mechanisms
In several in vitro studies, chloroquine [3, 5] and hydroxychloroquine [2, 4] were found to be able to inhibit SARS-CoV-2 infection and show clinical and virologic benefits in COVID-19 patients [6,7,8,9,10]. The two drugs inhibited SARS-CoV-2 at low-micromolar concentrations with 50% cytotoxic concentration (CC50) values over 100 μM, implying the efficacy and safety of these drugs [2,3,4,5]. Although these in vitro and anecdotal data are still limited and the mechanisms of the anti-COVID-19 effects remain unclear, the apparent efficacy and safety of chloroquine phosphate or hydroxychloroquine have attracted much attention for application of these drugs as potential therapies for COVID-19. Chloroquine phosphate was therefore recommended in the Guidelines for the Prevention, Diagnosis, and Treatment of COVID-19 by the National Health Commission (NHC) of China. An open-label nonrandomized study by Gautret et al. [7] that used hydroxychloroquine in combination in some patients with azithromycin, an azalide antibiotic with putative antiviral properties, has garnered unusual attention. Hydroxychloroquine in combination with azithromycin was granted limited emergency-use authorization by the USA FDA to treat COVID-19 patients. Preliminary clinical data showed that chloroquine or hydroxychloroquine prevented the exacerbation of pneumonia, promoted viral clearance and shortened the disease course in SARS-CoV-2-infected patients [7,8,9,10]. Until May 13th, 2020, a total of 182 clinical trials proposed using hydroxychloroquine, and 58 studies that proposed using chloroquine in the treatment of COVID-19 were registered with the FDA (Tables 2 and 3). For patients diagnosed with mild, moderate and severe cases of SARS-CoV-2 pneumonia, 500 mg chloroquine phosphate tablets were given twice a day for 10 days according to the sixth and seventh editions of the Guidelines for the Prevention, Diagnosis, and Treatment of Pneumonia Caused by COVID-19 recommended by the NHC of China. In several other countries, including the United States, hydroxychloroquine has been administered to hospitalized COVID-19 patients due to its higher in vitro activity against SARS-CoV-2 and wider availability in these countries compared with chloroquine phosphate.
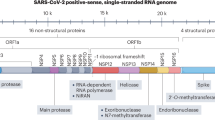
Chloroquine and hydroxychloroquine are oral prescription drugs that have been used for several decades in the treatment of malaria and autoimmune diseases. Currently, it is not clear why they can inhibit SARS-CoV-2 and have therapeutic effects on COVID-19. Since both of these drugs contain an amino group attached to a quinoline ring (Fig. 1), they are weak diprotic bases that probably accumulate within intracellular acidic compartments such as lysosomes [11]. They increase the pH of lysosomes and lead to the expansion, vacuolization and dysfunction of lysosomes [12]. As a result, these drugs interfere with the fusion process and prevent RNA release by the viruses, thus inhibiting the growth of these intracellular pathogens [3, 13, 14]. In addition, these drugs can alter the terminal glycosylation of ACE2, the cellular receptor of the coronaviruses SARS-CoV and SARS-CoV-2, preventing virus-receptor binding and abrogating the infection [3]. This might help to explain the broad-spectrum antiviral effects of chloroquine and hydroxychloroquine. Indeed, both drugs have been previously demonstrated to have profound antiviral activity against various viruses, including SARS-CoV [3], dengue virus [15, 16], HIV-1 [17, 18], influenza A [19], Ebola virus [20], and human coronavirus OC43 [21].

Left: chloroquine; right: hydroxychloroquine.
Anti-inflammatory and immunomodulatory effects
Chloroquine and hydroxychloroquine are regarded as potent anti-inflammatory agents and immunomodulators. These drugs have been widely used for many years in the treatment of malaria and autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus and Sjögren’s syndrome. Both drugs impair the production of proinflammatory cytokines, including tumor necrosis factor α (TNFα) [22,23,24], interleukin (IL)-1 [22, 25], IL-6 [22, 24, 25], and interferon-γ [24]. Additionally, they prevent innate immune activation by blocking the interaction between cytosolic DNA and the nucleic acid sensor cyclic GMP-AMP (cGAMP) synthase (cGAS) [26] and the interaction of Toll-like receptors (TLRs) with nucleic acid ligands [27,28,29].
Considering that inflammatory reactions and cytokine storms occur commonly in severe COVID-19 patients, the anti-inflammatory properties of chloroquine and hydroxychloroquine might provide important clinical benefits in the treatment of COVID-19 as well.
Adverse effects
Although chloroquine and hydroxychloroquine are generally well tolerated and usually considered safe even during pregnancy, we should be aware of their serious adverse effects, even with a short duration of treatment. These adverse effects include gastrointestinal and cutaneous manifestations, hypoglycemia, neuropsychiatric effects, drug–drug interactions, idiosyncratic hypersensitivity reactions, myopathy, peripheral neuropathy, and cardiac toxicity [30]. According to a systematic review of case series concerning cardiac complications attributed to chloroquine and hydroxychloroquine, conduction disorders are the main reported side effect, affecting 85% of patients; other nonspecific adverse cardiac events include ventricular hypertrophy (22%), heart failure (26.8%), hypokinesia (9.4%), pulmonary arterial hypertension (3.9%), and valvular dysfunction (7.1%) [31]. For patients reported to have been withdrawn from treatment, only 44.9% recovered normal heart function, whereas 30.8% died and 12.9% reported irreversible damage [31]. It is not currently known, however, whether heart is more susceptible to chloroquine treatment than other organs. Chloroquine- or hydroxychloroquine-related cardiac disorders, such as prolongation of the QTc interval, are rare but severe and life-threatening. Both chloroquine and hydroxychloroquine interfere with ventricular repolarization and may lead to prolongation of the QT interval and increase the risk of Torsades de Pointes. The effect is dose-dependent: after receiving a dose of 600 mg, the mean QTc increases by 6.1 ms, and after a dose of 1200 mg, the mean QTc increases by 28 ms [32, 33]. However, the effect varies among individuals. Among 30 children given short courses of chloroquine for malaria, 1 experienced an increase in the QTc interval of 64 ms after just 1 day of treatment [34].
Potential mechanisms of the adverse effects
Blockade of K+ channels
The activities of K+ channels are the major determinants of the repolarization of cardiac myocytes. Chloroquine and hydroxychloroquine have been reported to inhibit multiple K+ currents, including the inward rectifier K+ (Kir) current (IK1) [35,36,37,38], the rapidly activating delayed rectifier K+ current (IKr) associated with the human eEther-a-go-go-go related gene (hERG) [39, 40], the fast transient outward K+ current (Ito) [41], and the ATP-sensitive inward rectifier K+ current (KATP) [42, 43].
-
(1)
IK1: Chloroquine, hydroxychloroquine and other quinines inhibit the cardiac IK1 current and can induce lethal ventricular arrhythmias. Chloroquine causes a dose- and voltage-dependent reduction in the IK1 current magnitude, action potential (AP) duration and effective refractory period [35,36,37,38]. The peak outward current elicited by the ventricular AP is inhibited by chloroquine at a half-maximal inhibitory concentration (IC50) of ~1.2–3 μM [35, 36, 38, 43, 44]. Chloroquine and related compounds can inhibit Kir channels by multiple potential mechanisms. First, chloroquine can bind to the cytoplasmic pore domain and block the cytoplasmic conduction pathway, which is stabilized by negatively charged and aromatic amino acids within a central pocket [37]. Unlike most ion channel blockers, chloroquine does not bind within the transmembrane pore and thus can reach its binding site even while polyamines remain deeper within the channel vestibule [37]. These binding features of chloroquine could explain its relatively low affinity for Kir channels but high effectiveness in blocking the IK1 current [37]. Second, chloroquine shows allosteric effects on channel gating. Comparative molecular modeling and ligand docking of chloroquine in the intracellular domains of Kir2.1, Kir3.1 and Kir6.2 suggest that chloroquine blocks K+ flow by interacting with negatively charged amino acids facing the ion permeation vestibule of the channels [38, 43]. In addition, chloroquine abrogates the cardiac protective effect of the IK1 channel activator zacopride, exacerbating myocardial infarction (MI) and post-MI cardiac remodeling [45, 46].
-
(2)
IhERG: Chloroquine inhibits IhERG in a concentration- and time-dependent manner at a relatively low IC50 of 2.5 μM, implying that it is a potent blocker of IhERG [40, 47]. Inhibition of the hERG current (IhERG) can lead to a prolongation of the QT interval, which may, under certain circumstances, lead to Torsade de Pointes [48]. However, not all hERG blockers have a high risk of Torsade de Pointes. There are drugs that block hERG and prolong the heart rate-corrected QT (QTc) interval but have a low risk of Torsade de Pointes (e.g., ranolazine, verapamil, and amiodarone) because they block other inward currents such as late Na+ and/or L-type Ca2+ currents [39]. Blocking these inward currents has antiarrhythmic effects by preventing early afterdepolarizations [39]. Thus, these drugs prolong QTc without heart rate-corrected J‐Tpeak (J‐Tpeakc) prolongation and are considered to be balanced ion channel blockers [39]. To further evaluate the torsade risk of chloroquine, a comprehensive in vitro proarrhythmia assay (CiPA) was conducted. Chloroquine prolongs both the QTc interval and the J‐Tpeakc interval, suggesting that chloroquine is a strong blocker of IhERG with a high torsade risk [39].
-
(3)
Ito: Chloroquine also blocks another important outward K+ current, the transient outward K+ current Ito, at an IC50 of 4.6 ± 0.9 mM [41]. Chloroquine accelerates the apparent inactivation time constant of Ito. Exposure to 1 mM chloroquine results in a rightward shift of the steady-state inactivation constant, whereas recovery from inactivation is only mildly affected. These data suggest that chloroquine is an open-channel blocker of Ito and has potential clinical proarrhythmic effects [41].
-
(4)
KATP: Chloroquine inhibits the KATP current via a fast-onset effect and a slow-onset voltage-independent effect. The fast-onset effect of chloroquine involves direct channel blocking by binding to the channel pore from the cytoplasmic side. In contrast, the slow-onset effect is regulated by the disruption of interactions between KATP and phosphatidylinositol 4,5-bisphosphate [42].
Inhibition of these K+ currents leads to delayed repolarization of cardiac myocytes, prolongation of the cardiac action potential duration (APD) and the QT interval of the electrocardiogram, which is a sensitive but nonspecific risk marker for the development of Torsade de Pointes - a potentially lethal polymorphic ventricular tachyarrhythmia [48]. Therefore, the inhibition of multiple K+ currents strongly suggests the proarrhythmic potential of chloroquine. The prominent clinical features of cardiac conduction disorders caused by chloroquine and hydroxychloroquine are prolongation of the QT interval, which increases the risk of sudden death due to the development of Torsade de Pointes, especially if the drug is coprescribed with other K+ channel blockers, such as azithromycin, or used in patients with preexisting cardiac diseases.
Inhibition of the autophagy-lysosome pathway
As an evolutionarily conserved cellular housekeeping and quality control mechanism essential for homeostasis and survival, autophagy is responsible for the removal of superfluous, aging, or damaged cytoplasmic proteins and organelles from cells and tissues [49,50,51,52,53,54,55]. The substrates of the autophagic response involve dedicated double-membrane organelles and are commonly known as autophagosomes. Autophagosomes are ultimately delivered to the lysosome for degradation [50]. Since chloroquine interferes with acidic hydrolases in lysosomes, it acts as an inhibitor of the autophagy-lysosome pathway [52]. Since dysregulation of autophagy is linked to the occurrence and development of chronic diseases, including cardiovascular diseases, impairing autophagic flux with chloroquine may play a role in the development of various cardiovascular diseases.
Chloroquine at a high dose (50 mg·kg−1·d−1, for 2 weeks) exacerbated pressure overload hypertrophy and impaired cardiac contractility in rats [56]. Ultrastructurally, chloroquine accentuates mitochondrial fragmentation and cristae destruction by producing a plethora of autophagosomes containing collapsed mitochondria and lysosomal lamellar bodies, suggesting the impairment of the autophagy-lysosome pathway [56]. In addition, high-dose chloroquine significantly impairs the mitochondrial antioxidant buffering capacity and accentuates oxidative stress and mitochondrial dysfunction. These observations highlight the risk of chloroquine administration in patients under high-oxidative stress conditions, such as pathological myocardial hypertrophy or heart failure [56].
During myocardial infarction, cardiomyocyte necroptosis results in the loss of functional cardiac cells, subsequently leading to left ventricular remodeling, cardiac dysfunction and heart failure [57]. By disrupting autophagic degradation, treatment with chloroquine aggravates cardiac myocyte necroptosis and cardiac dysfunction, contributing to adverse ventricular remodeling and progressive heart failure after myocardial infarction [57].
Although autophagy is regarded as a double-edged sword, it is well accepted that activation of autophagy promotes adaptation to stress and supports cellular viability in response to pathological stimulation [54, 58]. Thus, inhibition of autophagy generally accelerates the death of cells exposed to potentially lethal perturbations of homeostasis [54, 58]. Numerous studies have reported that the activation of autophagy might contribute to ameliorating myocardial infarction/reperfusion and myocardial ischemia, protecting cardiomyocytes against apoptosis and necroptosis, and preventing cardiac hypertrophy and cardiac fibrosis [59,60,61,62,63,64,65,66,67]. However, treatment with chloroquine might reverse the cardioprotective effects by inhibiting autophagic flux [59,60,61,62,63,64,65,66,67].
Furthermore, chloroquine could influence Ca2+ channels through repression of autophagy [68]. Blocking autophagy with chloroquine results in depolarization of the mitochondrial membrane potential and increased production of mito-ROS, finally facilitating the oxidation of ryanodine receptor 2 (RyR2) [46, 68]. RyR2 is a Ca2+-releasing channel located on the sarcoplasmic reticulum of cardiomyocytes. The oxidation and dysfunction of RyR2 enhance pro-arrhythmic spontaneous Ca2+ release under β-adrenergic stimulation and disturb intracellular Ca2+ homeostasis, finally resulting in cardiac disorders such as arrhythmia, cardiac hypertrophy and heart failure [46, 68].
Immunologically mediated adverse reactions
Although rare, several severe cutaneous adverse reactions, including Stevens–Johnson syndrome, toxic epidermal necrolysis [69], and drug reaction with eosinophilia and systemic symptoms (DRESS) [70], have been reported to be associated with the use of chloroquine and hydroxychloroquine. Patients may manifest with clinical symptoms of new-onset fever and mucositis, particularly when presenting with new hematologic abnormalities (such as lymphopenia, eosinophilia or atypical lymphocytosis) or unexplained liver or kidney injury after the start of treatment with chloroquine or hydroxychloroquine.
Drug combination-related adverse effects
Hydroxychloroquine-azithromycin combination
Azithromycin is a macrolide antibiotic agent primarily used for the treatment of respiratory infections and some sexually transmitted infections. Since azithromycin also showed antivirus [71, 72] and anti-inflammatory [73, 74] activities and might have synergistic effects when combined with chloroquine or derivatives, a French group used the combination of hydroxychloroquine and azithromycin to treat COVID-19 and observed clinical benefits [7, 8]. However, it should be highlighted that the cardiovascular risk would probably be increased. In fact, the tendency of azithromycin to induce arrhythmia and the risk of prolonged cardiac repolarization and QT interval, leading to fatal Torsade de Pointes, have been well documented [75,76,77]. Recent data from some cohort studies and retrospective studies demonstrated that chloroquine, hydroxychloroquine and concurrent treatment with azithromycin significantly prolonged the QTc interval in a clinically relevant matter [78,79,80]. More than 20% of patients receiving chloroquine, hydroxychloroquine or the hydroxychloroquine and azithromycin combination developed QTc interval prolongation of >500 ms, which is a known marker of a high risk of Torsade de Pointes [78,79,80]. Compared with monotherapy, the hydroxychloroquine/azithromycin combination was associated with greater changes in QTc [79]. The sporadic incidence of Torsade de Pointes has also been reported [78,79,80]. Thus, additional caution is advised when administering this combination in patients with cardiac dysfunction and renal and hepatic diseases.
Chloroquine-propranolol combination
Propranolol is widely used for cardiovascular diseases, including angina, hypertension, tachyarrhythmia, cardiac hypertrophy, and prophylaxis of myocardial infarction. However, a study in anesthetized dogs reported that administration of propranolol potentiates the proarrhythmic effects of chloroquine on prolongation of ECG intervals and bradycardia [81], suggesting the increased proarrhythmic potential of the chloroquine-propranolol combination. In view of the common use of propranolol in cardiovascular patients, the therapeutic use of chloroquine or hydroxychloroquine in COVID-19 patients under propranolol treatment should be carefully considered.
Chloroquine-diclofenac combination
Although this combination is unlikely to be used in COVID-19 patients, combined therapy with these two drugs for rheumatoid arthritis is not rare. The combination could shift the balance between pro- and anti-apoptotic proteins in the left ventricule towards myocardial apoptosis, thus indicating a risk of myocardial damage [82].
Drug–drug interactions
Both chloroquine and hydroxychloroquine are metabolized by the hepatic cytochrome P450 enzyme 2D6 (CYP2D6), and they also competitively inhibit CYP2D6 activity [83]. This has the potential to influence the fate of other drugs reliant on CYP2D6 for metabolism. Azithromycin exhibits little inhibition of cytochrome P450 enzymes or drug-transport proteins such as P-glycoprotein. As such, azithromycin is unlikely to precipitate clinically important drug–drug interactions. The expression of CYP2D6 varies among individuals as a result of genetic polymorphisms. Genetic variability in the metabolism of these drugs is considerable and may also influence their safety and effectiveness when used in combination for COVID-19.
Overdose
Overdoses of chloroquine and hydroxychloroquine are extremely toxic. Rapid onset of central nervous system toxicity, such as seizures and coma, cardiovascular collapse, including QRS widening and QT interval prolongation, and hypokalemia resulting from intracellular changes may occur.
Therapeutic dosage of chloroquine and hydroxychloroquine for COVID-19
Since overdose is the major cause of life-threatening toxicity and adverse events, an appropriate therapeutic dosage within the safety window is extremely important. The strategies used for the treatment of COVID-19 with chloroquine and hydroxychloroquine in clinical applications vary in different countries or hospitals. Some of the currently available treatment strategies and the efficacy of chloroquine, hydroxychloroquine and combination therapy for treatment of COVID-19 in clinical studies are summarized in Table 4. We checked the guidelines from the WHO for drug therapy of COVID-19 and failed to find any guidelines or suggestions on the usage of chloroquine or hydroxychloroquine for COVID-19. In fact, this is true for any drugs used for COVID-19. Instead, WHO released advice for “off-label” use. As stated by the WHO, “No pharmaceutical products have yet been shown to be safe and effective for the treatment of COVID-19. However, a number of medicines have been suggested as potential investigational therapies, many of which are now being or will soon be studied in clinical trials, including the SOLIDARITY trial cosponsored by WHO and participating countries. In many countries, doctors are giving COVID-19 patients medicines that have not been approved for this disease. The use of licensed medicines for indications that have not been approved by a national medicine regulatory authority is considered “off-label” use. Further, such prescribing should be done on a case-by-case basis.” However, the NIH released a new guideline for the usage of chloroquine and hydroxychloroquine for COVID-19 in June 2020. The Panel recommends against the use of high-dose chloroquine (600 mg twice daily for 10 days) for the treatment of COVID-19 (AI). High-dose chloroquine (600 mg twice daily for 10 days) has been associated with more severe toxicities than lower-dose chloroquine (450 mg twice daily for 1 day, followed by 450 mg once daily for 4 days). The steady-state plasma concentration of chloroquine might reach at least 3.125 μM in the treatment of viral infectious diseases [14]. According to in vitro studies, the EC50 of chloroquine in inhibiting SAR-CoV-2 ranges from 2.71 to 23.9 μM, whereas that of hydroxychloroquine ranges from 4.06 to 17.31 μM [2, 4, 5]. However, these concentrations exceed the IC50 of chloroquine used for blockade of Kir (1.2–3 μM) [35, 36, 43, 44] and hERG channels (2.5 μM) [40] and might possibly result in increased proarrhythmic risk. At the recommended dosage of chloroquine phosphate in China, the steady-state plasma concentration can reach the therapeutic concentration (3.125 μM), which also exceeds the IC50 necessary for Kir and hERG blockade and is sufficient to result in adverse effects and cause myocardial dysfunction. The safety margin of chloroquine and hydroxychloroquine is narrow. Thus, attention should be paid to the proarrhythmic risk of the use of chloroquine or hydroxychloroquine, especially in patients with hepatic or renal dysfunction, which might cause accumulation of drugs. Patients with chloroquine or hydroxychloroquine administration should undergo electrocardiogram monitoring.
Comorbidities of COVID-19 patients with cardiovascular disease or risks
A large number of patients with a history of cardiovascular disease or cardiovascular risk factors appeared to have heightened vulnerability to developing COVID-19 with more severe symptoms and worse clinical outcomes. In some confirmed SARS-CoV-2 infection cases, cardiovascular symptoms were one of the most important presentations. Clinical data reported that ~15%–30% of COVID-19 patients have hypertension, and ~2.5%–15% have coronary heart disease [84,85,86,87]. In a report of 72,314 cases from China’s CDC, COVID-19 patients who required admission to an intensive care unit were more likely to have comorbidities, the majority of which were cardiovascular disease [87]. According to the NHC, among the patients who died from COVID-19, 11.8% had substantial heart damage with elevated troponin I levels or cardiac arrest during hospitalization. Another large-scale analysis of 44,672 confirmed cases indicated an increased mortality risk for those with diabetes (7.3%), hypertension (6%), and cardiovascular disease (10.5%), although the mortality rate remains highly variable depending on the region (currently less than 4% in Germany and China and more than 12% in France and Italy) [84,85,86,87,88]. The case fatality rate for underlying cardiovascular disease (10.5%) is larger than that for patients with underlying chronic respiratory disease (6.3%) [84,85,86,87,88].
For COVID-19 patients in weak cardiac condition, increased oxygen and energy demand imposed by viral illnesses is poorly tolerated. Therefore, cardiac involvement is an important feature of COVID-19 and is associated with a poor prognosis. Additional evidence that aids in understanding the interplay between COVID-19 and cardiovascular disease from clinical studies may help to attenuate the risk for patients with preexisting cardiovascular diseases or cardiovascular risk factors. COVID-19 patients suffer from cardiovascular injury, and it is necessary to revisit the potential mechanisms underlying these cardiac adverse effects to reexamine the possible drug–drug interactions that might increase cardiovascular risk and to reassess the therapeutic dosage of chloroquine or hydroxychloroquine in the treatment of COVID-19.
SARS-CoV-2-induced cardiovascular injuries
Cardiovascular complications of COVID-19
With the increasing number of confirmed cases and the accumulating clinical data, it has become clearer that many COVID-19 patients suffer from cardiovascular injury, which is closely associated with worse disease progression in addition to the typical respiratory symptoms caused by SARS-CoV-2 infection. The cardiovascular complications of COVID-19 have drawn more attention from both clinicians and investigators, although the exact mechanism for the myocardial injuries caused by SARS-CoV-2 is not completely understood [89,90,91]. Recently, new clinical syndromes associated with coagulopathy and vasculopathy have emerged as a cause of sudden death and other serious clinical manifestations in younger COVID-19 patients [90]. Angiotensin-converting enzyme 2 (ACE2), the receptor for SARS-CoV-2 and other coronaviruses, is a transmembrane protein expressed by lung alveolar epithelial cells, enterocytes, and vascular endothelial cells whose physiological role is to induce the maturation of angiotensin I to generate angiotensin 1-7, a peptide hormone that controls vasoconstriction and blood pressure [92, 93].
The common cardiac manifestations of COVID-19 include acute myocardial injury and arrhythmias. The overall incidence of acute cardiac injury has been variable, but ~8%–12% of positive cases are known to develop significant elevation of cTnI [94,95,96]. Both tachy- and brady-arrhythmias have occurred in COVID-19 patients. The incidence was much higher (44.4%) in patients who required intensive care unit (ICU) admission than in those who did not require ICU admission (8.9%) [86]. An estimated 11.8% of COVID-19 deaths in China were associated with substantial heart damage with elevated levels of cTnI or cardiac arrest during hospitalization, although the deceased patients had no pre-existing cardiovascular disease [87]. In another case study of 187 patients with COVID-19, 27.8% of patients had myocardial injury that caused cardiac dysfunction and arrhythmias. Myocardial injury is significantly associated with a fatal outcome for COVID-19 [97]. The prognosis of patients with underlying cardiovascular disease but without myocardial injury was relatively improved [97]. Another recent study found that 16.7% of the 138 hospitalized patients with COVID-19 had arrhythmias, and 7.2% of them had acute myocardial injury [86].
Collectively, COVID-19 patients with severe symptoms often have complications involving acute myocardial injury. Cardiac injury had an increased prevalence among COVID-19 patients requiring ICU admission. Myocardial injury-associated biomarker levels were significantly higher in patients requiring ICU admission than those in patients not treated in the ICU. Therefore, a full understanding of the cardiovascular complications of COVID-19 may help in making more precise and effective therapeutic decisions and improve outcomes by reducing mortality.
Cardiovascular complications after recovery from COVID-19
Although COVID-19 has been spreading rapidly and has caused a global pandemic for several months, it is still too early to predict the long-term outcomes for patients who recover from this illness. However, there are reports of complications that occurred soon after the resolution of the acute symptoms [84,85,86,87,88]. Case reports from Italy showed fulminant myocarditis in a convalescent patient one week after respiratory symptoms were resolved [98]. Moreover, sudden cardiac death (SCD) was found to be likely to occur in many nonhospitalized patients with mild symptoms, who were found dead at their home during self-quarantine in the epicenter of the COVID-19 outbreak in Italy [98]. Insidious cardiovascular injury might be a potential trigger of SCD. Therefore, biomarkers for myocardial injury (cTnI and NT-proBNP) and coagulation (D-dimer) should be evaluated early after hospitalization in all COVID-19 patients to attenuate risk and promote intervention. The concern is that background inflammation may persist and evolve silently and manifest later in an insidious manner. There may be chronic sequelae even after an apparent “complete recovery”. Even after hospital discharge, myocardial injury might result in ventricular fibrosis, which may lead to cardiac arrhythmias and SCD. Important lessons should be learned from previous experiences with SARS, a disease caused by SARS-CoV that shares considerable similarity with SARS-CoV-2. Many survivors from the epidemic develop avascular necrosis, pulmonary fibrosis, and dyslipidemia [99,100,101]. Thus, cardiovascular involvement may persist long after the resolution of the acute illness. These latter manifestations are particularly important because they represent cardiovascular risk factors that may lead to further cardiovascular injury. Therefore, careful follow-up of cured COVID-19 patients would be critical for understanding the long-term impact of this illness and the protection of these patients from future cardiovascular disease.
Potential mechanisms for SARS-CoV-2-induced cardiovascular injuries
Understanding the effects of SARS-CoV-2 infection on the cardiovascular system is essential to provide comprehensive medical care for cardiac patients. Several studies have shown that high expression of ACE2 is key for SARS-CoV-2 to enter into human lung, heart, kidney, and liver cells. Single-cell RNA-seq analysis of the ACE2 receptor in major human physiological systems revealed that the lung and heart are vulnerable to SARS-CoV-2 infection [102]. It was reported that the blood concentration of Ang II in COVID-19 patients is higher than that in healthy controls and positively correlated with SARS-CoV-2 virus loads [103]. Downregulation of ACE2 by SARS-CoV-2 infection may cause an increase in Ang II and vascular endothelial injury, increased blood pressure, and reduced contraction function. Clinical evidence has also shown that increased viral loads are linked to an increase in cardiac enzymes such as hs-TnI and CK-MB and myocardial injury [103]. Pathological manifestations also included interstitial mononuclear inflammatory infiltrates in the hearts of COVID-19 patients [84]. The cytokine storm caused by SARS-CoV-2 infection may lead to further cardiovascular injury. COVID-19 patients had increased blood concentrations of interleukin (IL)-2, IL-7, granulocyte-colony stimulating factor (G-CSF), interferon-γ-inducible protein 10 (IP-10), monocyte chemoattractant protein 1 (MCP1), macrophage inflammatory protein 1-α (MIP-1α), and tumor necrosis factor-α (TNF-α) [86, 88]. Unbalanced reactions of Th1 and Th2 cells, increases in CCR6+ Th17 cells and decreases in CD4+/CD8+ T cells are also observed in COVID-19 patients [86, 88]. A similar cytokine profile was found to be associated with the severity of COVID-19 disease. Increased inflammatory reactions and cytokine storms lead to tissue swelling, ischemia-reperfusion injury, and heart failure in COVID-19 patients. Severe hypoxia from acute respiratory damage caused by SARS-CoV-2 infection may impair the myocardial oxygen demand-supply relationship and result in myocardial injury from increased myocardial oxygen demand in the presence of severe hypoxia due to acute lung injury. In addition to the altered myocardial demand-supply ratio, increased coronary blood flow due to systemic inflammation and increased shear stress may lead to plaque rupture and result in acute myocardial infarction. The prothrombotic milieu associated with exaggerated systemic inflammation may further aggravate the risk. Cerebrovascular disease and ischemic stroke associated with COVID-19 were reviewed, and it seems that SARS-CoV-2 infection was more likely to induce ischemic stroke than influenza A infection [104,105,106].
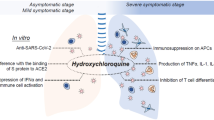
Therefore, as shown in Fig. 2, the potential mechanisms underlying SARS-CoV-2-induced cardiovascular effects may be complex, with the involvement of (1) direct viral infection of cardiac myocytes by SARS-CoV-2; (2) systemic inflammation and immune overreactions, including cytokine storm and imbalances in T cells, helper T cells and regulatory T cells; (3) acute cardiac stress, including an altered myocardial demand-supply ratio, plaque rupture and coronary thrombosis due to respiratory failure and hypoxemia; and (4) endothelial cell involvement across vascular beds of different organs in COVID-19 and direct viral infection of endothelial cells and diffuse endothelial inflammation (endotheliitis), which could explain the systemic impaired microcirculatory function in different vascular beds and the resulting clinical sequelae in patients with COVID-19 [107].

Blue arrows indicate the actions of SARS-COV-2. ACE2 is key for SARS-CoV-2 to enter into cells in human organs such as the lung and heart. The injuries of organs and cells are caused by (1) direct SARS-CoV-2 viral infection of cells; (2) systemic inflammation and immune overreactions, including cytokine storms and imbalances in T cells, helper T cells and regulatory T cells; (3) acute respiratory and hypoxia stress, including an altered myocardial demand-supply ratio, plaque rupture and coronary thrombosis due to respiratory failure and hypoxemia; and (4) endothelial cell involvement across vascular beds of different organs in COVID-19 and direct viral infection of endothelial cells and diffuse endothelial inflammation (endotheliitis), which could explain the systemic impaired microcirculatory function in different vascular beds and the resulting clinical sequelae in patients with COVID-19. Red dashed lines indicate the potential mechanisms of the therapeutic and toxic effects of hydroxychloroquine on SARS-CoV-2 and the organs in COVID-19 patients.
Conclusions
From both the pharmacological and cardiovascular points of view, it is urgent to perform a systematic evaluation of the efficacy and safety of the current therapy of COVID-19 with chloroquine and hydroxychloroquine and associated combination therapies. It is extremely important to show caution and to closely monitor cardiovascular function with electrocardiography because the adverse effects of these drugs on the cardiovascular system, especially in COVID-19 patients with cardiovascular conditions and injury as well as kidney and liver diseases, these effects are potentially fatal.
References
Song Y, Zhang M, Yin L, Wang K, Zhou Y, Zhou M, et al. COVID-19 treatment: close to a cure? - A rapid review of pharmacotherapies for the novel coronavirus. Int J Antimicrob Agents. 2020;56:106080. https://doi.org/10.1016/j.ijantimicag.2020.106080.
Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;6:16. https://doi.org/10.1038/s41421-020-0156-0.
Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005;2:69.
Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2). Clin Infect Dis. 2020;71:732–9.
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–71.
Chen X, Geiger JD. Janus sword actions of chloroquine and hydroxychloroquine against COVID-19. Cell Signal. 2020;109706;1–8. https://doi.org/10.1016/j.cellsig.2020.109706.
Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020;105949:1–6. https://doi.org/10.1016/j.ijantimicag.2020.105949.
Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Sevestre J, et al. Clinical and microbiological effect of a combination of hydroxychloroquine and azithromycin in 80 COVID-19 patients with at least a six-day follow up: a pilot observational study. Travel Med Infect Dis. 2020;34:101663.
Gao J, Hu S. Update on use of chloroquine/hydroxychloroquine to treat coronavirus disease 2019 (COVID-19). Biosci Trends. 2020;14:156–8.
Gao J, Tian Z, Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci Trends. 2020;14:72–3.
Kaufmann AM, Krise JP. Lysosomal sequestration of amine-containing drugs: analysis and therapeutic implications. J Pharm Sci. 2007;96:729–46.
Yoon YH, Cho KS, Hwang JJ, Lee SJ, Choi JA, Koh JY. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest Ophthalmol Vis Sci. 2010;51:6030–7.
Al-Bari MAA. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol Res Perspect. 2017;5:e00293. https://doi.org/10.1002/prp2.293.
Akpovwa H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem Funct. 2016;34:191–6.
Farias KJ, Machado PR, Muniz JA, Imbeloni AA, da Fonseca BA. Antiviral activity of chloroquine against dengue virus type 2 replication in Aotus monkeys. Viral Immunol. 2015;28:161–9.
Wang LF, Lin YS, Huang NC, Yu CY, Tsai WL, Chen JJ, et al. Hydroxychloroquine-inhibited dengue virus is associated with host defense machinery. J Interferon Cytokine Res. 2015;35:143–56.
Romanelli F, Smith KM, Hoven AD. Chloroquine and hydroxychloroquine as inhibitors of human immunodeficiency virus (HIV-1) activity. Curr Pharmacol Des. 2004;10:2643–8.
Savarino A, Gennero L, Sperber K, Boelaert JR. The anti-HIV-1 activity of chloroquine. J Clin Virol. 2001;20:131–5.
Paton NI, Lee L, Xu Y, Ooi EE, Cheung YB, Archuleta S, et al. Chloroquine for influenza prevention: a randomised, double-blind, placebo controlled trial. Lancet Infect Dis. 2011;11:677–83.
Dowall SD, Bosworth A, Watson R, Bewley K, Taylor I, Rayner E, et al. Chloroquine inhibited Ebola virus replication in vitro but failed to protect against infection and disease in the in vivo guinea pig model. J Gen Virol. 2015;96:3484–92.
Keyaerts E, Li S, Vijgen L, Rysman E, Verbeeck J, Van RM, et al. Antiviral activity of chloroquine against human coronavirus OC43 infection in newborn mice. Antimicrob Agents Chemother. 2009;53:3416–21.
Jang CH, Choi JH, Byun MS, Jue DM. Chloroquine inhibits production of TNF-alpha, IL-1beta and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology (Oxf). 2006;45:703–10.
Picot S, Peyron F, Vuillez JP, Polack B, Ambroise-Thomas P. Chloroquine inhibits tumor necrosis factor production by human macrophages in vitro. J Infect Dis. 1991;164:830. https://doi.org/10.1093/infdis/164.4.830.
van den Borne BE, Dijkmans BA, de Rooij HH, le CS, Verweij CL. Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J Rheumatol. 1997;24:55–60.
Sperber K, Quraishi H, Kalb TH, Panja A, Stecher V, Mayer L. Selective regulation of cytokine secretion by hydroxychloroquine: inhibition of interleukin 1 alpha (IL-1-alpha) and IL-6 in human monocytes and T cells. J Rheumatol. 1993;20:803–8.
Chandler LC, Barnard AR, Caddy SL, Patricio MI, McClements ME, Fu H, et al. Enhancement of adeno-associated virus-mediated gene therapy using hydroxychloroquine in murine and human tissues. Mol Ther Methods Clin Dev. 2019;14:77–89.
Hong Z, Jiang Z, Liangxi W, Guofu D, Ping L, Yongling L, et al. Chloroquine protects mice from challenge with CpG ODN and LPS by decreasing proinflammatory cytokine release. Int Immunopharmacol. 2004;4:223–34.
Zhu X, Pan Y, Li Y, Jiang Y, Shang H, Gowda DC, et al. Targeting Toll-like receptors by chloroquine protects mice from experimental cerebral malaria. Int Immunopharmacol. 2012;13:392–7.
Yasuda H, Leelahavanichkul A, Tsunoda S, Dear JW, Takahashi Y, Ito S, et al. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am J Physiol Ren Physiol. 2008;294:F1050–8.
Plantone D, Koudriavtseva T. Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review. Clin Drug Investig. 2018;38:653–71.
Chatre C, Roubille F, Vernhet H, Jorgensen C, Pers YM. Cardiac complications attributed to chloroquine and hydroxychloroquine: a systematic review of the literature. Drug Saf. 2018;41:919–31.
Pukrittayakamee S, Tarning J, Jittamala P, Charunwatthana P, Lawpoolsri S, Lee SJ, et al. Pharmacokinetic interactions between primaquine and chloroquine. Antimicrob Agents Chemother. 2014;58:3354–9.
Mzayek F, Deng H, Mather FJ, Wasilevich EC, Liu H, Hadi CM, et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin Trials. 2007;2:e6.
Ursing J, Rombo L, Eksborg S, Larson L, Bruvoll A, Tarning J, et al. High-dose chloroquine for uncomplicated plasmodium falciparum malaria is well tolerated and causes similar QT interval prolongation as standard-dose chloroquine in children. Antimicrob Agents Chemother. 2020;64:e01846–19.
Sanson C, Schombert B, Filoche-Romme B, Partiseti M, Bohme GA. Electrophysiological and pharmacological characterization of human inwardly rectifying Kir2.1 channels on an automated patch-clamp platform. Assay Drug Dev Technol. 2019;17:89–99.
El HA, McPate MJ, Zhang Y, Zhang H, Hancox JC. Action potential clamp and chloroquine sensitivity of mutant Kir2.1 channels responsible for variant 3 short QT syndrome. J Mol Cell Cardiol. 2009;47:743–7.
Rodriguez-Menchaca AA, Navarro-Polanco RA, Ferrer-Villada T, Rupp J, Sachse FB, Tristani-Firouzi M, et al. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc Natl Acad Sci U S A. 2008;105:1364–8.
Noujaim SF, Stuckey JA, Ponce-Balbuena D, Ferrer-Villada T, Lopez-Izquierdo A, Pandit S, et al. Specific residues of the cytoplasmic domains of cardiac inward rectifier potassium channels are effective antifibrillatory targets. FASEB J. 2010;24:4302–12.
Vicente J, Zusterzeel R, Johannesen L, Ochoa-Jimenez R, Mason JW, Sanabria C, et al. Assessment of multi-ion channel block in a phase I randomized study design: results of the CiPA phase I ECG biomarker validation study. Clin Pharmacol Ther. 2019;105:943–53.
Traebert M, Dumotier B, Meister L, Hoffmann P, Dominguez-Estevez M, Suter W. Inhibition of hERG K+ currents by antimalarial drugs in stably transfected HEK293 cells. Eur J Pharmacol. 2004;484:41–8.
Wagner M, Riepe KG, Eberhardt E, Volk T. Open channel block of the fast transient outward K+ current by primaquine and chloroquine in rat left ventricular cardiomyocytes. Eur J Pharmacol. 2010;647:13–20.
Ponce-Balbuena D, Rodriguez-Menchaca AA, Lopez-Izquierdo A, Ferrer T, Kurata HT, Nichols CG, et al. Molecular mechanisms of chloroquine inhibition of heterologously expressed Kir6.2/SUR2A channels. Mol Pharmacol. 2012;82:803–13.
Noujaim SF, Stuckey JA, Ponce-Balbuena D, Ferrer-Villada T, Lopez-Izquierdo A, Pandit SV, et al. Structural bases for the different anti-fibrillatory effects of chloroquine and quinidine. Cardiovasc Res. 2011;89:862–9.
Lopez-Izquierdo A, Ponce-Balbuena D, Ferrer T, Sachse FB, Tristani-Firouzi M, Sanchez-Chapula JA. Chloroquine blocks a mutant Kir2.1 channel responsible for short QT syndrome and normalizes repolarization properties in silico. Cell Physiol Biochem. 2009;24:153–60.
Liu CF, Liu QH, Liu EL, Zhai XW, Zhang L, Luo TE, et al. Activation of IK1 channel by zacopride attenuates left ventricular remodeling in rats with myocardial infarction. J Cardiovasc Pharmacol. 2014;64:345–56.
Liu QH, Qiao X, Zhang LJ, Wang J, Zhang L, Zhai XW, et al. IK1 channel agonist zacopride alleviates cardiac hypertrophy and failure via alterations in calcium dyshomeostasis and electrical remodeling in rats. Front Pharmacol. 2019;10:929. https://doi.org/10.3389/fphar.2019.00929.
Borsini F, Crumb W, Pace S, Ubben D, Wible B, Yan GX, et al. In vitro cardiovascular effects of dihydroartemisin-piperaquine combination compared with other antimalarials. Antimicrob Agents Chemother. 2012;56:3261–70.
Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–80.
Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and Autophagy in the Heart. Circ Res. 2016;118:1563–76.
Ryter SW, Bhatia D, Choi ME. Autophagy: a lysosome-dependent process with implications in cellular redox homeostasis and human disease. Antioxid Redox Signal. 2019;30:138–59.
Abdellatif M, Ljubojevic-Holzer S, Madeo F, Sedej S. Autophagy in cardiovascular health and disease. Prog Mol Biol Transl Sci. 2020;172:87–106.
Bonam SR, Muller S, Bayry J, Klionsky DJ. Autophagy as an emerging target for COVID-19: lessons from an old friend, chloroquine. Autophagy. 2020;1–7
Yan Y, Finkel T. Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic Biol Med. 2017;109:108–13.
Jimenez RE, Kubli DA, Gustafsson AB. Autophagy and mitophagy in the myocardium: therapeutic potential and concerns. Br J Pharmacol. 2014;171:1907–16.
Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:252–61.
Chaanine AH, Gordon RE, Nonnenmacher M, Kohlbrenner E, Benard L, Hajjar RJ. High-dose chloroquine is metabolically cardiotoxic by inducing lysosomes and mitochondria dysfunction in a rat model of pressure overload hypertrophy. Physiol Rep. 2015;3:e12413. https://doi.org/10.14814/phy2.12413.
Zhang H, Yin Y, Liu Y, Zou G, Huang H, Qian P, et al. Necroptosis mediated by impaired autophagy flux contributes to adverse ventricular remodeling after myocardial infarction. Biochem Pharmacol. 2020;175:113915.
Bartlett JJ, Trivedi PC, Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol. 2017;104:1–8.
Fu S, Chen L, Wu Y, Tang Y, Tang L, Zhong Y, et al. Gastrodin pretreatment alleviates myocardial ischemia/reperfusion injury through promoting autophagic flux. Biochem Biophys Res Commun. 2018;503:2421–8.
Liu S, Chen S, Li M, Zhang B, Shen P, Liu P, et al. Autophagy activation attenuates angiotensin II-induced cardiac fibrosis. Arch Biochem Biophys. 2016;590:37–47.
Zhang J, Nadtochiy SM, Urciuoli WR, Brookes PS. The cardioprotective compound cloxyquin uncouples mitochondria and induces autophagy. Am J Physiol Heart Circ Physiol. 2016;310:H29–38.
Zuo Z, Zuo PF, Sheng ZL, Wang X, Ding JD, Ma GS. Tetramethylprazine attenuates myocardial ischemia/reperfusion injury through modulation of autophagy. Life Sci. 2019;239:117016.
Qiao SG, Sun Y, Sun B, Wang A, Qiu J, Hong L, et al. Sevoflurane postconditioning protects against myocardial ischemia/reperfusion injury by restoring autophagic flux via an NO-dependent mechanism. Acta Pharmacol Sin. 2019;40:35–45.
Yan J, Yan JY, Wang YX, Ling YN, Song XD, Wang SY, et al. Spermidine-enhanced autophagic flux improves cardiac dysfunction following myocardial infarction by targeting the AMPK/mTOR signalling pathway. Br J Pharmacol. 2019;176:3126–42.
Yu T, Liu D, Gao M, Yang P, Zhang M, Song F, et al. Dexmedetomidine prevents septic myocardial dysfunction in rats via activation of alpha7nAChR and PI3K/Akt- mediated autophagy. Biomed Pharmacother. 2019;120:109231.
Guo L, Xu JM, Mo XY. Ischemic postconditioning regulates cardiomyocyte autophagic activity following ischemia/reperfusion injury. Mol Med Rep. 2015;12:1169–76.
Wu X, Zheng D, Qin Y, Liu Z, Zhang G, Zhu X, et al. Nobiletin attenuates adverse cardiac remodeling after acute myocardial infarction in rats via restoring autophagy flux. Biochem Biophys Res Commun. 2017;492:262–8.
Murphy KR, Baggett B, Cooper LL, Lu Y, Uchi J, Sedivy JM, et al. Enhancing autophagy diminishes aberrant Ca2+ homeostasis and arrhythmogenesis in aging rabbit hearts. Front Physiol. 2019;10:1277.
Cameron MC, Word AP, Dominguez A. Hydroxychloroquine-induced fatal toxic epidermal necrolysis complicated by angioinvasive rhizopus. Dermatol Online J. 2014;20:13030.
Volpe A, Marchetta A, Caramaschi P, Biasi D, Bambara LM, Arcaro G. Hydroxychloroquine-induced DRESS syndrome. Clin Rheumatol. 2008;27:537–9.
Li C, Zu S, Deng YQ, Li D, Parvatiyar K, Quanquin N, et al. Azithromycin protects against Zika virus infection by upregulating virus-induced type I and III interferon responses. Antimicrob Agents Chemother. 2019;63:e00394–19.
Tran DH, Sugamata R, Hirose T, Suzuki S, Noguchi Y, Sugawara A, et al. Azithromycin, a 15-membered macrolide antibiotic, inhibits influenza A(H1N1)pdm09 virus infection by interfering with virus internalization process. J Antibiot. 2019;72:759–68.
Ersoy B, Aktan B, Kilic K, Sakat MS, Sipal S. The anti-inflammatory effects of erythromycin, clarithromycin, azithromycin and roxithromycin on histamine-induced otitis media with effusion in guinea pigs. J Laryngol Otol. 2018;132:579–83.
Mosquera RA, De Jesus-Rojas W, Stark JM, Yadav A, Jon CK, Atkins CL, et al. Role of prophylactic azithromycin to reduce airway inflammation and mortality in a RSV mouse infection model. Pediatr Pulmonol. 2018;53:567–74.
Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med. 2012;366:1881–90.
Zhang M, Xie M, Li S, Gao Y, Xue S, Huang H, et al. Electrophysiologic studies on the risks and potential mechanism underlying the proarrhythmic nature of azithromycin. Cardiovasc Toxicol. 2017;17:434–40.
Maisch NM, Kochupurackal JG, Sin J. Azithromycin and the risk of cardiovascular complications. J Pharm Pract. 2014;27:496–500.
van den Broek MPH, Mohlmann JE, Abeln BGS, Liebregts M, van Dijk VF, van de Garde EMW. Chloroquine-induced QTc prolongation in COVID-19 patients. Neth Heart J. 2020;28:406–9.
Mercuro NJ, Yen CF, Shim DJ, Maher TR, McCoy CM, Zimetbaum PJ, et al. Risk of QT interval prolongation associated with use of hydroxychloroquine with or without concomitant azithromycin among hospitalized patients testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;e201834. https://doi.org/10.1001/jamacardio.2020.1834.
Chorin E, Dai M, Shulman E, Wadhwani L, Bar-Cohen R, Barbhaiya C, et al. The QT interval in patients with COVID-19 treated with hydroxychloroquine and azithromycin. Nat Med. 2020;26:808–9.
Sofola OA, Olude IO, Adegoke F. The effect of chronic chloroquine toxicity on blood pressure of rats. J Trop Med Hyg. 1981;84:249–52.
Arafa MH, Mohammad NS, Atteia HH. Rho-Kinase inhibitors ameliorate diclofenac-induced cardiotoxicity in chloroquine-treated adjuvant arthritic rats. Life Sci. 2020;254:117605.
Somer M, Kallio J, Pesonen U, Pyykko K, Huupponen R, Scheinin M. Influence of hydroxychloroquine on the bioavailability of oral metoprolol. Br J Clin Pharmacol. 2000;49:549–54.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506.
Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17:259–60.
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–9.
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in china: summary of a report of 72314 cases from the chinese center for disease control and prevention. JAMA. 2020. https://doi.org/10.1001/jama.2020.2648.
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708–20.
Boukhris M, Hillani A, Moroni F, Annabi MS, Addad F, Ribeiro MH, et al. Cardiovascular implications of the COVID-19 pandemic: a global perspective. Can J Cardiol. 2020;36:1068–80.
Labo N, Ohnuki H, Tosato G. Vasculopathy and coagulopathy associated with SARS-CoV-2 infection. Cells. 2020;9:1583. https://doi.org/10.3390/cells9071583.
Mondal R, Lahiri D, Deb S, Bandyopadhyay D, Shome G, Sarkar S, et al. COVID-19: are we dealing with a multisystem vasculopathy in disguise of a viral infection? J Thromb Thrombolysis. 2020; 1–13. https://doi.org/10.1007/s11239-020-02210-8.
Albini A, Di GG, Noonan DM, Lombardo M. The SARS-CoV-2 receptor, ACE-2, is expressed on many different cell types: implications for ACE-inhibitor- and angiotensin II receptor blocker-based cardiovascular therapies. Intern Emerg Med. 2020;15:759–66.
Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. 2020;116:1097–100.
Lippi G, Wong J, Henry BM. Hypertension and its severity or mortality in Coronavirus Disease 2019 (COVID-19): a pooled analysis. Pol Arch Intern Med. 2020;130:304–9.
Lippi G, Lavie CJ, Sanchis-Gomar F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): evidence from a meta-analysis. Prog Cardiovasc Dis. 2020;63:390–1.
Lippi G, Plebani M. Laboratory abnormalities in patients with COVID-2019 infection. Clin Chem Lab Med. 2020;58:1131–4.
Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5:1–8. https://doi.org/10.1001/jamacardio.2020.1017.
Inciardi RM, Lupi L, Zaccone G, Italia L, Raffo M, Tomasoni D, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5:1–6. https://doi.org/10.1001/jamacardio.2020.1096.
Wu Q, Zhou L, Sun X, Yan Z, Hu C, Wu J, et al. Altered lipid metabolism in recovered SARS patients twelve years after infection. Sci Rep. 2017;7:9110.
Antonio GE, Wong KT, Hui DS, Wu A, Lee N, Yuen EH, et al. Thin-section CT in patients with severe acute respiratory syndrome following hospital discharge: preliminary experience. Radiology. 2003;228:810–5.
Hong N, Du XK. Avascular necrosis of bone in severe acute respiratory syndrome. Clin Radiol. 2004;59:602–8.
Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020;14:185–92.
Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63:364–74.
Liang JW, Reynolds AS, Reilly K, Lay C, Kellner CP, Shigematsu T, et al. COVID-19 and decompressive hemicraniectomy for acute ischemic stroke. Stroke. 2020;51:e215–18.
Reddy ST, Garg T, Shah C, Nascimento FA, Imran R, Kan P, et al. Cerebrovascular disease in patients with COVID-19: a review of the literature and case series. Case Rep. Neurol. 2020;12:199–209.
Saggese CE, Del BC, Di Ruzza MR, Magarelli M, Gandini R, Plocco M. COVID-19 and stroke: casual or causal role? Cerebrovasc Dis. 2020;49:341–4.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–8.
Du RH, Liang LR, Yang CQ, Wang W, Cao TZ, Li M, et al. Predictors of mortality for patients with COVID-19 pneumonia caused by SARS-CoV-2: a prospective cohort study. Eur Respir J. 2020;55:1–8.
Huang I, Lim MA, Pranata R. Diabetes mellitus is associated with increased mortality and severity of disease in COVID-19 pneumonia – a systematic review, meta-analysis, and meta-regression. Diabetes Metab Syndr. 2020;14:395–403.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62.
Henry BM, Aggarwal G, Wong J, Benoit S, Vikse J, Plebani M, et al. Lactate dehydrogenase levels predict coronavirus disease 2019 (COVID-19) severity and mortality: a pooled analysis. Am J Emerg Med. 2020;38:1722–6.
Liu Y, Du X, Chen J, Jin Y, Peng L, Wang HHX, et al. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J Infect. 2020;81:e6–12.
Chen ZHJ, Zhang Z, Jiang S, Han S, Yan D, Zhuang R, et al. Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial. medRxiv. 2020; 2020.03.22.20040758.
Chen J, Liu D, Liu L, Liu P, Xu Q, Xia L. A pilot study of hydroxychloroquine in treatment of patients with moderate COVID-19. J Zhejing Univ (Med Sci). 2020;49:215–9.
Molina JM, Delaugerre C, Le Goff J, Mela-Lima B, Ponscarme D, Goldwirt L, et al. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med Mal Infect. 2020;50:384. https://doi.org/10.1016/j.medmal.2020.03.006.
Acknowledgements
This work was supported by grants from the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093), National Engineering and Technology Research Center for New Druggability Evaluation (Seed Program of Guangdong Province, 2017B090903004), Guangdong Provincial Key Laboratory of Construction Foundation (No. 2017B030314030), and the Science and Technology Strategic Cooperation Programs of Luzhou Municipal People’s Government and Southwest Medical University (No. 2017LZXNYD-P01 and 2019 LZXNYD-P01DUAN to Dayue Darrel Duan).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Zhang, Xl., Li, Zm., Ye, Jt. et al. Pharmacological and cardiovascular perspectives on the treatment of COVID-19 with chloroquine derivatives. Acta Pharmacol Sin 41, 1377–1386 (2020). https://doi.org/10.1038/s41401-020-00519-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00519-x
Keywords
This article is cited by
-
Phenome-wide association study and precision medicine of cardiovascular diseases in the post-COVID-19 era
Acta Pharmacologica Sinica (2023)
-
Treating diabetes with combination of phosphodiesterase 5 inhibitors and hydroxychloroquine—a possible prevention strategy for COVID-19?
Molecular and Cellular Biochemistry (2023)
-
Inhibition of drug-metabolizing enzymes by Jingyin granules: implications of herb–drug interactions in antiviral therapy
Acta Pharmacologica Sinica (2022)
-
Reviews on Biological Activity, Clinical Trial and Synthesis Progress of Small Molecules for the Treatment of COVID-19
Topics in Current Chemistry (2021)
