
Abstract
Nicotinamide phosphoribosyltransferase (Nampt) is the rate-limiting enzyme of nicotinamide adenine dinucleotide (NAD) salvage biosynthesis in mammals, and is involved in fundamental physiological processes and pathophysiology of many diseases. Thus far, however, the role of Nampt in atherosclerosis development is still in debate. In this study, we crossed global Nampt transgenic mice (Nampt-Tg) with a well-established atherosclerosis animal model (ApoE knockout mice, ApoE−/−) to generate ApoE−/−;Nampt-Tg mice and investigated the effects of Nampt overexpression on atherosclerosis development in ApoE−/− mice. Both ApoE−/− and ApoE−/−;Nampt-Tg mice were fed with a pro-atherosclerotic high-fat diet (HFD) for 16 weeks. Their serum lipid contents and atherosclerotic lesion were assessed. The results showed that there was no significant difference in body weight or serum levels of glucose, total cholesterol, triglycerides, high-density lipoprotein cholesterol, and low-density lipoprotein cholesterol between the two strains of mice, but ApoE−/−;Nampt-Tg mice had a significantly higher level of serum non-esterified fatty acid. Compared with ApoE−/− mice, ApoE−/−;Nampt-Tg mice displayed significantly increased atherosclerotic lesion area and thickness, lower collagen content, decreased collagen I/III ratio (collagen immaturation), increased number of apoptotic cells, and enhanced activities of caspase-3, caspase-8, and caspase-9. Moreover, macrophage infiltration (F4/80 staining), tumor necrosis factor signaling, and chemokines expression (ICAM-1 and CXCR-4) were all activated in aortic atherosclerotic plaque of ApoE−/−;Nampt-Tg mice compared with ApoE−/− mice. Our results provide in vivo evidence that Nampt transgene aggravates atherosclerotic inflammation and promotes atherosclerosis development in ApoE−/− mice.
Similar content being viewed by others


Introduction
Nicotinamide adenine dinucleotide (NAD) is currently recognized as more than an essential coenzyme that is involved in intracellular redox reactions; it is also a multifunctional metabolite that regulates NAD-dependent enzymes, such as sirtuins and poly [ADP-ribose] polymerases (PARPs) [1]. Nicotinamide phosphoribosyltransferase (Nampt) has two different forms: intracellular and extracellular Nampt. Intracellular Nampt is the rate-limiting enzyme of NAD salvage biosynthesis in mammals [1]. Due to this fundamental role of Nampt for life, knockout of Nampt is lethal in mice. Nampt is ubiquitously detected in all tissues, with the highest levels of expression in the bone marrow, liver, muscles, and adipose [1]. Extracellular Nampt, which is also referred to as visfatin or pre-B cell colony-enhancing factor (PBEF), has been considered to be a circulating hormone/cytokine released by several tissues, such as adipose and liver tissues [2,3,4]. Many investigations have shown the key roles of intracellular or extracellular Nampt in cell survival [5, 6], metabolism [2, 7, 8], development [9], circadian clock [10], inflammation [11, 12], oncogenesis [13, 14], and the lifespan [15, 16].
Atherosclerosis is one of the most common causes of cardio- and cerebrovascular diseases, causing progressive narrowing of the arteries, which results in the blockage of the vessel lumen and impairment of the vascular supply and ultimately induces arterial thrombosis or sudden occlusion of blood flow [17]. The pathophysiological role of Nampt in atherosclerosis has drawn substantial attention in recent years. Higher Nampt levels in serum are found in patients with carotid atherosclerosis [18]. High Nampt serum levels are significantly associated with advanced carotid atherosclerosis in patients with type 2 diabetes mellitus [19] or chronic kidney disease [20]. Extracellular Nampt stimulates vascular smooth muscle cell (VSMC) proliferation [21] and activates pro-inflammatory signaling in VSMCs [22]. Moreover, Nampt was found to localize in areas that were rich in lipid-loaded macrophages and was markedly enhanced in unstable plaque [23]. Pharmacological inhibition of Nampt with a chemical agent FK866 inhibits inflammation in atherosclerotic plaques [24]. These results suggest that Nampt may be an unfavorable factor for atherosclerosis.
However, there is also contradictory evidence. Nampt significantly extends the lifespan in endothelial cells and VSMCs [15, 16]. Nampt maintains the VSMC genome integrity and suppresses human thoracic aortic aneurysm disease by resisting aortic medial degeneration [25]. We previously showed that Nampt stimulated proliferation and conferred a protection against oxidative-induced apoptosis in VSMCs [21]. Notably, a recent study showed that the injection of bone marrow cells that overexpressed Nampt by lentivirus in low-density lipoprotein receptor (LDLR)-deficient mice increased lesion stabilization and inhibited atherosclerotic development [26]. Nampt or its substrates displayed protective actions in various cardio-cerebral vascular diseases, such as cerebral ischemia/hemorrhage [27,28,29,30], myocardial ischemia and reperfusion [5, 31], post-ischemic vascular repair [32], diabetic vascular malfunction [33], and vascular aging [34].
Thus, the role of Nampt in atherosclerosis remains under debate. In the present study, we crossed global Nampt transgenic mice (Nampt-Tg) with a well-established atherosclerosis animal model (ApoE knockout mice, ApoE−/−) to generate ApoE−/−;Nampt-Tg mice. Using this animal model, we investigated the effects of Nampt overexpression on atherosclerosis development.
Methods
Animals and pro-atherosclerotic diet
Eight-week-old C57BL/6J mice were purchased from the Shanghai Laboratory Animal Center, Chinese Academy of Science, China. ApoE knockout (ApoE−/−) mice on the C57BL/6 background were purchased from the Jackson Laboratory (#002052; Bar Harbor, ME, USA) and bred in house. Mice were maintained under a 12 h dark–light cycle. Atherosclerosis was induced using a pro-atherosclerotic high-fat diet (HFD, Harlan Teklad Diet TD88137; 42% fat calories and 0.15% cholesterol) for 16 weeks. All animal experiments were performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the ethical committee for animal experiments of the Second Military Medical University.
Generation of ApoE −/−;Nampt-Tg mice
Nampt transgenic mice (Nampt-Tg), described in our previous studies [32, 35], were backcrossed 10 times to C67BL/6 mice. ApoE−/− mice were crossed with Nampt-Tg mice to generate the F1 ApoE+/−;Nampt-Tg mice. The F1 progeny ApoE+/−;Nampt-Tg mice were mated with ApoE−/− mice to produce ApoE−/−;Nampt-Tg mice. Genomic DNA was obtained from tail tissue for the determination of Nampt transgenic using the following primer: forward, 5′-CCC GCC TCG AGA GGA GAT ATA CCA TGG GCA G-3′; reverse, 5′-GCA ACT GCA GCC TAA TGA GGT GCC ACG TCC T-3′.
Plasma cholesterol and triglyceride analyses
After an overnight fasting period, 500–800 μL of peripheral blood was obtained from the mice through the inferior vena cava prior to perfusion. Whole blood was allowed to clot at room temperature for 1 h to obtain serum. The serum levels of glucose, total cholesterol (TC), triglycerides (TG), high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, and non-esterified fatty acid (NEFA) were measured using an auto-analyzer (Hitachi, Tokyo, Japan) at Changhai Hospital, Second Military Medical University.
Atherosclerotic lesion analysis
On the day of surgery, the mice were fasted for 4 h and then deeply anesthetized with 40% chloral hydrate (0.6 g/kg, i.p.). The aortas were isolated, opened longitudinally, and then pinned flat on a black wax surface with 0.2-mm-diameter stainless-steel pins. The aortic tree was subsequently stained with Oil Red O. The pinned aortas were stained with Oil Red O, images were captured with a digital camera, and the aortic plaque area was measured using Image J software (NIH) as previously described [21, 36]. For the evaluation of the aortic sinus atherosclerosis lesions under microscopy, hearts with an aortic root were fixed in 4% paraformaldehyde and cryopreserved in 30% sucrose plus 4% paraformaldehyde for an additional 12 h. The aortic sinus was then cryo-sectioned to 8-μm sections and stained for lesion areas with Oil Red O. The plaque lesion area (in μm2) was calculated from consecutive sections of the aortic root for 1 cm, starting at the appearance of the tricuspid valves.
Histology analysis
The aortas were serially sectioned and stained with hematoxylin and eosin (H&E). The collagen content was determined with Masson’s trichrome staining and Picrosirius red staining. Images of Masson’s trichrome staining were acquired via Leica microscopy and used to quantify the number of necrotic core and the collagen content (blue staining area). Picrosirius red-stained sections were examined under bright-field illumination for the general collagen content or polarized light for fibrillary collagen observations, in which collagen I appears red-orange and collagen III appears green [37].
TdT-mediated dUTP nick end-labeling (TUNEL) staining
A TUNEL staining kit (DeadEnd Colorimetric TUNEL system; Promega) was used to detect apoptosis in lesion areas. The main procedure of the TUNEL assay has been described in detail in our previous studies [21, 29]. For the quantification of TUNEL staining, five sections per animal and five random microscope fields per section were chosen. The average number was then calculated.
Caspase activities
The activities of caspase-3, caspase-8, and caspase-9 were determined using commercial kits purchased from Beyotime Institute of Biotechnology (Haimen, China) according to the manufacturer’s instructions. These kits were generated based on the specific tetrapeptide substrates Ac-DEVD-pNA, Ac-IETD-pNA, and Ac-LEHD-pNA, respectively. The activities of caspase-3, caspase-8, and caspase-9 were quantified by spectrophotometric detection of free pNA derived from Ac-DEVD-pNA, Ac-IETD-pNA, and Ac-LEHD-pNA using an M200 microplate reader (Tecan, Group Ltd., Switzerland).
Immunochemistry
Immunochemistry was performed as previously described [30, 38]. The frozen 8-μm-thick sections fixed in 4% paraformaldehyde were blocked by 8% normal goat serum for 4 h and then incubated in specific primary antibodies. After being washed three times by phosphate-buffered saline (PBS), the sections were incubated with horseradish peroxidase-conjugated secondary antibodies. Staining was visualized using the substrate diaminobenzidine. The following antibodies were used: F4/80 (#ab6640, Abcam, Cambridge, UK, 1:500 dilution), tumor necrosis factor-α (TNF-α, #ab9635, Abcam, Cambridge, UK, 1:1000 dilution), interleukin-1β (IL-1β, #MAB4012, R&D Systems, Minneapolis, MN, USA, 1:1000 dilution), ICAM-1 (#ab171123, Abcam, Cambridge, UK, 1:500 dilution), and VCAM-1 (#ab134047, Abcam, Cambridge, UK, 1:500 dilution). In our immunohistochemistry study, the negative control sections were incubated with normal immunoglobulin G (IgG) without primary antibodies to confirm the specific staining.
Immunoblotting
Immunoblotting was performed in an Odyssey Infrared Fluorescence Imaging System (Li-Cor) as previously described [39]. Aortic tissues were washed in PBS (0.1 mM) and homogenized with RIPA buffer and protease inhibitors (Pierce). The total protein concentration was determined by the Bradford assay. Approximately 30 µg samples were run on 10% SDS-PAGE. The proteins were electrotransferred to nitrocellulose membranes, probed with primary antibody against CXCR-4 (Abcam, 1:1500 dilution) overnight, and then incubated with Infrared-Dyes-conjugated secondary antibodies (Li-Cor). The images were obtained using an Odyssey Infrared Fluorescence Imaging System. All immunoblotting experiments were repeated at least three times.
Statistical analysis
Data were analyzed with GraphPad Prism-5 statistical software (La Jolla, CA, USA). All values were presented as the mean ± SEM and were analyzed by Student’s t test or ANOVA followed by Tukey post-hoc test. P < 0.05 was considered statistically significant.
Results
Body weight and serum parameters of ApoE −/−;Nampt-Tg mice
We initially compared body weight and serum parameters between the ApoE−/− mice and ApoE−/−;Nampt-Tg mice. There were no differences between the ApoE−/− mice and ApoE−/−;Nampt-Tg mice under normal chow (Supplementary Table 1). Under a 16-week challenge of the pro-atherosclerotic HFD, body weight did not significantly differ between the ApoE−/− mice and ApoE−/−;Nampt-Tg mice (Fig. 1a). We isolated the serum that was rich in lipid content and measured the glucose and lipid levels. As shown in Fig. 1b, there was no significant difference in the serum glucose, TG, TC, LDL, and HDL levels between the ApoE−/− mice and ApoE−/−;Nampt-Tg mice. However, the serum NEFA level in the ApoE−/−;Nampt-Tg mice was slightly and significantly higher than that in the ApoE−/− mice.

Body weight and serum parameters of mice fed HFD. a Body weight curve of ApoE−/− mice and ApoE−/−;Nampt-Tg mice fed HFD for 16 weeks. b Serum glucose, TG, TC, LDL, HDL, and NEFA levels in ApoE−/− mice and ApoE−/−;Nampt-Tg mice. *P < 0.05 vs. ApoE−/−. TC total cholesterol, TG triglycerides, HDL high-density lipoproteins, LDL low-density lipoproteins, NEFA non-esterified fatty acid, NS no significance
ApoE −/−;Nampt-Tg mice display aggravated atherosclerotic lesion
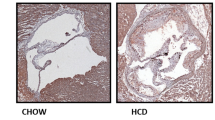
Oil Red O staining of the aortic tree showed that the atherosclerotic lesion size in the ApoE−/−;Nampt-Tg mice was significantly greater than that in the ApoE−/− mice (Fig. 2a). In addition, oil red O staining demonstrated that the lipid-enriched lesion in the aortic root of the ApoE−/−;Nampt-Tg mice was greater than that in the ApoE−/− mice (Fig. 2b). Morphological analysis demonstrated that the atheromatous plaque area in the cephalic brachial trunk of the ApoE−/−;Nampt-Tg mice was greater than that in the ApoE−/− mice (Fig. 2c). H&E staining of the aortae analysis confirmed that the thickness of the aortic atherosclerosis plaque in the ApoE−/−;Nampt-Tg mice was greater than that in the ApoE−/− mice (Fig. 2d).

Comparison of atherosclerotic lesion between ApoE−/− and ApoE−/−;Nampt-Tg mice. a Representative images and statistical data of Oil Red O staining on aortic tree. **P < 0.01 vs. ApoE−/−. b Representative images and statistical data of Oil Red O staining on aortic root section. **P < 0.01 vs. ApoE−/−. Scale bar, 100 μm. c Morphological images and statistical data of atherosclerotic lesion in cephalic brachial trunk. **P < 0.01 vs. ApoE−/−. d H&E staining and statistical data of atherosclerotic lesion thickness. **P < 0.01 vs. ApoE−/−. Scale bar, 100 μm
Transgene of Nampt modulates the collagen content in atherosclerotic plaque in ApoE −/− mice
We subsequently compared the collagen content of the atherosclerotic plaque between the ApoE−/− mice and ApoE−/−;Nampt-Tg mice by measuring the collagen content with Masson trichrome staining and PicroSirius Red staining. Masson trichrome staining showed that the blue stained area, which indicates the collagen content, was reduced by ~50% in the ApoE−/−;Nampt-Tg mice compared with the ApoE−/− mice (Fig. 3a). Moreover, the number of necrotic core (indicated as #) in the atherosclerotic plaque of the ApoE−/−;Nampt-Tg mice was ~1.8-fold higher than that in the ApoE−/− mice (Fig. 3a). The collagen fibers (red stained area) in the vascular wall and atherosclerosis plaque were further analyzed using PicroSirius Red staining. Under light microscopy, the total collagen content in the ApoE−/−;Nampt-Tg mice was significantly decreased compared with the ApoE−/− mice (Fig. 3b). The proportions of the different fibrillar collagen types in the mice were further assessed using polarized light microscopy. Collagen I was mature and stained red-orange, while collagen III was immature and stained green [37]. The collagen maturation (collagen I/III ratio) was reduced by ~50% in the ApoE−/−;Nampt-Tg mice compared with that in the ApoE−/− mice (Fig. 3c). These results suggest that Nampt overexpression could modulate the collagen content in the atherosclerotic plaque in ApoE−/− mice.

Evaluation of collagen content in atherosclerotic lesion of ApoE−/− and ApoE−/−;Nampt-Tg mice. a Masson trichrome staining images and statistical data of collagen content (blue) and necrosis core in aortic atherosclerotic plaque. **P < 0.01 vs. ApoE−/−. Scale bar, 200 μm. b PicroSirius Red staining images under light microscopy and statistical data of collagen fibers (red). *P < 0.05 vs. ApoE−/−. Scale bar, 50 μm. c Different fibrillar collagen types in plaque were assessed using polarized light microscopy with PicroSirius Red staining. Collagen I is mature and stained red-orange, and collagen III is immature and stained green. **P < 0.01 vs. ApoE−/−. Scale bar, 50 μm
Apoptosis in atherosclerotic plaque is aggravated in ApoE −/−;Nampt-Tg mice
The TUNEL staining showed that there were many apoptotic cells in the atherosclerotic plaque in the ApoE−/− mice, while the number of TUNEL-positive apoptotic cells in the ApoE−/−;Nampt-Tg mice was substantially higher than that in the ApoE−/− mice (Fig. 4a). In support of this finding, the activities of caspase-3, caspase-8, and caspase-9 in the aortic samples of the ApoE−/−;Nampt-Tg mice were higher than those in the ApoE−/− mice (Fig. 4b–d). These data indicate that the apoptosis in the atherosclerotic plaque is aggravated in ApoE−/−;Nampt-Tg mice compared with ApoE−/− mice.

Determination of apoptosis in atherosclerotic plaque of ApoE−/− and ApoE−/−;Nampt-Tg mice. a Representative images of TUNEL staining in atherosclerosis plaque and quantitative analysis. **P < 0.01 vs. ApoE−/−. Scale bar, 100 μm. b–d Activities of caspase-3 (b), caspase-8 (c) and caspase-9 (d) were measured. **P < 0.01 vs. ApoE−/−
TNF-α signaling in atherosclerotic plaque is stimulated in ApoE −/−;Nampt-Tg mice
We subsequently compared the inflammation in the atherosclerotic plaque between the ApoE−/− and ApoE−/−;Nampt-Tg mice. F4/80 immunohistochemistry staining demonstrated a significantly increased F4/80-positive area in the aortic atherosclerotic plaque in the ApoE−/−;Nampt-Tg mice compared with the ApoE−/− mice (Fig. 5a), which suggests that the plaque of the ApoE−/−;Nampt-Tg mice has more macrophage content. TNF-α immunohistochemistry staining also showed that the TNF-α-positive area in the ApoE−/−;Nampt-Tg mice was significantly greater than that in the ApoE−/− mice (Fig. 5b). However, the IL-1β-positive area in the ApoE−/−;Nampt-Tg mice was comparable with that in the ApoE−/− mice (Fig. 5c). Moreover, there was no difference in the TNF-α concentration between the ApoE−/− and ApoE−/−;Nampt-Tg mice (Fig. 5d). These results imply that Nampt transgene could further stimulate the pro-inflammatory TNF-α signaling in the atherosclerotic plaque in ApoE−/− mice.

TNF-α, but not IL-1β, is activated in atherosclerotic plaque of ApoE−/−;Nampt-Tg mice. a Infiltrated macrophages were evaluated by F4/80 staining. **P < 0.01 vs. ApoE−/−. b TNF-α immunohistochemistry staining and quantitative analysis. *P < 0.05 vs. ApoE−/−. c IL-1β immunohistochemistry staining and quantitative analysis. Scale bar, 100 μm. d Serum TNF-α level in ApoE−/− and ApoE−/−;Nampt-Tg mice. NS no significance
ICAM-1 and CXCR-4 are activated in atherosclerotic plaque of ApoE −/−;Nampt-Tg mice
Finally, we measured the chemokines and chemokine receptors in the atherosclerotic plaques of the ApoE−/− and ApoE−/−;Nampt-Tg mice. Compared with the ApoE−/− mice, the ICAM-1 expression in the atherosclerotic plaque was higher in the ApoE−/−;Nampt-Tg mice (Fig. 6a). However, we did not observe a significant difference in the VCAM-1 expression between the ApoE−/− and ApoE−/−;Nampt-Tg mice (Fig. 6b). The protein level of C-X-C chemokine receptor type 4 (CXCR-4) was determined using immunoblotting. CXCR4 is a chemokine receptor specific for stromal-derived factor-1 (SDF-1, also referred to as CXCL12), a molecule endowed with potent chemotactic activity for lymphocytes [40, 41]. Vascular CXCR4 limits atherosclerosis by maintaining the arterial integrity [40], while CXCR4 blockade induces atherosclerosis [41]. In our experiment, we found that the CXCR4 expression in the atherosclerotic aortic sample of the ApoE−/−;Nampt-Tg mice was significantly lower than that in the ApoE−/− mice (Fig. 6c).

Measurement of chemokines in atherosclerotic plaque of ApoE−/− and ApoE−/−;Nampt-Tg mice. a, b Representative immunohistochemistry staining images and quantitative analyses of ICAM-1 (a) and VCAM-1 (b) in atherosclerotic plaques. *P < 0.05 vs. ApoE−/−. Scale bar, 100 μm. (c) Representative immunoblotting images and quantitative analysis of CXCR-4 in atherosclerotic plaques. *P < 0.05 vs. ApoE−/−. NS no significance
Discussion
There is no doubt Nampt represents an essential factor for all cells in energy metabolism, circadian clock, and cell survival through its fundamental enzymatic activity for NAD+ biosynthesis. Although several previous reports have described the relevance of Nampt in atherosclerosis, it seems that they do not reach a consensus and are even contradictory. Moreover, there are many uncertainties regarding the molecular mechanisms that mediate the actions of Nampt in atherosclerosis. The current study aimed to evaluate the role of Nampt in atherosclerosis development in ApoE−/− mice. Our experimental evidence demonstrates that the overexpression of Nampt accelerates atherosclerotic lesion formation in ApoE−/− mice.
Lipid accumulation in the vascular wall is the first key step of atherosclerosis. Nampt and its enzymatic product nicotinamide mononucleotide (NMN), as well as the compound nicotinamide ribose (NR), critically regulate lipid homeostasis. Nampt was found to be required for de novo lipogenesis in tumor cells [42]. The overexpression of Nampt decreased the whole-body lipid profile in rats via upregulation of the tyrosine phosphorylation of IRS-1 protein and the mRNA levels of PPARγ and SREBP-2 [43]. Both NMN and NR exhibited potent improvements in glucose intolerance and lipid profiles in high-fat diet-induced metabolic abnormalities [44, 45]. Our previous work also found enzymatically inactive NAMPT transgenic mice had moderate lipid accumulation in the liver, and NR substantially reduced the hepatic liver content [38]. Therefore, it seems to be safe to consider that Nampt overexpression might improve the blood lipid profile in ApoE−/− mice. However, our data in this study showed that the overexpression of Nampt in ApoE−/− mice failed to reduce serum lipid concentrations. By contrast, the serum NEFA level was even higher in ApoE−/−;Nampt-Tg mice than that in ApoE−/− mice, which suggests the effect of enforced Nampt overexpression may differ from the administration of NMN or NR. These unexpected results are difficult to interpret. We speculate that the overexpression of Nampt in the whole body may affect lipid absorption and consumption not only by prompting NAD+ biosynthesis but also influencing other unknown biological events associated with lipid homeostasis.
As a key feature of atherosclerosis, atherosclerotic inflammation contributes to a group of complex procedures during atherosclerosis from foam-cell formation to plaque rupture and thrombosis. Both the innate and adaptive immune systems govern the inflammatory process in the arterial wall. Monocytes and macrophages act together to fuel the inflammatory cascade in atherosclerosis development. To investigate the effect of Nampt in atherosclerosis, early investigations used in vitro cultured macrophages following stimuli under oxidized LDL cholesterol (ox-LDL). Dahl et al. reported that Nampt was markedly enhanced in carotid plaques from symptomatic individuals compared with plaques from asymptomatic individuals and localized in areas that were rich in lipid-loaded macrophages [23]. Moreover, the Nampt expression in THP-1 monocytes was increased by ox-LDL and TNF-α [23]. These results implied that Nampt might be an inflammatory mediator localized within unstable atherosclerotic lesions and seemed to play a role in plaque destabilization. Intriguingly, in another investigation, the same group showed that Nampt knockdown increased lipid accumulation in macrophages [46], which suggests Nampt might not be a completely undesirable factor in atherosclerosis. However, as other cells such as VSMCs and T-lymphocyte cells also contributed to the atherosclerotic inflammation, evaluating the effects of Nampt in atherosclerosis development in vivo might be more appropriate. Furthermore, the potential effects of Nampt on the adhesion of monocytes to endothelial cells should be considered. Several previous studies had provided strong evidence for this possibility. For example, Kim et al. reported that Nampt enhanced ICAM-1 and VCAM-1 expression through ROS-dependent NF-κB activation in endothelial cells [47]. Adya et al. showed that Nampt induced VEGF and MMP-2/9 production in endothelial cells [48]. Thus, the changed adhesion molecules in endothelial cells might promote the recruitment of immune cells and thus trigger inflammation.
Furthermore, the effects of Nampt in atherosclerosis in vivo have been explored by several investigations. Nencioni et al. found the Nampt inhibitor FK866 mitigated inflammation in atherosclerotic plaques by reducing CXCL1-mediated activities on neutrophils in ApoE−/− mice with a HFD [24]. Li et al. also reported that Nampt knockdown by adenovirus injection via the tail vein attenuated atherosclerosis and promoted reverse cholesterol transport in ApoE−/− mice with a HFD [49]. By contrast, Bermudez et al. recently provided evidence that hematopoietic overexpression of human Nampt attenuated the plaque burden and stabilized lesions in LDL-deficient mice fed a western-type diet [26]. The authors considered that the seemingly paradoxical results of their study compared with those of previous studies can be attributed to the diverging activities of intracellular and extracellular Nampt because extracellular Nampt was commonly thought to be a pro-inflammation factor [22]. There are several limitations in these studies. First, the chemical inhibitor FK866, siRNA-mediated knockdown and lentivirus-mediated overexpression were applied to modulate Nampt activity or expression in these studies. As chemical inhibitors always have a non-specific action in vivo and virus-mediated gene delivery is rather challenging, particularly in a long-term animal study, we aimed to test the assumption more specifically. Thus, we used a transgenic mouse model in an ApoE gene knockout background with hereditary overexpression of Nampt (ApoE−/−;Nampt-Tg mice) in the present study and demonstrated that Nampt transgene promoted atherosclerosis inflammation and enlarged the atherosclerotic lesion. Specifically, we observed that the plaque of ApoE−/−;Nampt-Tg mice displayed promoted TNF-α expression, but not IL-1β. This result is clearly in line with the previous study that showed the intracellular NAD+ pool facilitates TNF-α release [11]. It should be noted that our data in this study only suggest Nampt systemic overexpression may have a pro-atherosclerosis property under the HFD condition. To clarify the exact role of Nampt in atherosclerosis, additional investigations are warranted.
The change of the NAD+ level in a cell may not only affect apoptosis but also other types of cell death, such as necroptosis [50, 51], autophagy [52, 53], and parthanatos [54, 55]. As NAD+ and NADH play crucial roles in a number of fundamental biological processes, such as energy metabolism, mitochondrial functions, and gene regulation, the fluctuation in the intracellular NAD+ pool may serve as a potential therapeutic target for treating several disorders. Specifically, our group previously reported that the Nampt-NAD+ axis modulated neuronal autophagy following ischemic stress in vivo and in vitro [27]. More importantly, we demonstrated the induction of autophagy contributed to the neuroprotection of Nampt in cerebral ischemia [27]. However, in this study, our results suggested that the Nampt transgene promoted apoptosis in atherosclerotic lesions. Complex mechanisms must underlie this discrepancy and should be explored in the future.
In conclusion, our results illustrate that the transgene of Nampt potentiates inflammation and promotes atherosclerosis in ApoE−/− mice. Moreover, these findings support further assessments of Nampt as a therapeutic target for atherosclerosis prevention and treatment.
References
Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 2015;11:535–46.
Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–75.
Li Y, Zhang Y, Dorweiler B, Cui D, Wang T, Woo CW, et al. Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signaling mechanism. J Biol Chem. 2008;283:34833–43.
Garten A, Petzold S, Barnikol-Oettler A, Korner A, Thasler WE, Kratzsch J, et al. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes. Biochem Biophys Res Commun. 2010;391:376–81.
Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+synthesis in cardiac myocytes. Circ Res. 2009;105:481–91.
Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+levels dictate cell survival. Cell. 2007;130:1095–107.
Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531:528–32.
Kieswich J, Sayers SR, Silvestre MF, Harwood SM, Yaqoob MM, Caton PW. Monomeric eNAMPT in the development of experimental diabetes in mice: a potential target for type 2 diabetes treatment. Diabetologia. 2016;59:2477–86.
Skokowa J, Lan D, Thakur BK, Wang F, Gupta K, Cario G, et al. NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat Med. 2009;15:151–8.
Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+biosynthesis. Science. 2009;324:651–4.
Van Gool F, Galli M, Gueydan C, Kruys V, Prevot PP, Bedalov A, et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med. 2009;15:206–10.
Gerner RR, Klepsch V, Macheiner S, Arnhard K, Adolph TE, Grander C, et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut. 2018;67:1813–23.
Audrito V, Serra S, Brusa D, Mazzola F, Arruga F, Vaisitti T, et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood. 2015;125:111–23.
Takao S, Chien W, Madan V, Lin DC, Ding LW, Sun QY, et al. Targeting the vulnerability to NAD(+) depletion in B-cell acute lymphoblastic leukemia. Leukemia. 2018;32:616–25.
van der Veer E, Ho C, O’Neil C, Barbosa N, Scott R, Cregan SP, et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J Biol Chem. 2007;282:10841–5.
Borradaile NM, Pickering JG. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell. 2009;8:100–12.
Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–25.
Zhong M, Tan HW, Gong HP, Wang SF, Zhang Y, Zhang W. Increased serum visfatin in patients with metabolic syndrome and carotid atherosclerosis. Clin Endocrinol (Oxf). 2008;69:878–84.
Kadoglou NP, Sailer N, Moumtzouoglou A, Kapelouzou A, Tsanikidis H, Vitta I, et al. Visfatin (nampt) and ghrelin as novel markers of carotid atherosclerosis in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2010;118:75–80.
Mu J, Feng B, Ye Z, Yuan F, Zeng W, Luo Z, et al. Visfatin is related to lipid dysregulation, endothelial dysfunction and atherosclerosis in patients with chronic kidney disease. J Nephrol. 2011;24:177–184.
Wang P, Xu TY, Guan YF, Su DF, Fan GR, Miao CY. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res. 2009;81:370–80.
Romacho T, Azcutia V, Vazquez-Bella M, Matesanz N, Cercas E, Nevado J, et al. Extracellular PBEF/NAMPT/visfatin activates pro-inflammatory signalling in human vascular smooth muscle cells through nicotinamide phosphoribosyltransferase activity. Diabetologia. 2009;52:2455–63.
Dahl TB, Yndestad A, Skjelland M, Oie E, Dahl A, Michelsen A, et al. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation. 2007;115:972–80.
Nencioni A, da Silva RF, Fraga-Silva RA, Steffens S, Fabre M, Bauer I, et al. Nicotinamide phosphoribosyltransferase inhibition reduces intraplaque CXCL1 production and associated neutrophil infiltration in atherosclerotic mice. Thromb Haemost. 2014;111:308–22.
Watson A, Nong Z, Yin H, O’Neil C, Fox S, Balint B, et al. Nicotinamide phosphoribosyltransferase in smooth muscle cells maintains genome integrity, resists aortic medial degeneration, and is suppressed in human thoracic aortic aneurysm disease. Circ Res. 2017;120:1889–902.
Bermudez B, Dahl TB, Medina I, Groeneweg M, Holm S, Montserrat-de la Paz S, et al. Leukocyte overexpression of intracellular NAMPT attenuates atherosclerosis by regulating PPARgamma-dependent monocyte differentiation and function. Arterioscler Thromb Vasc Biol. 2017;37:1157–67.
Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 2012;8:77–87.
Wei CC, Kong YY, Li GQ, Guan YF, Wang P, Miao CY. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci Rep. 2017;7:717.
Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC, et al. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann Neurol. 2011;69:360–74.
Wei CC, Kong YY, Hua X, Li GQ, Zheng SL, Cheng MH, et al. NAD replenishment with nicotinamide mononucleotide protects blood-brain barrier integrity and attenuates delayed tissue plasminogen activator-induced haemorrhagic transformation after cerebral ischaemia. Br J Pharmacol. 2017;174:3823–36.
Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S, Sadoshima J. Nicotinamide mononucleotide, an intermediate of NAD+synthesis, protects the heart from ischemia and reperfusion. PLoS One. 2014;9:e98972.
Wang P, Du H, Zhou CC, Song J, Liu X, Cao X, et al. Intracellular NAMPT-NAD+-SIRT1 cascade improves post-ischaemic vascular repair by modulating Notch signalling in endothelial progenitors. Cardiovasc Res. 2014;104:477–88.
Wang P, Yang X, Zhang Z, Song J, Guan YF, Zou DJ, et al. Depletion of NAD pool contributes to impairment of endothelial progenitor cell mobilization in diabetes. Metabolism. 2016;65:852–62.
de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, et al. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016;15:522–30.
Zhao Y, Guan YF, Zhou XM, Li GQ, Li ZY, Zhou CC, et al. Regenerative neurogenesis after ischemic stroke promoted by nicotinamide phosphoribosyltransferase-nicotinamide adenine dinucleotide cascade. Stroke. 2015;46:1966–74.
Miao CY, Xie HH, Zhan LS, Su DF. Blood pressure variability is more important than blood pressure level in determination of end-organ damage in rats. J Hypertens. 2006;24:1125–35.
Arnaud C, Bouyon S, Recoquillon S, Brasseur S, Lemarie E, Briancon-Marjollet A, et al. Nonmuscle myosin light chain kinase: a key player in intermittent hypoxia-induced vascular alterations. J Am Heart Assoc. 2018;7:e007893.
Zhou CC, Yang X, Hua X, Liu J, Fan MB, Li GQ, et al. Hepatic NAD(+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br J Pharmacol. 2016;173:2352–68.
Wang P, Zhang RY, Song J, Guan YF, Xu TY, Du H, et al. Loss of AMP-activated protein kinase-alpha2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes. 2012;61:1051–61.
Doring Y, Noels H, van der Vorst EPC, Neideck C, Egea V, Drechsler M, et al. Vascular CXCR4 limits atherosclerosis by maintaining arterial integrity: evidence from mouse and human studies. Circulation. 2017;136:388–403.
Bot I, Daissormont IT, Zernecke A, van Puijvelde GH, Kramp B, de Jager SC, et al. CXCR4 blockade induces atherosclerosis by affecting neutrophil function. J Mol Cell Cardiol. 2014;74:44–52.
Bowlby SC, Thomas MJ, D’Agostino RB Jr., Kridel SJ. Nicotinamide phosphoribosyl transferase (Nampt) is required for de novo lipogenesis in tumor cells. PLoS One. 2012;7:e40195.
Sun Q, Li L, Li R, Yang M, Liu H, Nowicki MJ, et al. Overexpression of visfatin/PBEF/Nampt alters whole-body insulin sensitivity and lipid profile in rats. Ann Med. 2009;41:311–20.
Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–36.
Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–47.
Dahl T, Ranheim T, Holm S, Berge R, Aukrust P, Halvorsen B. Nicotinamide phosphoribosyltransferase and lipid accumulation in macrophages. Eur J Clin Invest. 2011;41:1098–104.
Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH, Koo TH, et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim Biophys Acta. 2008;1783:886–95.
Adya R, Tan BK, Punn A, Chen J, Randeva HS. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc Res. 2008;78:356–65.
Li S, Wang C, Li K, Li L, Tian M, Xie J, et al. NAMPT knockdown attenuates atherosclerosis and promotes reverse cholesterol transport in ApoE KO mice with high-fat-induced insulin resistance. Sci Rep. 2016;6:26746.
Pajuelo D, Gonzalez-Juarbe N, Tak U, Sun J, Orihuela CJ, Niederweis M. NAD(+) depletion triggers macrophage necroptosis, a cell death pathway exploited by Mycobacterium tuberculosis. Cell Rep. 2018;24:429–40.
Preyat N, Rossi M, Kers J, Chen L, Bertin J, Gough PJ, et al. Intracellular nicotinamide adenine dinucleotide promotes TNF-induced necroptosis in a sirtuin-dependent manner. Cell Death Differ. 2016;23:29–40.
Cea M, Cagnetta A, Fulciniti M, Tai YT, Hideshima T, Chauhan D, et al. Targeting NAD+salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood. 2012;120:3519–29.
Hsu CP, Hariharan N, Alcendor RR, Oka S, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through autophagy in cardiomyocytes. Autophagy. 2009;5:1229–31.
Preyat N, Leo O. Reassessing the role of NAD as a prosurvival factor. Mol Cell Oncol. 2016;3:e1062591.
Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagne JP, et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111:10209–14.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81473208, No. 81673485, No. 81422049, and No. 81373414), National Key R&D Program of China (2018YFA0108301) and Yantai University Key Laboratory of Molecular Pharmacology and Drug Evaluation of Ministry of Education (P201603).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Kong, Yy., Li, Gq., Zhang, Wj. et al. Nicotinamide phosphoribosyltransferase aggravates inflammation and promotes atherosclerosis in ApoE knockout mice. Acta Pharmacol Sin 40, 1184–1192 (2019). https://doi.org/10.1038/s41401-018-0207-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0207-3
Keywords
This article is cited by
-
Research Progress on Histone Deacetylases Regulating Programmed Cell Death in Atherosclerosis
Journal of Cardiovascular Translational Research (2024)
-
NAD+ salvage governs the immunosuppressive capacity of mesenchymal stem cells
Cellular & Molecular Immunology (2023)
