
Abstract
Anandamide is a lipid mediator that acts as an endogenous ligand of CB1 receptors. These receptors are also the primary molecular target responsible for the pharmacological effects of Δ9-tetrahydrocannabinol, the psychoactive ingredient in Cannabis sativa. Several studies demonstrate that anandamide exerts an overall modulatory effect on the brain reward circuitry. Several reports suggest its involvement in the addiction-producing actions of other abused drugs, and it can also act as a behavioral reinforcer in animal models of drug abuse. Importantly, all these effects of anandamide appear to be potentiated by pharmacological inhibition of its metabolic degradation. Enhanced brain levels of anandamide after treatment with inhibitors of fatty acid amide hydrolase, the main enzyme responsible for its degradation, seem to affect the rewarding and reinforcing actions of many drugs of abuse. In this review, we will provide an overview from a preclinical perspective of the current state of knowledge regarding the behavioral pharmacology of anandamide, with a particular emphasis on its motivational/reinforcing properties. We will also discuss how modulation of anandamide levels through inhibition of enzymatic metabolic pathways could provide a basis for developing new pharmaco-therapeutic tools for the treatment of substance use disorders.
Similar content being viewed by others


Introduction
Cannabis sativa and its derivatives, i.e., marijuana, are among the best-known mind-altering substances used by man in ancient times and are still among the most abused substances worldwide. The major psychoactive component of marijuana, Δ9-tetrahydrocannabinol (THC), was isolated and its chemical structure clarified by Gaoni and Mechoulam in 1964 [1]. Since then, THC has been synthesized, and many studies have been conducted on its activity [2]. Nevertheless, until the discovery of CB1 receptors in 1988 [3], the mechanisms involved in the action of THC on the brain were elusive and were suggested to consist of non-specific activity on the neuronal cell membrane [4, 5]. The discovery of CB1 receptors [6], the primary pharmacological target of THC, prompted research to find the endogenous ligands for these receptors. The first endocannabinoid to be discovered was anandamide (AEA), which was isolated from the pig brain by William Devane and co-workers in 1992 [7]. Its name comes from the Sanskrit word “ananda” (internal bliss), which emphasizes the interesting role of AEA as an endogenous marijuana-like substance self-delivered by the brain. 2-Arachidonoylglycerol (2-AG) was the second endocannabinoid to be identified; it was isolated from the rat brain and the canine gut [8, 9]. Since the discovery of these first two endogenous cannabinoids, numerous studies have been carried out demonstrating that their behavioral and molecular effects only partially overlap between compounds, as well as between each compound and the most studied CB1 agonist, THC [10]. The discovery of CBRs and endocannabinoids led to the identification of other components of what was then called the “endocannabinoid system”, which also includes the enzymes that are responsible for both the biosynthesis and the metabolism of endocannabinoids [11]. The endocannabinoid system is an important lipid signaling system that emerged before the evolution of vertebrates and it is well preserved across species [12]. In mammals, it has been recently recognized as a modulator of a large variety of physiological processes, including inflammation and pain [13, 14], appetite [15], mood [16], and pre-and postnatal development [17]. Furthermore, the endocannabinoid system is an important constituent of the neuronal substrates involved in the reinforcement and reward processes of the brain [18, 19]. It modulates the rewarding and pharmacological responses induced by cannabinoids [20], as well as those induced by other drugs of abuse [19, 21, 22].
With regard to its involvement in the mechanisms of reward, AEA has been the most studied among the endocannabinoids. Indeed, several studies indicate that AEA exerts an overall modulatory effect on the reward circuitry. It may possess reinforcing effects of its own, and it is implicated in the reinforcing and addictive effects of various drugs of abuse, including non-cannabinoids [19, 21, 23]. For instance, AEA may play important roles in the sequence of events from experiencing the euphoric effects of a drug to abuse and drug use disorders. The aim of the present manuscript is to explore and discuss preclinical behavioral studies about the best-known brain endocannabinoid, AEA, and how its direct effects or changes in its abundance in the brain might (or might not) suggest a role in drug use disorders. We have reviewed results obtained through specific behavioral pharmacology methodologies such as brain stimulation reward place conditioning, self-administration, reinstatement, and relapse in order to provide discussion material about the potential of AEA as a reinforcer and/or as a treatment for drug addiction. Then, we briefly introduce some notions about the endocannabinoid system. The last part of the review focuses on specific preclinical brain/behavioral activities of AEA as a reinforcer or as a modulator of rewarding/reinforcing effects of non-cannabinoid drugs of abuse.
Endocannabinoids
As mentioned in the introduction, discoveries regarding candidate endogenous ligands of the CBRs started in the early 1990s, soon after the identification of CBRs as specific receptors for THC. Both AEA and 2-AG are bioactive lipids, belonging to the subclasses known as N-acylethanolamines (NAEs) and monoacylglycerols (MAGs), respectively [24]. Numerous studies have shown that these compounds play key roles in several biological activities within the central nervous system (CNS) and the periphery [25]. Although similar in structure (AEA and 2-AG are arachidonic acid derivatives conjugated with ethanolamine or glycerol, respectively) (Fig. 1), these compounds show some crucial distinctions that are responsible for their different physiological and pathophysiological roles: (i) they are regulated by different biosynthetic and degradative pathways [26]; (ii) brain tissue levels of AEA are 10–100 times lower than those of 2-AG [27]; (iii) AEA (as well as THC) activates CBRs with low intrinsic efficacy (partial agonist), whereas 2-AG is an agonist with high intrinsic efficacy (full agonist) [28]. In addition to AEA and 2-AG, several other lipids with endocannabinoid-like activity have been isolated [29], including 2-arachidonylglyceryl ether (2-AGE, noladin), O-arachidonylethanolamine (virodhamine), and N-arachidonyldopamine (NADA) [30,31,32]. However, their physiological role is not clear, and thus it remains to be ascertained whether they are true endocannabinoids.

Structures of AEA and 2-AG
Endocannabinoid biosynthesis and metabolism
Given their hydrophobic nature, endocannabinoids are precluded from stable uptake and storage into synaptic vesicles, and there are no known cannabinoid neurons or cannabinoid neuronal pathways. However, virtually all cells possess AEA/2-AG precursors, which are usually found as components of the cell membrane, and the end products themselves can be synthesized on demand in a Ca2+-dependent manner after cellular depolarization or receptor stimulation [24, 33]. Several pathways exist for the biosynthesis of AEA and 2-AG; these pathways, together with the catabolic pathways, represent the key points in the regulation of their tissue levels (Fig. 2). AEA is derived mainly from the cleavage of a phospholipid precursor, N-arachidonoyl phosphatidylethanolamine (NAPE); NAPE, in turn, is derived from the enzymatic transfer, catalyzed by N-acyltransferase (NAT), of an acyl group from the sn-1 position of arachidonic acid to the amino group of a phosphatidylethanolamine (PE). NAPE is hydrolyzed to AEA and phosphatidic acid by a phosphodiesterase, a substrate-specific phospholipase D (NAPE-PLD). In addition to NAPE-PLD, NAPE can also be hydrolyzed by other enzymes such as phospholipase A2 (PA2), phospholipase C (PLC) and α/β-hydrolase 4 (Abh4) [34]. The formation of 2-AG proceeds mainly from inositol phospholipids (PI) via diacylglycerol (DAG), by the phospholipase C (PLC)/DAG lipase pathway [35,36,37]. In addition, 2-AG may be synthesized from PI by sequential hydrolysis catalyzed by PI-specific phospholipase A1 (PLA1) and lyso-PI-specific PLC [38]. As already mentioned above, endocannabinoids are not stored in vesicles, and their half-life is very short, as they undergo rapid metabolic deactivation by specific enzymes after being taken up into the cell. Degradation of AEA occurs mainly through the action of the intracellular enzyme fatty acid amide hydrolase (FAAH), which breaks it down into free arachidonic acid and ethanolamine [26, 39]. In addition to FAAH, AEA is also degraded by N-acylethanolamine acid amidase (NAAA) [40]. Both FAAH (which most prefers AEA as a substrate) and NAAA also hydrolyze oleoylethanolamide (OEA) and palmitoylethanolamide (PEA), two endogenous fatty acid amides that bind primarily to the α-type of peroxisome proliferator-activated receptors (PPARα) but that seem to enhance AEA activity through an entourage effect [41] . In addition to hydrolytic pathways, AEA can be oxygenated by cyclooxygenase-2 (COX-2), lipoxygenase (LOX) isoenzymes, and by cytochrome P-450 [25].

Schematic representation of AEA and 2-AG biosynthesis and degradation
FAAH may represent an attractive therapeutic target for the treatment of pain, depression and other CNS conditions [42]. For example, knocking out the FAAH enzyme in mice leads to more than 10-fold elevation of AEA levels within many brain regions [43] and gives rise to a CBR-mediated analgesic phenotype [44]. The discovery of this FAAH knockout phenotype led researchers to focus on FAAH inhibitors, such as URB597 ([3-(3-carbamoylphenyl)phenyl] N-cyclohexylcarbamate). Administration of an FAAH inhibitor prolongs the half-life and increases the activity of AEA [45]. Furthermore, URB597 has been demonstrated to be active in neuropathic [46], inflammatory [47], and acute pain [48]; depression [49]; and anxiety [48]. Several α-keto-heterocycles, such as OL-135, have also been found to increase AEA levels and display in vivo activity, producing analgesic effects in acute thermal and noxious chemical pain assays in mice [50]. While degradative enzymatic pathways for AEA have been evidenced during the last 20 years, AEA transport through the neuronal cell membrane is still a debated issue in the scientific community [25, 51,52,53]. To date, molecular identification and cloning of the AEA transporter has not been achieved. Nevertheless, a model has been proposed claiming that AEA uptake is also related to FAAH [54]. For this reason, recently identified AEA transport inhibitors, such as N-arachidonoyl-aminophenol (AM404) [55], N-arachidonyl-2-methyl,4-hydroxyphenylamine (VDM11) [56] and LY218240 [51], which prevent the recycling of AEA back into the cells from the synaptic cleft, also lead to FAAH inhibition. In contrast, other transport inhibitors, such as N-(5Z, 8Z, 11Z, 14Z eicosatetraenyl)-4-hydroxybenzamide (AM1172), N-arachidonyl-3-furylmethylamine (UCM707), and (R)-N-(1-(4-hydroxyphenyl)-2-hydroxyethyl)oleamide (OMDM2), inhibit FAAH weakly or not at all in vitro [57].
Similar to AEA, 2-AG is rapidly degraded by different enzymes to yield arachidonic acid and glycerol. The most ubiquitous mechanism of degradation of 2-AG is by monoacylglycerol lipase [34]. However, some studies have provided evidence that 2-AG is also degraded by FAAH [58, 59]. Finally, 2-AG can be degraded by a series of serine hydrolase α-β-hydrolase domain 6 or 12 that accounts for approximately 4%–9% of total brain 2-AG hydrolysis [60, 61]. Similar to FAAH inhibitors, the irreversible MAGL inhibitor N-arachidonyl maleimide (NAM) decreases 2-AG hydrolysis by ~85% [62]. Despite conflicting results [63], other MAGL inhibitors such as URB602 and URB754 have been reported to increase 2-AG brain levels and produce anti-hyperalgesia in rodents [64, 65].
Endocannabinoid signaling
It is well established that endocannabinoids retrogradely regulate synaptic neurotransmission, controlling the activity of a range of different neurotransmitters such as glutamate and gamma-aminobutyric acid (GABA) and thus controlling both excitatory and inhibitory inputs [66, 67]. Once synthesized from cell membrane phospholipids, endocannabinoids are immediately released by the postsynaptic terminal, and after traveling through the synaptic cleft, they bind to and activate CBRs in the presynaptic membrane, inhibiting neurotransmission release by activating presynaptic K+ channels and inhibiting N- and P/Q-type Ca2+ channels [66, 68, 69] (Fig. 3). Presynaptic inhibition of neurotransmitter release by endocannabinoids can lead to two different forms of synaptic plasticity depending on the involvement of GABA or glutamate transmission: activation of CBRs on axon terminals of GABAergic neurons mediates depolarization-induced suppression of inhibition (DSI), whereas activation of CBRs on axon terminals of glutamatergic neurons mediates depolarization-induced suppression of excitation (DSE) [70]. Moreover, the suppression of neurotransmitter release can be either transient (endocannabinoid-mediated short-term depression) or persistent (endocannabinoid-mediated long-term depression) [66]. This retrograde signaling function of endocannabinoids appears to be widely distributed throughout the CNS [71, 72].

Schematic representation of the “retrograde” mechanism of action of endocannabinoids: after depolarization, endocannabinoids are released by the postsynaptic terminal and activate CBRs in the presynaptic terminal, inhibiting neurotransmission release
Cannabinoid receptors
The pharmacological actions of marijuana and of its principal psychoactive ingredient THC were attributed in early studies to the ability of THC to penetrate cell membranes and to alter specific membrane properties due to its very high lipophilicity [4, 5]. This was the best scientific explanation possible before the discovery of specific receptors for this compound. The existence of CBRs was initially suggested in the late 1980s by ligand-binding studies and then confirmed by cloning the receptors, demonstrating unequivocally that CBRs were the receptors mediating the behavioral-pharmacological effects of marijuana [3, 6]. Thus, CBRs, revealed years after the discovery of most of the other known neurotransmitter receptors, were shown to be the most expressed receptors in the brain. However, there are no cannabinoid neurons that release endogenous cannabinoids to activate these receptors. Indeed, endogenous ligands are released on demand by cleavage of lipid precursors. It is interesting to note that while most of the classical neurotransmitter-neuromodulator receptor systems have only one endogenous ligand, CBRs have at least 2, likely more, endogenous ligands [29]. It is also interesting to note that another receptor system showing more than one endogenous ligand is the opioid system, and members of these two families of endogenous ligands show reinforcing effects in self-administration studies in animals.
To date, two CBRs have been characterized: the CB1 receptor (CB1R) identified in 1988 [3], and cloned in the early 1990s from rat cerebral cortex [6], and later from human [73] and mouse brain [74]; the second CBR subtype, CB2 receptor (CB2R), was derived from human promyelocytic-leukemia cells (HL-60 cells) [75]. Both CB1R and CB2R are members of the G-protein-coupled receptor (GPCRs) that belong to the rhodopsin GPCR family (Class A). AEA, 2-AG, and THC, as well as synthetic cannabinoids of other structural classes, such as CP55940,5 and WIN 55,212-2 are able to bind with high affinity to CB1R. CB1R represents the most abundant G-protein-coupled receptor in the brain: it has been identified at extremely high density in the cingulate gyrus, frontal cortex, hippocampus, cerebellum and basal ganglia [76,77,78]; at moderate density in the basal forebrain, amygdala, nucleus accumbens, periaqueductal gray and hypothalamus; and at low density in the midbrain, pons, medulla, primary motor cortex and thalamus [79]. CB1R is expressed mostly on axons and axon terminals of neurons but also on interneurons and astrocytes [80, 81]. Once CB1R is activated, it couples predominantly with G proteins of the αi and αo subtypes (Fig. 4). Its general mechanism comprises a signaling cascade that leads to inhibition of adenylyl cyclase [82], inhibition of the opening of voltage gated calcium channels [83], an increase in potassium channel conductance [83, 84], activation of the mitogen-activated protein kinases (MAPKs) [85], and overall suppression of neurotransmitter release. Nevertheless, the endocannabinoid system is characterized by a great complexity such that the signaling pathways and interacting proteins used by CBRs are much more various than the originally established ones [86, 87]. Furthermore, CB1R have also been described in peripheral sympathetic axon terminals, where they inhibit norepinephrine release [88, 89], and throughout the enteric nervous system, where they act inhibiting both intestinal motility and secretion [90]. In contrast to the view that CB2R is restrictively expressed in the immune system, such as in the marginal zone of the spleen, the thymus, the tonsils, and the surface of immune cells [75], various evidence has more recently shown that CB2Rs are also present within the brain, especially in microglial cells [91]. Despite controversy surrounding the data, they may also be involved in functions regulating substance abuse [92, 93]. Recent reports also demonstrated that CB2Rs are expressed in midbrain DA neurons [94, 95], where they modulate alcohol preference and the reinforcing and neurochemical effects of cocaine [92, 96, 97]. However, it appears that species differences related to CB2R genes splicing and their functional expression might produce different behavioral outcomes related to cocaine self-administration. Indeed, brain gene-expression studies on mice and rats have shown rat-specific CB2R isoforms, together with different regional levels of CB2R mRNA and cell-type localization [98, 99]. In humans, CB2R, encoded by the CNR2 gene, shares 68% identity with human CB1R within the transmembrane regions and only 44% homology throughout the total protein [75]. Despite belonging to the same family of G proteins and sharing some ligands, CB1R and CB2R differ significantly from one another in their signaling: CB2R poorly modulates calcium channels and inwardly rectifying potassium channels [100]. Moreover, CB2Rs from different species have often shown distinct pharmacological results in response to activation by identical drugs [101, 102].

Schematic representation of CBR stimulation: activation of CBR leads to inhibition of adenylyl cyclase, inhibition of Ca2+ channels, an increase in K+ channel conductance and activation of the mitogen-activated protein kinases (MAPKs)
Although AEA and 2-AG are widely accepted as endocannabinoids due to their interactions with CB1R and CB2R, they also stimulate other receptor types [31, 103]. A large body of evidence demonstrated that lipid-based molecules, in particular AEA (but not 2-AG), can bind TRPV1 vanilloid receptors, the molecular target of capsaicin [104]. Indeed, Zygmunt et al. [105] reported that AEA induces vasodilation by activating TRPV1 receptors on perivascular sensory nerves. The interaction of AEA with TRPV1 receptors is specific and depends on the ability of AEA to reach the intracellular binding site [34]. While AEA and capsaicin have similar affinity for TRPV1, the former has significantly lower potency, and higher concentrations of AEA are necessary to induce the typical TRPV1 responses than are required for CB1 activation [106, 107]. Furthermore, the activity of AEA at TRPV1 receptor may be modulated by molecular mechanisms that regulate its internalization and degradation [108]. AEA seems to act as a partial agonist when the TRPV1 receptor expression is low, while it is a full agonist when receptor expression is high [104, 106]. Additionally, TRPV1 sensitivity to AEA is enhanced by entourage effects of other endogenous lipids and by phosphorylation of TRPV1 itself. The activation of TRPV1 receptors by AEA has potential implications in the treatment of inflammatory, respiratory, and cardiovascular disorders [106].
Different studies have established the orphan G-protein-coupled receptor GPR55 as a cannabinoid receptor with signaling distinct from that of the CB1 and CB2 receptors [109,110,111]. Specifically, recent studies revealed that GPR55 could be a pharmacological target for AEA [112]. GPR55 is highly expressed in large dorsal root ganglion neurons and, upon activation, increases intracellular calcium in these neurons. GPR55 possesses numerous signaling pathways through which it exerts its physiological effects. Examination of its signaling pathway in HEK293 cells transiently expressing GPR55 indicated that the calcium increase involves G(q), G(12), RhoA, actin, phospholipase C, and calcium release from IP(3)R-gated stores [112, 113]. GPR55 activation also inhibits M currents [114].
A growing body of evidence identifies PPARs as cannabinoid targets and suggests that this interaction could be responsible for some effects of endogenous cannabinoid-like molecules [115, 116]. PPARs are a family of nuclear receptors that regulate the transcription and expression of different genes [117] and control numerous physiological functions such as inflammation, cell differentiation and homeostasis [115, 118, 119]. Recently, several studies have produced evidence supporting the structural and functional analogy between endogenous PPAR ligands and AEA, demonstrating the capacity of the latter to activate some members of this family directly [116, 120]. Specifically, it was found that AEA is a weak PPARα ligand and is also able to activate PPARγ [115, 120]. Regarding PPARγ, AEA has anti-inflammatory effects inhibiting the release of the pro-inflammatory cytokine IL-2 in a CB1R/CB2R-independent manner [121]. Furthermore, the possibility that AEA causes PPARγ and CB1 receptor upregulation has been suggested [122].
The endocannabinoid system in brain reward processes
Neurochemistry
The mesocorticolimbic system of the CNS consists of subpopulations of dopaminergic neurons, originating in the ventral tegmental area (VTA) and pars compacta of the substantia nigra, which project to the nucleus accumbens, as well as to other limbic structures, such as the prefrontal cortex (PFC), amygdala and hippocampus. The mesocorticolimbic system has multifaceted functions that are among the hallmarks of reward processing and motivated behavior [123,124,125].
It is well established that dopaminergic neurons of the mesocorticolimbic pathway are under the control of excitatory (primarily glutamatergic) and inhibitory (primarily GABAergic) inputs that regulate their neuronal activity [126,127,128,129]. Several findings support the hypothesis that the endocannabinoid system also contributes to the brain reward processes modulating the activity of dopaminergic neurons, even if indirectly [130,131,132]. CB1Rs are present in the VTA and in the accumbens, as well as in the PFC, central amygdala, and hippocampus, where they are mainly located at the presynaptic terminal [133]. An important functional consequence of their activation is that they inhibit neurotransmitter release by acting as retrograde messengers [134, 135]. Thus, endocannabinoids can be released in both the accumbens and the VTA following depolarization [136, 137]. Activation of CB1Rs on axon terminals of GABAergic neurons in the VTA and glutamatergic neurons in both the VTA and accumbens was shown to inhibit GABAergic and glutamatergic neurotransmission [136, 138]. The final effect on VTA dopaminergic activity depends on the relative level of input activation under distinct behavioral circumstances [138].
A shared feature of most drugs abused by humans is their ability to activate dopaminergic neurotransmission, resulting in increased extracellular levels of dopamine (DA) in the accumbens and particularly in its ventromedial portion, the shell [139,140,141]. Moreover, more natural rewards such as palatable food increase DA release in the accumbens shell [142,143,144]. It is well established that THC, as well as AEA and 2-AG, also produce increases in the extracellular levels of DA in the shell of the nucleus accumbens, suggesting that they themselves could have either rewarding or reinforcing effects [131, 145, 146]. Moreover, this effect is blocked by the CB1 antagonist rimonabant, indicating that the dopaminergic effects of endocannabinoids involve CB1Rs [131, 146]. It should be emphasized that pharmacological blockade of CB1Rs also prevents or reduces the transient increases in DA in the shell of the accumbens produced by the administration of several drugs of abuse such as nicotine, ethanol and cocaine [147]. Moreover, CB1R blockade reduces the increase in DA release in the rat accumbens induced by a novel highly palatable food [148].
Finally, it is interesting to note that brain levels of AEA and 2-AG have been found to be altered by activation of reward processes after exposure to different drugs of abuse. For example, Gonzàlez et al. [149] found that chronic exposure to nicotine or ethanol did produce a significant increase in AEA content in the limbic forebrain, a brain structure that, among other nuclei, includes the accumbens. In contrast, the same authors found that chronic alcohol exposure caused a decrease in the contents of both AEA and 2-AG in the midbrain, as well as within the hippocampus, the striatum and the cerebral cortex. A decrease in AEA and/or 2-AG after chronic nicotine exposure has also been observed [149]. Changes in AEA and 2-AG content were also found in different brain areas, including the hippocampus and PFC, of morphine-sensitized rats [150]. Furthermore, ethanol self-administration causes an increase in dialysate 2-AG levels from the accumbens of rats with no concomitant changes in dialysate AEA concentrations. On the other hand, heroin self-administration was found to increase dialysate AEA levels and to induce a subtle but significant decrease in dialysate 2-AG levels [151]. Finally, various studies have shown that psychostimulants, such as cocaine or amphetamine, also alter AEA and 2-AG contents in the striatum [152, 153].
Behavioral studies in animals
Several animal models have been used to study and characterize the effects of AEA and its potential role in the reinforcing/rewarding actions of drugs. Here, we review the discoveries yielded by the most commonly used behavioral tests over the past few decades.
These behavioral tests include (1) the brain stimulation reward procedure, which evaluates the effects of the drug in the brain reward circuits; (2) conditioned place preference, which assesses conditioned responses related to the rewarding effects of drugs; (3) self-administration procedures, which directly measures the reinforcing properties of drugs; and (4) the reinstatement model of relapse, used to investigate mechanisms underlying drug-seeking behavior.
Brain stimulation reward
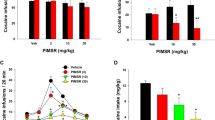
This procedure [154], called also intracranial self-stimulation (ICSS), allows an indirect preclinical assessment of the potential reinforcing effects of drugs in vivo (see for review: [155]). Indeed, when lever pressing causes the delivery of electrical stimulation to selected brain regions, the stimulation may produce reward-like feelings, which, in turn, maintain the lever-pressing behavior. In general, systemic administration of drugs of abuse reduces the electrical threshold at which the behavior is maintained, thus facilitating the reward-like experience. However, the same method also allows the testing substances that might interfere negatively with the reward-like feelings, thus increasing the threshold for reward. As discussed by Tanda [156], cannabinoids tested with ICSS procedures have provided inconsistent, sometimes opposite, outcomes. Indeed, administration of cannabinoid CB1 receptor agonists such as THC, WIN-55212,2 or CP55940 have produced mixed results [156,157,158]. For example, it has been shown that THC, like other drugs of abuse, might facilitate reward mechanism(s) under the ICSS procedure [159,160,161]. In contrast, a lack of effect or even an increase in brain stimulation thresholds by THC and other cannabinoid agonists has also been reported [160, 162, 163]. Moreover, these effects were all reversed by pretreatments with CB1 antagonists, indicating the involvement of CB1Rs in these actions [160, 162, 163]. Thus, as with many behavioral assessments, THC shows an inverted U-shaped dose-response function for behaviors maintained by electrical brain self-stimulation, with positive and negative modulation of the stimulation threshold depending on dose and experimental conditions. To the best of our knowledge, AEA and its more metabolically stable analog meth-anandamide (methAEA) have never been tested in the ICSS procedure. However, the effects of administration of drugs able to increase the circulating brain levels of AEA have been assessed with this procedure. For example, ICSS testing of cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597), N-3-pyridinyl-4-[[3-[[5-(trifluoromethyl)-2-pyridinyl]oxy]phenyl]methyl]-1-piperidinecarboxamide(PF-3845) and phenylmethyl-sulfonyl-fluoride (PMSF), which block the FAAH enzyme, thus inhibiting the degradation of AEA, and OMDM2, an inhibitor of the cellular reuptake of AEA, have been reported [164,165,166]. URB-597 significantly increased the ICSS electrical threshold in rats at doses of 1, 3, and 10 mg/kg, but not at the 0.3 mg/kg dose that has already been shown to be already very effective in increasing the levels of AEA in rats and efficacious in several behavioral tests [48, 49, 167, 168]. However, the selectivity of URB-597 effects on CB1Rs in this paradigm was confirmed by blunting the effects with administration of a low dose of rimonabant, an antagonist of CB1 receptors [165], which by itself does not affect brain stimulation thresholds [163]. PMFS increased the threshold for ICSS at all doses tested, but its effects were not blunted by antagonism at CB1 receptors, suggesting that PMSF would increase levels of endogenous compounds, other than endocannabinoids, that activate non-cannabinoid receptors to produce behavioral effects in the ICSS procedure. Only the highest doses of OMDM2 and PF-3845 (30 mg/kg) enhanced the threshold for ICSS in rats. The effects of OMDM2 were blocked by a low dose of rimonabant. Blockade of FAAH in rats would increase the endogenous levels of AEA but also the levels of other endogenous substances, such as OEA and PEA, which, as mentioned before, are PPARα agonists. This effect would raise a question about the effects obtained in the ICSS after FAAH blockade: are those effects the result of a cooperative/synergistic action of AEA/OEA/PEA on CB1 and/or PPARα and/or TRPV1? Thus, the increased ICSS threshold observed in these published reports indicates a possible anhedonic effect of AEA due to its increased levels after FAAH blockade. This would be in agreement with other reports showing, for example, that AEA elicits place aversion in rats [169]. However, blockade of FAAH by URB597 has been shown to elicit anti-anxiety effects in rats [170,171,172,173,174,175]. Furthermore, to complicate this picture, AEA has been shown to be intravenously self-administered in squirrel monkeys, suggesting that under some circumstances it works as a behavioral reinforcer. Clearly, species differences might be a factor in the effects of cannabinoids, although more studies would be required to better understand the neurobiology underlying the effects of AEA in brain stimulation reward procedures in rodents.
Place conditioning
Conditioned place preference (CPP) is an experimental protocol largely used for studying drug reward in animal models [176, 177]. In the context of reward and addiction, CPP measures the motivational effects of drug and non-drug stimuli in animals, typically mice or rats, as subjects [178]. In such experiments, conditioning sessions start with administering a drug or its vehicle to animals in one of two distinct, contiguous environments at separate times. Then, after one or more conditioning sessions, animals are allowed to freely explore the two environments. Depending on the amount of time the animal spends in the drug-conditioned versus vehicle-conditioned environment, it is possible to infer the animals’ preference for the drug or vehicle stimulus [179]. This paradigm is valid for aversive stimuli as well, in which use it is known as conditioned place aversion (CPA). It is well established that virtually all drugs abused by humans increase, at specific doses, the time spent by the animals in the drug-conditioned environment, thus producing robust CPP. In contrast, as in the test reported for the ICSS procedures, administration of cannabinoids agonists such as THC or WIN55,212-2 did not produce consistent results in place-conditioning procedures: both CPP and CPA have been reported in different studies (see, for review: [156, 157]). For example, dose- and injection-time-dependent effects of THC on place-conditioning procedures have been reported in rats and mice [180,181,182,183], providing evidence for both CPP and CPA, thus also suggesting the biphasic effects of this drug. However, other research groups have reported only CPA induced by administration of CB1R agonists [184,185,186]. Although AEA has been scarcely investigated in the CPP paradigm, interesting insights have been provided in the literature. Mallet and Beringer found neither CPP nor CPA for AEA in rats when the endogenous agonist was delivered intraperitoneally (i.p.) across a large dose range (0.031–16 mg/kg) [187]. It should be noted that the latter authors administered AEA in combination with the protease inhibitor PMSF, capable of blocking AEA metabolism and thus prolonging its half-life. In the same report, different results were obtained with a non-endogenous cannabinoid, THC, which produced significant CPA at doses of 1.0 and 1.5 mg/kg. In our recent study, we reported that intravenously administered AEA (0.03–3 mg/kg), by itself, produced neither CPP nor CPA [169]. However, in contrast to Mallet and Beringer, we found that AEA produced dose-related CPA when rats were pretreated with URB597 [169], which, similar to PMSF in the experiments by Mallet and Beringer, prolonged the half-life of AEA by blocking its FAAH-driven enzymatic metabolism. The aversive effects of AEA during FAAH inhibition appeared to be mediated mainly by CB1Rs, since pretreatment with the CB1R antagonist AM251 blocked the development of CPA. Consistent with previous findings by Gobbi et al.[49], we also found that URB597 by itself, at a dose (0.3 mg/kg) that almost completely inhibits FAAH activity, produced neither CPP nor CPA. Finally, similarly to what reported by Mallet and Beringer, we observed that WIN 55,212-2 (50–300 μg/kg, intravenous) produced dose-dependent CPA [169].
Unlike URB597, the AEA transport inhibitor AM404, at the dose of 2.5 mg/kg given i.p., has been reported to produce CPP in rats, but only when they are housed under enriched conditions [188], suggesting that environmental factors might play a pivotal role. In contrast, we found that when administered at a higher dose of 10 mg/kg, AM404 induced significant CPP in rats housed under standard conditions [189].
Drug self-administration
As mentioned in the previous sections, AEA acts as a reinforcer in squirrel monkeys, and it is one of the few endogenous substances that maintain self-administration behavior in animal models. While a few CB1 receptor agonists are self-administered in rats and mice, for example, WIN55-212,2, JWH-018 and 2-AG [156], other CBR agonists, such as THC, do not act as behavioral reinforcers in rats when administered systemically. However, it should be mentioned that rats learned to lever-press to receive local THC injections into brain areas known to play important roles in drug abuse and addiction, such as the VTA and the shell of the nucleus accumbens [182, 190]. Intra-VTA injection of THC was effective only in the posterior region and not in the anterior region of the VTA. Intra-accumbens delivery of THC maintained self-administration behavior only when injections were directed to the shell rather than the core subdivision of this nucleus [190]. Additionally, THC-maintained behavior was dependent on CB1 receptor activation, as demonstrated by CB1 antagonist pretreatments [182, 190]. However, even recent attempts to obtain intravenous AEA or methAEA self-administration behavior in rats in our laboratories have been unsuccessful, as with intravenous self-administration of THC [156]. It seems clear that species differences play an important role in self-administration behavior, which is maintained by THC and AEA in squirrel monkeys but not in other monkeys or in rodents (see for review: [156, 158]). Thus, specific differences in reward-related circuitry in the brains of different species might play a larger role in the rewarding effects of cannabinoids than of other abused drugs. Indeed, cannabinoid receptors are highly expressed throughout the brain in all species, but differences in their specific expression in selected areas/pathways could be among the reasons for such differences in response. AEA has been successfully demonstrated to act as a behavioral reinforcer in drug-naïve squirrel monkeys [191]. Rates of responding for AEA and its analog methAEA were comparable to those obtained under similar conditions for THC or cocaine. However, when compared to THC (highest rate of responding obtained at 4 µg/kg), it appeared that significantly increased doses of intravenous AEA or methAEA (for both of these drugs, the highest rate of responding was obtained at 40 µg/kg) were necessary to maintain a similar pattern of behavior. While the higher dose of AEA might reflect its short half-life because of its metabolic instability, methAEA is a metabolically stable analog of anandamide; thus, its circulating levels will not be reduced quickly by metabolism. As discussed by Justinova and colleagues [191], it is likely that potential pharmacokinetic differences (for example, hepatic first-pass metabolism) are reduced by administering drugs intravenously. It is also important to note that AEA and methAEA self-administration behavior was blunted by rimonabant administered before the session, which also blocked THC-maintained self-administration [192]. Thus, the efficacy of plant-derived (THC), endogenous (AEA) and synthetic (methAEA) cannabinoid drugs as behavioral reinforcers in the squirrel monkey is mediated by activation of CB1 receptors. In a different series of experiments, reinstatement of extinguished anandamide self-administration behavior was not obtained by pretreatment doses of URB597 fully capable of blocking the enzyme FAAH. Indeed, after URB597 administration, it was found that anandamide levels were higher and 2-AG levels were lower than their basal levels. Additionally, URB597 alone did not maintain self-administration behavior in squirrel monkeys, but it potentiated the effects of anandamide when given in combination. On the other hand, in contrast to URB597, the anandamide transporter inhibitor AM404 was shown to work as a behavioral reinforcer in squirrel monkeys [193]. In the same series of experiments, AM404 pretreatment potentiated anandamide- but not cocaine-maintained self-administration behavior, suggesting that activation of cannabinoid signaling is not involved in the self-administration of cocaine. In the same report, anandamide and AM404 pretreatments could reinstate extinguished self-administration behavior previously maintained by intravenous injections of anandamide, THC, or cocaine. This might not be surprising, since the CB1R antagonist/inverse agonist rimonabant blocked reinstatement of cocaine-seeking behavior in rats induced by cocaine itself or by cocaine-associated cues. Rimonabant also blocked the effects of AM404 on relapse to both anandamide- and cocaine-seeking behavior. Thus, while cannabinoid receptor activation might not be a factor influencing cocaine-reinforced versus AEA-reinforced self-administration behavior, CB1R seems to be involved in the reinstatement of seeking behavior for both drugs. A strong suggestion about the potential involvement of increased anandamide levels in these behaviors elicited by AM404 came from the potentiation of its effects by pretreatment with URB597 before self-administration sessions maintained by AM404, similar to what occurs when self-administration behavior is maintained by AEA.
Reinstatement of drug-seeking behavior
Relapse is defined as a setback that occurs during the process of behavioral change, such that progress toward the initiation or maintenance of a behavioral change goal (e.g., abstinence from drug use) is interrupted by a reversion to the target behavior [194]. The reinstatement model is currently the foremost method used in research to investigate the mechanisms underlying relapse to drug seeking. Reinstatement is typically studied using animals with a history of self-administration. Subsequent to the self-administration procedure and different periods of drug withdrawal, extinction training and tests for reinstatement are conducted [195]. The test for reinstatement for drug-seeking behavior can be achieved with a priming injection of a compound (drug-induced reinstatement) or with exposure to other experimental manipulations, such as a cue or context previously associated with the self-administration of the drug (cue- or context-induced reinstatement), or a stressor (stress-induced reinstatement) [196]. Using the discontinued delivery of THC and/or THC-associated cues as reinstatement model of relapse, interesting insights have been recently provided about the effects of AEA on cannabinoid reinstatement. For example, Justinova and colleagues [197] found that priming administrations of AEA or methAEA reinstate THC seeking [197].
Cannabinoids can produce reinstatement of drug-seeking behaviors for agonists of CB1Rs [198] and for other drugs of abuse [22, 198,199,200,201]. For instance, in the study by Spano and colleagues [198], rats previously trained to intravenously self-administer the CB1R agonist WIN 55,212–2 were found to be susceptible to intraperitoneal priming injections of the same drug, which reinstated cannabinoid-seeking behavior following extinction [198]. Furthermore, interesting insights have been provided recently about the effects of AEA in cannabinoid-reinstatement models of relapse using the discontinued delivery of THC and/or THC-associated cues. On this subject, Justinova and co-workers [197] found that priming administration of AEA (30–560 μg/kg) or its longer-acting congener methAEA (3–100 μg/kg) in monkeys produced dose-dependent reinstatement of THC seeking [197].
Modulation of anandamide brain levels and its potential therapeutic efficacy on substance use disorders
FAAH inhibition as a new therapeutic tool for drug use disorders
Although the use of cannabis for medicinal purposes remains controversial [202], the discovery of the endocannabinoids and the characterization of their degradative enzymes, FAAH and MAGL, has opened new horizons for cannabinoid-based pharmacological strategies for treating pain and several brain disorders. Cannabis has been used for centuries as a therapeutic drug, and some of its main components, THC and cannabidiol, are widely demonstrated to exert strong analgesic actions [203, 204]. In human subjects, repeated “global” activation of brain CB1Rs by cannabis derivatives containing THC may result in cannabis use disorders. AEA, similar to THC, is a partial agonist of CB1Rs. As such, its systemic administration could mimic several behavioral actions of typical agonists of CB1Rs. Among these actions, AEA may elicit potential reinforcing effects, likely due to its “global” activation of brain systems, as demonstrated by Justinova and colleagues [191]. One of the more convenient ways to elicit the activation of brain CB1Rs in specific areas without the “global” activation could be obtained by administration of AEA level modulators, such as FAAH inhibitors or putative blockers of AEA neuronal membrane uptake.
Taking into account the aforementioned considerations about the crucial importance of FAAH inhibition in developing novel pharmacological solutions, we will now review the role of AEA in the context of reward and drug use disorders and the potential use of AEA level enhancers as potential therapeutic strategies against substance use disorders.
Nicotine use disorders
CB1R activation increases the rewarding effects of nicotine [199], while decreasing CB1R function has the opposite effect [205]. Accordingly, Merritt and her group found that FAAH knockout or inhibition enhanced nicotine CPP [206]. However, several works contradict the results obtained by Merritt and colleagues. For example, the FAAH inhibitor URB597 has been found to alter the abuse-related effects of nicotine in rats. When administered at a certain dose (0.3 mg/kg, ip), which does not give rise to behavioral side effects, it is able to prevent the development of nicotine-induced CPP and the acquisition of nicotine self-administration [207]. URB597 is also able to reduce nicotine-induced reinstatement in both CPP and self-administration models of drug abuse, as well as nicotine-induced elevation of DA outflow in the accumbens shell and nicotine-induced excitation of mesolimbic dopaminergic neurons in rodents [207, 208]. Similarly, URB597 decreased the reinstatement of nicotine seeking induced either by presentation of nicotine-associated stimuli or by nicotine priming, although it failed to reduce the breakpoint of nicotine self-administration on a progressive-ratio schedule in rats [209]. Not only URB597 but also VDM11 was found to attenuate nicotine reinstatement induced by nicotine-associated cues and nicotine priming [210], and finally, AM404 was reported to prevent nicotine CPP as well as the reinstatement of nicotine CPP [189]. It is well established that chronic administration of nicotine in rodents can result in a state of “physical dependence” that can be unmasked and characterized by abruptly interrupting the administration of nicotine or by administering a nicotine receptor antagonist such as mecamylamine, resulting in the occurrence of a nicotine withdrawal syndrome consisting of somatic signs as well as aversive motivational and affective states [167]. AEA has also been implicated in alleviating the somatic and motivational effects of nicotine withdrawal.
Indeed, pharmacological inhibition of FAAH mediated by URB597 (0.1 and 0.3 mg/kg, i.p.) has been found to reduce the aversive effects of nicotine-withdrawal-induced anxiety as demonstrated in the elevated plus maze test and in the shock-probe defensive burying paradigm, suggesting that an increase in the endocannabinoid signal could be useful to treat the negative affective state produced by nicotine withdrawal [167]. In a recent report, blockade of the enzyme FAAH by administration of URB597 or URB694 reduced the reinforcing actions of nicotine in squirrel monkeys [211]. The FAAH blockers produced a rightward shift in the dose-response curve of nicotine self-administration under fixed-ratio schedules, suggesting an attenuation of nicotine-induced reward. Additionally, FAAH blockade counteracted the reinstatement of nicotine-seeking behaviors elicited by priming administration of nicotine or by cues previously associated with nicotine self-administration. These effects of FAAH on nicotine were attenuated but not completely blocked by an antagonist of PPARα, suggesting the involvement of this receptor in the actions of FAAH [211]. Indeed, blockade of FAAH increases the endogenous levels of AEA, OEA, and PEA, which are endogenous ligands for PPARα. In a recent study, blockade of the enzyme MAGL by JZL184 treatment, which increases brain levels of 2-AG, potentiated the reinstatement of nicotine-seeking behavior induced in mice by extinguished nicotine-associated cues, without alteration of behaviors maintained by fixed-ratio or progressive-ratio schedules of nicotine self-administration [212]. These effects are likely due to activation of CB1Rs, since 2-AG is a full agonist of those receptors. AEA is a partial agonist of CB1Rs. Thus, differences in the activity of increased levels of AEA and 2-AG could result from their differential activation of CB1Rs, where changes in tonic levels of AEA and/or 2AG might shift the activation and thus the functionality of CB1R. Taken together, these studies of FAAH blockade under the reported experimental conditions in rodents and squirrel monkeys provide a significant contribution to the search for potential pharmacological therapies for nicotine use disorders.
Alcohol use disorders
The relation between FAAH inhibition and drug dependence is not exclusive to nicotine abuse but is also a matter of discussion relevant to a wider variety of abuse behaviors. For example, Cippitelli and co-workers found that AM404 reduced alcohol self-administration in rats [213]. In contrast, AM404 did not potentiate alcohol-induced hypothermia or hypolocomotion and did not affect cue-induced reinstatement of alcohol seeking [214]. However, the same authors reported that administration of URB597 failed to decrease the risk of alcohol abuse in voluntary home-cage drinking and self-administration models [214]. In contrast, Blednov et al. [215] reported that FAAH knockout mice consumed more alcohol than wild-type (WT) littermates in self-administration studies. Additionally, treatment with URB597 increased alcohol intake in WT mice [215]. Similarly, Hansson and colleagues showed that intra-PFC administration of URB597 increased operant alcohol self-administration in non-selected Wistar rats [216]. More recently, Stopponi et al. [175] investigated the blockade of FAAH by URB597 in the central (CeA) and basolateral (BLA) amygdala in Marchigian Sardinian alcohol-preferring (msP) and control rats. They reported, partially in contrast to Hansson and co-workers, that intra-CeA URB597 as well as intra-BLA URB597 (in a less pronounced manner) leads to a reduction of the anxiogenic effects of restraint stress and alcohol self-administration in msP but not in control rats [175]. Recently, URB597 effects were tested in a voluntary alcohol drinking model in mice.[217] Dependence-induced anxiety is a significant risk factor for relapse to alcohol use during withdrawal. As already shown for nicotine, URB597 prevents the anxiogenic response measured during withdrawal after acute administration of alcohol [217]. Moreover, pretreatment with URB597 reduced alcohol intake and preference after acute withdrawal, and such reduction was reversed by the CB1R antagonist AM251. URB597 prevented relapse to alcohol drinking after 1-week withdrawal when administered in single or multiple doses. Furthermore, AEA levels increased in several brain regions, including the accumbens and basolateral amygdala, as a result of acute (1-day) alcohol withdrawal. Based on the authors’ interpretation, the increased levels of AEA may provide a kind of protection from stress during alcohol withdrawal [217]. Together, these results demonstrate that activation of AEA signaling by selective inhibition of FAAH reduces anxiety associated with alcohol withdrawal. On this issue, a study on a human single-nucleotide polymorphism of the FAAH gene that would decrease the enzymatic rate of FAAH [218] has been recently reported. This polymorphism, when transferred into genetically modified knockin mice, increased the endocannabinoid tone, leading to increased alcohol intake and preference compared to control WT mice. These experiments suggest that this FAAH gene polymorphism might be involved in alcohol binge drinking. Additionally, an altered endocannabinoid tone at CB1Rs, caused by increased levels of AEA, could be mediating those effects, which were indeed blocked by the CB1R antagonist AM251 [218].
From the aforementioned considerations, it is clear (in rodents, at least) that the upregulation of CB1 transmission gives rise to an increase in alcohol drinking, whereas downregulation of CB1 transmission causes a decrease in this behavior [219]. Nevertheless, this assessment is more complicated in humans and has yet to be ascertained. For example, while rimonabant and other CB1 antagonists suppress rodent alcohol consumption, no significant effect of rimonabant was reported in clinical trials [220], and there were no effects of rimonabant on alcohol self-administration [221]. Recently, a clinical study of the association between FAAH functional genetic polymorphisms (FAAH Pro129Thr, rs324420) and alcohol dependence and severity was conducted in European-American and African-American subjects [222]. Results show that, in European-American but not in African-American subjects, there was a higher frequency of the Thr129 allele in the alcohol-dependent group than in the non-dependent controls. Thus, although the clinical trials cited above did not report significant interactions between CB1 antagonism and alcohol consumption, there is a possibility that alterations in endocannabinoid levels might play a role in alcohol use disorders and in their pharmacological treatment.
Opioid use disorders
In a series of experiments by Solinas et al. [223], the CB1R agonists THC and WIN55,212-2 did not modify the fixed-ratio 1 self-administration of heroin but significantly increased the breakpoint for heroin injections at all doses tested. In contrast, blockade of FAAH or AEA transport by URB597 and AM404, respectively, or their combination did not alter the efficacy of heroin as a reinforcer. Thus, it seems that an interaction between CB1Rs and opioid receptors facilitates the reinforcing action of heroin, while the lack of effects of AM404 and URB597 might be explained by lack of effect of heroin on the release of AEA [223]. On the other hand, endocannabinoid catabolic enzyme inhibitors have been shown to reduce abrupt withdrawal in morphine-dependent mice, as reported by Ramesh et al. [224]. They used the FAAH inhibitor PF-3845, the MAGL inhibitor JZL184 (at a low dose and in combination with PF-3845), and the dual FAAH-MAGL inhibitor SA-57. The combination of JZL184 and PF-3845, as well as SA-57 by itself, increased brain AEA levels (SA-57 more than the others) compared to those of controls and reduced all of the examined opioid withdrawal symptoms (i.e., platform jumping, paw flutters, head shakes, diarrhea, and total body weight loss) [224]. These results suggest a very interesting interaction between the cannabinoid and opioid systems, which might be important at the translational level. Thus, endocannabinoid level enhancers could open a new way to potential pharmacotherapies for alleviating withdrawal symptoms. These symptoms related to pain and stress are likely among the main reasons that prevent individuals from starting on the road to recovery from addiction. Attenuation of spontaneous opiate withdrawal has also been reported after administration of AM404 [225], and the direct action of AEA administration has been shown to attenuate naloxone-precipitated withdrawal in mice chronically treated with morphine [226]. However, URB597 was reported ineffective in modifying morphine-induced reinstatement of conditioned floor preference and naloxone-precipitated withdrawal-induced conditioned floor avoidance [227]. In a more recent study, SA57 dose-dependently reduced heroin drug-seeking behavior in mice.[228], Additionally, when mice were assessed in the progressive-ratio self-administration procedure, SA57-treated mice showed significantly lower heroin breakpoints than control mice. Since the breakpoint in a progressive-ratio schedule of self-administration could be a measure of the willingness of the animals to self-administer a certain dose of a drug, this experiment suggests that mice treated with SA57 had a decreased level of motivation to self-inject heroin. However, SA57 failed to significantly modify heroin-induced reinstatement of drug-seeking in mice under behavioral extinction [228].
Psychostimulant use disorders
In contrast to other drugs of abuse, psychostimulants produce direct effects on the mesolimbic dopaminergic neurons by acting on the reuptake of monoamines, thus enhancing the overall activity of DA neurons [229, 230]. The endocannabinoid system is implicated in different mechanisms and relative behavioral outcomes, such as rewarding, motivational and seeking-related effects of diverse psychostimulants. In fact, it has been shown by various authors that some of the effects of both acute and chronic administration of psychostimulants are absent in CB1 knockout mice [21].
Other interesting insights come from studies of FAAH inhibition and behavioral sensitization to cocaine, one of the most abused psychostimulants worldwide. Cocaine sensitization has been described as one of the steps occurring during the transition from simple taking of the drug to abuse and dependence [231,232,233]. Several different methodologies have been tested to induce cocaine behavioral sensitization [234,235,236,237,238,239]. A role of endocannabinoids in the neuroplasticity induced by repeated injections of cocaine has been shown [23, 234, 235]. However, the role of cannabinoids in cocaine sensitization is still debated [240], as is the evidence that cocaine administration induces the release of endocannabinoids [149, 151,152,153, 241, 242]. In a recent study, a single injection of cocaine was shown to induce behavioral sensitization that was related to increased stimulation of extracellular DA levels in the core, but not in the shell, of the accumbens in mice [23]. These effects were significantly reversed by pretreatment with the CB1R antagonist rimonabant, suggesting the possibility that an increase in endocannabinoid tone could be involved in the neuroadaptation induced by cocaine. Indeed, a dose of cocaine lower than the dose able to induce behavioral sensitization had no effect unless the animals were pretreated with the FAAH inhibitor URB597. Thus, blockade of anandamide metabolism was likely magnifying the cocaine-induced release of AEA, suggesting its involvement in the neuroadaptation that occurs during the early stages of cocaine use [23]. The involvement of the endocannabinoid system in cocaine use disorders has also been shown in studies where URB597 reduced cocaine-induced seizures and cocaine-induced activation and death of hippocampal neurons, both in primary cell culture and in mice [243]. Furthermore, Luchicchi and colleagues found that URB597 blocked the inhibitory effects of nicotine and cocaine in medium spiny neurons of the accumbens shell in rats by activating both CB1Rs and PPARα or only PPARα, respectively. Additionally, URB597 did not alter the effects of either cocaine or morphine on VTA DA neurons [244]. Finally, in the monkey brain, it has been found that pretreatment with URB597 did not modify self-administration of cocaine but significantly potentiated the behavior maintained by AEA [245]. In the same experiments, it was also shown that URB597 enhanced AEA levels while causing a compensatory downregulation in 2-AG levels [245]. The effects of FAAH inhibitors on cocaine self-administration and reinstatement have also been tested in rodents [246]. Intravenous cocaine self-administration, 0.5 mg/kg/inj, fixed ratio 5 in rats was not altered by pretreatments with the AEA metabolism blockers PMSF and URB597 at any dose tested. Both drugs attenuated cocaine- and cue-induced reinstatement of cocaine-seeking behavior, but URB597 did so without behavioral disruptions. Based on the authors’ interpretation, enhancing endocannabinoid levels in the brain would modulate the motivational and conditioned aspects of cocaine-directed behaviors, while the consummatory behaviors are not under endocannabinoid control [246].
Conclusions
In the present manuscript, we have reviewed scientific evidence regarding how AEA works as a behavioral reinforcer under certain conditions and how it could elicit aversive behaviors or be anxiogenic under different conditions. Such opposite effects are also shown by many other drugs and very often depend on the dose of the drug under investigation. However, with cannabinoids, doses are not always the explanation for opposite outcomes. In several animal models, experimental conditions other than drug doses may have a more important impact test results for cannabinoids than for other drug classes [156]. For example, drug pre-exposure, timing between drug injections, and species differences are among the most likely candidates to explain some of the contradictory results of behavioral tests. For example, THC, similar to AEA and methAEA, is self-administered by squirrel monkeys but not by other monkeys or by rodents. Furthermore, similar to THC, AEA does not show a clear “drug of abuse” profile when tested in animal paradigms such as ICSS or conditioned place preference. To understand the neurobiology underlying these interesting discrepancies would certainly require many more years of study. On the other hand, AEA and drugs that modulate its circulating brain levels have been shown not only to be involved in mediating the effects of several drugs of abuse but also, in many circumstances, to create potential positive interference with the dependence-producing actions of several drugs of abuse. Indeed, blockers of AEA metabolism have been shown to efficiently counteract some of the behavioral/reinforcing effects of nicotine, alcohol, opiates, and psychostimulants, suggesting potential therapeutic activity against substance use disorders. For example, the effects of cannabinoids on nicotine response were studied initially by trying to block CB1Rs with antagonists such rimonabant. However, full antagonism of CB1Rs could severely compromise brain physiological functions, and indeed, significant psychiatric side effects of rimonabant use have led the European Medicines Agency to suspend the marketing of this drug [247]. In contrast to the potential therapeutic effects of rimonabant mediated by CB1R antagonism [248], FAAH blockade seems to exert its own potential therapeutic effects on nicotine use disorders by merely increasing the levels of AEA, which is a partial agonist of CB1Rs. This interesting discrepancy could be the result of inverse-agonist effects of rimonabant, in addition to its antagonist effects, or it could result from partial activation of CB1 receptors or from their activation exclusively in selected brain areas. Indeed, after FAAH blockade, AEA levels are enhanced only in those brain areas where AEA is released. Another explanation might come from potential interactions between CB1Rs and/or CB2Rs and PPARα, all of which are pharmacological targets of AEA. Moreover, OEA and PEA, which are substrates of the enzyme FAAH and whose circulating brain levels increase after FAAH blockade, might be a potential factor in the therapeutic actions of cannabinoid-level enhancers. Potential therapeutic intervention based on modulation of endocannabinoid levels has also been suggested for pain [42] and for several brain disorders [16, 249] other than substance use disorders, for example, depression and anxiety. To this end, cannabinoid drugs that are currently under investigation in clinical trials for psychiatric disorders might open new avenues for treatments related to substance use disorders [219, 249]. Translational preclinical research in the endocannabinoid field has greatly accelerated in recent years. We believe that the same acceleration should be facilitated for clinical studies aimed at the development of potential pharmacotherapies for substance use disorders based on alteration of brain endocannabinoids.
References
Gaoni Y, Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of Hashish. J Am Chem Soc. 1964;86:1646–7.
Mechoulam R. Interview with Prof. Raphael Mechoulam, codiscoverer of THC. Interview by Stanley Einstein. Int J Addict. 1986;21:579–87.
Devane WA, Dysarz FA 3rd, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–13.
Leuschner JT, Wing DR, Harvey DJ, Brent GA, Dempsey CE, Watts A, et al. The partitioning of delta 1-tetrahydrocannabinol into erythrocyte membranes in vivo and its effect on membrane fluidity. Experientia. 1984;40:866–8.
Paton WD. Pharmacology of marijuana. Annu Rev Pharmacol. 1975;15:191–220.
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–4.
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–9.
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90.
Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97.
Luchicchi A, Pistis M. Anandamide and 2-arachidonoylglycerol: pharmacological properties, functional features, and emerging specificities of the two major endocannabinoids. Mol Neurobiol. 2012;46:374–92.
Mechoulam R, Hanus LO, Pertwee R, Howlett AC. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat Rev Neurosci. 2014;15:757–64.
Elphick MR, Satou Y, Satoh N. The invertebrate ancestry of endocannabinoid signalling: an orthologue of vertebrate cannabinoid receptors in the urochordate Ciona intestinalis. Gene. 2003;302:95–101.
Jhaveri MD, Richardson D, Chapman V. Endocannabinoid metabolism and uptake: novel targets for neuropathic and inflammatory pain. Br J Pharmacol. 2007;152:624–32.
Woodhams SG, Chapman V, Finn DP, Hohmann AG, Neugebauer V. The cannabinoid system and pain. Neuropharmacology. 2017;124:105–20.
Kirkham TC. Endocannabinoids in the regulation of appetite and body weight. Behav Pharmacol. 2005;16:297–313.
Ashton CH, Moore PB. Endocannabinoid system dysfunction in mood and related disorders. Acta Psychiatr Scand. 2011;124:250–61.
Fride E. The endocannabinoid-CB(1) receptor system in pre- and postnatal life. Eur J Pharmacol. 2004;500:289–97.
Gardner EL. Endocannabinoid signaling system and brain reward: emphasis on dopamine. Pharmacol Biochem Behav. 2005;81:263–84.
Parsons LH, Hurd YL. Endocannabinoid signalling in reward and addiction. Nat Rev Neurosci. 2015;16:579–94.
Gonzalez S, Cebeira M, Fernandez-Ruiz J. Cannabinoid tolerance and dependence: a review of studies in laboratory animals. Pharmacol Biochem Behav. 2005;81:300–18.
Maldonado R, Valverde O, Berrendero F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci. 2006;29:225–32.
Tanda G. Modulation of the endocannabinoid system: therapeutic potential against cocaine dependence. Pharmacol Res. 2007;56:406–17.
Mereu M, Tronci V, Chun LE, Thomas AM, Green JL, Katz JL, et al. Cocaine-induced endocannabinoid release modulates behavioral and neurochemical sensitization in mice. Addict Biol. 2015;20:91–103.
De Petrocellis L, Di Marzo V. An introduction to the endocannabinoid system: from the early to the latest concepts. Best Pract Res Clin Endocrinol Metab. 2009;23:1–15.
Maccarrone M. Metabolism of the endocannabinoid anandamide: open questions after 25 Years. Front Mol Neurosci. 2017;10:166.
Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–707.
Shen M, Thayer SA. Delta9-tetrahydrocannabinol acts as a partial agonist to modulate glutamatergic synaptic transmission between rat hippocampal neurons in culture. Mol Pharmacol. 1999;55:8–13.
Mackie K. Cannabinoid receptors: where they are and what they do. J Neuroendocrinol. 2008;20(Suppl 1):10–4.
Di Marzo V, De Petrocellis L. Why do cannabinoid receptors have more than one endogenous ligand? Philos Trans R Soc Lond B Biol Sci. 2012;367:3216–28.
Hanus L, Abu-Lafi S, Fride E, Breuer A, Vogel Z, Shalev DE, et al. 2-arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc Natl Acad Sci U S A. 2001;98:3662–5.
Pertwee RG. Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond. Addict Biol. 2008;13:147–59.
Porter AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J, et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther. 2002;301:1020–4.
Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, et al. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–91.
Bisogno T, Ligresti A, Di Marzo V. The endocannabinoid signalling system: biochemical aspects. Pharmacol Biochem Behav. 2005;81:224–38.
Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–84.
Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–8.
Sugiura T, Waku K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem Phys Lipids. 2000;108:89–106.
Ueda N, Tsuboi K, Uyama T, Ohnishi T. Biosynthesis and degradation of the endocannabinoid 2-arachidonoylglycerol. Biofactors. 2011;37:1–7.
Di Marzo V, Maccarrone M. FAAH and anandamide: is 2-AG really the odd one out? Trends Pharmacol Sci. 2008;29:229–33.
Ueda N, Yamanaka K, Terasawa Y, Yamamoto S. An acid amidase hydrolyzing anandamide as an endogenous ligand for cannabinoid receptors. FEBS Lett. 1999;454:267–70.
Ho WS, Barrett DA, Randall MD. 'Entourage' effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol. 2008;155:837-46.
Ulugol A. The endocannabinoid system as a potential therapeutic target for pain modulation. Balk Med J. 2014;31:115–20.
Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–6.
Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004;109:319–27.
Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, et al. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006;12:21–38.
Russo R, Loverme J, La Rana G, Compton TR, Parrott J, Duranti A, et al. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3’-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322:236–42.
Holt S, Comelli F, Costa B, Fowler CJ. Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br J Pharmacol. 2005;146:467–76.
Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81.
Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, et al. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A. 2005;102:18620–5.
Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, et al. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004;311:441–8.
Alexander JP, Cravatt BF. The putative endocannabinoid transport blocker LY2183240 is a potent inhibitor of FAAH and several other brain serine hydrolases. J Am Chem Soc. 2006;128:9699–704.
Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–7.
Glaser ST, Kaczocha M, Deutsch DG. Anandamide transport: a critical review. Life Sci. 2005;77:1584–604.
Glaser ST, Abumrad NA, Fatade F, Kaczocha M, Studholme KM, Deutsch DG. Evidence against the presence of an anandamide transporter. Proc Natl Acad Sci U S A. 2003;100:4269–74.
Costa B, Siniscalco D, Trovato AE, Comelli F, Sotgiu ML, Colleoni M, et al. AM404, an inhibitor of anandamide uptake, prevents pain behaviour and modulates cytokine and apoptotic pathways in a rat model of neuropathic pain. Br J Pharmacol. 2006;148:1022–32.
Vandevoorde S, Fowler CJ. Inhibition of fatty acid amide hydrolase and monoacylglycerol lipase by the anandamide uptake inhibitor VDM11: evidence that VDM11 acts as an FAAH substrate. Br J Pharmacol. 2005;145:885–93.
Kaczocha M, Hermann A, Glaser ST, Bojesen IN, Deutsch DG. Anandamide uptake is consistent with rate-limited diffusion and is regulated by the degree of its hydrolysis by fatty acid amide hydrolase. J Biol Chem. 2006;281:9066–75.
Bisogno T, Melck D, De Petrocellis L, Bobrov M, Gretskaya NM, Bezuglov VV, et al. Arachidonoylserotonin and other novel inhibitors of fatty acid amide hydrolase. Biochem Biophys Res Commun. 1998;248:515–22.
Goparaju SK, Ueda N, Yamaguchi H, Yamamoto S. Anandamide amidohydrolase reacting with 2-arachidonoylglycerol, another cannabinoid receptor ligand. FEBS Lett. 1998;422:69–73.
Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–7.
Savinainen JR, Saario SM, Laitinen JT. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol (Oxf). 2012;204:267–76.
Saario SM, Salo OM, Nevalainen T, Poso A, Laitinen JT, Jarvinen T, et al. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem Biol. 2005;12:649–56.
Saario SM, Palomaki V, Lehtonen M, Nevalainen T, Jarvinen T, Laitinen JT. URB754 has no effect on the hydrolysis or signaling capacity of 2-AG in the rat brain. Chem Biol. 2006;13:811–4.
Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–12.
Makara JK, Mor M, Fegley D, Szabo SI, Kathuria S, Astarita G, et al. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci. 2005;8:1139–41.
Ohno-Shosaku T, Kano M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr Opin Neurobiol. 2014;29:1–8.
Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–7.
Alger BE. Endocannabinoids at the synapse a decade after the dies mirabilis (29 March 2001): what we still do not know. J Physiol. 2012;590:2203–12.
Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–80.
Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–82.
Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–75.
Maejima T, Ohno-Shosaku T, Kano M. Endogenous cannabinoid as a retrograde messenger from depolarized postsynaptic neurons to presynaptic terminals. Neurosci Res. 2001;40:205–10.
Gerard CM, Mollereau C, Vassart G, Parmentier M. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem J. 1991;279:129–34.
Chakrabarti A, Onaivi ES, Chaudhuri G. Cloning and sequencing of a cDNA encoding the mouse brain-type cannabinoid receptor protein. DNA Seq. 1995;5:385–8.
Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–5.
Mackie K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handbook Exp Pharmacol. 2005;168:299–325.
Tsou K, Nogueron MI, Muthian S, Sanudo-Pena MC, Hillard CJ, Deutsch DG, et al. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett. 1998;254:137–40.
Glass M, Dragunow M, Faull RL. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77:299–318.
Hu SS, Mackie K. Distribution of the Endocannabinoid system in the central nervous system. Handb Exp Pharmacol. 2015;231:59–93.
Breivogel CS, Selley DE, Childers SR. Cannabinoid receptor agonist efficacy for stimulating [35S]GTPgammaS binding to rat cerebellar membranes correlates with agonist-induced decreases in GDP affinity. J Biol Chem. 1998;273:16865–73.
Breivogel CS, Childers SR. The functional neuroanatomy of brain cannabinoid receptors. Neurobiol Dis. 1998;5:417–31.
Howlett AC. Cannabinoid inhibition of adenylate cyclase. Biochemistry of the response in neuroblastoma cell membranes. Mol Pharmacol. 1985;27:429–36.
Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A. 1992;89:3825–9.
Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharmacol Exp Ther. 1995;273:734–43.
Bouaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, et al. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995;312:637–41.
Demuth DG, Molleman A. Cannabinoid signalling. Life Sci. 2006;78:549–63.
Howlett AC, Blume LC, Dalton GD. CB(1) cannabinoid receptors and their associated proteins. Curr Med Chem. 2010;17:1382–93.
Ishac EJ, Jiang L, Lake KD, Varga K, Abood ME, Kunos G. Inhibition of exocytotic noradrenaline release by presynaptic cannabinoid CB1 receptors on peripheral sympathetic nerves. Br J Pharmacol. 1996;118:2023–8.
Vizi ES, Katona I, Freund TF. Evidence for presynaptic cannabinoid CB(1) receptor-mediated inhibition of noradrenaline release in the guinea pig lung. Eur J Pharmacol. 2001;431:237–44.
Izzo AA, Sharkey KA. Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther. 2010;126:21–38.
Nunez E, Benito C, Pazos MR, Barbachano A, Fajardo O, Gonzalez S, et al. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: an immunohistochemical study. Synapse. 2004;53:208–13.
Ishiguro H, Iwasaki S, Teasenfitz L, Higuchi S, Horiuchi Y, Saito T, et al. Involvement of cannabinoid CB2 receptor in alcohol preference in mice and alcoholism in humans. Pharm J. 2007;7:380–5.
Onaivi ES, Ishiguro H, Gong JP, Patel S, Meozzi PA, Myers L, et al. Brain neuronal CB2 cannabinoid receptors in drug abuse and depression: from mice to human subjects. PLoS One. 2008;3:e1640.
Liu QR, Canseco-Alba A, Zhang HY, Tagliaferro P, Chung M, Dennis E, et al. Cannabinoid type 2 receptors in dopamine neurons inhibits psychomotor behaviors, alters anxiety, depression and alcohol preference. Sci Rep. 2017;7:17410.
Zhang HY, Gao M, Shen H, Bi GH, Yang HJ, Liu QR, et al. Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict Biol. 2017;22:752–65.
Xi ZX, Peng XQ, Li X, Song R, Zhang HY, Liu QR, et al. Brain cannabinoid CB(2) receptors modulate cocaine’s actions in mice. Nat Neurosci. 2011;14:1160–6.
Zhang HY, Gao M, Liu QR, Bi GH, Li X, Yang HJ, et al. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci U S A. 2014;111:E5007–15.
Liu QR, Pan CH, Hishimoto A, Li CY, Xi ZX, Llorente-Berzal A, et al. Species differences in cannabinoid receptor 2 (CNR2 gene): identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Genes Brain Behav. 2009;8:519–30.
Zhang HY, Bi GH, Li X, Li J, Qu H, Zhang SJ, et al. Species differences in cannabinoid receptor 2 and receptor responses to cocaine self-administration in mice and rats. Neuropsychopharmacology. 2015;40:1037–51.
Felder CC, Nielsen A, Briley EM, Palkovits M, Priller J, Axelrod J, et al. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996;393:231–5.
Bingham B, Jones PG, Uveges AJ, Kotnis S, Lu P, Smith VA, et al. Species-specific in vitro pharmacological effects of the cannabinoid receptor 2 (CB2) selective ligand AM1241 and its resolved enantiomers. Br J Pharmacol. 2007;151:1061–70.
Yao BB, Mukherjee S, Fan Y, Garrison TR, Daza AV, Grayson GK, et al. In vitro pharmacological characterization of AM1241: a protean agonist at the cannabinoid CB2 receptor? Br J Pharmacol. 2006;149:145–54.
Pertwee RG, Ross RA. Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fat Acids. 2002;66:101–21.
Toth A, Blumberg PM, Boczan J. Anandamide and the vanilloid receptor (TRPV1). Vitam Horm. 2009;81:389–419.
Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–7.
Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003;140:790–801.
Szolcsanyi J. Anandamide and the question of its functional role for activation of capsaicin receptors. Trends Pharmacol Sci. 2000;21:203–4.
De Petrocellis L, Bisogno T, Maccarrone M, Davis JB, Finazzi-Agro A, Di Marzo V. The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J Biol Chem. 2001;276:12856–63.
Johns DG, Behm DJ, Walker DJ, Ao Z, Shapland EM, Daniels DA, et al. The novel endocannabinoid receptor GPR55 is activated by atypical cannabinoids but does not mediate their vasodilator effects. Br J Pharmacol. 2007;152:825–31.
Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–101.
Sharir H, Console-Bram L, Mundy C, Popoff SN, Kapur A, Abood ME. The endocannabinoids anandamide and virodhamine modulate the activity of the candidate cannabinoid receptor GPR55. J Neuroimmune Pharmacol. 2012;7:856–65.
Brown AJ, Robin Hiley C. Is GPR55 an anandamide receptor? Vitam Horm. 2009;81:111–37.
Sharir H, Abood ME. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol Ther. 2010;126:301–13.
Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A. 2008;105:2699–704.
O’Sullivan SE, Kendall DA. Cannabinoid activation of peroxisome proliferator-activated receptors: potential for modulation of inflammatory disease. Immunobiology. 2010;215:611–6.
Sun Y, Alexander SP, Garle MJ, Gibson CL, Hewitt K, Murphy SP, et al. Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br J Pharmacol. 2007;152:734–43.
Michalik L, Wahli W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J Clin Invest. 2006;116:598–606.
Ferre P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53(Suppl 1):S43–50.
Glass CK. Going nuclear in metabolic and cardiovascular disease. J Clin Invest. 2006;116:556–60.
Bouaboula M, Hilairet S, Marchand J, Fajas L, Le Fur G, Casellas P. Anandamide induced PPARgamma transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur J Pharmacol. 2005;517:174–81.
Rockwell CE, Kaminski NE. A cyclooxygenase metabolite of anandamide causes inhibition of interleukin-2 secretion in murine splenocytes. J Pharmacol Exp Ther. 2004;311:683–90.
Karaliota S, Siafaka-Kapadai A, Gontinou C, Psarra K, Mavri-Vavayanni M. Anandamide increases the differentiation of rat adipocytes and causes PPARgamma and CB1 receptor upregulation. Obesity (Silver Spring). 2009;17:1830–8.
Fields HL. Understanding how opioids contribute to reward and analgesia. Reg Anesth Pain Med. 2007;32:242–6.
Wise RA. Roles for nigrostriatal—not just mesocorticolimbic—dopamine in reward and addiction. Trends Neurosci. 2009;32:517–24.
Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol. 1989;40:191–225.
Kalivas PW. Neurotransmitter regulation of dopamine neurons in the ventral tegmental area. Brain Res Brain Res Rev. 1993;18:75–113.
Korotkova TM, Ponomarenko AA, Brown RE, Haas HL. Functional diversity of ventral midbrain dopamine and GABAergic neurons. Mol Neurobiol. 2004;29:243–59.
Morales M, Root DH. Glutamate neurons within the midbrain dopamine regions. Neuroscience. 2014;282:60–8.
Wise RA, Morales M. A ventral tegmental CRF-glutamate-dopamine interaction in addiction. Brain Res. 2010;1314:38–43.
Bloomfield MA, Ashok AH, Volkow ND, Howes OD. The effects of Delta(9)-tetrahydrocannabinol on the dopamine system. Nature. 2016;539:369–77.
Solinas M, Justinova Z, Goldberg SR, Tanda G. Anandamide administration alone and after inhibition of fatty acid amide hydrolase (FAAH) increases dopamine levels in the nucleus accumbens shell in rats. J Neurochem. 2006;98:408–19.
Solinas M, Scherma M, Tanda G, Wertheim CE, Fratta W, Goldberg SR. Nicotinic facilitation of delta9-tetrahydrocannabinol discrimination involves endogenous anandamide. J Pharmacol Exp Ther. 2007;321:1127–34.
Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267–74.
Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci. 2001;22:565–72.
Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–92.
Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62.
Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8.
Lupica CR, Riegel AC. Endocannabinoid release from midbrain dopamine neurons: a potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology. 2005;48:1105–16.
Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–8.
Pontieri FE, Tanda G, Di Chiara G. Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc Natl Acad Sci U S A. 1995;92:12304–8.
Pontieri FE, Tanda G, Orzi F, Di Chiara G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature. 1996;382:255–7.
Bassareo V, Di Chiara G. Differential influence of associative and nonassociative learning mechanisms on the responsiveness of prefrontal and accumbal dopamine transmission to food stimuli in rats fed ad libitum. J Neurosci. 1997;17:851–61.
Martel P, Fantino M. Mesolimbic dopaminergic system activity as a function of food reward: a microdialysis study. Pharmacol Biochem Behav. 1996;53:221–6.
Tanda G, Di Chiara G. A dopamine-mu1 opioid link in the rat ventral tegmentum shared by palatable food (Fonzies) and non-psychostimulant drugs of abuse. Eur J Neurosci. 1998;10:1179–87.
De Luca MA, Valentini V, Bimpisidis Z, Cacciapaglia F, Caboni P, Di Chiara G. Endocannabinoid 2-arachidonoylglycerol self-administration by Sprague-Dawley rats and stimulation of in vivo dopamine transmission in the nucleus accumbens shell. Front Psychiatry. 2014;5:140.
Tanda G, Pontieri FE, Di Chiara G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science. 1997;276:2048–50.
Cheer JF, Wassum KM, Sombers LA, Heien ML, Ariansen JL, Aragona BJ, et al. Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci. 2007;27:791–5.
Melis T, Succu S, Sanna F, Boi A, Argiolas A, Melis MR. The cannabinoid antagonist SR 141716A (Rimonabant) reduces the increase of extra-cellular dopamine release in the rat nucleus accumbens induced by a novel high palatable food. Neurosci Lett. 2007;419:231–5.
Gonzalez S, Cascio MG, Fernandez-Ruiz J, Fezza F, Di Marzo V, Ramos JA. Changes in endocannabinoid contents in the brain of rats chronically exposed to nicotine, ethanol or cocaine. Brain Res. 2002;954:73–81.
Vigano D, Valenti M, Cascio MG, Di Marzo V, Parolaro D, Rubino T. Changes in endocannabinoid levels in a rat model of behavioural sensitization to morphine. Eur J Neurosci. 2004;20:1849–57.
Caille S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH. Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J Neurosci. 2007;27:3695–702.
Centonze D, Battista N, Rossi S, Mercuri NB, Finazzi-Agro A, Bernardi G, et al. A critical interaction between dopamine D2 receptors and endocannabinoids mediates the effects of cocaine on striatal gabaergic transmission. Neuropsychopharmacology. 2004;29:1488–97.
Thiemann G, van der Stelt M, Petrosino S, Molleman A, Di Marzo V, Hasenohrl RU. The role of the CB1 cannabinoid receptor and its endogenous ligands, anandamide and 2-arachidonoylglycerol, in amphetamine-induced behavioural sensitization. Behav Brain Res. 2008;187:289–96.
Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol. 1954;47:419–27.
Negus SS, Miller LL. Intracranial self-stimulation to evaluate abuse potential of drugs. Pharmacol Rev. 2014;66:869–917.
Tanda G. Preclinical studies on the reinforcing effects of cannabinoids. A tribute to the scientific research of Dr. Steve Goldberg. Psychopharmacology (Berl). 2016;233:1845–66.
Panagis G, Mackey B, Vlachou S. Cannabinoid regulation of brain reward processing with an emphasis on the role of CB1 receptors: a step back into the future. Front Psychiatry. 2014;5:92.
Tanda G, Goldberg SR. Cannabinoids: reward, dependence, and underlying neurochemical mechanisms—a review of recent preclinical data. Psychopharmacology (Berl). 2003;169:115–34.
Gardner EL, Paredes W, Smith D, Donner A, Milling C, Cohen D, et al. Facilitation of brain stimulation reward by delta 9-tetrahydrocannabinol. Psychopharmacology (Berl). 1988;96:142–4.
Katsidoni V, Kastellakis A, Panagis G. Biphasic effects of Delta9-tetrahydrocannabinol on brain stimulation reward and motor activity. Int J Neuropsychopharmacol. 2013;16:2273–84.
Lepore M, Liu X, Savage V, Matalon D, Gardner EL. Genetic differences in delta 9-tetrahydrocannabinol-induced facilitation of brain stimulation reward as measured by a rate-frequency curve-shift electrical brain stimulation paradigm in three different rat strains. Life Sci. 1996;58:PL365–72.
Vlachou S, Nomikos GG, Panagis G. CB1 cannabinoid receptor agonists increase intracranial self-stimulation thresholds in the rat. Psychopharmacology (Berl). 2005;179:498–508.
Vlachou S, Nomikos GG, Stephens DN, Panagis G. Lack of evidence for appetitive effects of Delta 9-tetrahydrocannabinol in the intracranial self-stimulation and conditioned place preference procedures in rodents. Behav Pharmacol. 2007;18:311–9.
Kwilasz AJ, Abdullah RA, Poklis JL, Lichtman AH, Negus SS. Effects of the fatty acid amide hydrolase inhibitor URB597 on pain-stimulated and pain-depressed behavior in rats. Behav Pharmacol. 2014;25:119–29.
Vlachou S, Nomikos GG, Panagis G. Effects of endocannabinoid neurotransmission modulators on brain stimulation reward. Psychopharmacology (Berl). 2006;188:293–305.
Wiebelhaus JM, Grim TW, Owens RA, Lazenka MF, Sim-Selley LJ, Abdullah RA, et al. Delta9-tetrahydrocannabinol and endocannabinoid degradative enzyme inhibitors attenuate intracranial self-stimulation in mice. J Pharmacol Exp Ther. 2015;352:195–207.
Cippitelli A, Astarita G, Duranti A, Caprioli G, Ubaldi M, Stopponi S, et al. Endocannabinoid regulation of acute and protracted nicotine withdrawal: effect of FAAH inhibition. PLoS One. 2011;6:e28142.
Solinas M, Tanda G, Wertheim CE, Goldberg SR. Dopaminergic augmentation of delta-9-tetrahydrocannabinol (THC) discrimination: possible involvement of D(2)-induced formation of anandamide. Psychopharmacology (Berl). 2010;209:191–202.
Scherma M, Medalie J, Fratta W, Vadivel SK, Makriyannis A, Piomelli D, et al. The endogenous cannabinoid anandamide has effects on motivation and anxiety that are revealed by fatty acid amide hydrolase (FAAH) inhibition. Neuropharmacology. 2008;54:129–40.
Duan T, Gu N, Wang Y, Wang F, Zhu J, Fang Y, et al. Fatty acid amide hydrolase inhibitors produce rapid anti-anxiety responses through amygdala long-term depression in male rodents. J Psychiatry Neurosci. 2017;42:230–41.
Lisboa SF, Resstel LB, Aguiar DC, Guimaraes FS. Activation of cannabinoid CB1 receptors in the dorsolateral periaqueductal gray induces anxiolytic effects in rats submitted to the Vogel conflict test. Eur J Pharmacol. 2008;593:73–8.
Moise AM, Eisenstein SA, Astarita G, Piomelli D, Hohmann AG. An endocannabinoid signaling system modulates anxiety-like behavior in male Syrian hamsters. Psychopharmacology (Berl). 2008;200:333–46.
Moreira FA, Kaiser N, Monory K, Lutz B. Reduced anxiety-like behaviour induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacology. 2008;54:141–50.
Rubino T, Realini N, Castiglioni C, Guidali C, Vigano D, Marras E, et al. Role in anxiety behavior of the endocannabinoid system in the prefrontal cortex. Cereb Cortex. 2008;18:1292–301.
Stopponi S, Fotio Y, Domi A, Borruto AM, Natividad L, Roberto M, et al. Inhibition of fatty acid amide hydrolase in the central amygdala alleviates co-morbid expression of innate anxiety and excessive alcohol intake. Addict Biol. 2017. doi: 10.1111/adb.12573
Mackintosh NJ. The psychology of animal learning. London: Academic; 1974. p. 730.
Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berl). 2000;153:31–43.
Tzschentke TM. Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol. 2007;12:227–462.
Huston JP, Silva MA, Topic B, Muller CP. What’s conditioned in conditioned place preference? Trends Pharmacol Sci. 2013;34:162–6.
Lepore M, Vorel SR, Lowinson J, Gardner EL. Conditioned place preference induced by delta 9-tetrahydrocannabinol: comparison with cocaine, morphine, and food reward. Life Sci. 1995;56:2073–80.
Valjent E, Maldonado R. A behavioural model to reveal place preference to delta 9-tetrahydrocannabinol in mice. Psychopharmacology (Berl). 2000;147:436–8.
Braida D, Iosue S, Pegorini S, Sala M. Delta9-tetrahydrocannabinol-induced conditioned place preference and intracerebroventricular self-administration in rats. Eur J Pharmacol. 2004;506:63–9.
Le Foll B, Wiggins M, Goldberg SR. Nicotine pre-exposure does not potentiate the locomotor or rewarding effects of Delta-9-tetrahydrocannabinol in rats. Behav Pharmacol. 2006;17:195–9.
McGregor IS, Issakidis CN, Prior G. Aversive effects of the synthetic cannabinoid CP 55,940 in rats. Pharmacol Biochem Behav. 1996;53:657–64.
Parker LA, Gillies T. THC-induced place and taste aversions in Lewis and Sprague-Dawley rats. Behav Neurosci. 1995;109:71–8.
Sanudo-Pena MC, Tsou K, Delay ER, Hohman AG, Force M, Walker JM. Endogenous cannabinoids as an aversive or counter-rewarding system in the rat. Neurosci Lett. 1997;223:125–8.
Mallet PE, Beninger RJ. Delta9-tetrahydrocannabinol, but not the endogenous cannabinoid receptor ligand anandamide, produces conditioned place avoidance. Life Sci. 1998;62:2431–9.
Bortolato M, Campolongo P, Mangieri RA, Scattoni ML, Frau R, Trezza V, et al. Anxiolytic-like properties of the anandamide transport inhibitor AM404. Neuropsychopharmacology. 2006;31:2652–9.
Scherma M, Justinova Z, Zanettini C, Panlilio LV, Mascia P, Fadda P, et al. The anandamide transport inhibitor AM404 reduces the rewarding effects of nicotine and nicotine-induced dopamine elevations in the nucleus accumbens shell in rats. Br J Pharmacol. 2012;165:2539–48.
Zangen A, Solinas M, Ikemoto S, Goldberg SR, Wise RA. Two brain sites for cannabinoid reward. J Neurosci. 2006;26:4901–7.
Justinova Z, Solinas M, Tanda G, Redhi GH, Goldberg SR. The endogenous cannabinoid anandamide and its synthetic analog R(+)-methanandamide are intravenously self-administered by squirrel monkeys. J Neurosci. 2005;25:5645–50.
Tanda G, Munzar P, Goldberg SR. Self-administration behavior is maintained by the psychoactive ingredient of marijuana in squirrel monkeys. Nat Neurosci. 2000;3:1073–4.
Schindler CW, Scherma M, Redhi GH, Vadivel SK, Makriyannis A, Goldberg SR, et al. Self-administration of the anandamide transport inhibitor AM404 by squirrel monkeys. Psychopharmacology (Berl). 2016;233:1867–77.
Hendershot CS, Witkiewitz K, George WH, Marlatt GA. Relapse prevention for addictive behaviors. Subst Abus Treat Prev Policy. 2011;6:17.
Tran-Nguyen LT, Fuchs RA, Coffey GP, Baker DA, O’Dell LE, Neisewander JL. Time-dependent changes in cocaine-seeking behavior and extracellular dopamine levels in the amygdala during cocaine withdrawal. Neuropsychopharmacology. 1998;19:48–59.
Shaham Y, Shalev U, Lu L, de Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl). 2003;168:3–20.
Justinova Z, Munzar P, Panlilio LV, Yasar S, Redhi GH, Tanda G, et al. Blockade of THC-seeking behavior and relapse in monkeys by the cannabinoid CB(1)-receptor antagonist rimonabant. Neuropsychopharmacology. 2008;33:2870–7.
Spano MS, Fattore L, Cossu G, Deiana S, Fadda P, Fratta W. CB1 receptor agonist and heroin, but not cocaine, reinstate cannabinoid-seeking behaviour in the rat. Br J Pharmacol. 2004;143:343–50.
Gamaleddin I, Wertheim C, Zhu AZ, Coen KM, Vemuri K, Makryannis A, et al. Cannabinoid receptor stimulation increases motivation for nicotine and nicotine seeking. Addict Biol. 2012;17:47–61.
Lopez-Moreno JA, Gonzalez-Cuevas G, Rodriguez de Fonseca F, Navarro M. Long-lasting increase of alcohol relapse by the cannabinoid receptor agonist WIN 55,212-2 during alcohol deprivation. J Neurosci. 2004;24:8245–52.
McGregor IS, Dam KD, Mallet PE, Gallate JE. Delta9-THC reinstates beer- and sucrose-seeking behaviour in abstinent rats: comparison with midazolam, food deprivation and predator odour. Alcohol Alcohol. 2005;40:35–45.
Mack A, Joy J Marijuana as medicine? The science beyond the controversy. Washington (DC): National Academies Press (US); 2000.
Gunduz O, Oltulu C, Ulugol A. Role of GLT-1 transporter activation in prevention of cannabinoid tolerance by the beta-lactam antibiotic, ceftriaxone, in mice. Pharmacol Biochem Behav. 2011;99:100–3.
Ulugol A, Ozyigit F, Yesilyurt O, Dogrul A. The additive antinociceptive interaction between WIN 55,212-2, a cannabinoid agonist, and ketorolac. Anesth Analg. 2006;102:443–7.
Cohen C, Kodas E, Griebel G. CB1 receptor antagonists for the treatment of nicotine addiction. Pharmacol Biochem Behav. 2005;81:387–95.
Merritt LL, Martin BR, Walters C, Lichtman AH, Damaj MI. The endogenous cannabinoid system modulates nicotine reward and dependence. J Pharmacol Exp Ther. 2008;326:483–92.
Scherma M, Panlilio LV, Fadda P, Fattore L, Gamaleddin I, Le Foll B, et al. Inhibition of anandamide hydrolysis by cyclohexyl carbamic acid 3’-carbamoyl-3-yl ester (URB597) reverses abuse-related behavioral and neurochemical effects of nicotine in rats. J Pharmacol Exp Ther. 2008;327:482–90.
Melis M, Pillolla G, Luchicchi A, Muntoni AL, Yasar S, Goldberg SR, et al. Endogenous fatty acid ethanolamides suppress nicotine-induced activation of mesolimbic dopamine neurons through nuclear receptors. J Neurosci. 2008;28:13985–94.
Forget B, Coen KM, Le Foll B. Inhibition of fatty acid amide hydrolase reduces reinstatement of nicotine seeking but not break point for nicotine self-administration--comparison with CB(1) receptor blockade. Psychopharmacology (Berl). 2009;205:613–24.
Gamaleddin I, Guranda M, Goldberg SR, Le Foll B. The selective anandamide transport inhibitor VDM11 attenuates reinstatement of nicotine seeking behaviour, but does not affect nicotine intake. Br J Pharmacol. 2011;164:1652–60.
Justinova Z, Panlilio LV, Moreno-Sanz G, Redhi GH, Auber A, Secci ME, et al. Effects of Fatty Acid Amide Hydrolase (FAAH) Inhibitors in Non-Human Primate Models of Nicotine Reward and Relapse. Neuropsychopharmacology. 2015;40:2185–97.
Trigo JM, Le Foll B. Inhibition of monoacylglycerol lipase (MAGL) enhances cue-induced reinstatement of nicotine-seeking behavior in mice. Psychopharmacology (Berl). 2016;233:1815–22.
Cippitelli A, Cannella N, Braconi S, Duranti A, Tontini A, Bilbao A, et al. Increase of brain endocannabinoid anandamide levels by FAAH inhibition and alcohol abuse behaviours in the rat. Psychopharmacology (Berl). 2008;198:449–60.
Cippitelli A, Bilbao A, Gorriti MA, Navarro M, Massi M, Piomelli D, et al. The anandamide transport inhibitor AM404 reduces ethanol self-administration. Eur J Neurosci. 2007;26:476–86.
Blednov YA, Cravatt BF, Boehm SL 2nd, Walker D, Harris RA. Role of endocannabinoids in alcohol consumption and intoxication: studies of mice lacking fatty acid amide hydrolase. Neuropsychopharmacology. 2007;32:1570–82.
Hansson AC, Bermudez-Silva FJ, Malinen H, Hyytia P, Sanchez-Vera I, Rimondini R, et al. Genetic impairment of frontocortical endocannabinoid degradation and high alcohol preference. Neuropsychopharmacology. 2007;32:117–26.
Zhou Y, Schwartz BI, Giza J, Gross SS, Lee FS, Kreek MJ. Blockade of alcohol escalation and “relapse” drinking by pharmacological FAAH inhibition in male and female C57BL/6J mice. Psychopharmacology (Berl). 2017;234:2955–70.
Zhou Y, Huang T, Lee F, Kreek MJ. Involvement of endocannabinoids in alcohol “Binge” drinking: studies of mice with human fatty acid amide hydrolase genetic variation and after CB1 receptor antagonists. Alcohol Clin Exp Res. 2016;40:467–73.
Sloan ME, Gowin JL, Ramchandani VA, Hurd YL, Le Foll B. The endocannabinoid system as a target for addiction treatment: trials and tribulations. Neuropharmacology. 2017;124:73–83.
Soyka M, Koller G, Schmidt P, Lesch OM, Leweke M, Fehr C, et al. Cannabinoid receptor 1 blocker rimonabant (SR 141716) for treatment of alcohol dependence: results from a placebo-controlled, double-blind trial. J Clin Psychopharmacol. 2008;28:317–24.
George DT, Herion DW, Jones CL, Phillips MJ, Hersh J, Hill D, et al. Rimonabant (SR141716) has no effect on alcohol self-administration or endocrine measures in nontreatment-seeking heavy alcohol drinkers. Psychopharmacology (Berl). 2010;208:37–44.
Sloan ME, Gowin JL, Yan J, Schwandt ML, Spagnolo PA, Sun H, et al. Severity of alcohol dependence is associated with the fatty acid amide hydrolase Pro129Thr missense variant. Addict Biol. 2018;23:474–84.
Solinas M, Panlilio LV, Tanda G, Makriyannis A, Matthews SA, Goldberg SR. Cannabinoid agonists but not inhibitors of endogenous cannabinoid transport or metabolism enhance the reinforcing efficacy of heroin in rats. Neuropsychopharmacology. 2005;30:2046–57.
Ramesh D, Gamage TF, Vanuytsel T, Owens RA, Abdullah RA, Niphakis MJ, et al. Dual inhibition of endocannabinoid catabolic enzymes produces enhanced antiwithdrawal effects in morphine-dependent mice. Neuropsychopharmacology. 2013;38:1039–49.
Del Arco I, Navarro M, Bilbao A, Ferrer B, Piomelli D, Rodriguez De, Fonseca F. Attenuation of spontaneous opiate withdrawal in mice by the anandamide transport inhibitor AM404. Eur J Pharmacol. 2002;454:103–4.
Vela G, Ruiz-Gayo M, Fuentes JA. Anandamide decreases naloxone-precipitated withdrawal signs in mice chronically treated with morphine. Neuropharmacology. 1995;34:665–8.
McCallum AL, Limebeer CL, Parker LA. Reducing endocannabinoid metabolism with the fatty acid amide hydrolase inhibitor, URB597, fails to modify reinstatement of morphine-induced conditioned floor preference and naloxone-precipitated morphine withdrawal-induced conditioned floor avoidance. Pharmacol Biochem Behav. 2010;96:496–500.
Wilkerson JL, Ghosh S, Mustafa M, Abdullah RA, Niphakis MJ, Cabrera R, et al. The endocannabinoid hydrolysis inhibitor SA-57: Intrinsic antinociceptive effects, augmented morphine-induced antinociception, and attenuated heroin seeking behavior in mice. Neuropharmacology. 2017;114:156–67.
Tanda G, Newman AH, Katz JL. Discovery of drugs to treat cocaine dependence: behavioral and neurochemical effects of atypical dopamine transport inhibitors. Adv Pharmacol. 2009;57:253–89.
Rothman RB, Baumann MH. Monoamine transporters and psychostimulant drugs. Eur J Pharmacol. 2003;479:23–40.
Bowers MS, Chen BT, Bonci A. AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron. 2010;67:11–24.
Pierce RC, Vanderschuren LJ. Kicking the habit: the neural basis of ingrained behaviors in cocaine addiction. Neurosci Biobehav Rev. 2010;35:212–9.
Robinson TE, Berridge KC. Incentive-sensitization and addiction. Addiction. 2001;96:103–14.
Corbille AG, Valjent E, Marsicano G, Ledent C, Lutz B, Herve D, et al. Role of cannabinoid type 1 receptors in locomotor activity and striatal signaling in response to psychostimulants. J Neurosci. 2007;27:6937–47.
Gerdeman GL, Schechter JB, French ED. Context-specific reversal of cocaine sensitization by the CB1 cannabinoid receptor antagonist rimonabant. Neuropsychopharmacology. 2008;33:2747–59.
Kalivas PW, Duffy P. Time course of extracellular dopamine and behavioral sensitization to cocaine. I. Dopamine axon terminals. J Neurosci. 1993;13:266–75.
Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–82.
Scheggi S, Marchese G, Grappi S, Secci ME, De Montis MG, Gambarana C. Cocaine sensitization models an anhedonia-like condition in rats. Int J Neuropsychopharmacol. 2011;14:333–46.
Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–7.
Lesscher HM, Hoogveld E, Burbach JP, van Ree JM, Gerrits MA. Endogenous cannabinoids are not involved in cocaine reinforcement and development of cocaine-induced behavioural sensitization. Eur Neuropsychopharmacol. 2005;15:31–7.
Ferrer B, Asbrock N, Kathuria S, Piomelli D, Giuffrida A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: implications for the treatment of levodopa-induced dyskinesias. Eur J Neurosci. 2003;18:1607–14.
Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–63.
Vilela LR, Gobira PH, Viana TG, Medeiros DC, Ferreira-Vieira TH, Doria JG, et al. Enhancement of endocannabinoid signaling protects against cocaine-induced neurotoxicity. Toxicol Appl Pharmacol. 2015;286:178–87.
Luchicchi A, Lecca S, Carta S, Pillolla G, Muntoni AL, Yasar S, et al. Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: involvement of PPAR-alpha nuclear receptors. Addict Biol. 2010;15:277–88.
Justinova Z, Mangieri RA, Bortolato M, Chefer SI, Mukhin AG, Clapper JR, et al. Fatty acid amide hydrolase inhibition heightens anandamide signaling without producing reinforcing effects in primates. Biol Psychiatry. 2008;64:930–7.
Adamczyk P, McCreary AC, Przegalinski E, Mierzejewski P, Bienkowski P, Filip M. The effects of fatty acid amide hydrolase inhibitors on maintenance of cocaine and food self-administration and on reinstatement of cocaine-seeking and food-taking behavior in rats. J Physiol Pharmacol. 2009;60:119–25.
E.M.A. The European Medicines Agency recommends suspension of the marketing authorisation of Acomplia. Press Office of the European Medicines Agency, Lodon, UK, 2008. http://www.ema.europa.eu/humandocs/PDFs/EPAR/acomplia/53777708en.pdf.
Le Foll B, Gorelick DA, Goldberg SR. The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology (Berl). 2009;205:171–4.
Micale V, Di Marzo V, Sulcova A, Wotjak CT, Drago F. Endocannabinoid system and mood disorders: priming a target for new therapies. Pharmacol Ther. 2013;138:18–37.
Acknowledgements
This work was supported in part by Medication Development Program funds, NIDA-IRP, NIH, DHHS, to GT (ZIA DA000569), and in part by funds from the Department of Biomedical Sciences Project (RICDIP_2012_Fratta_01), University of Cagliari.
Author contributions
All authors have contributed to the writing of the present manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Scherma, M., Masia, P., Satta, V. et al. Brain activity of anandamide: a rewarding bliss?. Acta Pharmacol Sin 40, 309–323 (2019). https://doi.org/10.1038/s41401-018-0075-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0075-x
Keywords
This article is cited by
-
Oral Cannabis consumption and intraperitoneal THC:CBD dosing results in changes in brain and plasma neurochemicals and endocannabinoids in mice
Journal of Cannabis Research (2024)
-
Inhibition of fatty acid binding protein-5 in the basolateral amygdala induces anxiolytic effects and accelerates fear memory extinction
Psychopharmacology (2024)
-
Anandamide Attenuates Neurobehavioral Deficits and EEG Irregularities in the Chronic Sleep Deprivation Rats: The Role of Oxidative Stress and Neuroinflammation
Neurochemical Research (2023)
-
Combined alcohol and cannabinoid exposure leads to synergistic toxicity by affecting cerebellar Purkinje cells
Nature Metabolism (2022)
-
Examining the role of cannabinoids on osteoporosis: a review
Archives of Osteoporosis (2022)
