
Abstract
Perivascular adipose tissue (PVAT), a special type of adipose tissue, closely surrounds vascular adventitia and produces numerous bioactive substances to maintain vascular homeostasis. PVAT dysfunction has a crucial role in regulating vascular remodeling, but the exact mechanisms remain unclear. In this study, we investigated whether and how obesity-induced PVAT dysfunction affected adventitia remodeling in early vascular injury stages. Mini pigs were fed a high sugar and fat diet for 6 months to induce metabolic syndrome and obesity. In the mini pigs, left carotid vascular injury was then generated using balloon dilation. Compared with normal mini pigs, obese mini pigs displayed significantly enhanced vascular injury-induced adventitial responses, evidenced by adventitia fibroblast (AF) proliferation and differentiation, and adventitia fibrosis, as well as exacerbated PVAT dysfunction characterized by increased accumulation of resident macrophages, particularly the M1 pro-inflammatory phenotype, increased expression of leptin and decreased expression of adiponectin, and production of pro-inflammatory cytokines interleukin (IL)-1β and IL-18. Primary AFs cultured in PVAT-conditioned medium from obese mini pigs also showed significantly increased proliferation and differentiation. We further revealed that activated nod-like receptor protein 3 (NLRP3) inflammasome and its downstream products, i.e., IL-1 family members such as IL-1β and IL-18 were upregulated in the PVAT of obese mini pigs; PVAT dysfunction was also demonstrated in preadipocytes treated with palmitic acid. Finally, we showed that pretreatment with IL-1 receptor (IL-1R) antagonist or IL-1R knockdown blocked AF proliferation and differentiation in AFs cultured in PVAT-conditioned medium. These results demonstrate that obesity-induced PVAT dysfunction aggravates adventitial remodeling after early vascular injury with elevated AF proliferation and differentiation via activating the NLRP3/IL-1 signaling pathway.
Similar content being viewed by others


Introduction
Obesity, one of the biggest health problems in the world, is considered a crucial stimulus for vascular remodeling after injury that is an essential pathological process in the development of cardiovascular diseases (CVDs), including atherosclerosis and restenosis after endovascular intervention [1]. Adventitia, mainly comprising adventitia fibroblasts (AFs) and connective tissue, has recently been confirmed to be closely related to the process of vascular remodeling [2], which has been found to be activated before neointimal hyperplasia formation in a vascular injury animal model [3]. Perivascular adipose tissue (PVAT), a special type of adipose tissue (AT), closely surrounds vascular adventitia and produces numerous bioactive substances to maintain vascular homeostasis [4]. In response to sustained overnutrition, PVAT undergoes dysfunction to accommodate obesity-triggered uncontrolled chronic inflammatory responses, including inflammatory cell infiltration, increased pro-inflammatory cytokine secretion, and decreased anti-inflammatory adipokine secretion, which further aggravate vascular remodeling after vascular injury [2, 5, 6]. The exact mechanisms of this process, however, remain to be explored.
The nod-like receptor protein 3 (NLRP3) inflammasome is an intracellular multiprotein complex consisting of NLRP, adaptor protein ASC and pro-caspase-1. The activation of NLRP3 contributes to the maturation of caspase-1 and subsequently promotes the processing of interleukin (IL)-1 family members, including IL-1β and IL-18, both of which have been shown to have crucial roles in AT inflammation [7]. Suppressing the activation of the NLRP3 inflammasome decreases IL-1β levels and produces an AT protective function [7, 8]. The excessive expression of the NLRP3 inflammasome increases aortic wall thickness and fibrosis during abdominal aortic aneurysm development [9]. Moreover, IL-1β has been demonstrated to participate in the progression of adventitia fibrosis [10]. However, the roles of the NLRP3 inflammasome/IL-1 pathway in PVAT dysfunction and adventitia remodeling after vascular injury in the obese state have not been fully elucidated.
In this study, we investigated the role of the NLRP3 inflammasome and its downstream products in obesity-induced PVAT dysfunction and adventitia remodeling in an obese mini pig model. We observed that obesity-induced PVAT dysfunction aggravated adventitia fibroblast proliferation and differentiation in a mini pig model of early vascular injury, characterized by macrophage polarization, NLRP3 inflammasome activation and the production of downstream pro-inflammatory cytokines IL-1β and IL-18. Moreover, NLRP3-induced IL-1β and IL-18 activation contributed to AF proliferation and differentiation via binding to the IL-1 receptor (IL-1R). These results suggest that the NLRP3 inflammasome/IL-1 pathway in preadipocytes has a crucial role in controlling PVAT function and adventitia remodeling under obesity conditions.
Materials and methods
Antibodies and reagents
P4H, F4/80, adiponectin ELISA kit, Ki67, NLRP3, caspase-1, and IL-1R1 antibodies were purchased from Abcam (Cambridge, UK). The α-SMA and ASC antibodies and lipopolysaccharide (LPS), palmitic acid (PA), ATP, and MCC950 were purchased from Sigma (St. Louis, MO, USA). ELISA kits for leptin, TNF-α, IL-1β, IL-6, and IL-18; the IL-1β and IL-18 antibodies; and recombinant porcine IL-1R antagonist (IL-1Ra) protein were purchased from R&D Systems (Minneapolis, MN, USA). IL-1R1 siRNA was purchased from SCBT (Santa Cruz, CA, USA).
Animals
All protocols in the present study were approved by the Institutional Animal Care and Use Committee of University of South China. Bama mini pigs were purchased from Third Military Medical University of China and randomly divided into two groups: the normal diet group (n = 6) and the high sugar (33% sugar) and fat (10% lard) diet (HSFD) group (n = 6). Animals were housed in the Animal Facility of University of South China (Temperature 20–24 °C, under relative humidity of 30–70%). Body mass was measured at the end of each month.
Lipid, glucose, and insulin measurements
Before balloon dilatation, the serum total cholesterol, triglycerides, high-density lipoprotein cholesterol (HDL-C), low density lipoprotein cholesterol (LDL-C), glucose, and insulin levels of the mini pigs were measured every 2 months. Blood samples were collected from ear veins and detected with an automatic blood analyzer produced from Roche (Basel, Switzerland).
Oral glucose tolerance test
After 6 months, mini pigs were subjected to an oral glucose tolerance test. Mini pigs were fasted for 12 h, weighed and treated with glucose (2 g/kg weight) and 25 g of mixed feed (dissolved in 100 mL of water). Then, blood samples were collected and detected at 0, 30, 60, 90, 120, and 150 min.
Balloon dilatation
After 6 months, mini pigs were anaesthetized with ketamine (10 mg/kg) combined with acepromazine (0.2 mg/kg) via muscle injection for induction, followed by continuous intravenous infusion with ketamine (3 mg/kg/h) for maintenance. A ventilator and ECG were used to check the adequacy of the anesthesia and physiological function. The operative area was prepared by removing hair and disinfecting with 70% ethanol. Then, the femoral artery was punctured, and an arterial sheath was placed. The area was heparinized (100 U/kg sodium heparin), and angiography was used for the determination of the left carotid artery diameter. According to the diameter size, balloon dilatation was performed using a 5-Fr balloon. The balloon was inflated to 680–892 kPa for 10 s, which was repeated for three times at an interval of 30 s. Then, the balloon pressure was kept at 187–465 kPa and the balloon was pulled back and forth for three times to injure the left carotid artery for 3–4 cm. The surgeries were performed in the Department of Interventional Medicine of the Affiliated Hospital of University of South China.
Tissue isolation
After 2 weeks of balloon dilatation, mini pigs were deeply anesthetized and then killed with ketamine (40 mg/kg) combined with acepromazine (4 mg/kg). The killed mini pigs were placed in the supine position, and their left carotid arteries were carefully exposed. The injured left carotid arteries with PVAT were then isolated from the normal and HSFD group. The separated artery was divided into two parts, one of which was fixed in 10% buffered formalin and the other of which was used for the isolation of PVAT and primary fibroblasts.
Hematoxylin–eosin staining
The extracted injured left carotid artery was fixed in 10% buffered formalin for 24 h. Afterward, the artery was sliced into 2 to 3-mm portions and embedded in paraffin after treatment with ethanol and xylene. The paraffin blocks were sliced and dried. After processing with xylene and a graded alcohol series, the sections were washed with double-distilled water and stained with hematoxylin and 5% eosin. The pathological changes in the left carotid artery were examined by light microscopy.
Immunofluorescence
Immunofluorescence was performed according to previous methods [11]. Briefly, paraffin-embedded left carotid arteries were treated by steam heating for antigen retrieval (15–20 min) after deparaffinization and rehydration. After blocking in 2% donkey serum for 1 h at room temperature, the slides were washed in phosphate buffer saline (PBS) three times for 5 min and then incubated with 1:100 anti-P4H and 1:100 anti-F4/80 at 4 °C overnight. They were then washed with PBS three times for 5 min. After incubation with CY3-conjugated (red) secondary antibody at room temperature for 2 h, the slides were washed with PBS three times for 5 min and stained with DAPI (blue) for 5 min at room temperature. They were then washed with PBS three times for 5 min. Fluorescence images were obtained using a fluorescence microscope.
Western blotting
Total proteins were isolated according to previous methods [12, 13]. The total protein concentration was determined using the Bio-Rad protein assay (California, USA), before separation with SDS-PAGE for 30 min at 80 V and 90 min at 120 V in buffer containing 25 mM Tris base, 250 mM glycine, and 0.1% SDS. After electrophoresis, the proteins were electrically transferred to the immobilon-P transfer membrane in buffer containing 25 mM Tris, 192 mM glycine and 20% methanol. After transfer, the membrane was blocked in TBST (20 mM Tris base pH 7.6, 15 M NaCl, 0.1% Tween-20) containing 5% skimmed milk for 2 h at room temperature. After that, membranes were incubated with primary antibodies (4 °C, overnight) at the following concentrations: 1:1000 anti-P4H, 1:2000 anti-α-SMA, 1:5000 anti-GAPDH, 1:2000 anti-Ki67, 1:1000 anti-NLRP3, 1:1000 anti-ASC, 1:1000 anti-pro-caspase-1, 1:2000 anti-caspase-1, 1:1000 anti-IL-1β, 1:1000 anti-IL-18 in TBST dilution buffer. Thereafter, the membrane was washed three times with TBST for 10 min and incubated with secondary antibody in the blocking solution for 2 h at room temperature and washed three times with TBST for 10 min. The proteins were visualized using the chemiluminescence method. Densitometric analysis was then completed to quantify the amount of protein.
Relative quantitative real-time PCR
The total RNA of the cells was extracted from PVAT by TRIzol reagent produced from Invitrogen (California, USA) and reverse-transcribed by qScript cDNA SuperMix produced from Quanta BioSciences (Maryland, USA) in accordance with the manufacturer’s instructions. cDNA was synthesized by iScript cDNA Synthesis kit (Bio-Rad). Subsequently, cDNA was used for quantitative PCR in triplicate in a 7500 Fast Real-Time PCR System. RNA content of all samples was normalized to the expression of GAPDH. The primers used for the RT-PCR are shown in Supplementary Table 1.
Immunohistochemistry
The slices of the injured left carotid arteries samples were deparaffinized with xylene and rehydrated, after which the antigen was repaired in 10 mM of citrate buffer for 2–3 min at 37 °C and washed with distilled water three times for 3 min. At the cell level, the primary adventitial fibroblasts were co-cultured with PVAT-conditioned medium and fixed with 4% paraformaldehyde (4 °C, overnight), washed three times with PBS for 5 min, then incubated with 3% H2O2 (room temperature, 20 min) and washed with PBS. Thereafter, the slices and fibroblasts were blocked for non-specific staining with a buffer containing 5% goat serum (37 °C, 20 min) and incubated with primary anti-α-SMA (1:200) and anti-Ki67 (1:100) overnight at 4 °C. After washing with PBS three times for 5 min, samples were incubated with biotinylated anti-mouse secondary antibodies. IgG-conjugated horseradish peroxidase and 3,3′-diaminobenzidine tetrachloride were used to detect antibody binding.
ELISA
Adipokines (adiponectin and leptin) and inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-18) were detected in the PVAT samples from the normal and HSFD groups (all samples were normalized with the weight of PVAT); preadipocytes were detected by an ELISA kit according to the guiding manual.
Preadipocytes culture
Preadipocytes were extracted from PVAT according to previous methods [14]. Briefly, the minced PVAT sample were washed three times with PBS (adding penicillin/streptomycin), then placed in DMEM/F12-conditioned medium containing type 1 collagenase (0.25%) and incubated on a shaker (250 rpm, 37 °C, 60 min). The undigested PVAT sample was filtered with 100-μm mesh fabric, and the matured adipose cells were isolated (500×g, 4 °C, 10 min). Finally, the stromal vascular fraction was resuspended in adipocyte medium. After filtration through a 20-μm nylon mesh, the cells were dispensed onto a 24-well plate for confluency. After an overnight rest, adherent preadipocytes were washed three times with FBS-free DMEM/F12 medium and subsequently cultured for 24 h in the same medium. After that, the preadipocytes were cultured in DMEM/F12-conditioned medium with 0.2% FBS to induce growth arrest.
Immunoprecipitation
The assembly of NLRP3–ASC was assayed through previous methods [15]. After experiments factor treatment, preadipocytes were lysed in RIPA buffer, and the isolated lysates (400 μg) were incubated with anti-NLRP3 antibody (4 °C, overnight). Then, co-incubation with protein A agarose beads (40 μL) (4 °C, 3 h) was completed on a horizontal shaker (4 °C, 5 min), after which the supernatant was discarded. Protein complexes that linked with protein A were washed five times at least with RIPA buffer and centrifuged on horizontal shaker (4 °C, 5 min) before supernatant collection. Finally, the collection buffer was mixed with 5× loading buffer, heated (95 °C, 10 min), and used for Western blot analysis with an anti-ASC and anti-NLRP3 antibody. Whole preadipocyte lysate prepared for immunoprecipitation (60 μg) was used as an input sample.
Preparation of PVAT-conditioned medium
The preparation of PVAT-conditioned medium was conducted as previously described [16, 17]. Briefly, PVAT samples were dissected along the injured left carotid artery, placed in petri dishes and repeatedly washed with 1× PBS buffer (pH 7.4). Then, samples were minced into fine pieces carefully under aseptic conditions and cultured in serum-free DMEM medium, with a volume normalized to the weight of PVAT (37 °C, 48 h). For follow-up experiments, the PVAT-conditioned medium was collected.
Fibroblast co-culture
Primary fibroblasts were isolated and cultured as described previously [17, 18]. In brief, the PVAT media as well as endothelium of injured left carotid artery from normal and HSFD mini pigs were removed using fine forceps. The adventitia of the injured vasculature was minced into fine pieces and cultured in DMEM containing 15% FBS, 100 U/mL penicillin, and streptomycin. After reaching confluence, primary fibroblasts were induced to undergo growth arrest by DMEM supplemented with 0.2% FBS before treatment. Fibroblasts were used from passage 3 to 6 and then cultured in PVAT-conditioned medium at 37 °C in 5% CO2 incubator.
siRNA experiment
Primary fibroblasts were cultured in a 24-well plate after reaching confluence (40–50%), and cells were transfected with IL-1R1 siRNAs (120 pmol) using Lip2000 (10 µL), then cultured at 37 °C in a 5% CO2 incubator. After 48 h, the effect of the interference on cells was detected by Western blot analysis. The primary fibroblasts were then co-cultured with PVAT-conditioned medium for 48 h.
Statistical analysis
Quantitative data are presented as the means ± SD. Curve-fitting was completed using GraphPad Prism version 6.0 software. Statistical differences were analyzed by a one-way ANOVA and t-test. The results were considered significant when P values were <0.05.
Results
Obesity aggravates vascular injury-induced adventitia fibroblast proliferation and differentiation
To investigate the effects of obesity on the progression of adventitia remodeling, mini pigs were first fed a normal diet (n = 6) or an HSFD with 33% sugar and 10% lard based on a normal diet (n = 6) for 6 months. When HSFD was compared with the normal diet, we confirmed that HSFD induced an obvious increase in body mass and the levels of glycolipids, including total cholesterol, LDL-C, HDL-C, free fatty acids (FFA), glucose, and insulin within 4 to 6 months in the pigs, though the content of triglycerides showed no significant difference between the two groups (Supplementary Fig. 1A to C and Supplementary Table 2), suggesting that HSFD induced metabolic syndrome and obesity in 6 months. Subsequently, the mini pigs underwent balloon dilation in the left carotid artery (Supplementary Fig. 1D to I) and were killed at 2 weeks after surgery. We observed that the lesion area and media-lumen ratio between HSFD and the normal diet group had no significant difference (Fig. 1a–c). However, the thickness of the adventitia was obviously increased in the HSFD group compared to the normal diet group (Fig. 1a, d). AF, a major cell type of vascular adventitia, has been shown to be induced to participate in adventitia remodeling via proliferation and differentiation to myofibroblasts [3]. Furthermore, we found that in the injured left carotid artery adventitia, HSFD induced the upregulation of Ki67 (Fig. 1e, f), a marker of proliferation; P4H (Fig. 1g, h), and α-SMA (Fig. 1i, j), markers of AF differentiation. Furthermore, the collagen content of adventitia in HSFD also increased (Fig. 1j, k). These results suggested that HSFD promoted AF proliferation and differentiation and adventitial fibrosis in response to early vascular injury.

Obesity aggravates vascular injury-induced AF proliferation and differentiation. The intimal hyperplasia area (a, b), media-lumen ratio (a, c), and adventitia thickness (a, d) in the left carotid artery were visualized by hematoxylin–eosin staining and measured by Image-Pro plus at 2 weeks after the balloon dilation. e, f Immunohistochemistry staining of Ki67 as a marker of AF proliferation was quantified by Image-Pro Plus 6.0 in the normal and HSFD diet mini pigs. Image-Pro Plus 6.0 was used to measure the number of hematoxylin staining nuclei (blue) and Ki67+ nuclei (dark brown). The ratio of Ki67 staining was defined as the percentage of positive nuclei (brown) within the total (hematoxylin staining plus Ki67+ staining) number of nuclei. g Immunofluorescence staining of P4H (red), DAPI (blue) in the adventitia of mini pigs. h Western blot analysis was used to detect the expression of P4H in the adventitia of mini pigs. i, j The level of α-SMA in the adventitia was detected by immunohistochemistry and Western blot analysis, which was quantified by Image-Pro Plus 6.0 (dashed line in black indicates the border of adventitia and media; dashed line in red indicates the border of adventitia and perivascular adipose tissue; the area between the two dashed lines is the region of interest to quantify α-SMA level). j, k The fibrosis of adventitia was detected by Masson staining and quantified by Image-Pro Plus 6.0. Means ± SD. n = 6. *P < 0.05 vs. normal diet
Obesity-induced PVAT dysfunction promotes adventitia fibroblast proliferation and differentiation
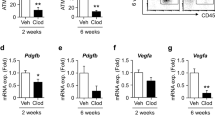
PVAT has a crucial role in regulating vascular function, whereas its phenotype and function undergo a significant change in the obese state [6, 19]. To investigate the role of obesity in PVAT dysfunction, we first analyzed the anti-/pro-inflammatory phenotype of PVAT surrounding the injured left carotid artery. Immunofluorescence staining showed a great increase in F4/80+ macrophages in obese PVAT animals (Fig. 2a). Interestingly, the levels of the pro-inflammatory M1 macrophage markers CCL2 and CD11c were markedly increased, while the levels of anti-inflammatory M2 markers, such as Arg1 and CD206, in obese PVAT were significantly decreased (Fig. 2b), suggesting that HSFD promoted the pro-inflammatory phenotype of PVAT. Since PVAT was considered to be an important endocrine organ that regulated vascular function via the secretion of cytokines, we examined the expression of cytokines in PVAT and determined that protective cytokines, such as adiponectin, were significantly decreased. In contrast, pro-inflammatory factors, such as leptin, TNF-α, IL-6, and especially IL-1β and IL-18, were significantly increased in HSFD (Fig. 2c, d). We next investigated whether PVAT dysfunction had a role in regulating adventitia remodeling. Primary AFs were isolated from injured left carotid artery adventitia and co-cultured with PVAT culture medium. It was identified that AFs in the HSFD culture medium expressed higher levels of P4H and α-SMA than AFs cultured in the control medium (Fig. 2e, f). Furthermore, Ki67+ fibroblasts showed significant increases in PVAT from HSFD co-cultured medium (Fig. 2g). This suggested that obesity-induced PVAT dysfunction aggravated adventitial remodeling by promoting AF proliferation and differentiation.

Obesity-induced PVAT dysfunction promotes AF proliferation and differentiation. a Immunofluorescence staining of F4/80+ (red) macrophages showed that the infiltration of F4/80+ macrophages was significantly increased in HSFD PVAT. b The mRNA expression levels of macrophage polarization markers were determined by quantitative real-time PCR. c The expression levels of adiponectin and leptin in PVAT of mini pigs were determined by ELISA. d The expression levels of TNF-α, IL-1β, IL-6, IL-18 in PVAT were determined by ELISA. e, f The expression levels of P4H and α-SMA in AFs co-cultured with PVAT culture medium from normal and HSFD mini pigs. g Ki67 staining as a marker of AF proliferation was quantified in PVAT-conditioned medium from the normal and HSFD group. Means ± SD. n = 6. *P < 0.05 vs. normal diet
Obesity-induced NLRP3 inflammasome activation in PVAT
After 6 months of HSFD, the level of FFA in obese mini pigs was increased (Supplementary Table 2). FFA, especially in its saturated form, has been shown to promote NLRP3 inflammasome activation, which has an important role in regulating the AT inflammatory state [20, 21]. However, the exact role of FFA in the activation of the NLRP3 inflammasome in PVAT remains unknown. We thus explored the expression of NLRP3 and observed that NLRP3, ASC, pro-caspase-1 and its downstream signaling products, including caspase-1, were significantly increased in PVAT from HSFD mini pigs (Fig. 3a), indicating the effect of obesity on the activation of NLRP3 inflammasome.

Obesity-induced NLRP3 inflammasome activation in PVAT. a HSFD promotes the expression of NLRP3, ASC, pro-caspase-1, and caspase-1 in PVAT, which was presented as relative protein expression to GAPDH (means ± SD; *P < 0.05 vs. normal diet; n = 6). b Isolated primary preadipocytes from PVAT were treated with control media PBS, PA (250 μM, 5 h), LPS (10 μg/mL, 4 h) plus ATP (10 mM, 1 h) and PA (250 μM, 4 h) plus MCC950 (2 μM, 1 h). Furthermore, the formation of the NLRP3–ASC complex in preadipocytes was detected by immunoprecipitation. c The activation of caspase-1, and the expression and maturation of pro-IL-1β and pro-IL-18 in preadipocytes were detected by Western bolt analysis, GAPDH expression was used as a control. d The expression of IL-1β and IL-18 in preadipocytes were detected by ELISA (means ± SD; *P < 0.05 vs. PBS; #P < 0.05 vs. PA; n = 3). PBS phosphate buffer saline, PA palmitic acid, LPS lipopolysaccharide, MCC950 an inhibitor of NLRP3 inflammasome activation
Previous studies have confirmed that NLRP3 inflammasome activation is essential for the activation of IL-1β and IL-18 [22]. Therefore, we next investigated whether the upregulation of IL-1β and IL-18 in the PVAT of HSFD mini pigs depended on NLRP3 inflammasome activation. Since preadipocytes exhibited higher inflammatory responses after exposure to saturated FFA [23], the primary preadipocytes extracted from PVAT were treated with PBS, palmitic acid (PA), LPS + ATP (an NLRP3 inflammasome agonist) and PA + MCC950 (an inhibitor of NLRP3 inflammasome activation). We observed that the NLRP3–ASC complex in preadipocytes was necessary for NLRP3 inflammasome activation and was significantly increased in PA and LPS + ATP compared with PBS alone. In contrast, it was significantly decreased in the PA + MCC950 group (Fig. 3b). Consistent with NLRP3–ASC complex formation, the activation of caspase-1 and the expression and maturation of pro-IL-1β and pro-IL-18 were shown to be increased in preadipocytes after stimulation with PA and LPS + ATP (Fig. 3c, d). These results suggest that obesity induces the activation of the NLRP3 inflammasome and the release of its downstream products, including IL-1β and IL-18, in PVAT, which may have a role in early vascular injury following balloon dilatation.
Obesity promotes adventitia fibroblast proliferation and differentiation via IL-1 in PVAT
To clarify the exact mechanism of obesity-induced PVAT dysfunction in adventitial changes, we investigated whether NLRP3 inflammasome-mediated IL-1 signaling in PVAT contributed to AF proliferation and differentiation. Primary AFs were pretreated with PBS or recombinant porcine IL-1Ra, an inhibitor of IL-1 signaling, and were then co-cultured with PVAT-conditioned medium from HSFD mini pigs. We confirmed that the expression of P4H and α-SMA and Ki67+ staining in primary AFs were obviously inhibited in the IL-1Ra group (Fig. 4a–c). To further verify the role of IL-1R in this process, the primary AFs were pretreated with IL-1R1 siRNA. Our analysis indicated a similar tendency of P4H, α-SMA and Ki67 expression following treatment with IL-1Ra (Fig. 4d–g). Thus, AF proliferation and differentiation were antagonized. These results indicated that obese PVAT promoted AF proliferation and differentiation via IL-1, a crucial pro-inflammatory cytokine whose secretion depends on NLRP3 inflammasome activation.

Obesity promotes AF proliferation and differentiation via the activation of NLRP3 inflammasome/IL-1 in PVAT. a–c Primary AFs were isolated and pretreated with PBS and IL-1Ra (75 ng/mL, 4 h), then co-cultured with PVAT-conditioned medium of HSFD, Western bolt was used to detect the expression of P4H and α-SMA, Ki67 staining was used for analysis of AF proliferation (means ± SD; *P < 0.05 vs. PBS; n = 3). The primary AFs were pretreated with control siRNA or IL-1R1 siRNA (120 pmol, 48 h), and the protein samples were immunoblotted with anti-IL-1R1 antibody (d). Then, the transfected primary AFs were co-cultured with PVAT-conditioned medium of HSFD, and Western bolt analysis was used to detect the expression levels of P4H and α-SMA (e, f). g Ki67 staining was used for analysis of AF proliferation (means ± SD; *P < 0.05 vs. control; n = 3)
Discussion
Epidemiological studies have verified that obesity is closely related to the CVD incidence, including atherosclerosis and restenosis after percutaneous coronary intervention [24]. Although recent studies have shown that obesity exacerbates vascular remodeling after injury [2, 25], the exact mechanism through which this occurs is still unclear. The adventitia has been verified to participate in the vascular repair process. Changes in the adventitia rapidly occur in coronary arteries in the early stages after balloon-induced medial injury but decline by 28 days after injury [26]. In this study, we observed that HSFD-induced obesity in mini pigs aggravated early vascular injury, characterized by obvious changes in adventitia, including the increased proliferation and differentiation of AFs. PVAT is an endocrine and metabolic organ, which has no anatomical barrier to vessel walls. In addition to supporting blood vessels, PVAT produces a large number of beneficial active substances that maintain vascular function under physiological conditions [27,28,29]. Nevertheless, the function of PVAT is significantly altered when protective adipokines are significantly downregulated, while pro-inflammatory factors, such as leptin and IL-1β, are significantly upregulated, eventually impairing vascular function in the obese state [27,28,29]. In the present study, we determined that HSFD-induced obesity significantly promoted PVAT dysfunction as characterized by an increased accumulation of resident macrophages, particularly the M1 pro-inflammatory phenotype, with the increased expression of leptin and the decreased expression of adiponectin. Moreover, the production of pro-inflammatory cytokines, especially IL-1β and IL-18, in PVAT was significantly elevated in HSFD mini pigs.
Previous studies have demonstrated that obesity-induced PVAT dysfunction accelerates vascular injury by regulating the activation of endothelial cells and vascular smooth muscle cells [30,31,32]. Recently, PVAT dysfunction was confirmed to aggravate the pathogenesis of vascular remodeling by targeting the adventitial response in deoxycorticosterone acetate-salt mice [33]. Interestingly, our study revealed that AFs co-cultured with PVAT-conditioned medium from HSFD mini pigs showed a significant elevation in AF proliferation and differentiation compared to the normal group, suggesting that adventitial remodeling after vascular injury was induced by PVAT dysfunction in obese mini pigs. Studies have shown that restoring the impaired function of PVAT by drugs [34, 35] promoted weight loss, [6] which led to an increase in AT browning [36] and a delay in the development of vascular dysfunction, further suggesting that PVAT may be a novel treatment target for obesity-related CVDs.
The NLRP3 inflammasome has been verified to induce innate and immune inflammatory responses by promoting IL-1β and IL-18 maturation [37], which has been found to increase the pro-inflammatory states of the PVAT, while the inhibition of NLPR3 significantly alleviates AT inflammation in obese mice [38, 39]. In this mini pig model, we have shown for the first time that the activity of the NLRP3 inflammasome in PVAT is obviously unregulated under HSFD treatment. Since the NLRP3 inflammasome is a major stimulator of IL-1β and IL-18 secretion, we proceeded to determine whether the increased expression of IL-1β and IL-18 in PVAT relied on NLRP3 inflammasome activation. In preadipocytes, we determined that NLRP3 inflammasome activation was induced by PA, which was responsible for the increased expression of IL-1β and IL-18. Recently, IL-1β, one of the most important pro-inflammatory cytokines, was confirmed to be a crucial regulator of adventitia changes that functioned via increasing collagen accumulation in AFs [10]. Consistent with this finding, our study showed that IL-1 family in PVAT from HSFD has a significant effect on regulating AF proliferation and differentiation. Autophagy, the process of degrading damaged DNA, proteins and cell debris, has been implicated in the maintenance of cellular homeostasis. Several recent studies have shown that a homeostatic role of autophagy is impaired in obese AT, leading to an inflammatory response via the upregulation of NLRP3 in AFs [40]. These data suggest that the regulation of the autophagic function of PVAT may provide novel insight in controlling vascular remodeling, although the exact roles of the autophagy-mediated NLRP3 inflammasome pathway in AF proliferation and differentiation must be further studied.
In conclusion, we have for the first time demonstrated that PVAT dysfunction that is mediated in a NLRP3 inflammasome/IL-1-dependent manner has an important role in promoting AF proliferation and differentiation in response to vascular injury in an obese mini pig model. Systemic HFSD influences not only PVAT but also the vessel wall. However, our study observed a significant difference in the adventitia, but not the intima, during the early stages after balloon-induced injury, suggesting that HFSD-induced PVAT dysfunction has a crucial role at least in the early stages of vascular adventitia remodeling. Because the anatomy and physiology of the arteries and metabolic syndrome in mini pigs are more similar to those of humans, it seemed appropriate to test clinical procedures after injury by endovascular injury, and eventually develop an understanding of human pathophysiology under obesity conditions [14]. Our findings suggest that the intervention of the NLRP3 inflammasome/IL-1 axis in PVAT under obesity may provide a novel strategy for anti-atherosclerosis and anti-restenosis therapies.
References
Martinez-Martinez E, Rodriguez C, Galan M, Miana M, Jurado-Lopez R, Bartolome MV, et al. The lysyl oxidase inhibitor (beta-aminopropionitrile) reduces leptin profibrotic effects and ameliorates cardiovascular remodeling in diet-induced obesity in rats. J Mol Cell Cardiol. 2016;92:96–104.
Krueger F, Kappert K, Foryst-Ludwig A, Kramer F, Clemenz M, Grzesiak A, et al. AT1-receptor blockade attenuates outward aortic remodeling associated with diet-induced obesity in mice. Clin Sci. 2017;131:1989–2005.
Sun M, Ji J, Guo X, Liu W, Wang Y, Ma S, et al. Early adventitial activation characterized by NADPH oxidase expression and neovascularization in an aortic transplantation model. Exp Mol Pathol. 2016;100:67–73.
Brown NK, Zhou Z, Zhang J, Zeng R, Wu J, Eitzman DT, et al. Perivascular adipose tissue in vascular function and disease: a review of current research and animal models. Arterioscler Thromb Vasc Biol. 2014;34:1621–30.
Patel VB, Mori J, McLean BA, Basu R, Das SK, Ramprasath T, et al. ACE2 Deficiency worsens epicardial adipose tissue inflammation and cardiac dysfunction in response to diet-induced obesity. Diabetes. 2016;65:85–95.
Bussey CE, Withers SB, Aldous RG, Edwards G, Heagerty AM. Obesity-related perivascular adipose tissue damage is reversed by sustained weight loss in the rat. Arterioscler Thromb Vasc Biol. 2016;36:1377–85.
Yuan C, Liu C, Wang T, He Y, Zhou Z, Dun Y, et al. Chikusetsu saponin IVa ameliorates high fat diet-induced inflammation in adipose tissue of mice through inhibition of NLRP3 inflammasome activation and NF-kappaB signaling. Oncotarget. 2017;8:31023–40.
Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17:250–8.
Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, et al. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35:127–36.
Zhang G, Li X, Sheng C, Chen X, Chen Y, Zhu D, et al. Macrophages activate iNOS signaling in adventitial fibroblasts and contribute to adventitia fibrosis. Nitric Oxide. 2016;61:20–8.
Yin K, You Y, Swier V, Tang L, Radwan MM, Pandya AN, et al. Vitamin D protects against atherosclerosis via regulation of cholesterol efflux and macrophage polarization in hypercholesterolemic swine. Arterioscler Thromb Vasc Biol. 2015;35:2432–42.
Carswell KA, Lee MJ, Fried SK. Culture of isolated human adipocytes and isolated adipose tissue. Methods Mol Biol. 2012;806:203–14.
McTernan PG, Anderson LA, Anwar AJ, Eggo MC, Crocker J, Barnett AH, et al. Glucocorticoid regulation of p450 aromatase activity in human adipose tissue: gender and site differences. J Clin Endocrinol Metab. 2002;87:1327–36.
Chen S, Swier VJ, Boosani CS, Radwan MM, Agrawal DK. Vitamin D deficiency accelerates coronary artery disease progression in swine. Arterioscler Thromb Vasc Biol. 2016;36:1651–9.
Yue N, Huang H, Zhu X, Han Q, Wang Y, Li B, et al. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J Neuroinflamm. 2017;14:102.
Barandier C, Montani JP, Yang Z. Mature adipocytes and perivascular adipose tissue stimulate vascular smooth muscle cell proliferation: effects of aging and obesity. Am J Physiol Heart Circ Physiol. 2005;289:H1807–13.
Ruan CC, Zhu DL, Chen QZ, Chen J, Guo SJ, Li XD, et al. Perivascular adipose tissue-derived complement 3 is required for adventitial fibroblast functions and adventitial remodeling in deoxycorticosterone acetate-salt hypertensive rats. Arterioscler Thromb Vasc Biol. 2010;30:2568–74.
Zhu DL, Herembert T, Marche P. Increased proliferation of adventitial fibroblasts from spontaneously hypertensive rat aorta. J Hypertens. 1991;9:1161–8.
Salim HM, Fukuda D, Higashikuni Y, Tanaka K, Hirata Y, Yagi S, et al. Teneligliptin, a dipeptidyl peptidase-4 inhibitor, attenuated pro-inflammatory phenotype of perivascular adipose tissue and inhibited atherogenesis in normoglycemic apolipoprotein-E-deficient mice. Vasc Pharmacol. 2017;96-98:19–25.
Ahmad F, Chung YW, Tang Y, Hockman SC, Liu S, Khan Y, et al. Phosphodiesterase 3B (PDE3B) regulates NLRP3 inflammasome in adipose tissue. Sci Rep. 2016;6:28056.
Robblee MM, Kim CC, Porter Abate J, Valdearcos M, Sandlund KL, Shenoy MK, et al. Saturated fatty acids engage an IRE1alpha-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep. 2016;14:2611–23.
Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125:665–80.
Umemoto T, Han CY, Mitra P, Averill MM, Tang C, Goodspeed L, et al. Apolipoprotein AI and high-density lipoprotein have anti-inflammatory effects on adipocytes via cholesterol transporters: ATP-binding cassette A-1, ATP-binding cassette G-1, and scavenger receptor B-1. Circ Res. 2013;112:1345–54.
Mongraw-Chaffin M, Foster MC, Kalyani RR, Vaidya D, Burke GL, Woodward M, et al. Obesity severity and duration are associated with incident metabolic syndrome: Evidence against metabolically healthy obesity from the multi-ethnic study of atherosclerosis. J Clin Endocrinol Metab. 2016;101:4117–24.
Wang L, Han J, Shan P, You S, Chen X, Jin Y, et al. MD2 Blockage protects obesity-induced vascular remodeling via activating AMPK/Nrf2. Obesity. 2017;25:1532–9.
Shi Y, Pieniek M, Fard A, O’Brien J, Mannion JD, Zalewski A. Adventitial remodeling after coronary arterial injury. Circulation. 1996;93:340–8.
Lian X, Gollasch M. A clinical perspective: contribution of dysfunctional perivascular adipose tissue (PVAT) to cardiovascular risk. Curr Hypertens Rep. 2016;18:82.
Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G, et al. Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circ Res. 2009;104:541–9.
Police SB, Thatcher SE, Charnigo R, Daugherty A, Cassis LA. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2009;29:1458–64.
Owen MK, Witzmann FA, McKenney ML, Lai X, Berwick ZC, Moberly SP, et al. Perivascular adipose tissue potentiates contraction of coronary vascular smooth muscle: influence of obesity. Circulation. 2013;128:9–18.
Noblet JN, Owen MK, Goodwill AG, Sassoon DJ, Tune JD. Lean and obese coronary perivascular adipose tissue impairs vasodilation via differential inhibition of vascular smooth muscle K+ channels. Arterioscler Thromb Vasc Biol. 2015;35:1393–400.
Payne GA, Borbouse L, Kumar S, Neeb Z, Alloosh M, Sturek M, et al. Epicardial perivascular adipose-derived leptin exacerbates coronary endothelial dysfunction in metabolic syndrome via a protein kinase C-beta pathway. Arterioscler Thromb Vasc Biol. 2010;30:1711–7.
Ruan CC, Ge Q, Li Y, Li XD, Chen DR, Ji KD, et al. Complement-mediated macrophage polarization in perivascular adipose tissue contributes to vascular injury in deoxycorticosterone acetate-salt mice. Arterioscler Thromb Vasc Biol. 2015;35:598–606.
Agabiti-Rosei C, Favero G, De Ciuceis C, Rossini C, Porteri E, Rodella LF, et al. Effect of long-term treatment with melatonin on vascular markers of oxidative stress/inflammation and on the anticontractile activity of perivascular fat in aging mice. Hypertens Res. 2016;40:41–50.
Pelham CJ, Drews EM, Agrawal DK. Vitamin D controls resistance artery function through regulation of perivascular adipose tissue hypoxia and inflammation. J Mol Cell Cardiol. 2016;98:1–10.
Liu PS, Lin YW, Lee B, McCrady-Spitzer SK, Levine JA, Wei LN. Reducing RIP140 expression in macrophage alters ATM infiltration, facilitates white adipose tissue browning, and prevents high-fat diet-induced insulin resistance. Diabetes. 2014;63:4021–31.
Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213:617–29.
Rossi C, Santini E, Chiarugi M, Salvati A, Comassi M, Vitolo E, et al. The complex P2X7 receptor/inflammasome in perivascular fat tissue of heavy smokers. Eur J Clin Invest. 2014;44:295–302.
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88.
Wang WR, Li TT, Jing T, Li YX, Yang XF, He YH, et al. SIRT1 regulates the inflammatory response of vascular adventitial fibroblasts through autophagy and related signaling pathway. Cell Physiol Biochem. 2017;41:569–82.
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Sciences Foundation of China (81100213, 81470569), the Natural Science Foundation of Hunan Province (2018JJ2341), the Innovation Foundation for Postgraduate of Hunan Province (CX2017B550), the Innovation Foundation for Undergraduates of Hunan Province (2017-336), the Scientific Research Foundation for Doctor in University of South China (2015XQD49), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars in University of South China (2015XQD55).
Author contributions
XZ and KY designed the research; XZ, H-wZ, X-jD, and KY performed research; XZ, H-nC, Y-xT, AOJ, J-nQ, and KY analyzed data; A-pW and VP contributed new reagents or analytic tools; XZ and KY wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Zhu, X., Zhang, Hw., Chen, Hn. et al. Perivascular adipose tissue dysfunction aggravates adventitial remodeling in obese mini pigs via NLRP3 inflammasome/IL-1 signaling pathway. Acta Pharmacol Sin 40, 46–54 (2019). https://doi.org/10.1038/s41401-018-0068-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0068-9
Keywords
This article is cited by
-
Impact of Perivascular Adipose Tissue on Neointimal Formation Following Endovascular Placement
Journal of Cardiovascular Translational Research (2024)
-
Bergapten inhibits NLRP3 inflammasome activation and pyroptosis via promoting mitophagy
Acta Pharmacologica Sinica (2023)
-
Extracellular vesicles in vascular remodeling
Acta Pharmacologica Sinica (2022)
-
Influence of reservoir lithology on porous flow resistance of gas-bearing tight oil reservoirs and production forecast
Journal of Petroleum Exploration and Production Technology (2022)
-
Involvement of the secreted protein Metrnl in human diseases
Acta Pharmacologica Sinica (2020)
