
Abstract
We previously linked TSHZ3 haploinsufficiency to autism spectrum disorder (ASD) and showed that embryonic or postnatal Tshz3 deletion in mice results in behavioral traits relevant to the two core domains of ASD, namely social interaction deficits and repetitive behaviors. Here, we provide evidence that cortical projection neurons (CPNs) and striatal cholinergic interneurons (SCINs) are two main and complementary players in the TSHZ3-linked ASD syndrome. In the cerebral cortex, TSHZ3 is expressed in CPNs and in a proportion of GABAergic interneurons, but not in cholinergic interneurons or glial cells. In the striatum, TSHZ3 is expressed in all SCINs, while its expression is absent or partial in the other main brain cholinergic systems. We then characterized two new conditional knockout (cKO) models generated by crossing Tshz3flox/flox with Emx1-Cre (Emx1-cKO) or Chat-Cre (Chat-cKO) mice to decipher the respective role of CPNs and SCINs. Emx1-cKO mice show altered excitatory synaptic transmission onto CPNs and impaired plasticity at corticostriatal synapses, with neither cortical neuron loss nor abnormal layer distribution. These animals present social interaction deficits but no repetitive patterns of behavior. Chat-cKO mice exhibit no loss of SCINs but changes in the electrophysiological properties of these interneurons, associated with repetitive patterns of behavior without social interaction deficits. Therefore, dysfunction in either CPNs or SCINs segregates with a distinct ASD behavioral trait. These findings provide novel insights onto the implication of the corticostriatal circuitry in ASD by revealing an unexpected neuronal dichotomy in the biological background of the two core behavioral domains of this disorder.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) includes a heterogeneous group of neurodevelopmental pathologies the diagnosis of which is based exclusively on behavioral criteria. The two behavioral domains that are selected by the DSM-5 are: (i) deficit in social communication and (ii) restrictive, repetitive patterns of behavior, interests, or activities [1]. These domains also emerge from factor analyses of the 13 available diagnostic instruments in patients [2] and in a model that aligns mouse and patient features [3]. More than 900 genes have been liked to ASD [4], among which >100 impact synaptic functions or interact with genes involved in neuronal development [5]. As a possible neurobiological substrate, clinical and animal studies point to molecular, neurodevelopmental and functional changes of deep-layer cortical projection neurons (CPNs), in particular those of layer 5 (L5) forming the corticostriatal pathway [6,7,8,9]. In this context, we have linked heterozygous TSHZ3 gene deletion to a syndrome characterized by neurodevelopmental disorders including autistic behavior, cognitive disabilities and language disturbance, with some patients also showing renal tract abnormalities [10]. TSHZ3 encodes the highly conserved, zinc-finger homeodomain transcription factor TSHZ3, and has been identified in networks of human neocortical genes highly expressed during late fetal development, which are involved in neurodevelopmental and neuropsychiatric disorders [9, 10]. It is now ranked as a high-confidence risk gene for ASD (https://gene.sfari.org/database/human-gene/TSHZ3#reports-tab). In human and mouse, high TSHZ3 gene or protein expression is detectable in the cortex during pre- and postnatal development [11]. We showed that heterozygous deletion of Tshz3 (Tshz3+/lacZ) and early postnatal conditional knockout (KO) using the Camk2a-Cre promoter (Camk2a-cKO mice) lead to ASD-relevant behavioral deficits paralleled by changes in cortical gene expression and corticostriatal synaptic abnormalities [10, 12]. These data suggest that Tshz3 plays a crucial role in both pre- and postnatal brain development and functioning, and point to CPNs, and in particular to the corticostriatal pathway, as a main player in the Tshz3-linked ASD syndrome. In the mouse striatum, TSHZ3 is not expressed in striatal spiny projection neurons (SSPNs), which represent >90% of striatal neurons, but in a small population of cells that are likely interneurons [10]. We [13] and others [14, 15] identified these cells as being mainly striatal cholinergic interneurons (SCINs), whose implication in ASD has been suggested by some studies [16, 17]. We also showed that the Camk2a-Cre transgene is unexpectedly expressed in the SCIN lineage, where it efficiently elicits the deletion of Tshz3 in Camk2a-Cre mice [13]. Together, these data demonstrate that, within the corticostriatal circuitry, Tshz3 is deficient in both CPNs and SCINs, in Tshz3+/lacZ heterozygous [10] as well as in Camk2a-cKO mice [12], which both show the full repertoire of ASD-like behavioral defects. Here, we aimed at investigating the respective contribution of CPNs and SCINs to the pathophysiology of Tshz3-linked ASD using targeted conditional deletion of this gene, and provided evidence for the complementary implication of these two neuronal populations in the ASD-related core features.
Results
Conditional deletion of Tshz3 in CPNs
High levels of Tshz3 gene or TSHZ3 protein expression are detectable in the mouse cortex during pre- and postnatal development [10, 11]. In the adult cerebral cortex, TSHZ3 is detected in the great majority of CPNs [10]. Here, performing immunostaining for beta-galactosidase (ß-Gal) to report the expression of Tshz3, we show that Tshz3 is also expressed in 26.8 ± 1.3% (n = 20 sections from 3 mice) of cortical GABAergic interneurons, as evidenced using Tshz3+/lacZ;GAD67-GFP mice (Fig. S1a); the percentage of dually labeled cells is significantly higher in the deep vs. upper cortical layers (36.5 ± 1.6% vs. 20.0 ± 1.0%, respectively; P < 0.0001, Student’s t test). In contrast, ß-Gal is not detectable in cortical choline acetyltransferase (CHAT) positive neurons (Fig. S1b), Olig2-positive oligodendrocytes (Fig. S1c) and GFAP-positive astrocytes (Fig. S1d, e). To address the role of Tshz3 in CPNs, Tshz3flox/flox mice were crossed with Emx1-Cre (empty spiracle homeobox 1) mice (Emx1-cKO). The Emx1-Cre mouse expresses the Cre-recombinase in the progenitors of cortical glutamatergic projection neurons (i.e., CPNs) and glial cells from embryonic day 9 (E9), but neither in those of cortical GABAergic neurons, nor of striatal interneurons, including cholinergic ones [18]. Therefore, in the corticostriatal circuitry of Emx1-cKO mice, Tshz3 should be specifically lost in CPNs. Compared to control, Emx1-cKO mice show a drastic reduction of Tshz3 mRNA levels and of the density of TSHZ3-positive cells in the cerebral cortex, showing the efficacy of the deletion, while the density of striatal cells expressing TSHZ3 is unchanged (Fig. 1a–c). Despite the loss of Tshz3 expression in the vast majority of CPNs, the density of NeuN-positive cells is unchanged (Fig. S2a, b), showing no neuronal loss; in addition, neither the pattern of expression of layer-specific CPN markers, namely CUX1 for L2-4 and BCL11B for L5-6, nor the density of cells expressing these markers is affected (Fig. S2c, d), indicating no major alteration in cortical layering. However, spine density of L5 CPNs from Thy1-GFP-M;Emx1-cKO mice is significantly reduced compared to Thy1-GFP-M control mice (Fig. 1d, e). By crossing Emx1-cKO with GAD67-GFP mice, we show that cortical GABAergic neurons still express TSHZ3 (Fig. 1f), confirming the specificity of Tshz3 deletion in CPNs. To study whether Tshz3 loss in CPNs could indirectly affect cortical GABAergic interneurons, we compared GAD67-GFP control mice (Control-GAD67-GFP) to Emx1-cKO-GAD67-GFP mutant mice. No significant changes in the number of GABAergic interneurons (Control-GAD67-GFP: 140.7 ± 4.9, n = 37 sections from 5 mice; Emx1-cKO-GAD67-GFP: 144.6 ± 6.1, n = 41 sections from 7 mice; P = 0.624, Student’s t test) and in their distribution are found (Fig. S3a, b). CHAT immunostaining on striatal slices in Emx1-cKO mice also shows no significant modification of the density of SCINs (Fig. S3c, d). Overall, these data show that Tshz3 is specifically lost in CPNs of Emx1-cKO mice, with no major consequences on the number and layer distribution of CPNs and GABAergic neurons, but with a significant reduction of L5 CPN dendritic spine density, suggesting altered synaptic communication.

a Coronal brain sections from control and Emx1-cKO mice immunostained for TSHZ3. Scale bar 250 µm. b Tshz3 mRNA relative expression in the cortex of control and Emx1-cKO mice measured by RT-qPCR (4 cortices per group; *P < 0.05, Mann–Whitney test). c TSHZ3-positive cell density in control and Emx1-cKO mice in cortical layers (cell counts performed using frames of 400 μm width spanning from L1 to L6 in 9 sections from 3 control mice and 18 sections from 3 Emx1-cKO mice; **P < 0.01, Mann–Whitney test) and in the whole striatal surface (cell counts performed in the whole dorsal striatum in 6 sections from 3 control mice and 7 sections from 3 Emx1-cKO mice; P = 0.1496, Mann–Whitney test). d Representative confocal images showing dendritic spines of GFP-positive L5 neurons from control (Thy1-GFP-M) and Emx1-cKO (Thy1-GFP-M;Emx1-cKO) mice. Scale bar 5 µm. e Density of different classes of dendritic spines in control (1688 spines/1135 µm) and Emx1-cKO (1308 spines/1220 µm) mice. f Coronal brain sections from GAD67-GFP control and Emx1-cKO-GAD67-GFP mice immunostained for TSHZ3. Lower panels are magnifications of the framed areas in the upper images. Scale bars 100 µm. *P < 0.02, ***P < 0.001 and ****P < 0.0001, Student’s t test. Data in (b) and (c) are expressed as medians with interquartile range; data in (e) are expressed as means + SEM.
Cortical excitatory synaptic transmission and corticostriatal synaptic plasticity in Emx1-cKO mice
We then examined whether the loss of Tshz3 in CPNs affects their electrophysiological properties and synaptic transmission. We recorded L5 CPNs, which are at the origin of the corticostriatal pathway, in slices from Emx1-cKO mice. They show no significant changes in their membrane properties and excitability compared to control (Fig. S4a–e). Action potential (AP)-dependent glutamate release onto L5 CPNs, evaluated by measuring paired-pulse ratio, is also unaffected (Fig. S4f). However, both NMDA/AMPA ratio (Fig. S4g) and NMDA-induced currents (Fig. S4h) are significantly reduced, suggesting decreased NMDA receptor-mediated signaling in Emx1-cKO mice. The amplitude of AMPA receptor-mediated miniature excitatory postsynaptic currents (mEPSCs) is similar in control and Emx1-cKO mice (Fig. S4i), further arguing for the implication of NMDA but not AMPA receptors. Conversely, mEPSC frequency is reduced (Fig. S4i), suggesting decreased AP-independent glutamate release onto L5 CPNs and/or reduced number of active excitatory synapses in Emx1-cKO mice, consistent with the decreased spine density on L5 CPNs (Fig. 1d, e).
SSPNs recorded in slices from Emx1-cKO mice show electrophysiological properties (Fig. S5a–d) and basal corticostriatal synaptic transmission (Fig. S5e–g) similar to control. However, both long-term potentiation (LTP) and long-term depression (LTD) at corticostriatal synapses are abolished in Emx1-cKO mice (Fig. 2). These findings suggest that the loss of Tshz3 in CPNs does not impact their electrophysiological properties, but profoundly affects cortical synaptic transmission and corticostriatal synaptic plasticity, confirming a critical role of Tshz3 in the functioning of the corticostriatal circuit.

LTP (a) and LTD (b) are lost in Emx1-cKO mice. Left graphs: time-course (normalized EPSC amplitude expressed as means ± SEM; gray bars represent induction protocols; 2-way ANOVA from 15 to 30 min; LTP: F(1,211) = 44.8, P < 0.0001; LTD: F(1,216) = 153.2, P < 0.0001). Traces show EPSCs before (black) and after (gray) LTP and LTD induction protocols. Right graphs: EPSC amplitude at 15–30 min (medians with interquartile range; Wilcoxon matched-pairs signed rank test vs. baseline: *P < 0.05, **P < 0.01, ns non-significant; Mann–Whitney test: $$P < 0.01). Data obtained from 17 SSPNs of control and 14 of Emx1-cKO mice.
Conditional deletion of Tshz3 in cholinergic neurons
Dual immunodetection of CHAT and ß-Gal in Tshz3+/lacZ mice was performed to analyze the expression of Tshz3 in brain cholinergic neuron populations. This was preferred to dual immunodetection of CHAT and TSHZ3 since the tissue fixation conditions for obtaining optimal detection of each protein are different, and also because TSHZ3 immunodetection provides weaker labeling and higher background than ß-Gal immunodetection. As reported previously [13], virtually all SCINs, both in the dorsal striatum and the nucleus accumbens, express Tshz3 (Fig. S6a, h). In contrast, there are no or a little proportion (<30%) of ß-Gal-positive cells within CHAT-positive neurons in the components of the basal forebrain cholinergic system (medial septal nucleus, diagonal band nuclei, nucleus basalis of Meynert and substantia innominata) (Fig. S6a–d, h). SCINs thus represent the major population of Tshz3-expressing cells among the forebrain cholinergic neurons. In addition, there is almost no co-expression of ß-Gal and CHAT in the pedunculopontine (Fig. S6e, f, h) and laterodorsal tegmental nuclei (Fig. S6g, h), which are known to provide cholinergic afferents to several brain areas including the striatum [19]. Among the other brainstem nuclei, co-expression ranges from poor to extensive, as illustrated in the parabigeminal nucleus and the oculomotor nucleus, respectively (Fig. S6e, f, h).
We previously reported that around 90% of the Tshz3-expressing cells in the striatum are SCINs [13]. Using Tshz3+/lacZ;GAD67-GFP mice, we found here that GABAergic neurons constitute 9.7 ± 1.3% (n = 21 sections from 3 mice) of the striatal Tshz3 population. Therefore, in the striatum, Tshz3 is expressed mainly in SCINs and in a very small fraction of GABAergic neurons; the latter are likely to be interneurons, as we previously reported that SSPNs do not express Tshz3 [10]. To address the role of Tshz3 in cholinergic neurons, Tshz3flox/flox mice were crossed with Chat-Cre mice (Chat-cKO model). CHAT is expressed in the brain from early embryonic development and as soon as E18.5 in the striatum [20]. Chat-cKO mice show a marked decrease in the density of TSHZ3-positive cells, which confirms the loss of Tshz3 in SCINs (Fig. 3a, b). The 20% TSHZ3-positive cells still observed in these mice may include a fraction of SCINs in which the Cre was inefficient and the above-mentioned GABAergic interneurons. The loss of Tshz3 expression in Chat-cKO mice does not affect the number of striatal CHAT-positive cells either in the dorsal striatum (Fig. 3c, d) or in the nucleus accumbens (35.8 ± 1.1 vs. 33.8 ± 1.4 CHAT-positive cells/µm2 in control vs. Chat-cKO, respectively; 3 mice per genotype, 15 and 17 sections, respectively; P = 0.285, Student’s t test). This result was confirmed using Chat-Cre;Ai14Flox/+ control mice (Chat-Cre;Rosa26-STOP-Tomato) to visualize SCINs in the presence or absence of Tshz3 (Fig. 3e, f).

a Coronal brain sections from control and Chat-cKO mice immunostained for TSHZ3 and counterstained with DAPI. Scale bar 500 µm. b Number of TSHZ3-positive cells in the striatum of control and Chat-cKO mice (15 sections from 3 control mice; 11 sections from 3 Chat-cKO mice; ****P < 0.0001, Student’s t test). c Coronal brain sections from control and Chat-cKO mice stained for CHAT. Scale bar 200 µm. d Number of CHAT-positive SCINs in the striatum of control and Chat-cKO mice (40 sections from 9 control mice; 53 sections from 11 Chat-cKO mice; P = 0.6373, Student’s t test). e Representative images showing tdTomato fluorescence detection (red) in SCINs of Chat-Cre;Ai14Flox/+ control and Chat-cKO;Ai14Flox/+ mutant mice (coronal sections). cx, cerebral cortex; st, striatum. Nuclei are counterstained with DAPI. Scale bar 500 µm. f Number of tdTomato-positive cells in the striatum of Chat-Cre;Ai14Flox/+ control and Chat-cKO;Ai14Flox/+ mutant mice (14 sections from 3 control mice; 12 sections from 3 Chat-cKO;Ai14Flox/+ mice; P = 0.7773, Student’s t test). Data in (b), (d), and (f) are expressed as percent of mean control value and represented as means + SEM.
Tshz3 loss and SCIN electrophysiological properties
We then determined the effect of Tshz3 loss in cholinergic neurons on the electrophysiological properties of SCINs in the dorsal striatum. In acute brain slices, SCINs are easily recognizable among the other striatal neurons due to their larger soma [21]. Moreover, they are the only autonomously active cells, firing APs with either a regular, irregular, or bursting pattern [22, 23]. SCINs also show a characteristic depolarizing voltage sag in response to the injection of negative current pulses due to the activation of the nonspecific Ih cation current mediated by HCN channels, which largely contributes to the spontaneous AP discharge characterizing these neurons [23,24,25]. To test a possible effect of Tshz3 loss on these SCIN properties, we measured the mean frequency of spontaneous AP discharge, its regularity [expressed as the coefficient of variation (CV) of the inter-AP intervals], and the amplitude of the sag [expressed as voltage sag ratio (VSR)] in SCINs from Chat-cKO mice and control littermates (Fig. 4a–c). We found that SCINs recorded from Chat-cKO mice show a significant reduction of both VSR (Fig. 4d) and spontaneous firing frequency (Fig. 4e), as well as an increased CV of inter-AP intervals that suggests a less regular discharge activity (Fig. 4f). The resting membrane potential at steady state is similar between control vs. Chat-cKO SCINs (46.64 ± 0.68 vs. 45.65 ± 0.64 mV, 56 vs. 86 SCINs, respectively; P = 0.305, Student’s t test), while the current–voltage relationship reveals a slight but significant increase of input resistance in Chat-cKO SCINs vs. control, calculated as the slope of the linear best fit (Fig. 4g; 125.7 ± 4.5 vs. 107.5 ± 4.0 MΩ, respectively; F(1,911) = 8.816, P = 0.0031). Overall, SCINs in Chat-cKO mice have a lower frequency and less regular AP discharge activity possibly due to a reduced Ih, which could impair the physiological cholinergic tone and affect the role these neurons play in modulating striatal function.

a Simplified scheme of the corticostriatal circuitry with the recording patch-clamp pipette placed on a SCIN. TSHZ3-expressing neurons are blue (L1-6, cortical layers 1–6; cc, corpus callosum; st, striatum). b Sample traces obtained from a representative control SCIN: note the prominent voltage sag in response to −200 and −120 pA hyperpolarizing currents, and the AP firing during a + 100 pA depolarizing current (1st line), as well as the sustained and regular firing in cell-attached (CA) and whole-cell (WC) configuration (2nd and 3rd line, respectively). c Sample traces obtained from a representative Chat-cKO SCIN: compared to (b), note the smaller voltage sag as well as the less regular, lower frequency spontaneous firing. b, c The values of voltage sag ratio (VSR) of the response to −120 pA current injection (arrowhead), as well as the frequency and coefficient of variation (CV) of spontaneous firing of these samples, are reported; spikes have been cut; calibration bars are the same in (b) and (c). Compared to control, SCINs from Chat-cKO mice show a significant reduction of mean voltage sag ratio (d) and frequency of spontaneous discharge (e), while the CV of their inter-AP interval is increased (f) meaning that their spontaneous firing is more irregular. The number of recorded SCINs in (d)–(f) is reported in the graphs. g Current–voltage relationship obtained from 51 control and 62 Chat-cKO SCINs, and the linear best fit to calculate input resistance (see Results). *P < 0.05, ***P < 0.001, Student’s t test; data in (d–f) are expressed as box and whiskers (25th–75th and 5th–95th percentiles, respectively), where bar = median and cross = mean; data in (g) are expressed as means ± SEM.
Conditional deletion of Tshz3 in CPNs or in cholinergic neurons segregates the two core behavioral domains of ASD
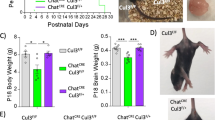
As altered physiology of the corticostriatal circuit is postulated to play a central role in the pathophysiology of ASD, we characterized Emx1-cKO and Chat-cKO mice for ASD-relevant phenotype using a battery of behavioral tests [3] after having verified that these mice do not present visual, auditory and olfactory impairment (Fig. S7). They were tested for deficits in social behavior, the first core feature of ASD, as well for stereotyped/repetitive patterns of behavior and for restricted field of interests, which are subcategories of the second ASD core feature. During the habituation phase in the two-chamber test, both Emx1-cKO and Chat-cKO mice show no significant differences in their exploration of the lured boxes as compared to their respective controls (P = 0.14, η2 = 0.12, P = 0.84, η2 = 0.002, respectively Fig. 5a). However, Emx1-cKO but not Chat-cKO mice show impaired social relationships (Fig. 5). Emx1-cKO mice have less preference than their controls for a conspecific (sociability, Fig. 5b) and for an unfamiliar male (social novelty, Fig. 5c), the interaction between genotype and box content being large in each case, as shown by the effect size that exceeds the typical range of variation (Fig. 5d). Conversely, Chat-cKO but not Emx1-cKO mice present more stereotyped or repetitive patterns of behavior than their controls, as shown by the marble-burying score, time burrowing in a new cage, stereotyped dips on a hole board, and number of leanings in an open field (Fig. 6a–d), with a large effect size (Fig. 6e). Restricted field of interest is impacted neither in Emx1-cKO nor in Chat-cKO mice (Fig. S8a–c).

a Nose pokes during habituation, used as covariate for mixed-design ANCOVAs in (b) and (c). b Sociability measured as the number of nose pokes against a C57BL/6J male mouse or a lure. Emx1-cKO mice (n = 9) vs. control (n = 8): Finteraction(1,14) = 18.59, P < 0.001. Chat-cKO mice (n = 12) vs. control (n = 9): Finteraction(1,18) = 0.55, P = 0.47. c Interest in social novelty measured as the number of nose pokes against the same C57BL/6J or a SWR mouse. Emx1-cKO vs. control: Finteraction(1,14) = 19.70, P < 0.001. Chat-cKO vs. control: Finteraction(1,18) = 0.02, P = 0.89. d Sizes of the difference for Emx1-cKO (partial η2 = 0.57 and 0.59 for (b) and (c), respectively) and Chat-cKO mice (partial η2 = 0.03 and 0.001, respectively) vs. their respective control. Data in (a–c) are expressed as means ± SEM. ***P < 0.001.

a Marble-burying, Emx1-cKO, Student’s-t(15) = 1.0, P = 0.33; Chat-cKO, t(19) = 3.97, P = 0.001. b Time burrowing, Emx1-cKO, t(15) = 1.16, P = 0.13); Chat-cKO, t(19) = 3.225, P = 0.004. c Stereotyped dips, Emx1-cKO, Finteraction(1,15) = 0.08, P = 0.87 (with non-stereotyped dips as covariate, P = 0.76); Chat-cKO, Finteraction(1,19) = 32.69, P = 0.00001 (with non-stereotyped dips as covariate, P = 0.24). d Number of leanings, Emx1-cKO, t(15) = 1.51, P = 0.15; Chat-cKO, t(18) = 4.35, P = 0.0003. e Sizes of the difference in Emx1-cKO (η2 = 0.06, 0.08, 0.13 in (a), (b) and (d), respectively, and partial η2 = 0.01 in (c) and in Chat-cKO (η2 = 0.45, 0.35, 0.51 in (a), (b) and (d), respectively, and partial η2 = 0.63 in (c). Sample size of (a–d) were: 9, 9, 9, and 12 for Emx1-cKO; 8, 8, 9, and 11 for their controls; 12, 12, 12, and 11 for Chat-cKO; 9, 9, 11, and 8 for their controls. Data in (a–d) are expressed as means + SEM. **P < 0.01 ***P < 0.001.
Finally, since impairment of motor control and learning have been reported in children with ASD [26, 27], we checked Emx1-cKO and Chat-cKO mice for motor and cognitive deficits. Hind paw coordination is impaired in Chat-cKO but not in Emx1-cKO mice (Fig. S8d, e), while spatial learning ability is unaffected in both models (Fig. S8f-i).
Discussion
Previous studies showed that haploinsufficiency or postnatal deletion of Tshz3 results in ASD-relevant behavioral deficits and suggested altered function of the corticostriatal circuitry as a possible substrate [10, 12]. The present findings point to SCINs as an additional player in the Tshz3-linked ASD syndrome. They also provide evidence that targeted conditional deletion of Tshz3 in either CPNs (Emx1-cKO) or cholinergic neurons (Chat-cKO) segregates the two core behavioral traits used to diagnose ASD, respectively social behavior deficits and repetitive behavioral patterns, suggesting that alterations in CPNs and in SCINs contribute in a complementary manner to the repertoire of behavioral deficits linked to Tshz3 deficiency. Restricted field of interest, which defines a sub-category of the second ASD domain, was observed neither in Emx1-cKO nor in Chat-cKO mice. This suggests that the expression of this deficit in the previously characterized models of Tshz3 deletion may involve additional players, such as the cortical and striatal GABAergic interneurons expressing Tshz3 whose specific role remains to be determined, and/or result from the combined dysfunction of CPNs and SCINs due to the loss of Tshz3 in both these neuronal types. Learning ability was impacted neither by Tshz3 postnatal deletion [12], nor in Emx1-cKO and Chat-cKO models.
Among the multiplicity of circuits involved in social behavior, the literature points out the crucial role of the cortex [28, 29]. Here we focused on the sensorimotor cortex and the dorsal striatum as a model circuit that has been characterized in several ASD mouse models. However, we cannot exclude that dysfunction in the prefrontal cortex-nucleus accumbens circuitry may also be implicated in the described ASD-related phenotype, since Tshz3 is expressed in all cortical areas and in SCINs of both the dorsal striatum and the nucleus accumbens. Corticostriatal and striatal circuit dysfunctions are associated with ASD features, both in patients and in mouse models, with CPNs and SSPNs being highly impacted by mutations of ASD-linked genes [7, 8, 10, 12, 30, 31]. There is however increasing evidence incriminating interneuron populations of the cortex and the striatum in ASD [32]. Here, we show that, in the cortex, the ASD-related Tshz3 gene is expressed not only in CPNs but also in about a third of GABAergic interneurons, while not in cholinergic interneurons. In contrast, in the striatum, Tshz3-expressing cells are for their vast majority cholinergic interneurons [13] and comprise a minority of GABAergic interneurons. To disentangle the role of CPNs from that of interneurons in the ASD symptoms linked to Tshz3 deficiency, we generated and characterized Emx1-cKO mice. We confirmed the specificity of Tshz3 deletion in CPNs within the corticostriatal circuit in this model, Tshz3 expression in cortical and striatal interneurons being maintained. In addition, no change in the numbers and positioning of these interneurons were detected. Interestingly, we found that Emx1-cKO mice specifically exhibit impaired social behavior and that this deficit co-segregates with altered NMDA receptor-mediated transmission in the cortex and loss of plasticity at corticostriatal synapses. Corticostriatal synaptic plasticity has been deeply characterized, but discrepancies concerning its induction protocols and the underlying molecular and cellular mechanisms [33] make it difficult to univocally interpret our results. However, since both LTD and LTP expression require pre- and postsynaptic changes, their disruption in Emx1-cKO mice could be attributable to cortical circuitry defects, such as the observed decrease of NMDA receptor activity in L5 CPNs [34,35,36]. Our findings are also in line with studies substantiating the involvement of NMDA receptor dysfunction in social deficits associated with ASD in rodent models as well as in patients [37, 38]. Finally, consistent with the literature linking ASD with changes of dendritic spine density [39], we evidence decreased spine density in L5 CPNs of Emx1-cKO mice, as in our previous model [12]. Overall, these data indicate that the loss of Tshz3 in CPNs induces morphofunctional changes in these neurons and deeply affects corticostriatal plasticity, which might result in altered processing of cortical information and account for the observed social behavior deficits.
We also investigated the contribution of cholinergic neurons in the pathophysiology of Tshz3-linked ASD. We show that TSHZ3 is expressed in virtually 100% of SCINs of both the dorsal striatum and the nucleus accumbens, while its expression is absent or partial in the other main brain cholinergic systems. Despite their low number, SCINs have morphofunctional features that place them as key modulators of striatal microcircuits. They play a crucial role in movement control, attentional set-shifting, habit-mediated and goal-directed behavior, and selection of appropriate behavioral responses to changes in environmental contingencies, conferring behavioral flexibility [40,41,42,43,44]. These interneurons are also involved in basal ganglia-related pathologies such as dystonia, Parkinson’s and Huntington’s disease, Tourette’s syndrome, obsessive compulsive disorder and drug addiction [45,46,47,48,49,50]. In contrast, despite the array of data pointing to basal ganglia and to cholinergic transmission abnormalities in ASD and in ASD models [16, 51,52,53,54,55], to date there is little evidence showing the specific involvement of SCINs: the partial depletion of both SCINs and fast-spiking GABAergic interneurons produces stereotypy and impaired social behavior in male mice [17], while total elimination of SCINs results in perseverative behavior that extends to social behavior, rather reminiscent of neuropsychiatric conditions as Tourette’s syndrome or obsessive-compulsive disorder [56]. The present work reveals that targeted Tshz3 deletion in CHAT-expressing neurons leads to robust stereotyped and repetitive patterns of behavior without impacting social behavior. Given the literature associating drug-induced stereotypies with abnormalities in striatal cholinergic signaling [57,58,59], and the co-expression of CHAT and TSHZ3 in SCINs but not in brainstem cholinergic neurons that are known to project to the striatum [19], this behavioral deficit is likely attributable to SCINs. The lack of social behavior impairment is surprising, as altered striatal physiology is assumed to be a central node mediating repetitive motor behaviors and also a range of ASD-associated behaviors, including social deficit [30]. However, the studies examining the specific involvement of SCINs in several neurodevelopmental disorders have associated altered sociability with the depletion of this interneuron population [17, 56, 60], which is not observed in Chat-cKO mice. Whereas the number of SCINs in these mice is unchanged, suggesting that their generation and viability are not affected, we evidenced modifications in their firing activity and electrophysiological membrane properties. This finding is an addition to the increasing amount of data stressing the complex implication of SCINs in health and diseases [61]. How the selective loss of Tshz3 in SCINs leads to these electrophysiological changes, what are their molecular bases and what are the consequences on striatal cholinergic signaling still need to be determined. However, SCINs are important modulators of the two populations of SSPNs forming the ”direct” and ”indirect” pathways by which the striatum regulates basal ganglia outflow, whose balanced activity is determinant for appropriate action selection [42, 62]. Thus, the changes in SCIN properties observed here could alter the way they normally respond to salient stimuli and/or reward-associated cues, thereby the way they modulate the transfer of cortical information through the striatum [40, 41, 63], as observed for example after targeted deletion of the transcription factor Er81 in SCINs [44]. This could underlie the increased stereotyped behaviors observed in Chat-cKO mice and, possibly, also in Tshz3+/lacZ [10], as well as in CamK2a-cKO [12] in which we recently showed that Tshz3 is lost also in SCINs [13]. Finally, Chat-cKO mice do not show basal exploration deficit, similarly to Emx1-cKO mice, but present impaired hind paw coordination, which is in line with motor deficiencies frequently associated with ASD [64] and with a study linking partial SCIN ablation with motor incoordination [65]. Although TSHZ3 is expressed in about 25% of cholinergic neurons of the nucleus basalis of Meynert and the substantia innominata, the similarity of spatial learning curves of control and Chat-cKO mice suggests minor impact of Tshz3 deletion on the function of the basal forebrain cholinergic system, which is deeply involved in learning and memory processes [66].
In conclusion, this study shows that the conditional loss of the ASD-related gene Tshz3 in CPNs and SCINs does not affect the numbers of these neurons but induces changes in their electrophysiological and synaptic properties, paralleled by specific ASD-like behavioral defects. It provides new experimental evidence that the two behavioral domains used to diagnose ASD are independent domains that can be triggered by dysfunction in distinct neuronal subtypes. These findings may open the road to domain-specific pharmacological and behavioral therapies.
Materials and methods
Mouse strains and genotyping
The Tshz3lacZ, Tshz3flox/flox, Emx1-Cre, Chat-Cre, Rosa26-STOP-lacZ and Ai14 (Rosa26-STOP-Tomato), GAD67-GFP, and Thy1-GFP mouse lines have been described previously [10, 12, 18, 67,68,69,70,71,72]. Male heterozygous Cre mice were crossed with female Tshz3flox/flox to generate the two Tshz3 conditional knockout (cKO) mice models: Emx1-cKO and Chat-cKO [18, 70]. Littermate Emx1-Cre−/− and Chat-Cre−/− mice were used as respective controls. Animals carrying the Tshz3flox allele and Tshz3∆ allele were genotyped as described previously [12]. Experimental procedures were in agreement with the recommendations of the European Communities Council Directive (2010/63/EU). They have been approved by the “Comité National de Réflexion Ethique sur l’Expérimentation Animale n°14” and the project authorization delivered by the French Ministry of Higher Education, Research and Innovation. (ID numbers 57-07112012, 2019020811238253-V2 #19022 and 2020031615241974-V5 #25232). No randomization was used and no animals or samples were excluded from the different analyses performed.
Immunohistochemistry and histology
All stains were processed on coronal brain sections of postnatal day (P) 28–34 mice. Immunostaining for TSHZ3 alone was performed on cryostat sections of brains immediately removed after anesthesia (ketamine + xylazine, 100 + 10 mg/kg, respectively, i.p.) and frozen in dry ice until use. Before incubation with the antibodies, sections were fixed with 4% paraformaldehyde (PFA) for 15 min, then washed twice for 5 min in PBS. For TSHZ3 immunostaining and GFP detection, GAD67-GFP mice were anesthetized (see above) and transcardially perfused with PBS. Brains were immediately dissected out, post-fixed by immersion 2 h in 4% PFA in PBS, placed in 30% sucrose in PBS overnight and frozen in dry ice until sectioning. For the other stains, mice were anesthetized (see above) and transcardially perfused with 4% PFA in 0.1 M phosphate buffer. Brains were removed and post-fixed in 4% PFA for at least 2 h before cryostat sectioning (40 µm-thick). Brain sections were washed with PBS and blocked in PBST (0.3% Triton X-100 in PBS) with 5% BSA for 1 h at room temperature. Sections were then incubated in primary antibody diluted in blocking solution (PBST, 1% BSA) overnight at 4 °C with the following primary antibodies: mouse anti-NeuN (1:500, Millipore, Mab377), rat anti-BCL11B (1:1,000, Abcam, ab18465), goat anti-CHAT (1:100, Millipore, AB144P), rabbit anti-ß-Galactosidase (1:1,000, Cappel, 599762), goat anti-CDP/CUX1 (1:200, Santa Cruz Biotechnology, C20, SC6327) and guinea-pig anti-TSHZ3 (1:2,000; ref. [67]). Sections were then washed with PBS three times and incubated overnight at 4 °C in secondary antibodies diluted 1:1,000 in blocking solution: donkey anti-rabbit Cy3, donkey anti-guinea pig Cy3 and donkey anti-goat Cy3 (Jackson ImmunoResearch Laboratories) and goat anti-mouse Alexa Fluor 488, goat anti-rat Alexa Fluor 555 and donkey anti-goat Alexa Fluor 488 (Life Technologies). Sections were counterstained by 5 min incubation in 300 µM DAPI intermediate solution (1:1,000, Molecular Probes, Cat# B34650). Section were then washed with PBS three times, mounted on Superfrost Plus slides (Fischer Scientific) and coverslipped for imaging on a laser scanning confocal microscope (Zeiss LSM780 with Quasar detection module). Spectral detection bandwidths (nm) were set at 411–473 for DAPI, 498–568 for GFP and 568–638 for Cy3; pinhole was set to 1 Airy unit. Unbiased stereological counting of NeuN, TSHZ3, CUX1, BCL11B, CHAT, ß-Gal, and GAD67-GFP-positive neurons were done from confocal images using ImageJ software (see Figure legends for frame details). Images were assembled using Photoshop 21.2.3.
Cell counts were performed blind to the genotype in the striatum (dorsal striatum and nucleus accumbens) and in the surrounding motor and sensorimotor cortex on sections spanning from bregma 0 to +1.18 mm, AP. The whole surface was analyzed for the dorsal striatum and the nucleus accumbens. For the cortex, counts were performed in frames of 400-μm width either considering the total thickness of the cortex (NeuN, ß-Gal, GAD67-GFP), the thickness of specific layers (TSHZ3, CUX1, BCL11B) or division into 10 bins of equal size for the analysis of the distribution of GAD67-GFP-positive cells and the quantification of GAD67-GFP;ß-Gal-positive cells. For the different cholinergic nuclei, the analyses were performed on sections spanning from bregma +0.5 to +1.6 mm for the nac, +0.62 to +0.38 for ms and hdb, −0.34 to −0.8 for si and nbm, +3.8 to −4.16 for 3 N, −4.16 to −4.6 for pbg and pptg and −4.72 to −5.2 for ldtg (see Fig. S6 for abbreviations).
Morphometric and dendritic spine analysis Of L5 CPNS
We used transgenic mouse lines (P28) expressing Thy1-GFP (green fluorescent protein) in L5 CPNs [72]. Thy1-GFP-M;Emx1-cKO were obtained by crossing Emx1-Cre;Tshz3flox/flox males with Tshz3flox/flox females heterozygous for Thy1-GFP. Analysis of spine density and morphology was performed on stacks from 100 µm-thick vibratome sections (1 µm z-step) on 4 littermate pairs using a Zeiss LSM780 (Oberkochen, Germany) laser scanning confocal microscope (×63 objective NA 1.4, 0.03 µm/pixel, voxel size 0.033 µm2 × 0.37 µm). Spine counts were performed blind to the genotype. They were obtained from second or third-order basal dendritic branches of randomly selected L5 CPNs. Dendrites from 5 to 7 cells were analyzed per animal, providing a cumulated dendrite length > 750 µm for each genotype. Spine identification and density measures were done using NeuronStudio [73].
RT-qPCR
Total RNA from control and Tshz3 mutant (P28) cerebral cortex was prepared using RNeasy Plus Universal Mini Kit gDNA eliminator (Qiagen™) and first-strand cDNA was synthesized using iScript Reverse Transcription Supermix kit (Bio-RAD™). No blinding was done. Real-time quantitative PCR (RT-qPCR) was performed on a CFX96 qPCR detection system (Bio-RAD™) using SYBR® GreenER™ qPCR SuperMixes (Life Technologies™). RT-qPCR conditions: 40 cycles of 95 °C for 15 s and 60 °C for 60 s. Analyses were performed in triplicate. Transcript levels were first normalized to the housekeeping gene Gapdh. Primer sequences used for RT-qPCR: Gapdh Forward: 5′ GTCTCCTGCGACTTCAACAGCA 3′; Gapdh Reverse: 5′ ACCACCCTGTTGCTGTAGCCGT 3′. Tshz3 Forward: 5′ CACTCCTTCCAGCATCTCTGAG 3′; Tshz3 Reverse: 5′ TAGCAGGTGCTGAGGATTCCAG 3′.
Electrophysiology
Electrophysiological data were obtained from 57 Emx1-cKO and 44 Emx1-Cre−/− control littermates, and from 16 Chat-cKO and 16 Chat-Cre−/− control littermates, aged P21–28. No blinding was done. Procedures were similar to those described previously [10, 12, 74]. Briefly, acute coronal slices (250 µm-thick) containing cortex and striatum were cut using a S1000 Vibratome (Leica) in ice-cold solution containing (in mM): 110 choline, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 25 NaHCO3, 7 glucose, pH 7.4. Slices were kept at room temperature in oxygenated artificial cerebrospinal fluid (ACSF), whose composition was (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose and 25 NaHCO3, pH 7.4. Electrophysiological recordings were performed in oxygenated artificial cerebrospinal fluid (ACSF) at 34–35 °C, flowing at ~2 ml/min. L5 CPNs of the primary motor and somatosensory cortex, and SSPNs and SCINs of the dorsolateral striatum were identified by infrared video microscopy and by their electrophysiological properties [75, 76]. They were recorded by whole-cell patch-clamp using borosilicate micropipettes (5–6 MΩ) filled with an internal solution containing (in mM): 125 K-gluconate, 10 NaCl, 1 CaCl2, 2 MgCl2, 0.5 BAPTA, 19 HEPES, 0.3 Na-GTP, and 1 Mg-ATP, pH 7.3 (except for NMDA/AMPA ratio experiments, see below). Electrophysiological data were acquired by an AxoPatch 200B amplifier and pClamp 10.7 software (Molecular Devices, Wokingham, UK). Series and input resistance were continuously monitored by sending 5 mV pulses, and neurons showing ≥20% change in these parameters were discarded from the analysis.
Characterization of CPNs, SSPNs, and synaptic transmission
A stimulating bipolar electrode was placed either in the cortex at the level of L4 to activate local fibers mainly arising from L2-3 and evoke excitatory postsynaptic currents (EPSCs) in L5 CPNs, or in the corpus callosum to activate corticostriatal fibers and evoke EPSCs in SSPNs [12]. We did not distinguish the two main L5 CPN subtypes, i.e. intratelencephalic and pyramidal tract neurons, because they do not differ in the electrophysiological properties analyzed here [12, 77]. Glutamatergic EPSCs were recorded in the presence of 50 µM picrotoxin at a holding potential of −60 mV (CPNs) or −80 mV (SSPNs). Spontaneous miniature EPSCs (mEPSCs) were recorded in the presence of 50 µM picrotoxin and 1 µM tetrodotoxin. Current–voltage (I–V) relationship was obtained in current-clamp mode by injecting hyperpolarizing and depolarizing current steps (ΔI = ± 50 pA, 800 ms), and input resistance was calculated by linear regression analysis, i.e. as the slope of the linear best fit of the I–V relationship of each recorded neuron. Rheobase was measured as the minimal injected current (+5 pA increments) capable of eliciting an action potential (AP). For paired-pulse ratio (PPR), EPSC amplitude was measured on 6 averaged traces at each inter-pulse interval. For analyzing mEPSCs, the detection threshold (around 3–4 pA) was set to twice the noise after trace filtering (Boxcar low-pass), and only cells exhibiting stable activity and baseline were considered. For NMDA/AMPA ratio experiments, the internal solution contained (in mM): 140 CsCl, 10 NaCl, 0.1 CaCl2, 10 HEPES, 1 EGTA, 2 Mg-ATP and 0.5 Na-GTP, pH 7.3. The AMPA component of the EPSC was measured at the peak at a holding potential of −60 mV, while the NMDA component was measured at +40 mV and 40 ms after the stimulation artifact, when the AMPA component is negligible, as previously described [12]. Tonic NMDA currents were elicited by bath application of 50 µM NMDA for 60 s, after a stable baseline of at least 120 s; their amplitude was measured by averaging the current values of a 5 s window around the negative peak, compared to baseline; only neurons that were capable of returning to their baseline after washout were considered. EPSC amplitude for monitoring corticostriatal long-term depression and potentiation (LTD and LTP, respectively) was measured on averaged traces (6 per minute) to obtain time-course plots and to compare this parameter before (baseline) and after induction protocols. The induction protocol for corticostriatal LTD consisted of 3 trains at 100 Hz, 3 s duration, 20 s interval, at half intensity compared to baseline [78]. LTP induction protocol was identical but, during each train, neurons were depolarized to −10 mV to allow strong activation of NMDA receptors [10, 12, 79]. For a review about corticostriatal LTD and LTP see [36].
Characterization of SCINs
The resting membrane potential (RMP) was measured at the steady state between two consecutive APs. The current–voltage relationship was calculated from the membrane response at the end of current steps from −200 to −20 pA (20 pA steps lasting 800 ms). The voltage sag ratio (VSR) was calculated from the response to a −120 pA current step as the peak voltage drop (sag) against the voltage at the end of the current pulse [80, 81]. Such relatively small current step was chosen because, with larger steps, the sag amplitude was extremely variable between different SCINs. Spontaneous AP firing was analyzed in terms of discharge frequency (expressed in Hz) and regularity; to quantify this latter parameter, we calculated the coefficient of variation (CV) of the inter-AP intervals. Note that spontaneous AP firing was analyzed only from cell-attached recordings, which were done before switching to whole-cell; in some cases, spontaneous firing was not detectable in cell-attached configuration, thus the number of samples for AP firing analyses is smaller than the whole number of recorded SCINs.
Behavioral analysis
Housing conditions
Experiments were conducted blind for the genotypes in P71-87 male Emx1-cKO and Chat-cKO mice and their respective Emx1-Cre−/− and Chat-Cre−/− control littermates. We used males and not female mice because the ambulatory activity of females is impacted by the estrous cycle phases in rodents [82] and may bias the results of repetitive behavior measures that are partly dependent on motor activity.
Mice used in studies on social behavior are generally reared in groups of variable size and more rarely in isolation. The choice of our rearing strategy was based on the fact that the measures of social behavior in adult mice depends on the characteristics of the previous interactions that the observed male has experienced with its peers [3, 83,84,85]. In the rearing in group strategy, the social behaviors directed towards the tested male can vary according to the genotypes, the androgen levels and the neurotransmitter profiles of the individuals in the groups [86]. Consequently, the social behavior measured in an individual is the resultant of the individual social ability plus a component corresponding to the interactions of the individual with the other members of the group; this effect varies with the size of the group. In addition, behavioral “contamination” resulting in an impairment of sociability in wild-type mice by cohabitant KO modeling ASD was described [85]. Such undesirable effect plus the heterogeneity of the measures in mice reared in the group should contribute to avoid this strategy for testing social behavior. On the other hand, maintaining the mice socially deprived generates a specific set of agonistic reactions that prevent the measures of social abilities. To circumvent such biases, we have developed an alternative solution for years: each tested male is housed with one female mouse belonging to a single inbred strain [86]. Here, a cKO or a control male mouse was reared and maintained with CBA/H/Gnc female mice [3]. Housing was done in transparent 35 × 20 × 18 cm cages with 1-liter poplar woodchip bedding and weekly renewed enrichment (cardboard shelter). The light (07:00–19:00) was 60 lux on the ground of the cages. The temperature was 21.5 ± 0.5 °C. Behavioral tests were performed in a dedicated room, the housing cage having been transferred one hour before the beginning of the observations.
Assessment of sensory function
Visual, auditory, and olfactory integrity is required to ensure the validity of the behavioral data. These sensorial capacities were tested according to previously described protocols [3, 10, 12, 87].
Visual capacities
The mouse was raised, taken by the tail, and a thin stick was approached to its eyes, without touching the vibrissae. Raising the head was scored 1 and grasping or trying to grasp the pen was scored 2. The test was administered five times and the sum of the scores recorded. Swimming towards a distant shelf in the Morris Water Maze provided an additional assessment of the visual abilities.
Auditory capacities were measured using Preyer’s response. It consists of a pinna twitching and going flat backwards against the head as reaction to sound. It is correlated with the average evoked auditory potential and can be considered as an indicator of auditory acuity [88,89,90]. Mice emit vocalizations (less than 20 kHz) and ultrasounds (above 20 kHz) in the presence of a conspecific male. For this reason, we evaluated the responses to stimulations in the ultrasound bandwidth (50 ± 0.008 kHz and 35 ± 0.010 kHz) using commercial dog whistles. The mice received 5 stimulations with each sound. We scored 1 for ear twitching and 2 for a pinna going flat backwards against the head.
Olfactory ability to detect an odor was evaluated by an increased time in sniffing a new odor using an olfactory habituation/dishabituation test. Non-social aromas and social odors (urines from C57BL/6J and SWR male mice) were presented individually to each mouse [91]. The trial was renewed the following day. The individual score was the median time spent.
ASD core features
Behaviors modeling the ASD domains as defined by DSM-5 were assessed. The tests were selected based on their strong robustness (reliability from 0.77 to 0.92) and on their high loading scores in factor analysis [3].
Deficit in social behavior
A two-chamber test derived from Moy et al. [92] was used to assess sociability and interest in social novelty. The setup and the protocol were detailed previously [3, 10, 12]. We used a 550 × 550 mm Plexiglas box split in a 150 × 550 mm empty chamber and a 400 × 550 mm chamber containing the two boxes (43 mm diameter, distant from 340 mm) in which the mice or the lure were placed. Sociability is operationally defined as the higher number of visits towards the box containing a conspecific versus the one containing a lure (an adult mouse-sized oblong gray pebble), and the interest in social novelty as the higher number of visits towards a novel conspecific than towards the familiar one. Loss of social interest and poor interest in social novelty are expected in mice models of ASD. Briefly, the test consisted of a three-period observation, each lasting 10 min: (1) habituation (the two boxes containing lures), (2) sociability (one box containing a lure and the other a C57BL/6J male) and (3) interest in social novelty (one box containing the same C57BL/6J and the other a new SWR male). The behaviors were video-recorded (Viewpoint-Behavior technologies) and the number of nose pokes towards the boxes was counted as measure of the number of visits [93].
Repeated patterns of behavior
We selected four measures that were highly loaded on the “repetitive patterns of behavior” factor in a factor analysis:[3] marble-burying and time burrowing in a new cage, number of stereotyped dips in a hole-board device, and number of leanings in an open field. The protocols used have been previously detailed [3, 10, 12]. The marble-burying and time burrowing tests quantify perseverating behavior [94, 95]. Marble burying consists in scoring the amount of marbles buried by each mouse in a 30 min session, using a 40 × 18 cm cage with 45 cm-thick litter and containing 20 marbles (9 mm diameter) on the surface of 70 mm thick dust-free sawdust. Completely buried, 2/3 buried and 1/2 buried marbles were scored 3, 2, and 1, respectively. The time burrowing test leans on spontaneous digging and pushing behavior that rodents display when placed into a new home cage. The length of time each mouse spent digging plus pushing was measured. The number of stereotyped dips was counted in a hole-board device, consisting of a 40 × 40 cm board with 16 equidistant holes (3.5 cm diameter) each equipped with photo-beams for detecting head dipping. Exploratory head dipping occurs when a rodent is placed on a surface with holes: the mouse puts its head once into one hole of the board. Head dipping is considered stereotyped when the head dips at least twice in the same hole within 2 s. The open-field behavior was measured in a circular open field (100 cm diameter and 45 cm high walls) brightly lighted (210 lux on the ground). The ground was virtually divided into three concentric zones of the equal surface. The distances walked and the times spent in the open field in the zones were automatically measured via the Viewpoint-Behavior technologies system (http://www.viewpoint.fr/news.php). The observation lasted 20 min. The number of leanings (rearing while leaning) on the walls of the structure was previously validated as a measure of repetitive behavior [3, 96]. The number of zones crossed is a measure of the narrowness of the field of interest. The total distance walked during the observation period served as covariate for the comparison between cKO mice and their respective controls [3].
Additional behavioral measures
Motor abnormalities and intellectual disability are not included among the ASD core features while having a noticeable but incomplete prevalence in ASD patients (≤79% and ~45%, respectively [64]). In this connection, two additional tests were conducted.
Hind paw coordination
A mouse was first trained to cross a smooth bar (50 × 5 × 5 cm) with large platforms on each extremity. The trained mouse was then placed on the central platform (3 × 5 cm) of a notched bar (100 cm) formed of 1.5 cm deep carvings regularly spaced (2 cm). The task consisted of ten bar crossings from the central to an extremity platform. The experimenters on each side of the setup counted the left and right hind paw slips according to [97].
Spatial learning
The Morris water maze provides measures of the ability of rodents to solve spatial learning problems, namely the ability to find a submerged resting platform concealed beneath opaque water. The platform is a glass cylinder (66 mm diameter, 9 mm beneath the surface of the water) positioned 23 cm from the edge of a 100 cm diameter circular tank filled with water at 26 ± 1 °C and the light at 70 lux on the surface. Each mouse performed 7 blocks of 4 trials each: one block on day 1, and two blocks daily (one in the morning and one in the afternoon) for 3 successive days. A trial was stopped after 90 s if the mouse failed to reach the platform. We considered that the mouse had reached the platform when it stayed on the platform for 5 s at least. We presented a small metal shelf to the mouse 5 cm above the platform at the end of each trial of the first block (shaping). The mouse climbed on it and was transferred to a cage with dry sawdust for 120 s. We had previously assigned 4 virtual cardinal points to the tank, each being the starting point for a trial. The starting point for each trial was chosen randomly and within a block the mouse never started more than once from the same virtual cardinal point. We measured (1) the time to reach the hidden platform and (2) the cumulative distance to the center of the platform during swimming. The second measure eliminates possible bias resulting from floating during the trial. The time to reach the platform and the distance were automatically measured by a video tracking setup (Viewpoint-Behavior technologies), each over the 7 blocks. Strains can achieve different performance levels between blocks, but without a cumulative reduction in the time to reach the platform, which is the criterion to identify the learning process. We computed the slopes of the learning curves, a negative slope indicating learning behavior [98]. The strategy was used for both the time to reach the hidden platform and the cumulative distance to the center of the platform. The probe-test procedure, conducted after removing the platform, was done 24 h after block 7 to meet the requirements for reference memory [99] and lasted 90 s. The mouse was placed in the center of the tank, and we measured the time of first crossing the virtual annulus corresponding to the location of the platform. To check whether the differences in the time to reach the platform were due to vision and/or swimming abilities rather than learning ability, we also tested groups of naïve Emx1-cKO and ChAT-cKO mice, and their respective control, to the visible platform version of the test, in which the platform is 5 mm above non-opacified water.
Statistics
The sample size was based on previous experience and was similar to studies in the field.
Immunohistochemistry
Data were analyzed by Prism 7.05 (GraphPad Software, USA). Two-tailed Student’s t test or Mann–Whitney test was used for comparing two data sets when passing or not D’Agostino & Pearson’s normality test, respectively. Sample sizes, tests used, and P values are reported in Figure legends. The significance threshold was set at P < 0.05.
RT-qPCR
Statistical analysis was performed by unpaired Student’s t tests using the qbasePLUS software version 2 (Biogazelle). The significance threshold was set at P < 0.05.
Electrophysiology
Statistical analysis was performed by Prism 7.05 (GraphPad Software, USA). Two-tailed Student’s t test or Mann–Whitney test was used for comparing two data sets when passing or not D’Agostino & Pearson’s normality test, respectively. Two-way ANOVA was used to analyze the influence of 2 categorical variables. 2-samples Kolmogorov–Smirnov test was used to compare cumulative distributions. Sample sizes (n) reported in figure legends refer to the number of recorded neurons. The significance threshold was set at P < 0.05. The tests used, P values and sample sizes are indicated in the figures.
Behavior
Data were processed by Statistical Package for the Social Sciences [SPSS software, version 25 [100]]. The same statistical designs were used to compare Emx1-cKO and ChAT-cKO mice to their respective controls. Non-parametric statistics were chosen when the assumption of normality was rejected.
Impairment of social behavior
To analyze data from each social phase of the two-chamber test (sociability and interest for social novelty), a mixed-design analysis of covariance (ANCOVA) was used including the genotype as a fixed factor, the box content as repeated measure, with a measure of activity during habituation as covariate. A significant interaction between genotype and box content indicates that social behavior differs between the cKO and its control group.
Repetitive patterns of behavior and motor performance
The difference between two independent groups (cKO and its control group) was tested by an unpaired two-sample Student’s t test in each case where it was not necessary to include a covariate in the statistical design (i.e., stereotyped behavior: marble-burring score, time burrowing, number of leanings; motor behavior: number of hind paw slips). For measures of stereotyped dips, on which the activity level could have an impact, an analysis of covariance (ANCOVA) was performed, using the genotype as a fixed factor (cKO vs. respective control) and non-stereotyped dips as covariate.
Sensorial abilities
Comparison of the visual and auditory capacities of the cKO and their respective controls were conducted using a Student’s t test. Mixed repeated measures ANOVA, with genotype as a fixed factor and 15 odors as repeated measures, was used to compare cKO and their respective controls for olfactory capacities.
Spatial learning
The statistical design was the same for the time to reach the platform and the cumulative distance to the center of the platform in the Morris water maze test. Differences between the 7 blocks were tested either with Friedman’s ANOVA, a non-parametric version of one-way repeated measures ANOVA, or with two-way repeated measures mixed ANOVA design, with blocks as repeated measures variable and cKO vs. control as between-group variable. Learning may be deduced from within-bloc statistical difference and reduced time to reach the platform from one bloc to the next. The slope of the median values of the four trials in each of the seven blocks was calculated for each mouse. The median slopes for the cKO and their respective controls, as well as the time to reach the virtual platform (probe test) and the visible platform, were compared with a Student’s t test.
Effect size
Effect sizes are expressed as η2 or as partial η2 with 95% confidence interval [100, 101].
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Raw data (FastQ files) from the sequencing experiment (triplicates from wild-type and Tshz3-mutant striatum) and raw abundance measurements for genes (read counts) for each sample are available from Gene Expression Omnibus (GEO) under accession GSE157658, which should be quoted in any manuscript discussing the data.
Change history
23 May 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41398-022-01925-x
References
American Psychiatric Association. Diagnostic and statistical manual of mental disorders, DSM-5. 5th edn. Washington, DC: American Psychiatric Association; 2013.
Shuster J, Perry A, Bebko J, Toplak ME. Review of factor analytic studies examining symptoms of autism spectrum disorders. J Autism Dev Disord. 2014;44:90–110.
Roubertoux PL, Tordjman S, Caubit X, di Cristopharo J, Ghata A, Fasano L, et al. Construct validity and cross validity of a test battery modeling autism spectrum disorder (ASD) in mice. Behav Genet. 2020;50:26–40.
Banerjee-Basu S, Packer A. SFARI Gene: an evolving database for the autism research community. Dis Models Mech. 2010;3:133–5.
Bourgeron T. Current knowledge on the genetics of autism and propositions for future research. C R Biol. 2016;339:300–7.
Delmonte S, Gallagher L, O’Hanlon E, McGrath J, Balsters JH. Functional and structural connectivity of frontostriatal circuitry in autism spectrum disorder. Front Hum Neurosci. 2013;7:430.
Shepherd GM. Corticostriatal connectivity and its role in disease. Nat Rev: Neurosci. 2013;14:278–91.
Li W, Pozzo-Miller L. Dysfunction of the corticostriatal pathway in autism spectrum disorders. J Neurosci Res. 2020;98:2130–47.
Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362:eaat7615.
Caubit X, Gubellini P, Andrieux J, Roubertoux PL, Metwaly M, Jacq B, et al. TSHZ3 deletion causes an autism syndrome and defects in cortical projection neurons. Nat Genet. 2016;48:1359–69.
Caubit X, Tiveron MC, Cremer H, Fasano L. Expression patterns of the three Teashirt-related genes define specific boundaries in the developing and postnatal mouse forebrain. J Comp Neurol. 2005;486:76–88.
Chabbert D, Caubit X, Roubertoux PL, Carlier M, Habermann B, Jacq B, et al. Postnatal Tshz3 deletion drives altered corticostriatal function and autism spectrum disorder-like behavior. Biol Psychiatry. 2019;86:274–85.
Caubit X, Arbeille E, Chabbert D, Desprez F, Messak I, Fatmi A, et al. Camk2a-Cre and Tshz3 expression in mouse striatal cholinergic interneurons: implications for autism spectrum disorder. Front Genet. 2021;12:683959.
Munoz-Manchado AB, Bengtsson Gonzales C, Zeisel A, Munguba H, Bekkouche B, Skene NG, et al. Diversity of interneurons in the dorsal striatum revealed by single-cell RNA sequencing and PatchSeq. Cell Rep. 2018;24:2179–90. e2177
Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 2018;174:1015–30. e1016
Karvat G, Kimchi T. Acetylcholine elevation relieves cognitive rigidity and social deficiency in a mouse model of autism. Neuropsychopharmacology. 2014;39:831–40.
Rapanelli M, Frick LR, Xu M, Groman SM, Jindachomthong K, Tamamaki N, et al. Targeted interneuron depletion in the dorsal striatum produces autism-like behavioral abnormalities in male but not female mice. Biol Psychiatry. 2017;82:194–203.
Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–14.
Dautan D, Huerta-Ocampo I, Witten IB, Deisseroth K, Bolam JP, Gerdjikov T, et al. A major external source of cholinergic innervation of the striatum and nucleus accumbens originates in the brainstem. J Neurosci. 2014;34:4509–18.
Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000;20:6063–76.
Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–35.
Bennett BD, Wilson CJ. Spontaneous activity of neostriatal cholinergic interneurons in vitro. J Neurosci. 1999;19:5586–96.
Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci. 2000;20:8493–503.
Goldberg JA, Wilson CJ. The cholinergic interneurons of the striatum: intrinsic properties underlie multiple discharge patterns. In: Steiner H, Tseng KY (eds). Handbook of basal ganglia structure and function. Academic Press; 2010. p. 133–49.
Lozovaya N, Eftekhari S, Cloarec R, Gouty-Colomer LA, Dufour A, Riffault B, et al. GABAergic inhibition in dual-transmission cholinergic and GABAergic striatal interneurons is abolished in Parkinson disease. Nat Commun. 2018;9:1422.
Fournier KA, Hass CJ, Naik SK, Lodha N, Cauraugh JH. Motor coordination in autism spectrum disorders: a synthesis and meta-analysis. J Autism Dev Disord. 2010;40:1227–40.
Green D, Charman T, Pickles A, Chandler S, Loucas T, Simonoff E, et al. Impairment in movement skills of children with autistic spectrum disorders. Dev Med Child Neurol. 2009;51:311–6.
Bicks LK, Koike H, Akbarian S, Morishita H. Prefrontal cortex and social cognition in mouse and man. Front Psychol. 2015;6:1805.
Chen P, Hong W. Neural circuit mechanisms of social behavior. Neuron. 2018;98:16–30.
Fuccillo MV. Striatal circuits as a common node for autism pathophysiology. Front Neurosci. 2016;10:27.
Langen M, Schnack HG, Nederveen H, Bos D, Lahuis BE, de Jonge MV, et al. Changes in the developmental trajectories of striatum in autism. Biol Psychiatry. 2009;66:327–33.
Rapanelli M, Frick LR, Pittenger C. The role of interneurons in autism and tourette syndrome. Trends Neurosci.2017;40:397–407.
Berretta N, Nistico R, Bernardi G, Mercuri NB. Synaptic plasticity in the basal ganglia: a similar code for physiological and pathological conditions. Prog Neurobiol. 2008;84:343–62.
Zhou JJ, Li DP, Chen SR, Luo Y, Pan HL. The alpha2delta-1-NMDA receptor coupling is essential for corticostriatal long-term potentiation and is involved in learning and memory. J Biol Chem. 2018;293:19354–64.
Park H, Popescu A, Poo MM. Essential role of presynaptic NMDA receptors in activity-dependent BDNF secretion and corticostriatal LTP. Neuron. 2014;84:1009–22.
Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–61.
Marotta R, Risoleo MC, Messina G, Parisi L, Carotenuto M, Vetri L, et al. The neurochemistry of autism. Brain Sci. 2020;10:163.
Lee EJ, Choi SY, Kim E. NMDA receptor dysfunction in autism spectrum disorders. Curr Opin Pharm. 2015;20:8–13.
Forrest MP, Parnell E, Penzes P. Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci. 2018;19:215–34.
Apicella P. The role of the intrinsic cholinergic system of the striatum: What have we learned from TAN recordings in behaving animals? Neuroscience. 2017;360:81–94.
Tanimura A, Pancani T, Lim SAO, Tubert C, Melendez AE, Shen W, et al. Striatal cholinergic interneurons and Parkinson’s disease. Eur J Neurosci. 2018;47:1148–58.
Mallet N, Leblois A, Maurice N, Beurrier C. Striatal cholinergic interneurons: how to elucidate their function in health and disease. Front Pharm. 2019;10:1488.
Poppi LA, Ho-Nguyen KT, Shi A, Daut CT, Tischfield MA. Recurrent implication of striatal cholinergic interneurons in a range of neurodevelopmental, neurodegenerative, and neuropsychiatric disorders. Cells. 2021;10:907.
Ahmed NY, Ranjbar-Slamloo Y, Al Abed AS, Gao L, Sontani Y, RC-Hc-G A, et al. Er81 transcription factor fine-tunes striatal cholinergic interneuron activity and drives habit formation. J Neurosci. 2021;41:4392–409.
Gillan CM, Papmeyer M, Morein-Zamir S, Sahakian BJ, Fineberg NA, Robbins TW, et al. Disruption in the balance between goal-directed behavior and habit learning in obsessive-compulsive disorder. Am J Psychiatry. 2011;168:718–26.
Smith R, Chung H, Rundquist S, Maat-Schieman ML, Colgan L, Englund E, et al. Cholinergic neuronal defect without cell loss in Huntington’s disease. Hum Mol Genet. 2006;15:3119–31.
McKinley JW, Shi Z, Kawikova I, Hur M, Bamford IJ, Sudarsana Devi SP, et al. Dopamine deficiency reduces striatal cholinergic interneuron function in models of Parkinson’s disease. Neuron. 2019;103:1056–72. e1056
Eskow Jaunarajs KL, Bonsi P, Chesselet MF, Standaert DG, Pisani A. Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Prog Neurobiol. 2015;127-128:91–107.
Gonzales KK, Smith Y. Cholinergic interneurons in the dorsal and ventral striatum: anatomical and functional considerations in normal and diseased conditions. Ann N. Y Acad Sci. 2015;1349:1–45.
Abudukeyoumu N, Hernandez-Flores T, Garcia-Munoz M, Arbuthnott GW. Cholinergic modulation of striatal microcircuits. Eur J Neurosci. 2019;49:604–22.
Martin-Ruiz CM, Lee M, Perry RH, Baumann M, Court JA, Perry EK. Molecular analysis of nicotinic receptor expression in autism. Brain Res Mol Brain Res. 2004;123:81–90.
Perry EK, Lee ML, Martin-Ruiz CM, Court JA, Volsen SG, Merrit J, et al. Cholinergic activity in autism: abnormalities in the cerebral cortex and basal forebrain. Am J Psychiatry. 2001;158:1058–66.
Deutsch SI, Urbano MR, Neumann SA, Burket JA, Katz E. Cholinergic abnormalities in autism: is there a rationale for selective nicotinic agonist interventions? Clin Neuropharmacol. 2010;33:114–20.
Ragozzino ME, Mohler EG, Prior M, Palencia CA, Rozman S. Acetylcholine activity in selective striatal regions supports behavioral flexibility. Neurobiol Learn Mem. 2009;91:13–22.
Eissa N, Azimullah S, Jayaprakash P, Jayaraj RL, Reiner D, Ojha SK, et al. The dual-active histamine H3 receptor antagonist and acetylcholine esterase inhibitor E100 ameliorates stereotyped repetitive behavior and neuroinflammmation in sodium valproate induced autism in mice. Chem Biol Interact. 2019;312:108775.
Martos YV, Braz BY, Beccaria JP, Murer MG, Belforte JE. Compulsive social behavior emerges after selective ablation of striatal cholinergic interneurons. J Neurosci. 2017;37:2849–58.
Crittenden JR, Lacey CJ, Weng FJ, Garrison CE, Gibson DJ, Lin Y, et al. Striatal cholinergic interneurons modulate spike-timing in striosomes and matrix by an amphetamine-sensitive mechanism. Front Neuroanat. 2017;11:20.
Murray RC, Logan MC, Horner KA. Striatal patch compartment lesions reduce stereotypy following repeated cocaine administration. Brain Res. 2015;1618:286–98.
Aliane V, Perez S, Bohren Y, Deniau JM, Kemel ML. Key role of striatal cholinergic interneurons in processes leading to arrest of motor stereotypies. Brain. 2011;134:110–8.
Hanson KL, Lew CH, Hrvoj-Mihic B, Cuevas D, Greiner DMZ, Groeniger KM, et al. Decreased density of cholinergic interneurons in striatal territories in Williams syndrome. Brain Struct Funct. 2020;225:1019–32.
Ahmed NY, Knowles R, Dehorter N. New insights into cholinergic neuron diversity. Front Mol Neurosci. 2019;12:204.
Bariselli S, Fobbs WC, Creed MC, Kravitz AV. A competitive model for striatal action selection. Brain Res. 2019;1713:70–79.
Goldberg JA, Ding JB, Surmeier DJ. Muscarinic modulation of striatal function and circuitry. Handb Exp Pharm. 2012;208:223–41.
Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383:896–910.
Xu M, Kobets A, Du JC, Lennington J, Li L, Banasr M, et al. Targeted ablation of cholinergic interneurons in the dorsolateral striatum produces behavioral manifestations of Tourette syndrome. Proc Natl Acad Sci USA. 2015;112:893–8.
Blake MG, Boccia MM. Basal forebrain cholinergic system and memory. Curr Top Behav Neurosci. 2018;37:253–73.
Caubit X, Lye CM, Martin E, Core N, Long DA, Vola C, et al. Teashirt 3 is necessary for ureteral smooth muscle differentiation downstream of SHH and BMP4. Development. 2008;135:3301–10.
Mao X, Fujiwara Y, Orkin SH. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci USA. 1999;96:5037–42.
Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–40.
Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 2011;13:195–204.
Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79.
Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51.
Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS ONE. 2008;3:e1997.
Chassain C, Melon C, Salin P, Vitale F, Couraud S, Durif F, et al. Metabolic, synaptic and behavioral impact of 5-week chronic deep brain stimulation in hemiparkinsonian rats. J Neurochemistry. 2016;136:1004–16.
Hattox AM, Nelson SB. Layer V neurons in mouse cortex projecting to different targets have distinct physiological properties. J Neurophysiol. 2007;98:3330–40.
Jiang ZG, North RA. Membrane properties and synaptic responses of rat striatal neurones in vitro. J Physiol. 1991;443:533–53.
Chen L, Daniels S, Kim Y, Chu HY. Cell type-specific decrease of the intrinsic excitability of motor cortical pyramidal neurons in Parkinsonism. J Neurosci. 2021;41:5553–65.
Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–33.
Calabresi P, Pisani A, Mercuri NB, Bernardi G. Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of nmda receptor channels. Eur J Neurosci. 1992;4:929–35.
Haghdoust H, Janahmadi M, Behzadi G. Physiological role of dendrotoxin-sensitive K+ channels in the rat cerebellar Purkinje neurons. Physiol Res. 2007;56:807–13.
Maisano X, Litvina E, Tagliatela S, Aaron GB, Grabel LB, Naegele JR. Differentiation and functional incorporation of embryonic stem cell-derived GABAergic interneurons in the dentate gyrus of mice with temporal lobe epilepsy. J Neurosci. 2012;32:46–61.
Beau Yon de Jonage-Canonico M, Roubertoux PL, Lenoir V, Carlier M, Kerdelhué B. Long-term reduction in anxiety levels during the promotion phase of mammary adenocarcinoma induced by dimethylbenz (a) anthracene in female sprague-dawley rats. Open Neuroendocrinol J. 2010;3:52–58.
Ginsburg B, Allee W. Some effects of conditioning on social dominance and subordination in inbred strains of mice. Physiological Zool. 1942;15:485–506.
Maxson SC, Canastar A. Conceptual and methodological issues in the genetics of mouse agonistic behavior. Horm Behav. 2003;44:258–62.
Kalbassi S, Bachmann SO, Cross E, Roberton VH, Baudouin SJ. Male and female mice lacking neuroligin-3 modify the behavior of their wild-type littermates. eNeuro. 2017;4:ENEURO.0145-17.2017.
Roubertoux PL, Guillot PV, Mortaud S, Pratte M, Jamon M, Cohen-Salmon C, et al. Attack behaviors in mice: from factorial structure to quantitative trait loci mapping. Eur J Pharm. 2005;526:172–85.
Roubertoux PL, Ghata A, Carlier M. Measuring preweaning sensorial and motor development in the mouse. Curr Protoc Mouse Biol. 2018;8:54–78.
Ehret G, Romand R. Development of tone response thresholds, latencies and tuning in the mouse inferior colliculus. Brain Res Dev Brain Res. 1992;67:317–26.
Francis RL. The Preyer reflex audiogram of several rodents, and its relation to the “absolute” audiogram in the rat. J Aud Res. 1979;19:217–33.
Willott JF. The auditory psychobiology of the mouse. C.C. Thomas; 1983.
Yang M, Crawley JN. Simple behavioral assessment of mouse olfaction. Curr Protoc Neurosci. 2009;Chapter 8:Unit 8 24.
Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302.
Roubertoux PL, Carlier M, Tordjman S. Organism models of autism spectrum disorders. Springer New York; 2015. 487 pp.
Njung’e K, Handley SL. Effects of 5-HT uptake inhibitors, agonists and antagonists on the burying of harmless objects by mice; a putative test for anxiolytic agents. Br J Pharm. 1991;104:105–12.
Thomas A, Burant A, Bui N, Graham D, Yuva-Paylor LA, Paylor R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology (Berl). 2009;204:361–73.
Rizzo SJ. Repetitive behavioral assessments for compound screening in mouse models of autism spectrum disorders. Methods Mol Biol. 2016;1438:293–310.
Maarouf FD, Roubertoux PL, Carlier M. Is mitochondrial DNA involved in mouse behavioral laterality? Behav Genet. 1999;29:311–8.
Roubertoux PL, Baril N, Cau P, Scajola C, Ghata A, Bartoli C, et al. Differential brain, cognitive and motor profiles associated with partial trisomy. modeling down syndrome in mice. Behav Genet. 2017;47:305–22.
Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat Protoc. 2006;1:1671–9.
Field AP Discovering statistics using SPSS: (and sex, drugs and rock’n’roll). SAGE; 2005.
Cohen J Statistical power analysis for the behavioral sciences, 2nd edn. Hillsdale, NJ: L. Erlbaum Associates; 1988. xxi, 567pp.
Acknowledgements
Behavioral testing was performed at the mouse facility of the Marseille Medical Genetics (MMG) UMR1251 Aix Marseille Univ, INSERM. Microscopy was performed at the imaging platform of the IBDM and we acknowledge France-BioImaging/PiCsL infrastructure (ANR-10-INSB-04-01). This work has received support from the French government under the Programme “Investissements d’Avenir”, Initiative d’Excellence d’Aix-Marseille Université via A*Midex funding (NeuroMarseille Institute, AMX-19-IET-004; MarMaRa Institute, AMX-19-IET-007), and ANR (ANR-17-EURE-0029). We wish to thank the IBDM mouse facility. This work was supported by the French National Research Agency (ANR) “TSHZ3inASD” project grant n°ANR-17-CE16-0030-01 (to L.F. and L.K.-L.G.), the Fédération pour la Recherche sur le Cerveau (FRC) (to L.F.), the Centre National de la Recherche Scientifique (CNRS) and Aix-Marseille University. D.C. and J.M. were supported by PhD grants from the MESRI (Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation).
Author information
Authors and Affiliations
Contributions
X.C., J.M., and P.S. performed the histological experiments and the quantitative analyses; M.C. and P.L.R. conducted the behavioral experiments and analyzed the resulting data; Y.B., L.B., J.M., and D.C. performed patch-clamp experiments and P.G. analyzed electrophysiological data; M.M. performed dendritic spine imaging and counting; A.F. performed RT-qPCR; X.C. and J.M. generated and maintained transgenic mouse lines; X.C., L.F., P.G., and L.K.-L.G. conceived the project, supervised the work and wrote the paper with the contribution of M.C. and P.L.R; all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Caubit, X., Gubellini, P., Roubertoux, P.L. et al. Targeted Tshz3 deletion in corticostriatal circuit components segregates core autistic behaviors. Transl Psychiatry 12, 106 (2022). https://doi.org/10.1038/s41398-022-01865-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-01865-6
