
Abstract
Psychotic disorders affect 3% of the population at some stage in life, are a leading cause of disability, and impose a great economic burden on society. Major breakthroughs in the genetics of psychosis have not yet been matched by an understanding of its neurobiology. Biomarkers of perception and cognition obtained through non-invasive neurophysiological tools, especially EEG, offer a unique opportunity to gain mechanistic insights. Techniques for measuring neurophysiological markers are inexpensive and ubiquitous, thus having the potential as an accessible tool for patient stratification towards early treatments leading to better outcomes. In this paper, we review the literature on neurophysiological markers for psychosis and their relevant disease mechanisms, mainly covering event-related potentials including P50/N100 sensory gating, mismatch negativity, and the N100 and P300 waveforms. While several neurophysiological deficits are well established in patients with psychosis, more research is needed to study neurophysiological markers in their unaffected relatives and individuals at clinical high risk. We need to harness EEG to investigate markers of disease risk as key steps to elucidate the aetiology of psychosis and facilitate earlier detection and treatment.
Similar content being viewed by others

Introduction
Psychotic disorders, including schizophrenia and bipolar disorder, have a lifetime prevalence of ~3% [1] and are the leading cause of disability in young adults [2, 3]. They have a major economic impact, with an annual cost of €94 billion for psychotic disorders across Europe [4] and $156 billion for schizophrenia in the United States [5]. Major breakthroughs have been made in our understanding of the environmental and genetic origins of psychosis, with 270 loci associated with schizophrenia and several compelling rare genetic variants identified so far [6,7,8]. There are also a range of effective treatments for psychosis, including over 20 different drugs licensed for schizophrenia [9], as well as psychological therapies and rehabilitation interventions [10, 11]. However, the psychoses are highly heterogeneous in their clinical presentations and our understanding of underlying mechanisms remains limited. We face several challenges, including substantial delays in treatment access (typically of 6–12 months in the UK) [12], as well as low adherence to antipsychotics, with over 50% of people stopping their medication within two years for a variety of reasons, including adverse drug reactions [13, 14].
A potential solution to these challenges involves studying biomarkers for psychosis seeking to unravel its underlying mechanisms and develop new treatments. Biomarkers are quantitative traits that can be objectively and reliably measured [15], typically using laboratory, imaging, or standardised clinical assessment tools. Since psychosis is characterised by perceptual and cognitive deficits that often manifest prior to the onset or diagnosis of the illness [16], biomarkers of brain functions in relation to information processing are particularly attractive in psychosis research. Neurophysiological tools, especially electroencephalography (EEG) and event-related potentials (ERPs, also known as evoked potentials), which are changes in the EEG triggered by stimuli, are promising due to their exceptional temporal resolution that allows the study of perception and cognition safely in vivo. They are relatively inexpensive and ubiquitous, thus permitting the collection of large samples required for progress in understanding the aetiology of psychosis. Once validated in patients, such EEG/ERP biomarkers could pinpoint neurocognitive mechanisms most relevant to psychosis, and thus serve as targets for the development of new treatments.
There has been growing interest in neurophysiological biomarkers of psychosis risk, often studied in unaffected relatives of patients or individuals at clinical high risk. Consistent with the observation that heritability estimates for psychotic disorders are about 80% [17], some neurophysiological markers are also impaired in the unaffected relatives of patients with psychosis. Such biomarkers, also referred to as endophenotypes, can help as intermediate phenotypes to explain how the genetic risk of psychosis is conferred [18]. Several neurophysiological markers have also been found to be associated with increased psychosis risk amongst individuals at clinical high risk, or even predictive of transition to psychosis [19, 20]. Their implementation in assessments could help facilitate earlier and personalised treatment, thus leading to improved prognosis [21].
In this paper, we review the vast literature on neurophysiological markers that have been found to be impaired in patients with psychosis. In addition, we summarise the neuropsychological and clinical correlates of those biomarkers and discuss the insights they can offer into the underlying neurobiology of psychosis. We also review evidence for neurophysiological markers of disease risk reported in the unaffected relatives of patients and individuals at clinical high risk for psychosis, which are important for both mechanistic and clinical research in psychosis.
P50 and N100 sensory gating
The P50 waveform is a positive voltage ERP, a change in the EEG triggered by stimuli, which is used to measure sensory gating [22, 23]. The classic experiment to measure P50 sensory gating is the dual-click paradigm, where participants are presented with pairs of auditory stimuli (usually separated by 500 ms) and the amplitude of the ERP at approximately 50 ms after each stimulus constitutes the P50 [22]. The first stimulus is labelled the conditioning stimulus (C), the second stimulus is labelled the test stimulus (T), and P50 sensory gating is measured as the T/C ratio [24]. A ratio below 50% is usually considered a normal P50 sensory gating response [25]. Although studies on sensory gating in psychosis were primarily conducted on the P50, there has been increasing research recently on the N100 response. While the P50 reflects the early pre-attentive stage of sensory gating, the N100 involves later stages of information processing that share similar gating mechanisms.
Extensive evidence shows that individuals with psychotic disorders display larger responses to the second stimulus compared to healthy controls, with only a 20–50% suppression, resulting in increased P50 T/C ratios [22, 23, 26, 27]. The latest meta-analysis of P50 sensory gating reported that compared to controls (n = 3464), people with schizophrenia (n = 3666) and bipolar disorder (n = 656) had higher P50 ratios (Cohen’s d = 10.30 and 4.51, respectively) [24], indicative of impaired suppression. By contrast, there has been less research on N100 sensory gating in psychosis. A recent meta-analysis including 1027 patients with schizophrenia and 1131 controls found that the bigger T/C ratio of N100 in patients was likely due to their reduced amplitude to the conditioning stimuli (Hedges’ g = −0.61), instead of insufficient suppression of the test stimuli (Hedges’ g = −0.04) [28]. Although this meta-analysis revealed N100 reductions, these were not due to gating deficits in schizophrenia.
Reduced P50 gating reflects deficits in inhibitory processes and/or sensory adaptation, which could contribute to the sensory overload some patients experience. The few studies investigating the clinical correlates of P50 gating showed little evidence of association with psychotic symptoms but instead reported correlations between P50 gating and attention [29,30,31,32]. Thus, the research shows that sensory gating is not so much related to symptoms, but rather to the cognitive dysfunction that characterises psychosis.
P50 sensory gating has been associated with the CA3 region of the hippocampus in rodent models, as shown by studies using intracranial electrodes to investigate the analogous P20/N40 gating in rats [33]. In addition, human studies also suggested the involvement of the thalamus, the temporal cortex, and the frontal cortex in the generation of P50 sensory gating [34, 35]. Since P50 sensory gating is mainly mediated by neural oscillations in gamma and beta frequencies [36,37,38], it has been associated with the network of pyramidal neurons and GABAergic interneurons. It is hypothesised that when activated by auditory input, the interneurons release GABA that inhibits the activity of the pyramidal neurons, and thus diminishes the response to the second stimulus [39, 40]. P50 sensory gating has been proposed as a target for translational research, and the effect of pharmacological treatments on P50 sensory gating, especially drugs acting on nicotinic receptors, were found to be comparable across animal and human studies [39, 41].
P50 sensory gating impairment is also associated with the genetic risk of psychosis, making it a psychosis endophenotype. Twin studies of P50 sensory gating showed heritability estimates of about 68% [42], and increased P50 ratios have been found in the unaffected relatives of patients with psychosis (Cohen’s d = 2.26; 769 relatives and 3464 controls) [24]. Therefore, efforts have been made to explore the genetic basis of P50 sensory gating, as a strategy to elucidate biological processes relevant in psychosis. In line with the findings from pharmacological research, early candidate-gene and linkage analysis found that P50 sensory gating was associated with two α7 nicotinic acetylcholine receptor subunit genes (CHRNA7 and CHRNA7-like genes) [43,44,45]. While this association awaits replication by the latest genome-wide and sequencing approaches, the α7 nicotinic acetylcholine receptor could be a target for drug development for psychosis [41].
A few studies investigated P50 and N100 sensory gating in individuals at clinical high risk for psychosis, but the results were less consistent [20, 46,47,48]. While some studies reported impaired P50 and N100 sensory gating in those individuals [49, 50], others did not find such differences [51, 52]. A meta-analysis comparing high-risk individuals who developed psychosis with those who did not become ill found no differences in P50/N100 sensory gating between the two groups, but only three studies were available for analysis thus limiting statistical power [19].
In conclusion, P50 sensory gating abnormalities are prominent in patients with psychosis. The relatives of patients also display consistent if milder P50 suppression deficits, which supports its role as a biomarker of genetic predisposition to psychosis, especially in relation to the α7 nicotinic acetylcholine receptor genetic variants. We have less evidence supporting P50 sensory gating as a marker of the clinical risk for psychosis. There is also limited research on N100 sensory gating in psychosis, and it remains to be determined if the observed deficits were truly impairments in sensory gating or, instead, in sensory registration.
The N100 waveform as a marker of auditory processing
The N100 waveform, as a response to auditory stimuli, may also constitute a biomarker for psychosis. As the N100 can be easily elicited by any discernible auditory input regardless of task demand, various experimental designs have been used. The passive listening paradigm is perhaps the simplest way to measure the N100, where participants are presented with a series of identical auditory stimuli, and the N100 response is measured following each stimulus. Alternatively, in the auditory oddball task (widely used to measure the N100 and a range of ERPs), participants are asked to listen to a series of different auditory stimuli, including a small number of ‘deviant’ sounds randomly embedded in standard stimuli, which usually differ in pitch or duration [53]. The N100 can be observed following both the standard and the deviant stimuli, but the amplitude to the latter is consistently larger.
The literature comparing N100 between patients with psychosis and controls showed conflicting results [54]. However, N100 amplitude is strongly influenced by the interstimulus interval, with larger amplitudes observed in experiments using longer silent intervals [54, 55]. This reduces the statistical power of studies with short intervals to detect any group differences. Interestingly, this effect might also explain why the meta-analysis of N100 sensory gating only found decreased N100 amplitude to the first stimulus (C) but no changes to the second stimulus (T) in patients with schizophrenia, as only the first stimulus occurred after a long silent interval [28].
Although a review by Rosburg et al. concluded that the association between N100 amplitude and the general psychopathology of psychosis was weak [54], N100 deficits in patients could still reflect failures in specific domains of auditory processing related to the aetiology of psychosis. Given that the N1-P2 response complex can be used to measure hearing threshold, deficits in the N100 may well reflect subclinical hearing impairment. Indeed, hearing impairment is a risk factor for schizophrenia, with a pooled odds ratio of 3.15 in a meta-analysis of longitudinal studies [56]. Besides, some researchers suggested that N100 may reflect higher functions of the brain related to the detection of salient stimuli [57]. Patients with psychosis may have difficulty in detecting external stimuli due to the interference of internal stimuli [57], such as auditory hallucinations. Reduced N100 amplitude could also reflect a slower recovery of the N100 response in patients with psychosis compared to controls, which might explain why this impairment is more robustly detected at long interstimulus intervals [28].
The neurobiology of the N100 in relation to psychosis is not well understood. The N100 is mainly generated in the primary and association auditory cortices, as well as the frontal and motor cortices [58]. Some studies attempted to link the N100 to N-Methyl-d-Aspartate (NMDA) glutamate receptors function, which is implicated in the aetiology of psychosis [59]. However, the effect of NMDA receptor antagonists on the N100 is unclear, as previous studies investigating N40 in mice (the N100 analogous component in humans) reported mixed results [60, 61]. One study found that phencyclidine (an NMDA receptor antagonist) reduced N100 amplitude in monkeys at a long interstimulus interval [62], but another study reported that ketamine increased N100 amplitude in human participants [63].
In the oddball task, the heritability was estimated to be 60–70% for N100 amplitude [64, 65], and 56% for N100 latency at Cz [64]. However, it is unclear if the N100 response is impaired in the relatives of patients with psychosis due to limited and conflicting findings. While some studies reported reduced N100 amplitude or prolonged N100 latency in the unaffected relatives [66, 67], others did not find such differences [68]. Similarly, only a small number of studies have investigated N100 deficits amongst individuals at clinical high risk, with some reporting reduced N100 in clinical high-risk participants compared to controls [20, 50, 69]. In particular, one study found that a reduction in N100 amplitude between baseline and follow-up was only present in individuals at clinical high risk who made a transition to psychosis, but not in those without transition or controls [70].
The N100 remains under-researched in psychosis. Although reduced N100 amplitude at long interstimulus intervals has been reported in patients, whether or not the N100 is a marker for psychosis risk remains unclear, as there has been limited research in unaffected relatives and individuals at clinical high risk. Although N100 impairment is unlikely to be specific to psychosis [54], it offers a tool to elucidate the mechanisms through which deficits in auditory processing constitute a risk factor for psychosis. The literature covers a variety of experimental designs for the N100, usually obtained as a by-product in the oddball task, and we now need standardised paradigms targeted at a specific stage of auditory processing. An example of this is using N100 to measure corollary discharge, which is discussed in a later section in the review.
The mismatch negativity
The mismatch negativity (MMN) is elicited using the auditory oddball paradigm, and appears as an increased negativity in the ERP evoked by deviant relative to standard stimuli around 150–250 ms after stimulus onset. No actions or responses are required and the MMN occurs without participants’ cooperation, allowing the investigation of the early pre-attentive stages of auditory processing.
Reduced MMN amplitude is a well-established finding in patients with psychosis [71,72,73]. In a meta-analysis, Erickson et al. summarised 104 studies on the MMN in various clinical groups [71]. They found evidence that patients with schizophrenia (n = 3797) had reduced MMN amplitude compared to controls (n = 3960) with a large effect size (Hedges’ g = −0.95), as well as a medium effect size for reduced MMN amplitude in patients with bipolar disorder (n = 240; Hedges’ g = −0.37) [71]. The MMN might also reflect disease progression in psychosis, as the effect size of MMN reduction in first-episode schizophrenia (Hedges’ g = −0.42) is only a half of that in chronic schizophrenia (Hedges’ g = −0.81) [71].
The reductions of MMN amplitude in schizophrenia are interpreted as evidence of predictive coding deficits in the auditory system [74]. The predictive coding theory postulates that the brain compares the sensory input and its internal representation (model) inferred from the previous inputs while trying to minimise the discrepancy between the two [75]. The MMN is thought to reflect the prediction error of the model based on standard stimuli when a deviant stimulus occurs [76]. This theory has been supported by dynamic causal modelling of experimental EEG data [77, 78], and neuronal models that simulated MMN based on NMDA receptor neurotransmission [79]. Patients with psychosis are hypothesised to have a faulty prediction processing system.
Another meta-analysis pooling 3485 patients did not find evidence for an association between symptom severity and MMN in schizophrenia [80], indicating that although MMN deficits reflect liability for schizophrenia, they may not be symptom dependent. Although the MMN is often described as a pre-attentive component, Damaso et al. proposed that the MMN might serve as a call for the following automatic allocation of attentional resources [81]. Patients with psychosis are known to have attention and other cognitive deficits, although this paper did not find consistent correlations between performance in MMN and neuropsychological tests of attention [81]. MMN has also been associated with the functional outcomes of psychosis [82, 83], indicating its potential utility as a marker for recovery and a target for treatment.
The MMN is mainly generated by the auditory and prefrontal cortices [72], and thought to be a physiological marker of NMDA glutamate receptor neurotransmission. There is converging evidence that NMDA receptor antagonists, such as ketamine, decrease MMN amplitude and increase MMN latency in human subjects, akin to changes observed in patients with psychosis [84]. Although links with NMDA function have been established, the neural mechanisms of the MMN are still not well understood. Since the MMN mainly maps into the theta waves (4–7 Hz), the somatostatin-type GABAergic interneurons linked to lower-frequency oscillations, as well as their interplay with the glutamatergic pyramidal neurons, have been proposed to play a key role in MMN generation [85]. The leading candidate mechanism studied in animal models is a phenomenon called stimulus-specific adaptation, in which neurons show sensitivity to the probability of sensory stimuli over many seconds, for example in oddball sequences [86, 87]. Stimulus-specific adaptation is more pronounced in cortical than subcortical auditory areas [88], and at least in the auditory cortex, appears to be driven not only by sensory adaptation to frequent stimuli but also by heightened sensitivity to infrequent stimuli (true deviance detection) [89]. However, most studies of stimulus-specific adaptation in auditory cortical neurons have not found the combination of long latency, deviance detection, and NMDA dependence that is characteristic of the human MMN [90, 91]. Stimulus-specific adaptation may therefore be an early auditory precursor rather than a direct neural correlate of the MMN.
Reduced MMN amplitude might also be a marker of genetic risk for psychosis. MMN amplitude is a heritable trait, with heritability estimates ranging from 58% to 68% [42, 92]. Two meta-analyses both reported suggestive evidence of a decrease in MMN amplitude among the unaffected first-degree relatives of patients with schizophrenia (Hedges’ g = −0.26 [71] and −0.21 [93]). These were trends rather than clearly significant differences probably due to the limited number of studies available for inclusion. A recent transcriptome-wide association study from our group found that MMN is associated with the expression of two genes (FAM89A and ENGASE) in the cortex, and genes associated with MMN were overexpressed in the frontal cortex during prenatal development but under-expressed in adulthood [94]. This study also compared the genetic overlap between schizophrenia and the MMN as well as two other biomarkers (verbal memory and ventricular volume), and concluded that the MMN was a superior candidate endophenotype due to its greater genetic overlap with the disease [94].
The MMN is a promising marker for early detection of individuals at clinical high risk for psychosis. A significant reduction of MMN amplitude was reported amongst high-risk individuals (Hedges’ g = −0.40) in the meta-analysis by Erickson et al. [71]. Furthermore, Bodatsch et al. conducted a systematic review and meta-analysis that compared individuals at clinical high risk who developed psychosis and those who did not [19]. They found that the duration MMN amplitude had the largest pooled effect size (Hedges’ g = −0.71) among all investigated EEG markers (including duration/frequency MMN, N100 difference/ratio, and P50 difference/ratio), although duration MMN had the most studies included (n = 5) thus with the greatest statistical power [19].
In conclusion, research on the MMN has been fruitful and it is a well-established biomarker for psychosis. A promising area for MMN research focuses on its neurobiology, especially regarding the role of NMDA receptors in MMN generation, and its underlying neural circuits in the context of predictive coding. The MMN holds potential for clinical applicability towards detecting those individuals at clinical high risk most likely to develop psychosis, who would benefit from prompt access to treatments. There has been less research on the MMN among unaffected relatives with mixed results, and more research is needed to clarify its genetic basis.
The P300 waveform
The P300 is a positive voltage ERP, typically elicited by the auditory oddball paradigm [22, 95]. The P300 peak occurs ~300 ms following the presentation of a deviant (or target) stimulus which is randomly embedded in a series of frequent standard stimuli [20, 96, 97]. The P300 wave consists of two main sub-components occurring some 50 ms apart. The P3a is elicited by target stimuli that require no response from the participant and reflects attentional orienting and stimulus salience processing [20, 96, 97]. The following P3b is elicited by target stimuli requiring a response from the participant upon stimulus detection (for instance counting or pressing a button), and reflects working memory operations related to contextual updating [20, 96, 97].
Reduced P300 amplitude and prolonged P300 latency have consistently been found in patients with psychosis compared to healthy controls [22, 97,98,99]. A meta-analysis by our team comparing P300 between 1443 patients with schizophrenia and 1251 healthy controls reported a Cohen’s d of −0.85 for P300 amplitude and a Cohen’s d of 0.57 for P300 latency [22]. A more recent meta-analysis focusing on first-episode schizophrenia reported similar effect sizes (Cohen’s d = −0.83 for P300 amplitude and Cohen’s d = 0.48 for P300 latency) [99]. A meta-analysis of 30 studies with 1331 bipolar disorder patients (60% of whom had psychotic symptoms) and 1818 healthy controls reported that bipolar patients also exhibited reduced amplitude, and prolonged latency compared to the healthy subjects [97]. When examining only bipolar disorder with psychotic symptoms versus healthy controls, they found medium effect sizes with reduced amplitude (Hedges’ g = −0.58) and prolonged latencies (Hedges’ g = 0.52) [97].
The “context updating theory” postulates that the P300 amplitude reflects an attention-driven working memory comparison between new and repeated stimuli [20, 100]. Therefore, the more distinct the target is from the rest of the stimuli, the larger the P300 amplitude response will be. The P300 latency has been less precisely characterised but is thought to index processing speed, demonstrating how rapidly the individual responds to the stimulus, thus a neural correlate of reaction time [96]. Surprisingly, not many studies have reported the neuropsychological correlates of P300 in psychosis, except for some that found its association with memory performance [101, 102]. For clinical correlates, some studies reported negative correlations between P300 amplitude and symptom severity, especially for negative symptoms in psychosis [103, 104].
The P3a and P3b amplitudes show different topographic distributions, with P3a being prominent in the frontal lobes and P3b on the temporoparietal lobes [96, 97]. Given their different cortical sources, the P3a is hypothesised to be related to dopaminergic activity, while the P3b is related to norepinephrine activity [96, 100]. Indeed, some early pharmacological studies have found an effect of dopaminergic medications on the P300 (P3a) in humans [105, 106], although a later study using single-photon emission computerised tomography (SPECT) did not find any associations between dopamine transporter availability and P300 [107]. Besides, pharmacological studies using animal models also suggested an association between the locus coeruleus-norepinephrine system and the P300 (P3b) [108]. This is supported by a recent pilot study, which found that the P3b was influenced by transcutaneous vagus nerve stimulation (tVNS), a brain stimulation technique that may increase norepinephrine levels [109].
The P300 is one of the most well-established endophenotypes for psychosis, as similar deficits have been reported in the unaffected relatives of patients [93, 110, 111]. Heritability estimates are 68–80% for P300 amplitude and 21–56% for P300 latency [92]. When comparing unaffected relatives to controls, the latest meta-analyses reported a medium effect size (Hedges’ g) of −0.52 for P300 amplitude and 0.44 for P300 latency [93]. Malone et al. conducted a GWAS on P300 amplitude with 4211 participants, but found no hits that reached genome-wide significance, likely due to the limited sample size in a GWAS context [112]. Interestingly, their genome-wide analysis of all autosome genes identified an association with MYEF2 (myelin expression factor 2) [112], a promising finding awaiting replication.
Several studies described P300 deficits amongst individuals at clinical high risk compared to controls [95, 113,114,115]. A systematic review by Lepock et al. concluded that reduced P300 amplitude is a reliable finding in clinical high-risk individuals and thus has potential as a tool to identify people at risk [20]. However, fewer studies have compared the P300 between converters and non-converters within clinical high-risk individuals and the results were inconclusive [19, 70, 95]. In particular, a recent study by Tang et al. found that those people at clinical high risk who went on to develop psychosis had significantly lower P300 amplitude compared to healthy controls, and that high-risk cases who remitted had larger P300 amplitude compared to those remaining symptomatic or those who developed psychosis [95].
In summary, ample literature demonstrates that P300 deficits are evident not only in patients with psychosis, but also in their unaffected relatives and individuals at clinical high risk, thus, rendering P300 a correlate of genetic and clinical vulnerability to psychosis. However, despite the extensive literature on the P300 in psychosis, there has been limited research on its neuropsychological correlates and underlying neurobiology. Future pharmacological experiments across species, as well as studies employing other imaging methods and genomics, should help to address these gaps. The P300 could also have value for screening individuals at high risk of developing psychosis, although more data from longitudinal studies are needed to establish its potential for clinical applications.
Other neurophysiological markers for psychosis
N100 in corollary discharge
The N100 has also been used to measure deficits in corollary discharge in psychosis using the talk-listen paradigm. The corollary discharge theory contends that when individuals initiate an action, they also generate an ‘efferent copy’ of the same action in the brain [116]. The brain compares the actual and expected outcomes based on the efferent copy and suppresses the following response if the two outcomes are identical, which is referred to as ‘corollary discharge’ [116]. Patients with psychosis are hypothesised to have deficits in corollary discharge, and thus fail to suppress self-generated sensations. In the talk-listen paradigm, participants’ N100 responses are compared during listening to external stimuli and during their own speech. Several studies have found that patients with psychosis did not suppress N100 responses during their own speech to the same extent as controls [116,117,118]. As a theory-based approach, the N100 in the context of corollary discharge could illuminate the neurobiology of psychosis symptoms especially auditory hallucinations.
N400
The N400 is a negative voltage ERP elicited by stimuli that violate the semantic context, thus exploring language as a key skill frequently affected in psychosis. It is often measured using picture–word matching tasks [119]. Words that do not match preceding pictures trigger a larger N400 wave than the congruent word–picture pairs [119]. Previous studies have consistently found that patients with psychosis showed reduced N400 amplitude to semantically incongruent words compared to controls, as well as smaller priming effects (difference between N400 amplitude to congruent and incongruent words) [119, 120]. Similar deficits in N400 were also found in individuals at clinical high risk [121], but not in unaffected relatives of patients [122, 123], indicating that N400 might be a state marker for psychosis instead of an endophenotype of genetic predisposition.
Neural oscillations
In addition to ERPs, neural oscillations are also attractive biomarkers for psychosis due to their mapping into underlying local neural circuits and great transferability across species [124]. Deficits in gamma oscillations, measured by both evoked-activity paradigms and the auditory steady-state response paradigm, remain one of the most researched and well-replicated impairments in psychosis [125]. Abnormal gamma oscillations in psychosis are thought to be associated with the dysfunction of parvalbumin interneurons [126], which underlie gamma abnormalities in rodent models of NMDA receptor dysfunction [127]. By contrast, there has been conflicting evidence for abnormal neural oscillations in psychosis measured during resting state [125], and less research is available on lower-frequency oscillations. Future research should use animal models to elucidate the nature of such changes and help to identify new treatment targets.
Pre-pulse inhibition
Electromyography (EMG) has been used extensively to measure pre-pulse inhibition (PPI) in psychosis research. PPI is a marker of sensorimotor gating, referring to the attenuated amplitude of the startle response when a weak pre-stimulus is administered prior to a loud startling stimulus [128, 129]. Converging evidence in the literature shows that patients with psychosis exhibit impaired PPI compared to controls. A meta-analysis of 67 studies with a total of 4290 people with schizophrenia and 3685 healthy controls reported a lower level of PPI in the patient group with medium effect sizes (Hedges’ g) ranging from −0.50 to −0.44, depending on the interstimulus interval [130]. Two recent meta-analyses did not find evidence for PPI deficits in first-degree relatives of patients with psychosis [128], but found that individuals at clinical high risk had reduced PPI with an effect size (Cohen’s d) of −0.62 [129]. PPI deficits in psychosis are thought to share similar mechanisms with P50 and N100 sensory gating and are considered biomarkers of sensory information overload.
Discussion
The current review summarised the evidence for neurophysiological deficits in psychosis, as well as their potential research and clinical implications. There is strong evidence supporting deficits in P50 sensory gating, N100 amplitude (at long interstimulus intervals), MMN amplitude, and P300 amplitude/latency amongst patients with psychosis, rendering them neurophysiological biomarkers for the disease. Some of the neurophysiological markers may indicate the genetic or clinical risk of psychosis: although only deficits in P300 amplitude and latency have been established in both unaffected relatives and individuals at clinical high risk, there is also evidence supporting P50 gating impairment in unaffected relatives and MMN impairment in clinical high-risk individuals.
As EEG is a direct in vivo measure of neuronal electrical activity related to information processing, neurophysiological markers for psychosis are key to elucidate disease mechanisms. MMN and P300 are both related to the detection of deviant auditory stimuli. MMN explores the early pre-conscious stages of perception in the auditory cortex, while P300 captures higher-order cognitive processing including memory and attention. Although N100, MMN, and P300 can all be reliably measured by the auditory oddball paradigm, Rissling and Light proposed that they may represent three different underlying processes: sensory registration, automatic change detection, and attentional orientation/allocation [131]. Another group of neurophysiological markers, including P50 sensory gating, N100 sensory gating, and PPI, arise from other perceptual processes such as sensory adaptation and sensorimotor inhibition [132]. However, there has only been limited research investigating their clinical and neuropsychological correlates, and more work is needed to clarify the distinct and overlapping mechanisms between those biomarkers. Besides, while many hypotheses have been proposed, we need more compelling evidence to understand their neurobiology. Since many neurophysiological markers are comparable across species, translational and pharmacological studies using animal models are particularly useful to identify the underlying neural circuits and neurotransmitters. Other imaging methods, such as magnetoencephalography (MEG), SPECT, functional/structural magnetic resonance imaging (MRI), and non-invasive neurostimulation, should also be employed in conjunction with EEG to elucidate disease mechanisms in psychosis.
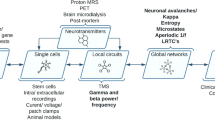
Figure 1 illustrates the utility of neurophysiological markers for psychosis in the multifactorial threshold model [133, 134]. In addition to deficits observed in patients, neurophysiological markers of genetic and clinical risk of psychosis can also improve our mechanistic understanding and potentially aid clinical practice. Genetic advances offer unprecedented insights into the aetiology of psychosis, yet imaging tools such as neurophysiological biomarkers of genetic risk (endophenotypes) are also key to elucidate disease mechanisms. As reviewed above, the classical method for testing the appropriateness of a potential endophenotype is through family studies that involve the unaffected relatives of people with psychosis [18], which are still needed for some markers, such as the N100. Another novel approach involves using polygenic risk scores to investigate if a neurophysiological marker is associated with common genetic variants increasing psychosis risk [135]. Once a biomarker has been established as an endophenotype, traditional methods such as candidate-gene and linkage analysis [43,44,45], as well as novel and hypothesis-free methods such as genome-wide or transcriptome-wide association studies [94, 112] and sequencing technologies can help identify its relevant genetic variants and biological pathways, which may serve as potential treatment targets. On the other hand, biomarkers of the clinical high-risk state could facilitate the early detection of those individuals, and thus accelerate access to timely treatments. While deficits in some markers have been well established in clinical high-risk populations (e.g. MMN amplitude, P300 amplitude/latency), others still await further research (e.g. P50 sensory gating, N100 amplitude). Previous research also suggests that about one in three individuals at clinical high risk will develop psychosis, while others remain at clinical high risk or even remit [136]. Therefore, identifying simple biological markers and tests that can help clinicians identify those people most likely to transition to psychosis before severe symptoms emerge is crucial. The neurophysiology literature in this field is promising but currently scarce.

Neurophysiological deficits found amongst unaffected relatives can serve as biomarkers of genetic risk to identify relevant biological mechanisms, while neurophysiological tests in high-risk individuals have potential to aid clinical practice in early detection and treatments for psychosis.
It is worth noting potential confounders, particularly antipsychotic medications, in case-control studies. The vast majority of neurophysiological studies involved participants taking antipsychotics. Although medication-free patients with first-episode psychosis exhibit similar neurophysiological deficits to chronic patients, such deficits are usually reported to be milder [71, 93]. Indeed, a systematic review and meta-analysis by Jackson and Seneviratne found that some antipsychotics, in particular clozapine, were associated with EEG slowing and epileptiform discharges (odds ratios = 16.9 and 6.2, respectively) [137]. Surprisingly, only a few studies have investigated the effect of antipsychotics on ERPs, mostly on small samples [138,139,140,141,142,143,144]. This highlights the importance of research in medication-free participants, as well as studies monitoring patients’ EEG or ERP changes before and after taking medication. Furthermore, there is substantial methodological heterogeneity in neurophysiological research in psychosis. The same EEG or ERP marker could be elicited using diverse paradigms and analysed by different methods. Methodological heterogeneity may also explain why there have been fewer systematic reviews or meta-analyses on certain markers, such as N100 and neural oscillations. Such evidence in previous literature still needs to be summarised systematically, and standardised paradigms and analysis pipelines would benefit future studies.
To conclude, EEG and ERPs are promising due to their ubiquity, temporal resolution and capacity for cross-species insights into neurobiology. Although there is evidence for deficits in P50 sensory gating, N100 amplitude, MMN amplitude, and P300 amplitude/latency in patients with psychosis, we need more studies to validate their role as markers for psychosis risk amongst unaffected relatives and individuals at clinical high risk. The combined use of neurophysiological markers and genetics holds great potential to illuminate the aetiology of psychosis and facilitate the development of new drugs, while research in high-risk individuals could allow more rapid access to psychological and medical treatments for people with psychosis.
References
Perälä J, Suvisaari J, Saarni SI, Kuoppasalmi K, Isometsä E, Pirkola S, et al. Lifetime prevalence of psychotic and bipolar I disorders in a general population. Arch Gen Psychiatry. 2007;64:19–28.
Charlson FJ, Ferrari AJ, Santomauro DF, Diminic S, Stockings E, Scott JG, et al. Global epidemiology and burden of schizophrenia: findings from the global burden of disease study 2016. Schizophr Bull. 2018;44:1195–203.
Ferrari AJ, Stockings E, Khoo JP, Erskine HE, Degenhardt L, Vos T, et al. The prevalence and burden of bipolar disorder: findings from the Global Burden of Disease Study 2013. Bipolar Disord. 2016;18:440–50.
Olesen J, Gustavsson A, Svensson M, Wittchen HU, Jönsson B. The economic cost of brain disorders in Europe. Eur J Neurol. 2012;19:155–62.
Cloutier M, Aigbogun MS, Guerin A, Nitulescu R, Ramanakumar AV, Kamat SA, et al. The economic burden of schizophrenia in the United States in 2013. J Clin Psychiatry. 2016;77:764–71.
Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35.
Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019;51:793–803.
Trubetskoy V, Pardiñas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. Nature (in the press).
Taylor DM, Barnes TRE, Young AH. The Maudsley prescribing guidelines in psychiatry. 13th ed. Wiley: Hoboken, NJ; 2018.
Killaspy H. Contemporary mental health rehabilitation. Epidemiol Psychiatr Sci 2019;28:1–3.
Peters E, Crombie T, Agbedjro D, Johns LC, Stahl D, Greenwood K, et al. The long-term effectiveness of cognitive behavior therapy for psychosis within a routine psychological therapies service. Front Psychol 2015;6. https://doi.org/10.3389/fpsyg.2015.01658.
Howes OD, Whitehurst T, Shatalina E, Townsend L, Onwordi EC, Mak TLA, et al. The clinical significance of duration of untreated psychosis: an umbrella review and random-effects meta-analysis. World Psychiatry. 2021;20:75–95.
Haddad P, Brain C, Scott J. Nonadherence with antipsychotic medication in schizophrenia: challenges and management strategies. Patient Relat Outcome Meas. 2014;5:43.
Fenton WS, Blyler CR, Heinssen RK. Determinants of medication compliance in schizophrenia: empirical and clinical findings. Schizophr Bull. 1997;23:637–51.
Strimbu K, Tavel JA. What are biomarkers? Curr Opin HIV AIDS. 2010;5:463–6.
Velthorst E, Mollon J, Murray RM, de Haan L, Germeys IM, Glahn DC, et al. Cognitive functioning throughout adulthood and illness stages in individuals with psychotic disorders and their unaffected siblings. Mol Psychiatry. 2021;10:1–15.
Cardno AG, Marshall EJ, Coid B, Macdonald AM, Ribchester TR, Davies NJ, et al. Heritability estimates for psychotic disorders: The Maudsley Twin psychosis series. Arch Gen Psychiatry. 1999;56:162–8.
Gottesman II, Gould TD. The endophenotype concept in psychiatry: Etymology and strategic intentions. Am J Psychiatry. 2003;160:636–45.
Bodatsch M, Brockhaus-Dumke A, Klosterkötter J, Ruhrmann S. Forecasting psychosis by event-related potentials—systematic review and specific meta-analysis. Biol Psychiatry. 2015;77:951–8.
Lepock JR, Mizrahi R, Korostil M, Bagby RM, Pang EW, Kiang M. Event-related potentials in the clinical high-risk (CHR) state for psychosis: a systematic review. Clin EEG Neurosci. 2018;49:215–25.
Fusar-Poli P, Borgwardt S, Bechdolf A, Addington J, Riecher-Rössler A, Schultze-Lutter F, et al. The psychosis high-risk state: a comprehensive state-of-the-art review. Arch Gen Psychiatry 2013;70:107–20.
Bramon E, Rabe-Hesketh S, Sham P, Murray RM, Frangou S. Meta-analysis of the P300 and P50 waveforms in schizophrenia. Schizophr Res. 2004;70:315–29.
Xia L, Wang D, Wang J, Xu H, Huo L, Tian Y, et al. Association of cognitive and P50 suppression deficits in chronic patients with schizophrenia. Clin Neurophysiol. 2020;131:725–33.
Atagun MI, Drukker M, Hall MH, Altun IK, Tatli SZ, Guloksuz S, et al. Meta-analysis of auditory P50 sensory gating in schizophrenia and bipolar disorder. Psychiatry Res—Neuroimaging. 2020;300:111078.
Adler LE, Pachtman E, Franks RD, Pecevich M, Waldo MC, Freedman R. Neurophysiological evidence for a defect in neuronal mechanisms involved in sensory gating in schizophrenia. Biol Psychiatry. 1982;17:639–54.
de Wilde OM, Bour LJ, Dingemans PM, Koelman JHTM, Linszen DH. A meta-analysis of P50 studies in patients with schizophrenia and relatives: Differences in methodology between research groups. Schizophr Res. 2007;97:137–51.
Shen CL, Chou TL, Lai WS, Hsieh MH, Liu CC, Liu CM, et al. P50, N100, and P200 auditory sensory gating deficits in schizophrenia patients. Front Psychiatry. 2020;11:868.
Rosburg T. Auditory N100 gating in patients with schizophrenia: a systematic meta-analysis. Clin Neurophysiol. 2018;129:2099–111.
Erwin RJ, Turetsky BI, Moberg P, Gur RC, Gur RE. P50 abnormalities in schizophrenia: relationship to clinical and neuropsychological indices of attention. Schizophr Res. 1998;33:157–67.
Cullum CM, Harris JG, Waldo MC, Smernoff E, Madison A, Nagamoto HT, et al. Neurophysiological and neuropsychological evidence for attentional dysfunction in schizophrenia. Schizophr Res. 1993;10:131–41.
Thoma RJ, Hanlon FM, Moses SN, Edgar JC, Huang M, Weisend MP, et al. Lateralization of auditory sensory gating and neuropsychological dysfunction in schizophrenia. Am J Psychiatry. 2003;160:1595–605.
Potter D, Summerfelt A, Gold J, Buchanan RW. Review of clinical correlates of P50 sensory gating abnormalities in patients with schizophrenia. Schizophr Bull. 2005;32:692–700.
Bickford-Wimer PC, Nagamoto H, Johnson R, Adler LE, Egan M, Rose GM, et al. Auditory sensory gating in hippocampal neurons: a model system in the rat. Biol Psychiatry. 1990;27:183–92.
Korzyukov O, Pflieger ME, Wagner M, Bowyer SM, Rosburg T, Sundaresan K, et al. Generators of the intracranial P50 response in auditory sensory gating. Neuroimage. 2007;35:814–26.
Williams TJ, Nuechterlein KH, Subotnik KL, Yee CM. Distinct neural generators of sensory gating in schizophrenia. Psychophysiology. 2011;48:470–8.
Nguyen AT, Hetrick WP, O’Donnell BF, Brenner CA. Abnormal beta and gamma frequency neural oscillations mediate auditory sensory gating deficit in schizophrenia. J Psychiatr Res. 2020;124:13–21.
Hall MH, Taylor G, Salisbury DF, Levy DL. Sensory gating event-related potentials and oscillations in schizophrenia patients and their unaffected relatives. Schizophr Bull. 2011;37:1187–99.
Hong LE, Summerfelt A, McMahon RP, Thaker GK, Buchanan RW. Gamma/beta oscillation and sensory gating deficit in schizophrenia. Neuroreport. 2004;15:155–9.
Smucny J, Stevens KE, Olincy A, Tregellas JR. Translational utility of rodent hippocampal auditory gating in schizophrenia research: a review and evaluation. Transl Psychiatry 2015;5:587.
Vlcek P, Bob P, Raboch J. Sensory disturbances, inhibitory deficits, and the P50 wave in schizophrenia. Neuropsychiatr Dis Treat. 2014;10:1309–15.
Hashimoto K. Targeting of α7 nicotinic acetylcholine receptors in the treatment of schizophrenia and the use of auditory sensory gating as a translational biomarker. Curr Pharm Des. 2015;21:3797–806.
Hall MH, Schulze K, Rijsdijk F, Picchioni M, Ettinger U, Bramon E, et al. Heritability and reliability of P300, P50 and duration mismatch negativity. Behav Genet. 2006;36:845–57.
Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, et al. Association of promoter variants in the α7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psychiatry. 2002;59:1085–96.
Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci USA. 1997;94:587–92.
Raux G, Bonnet-Brilhault F, Louchart S, Houy E, Gantier R, Levillain D, et al. The -2 bp deletion in exon 6 of the ‘alpha 7-like’ nicotinic receptor subunit gene is a risk factor for the P50 sensory gating deficit. Mol Psychiatry. 2002;7:1006–11.
Chang Q, Liu M, Tian Q, Wang H, Luo Y, Zhang J, et al. EEG-based brain functional connectivity in first-episode schizophrenia patients, ultra-high-risk individuals, and healthy controls during P50 suppression. Front Hum Neurosci. 2019;13:379.
Hsieh MH, Lin YT, Chien YL, Hwang TJ, Hwu HG, Liu CM, et al. Auditory event-related potentials in antipsychotic-free subjects with ultra-high-risk state and first-episode psychosis. Front Psychiatry. 2019;10:223.
Shaikh M, Dutt A, Broome MR, Vozmediano AG, Ranlund S, Diez A, et al. Sensory gating deficits in the attenuated psychosis syndrome. Schizophr Res. 2015;161:277–82.
Brockhaus-Dumke A, Schultze-Lutter F, Mueller R, Tendolkar I, Bechdolf A, Pukrop R, et al. Sensory gating in schizophrenia: P50 and N100 gating in antipsychotic-free subjects at risk, first-episode, and chronic patients. Biol Psychiatry. 2008;64:376–84.
Gonzalez-Heydrich J, Bosquet Enlow M, D’Angelo E, Seidman LJ, Gumlak S, Kim A, et al. Early auditory processing evoked potentials (N100) show a continuum of blunting from clinical high risk to psychosis in a pediatric sample. Schizophr Res. 2015;169:340–5.
Ziermans TB, Schothorst PF, Sprong M, MJCM Magnée, van Engeland H, Kemner C. Reduced prepulse inhibition as an early vulnerability marker of the psychosis prodrome in adolescence. Schizophr Res. 2012;134:10–15.
Van Tricht MJ, Nieman DH, Koelman JTM, Mensink AJM, Bour LJ, Van Der Meer JN, et al. Sensory gating in subjects at ultra high risk for developing a psychosis before and after a first psychotic episode. World J Biol Psychiatry. 2015;16:12–21.
Hillyard SA, Hink RF, Schwent VL, Picton TW. Electrical signs of selective attention in the human brain. Science (80-). 1973;182:177–80.
Rosburg T, Boutros NN, Ford JM. Reduced auditory evoked potential component N100 in schizophrenia—a critical review. Psychiatry Res. 2008;161:259–74.
Roth WT, Horvath TB, Pfefferbaum A, Kopell BS. Event-related potentials in schizophrenics. Electroencephalogr Clin Neurophysiol. 1980;48:127–39.
Linszen MMJ, Brouwer RM, Heringa SM, Sommer IE. Increased risk of psychosis in patients with hearing impairment: review and meta-analyses. Neurosci Biobehav Rev. 2016;62:1–20.
Joos K, Gilles A, Van de Heyning P, De Ridder D, Vanneste S. From sensation to percept: the neural signature of auditory event-related potentials. Neurosci Biobehav Rev 2014;42:148–56.
Näätänen R, Picton T. The N1 wave of the human electric and magnetic response to sound: a review and an analysis of the component structure. Psychophysiology. 1987;24:375–425.
Kantrowitz JT. N-methyl-d-aspartate-type glutamate receptor modulators and related medications for the enhancement of auditory system plasticity in schizophrenia. Schizophr Res. 2019;207:70–79.
Connolly PM, Maxwell C, Liang Y, Kahn JB, Kanes SJ, Abel T, et al. The effects of ketamine vary among inbred mouse strains and mimic schizophrenia for the P80, but not P20 or N40 auditory ERP components. Neurochem Res 2004;29:1179–88.
Ehrlichman RS, Maxwell CR, Majumdar S, Siegel SJ. Deviance-elicited changes in event-related potentials are attenuated by ketamine in mice. J Cogn Neurosci. 2008;20:1403–14.
Javitt DC, Jayachandra M, Lindsley RW, Specht CM, Schroeder CE. Schizophrenia-like deficits in auditory P1 and N1 refractoriness induced by the psychomimetic agent phencyclidine (PCP). Clin Neurophysiol. 2000;111:833–6.
Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 2000;57:1139–47.
O’Connor S, Morzorati S, Christian JC, Li TK. Heritable features of the auditory oddball event-related potential: peaks, latencies, morphology and topography. Electroencephalogr Clin Neurophysiol Evoked Potentials. 1994;92:115–25.
Ahveninen J, Jaaskelainen IP, Osipova D, Huttunen MO, Ilmoniemi RJ, Kaprio J, et al. Inherited auditory-cortical dysfunction in twin pairs discordant for schizophrenia. Biol Psychiatry. 2006;60:612–20.
Simons CJP, Sambeth A, Krabbendam L, Pfeifer S, van Os J, Riedel WJ, et al. Auditory P300 and N100 components as intermediate phenotypes for psychotic disorder: familial liability and reliability. Clin Neurophysiol. 2011;122:1984–90.
Foxe JJ, Yeap S, Snyder AC, Kelly SP, Thakore JH, Molholm S, et al. The N1 auditory evoked potential component as an endophenotype for schizophrenia: High-density electrical mapping in clinically unaffected first-degree relatives, first-episode, and chronic schizophrenia patients. Eur Arch Psychiatry Clin Neurosci. 2011;261:331–9.
Blackwood DHR, Clair DM, Muir WJ, Duffy JC. Auditory P300 and eye tracking dysfunction in schizophrenic pedigrees. Arch Gen Psychiatry. 1991;48:899–909.
del Re EC, Spencer KM, Oribe N, Mesholam-Gately RI, Goldstein J, Shenton ME, et al. Clinical high risk and first episode schizophrenia: auditory event-related potentials. Psychiatry Res - Neuroimaging. 2015;231:126–33.
van Tricht MJ, Nieman DH, Koelman JHTM, Bour LJ, van der Meer JN, van Amelsvoort TA, et al. Auditory ERP components before and after transition to a first psychotic episode. Biol Psychol. 2011;87:350–7.
Erickson MA, Ruffle A, Gold JM, Earls HA, Curran T, Mittal V, et al. A meta-analysis of mismatch negativity in schizophrenia: from clinical risk to disease specificity and progression. Biol Psychiatry. 2016;79:980–7.
Haigh SM, Coffman BA, Salisbury DF. Mismatch negativity in first-episode schizophrenia: a meta-analysis. Clin EEG Neurosci. 2017;48:3–10.
Avissar M, Xie S, Vail B, Lopez-Calderon J, Wang Y, Javitt DC. Meta-analysis of mismatch negativity to simple versus complex deviants in schizophrenia. Schizophr Res. 2018;191:25–34.
Heilbron M, Chait M. Great expectations: is there evidence for predictive coding in auditory cortex? Neuroscience 2018;389:54–73.
Friston K. A theory of cortical responses. Philos Trans R Soc B Biol Sci. 2005;360:815–36.
Randeniya R, Oestreich LKL, Garrido MI. Sensory prediction errors in the continuum of psychosis. Schizophr Res. 2018;191:109–22.
Ranlund S, Adams RA, Dutt A, Hall M, Carbayo AM, Mcdonald C, et al. Impaired prefrontal synaptic gain in people with psychosis and their relatives during the mismatch negativity. Hum Brain Mapp. 2016;37:351–65.
Garrido MI, Kilner JM, Kiebel SJ, Friston KJ. Dynamic causal modeling of the response to frequency deviants. J Neurophysiol. 2009;101:2620–31.
Wacongne C. A predictive coding account of MMN reduction in schizophrenia. Biol Psychol. 2016;116:68–74.
Erickson MA, Albrecht M, Ruffle A, Fleming L, Corlett P, Gold J. No association between symptom severity and MMN impairment in schizophrenia: a meta-analytic approach. Schizophr Res Cogn. 2017;9:13–17.
Damaso KAM, Michie PT, Todd J. Paying attention to MMN in schizophrenia. Brain Res. 2015;1626:267–79.
Lee SH, Sung K, Lee KS, Moon E, Kim CG. Mismatch negativity is a stronger indicator of functional outcomes than neurocognition or theory of mind in patients with schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry. 2014;48:213–9.
Wynn JK, Sugar C, Horan WP, Kern R, Green MF. Mismatch negativity, social cognition, and functioning in schizophrenia patients. Biol Psychiatry. 2010;67:940–7.
Rosburg T, Kreitschmann-Andermahr I. The effects of ketamine on the mismatch negativity (MMN) in humans-a meta-analysis. Clin Neurophysiol. 2016;127:1387–94.
Javitt DC, Lee M, Kantrowitz JT, Martinez A. Mismatch negativity as a biomarker of theta band oscillatory dysfunction in schizophrenia. Schizophr Res. 2018;191:51–60.
Ulanovsky N, Las L, Nelken I. Processing of low-probability sounds by cortical neurons. Nat Neurosci. 2003;6:391–8.
Nelken I. Stimulus-specific adaptation and deviance detection in the auditory system: experiments and models. Biol Cybern 2014;108:655–63.
Parras GG, Nieto-Diego J, Carbajal GV, Valdés-Baizabal C, Escera C, Malmierca MS. Neurons along the auditory pathway exhibit a hierarchical organization of prediction error. Nat Commun 2017;8. https://doi.org/10.1038/s41467-017-02038-6.
Harpaz M, Jankowski MM, Khouri L, Nelken I. Emergence of abstract sound representations in the ascending auditory system. Prog Neurobiol. 2021;202:102049.
Grimm S, Escera C, Nelken I. Early indices of deviance detection in humans and animal models. Biol Psychol. 2016;116:23–27.
Chen IW, Helmchen F, Lütcke H. Specific early and late oddball-evoked responses in excitatory and inhibitory neurons of mouse auditory cortex. J Neurosci. 2015;35:12560–73.
Hall MH, Schulze K, Rijsdijk F, Kalidindi S, McDonald C, Bramon E, et al. Are auditory P300 and duration MMN heritable and putative endophenotypes of psychotic bipolar disorder? A Maudsley Bipolar Twin and Family Study. Psychol Med. 2009;39:1277–87.
Earls HA, Curran T, Mittal V. A meta-analytic review of auditory event-related potential components as endophenotypes for schizophrenia: perspectives from first-degree relatives. Schizophr Bull. 2016;42:1504–16.
Bhat A, Irizar H, Thygesen JH, Kuchenbaecker K, Pain O, Adams RA, et al. Transcriptome-wide association study reveals two genes that influence mismatch negativity. Cell Rep. 2021;34:108868.
Tang Y, Wang J, Zhang T, Xu L, Qian Z, Cui H, et al. P300 as an index of transition to psychosis and of remission: data from a clinical high risk for psychosis study and review of literature. Schizophr Res. 2020;226:74–83.
Polich J. Updating P300: an integrative theory of P3a and P3b. Clin Neurophysiol 2007;118:2128–48.
Wada M, Kurose S, Miyazaki T, Nakajima S, Masuda F, Mimura Y, et al. The P300 event-related potential in bipolar disorder: a systematic review and meta-analysis. J Affect Disord 2019;256:234–49.
Jeon YW, Polich J. Meta-analysis of P300 and schizophrenia: patients, paradigms, and practical implications. Psychophysiology. 2003;40:684–701.
Qiu YQ, Tang YX, Chan RCK, Sun XY, He J. P300 aberration in first-episode schizophrenia patients: a meta-analysis. PLoS ONE. 2014;9:e97794.
Polich J. Neuropsychology of P300. In: The Oxford handbook of event-related potential components (eds Luck SJ, Kappenman ES). Oxford University Press: New York, NY; 2011.
Kim MS, Kang SS, Youn T, Kang DH, Kim JJ, Kwon JS. Neuropsychological correlates of P300 abnormalities in patients with schizophrenia and obsessive-compulsive disorder. Psychiatry Res-Neuroimaging. 2003;123:109–23.
Shajahan PM, O’Carroll RE, Glabus MF, Ebmeier KP, Blackwood DHR. Correlation of auditory ‘oddball’ P300 with verbal memory deficits in schizophrenia. Psychol Med. 1997;27:579–86.
Perlman G, Foti D, Jackson F, Kotov R, Constantino E, Hajcak G. Clinical significance of auditory target P300 subcomponents in psychosis: Differential diagnosis, symptom profiles, and course. Schizophr Res. 2015;165:145–51.
Kim DW, Shim M, Kim JI, Im CH, Lee SH. Source activation of P300 correlates with negative symptom severity in patients with schizophrenia. Brain Topogr. 2014;27:307–17.
Hansenne M, Pitchot W, Gonzalez Moreno A, Papart P, Timsit-Berthier M, Ansseau M. Catecholaminergic function and P300 amplitude in major depressive disorder (P300 and catecholamines). Electroencephalogr Clin Neurophysiol. 1995;96:194–6.
Wang L, Kuroiwa Y, Li M, Kamitani T, Wang J, Takahashi T, et al. The correlation between P300 alterations and regional cerebral blood flow in non-demented Parkinson’s disease. Neurosci Lett. 2000;282:133–6.
Chen KC, Lee IH, Yang YK, Landau S, Chang WH, Chen PS, et al. P300 waveform and dopamine transporter availability: a controlled EEG and SPECT study in medication-naive patients with schizophrenia and a meta-analysis. Psychol Med. 2014;44:2151–62.
Nieuwenhuis S, Aston-Jones G, Cohen JD. Decision making, the P3, and the locus coeruleus-norepinephrine system. Psychol Bull. 2005;131:510–32.
Ventura-Bort C, Wirkner J, Genheimer H, Wendt J, Hamm AO, Weymar M. Effects of transcutaneous vagus nerve stimulation (tVNS) on the P300 and alpha-amylase level: a pilot study. Front Hum Neurosci 2018;12:202.
Blakey R, Ranlund S, Zartaloudi E, Cahn W, Calafato S, Colizzi M, et al. Associations between psychosis endophenotypes across brain functional, structural, and cognitive domains. Psychol Med. 2018;48:1325–40.
Bramon E, McDonald C, Croft RJ, Landau S, Filbey F, Gruzelier JH, et al. Is the P300 wave an endophenotype for schizophrenia? A meta-analysis and a family study. Neuroimage. 2005;27:960–8.
Malone SM, Vaidyanathan U, Basu S, Miller MB, Mcgue M, Iacono WG. Heritability and molecular-genetic basis of the P3 event-related brain potential: a genome-wide association study. Psychophysiology. 2014;51:1246–58.
Bramon E, Shaikh M, Broome M, Lappin J, Bergé D, Day F, et al. Abnormal P300 in people with high risk of developing psychosis. Neuroimage. 2008;41:553–60.
Kim M, Lee TY, Lee S, Kim SN, Kwon JS. Auditory P300 as a predictor of short-term prognosis in subjects at clinical high risk for psychosis. Schizophr Res. 2015;165:138–44.
Oribe N, Hirano Y, Re E, Mesholam‐Gately RI, Woodberry KA, Ueno T, et al. Longitudinal evaluation of visual P300 amplitude in clinical high‐risk subjects: an event‐related potential study. Psychiatry Clin Neurosci. 2020;74:527–34.
Parlikar R, Bose A, Venkatasubramanian G. Schizophrenia and corollary discharge: a neuroscientific overview and translational implications. Clin Psychopharmacol Neurosci. 2019;17:170–82.
Ford JM, Gray M, Faustman WO, Roach BJ, Mathalon DH. Dissecting corollary discharge dysfunction in schizophrenia. Psychophysiology. 2007;44:522–9.
Perez VB, Ford JM, Roach BJ, Loewy RL, Stuart BK, Vinogradov S, et al. Auditory cortex responsiveness during talking and listening: early illness schizophrenia and patients at clinical high-risk for psychosis. Schizophr Bull. 2012;38:1216–24.
Mathalon DH, Faustman WO, Ford JM. N400 and automatic semantic processing abnormalities in patients with schizophrenia. Arch Gen Psychiatry. 2002;59:641–8.
Mathalon DH, Roach BJ, Ford JM. Automatic semantic priming abnormalities in schizophrenia. Int J Psychophysiol. 2010;75:157–66.
Lepock JR, Ahmed S, Mizrahi R, Gerritsen CJ, Maheandiran M, Drvaric L, et al. Relationships between cognitive event-related brain potential measures in patients at clinical high risk for psychosis. Schizophr Res. 2020;226:84–94.
Kiang M, Christensen BK, Zipursky RB. Event-related brain potential study of semantic priming in unaffected first-degree relatives of schizophrenia patients. Schizophr Res. 2014;153:78–86.
Sharma A, Sauer H, Hill H, Kaufmann C, Bender S, Weisbrod M. Abnormal N400 semantic priming effect may reflect psychopathological processes in Schizophrenia: a twin study. Schizophr Res Treat 2017;2017. https://doi.org/10.1155/2017/7163198.
Javitt DC, Siegel SJ, Spencer KM, Mathalon DH, Hong LE, Martinez A, et al. A roadmap for development of neuro-oscillations as translational biomarkers for treatment development in neuropsychopharmacology. Neuropsychopharmacology 2020;45:1411–22.
Reilly TJ, Nottage JF, Studerus E, Rutigliano G, Micheli AID, Fusar-Poli P, et al. Gamma band oscillations in the early phase of psychosis: a systematic review. Neurosci Biobehav Rev 2018;90:381–99.
Gonzalez-Burgos G, Cho RY, Lewis DA. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry 2015;77:1031–40.
Carlén M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2011;17:537–48.
Li W, Mao Z, Bo Q, Sun Y, Wang Z, Wang C. Prepulse inhibition in first-degree relatives of schizophrenia patients: a systematic review. Early Interv Psychiatry 2020. https://doi.org/10.1111/eip.13003.
Li W, Mao Z, Bo Q, Sun Y, Wang Z, Wang C. Pre-pulse inhibition deficits in individuals at clinical high-risk for psychosis: a systematic review and meta-analysis. Early Interv Psychiatry 2020. https://doi.org/10.1111/eip.13015.
San-Martin R, Castro LA, Menezes PR, Fraga FJ, Simões PW, Salum C. Meta-analysis of sensorimotor gating deficits in patients with schizophrenia evaluated by prepulse inhibition test. Schizophr Bull. 2020;46:1482–97.
Rissling AJ, Light GA. Neurophysiological measures of sensory registration, stimulus discrimination, and selection in schizophrenia patients. Curr Top Behav Neurosci. 2010;4:283–309.
Oranje B, Geyer MA, Bocker KBE, Leon Kenemans J, Verbaten MN. Prepulse inhibition and P50 suppression: commonalities and dissociations. Psychiatry Res. 2006;143:147–58.
McGue M, Gottesman II, Rao DC. The transmission of schizophrenia under a multifactorial threshold model. Am J Hum Genet. 1983;35:1161–78.
Gottesman II, Shields J. A polygenic theory of schizophrenia. Proc Natl Acad Sci USA. 1967;58:199–205.
Ranlund S, Calafato S, Thygesen JH, Lin K, Cahn W, Crespo-Facorro B, et al. A polygenic risk score analysis of psychosis endophenotypes across brain functional, structural, and cognitive domains. Am J Med Genet Part B. 2018;177:21–34.
Davies C, Cipriani A, Ioannidis JPA, Radua J, Stahl D, Provenzani U, et al. Lack of evidence to favor specific preventive interventions in psychosis: a network meta-analysis. World Psychiatry. 2018;17:196–209.
Jackson A, Seneviratne U. EEG changes in patients on antipsychotic therapy: a systematic review. Epilepsy Behav. 2019;95:1–9.
Umbricht D, Javitt D, Novak G, Bates J, Pollack S, Lieberman J, et al. Effects of clozapine on auditory event-related potentials in schizophrenia. Biol Psychiatry. 1998;44:716–25.
Gonul AS, Suer C, Coburn K, Ozesmi Ç, Oguz A, Yilmaz A. Effects of olanzapine on auditory P300 in schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry. 2003;27:173–7.
Zhou Z, Zhu H, Chen L. Effect of aripiprazole on mismatch negativity (MMN) in schizophrenia. PLoS ONE. 2013;8:e52186.
Blackwood DHR, Whalley LJ, Christie JE, Blackburn IM, St Clair DM, McInnes A. Changes in auditory P3 event-related potential in schizophrenia and depression. Br J Psychiatry. 1987;150:154–60.
Adler G, Gattaz WF. Auditory evoked potentials in schizophrenic patients before and during neuroleptic treatment—relationship to psychopathological state. Eur Arch Psychiatry Clin Neurosci. 1993;242:357–61.
Nagamoto HT, Adler LE, Hea RA, Griffith JM, McRae KA, Freedman R. Gating of auditory P50 in schizophrenics: unique effects of clozapine. Biol Psychiatry. 1996;40:181–8.
Nagamoto HT, Adler LE, McRae KA, Huettl P, Cawthra E, Gerhardt G, et al. Auditory P50 in schizophrenics on clozapine: improved gating parallels clinical improvement and changes in plasma 3-methoxy-4-hydroxyphenylglycol. Neuropsychobiology. 1999;39:10–17.
Acknowledgements
Elvira Bramon thanks the following funders: The NIHR (grants NIHR200756 and PDA/02/06/016); The Medical Research Council (G0901310, G1100583 and MR/W020238/1); Mental Health Research UK John Grace QC Scholarship 2018; The Wellcome Trust (085475/B/08/Z and 085475/Z/08/Z); The British Medical Association’s Margaret Temple Fellowship; NARSAD Young Investigator Awards 2005 and 2008; The NIHR Biomedical Research Centre at UCLH NHS Foundation Trust and University College London (Mental Health Theme). Jennifer F. Linden thanks the NIHR Biomedical Research Centre at UCLH NHS Foundation Trust and University College London (Deafness and Hearing Problems Theme). Baihan Wang thanks the China Scholarship Council-UCL Joint Research Scholarship.
Author information
Authors and Affiliations
Contributions
Conceptualisation, BW, EZ, and EB; Writing—Original Draft Preparation, BW, EZ, and EB; Writing—Review and Editing, BW, EZ, JFL, and EB; Supervision, EB.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, B., Zartaloudi, E., Linden, J.F. et al. Neurophysiology in psychosis: The quest for disease biomarkers. Transl Psychiatry 12, 100 (2022). https://doi.org/10.1038/s41398-022-01860-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-01860-x
