
Abstract
Cross-species translational approaches to human genomic analyses are lacking. The present study uses an integrative framework to investigate how genes associated with nicotine use in model organisms contribute to the genetic architecture of human tobacco consumption. First, we created a model organism geneset by collecting results from five animal models of nicotine exposure (RNA expression changes in brain) and then tested the relevance of these genes and flanking genetic variation using genetic data from human cigarettes per day (UK BioBank N = 123,844; all European Ancestry). We tested three hypotheses: (1) DNA variation in, or around, the ‘model organism geneset’ will contribute to the heritability to human tobacco consumption, (2) that the model organism genes will be enriched for genes associated with human tobacco consumption, and (3) that a polygenic score based off our model organism geneset will predict tobacco consumption in the AddHealth sample (N = 1667; all European Ancestry). Our results suggested that: (1) model organism genes accounted for ~5–36% of the observed SNP-heritability in human tobacco consumption (enrichment: 1.60–31.45), (2) model organism genes, but not negative control genes, were enriched for the gene-based associations (MAGMA, H-MAGMA, SMultiXcan) for human cigarettes per day, and (3) polygenic scores based on our model organism geneset predicted cigarettes per day in an independent sample. Altogether, these findings highlight the advantages of using multiple species evidence to isolate genetic factors to better understand the etiological complexity of tobacco and other nicotine consumption.
Similar content being viewed by others

Introduction
Contemporary thought on human genetic research is that large genome-wide association studies (GWAS) are required to identify reproducible single nucleotide polymorphism (SNP) associations that can lead to insights into biological systems that underpin a particular phenotype. The agnostic nature of GWAS (i.e., all SNPs being tested without bias) enables the identification of previously unrecognized biological underpinnings for human traits. However, GWASs are not without limitations. One limitation is the stiff penalty for multiple comparisons leading to the need for increasingly large sample sizes. The requirement of sample sizes in the 100’s of thousands to millions (i.e., mega-GWAS) exerts pressure on the depth of phenotyping that may be done (i.e., more intensive and costly phenotypes are untenable for mega-GWAS studies). Additionally, SNPs implicated by GWAS are not always readily associated with gene function. Specifically, the majority of GWAS associations reside in non-coding or intergenic regions1. To help make sense of these signals, studies rely upon arbitrarily defined gene regulatory regions (up/downstream of a gene). While GWAS findings have become increasingly reproducible as sample sizes increase, additional sources of data (e.g., gene regulatory and epigenetic data2) are needed to understand how SNP effects increase the risk for trait pathology3.
Association studies of tobacco consumption assume that variation in the biological sample collected (e.g., DNA extracted from blood and saliva) reflects the genetic influences in the brain that mediate the psychoactive properties of nicotine and other chemicals found in tobacco products. Nicotine causes changes in the neural organization, particularly in the brain’s reward systems, psychomotor, and cognitive processes via its ability to interact with nicotinic acetylcholine receptors (nAChRs)4,5. Nicotine takes on the properties of a reinforcer by altering neural circuits, in particular those comprising the dopaminergic systems of the midbrain6. Altogether, these properties of tobacco products highlight putative genetic mechanisms that may mediate consumption. The largest tobacco consumption meta-GWAS, to date, has identified 566 genetic variants in 406 loci associated with phenotypes related to tobacco consumption (i.e., initiation, cessation, and heaviness of use)7, yet how most of these gene variants contribute to specific tobacco behaviors is unknown.
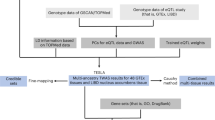
The lack of accessible human brain tissue for tobacco use has precluded understanding of how gene variants contribute to tissue-specific epigenetic and/or expression differences that arise from continued drug exposure. We used a novel and integrative framework that combined human GWAS data with high throughput model organism data to clarify how the genetic liability to human tobacco consumption relates to specific nicotine behaviors in particular brain regions. We hypothesized that genes associated with nicotine exposure paradigms in model organisms will: (1) contribute to the heritability to human tobacco consumption, (2) be enriched for genes associated with human tobacco consumption, and (3) aid in the prediction of a polygenic score of human tobacco consumption in an independent sample (see Fig. 1).

Schematic of analytical pipeline used for cross-species analysis. Panels indicate: 1) Derivation of gene list using genes associated with nicotine exposure from various animal paradigms. 2) Multicomponent SNP-heritability analysis in UKBiobank smokers using mixed linear models. 3) Examination of gene-list overlap using Jaccard similarity and Fisher exact tests. 4) Multicomponent polygenic score analysis using GWAS-LOCO summary statistics in an independent sample.
Materials and methods
Samples and phenotypes
To find genes associated with nicotine exposure, we used GeneWeaver8,9,10—a genomics and bioinformatics data repository and suite of tools. We created a model organism geneset that included brain-related RNA associations with animal paradigms of nicotine exposure from multiple species: Mus musculus, Rattus norvegicus, and Danio rerio (Table 1; GeneWeaver data gathered in October 2019). Aggregating across all GeneWeaver studies, we identified 786 orthologous genes with humans, which we dubbed “model organism genes”.
We used two independent human datasets from two different countries investigating the same trait—cigarettes per day (CPD; tobacco consumption). All individuals reported using tobacco (e.g., current or former smoker) and were of European ancestry. Our analyses used data from the UK BioBank11 (UKB; N = 123,844; Age = 58.1, SD = 7.8; Sex = 48.3% Female), which we used for partitioned heritability analyses and gene-based tests, and the National Study of Adolescent Health12 (Add Health, Wave IV; N = 1667, Age = 28.9, S.D = 1.70; Sex = 49% female) that was used for polygenic score prediction. Ancestry in both samples was determined using principal components analyses and multidimensional scaling13,14. This study was approved by the Institutional Review Board at Emory University (IRB00090295).
Genotype quality control
Human genomic analyses in UKBiobank samples focused on raw and imputed genotypes obtained using the Affymetrix UK BiLEVE Axiom and UK Biobank Axiom® arrays, which genotyped ~850,000 variants (details available here: https://www.ukbiobank.ac.uk/scientists-3/genetic-data/). Analyses in both samples focused on genotyped and imputed SNPs with good quality scores (r2 > 0.3). PLINK (version 1.9)15 was used to filter markers using the following criteria: genotyping rate >99%, minor allele frequency >0.01, Hardy–Weinberg equilibrium p value >0.0001, and missing genotype rate <0.10.
Partitioned heritability of cigarettes per day using nicotine genesets
Our study investigated whether model organism data on nicotine exposure was relevant to the genetics of human tobacco consumption via partitioned heritability analyses. To test this, we used Genome Complex Trait Analysis (GCTA; version 1.92)16 with multiple genetic components effects estimated via genome-based restricted maximum likelihood (GREML). To reduce the computational burden and to demonstrate the robustness of our partitioned heritability analyses, we separated the UKB into three separate subsets. Using the GCTA-GREML Power Calculator17 we estimated the sample sizes needed to provide at least 70% power to detect SNP-heritability estimates as small as one-third of 1% (i.e., 0.333%)2. Power was based on the previously reported SNP-heritability of CPD7 and the observed variance of the off-diagonal elements (~6.68 × 10−4) of the genomic relatedness matrix for individuals with smoking data in UKBiobank. We used this information to split the UKB sample into three approximately equal subsets (n1 = 41,263, n2 = 41,368, n3 = 41,213), each of which was made constitutionally equivalent by randomly sampling individuals from each quartile of the nicotine consumption distribution; individuals in each subset were no more related than second cousins.
We partitioned the SNP-heritability of tobacco consumption into three regions-of-interest (ROI; and thus set of three genetic relatedness matrices) based on the GeneWeaver model organism genes: (1) protein-coding regions, (2) surrounding regions, and (3) all other variants. Protein coding regions included all SNPs between the start and stop positions for each of the model organism genes that were orthologous in humans. The surrounding regions encompassed genomic loci up and downstream of the 5′ and 3′ ends of each gene, respectively, and captured potential regulatory DNA variants of genomic regions around the model organism genes (e.g., cis-expression quantitative trait loci; cis-eQTLs). The category “All other variants” reflected aggregate genetic effects from the remainder of the genome, given the corresponding size protein coding and surrounding regions. We investigated five differently sized surrounding regions around the model organism genes as the sizes vary substantially in the literature (5 kilobase pairs (kB), 10 kB, 25 kB, 35 kB, and 50 kB). In order to estimate the contribution of each of these regions to the genetic variance for CPD, we fitted six partitioned heritability models that included variance components including the effects of SNPs within the protein-coding regions identified by the model organism nicotine gene set + buffer around these genes of varying length + effects of all other variants in genome + error (i.e., (1) protein-coding regions + the effect of variants in the remainder of the genome, (2) protein-coding regions + SNP effects within a 5 kB buffer + all other variants in the genome, as well as subsequent models of varying buffer lengths, (3) 10 kB buffer, (4) 25 kB buffer, (5) 35 kB buffer, and (6) 50 kB buffer), which tested the role of the protein-coding regions and surrounding regions of the model organism genes, as well as all other variants and the total heritability of human tobacco consumption, simultaneously. Additional details are provided in the supplementary methods.
The significance of each variance component was assessed using a likelihood ratio test while accounting for covariates (sex, testing site location, age, and age2). Population stratification effects were controlled using strict selection for individuals of European Ancestry using genomic principal components and multidimensional scaling14,18. Enrichment was calculated to determine whether the observed component-heritability estimates were greater than what would be expected by chance given the observed total genetic variance and ~4.6 million SNPs used in the analysis (i.e., the variance explained that we would expect via a random selection of loci of the same size from the genome). As such, the statistical significance of enrichment was evaluated on whether the expected h2SNP fell within the 95% confidence interval of the observed h2SNP (i.e., enrichment >1.96).
Total SNP-heritability estimates were obtained by pooling results across folds and meta-analyzing using a standard weighted fixed-effect model. Heritability estimates across UKB subsets were combined using fixed-effects inverse-variance meta-analysis implemented in R using the “rmeta” package.
Gene-based associations
To investigate the overlap of individual genes associated with human tobacco consumption and animal paradigms of nicotine exposure, we compared our model organism genes with human findings from three gene-based tests. First, mixed linear model association analyses (MLMA) were performed in GCTA using the MLMA-leave one chromosome out approach (MLMA-LOCO)16. MLMA-LOCO analyses are powerful approaches to assess DNA associations with human traits (fixed effect) and assume a linear model while adjusting for population structure by estimating genomic relatedness matrices (random effect). Second, we used a conventional gene-based association approach: Multi-marker Analysis of GenoMic Annotation (MAGMA) via submitting GWAS summary statistics to Functional Mapping and Annotation (FUMA)19 of GWASs (using a 10 kb window to define a gene). Next, we used Hi-C coupled MAGMA (H-MAGMA)20, which investigates gene associations by encompassing regulatory SNPs based on chromatin interactions within a cell (cis-eQTLs and trans-eQTLs [long distance regulatory variants]). We collapsed findings across all cell data for H-MAGMA, including neuronal and astrocyte cell types as well as data from fetal and adult brains. Lastly, we used SMultiXcan21, which examines DNA to RNA relationships (cis-eQTLs) in specific tissues from GWAS summary statistics by training an elastic neural net algorithm on human donors from the Genotype-Tissue Expression database (GTEx)22. We elected to use 13 brain regions from GTEx and utilized multivariate regression from SMultiXcan to predict human brain-related gene expression associations with human cigarettes per day. Thus, H-MAGMA characterizes gene-based associations using specific cell-types and developmental stages whereas SMultiXcan reveals the direction of RNA expression in brain tissues for those at high genetic risk for tobacco consumption. All gene-based analyses utilized a Benjamini–Hochberg False Discovery Rate correction for multiple testing (padj < 0.05). To determine whether the overlap of gene-based associations and the model organism genes were more than we would expect by chance, we performed a Fisher’s Exact test—including the 20,809 homologous genes identified by biomaRt21.
Using two common negative controls from model organism research, we tested whether gene-based associations of human tobacco consumption were enriched for genes associated with mouse locomotor behavior23 and rat sucrose exposure24. Similar to our model organism geneset for nicotine exposure, these negative control studies examined RNA associations in similar brain regions and were also identified from GeneWeaver. Collapsing across locomotor behavior and sucrose exposure, our negative control geneset included 845 orthologous genes.
Polygenic prediction of CPD
To investigate the reproducibility of our model organism geneset in humans, we created polygenic scores from the full UKB sample and tested its predictive utility in AddHealth. We created polygenic scores using the summary-based Best Linear Unbiased Prediction (SBLUP25 implemented in GCTA16 method, which improves prediction accuracy26. These analyses adjusted genetic effect sizes based on linkage disequilibrium patterns using the European reference sample from the 1000 Genomes Project27. To adjust for population stratification, our polygenic scores co-varied for the first six principal components extracted from the genetic data of the Add Health sample. Similar to our partitioned heritability approach, we investigated whether polygenic scores were associated with tobacco consumption (1) within-protein coding regions of our nicotine geneset, (2) in the surrounding regions (using a 10 kB surrounding region window to maximize signal to noise ratio as indicated by enrichment analyses in UKB), and (3) all other genomic variants. We also examined whether a PGS using all variants (nSNPs = 4656938) was associated with CPD.
Results
Model organism nicotine genes
First, using GeneWeaver, we investigated our model organism genes and found that there was a small overlap across studies of the genes associated with nicotine exposure (Supplementary Table 1). For instance, some mouse and rat studies of nicotine use demonstrated significant overlap (Jaccard Similarity 0.01–0.02). Collapsing across the study, we found 21 replicated model organism genes (Supplementary Table 2). Subsequent analyses focused on the SNPs in and around the 786 orthologous model organism genes associated with nicotine exposure.
Partitioned heritability—human tobacco consumption
Additive genetic factors accounted for 7.6% to 9.5% of the variability in CPD across all subsets of UKB participants (see Table 2). Less than five percent of the SNP-heritability of CPD could be attributed to SNPs within protein-coding regions of the model organism genes, whereas up to 37% of the heritability was observed in the surrounding genomic regions of these genes. The enrichment of heritability started to decline after expanding the region to include a 10kB window of the surrounding genomic regions (directly up/downstream) of the model organism genes. The remaining regions of the genome (i.e., All other variants) were not significantly enriched across models 1 through 6 (see Table 2), indicating that the SNPs in and around genes associated with nicotine exposure in various animal paradigms pointed to important genomic regions underlying human tobacco consumption.
Nicotine/tobacco gene overlap across species
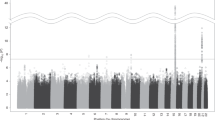
We then investigated the overlap of individual genes from model organisms and the gene-based tests associated with human tobacco consumption. Collapsing across MAGMA, H-MAGMA, and SMultiXcan methods, we identified 115 unique genes (with annotated HGNC gene symbols) associated with human cigarettes per day (all padj < 0.05; see Fig. 2; see Supplementary Files 1–3 for gene-based test results). Ten genes were significant across all gene-based tests (ADAMTS7, C19orf54, CHRNA3, CHRNA5, CYP2A7, GTF2I, HYKK, ITPKC, PSMA4, and SNRPA). Of all human gene-based associations, we found ten genes that were present in our model organism geneset (ADAR, CHRNA4, CHRNA5, CHRNB4, CTSL, CTSH, DNAJA4, NAA20, PSMC3, and RAB4B; see Fig. 3 for summary). This overlap was more than expected by chance (OR = 2.44, p = 0.012, 95% CI [1.13, 4.70]) and no enrichment was observed among our negative control geneset (locomotor behavior and sucrose exposure; OR = 0.458, p = 0.450, 95% CI [0.055, 1.70]). Eight out of ten of the overlapping genes from our model organism nicotine exposure genes came from a single study28 that investigated chronic nicotine self-administration from five brain regions using two strains of mice. When restricting our gene-based analyses to just the model organism nicotine exposure genes, we found three additional gene-based associations with tobacco consumption (NUP50, UCHL5, and SDC3; see Supplementary Fig. 1). Post-hoc examination of the gene-based association signals (–log10 p values) indicated that model organism genes from Mus musculus studies performed better than Rattus norvegicus, studies (all padj < 0.001) and better than random genes (via permutations, padj < 0.001; see Supplementary Fig. 2). Collapsing across gene-based tests, the 21 replicated nicotine genes (across studies in model organisms) were not more significant than random (permuted) genes (padj = 0.244).

Manhattan plot shows results of all three gene-based tests: MAGMA, H-MAGMA (neuron and astrocyte cell types; fetal and adult brain tissues), and S-MultiXcan (13 GTEx brain tissues) for human cigarettes per day. The labeled genes are those identified from our nicotine gene list derived from model organisms.

This figure shows a schematic representation for interpreting our cross-species genetic associations with A Human tobacco consumption, B Model organism nicotine exposure and inferring their effects in the C Human brain. Note: Nic means nicotine; Self-Admin refers to self-administration.
Polygenic score analysis
Partitioned polygenic scores for CPD were derived using the GWAS summary statistics from the full UKB sample and predicted CPD in Add Health (see Supplementary Table 3). Restricting polygenic scores to genomic regions of the nicotine genes resulted in significant prediction of cigarettes per day in an independent sample (within the protein coding regions, but not in the 10 kb surrounding regions of these genes). These results further highlight the utility of incorporating model organism data in human genetic studies of substance use.
Discussion
We found support for all three of our hypotheses. The genes associated with nicotine use in the brains of mice, fish, and rats (1) substantially contributed to the heritability of human tobacco consumption, (2) significantly overlapped with individual genes associated with this genetic predisposition, and (3) aided in polygenic score prediction of tobacco consumption in an independent sample. Our study applies a novel integrative framework for filling the translational space between human and animal genetics research. This line of research may enhance genomic discoveries, help interpret genetic associations with human traits and illuminate what, tissue, cell type and animal paradigm are best suited for genomic follow-up investigation.
Similar to previous research29, we found that up to a third of the heritability for the frequency of human tobacco use can be attributed to genomic regions stemming from RNA associations of specific nicotine behaviors in the brain (of model organisms). Our results suggest that the genetic proclivity to human tobacco use is mediated—in part—by RNA associations with voluntary nicotine use, nicotine preference and nicotine’s neuropharmacological properties (mostly) in the brain’s reward circuitry. Similarly, recent genome-wide research identified genome-wide significant loci in neurotransmission and reward learning genes for tobacco use and prioritized non-synonymous protein-coding variants7. By using approximately half of the sample size from Liu et al., (2019) our findings corroborated the importance of neurotransmission and reward-related genes underlying genetic susceptibility of tobacco consumption. Most cross-species findings appeared to be buried under the genomic significance threshold—demonstrating the strength of our partitioned heritability approach, which captures genes with small effect sizes peppered across the genome. Incorporating model systems allows for studies with small samples to be informative due to larger effect sizes and tighter experimental control and can be used to complement and contextualize human genetic findings.
Our study adds nuance to the genetic mechanisms underlying human tobacco consumption (see Fig. 3). We found cross-species associations with nicotine consumption with established nicotinic acetylcholine receptor genes (CHRNA4/B4/A5), as well as unconventional proteasome (PSMC3), heat-shock protein (DNAJA4), synaptic plasticity, and enzymatic genes (ADAR, CTSH, CTSL, NAA20, RAB4B). Most of these genes were contributing to molecular brain mechanisms of nicotine self-administration in C3H/HeJ and C57BL/6 J mice28, but Adar was associated with acute nicotine use in rats30 and naa20 was linked with nicotine preference in fish31. As a whole, genes from animal behavioral paradigms that best aligned with the human trait demonstrated the strongest gene-based effect sizes in humans. Our analyses suggest that humans at high genetic risk for frequent cigarette smoking had increased RNA expression of CTSH and CTSL in cortical and limbic regions, respectively (see Supplementary File 1). Corroborating this, nicotine-consuming mice had increased RNA expression of Ctsh and Ctsl in the pre-frontal cortex and limbic reward regions (NAc and hippocampus), respectively. Similarly, elevated pre-frontal cortex Psmc3 expression was associated with chronic nicotine exposure in mice28 and rats32 and was associated with human tobacco consumption via regulatory DNA variants in adult brain tissues and neuronal cell-types (see Supplementary File 2). The association of DNAJA4 with human tobacco consumption was mediated via long-distance gene regulation in neuronal cell-types and demonstrated increased expression in the VTA28 and cortical neurons33 of nicotine-exposed mice. Therefore, our integrative approach contextualizes otherwise puzzling genetic associations with human traits and characterizes potential mechanisms in relation to specific behaviors, tissues, developmental epochs, and cell-types.
Cross-species polygenic prediction illustrated a novel application for model organism data to be integrated with human GWAS data. In contrast to our partitioned heritability approach, we found significant prediction within protein-coding regions of the model organism genes—instead of the surrounding (potentially regulatory) regions of these genes. This approach furthers the line of research incorporating biologically relevant information for polygenic score approaches34,35. While novel, these were far from becoming clinically relevant and were limited in their predictive capacity, but this approach offered a way to replicate a priori gene lists in an independent sample.
We urge the reader to interpret the current findings with caution. Human and animal data are very different—ranging from their environments, genetic backgrounds, developmental stages, routes-of-administration, and data types (e.g., DNA versus RNA associations). The animal data was limited to microarray studies and a restricted pallet of behaviors, one tissue type (brain tissue), few samples, and three species. We sought to overcome these limitations by integrating across brain regions, behaviors, and model organisms, but future studies are needed to determine whether these effects are invariant and to determine what paradigms are most relevant to what human traits—especially as the volume of literature increases. The majority of human gene-based associations were not in our model organism nicotine exposure geneset potentially suggesting limited availability of targets for experimental follow-up. But we showed the specificity of this overlap (via our negative control) and also highlighted certain brain regions, behavioral paradigms, and species to follow-up individual genes that were anchored in human biology. Our analyses did not evaluate an exhaustive list of ‘omics data types (methylomics, proteomics, metabolomics, CHiP-seq, ATAC-seq, etc.) and focused on effects in the brain without considering other relevant tissue types or cell types.
Future research is warranted to determine whether our integrative framework generalizes across complex human traits. Traits with different genetic architectures, epigenetic landscapes, and animal models may yield disparate findings. We found that the bulk of our cross-species signal stemmed from mouse models of nicotine use, but it will be important for future research to be conducted across multiple smoking phenotypes and include additional species and studies, as well as incorporate findings from human tissues to benchmark findings with other model organisms. Ideally, integrative genomics comparisons would leverage equitable and minimally error-prone outcomes or endophenotypes across studies. Given the array of animal models for human traits, an inviting avenue of research should clarify the utility of specific tissues, cell types, and animal models in human genetics. With a large enough literature base, we may be able to better refine what tissues and specific mechanisms human genomic signals stem from and ultimately may better characterize the genetic make-up for complex traits. Future studies leveraging these approaches should consider strategies for examining heterogeneity across tissues and cell types, as well as whether the observed effects generalize across human populations (e.g., European, African, Asian, etc). Cross-species genetic research is a fertile territory for methodological innovations. This field is still in its infancy and thus is a ripe area for future research applications.
Conclusions
In sum, our study identifies biological overlap of nicotine use between human and animal research using integrative genomic models. Our study provides a proof-of-principle that model organism data can be used among standard methods used in human genetics research. Human researchers can take advantage of a rich array of model organism data to aid their interpretations with complex traits—even in small(er) GWASs—and animal researchers can assess the relevance of their findings to corresponding human traits. This study takes a step forward in cross-species research by incorporating a priori information into human genetics analyses and adds to the conversation regarding enhancing the utility of smaller GWASs. Our study suggests that cross-species genetics research is a worthwhile empirical avenue and that the intersection of human and animal biology can help unravel the genetic basis of complex traits.
References
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Wu, Y. et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 9, 918 (2018).
Vandiedonck, C. Genetic association of molecular traits: a help to identify causative variants in complex diseases. Clin. Genet. 93, 520–532 (2018).
Changeux, J.-P., Edelstein, S. & Edelstein, S. J. Nicotinic Acetylcholine Receptors: From Molecular Biology To Cognition (Odile Jacob Publishing Corp., 2005).
Besson, M. et al. Long-term effects of chronic nicotine exposure on brain nicotinic receptors. Proc. Natl Acad. Sci. 104, 8155–8160 (2007).
Grenhoff, J., Aston-Jones, G. & Svensson, T. H. Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol. Scand. 128, 351–358 (1986).
Liu, M. et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 51, 237–244 (2019).
Baker, E., Bubier, J. A., Reynolds, T., Langston, M. A. & Chesler, E. J. GeneWeaver: data driven alignment of cross-species genomics in biology and disease. Nucleic Acids Res. 44, D555–D559 (2016).
Baker, E. J., Jay, J. J., Bubier, J. A., Langston, M. A. & Chesler, E. J. GeneWeaver: a web-based system for integrative functional genomics. Nucleic Acids Res. 40(Database issue), D1067–D1076 (2012).
Baker, E. J. et al. Ontological discovery environment: A system for integrating gene-phenotype associations. Genomics 94, 377–387 (2009).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Harris K. M., Udry J. R. National Longitudinal Study of Adolescent to Adult Health (Add Health), 1994-2008 [Public Use] (Carolina Population Center, University of North Carolina-Chapel Hill [distributor], Inter-university Consortium for Political and Social Research [distributor], 2018).
1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015). et al.
Brick, L. A., Keller, M. C., Knopik, V. S., McGeary, J. E. & Palmer, R. H. C. Shared additive genetic variation for alcohol dependence among subjects of African and European ancestry. Addict. Biol. 24, 132–144 (2019).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Visscher, P. M. et al. Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet. 10, e1004269 (2014).
Brick, L. A., Micalizzi, L., Knopik, V. S. & Palmer, R. H. C. Characterization of DSM-IV opioid dependence among individuals of European ancestry. J. Stud. Alcohol Drugs 80, 319–330 (2019).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Sey, N. Y. A. et al. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci. 23, 583–593 (2020).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat.Commun. 9, 1825 (2018).
GTEx Consortium. Human genomics.The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Philip, V. M. et al. High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains. Genes Brain Behav. 9, 129–159 (2010).
Rodd, Z. A. et al. Differential gene expression in the nucleus accumbens with ethanol self-administration in inbred alcohol-preferring rats. Pharmacol. Biochem. Behav. 89, 481–498 (2008).
Robinson, M. R. et al. Genetic evidence of assortative mating in humans. Nat. Hum. Behav. 1, 0016 (2017).
Mignogna, K. M., Bacanu, S. A., Riley, B. P., Wolen, A. R. & Miles, M. F. Cross-species alcohol dependence-associated gene networks: co-analysis of mouse brain gene expression and human genome-wide association data. PLoS ONE 14, e0202063 (2019).
Kranzler, H. R. et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat. Commun. 10, 1499 (2019).
Wang, J. et al. Strain- and region-specific gene expression profiles in mouse brain in response to chronic nicotine treatment. Genes Brain Behav. 7, 78–87 (2008).
Evans, L. M. et al. The Role of A Priori-Identified Addiction and Smoking Gene Sets in Smoking Behaviors. Nicotine Tob. Res. 22, 1310–1315 (2020).
Polesskaya, O. O. et al. Nicotine causes age-dependent changes in gene expression in the adolescent female rat brain. Neurotoxicol. Teratol. 29, 126–140 (2007).
Kily, L. J. et al. Gene expression changes in a zebrafish model of drug dependency suggest conservation of neuro-adaptation pathways. J. Exp. Biol. 211, 1623–1634 (2008).
Kane, J. K., Konu, O., Ma, J. Z. & Li, M. D. Nicotine coregulates multiple pathways involved in protein modification/degradation in rat brain. Brain Res. Mol. Brain Res. 132, 181–191 (2004).
Wang, J. et al. Regulation of platelet-derived growth factor signaling pathway by ethanol, nicotine, or both in mouse cortical neurons. Alcohol. Clin. Exp. Res. 31, 357–375 (2007).
Neuner, S. M., Heuer, S. E., Huentelman, M. J., O’Connell, K. M. S. & Kaczorowski, C. C. Harnessing genetic complexity to enhance translatability of Alzheimer’s disease mouse models: a path toward precision medicine. Neuron 101, 399–411.e395 (2019).
Hari Dass, S. A. et al. A biologically-informed polygenic score identifies endophenotypes and clinical conditions associated with the insulin receptor function on specific brain regions. EBioMedicine 42, 188–202 (2019).
Acknowledgements
We acknowledge the National Institute on Drug Abuse award DP1DA042103 (to RHCP) and the National Institute on Alcohol Abuse and Alcohol (R01AA018776) (to EJC). We acknowledge the Wellcome Trust medical charity, Medical Research Council, Department of Health, Scottish Government, Northwest Regional Development Agency, Welsh Government, British Heart Foundation, Cancer Research UK and Diabetes UK, and the National Health Service (NHS) for their part in supporting the UK Biobank without which this study would not have been possible. The contents of this paper do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This research uses data from Add Health, a program project directed by Kathleen Mullan Harris and designed by J. Richard Udry, Peter S. Bearman, and Kathleen Mullan Harris at the University of North Carolina at Chapel Hill, and funded by grant P01-HD31921 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with cooperative funding from 23 other federal agencies and foundations. Special acknowledgment is due Ronald R. Rindfuss and Barbara Entwisle for assistance in the original design. Information on how to obtain the Add Health data files is available on the Add Health website. No direct support was received from grant P01-HD31921 for this analysis. Note that Figure 3 was created using BioRender.com
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Palmer, R.H.C., Benca-Bachman, C.E., Huggett, S.B. et al. Multi-omic and multi-species meta-analyses of nicotine consumption. Transl Psychiatry 11, 98 (2021). https://doi.org/10.1038/s41398-021-01231-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01231-y
This article is cited by
-
Improving the predictive power of mouse models
Nature Biotechnology (2024)
-
The Genetically Informed Neurobiology of Addiction (GINA) model
Nature Reviews Neuroscience (2023)
-
Gene expression genetics of the striatum of Diversity Outbred mice
Scientific Data (2023)
