
Abstract
Tourette syndrome (TS) is a neuropsychiatric disorder of complex genetic architecture involving multiple interacting genes. Here, we sought to elucidate the pathways that underlie the neurobiology of the disorder through genome-wide analysis. We analyzed genome-wide genotypic data of 3581 individuals with TS and 7682 ancestry-matched controls and investigated associations of TS with sets of genes that are expressed in particular cell types and operate in specific neuronal and glial functions. We employed a self-contained, set-based association method (SBA) as well as a competitive gene set method (MAGMA) using individual-level genotype data to perform a comprehensive investigation of the biological background of TS. Our SBA analysis identified three significant gene sets after Bonferroni correction, implicating ligand-gated ion channel signaling, lymphocytic, and cell adhesion and transsynaptic signaling processes. MAGMA analysis further supported the involvement of the cell adhesion and trans-synaptic signaling gene set. The lymphocytic gene set was driven by variants in FLT3, raising an intriguing hypothesis for the involvement of a neuroinflammatory element in TS pathogenesis. The indications of involvement of ligand-gated ion channel signaling reinforce the role of GABA in TS, while the association of cell adhesion and trans-synaptic signaling gene set provides additional support for the role of adhesion molecules in neuropsychiatric disorders. This study reinforces previous findings but also provides new insights into the neurobiology of TS.
Similar content being viewed by others
Introduction
Tourette syndrome (TS) is a chronic neurodevelopmental disorder characterized by several motor tics and at least one vocal tic that persist more than a year1. Its prevalence is between 0.6 and 1% in school-aged children2,3. Although TS is highly polygenic in nature, it is also highly heritable4. The population-based heritability is estimated at 0.75,6, with SNP-based heritability ranging from 21 to 58%4 of the total. The genetic risk for TS that is derived from common variants is spread throughout the genome4. The two genome-wide association studies (GWAS) conducted to date7,8 suggest that TS genetic variants may be associated, in aggregate, with tissues within the cortico-striatal and cortico-cerebellar circuits, and in particular, the dorsolateral prefrontal cortex. The GWAS results also demonstrated significant ability to predict tic severity using TS polygenic risk scores7,9. A genome-wide CNV study identified rare structural variation contributing to TS on the NRXN1 and CNTN6 genes10. De novo mutation analysis studies in trios have highlighted two high confidence genes, CELSR and WWC1, and four probable genes, OPA1, NIPBL, FN1, and FBN2 to be associated with TS11,12.
Investigating clusters of genes, rather than relying on single-marker tests is an approach that can significantly boost power in a genome-wide setting13. Common variant studies can account for a substantial proportion of additive genetic variance14 and have indeed produced a wealth of variants associated with neuropsychiatric disorders, which, however, lack strong predictive qualities, an issue commonly referred to as “missing heritability”15. Theoretical, as well as empirical, observations have long hinted toward the involvement of non-additive genetic variance into the heritability of common phenotypes. As such, pathway analyses could pave the way toward the elucidation of missing heritability in complex disease.
This approach has already proven useful in early genome-wide studies of TS. The first published TS GWAS, which included 1285 cases and 4964 ancestry-matched controls did not identify any genome-wide significant loci. However, by partitioning functional- and cell-type-specific genes into gene sets, an involvement of genes implicated in astrocyte carbohydrate metabolism was observed, with a particular enrichment in astrocyte-neuron metabolic coupling16. Here, we investigated further the pathways that underlie the neurobiology of TS, performing gene set analysis on a much larger sample of cases with TS and controls from the second wave TS GWAS. We employed both a competitive gene set analysis as implemented through MAGMA, as well as a self-contained analysis through a set-based association method (SBA). Besides highlighting a potential role for neuroimmunity, our work also provides further support for previously implicated pathways including signaling cascades and cell adhesion molecules.
Materials and methods
Samples and quality control
The sample collection and single variant analyses for the data we analyzed have been extensively described previously7,8. IRB approvals and consent forms were in place for all data collected and analyzed as part of this project. For the purposes of our analysis, we combined 1285 cases with TS and 4964 ancestry-matched controls from the first wave TS GWAS, with 2918 TS cases and 3856 ancestry-matched controls from the second wave TS GWAS. Standard GWAS quality control procedures were employed17,18. The data were partitioned first by genotyping platform and then by ancestry. The sample call rate threshold was set to 0.98, and the inbreeding coefficient threshold to 0.2. A marker call rate threshold was defined at 0.98, case-control differential missingness threshold at 0.02, and Hardy–Weinberg equilibrium (HWE) threshold to 10−6 for controls and 10−10 for cases. Before merging the partitioned datasets, we performed pairwise tests of association and missingness between the case-only and control-only subgroups to address potential batch effect issues. All SNPs with p-values ≤10−06 in any of these pairwise quality control analyses were removed. After merging all datasets, principal component analysis was utilized to remove samples that deviated more than 6 standard deviations and to ensure the homogeneity of our samples in the ancestry space of the first 10 principal components, through the use of the EIGENSOFT suite19. Identity-by-descent analysis with a threshold of 0.1875 was used to remove related samples, and thus to avoid confounding by cryptic relatedness. After quality control, the final merged dataset consisted of 3581 cases with TS and 7682 ancestry-matched controls on a total of 236,248 SNPs, annotated using dbSNP version 137 and the hg19 genomic coordinates.
We assessed the genomic variation in our data through PCA analysis to identify potential population structure (Supplementary Fig. 1 and Supplementary Table 1). The variation in our data was reduced to a triangular shape in the two-dimensional space of the first two principal components. One tip was occupied by Ashkenazi Jewish samples, the second by the Southern European samples, and the other by the North Europeans. Depicting geography, the Southern to Nothern axis was populated by European-ancestry samples. The first five principal components were deemed statistically significant (Tracy Widom test as implemented by EIGENSOFT, Supplementary Table 1) and were added to the association model as covariates, in order to avoid population structure influencing our results.
Gene sets
We collected neural-related gene sets from multiple studies on pathway analyses in neuropsychiatric disorders16,20,21,22,23,24. These studies relied on an evolving list of functionally-partitioned gene sets, focusing mainly on neural gene sets, including synaptic, glial sets, and neural cell-associated processes. We added a lymphocytic gene set also described in these studies23, in order to also investigate potential neuroimmune interactions.
In total, we obtained 51 gene sets, which we transcribed into NCBI Entrez IDs and subsequently filtered by removing gene sets that contained fewer than 10 genes. Forty-five gene sets fit our criteria and were used to conduct the analyses.
We examined two primary categories of pathway analysis methods, the competitive 25 and the self-contained test16,25. The competitive test compares the association signal yielded by the tested gene set to the association signals that do not reside in it26,27. In this type of test, the null hypothesis is that the tested gene set attains the same level of association with disease as equivalent random gene sets. In contrast, the self-contained test investigates associations of each tested gene set with the trait, and not with other gene sets, meaning that the null hypothesis in this case is that the genes in the gene set are not associated with the trait25,27. Therefore, for a competitive test, there should be data for the whole breadth of the genome, but this test cannot provide information regarding how strongly the gene set is associated with the trait28. We employ both methods for a comprehensive investigation into the neurobiological background of TS.
MAGMA on raw genotypes
We ran MAGMA26 on the individual-level genotype data using the aforementioned filtered gene set lists. MAGMA performs a three-step analytic process. First, it annotates the SNPs by assigning them to genes, based on their chromosomal location. Then it performs a gene prioritization step, which is used to perform the final gene set analysis step. We used a genomic window size of ±10 kb and the top 5 principal components as covariates to capture population structure. SNP-to-gene assignments were based on the NCBI 37.3 human gene reference build. The number of permutations required for the analysis was determined by MAGMA, using an adaptive permutation procedure leading to 11,263 permutations. MAGMA employs a family-wise error correction calculating a significance threshold of 0.00100496.
Set-based association (SBA) test
We conducted SBA tests on the raw individual genotype data, as described in PLINK25,29 and adapted in a later publication30. This test relies on the assignment of individual SNPs to a gene, based on their position, and thus to a pathway, according to the NCBI 37.3 human gene reference build. After single-marker association analysis, the top LD-independent SNPs from each set are retained and selected in order of decreasing statistical significance, and the mean of their association p-values is calculated. We permuted the case/control status, repeating the previous association and calculation steps described above, leading to the empirical p-value for each set. The absolute minimum number of permutations required for crossing the significance level is dictated by the number of gene sets tested. Testing for 45 gene sets requires at least 1000 permutations to produce significant findings. PLINK’s max(t) test recommends at least 64,000 permutations. We opted to increase the number of permutations to one million, the maximum that was computationally feasible, to maximize our confidence in the outcomes, given our large sample size.
We used logistic regression as the association model on the genotypes and the first five principal components as covariates on the genotype data to conduct the SBA test with the collected neural gene sets. Another repetition of this step was performed with a simple association test, to test for this method’s robustness to population structure. We proceeded to run the analysis on all samples, using all gene sets at a 10 kb genomic window size, the first five principal components as covariates, and one million permutations. Since the permutations were performed on the phenotypic status of the samples, and only served as a method of association of the trait with the gene sets, we also corrected the results by defining the significance threshold through Bonferroni correction at 1.1 × 10−3 (0.05/45).
Results
For the gene set association analysis, we ran PLINK’s self-contained set-based association method and MAGMA’s competitive association method, using the same 45 gene sets on the processed genotyped data of 3581 cases and 7682 ancestry-matched controls on a total of 236,248 SNPs. By performing both methods of analysis we aimed to obtain a global assessment of the gene sets’ relationship with TS.
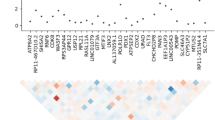
MAGMA analysis identified one significant gene set (Fig. 1), cell adhesion and trans-synaptic signaling (CATS), which achieved a nominal p-value of 6.2 × 10−5 (permuted p-value of 0.0032). While the CATS gene set is comprised of 83 genes, MAGMA’s annotation step prioritized 72 of its genes for the gene set analysis. It involves 3290 variants that were reduced to 1627 independent variants in our data. Results were mainly driven by associations in the CDH26, CADM2, and OPCML genes as indicated by MAGMA gene-based analysis (Table 1). In the gene-based tests, CDH26 attained a p-value of 8.9526 × 10−6, CADM2 a p-value of 4.6253 × 10−4, and OPCML a p-value of 7.9851 × 10−4, neither crossing the genome-wide significance threshold for gene tests (2.574 × 10−6 calculated on 19,427 genes contained in the NCBI 37.3 version of RefGene).

The gene set that crossed the significance threshold is depicted in red.
We next run SBA, which conducts an initial single-marker association step before performing permutations to calculate empirical p-values for the gene sets. This association step is performed on the total number of variants that are associated with the genes involved in the gene sets, leading to a subset of 25,630 variants in our data, which are then filtered based on their LD. Analysis identified three gene sets as significant (Table 2), the ligand-gated ion channel signaling (LICS) (P: 2.67 × 10−4), the lymphocytic (P: 3.5 × 10−4), and the cell adhesion and trans-synaptic signaling (CATS) (P: 1.07 × 10−3). Detailed results for all the tested gene sets are shown in Fig. 2.

The gene sets that crossed the significance threshold are depicted in red.
The LICS gene set was the top-scoring gene set, including 38 genes and involving 683 variants, 66 of which were associated with TS. The gene set’s signal was primarily driven by variants residing in the genes of the γ-aminobutyric acid receptors GABRG1 and GABBR2, the HCN1 channel gene and the glutamate receptor gene GRIK4. This signal was driven primarily by an association with SNP rs9790873, which is an eQTL for HCN1 in tibial nerve, according to GTEx31. GABBR2 is represented by two top SNPs, that are LD-independent, and removing either of those SNPs from the gene set did not cause the gene set to drop under the significance threshold.
The lymphocytic gene set was the next top-scoring gene set, including 143 genes that translated to 799 variants in our data, with 50 of these variants associated with TS. Its signal was driven by a missense variant inside the FLT3 gene and an intergenic variant between NCR1 and NLRP7, followed by IL12A, HDAC9, CD180. The rs1933437 SNP is the top variant for FLT3, and is a possibly damaging missense variant32, located in the sixth exon of the FLT3 gene leading to a p.Thr227Met mutation. It is a very common variant and the sixth exon appears to be less expressed than downstream exons. Given the tissues in which this eQTL affects FLT3’s expression, we tested the lymphocytic gene set by removing FLT3 from it, to identify whether the lymphocytic gene set association was biased by the presence of FLT3. After removing FLT3, the lymphocytic gene set association statistic decreased slightly (P: 0.00012), driven mainly by NCR1/NRLP7.
The third significant gene set, CATS, consisted of 83 genes, including multiple large genes. CATS was identified by both SBA and MAGMA in our analyses, and both gene set approaches identified CDH26 as the gene with the lowest p-value. Both SBA and MAGMA also identified NCAM2, NTM, and ROBO2 as strongly associated with TS, with NTM represented by two LD-independent SNPs. CATS’s top SNP, rs1002762, resides in the CDH26 gene on chromosome 20, and is the top associated SNP in our data (P: 2.031 × 10−6) with an odds ratio of 1.178.
Notable results from the SBA also include the Astrocyte small GTPase mediated signaling (ASGMS) and the Astrocyte-neuron metabolic coupling (ANMC) gene sets, with a p-values slightly under the significance thresholds. These gene sets attained a p-value of 0.00137 and 0.001504, respectively.
Discussion
Seeking to elucidate the neurobiology of TS, we present here the largest study to date aiming to interrogate the involvement of sets of genes that are related to neuronal and glial function in TS. We analyzed data from our recently performed TS GWAS and conducted two distinct types of testing, a competitive, regression-based test (MAGMA) and a self-contained, p-value combining test (SBA). Self-contained tests investigate for associations with a phenotype, while competitive tests compare a specific gene set against randomly generated gene sets. We employed both methods to perform a comprehensive investigation of the biological background of TS.
A potential problem in pathway analysis is false SNP assignment to genes, which in turn may increase false results. In order to address this issue, most studies in the literature use short window sizes (10–20 kb) when assigning SNPs to genes. Here, we used a 10 kb window, paired with excessive permutations to avoid false assignments, that would introduce false positive results. There is evidence that long-range SNP effects could play a role, mostly associated with large insertion/deletion events that are not in the scope of this study and would likely hamper the analysis33.
MAGMA’s regression-based algorithm has been reported to account for gene size biases, as can be also discerned by the variable sizes of the top genes. MAGMA’s top prioritized gene, CDH26, is represented by 4 SNPs in our data, CADM2 by 42, while OPCML is represented by 210 SNPs, as it covers an extensive genomic region. We addressed such issues in SBA by setting a low r2 threshold and conditioning on any LD-independent SNPs that resided on the same gene.
The gene sets used in our study come from a line of function-based analyses, aiming to investigate neurobiological mechanisms in neuropsychiatric disorders. A previous pathway analysis using individual-level genotype data of the first wave TS GWAS identified genes involved in astrocytic-neuron metabolic coupling, implicating astrocytes in TS pathogenesis16. In this study, we took advantage of the increased sample size of the second wave TS GWAS and the mechanics of the two distinct methods to identify gene sets associated with TS that provide a novel insight into the pathogenesis of TS, and substantiate the role of neural processes in this neuropsychiatric disorder.
The ANMC gene set that contains genes involved in carbohydrate metabolism in astrocytes was the single identified gene set in the previous pathway analysis study on TS16, raising a hypothesis on a potential mechanism that involves altered metabolism of glycogen and glutamate/γ-aminobutyric acid in the astrocytes. In our study, the ANMC gene set scored slightly under the significance threshold.
Here, analyzing a much larger sample size we identified three sets of genes as significantly associated to the TS phenotype. Among them the LICS gene set, which involves genes implicated in ion channel signaling through γ-aminobutyric acid and glutamate. Several genes in the LICS gene set have been previously implicated in neuropsychiatric phenotypes. HCN1, a hyperpolarization-activated cation channel involved in native pacemaker currents in neurons and the heart, has been significantly associated with schizophrenia and autism34,35,36. GABRG1, an integral membrane protein that inhibits neurotransmission by binding to the benzodiazepine receptor, has yielded mild associations with general cognitive ability37 and epilepsy38, while GABBR2, a g-protein-coupled receptor that regulates neurotransmitter release, with schizophrenia39 and post-traumatic stress disorder40 in multiple studies. The GABA-ergic pathway has been previously implicated in TS, and recent advances showcased the possibility that a GABA-ergic transmission deficit can contribute toward TS symptoms41. GRIK4, encoding a glutamate-gated ionic channel, has shown associations with mathematical ability and educational attainment42 and weaker associations with attention-deficit hyperactivity disorder43. The γ-aminobutyric acid receptors and the HCN channel, are features of inhibitory interneurons44 and also identified in the brain transcriptome of individuals with TS45, adding to the evidence that the phenotype of TS could be influenced by an inhibitory circuit dysfunction, as has previously been proposed46.
Individuals with TS are reported to present elevated markers of immune activation45,47. In addition, a number of studies have implicated neuroimmune responses with the pathogenesis of TS48,49,50. We investigated neuroimmune interactions by interrogating association to a gene set designed by Goudriaan et al.23 to study enrichment in lymphocytic genes. Indeed, our analysis yielded a statistically significant signal. The FLT3 association coincides with the results of the second wave TS GWAS, in which FLT3 was the only genome-wide significant hit7. FLT3 and its ligand, FLT3LG, have a known role in cellular proliferation in leukemia, and have been found to be expressed in astrocytic tumors51. The rs1933437 variant in FLT3 is an eQTL in the brain cortex and the cerebellum31, and has also been implicated in the age at the onset of menarche52. Variants in FLT3 have attained genome-wide significance in a series of studies focusing on blood attributes in populations of varying ancestry, and our current insights into its role are mostly based on these associations with blood cell counts, serum protein levels, hypothyroidism, and autoimmune disorders52,53,54,55.
FLT3 could play a role in neuroinflammation as supported by its intriguing association with peripheral neuropathic pain. The inhibition of FLT3 is reported to alleviate peripheral neuropathic pain (PNP)56, a chronic neuroimmune condition that arises from aberrations in the dorsal root ganglia. Cytokines and their receptors have been at the epicenter of the neuroimmune interactions, with microglia contributing significantly to chronic phenotypes of such states57. FLT3 is a critical component for neuroimmune interactions, especially in the case of the development and sustenance of the PNP phenotype. Interestingly, pain follows sex-specific routes, with glia having a prominent role for pain propagation in males, while females involve adaptive immune cells instead58. These, paired with previous evidence of glial involvement in TS16, raise an interesting hypothesis for TS symptom sustenance, since FLT3 has been shown to be critical for the chronicity of neuronal dysregulations56.
Notably, FLT3 has a prominent role in the hematologic malignancies, with one-third of adult acute myeloid leukemia (AML) patients presenting with activating mutations in FLT3, and wild-type FLT3 being found overexpressed in hematologic malignancies. FLT3 is implicated in apoptotic mechanisms, with its mutations being associated with59 Warburg effect promotion, inhibition of ceramide-dependent mitophagy60, and induction of pro-survival signals, through downstream signaling cascades, including PI3K-Akt-mTOR, Ras/MAPK, and JAK-STAT. This mitochondrial role of FLT3 has been further reinforced by findings that associate it with increased post-transcriptional methylation of mitochondrial tRNAs in cancer61. As such, FLT3 is regarded a molecular target for therapeutic intervention62.
FLT3 is expressed in the cerebellum and whole blood, while FLT3’s top variant, rs1933437, is an eQTL for FLT3 on GTEx31 in various brain tissues, such as the cortex, the cerebellum, the hypothalamus, the frontal cortex (BA9), and non-brain tissues, such as the skin, the pancreas, and adipose tissues. In order to test the robustness of the lymphocytic association in our findings, we repeated the analysis after removing FLT3 from the lymphocytic gene set. The p-value of the gene set decreased, but still remained significant, due to the association in the NCR1/NLRP7 locus. Besides FLT3, the other genes included in this gene set are also quite intriguing to consider as potential candidates that could underlie the pathophysiology of TS. In the same vein with FLT3, common variants in NCR1 have also been significantly associated with blood protein levels63. HDAC9 has been significantly associated with androgenetic alopecia52,64, hair color52, and ischemic stroke65. These seem to follow previous knowledge, given that genes involved in ischemic stroke have been identified as a common component between TS and ADHD66, and that TS, similar to other neuropsychiatric disorders, demonstrates a distinct preference for males. CD180 has shown associations with general cognitive ability37.
The CATS gene set involves many cell adhesion molecules, with the top signals found in CDH26. CDH26 is a cadherin that regulates leukocyte migration, adhesion, and activation, especially in the case of allergic inflammation67. Cell adhesion molecules have been consistently implicated in phenotypes related to brain function, with the latest addition of the high confidence TS gene CELSR3, a flamingo cadherin, that was identified in a large scale de novo variant study for TS12. Their relation to TS has been well documented, with the notable examples of neurexins, contactins, neuroligins, and their associated proteins10,68,69,70. These genes were present in the CATS gene set but did not reach a level of significance in our analysis. This hints toward their possible involvement in TS mostly through rare variants10,68,69, a notion reinforced by findings in other neuropsychiatric disorders71,72.
Most of the genes contained in the identified gene sets in this study are involved in cognitive performance, mathematical ability, and educational attainment42. OPCML, CADM2, and ROBO2 have been implicated in neuromuscular and activity phenotypes, such as grip strength73, physical activity74, and body mass index52. ROBO2 has been associated with depression75, expressive vocabulary in infancy76, while CADM2 is associated to a multitude of phenotypes, including anxiety75, risk-taking behavior, and smoking77. NTM displays similar patterns of pleiotropy, associated with smoking52, myopia64, hair color78, anxiety75, asperger’s syndrome79, bipolar disorder with schizophrenia80, and eating disorders81. NCAM2 and NTM, similarly to the lymphocytic genes, have been significantly associated with blood protein levels82 and leukocyte count52, respectively. Many of these phenotypes are known TS comorbidities, presenting themselves commonly or less commonly in TS cases, and others are related to functions that get impaired in TS symptomatology.
The CATS gene set was identified in both methods indicating the involvement of cell adhesion molecules in transsynaptic signaling. Using genotypes with both methods as a means of identifying pathways instead of summary statistics, gave our study the edge of sample-specific linkage disequilibrium rather than relying on an abstract linkage disequilibrium pattern reference. Our current understanding for regional structures of the genome and the cis-effects of genomic organization will aid the refinement of these associations as well as help shape our understanding of the pleiotropic mechanisms in the identified loci potentially responsible for disease pathogenesis.
In conclusion, our analysis reinforces previous findings related to TS neurobiology while also providing novel insights: We provide further support for the role of FLT3 in TS, as well as the possibility for the involvement of the GABA-ergic biological pathway in TS pathogenesis. At the same time, our study highlights the potential role of glial-derived neuroimmunity in the neurobiology of TS opening up intriguing hypotheses regarding the potential for gene-environment interactions that may underlie this complex phenotype.
References
Robertson, M. M., Cavanna, A. E. & Eapen, V. Gilles de la Tourette syndrome and disruptive behavior disorders: prevalence, associations, and explanation of the relationships. J. Neuropsychiatry Clin. Neurosci. 27, 33–41 (2015).
Scharf, J. M. et al. Population prevalence of Tourette syndrome: a systematic review and meta-analysis. Mov. Disord. 30, 221–228 (2015).
Robertson, M. M., Eapen, V. & Cavanna, A. E. he international prevalence, epidemiology, and clinical phenomenology of Tourette syndrome: a cross-cultural perspective. J. Psychosom. Res. 67, 475–483 (2009).
Davis, L. K. Partitioning the heritability of Tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS Genetics 9, e1003864 (2013).
Robertson, M. M. Gilles de la Tourette syndrome. Nat. Rev. Dis. Prim. 3, 16097 (2017).
Mataix-Cols, D. Familial risks of Tourette syndrome and chronic tic disorders: a population-based cohort study. JAMA Psychiatry 72, 787–793 (2015).
Yu, D. Interrogating the genetic determinants of Tourette syndrome and other tic disorders through genome-wide association studies. Am. J. Psychiatry 176, 217–227 (2019).
Scharf, J. M. Genome-wide association study of Tourette’s syndrome. Mol. Psychiatry 18, 721–728 (2013).
Abdulkadir, M. Polygenic risk scores derived from a Tourette syndrome genome-wide association study predict presence of tics in the Avon Longitudinal Study of Parents and Children cohort. Biol. Psychiatry 85, 298–304 (2019).
Huang, A. Rare copy number variants in NRXN1 and CNTN6 increase risk for Tourette syndrome. Neuron 94, 1101–1111 (2017).
Willsey, A. J. De novo coding variants are strongly associated with Tourette disorder. Neuron 94, 486–499 (2017).
Wang, S. De novo sequence and copy number variants are strongly associated with Tourette disorder and implicate cell polarity in pathogenesis. Cell Rep. 24, 3441–3454 (2018).
Ballard, D. H., Cho, J. & Zhao, H. Comparisons of multi-marker association methods to detect association between a candidate region and disease. Genet. Epidemiol. 34, 201–212 (2010).
Visscher, P. M. 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Zuk, O. Searching for missing heritability: designing rare variant association studies. Proc. Natl. Acad. Sci. USA 111, E455–E464 (2014).
de Leeuw, C. Involvement of astrocyte metabolic coupling in Tourette syndrome pathogenesis. Eur. J. Hum. Genet. 23, 1519–1522 (2015).
Turner, S. Quality control procedures for genome-wide association studies. Curr. Protoc. Hum. Genet. 68, 1.19.1–1.19.18 (2011).
Lam, M. RICOPILI: rapid imputation for COnsortias PIpeLIne. Bioinformatics 36, 930–933 (2020).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Ruano, D. Functional gene group analysis reveals a role of synaptic heterotrimeric g proteins in cognitive ability. Am. J. Hum. Genet. 86, 113–125 (2010).
Lips, E. S. Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol. Psychiatry 17, 996–1006 (2012).
Duncan, L. E. Pathway analyses implicate glial cells in schizophrenia. PLoS ONE 9, e89441 (2014).
Goudriaan, A. Specific glial functions contribute to Schizophrenia susceptibility. Schizophrenia Bull. 40, 925–935 (2014).
Jansen, A. Gene-set analysis shows association between FMRP targets and autism spectrum disorder. Eur. J. Hum. Genet. 25, 863–868 (2017).
Purcell, S. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Goeman, J. J. & Buhlmann, P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics 23, 980–987 (2007).
Mooney, M. A. & Wilmot, B. Gene set analysis: a step-by-step guide. Am. J. Med. Genet. Part B: Neuropsychiatr. Genet. 168, 517–527 (2015).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Skafidas, E. et al. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol. Psychiatry 19, 504–510 (2014).
Carithers, L. J. A novel approach to high-quality postmortem tissue procurement: the GTEx project. Biopreservation Biobanking 13, 311–319 (2015).
Karczewski, K. J. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Brodie, A., Azaria, J. R. & Ofran, Y. How far from the SNP may the causative genes be?”. Nucleic Acids Res. 44, 6046–6054 (2016).
Pardiñas, A. F. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 50, 381–389 (2018).
Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 8, 21 (2017).
Okbay, A. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–542 (2016).
Davies, G. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat. Commun. 9, 2098 (2018).
International League Against Epilepsy Consortium on Complex Epilepsies. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol. 13, 893–903 (2014).
Ikeda, M. Genome-Wide Association Study Detected Novel Susceptibility Genes for Schizophrenia and Shared Trans-Populations/Diseases Genetic Effect. Schizophr. Bull. (2018).
Xie, P. et al. Genome-wide association study identifies new susceptibility loci for posttraumatic stress disorder. Biol. Psychiatry 74, 656–663 (2013).
Puts, N. A. J. Reduced GABAergic inhibition and abnormal sensory symptoms in children with Tourette syndrome. J. Neurophysiol. 114, 808–817 (2015).
Lee, J. J. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018).
Yang, L. Polygenic transmission and complex neuro developmental network for attention deficit hyperactivity disorder: genome-wide association study of both common and rare variants. Am. J. Med. Genet. Part B, Neuropsychiatr. Genet. 162B, 419–430 (2013).
Kelsom, C. & Lu, W. Development and specification of GABAergic cortical interneurons. Cell Biosci. 3, 19 (2013).
Lennington, J. B. Transcriptome analysis of the human striatum in Tourette syndrome. Biol. Psychiatry 79, 372–382 (2016).
Rapanelli, M., Frick, L. R. & Pittenger, C. The role of interneurons in autism and Tourette syndrome. Trends Neurosci. 40, 397–407 (2017).
Krause, D. L. & Müller, N. The relationship between Tourette’s syndrome and infections. Open Neurol. J. 6, 124–128 (2012).
Ercan-Sencicek, A. G. L-histidine decarboxylase and Tourette’s syndrome. N. Engl. J. Med. 362, 1901–1908 (2010).
Castellan, L. C. Histidine decarboxylase deficiency causes Tourette syndrome: parallel findings in humans and mice. Neuron 81, 77–90 (2014).
Alexander, J. Targeted re-sequencing approach of candidate genes implicates rare potentially functional variants in Tourette syndrome etiology. Front. Neurosci. 10, 428 (2016).
Eßbach, C. Abundance of Flt3 and its ligand in astrocytic tumors. OncoTargets Ther. 6, 555–561 (2013).
Kichaev, G. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 104, 65–75 (2019).
Astle, W. J. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415–1429.e19 (2016).
Jain, D. Genome-wide association of white blood cell counts in Hispanic/Latino Americans: the Hispanic Community Health Study/Study of Latinos. Hum. Mol. Genet. 26, 1193–1204 (2017).
Kanai, M. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 50, 390–400 (2018).
Rivat, C. Inhibition of neuronal FLT3 receptor tyrosine kinase alleviates peripheral neuropathic pain in mice. Nat. Commun. 9, 1042 (2018).
Marchand, F., Perretti, M. & McMahon, S. B. Role of the immune system in chronic pain. Nat. Rev. Neurosci. 6, 521–532 (2005).
Sorge, R. E. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 18, 1081–1083 (2015).
Ju, H. Q. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia 31, 2143–2150 (2017).
Dany, M. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 128, 1944–1958 (2016).
Y. Idaghdour, Y. & Hodgkinson, A. Integrated genomic analysis of mitochondrial RNA processing in human cancers. Genome Med. 9, 36 (2017).
Daver, N., Schlenk, R. F., Russell, N. H. & Levis, M. J. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia 33, 299–312 (2019).
Sun, B. B. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
Malik, R. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 50, 524–537 (2018).
Tsetsos, F. Meta-analysis of tourette syndrome and attention deficit hyperactivity disorder provides support for a shared genetic basis. Front. Neurosci. 10, 340 (2016).
Caldwell, J. M. Cadherin 26 is an alpha integrin-binding epithelial receptor regulated during allergic inflammation. Mucosal Immunol. 10, 1190–1201 (2017).
Sun, N., Tischfield, J. A., King, R. A. & Heiman, G. A. Functional evaluations of genes disrupted in patients with Tourette’s disorder. Front. Psychiatry 7, 11 (2016).
Clarke, R. A., Lee, S. & Eapen, V. Pathogenetic model for Tourette syndrome delineates overlap with related neurodevelopmental disorders including autism. Transl. Psychiatry 2, e158–13 (2012).
Verkerk, A. J. M. H. CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics 82, 1–9 (2003).
Reissner, C., Runkel, F. & Missler, M. Neurexins. Genome Biol. 14, 213 (2013).
Chatterjee, M., Schild, D. & Teunissen, C. Contactins in the central nervous system: role in health and disease. Wolters Kluwer Medknow Publ. 14, 206–216 (2019).
Tikkanen, E. Biological insights into muscular strength: genetic findings in the UK Biobank. Sci. Rep. 8, 6451 (2018).
Klimentidis, Y. C. Genome-wide association study of habitual physical activity in over 377,000 UK Biobank participants identifies multiple variants including CADM2 and APOE. Int. J. Obes. 42, 1161–1176 (2018).
Nagel, M., Watanabe, K., Stringer, S., Posthuma, D. & van der Sluis, S. Item-level analyses reveal genetic heterogeneity in neuroticism. Nat. Commun. 9, 905 (2018).
St Pourcain, B. Common variation near ROBO2 is associated with expressive vocabulary in infancy. Nat. Commun. 5, 4831 (2014).
Clifton, E. A. D. Genome-wide association study for risk taking propensity indicates shared pathways with body mass index. Commun. Biol. 1, 36 (2018).
Morgan, M. D. Genome-wide study of hair colour in UK Biobank explains most of the SNP heritability. Nat. Commun. 9, 5271 (2018).
Salyakina, D. Variants in several genomic regions associated with Asperger disorder. Autism Res. 3, 303–310 (2010).
Wang, K.-S., Liu, X.-F. & Aragam, N. A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophrenia Res. 124, 192–199 (2010).
Cornelis, M. C. A genome-wide investigation of food addiction. Obesity 24, 1336–1341 (2016).
Emilsson, V. Co-regulatory networks of human serum proteins link genetics to disease. Science 361, 769–773 (2018).
Acknowledgements
This research is co-financed by Greece and the European Union (European Social Fund—ESF) through the Operational Programme «Human Resources Development, Education and Lifelong Learning» in the context of the project “Reinforcement of Postdoctoral Researchers - 2nd Cycle” (MIS-5033021), implemented by the State Scholarships Foundation (IKY). L.K.D. was supported by grants from the National Institutes of Health including U54MD010722-04, R01NS102371, R01MH113362, U01HG009086, R01MH118223, DP2HD98859, R01DC16977, R01NS105746, R56MH120736, R21HG010652, and RM1HG009034.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
I.M. has participated in research funded by the Parkinson Foundation, Tourette Association, Dystonia Coalition, AbbVie, Biogen, Boston Scientific, Eli Lilly, Impax, Neuroderm, Prilenia, Revance, Teva but has no owner interest in any pharmaceutical company. She has received travel compensation or honoraria from the Tourette Association of America, Parkinson Foundation, International Association of Parkinsonism and Related Disorders, Medscape, and Cleveland Clinic, and royalties for writing a book with Robert rose publishers. K.M.V. has received financial or material research support from the EU (FP7-HEALTH-2011 No. 278367, FP7-PEOPLE-2012-ITN No. 316978), the German Research Foundation (DFG: GZ MU 1527/3-1), the German Ministry of Education and Research (BMBF: 01KG1421), the National Institute of Mental Health (NIMH), the Tourette Gesellschaft Deutschland e.V., the Else-Kroner-Fresenius-Stiftung, and GW, Almirall, Abide Therapeutics, and Therapix Biosiences and has received consultant’s honoraria from Abide Therapeutics, Tilray, Resalo Vertrieb GmbH, and Wayland Group, speaker’s fees from Tilray and Cogitando GmbH, and royalties from Medizinisch Wissenschaftliche Verlagsgesellschaft Berlin, Elsevier, and Kohlhammer; and is a consultant for Nuvelution TS Pharma Inc., Zynerba Pharmaceuticals, Resalo Vertrieb GmbH, CannaXan GmbH, Therapix Biosiences, Syqe, Nomovo Pharma, and Columbia Care. B.M.N. is a member of the scientific advisory board at Deep Genomics and consultant for Camp4 Therapeutics, Takeda Pharmaceutical and Biogen. M.M.N. has received fees for memberships in Scientific Advisory Boards from the Lundbeck Foundation and the Robert-Bosch-Stiftung, and for membership in the Medical-Scientific Editorial Office of the Deutsches Ärzteblatt. M.M.N. was reimbursed travel expenses for a conference participation by Shire Deutschland GmbH. M.M.N. receives salary payments from Life & Brain GmbH and holds shares in Life & Brain GmbH. All this concerned activities outside the submitted work. M.S.O. serves as a consultant for the Parkinson’s Foundation, and has received research grants from NIH, Parkinson’s Foundation, the Michael J. Fox Foundation, the Parkinson Alliance, Smallwood Foundation, the Bachmann-Strauss Foundation, the Tourette Syndrome Association, and the UF Foundation. M.S.O.’s DBS research is supported by NIH R01 NR014852 and R01NS096008. M.S.O. is PI of the NIH R25NS108939 Training Grant. M.S.O. has received royalties for publications with Demos, Manson, Amazon, Smashwords, Books4Patients, Perseus, Robert Rose, Oxford and Cambridge (movement disorders books). M.S.O. is an associate editor for New England Journal of Medicine Journal Watch Neurology. M.S.O. has participated in CME and educational activities on movement disorders sponsored by the Academy for Healthcare Learning, PeerView, Prime, QuantiaMD, WebMD/Medscape, Medicus, MedNet, Einstein, MedNet, Henry Stewart, American Academy of Neurology, Movement Disorders Society, and by Vanderbilt University. The institution and not M.S.O. receives grants from Medtronic, Abbvie, Boston Scientific, Abbott and Allergan and the PI has no financial interest in these grants. M.S.O. has participated as a site PI and/or co-I for several NIH, foundation, and industry sponsored trials over the years but has not received honoraria. Research projects at the University of Florida receive device and drug donations. D.W. receives royalties for books on Tourette Syndrome with Guilford Press, Oxford University Press, and Springer Press. The rest of the authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsetsos, F., Yu, D., Sul, J.H. et al. Synaptic processes and immune-related pathways implicated in Tourette syndrome. Transl Psychiatry 11, 56 (2021). https://doi.org/10.1038/s41398-020-01082-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-020-01082-z
