
Abstract
Despite the substantial progress made in identifying genetic defects in autism spectrum disorder (ASD), the etiology for majority of ASD individuals remains elusive. Maternal exposure to valproic acid (VPA), a commonly prescribed antiepileptic drug during pregnancy in human, has long been considered a risk factor to contribute to ASD susceptibility in offspring from epidemiological studies in humans. The similar exposures in murine models have provided tentative evidence to support the finding from human epidemiology. However, the apparent difference between rodent and human poses a significant challenge to extrapolate the findings from rodent models to humans. Here we report for the first time the neurodevelopmental and behavioral outcomes of maternal VPA exposure in non-human primates. Monkey offspring from the early maternal VPA exposure have significantly reduced NeuN-positive mature neurons in prefrontal cortex (PFC) and cerebellum and the Ki67-positive proliferating neuronal precursors in the cerebellar external granular layer, but increased GFAP-positive astrocytes in PFC. Transcriptome analyses revealed that maternal VPA exposure disrupted the expression of genes associated with neurodevelopment in embryonic brain in offspring. VPA-exposed juvenile offspring have variable presentations of impaired social interaction, pronounced stereotypies, and more attention on nonsocial stimuli by eye tracking analysis. Our findings in non-human primates provide the best evidence so far to support causal link between maternal VPA exposure and neurodevelopmental defects and ASD susceptibility in humans.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) is a group of neurodevelopmental abnormalities characterized by persistent impairment in social communication and interaction, and restricted, repetitive patterns of behaviors, interests, or activities1. The prevalence of ASD increased steadily and significantly from one in 150 children during 2000–2002 to one in 59 in 2014 in the United States2. However, the etiology for the majority of ASD cases remains unclear, while 20–25% patients having a clear genetic determinant3,4. Genetic mutations in genes encoding the proteins involving synaptic development and epigenetic machinery are two major classes of the genes from recent autism genomics studies. Recent studies point to the contribution of non-genetic factors during pregnancy to the development of ASD, such as viral infection, autoimmune diseases, and exposure to teratogens and anticonvulsants such as valproic acid (VPA)5. VPA is a widely prescribed antiepileptic drug, but its use during pregnancy may have adverse effects on the offspring. For instance, impaired cognitive function and behavioral abnormalities have been reported in children after fetal exposure to antiepileptic drugs including VPA6,7. Several epidemiological studies have linked VPA usage during pregnancy to higher risks of ASD8,9,10, attention-deficit/hyperactivity disorder11, and a wide spectrum of malformations known as fetal valproate syndrome12,13. However, clinical studies are constrained by many factors to manipulate the system in order to establish causality, and hard to explore the molecular and cellular mechanisms of ASD.
Epigenetic dysregulation is an attractive hypothesis for ASD etiology, especially considering an alarming increase in the prevalence of ASD14. Recently, a histone-acetylome-wide association study revealed a characteristic histone acetylation signature in the brain of ASD patients, providing direct evidence for histone modifications, especially acetylation, in ASD-related neuropathology15. One known pharmacological action of VPA is inhibition of histone deacetylase (HDAC), and thus VPA might increase the risk of ASD through an epigenetic mechanism16,17.
Rodent models that recapitulate certain defects caused by fetal VPA exposure in humans have been established and described. The first VPA-induced rat model was generated with a single intraperitoneal injection to pregnant dams around the time of neural tube closure18. These VPA model rats exhibit lower exploratory activity, impaired social behavior, and increased repetitive movements (stereotypies), which are considered autism-related features19. Prenatal VPA exposure also enhances NMDA receptor-mediated synaptic plasticity20, and disrupts the normal excitatory-inhibitory shift of GABAergic currents during postnatal development21, changes that may help explain the emergence of autism-like behaviors. Mice exposed to VPA in utero also exhibit impaired cognitive function in adults, which is related to decreased hippocampal neurogenesis22. The ectopic localization of newborn neurons in the hippocampus increases seizure susceptibility in prenatally VPA-exposed adult mice23.
Despite many insights derived from rodent models, whether these neuronal defects are conserved between rodents and primates remains unknown. Non-human primates have the potential to validate the knowledge gained from rodent models and provide possible explanations for the effects of in utero VPA on humans24,25. Although rodent models are widely used to study human psychiatric disorders, the differences in brain anatomy and behavioral repertoires between rodents and humans preclude simple extrapolation24,25,26. Non-human primates have a much closer evolutional relationship with humans and thus are believed to have greater potential for translational research. For instance, monkeys are widely used for pharmacokinetic and toxicology studies preceding clinical trials27. In fact, the adverse effects of VPA have been examined in pregnant rhesus monkeys28. Craniofacial and skeletal defects were found in embryos from mothers treated at the dose that is 10 × human clinical dose (common dosage for children 20 mg/kg/day), while 100% of embryos were nonviable when mothers were treated at 30 × clinical dose28. However, the effect of VPA on brain development and behaviors in primates has not been addressed.
In this study, we investigated the molecular, cellular, and behavioral changes in cynomolgus monkeys (Macaca fascicularis) after early exposure to VPA in utero. We examined changes in gene expression and cellular development in the fetal brain, and aberrant behaviors in VPA-treated juvenile monkeys. Prominent neurogenesis defects and altered gene expression levels were found in the VPA-treated brain. The surviving juvenile offspring exhibited pronounced stereotypies and social interaction defects. Eye-tracking experiments revealed longer fixation towards nonsocial scenes by VPA-treated animals. The neuronal and behavioral defects found in prenatally VPA-exposed monkeys provide novel insights into the pathologies associated with VPA exposure in humans.
Materials and methods
Animals and drug administration
Healthy adult cynomolgus monkeys (Macaca fascicularis) were housed in cages at Guangdong Blooming-Spring Biological Technology Development Co., Ltd. All animals were housed in a controlled environment (room temperature at 25 °C, humidity 40–70%) with 10–h light/14–h dark cycle (lights on at 0800 hours). All animals were given regular diet twice a day with tap water ad libitum and were fed fruits and vegetables once daily. All animal procedures were approved by the Institutional Animal Care and Use Committee of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences (IGDB-2016-IRB-003).
Fifteen healthy and fertile female monkeys (body weight: 4.61 ± 0.26 kg; number of offspring produced: 5.67 ± 0.40; age: 9.33 ± 0.23 years old, data are presented as mean ± s.e.m., n = 15) were caged separately (cage size: 0.75 m L × 0.85 m W × 1 m H) for menstrual cycle observation over three months. Individual female was housed with one male (age: 8.75 ± 1.49 years old) for 5 days beginning 2 days before expected ovulation to facilitate mating. The day of ovulation was designated as gestational day (GD) 0. Pregnancy was confirmed in nine female monkeys by ultrasonographic examination (Vevo2100, VisualSonics, Toronto, CA) on GD 19–23 under general anesthesia by intraperitoneal injection with Zoletil 50 (4–6 mg/kg, Virbac S.A., France). Nine age- and gender-matched monkeys from control mothers without anesthesia and VPA injection were selected as controls. The controls were raised in conditions similar to that of VPA-treated group.
Two critical parts of the design are the time window and dosage of VPA exposure. The in utero rats were exposed to VPA once during E11.5–12.5 to induce autistic-like features18, a period around neural tube closure (E10–12)29. It has been speculated that VPA also disturbs the same stage to cause autism in humans18,30. We decided to administrate VPA twice on GD 26 and 29 based on the longer time of neural tube closure of monkey embryos. One group of five pregnant monkeys was intraperitoneally injected with 200 mg/kg NaVPA (Meilunbio, Dalian, China; dissolved in 0.9% sterile saline, pH 7.3), and another group of four pregnant monkeys was treated with 300 mg/kg NaVPA. The dose of 200 mg/kg in monkeys is equivalent to 500 mg/kg in rats20. The low pregnancy rate (9/15) and high abortion rate (4/9) in VPA-treated monkeys contributed to a small sample size.
All treated and untreated monkey offspring are listed in Table 1. Infant offspring were raised with their mothers in home cages (0.8 m L × 0.6 m W × 2 m H) and weaned at about 10 months of age. The juveniles were housed in home cages in groups before behavioral tests.
Immunohistochemistry
For immunostaining, the brains of aborted fetuses and untreated controls were removed and fixed for 48 h in 4% paraformaldehyde. Different brain regions including the PFC and cerebellum were dissected out and paraffin-embedded. Paraffin-embedded tissues were sliced into 4-μm-thick sections. The primary antibodies used in this study are listed in Supplementary Table 1. Samples were incubated with corresponding HRP-conjugated secondary antibodies (anti-mouse or anti-rabbit, 1:1000; Dako, USA). DAB (3, 3'-diaminobenzidine) staining was used for chemiluminescent detection and hematoxylin for nuclear staining. Images were acquired with a Leica SCN400 Slide Scanner (Leica Microsystems).
For cell density analysis, cells within specific areas ( > 0.2 × 0.1 mm2) across all layers of PFC were counted manually. The area of positive NeuN staining in the cerebellar internal granular layer and the thickness of the Ki67-positive (proliferating) external granular layer of cerebellum were measured by ImageJ.
Western blotting
Prefrontal cortex (PFC) was homogenized in RIPA buffer (Hua Xing Bo Chuang, with 1 × protease inhibitor cocktail) on ice. Supernatant protein was separated by SDS-PAGE and transferred onto PVDF membranes (Millipore). Carbonate blot buffer (10 mM NaHCO3, 3 mM Na2CO3, pH 9.9 and 20% methanol) was used for efficient electrophoretic transfer of histones to membranes. The primary antibodies used are listed in Supplementary Table 1. Specific bands were quantified by ImageJ and normalized to α-tubulin expression (the gel loading control).
RNA preparation and sequencing
Total RNA was extracted from the PFC of VPA-exposed (M1 and M2) and age-matched control monkeys (Ctl1 and Ctl2). Only samples with RNA integrity number (RIN) over 6.8 were used for cDNA library construction. Sequencing was performed on a single lane of an Illumina HiSeq 4000 to produce 150 bp paired-end reads. The clean reads were aligned to the cynomolgus monkey genome (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/364/345/GCF_000364345.1_Macaca_fascicularis_5.0/GCF_000364345.1_Macaca_fascicularis_5.0_genomic.fna.gz) using TopHat2 software (http://ccb.jhu.edu/software/tophat/index.shtml). Normalized transcript abundance was estimated by the expected fragments per kilobase of transcript per million fragments mapped (FPKM) using Cuffnorm (http://cufflinks.cbcb.umd.edu/). We performed three independent replicates from adjacent areas for each animal. Differentially expressed genes (DEGs) between VPA-treated and control monkeys were filtered using the DEseq package (http://www.bioconductor.org/packages/release/bioc/html/DESeq.html). DEGs defined by (1) fold change (FC) > 2 or < 0.5 and (2) false discovery rate (FDR) < 0.01 were grouped into different Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment pathways.
Quantitative real-time PCR (qRT-PCR)
Extracted RNA was reverse transcribed using a SuperScript III First Strand cDNA synthesis kit (Invitrogen), followed by qRT-PCR using SYBR Green PCR Master Mix (ABI) on a Real-Time QPCR System (Agilent). The relative mRNA expression levels were analyzed according to the ΔΔCt method31. GAPDH was used as the reference gene. The genes and primers used for qRT-PCR are listed in Supplementary Tables 2 and 3. For validation of RNA-seq results, we compared the qPCR results with RNA-seq data using Pearson correlation test.
Behavior analysis
For behavioral analysis, the surviving juvenile monkeys at 17–21 months of age (5 VPA-treated and 5 controls in total; Supplementary Table 4) were re-housed in observation cages (2 m L × 1 m W × 1 m H). All animals were divided into three groups (cage 1–3). In each cage, the VPA-treated monkeys of similar ages were grouped with the age- and sex-matched controls. In cage 3, two animals (ctl2 and t2) from cage 1 were re-used to ensure an equal group size.
Behaviors of all animals were video-recorded for 1 h daily at the same time (1000–1100 hours or 1500–1600 hours) of the day for 5 days within two weeks. A 20-min video in the middle of 1-h recording was selected for analysis. Five 20-min videos collected from 5 days were analyzed for each animal. Each animal wore a colored collar to be identified. We quantified the duration of active and passive social contact, stereotypical behaviors, and environmental exploration from videos using an established ethogram (see Supplementary Table 5). Each video-recording was analyzed by three trained technicians blinded to experimental conditions. An inter-observer reliability > 85% was established before scoring. During the scoring, the technician recorded the frequency and duration of a specific behavior by manually starting and stopping the video.
Noninvasive eye tracking
Eye-tracking analysis was performed largely following a previously published protocol32. Animals (5 VPA-treated and 5 control juvenile monkeys; Supplementary Table 4) were seated in a primate chair with their head fixed by a helmet 57 cm in front of a 27-inch 16:9 computer screen. Gaze positions were sampled at 1000 Hz using an infrared eye-tracker (SensoMotoric). Visual stimulus presentation and data collection were performed using the MonkeyLogic Toolbox of MATLAB (http://www.monkeylogic.net). We presented four types of visual stimuli to investigate animals’ attention and social processing. The four types of stimuli include (1) macaque faces (“face”, n = 20 images) (https://visiome.neuroinf.jp/primface), (2) natural complex scenes including monkeys (“social & other”; “social” refers to the monkeys while “other” indicates regions without monkeys, n = 25 images), (3) natural complex scenes without monkeys (“nonsocial”, n = 25 images), and (4) simple scenes such as grass land (“simple”, n = 10 images). Each stimulus was presented for 5 s followed by a 2-s blank gray screen. The image size was 17.8 × 17.8° of visual angle for “face” stimuli, and 17.8 × 13.3° for the remaining three types of stimuli. All animals were tested twice on two different days within one week. Data of five monkeys in each group from 2 days were averaged and each data point represents the results from an individual monkey.
Statistical analysis
Data analysis was conducted using SPSS IBM 19. Sample sizes were not pre-determined using statistical methods. The normality and variance of data distribution between two groups were analyzed by Kolmogorov–Smirnov test and Levene's test, respectively (P > 0.05). Unpaired Student’s t test (two-tailed) was performed for all experiments except for RNA-seq data analyses. P < 0.05 is considered indicative of significant differences between groups. No data points were excluded for any of the experiments. All data are presented as mean ± s.e.m.
Results
Increased abortion risk but largely normal infant development upon in utero VPA treatment
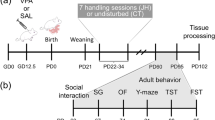
To investigate the effects of in utero VPA exposure in primates, we injected VPA intraperitoneally during neural tube closure. The detailed experiment schedule is shown in Fig. 1a. It has been reported that rats exposed to VPA during E11.5–12.5, a period around neural tube closure (E10–12), manifest autism-like behaviors18,19. Neural tube closure occurs rapidly over a two-day period in rodents, while a comparable process requires approximately one week in primates33. Therefore, VPA (200 or 300 mg/kg, double injections) was administered by intraperitoneal injection (i.p.) on GD 26 and 29 (Table 1), during which time neural tube closure is expected to occur in monkey embryos33. The dosage for i.p. route used was extrapolated from rat models19,20, about 10 times the human therapeutic dose of 20–30 mg/kg/day.

a Diagram of the experiment design. Pregnant monkeys were treated with 200 or 300 mg/kg VPA on G26 and 29. The full-term aborted fetuses were used for molecular and cellular analysis. Physical development, social interactions, and eye tracking were examined in the live offspring. b NeuN staining (brown) to label mature neurons in the prefrontal cortex (PFC). Nuclei were stained with hematoxylin (blue). Cell density and the ratio of NeuN-positive neurons appeared substantially reduced in each PFC layer of VPA-treated monkeys compared to age- and sex-matched controls. NeuN-positive cortical pyramidal neurons also showed aberrant dendritic morphology (insets). c, d Immunostaining for NeuN (brown) to mark mature neurons c and Ki67 (brown) to mark proliferating granule cell precursors d in cerebellum. Nuclei were stained with hematoxylin (blue). The density of NeuN-positive cells c and the thickness of EGL d appeared substantially lower in VPA-treated monkey cerebellum. EGL external granular layer; ML molecular layer; PCL Purkinje cell layer, IGL internal granular layer. e–h Quantitative analysis of total cell density e, the percentage of NeuN-positive cells in layers II–VI of the PFC f, the percentage of NeuN-positive area in the cerebellar granule cell layer g, and the thickness of the region stained by anti-Ki67 h. Scale bars: 100 μm for b and d and 20 μm for c; error bars represent s.e.m.; *P < 0.05, **P < 0.01, and ***P < 0.001 by Student’s t-test
After timed mating, we obtained nine pregnancies verified by ultrasonographic examination. Of these, four (t6, t7, M1 and M2) aborted after VPA administration. Among these abortions, one (t6) occurred on G29 shortly after VPA administration as evidenced by menstrual bleeding, while the other three were full-term abortions. Two fresh embryos, M1 and M2, were collected for further pathological analysis while the other (t7) was stillborn and not fresh enough for further analysis. In addition, we examined developmental and behavioral abnormalities in the five live VPA-treated monkey offspring (t1–t5). These VPA-treated monkeys, as well as age- and sex-matched controls were housed with their own mothers separately until 10 months old. Body weight, head circumference, and chest circumference of VPA-treated and age-matched control monkeys were measured each month starting from five months after birth. Compared to control monkeys, body weight, head circumference, and chest circumference of the VPA-treated monkeys appeared to be comparable to that of the controls (Supplementary Fig. 1).
Disrupted neurogenesis in the brain of VPA-treated monkeys
To examine whether VPA exposure may result in neuropathological changes, we performed immunochemical analysis with neuronal and glial markers in the brains of two aborted embryos, M1 (male) and M2 (female). NeuN is a neuron-specific nuclear protein commonly used to identify mature neurons, while GFAP is a known marker for astrocytes. We focused on two different brain regions, PFC and cerebellum, to indicate general changes across the primate brain. The layered structure of the PFC was compared between VPA-treated (M1 and M2) and two age- and sex-matched control embryos (Ctl1 with M1 and Ctl2 with M2). At the cellular level, we examined the morphology and number of neurons in the PFC. Typically, the apical dendrites of pyramidal neurons extend vertically above the soma toward the brain surface. In M1, however, the morphology of NeuN-positive cells exhibited smaller or absent dendritic extensions compared to the controls (Fig. 1b). Additionally, we found significantly fewer cells, based on hematoxylin staining, in the PFC of VPA-treated monkeys with largely normal structure of each layer (Fig. 1b, e). The cell density of cortical layers II to VI was significantly lower in VPA-treated monkeys than controls (2000 ± 20.69 cells/mm2 for Ctl1 vs. 1506.3 ± 46.45 cells/mm2 for M1, P < 0.001; 2079.2 ± 27.53 cells/mm2 for Ctl2 vs. 1525 ± 16.32 cells/mm2 for M2, P < 0.001; n = 4 regions for each layer; Fig. 1b, e). We further analyzed the percentage of NeuN-positive cells in layers II–VI of PFC (Fig. 1b, f) and the percentage of NeuN-positive area in the cerebellar granular layer (Fig. 1c, g). Both VPA-exposed brains exhibited a lower percentage of NeuN-positive cells in the PFC compared to controls (73.94% ± 1.42% NeuN+ cells/total cell counts for Ctl1 vs. 51.15% ± 0.95% for M1, P < 0.001; 73.44% ± 0.73% for Ctl2 vs. 22.1% ± 1.14% for M2, P < 0.001; n = 6 regions for each layer; Fig. 1b, f), as well as a smaller NeuN-positive area in the cerebellar internal granular layer (80.33% ± 2.11% area for Ctl1 vs. 50.85% ± 1.66% for M1, P < 0.001; 74.05% ± 2.22% for Ctl2 vs. 24.45% ± 1.19% for M2, P < 0.001; n = 6 regions for each individual; Fig. 1c, g). In contrast, the number of GFAP-positive astrocytes per square millimeter was higher in the PFC of M1 and M2 compared to their matched controls (Supplementary Fig. 2a, b).
The significantly lower cell density in the PFC and cerebellum of VPA-exposed brain suggests that neuronal production may be reduced. To study the effects of VPA exposure on neural cell proliferation directly, we stained the stem cell layer of cerebellum with the cell proliferation marker Ki67. The external granular layer (EGL) covering the cerebellar surface is mainly composed of granule cell precursors in the embryonic brain34. The Ki67-positive EGL was significantly thinner in VPA-exposed brains compared to controls (33.32 ± 1.08 μm for Ctl1 vs. 18.47 ± 0.97 μm for M1, P < 0.001; 36.03 ± 0.29 μm for Ctl2 vs. 8.31 ± 0.64 μm for M2, P < 0.001; n = 12 measurements from each individual; Fig. 1d, h). These results together demonstrate that prenatal VPA exposure disrupts normal neurogenesis in PFC and cerebellum of the monkey brain.
Prenatal VPA alters the expression of neural markers and synaptic proteins in the PFC
Prenatal VPA exposure has been reported to alter neural protein expressions in rodents20,35. We therefore compared the expression levels of neuronal markers and synaptic proteins in the PFC of VPA-treated and control monkeys by western blotting (Fig. 2a). VPA exposure altered the expression of neuronal and glial markers, consistent with results of immunohistochemistry (Fig. 1b and Supplementary Fig. 2a). In utero VPA reduced the expression levels of both the mature neuron marker NeuN (M1, 89.99% of Ctl1, P = 0.2939; M2, 25.07% of Ctl2, P = 0.0019; n = 3) and the immature neuron marker doublecortin (DCX) (M1, 39.35% of Ctl1, P = 0.0049; M2, 5.34% of Ctl2, P < 0.001; n = 3), but increased GFAP expression (M1, 809.05% of Ctl1, P = 0.0044; M2, 561.2% of Ctl2, P = 0.0065; n = 5) (Fig. 2a–c), again suggesting that VPA inhibits neurogenesis and promotes astrocyte production or activation during embryo development.

a Reduced expression of neuronal markers (VGLUT2, DCX, and NeuN) and glutamate receptors (mGluR5 and GluN2B), but increased expression of the GABAergic marker VGAT and glial marker GFAP in the PFC of VPA-treated monkey brain. Ctl1 and Ctl2 are age- and sex-matched controls. b–d Quantification of the relative expression levels of neuronal markers b, the glial marker GFAP c, and synaptic proteins d. Western blotting of independently prepared samples was repeated at least three times; error bars represent s.e.m.; *P < 0.05, **P < 0.01, and ***P < 0.001 by Student’s t-test
While there were no differences in the levels of PSD95 and Homer1b/c between VPA-treated and control samples (Fig. 2a, d), VPA-treated PFC expressed significantly lower levels of metabotropic glutamate receptor 5 (mGluR5) (M1, 53.74% of Ctl1, P < 0.001; M2, 10.07% of Ctl2, P < 0.001; n = 3), NMDA receptor subunit GluN2B (M1, 60.5% of Ctl1, P < 0.001; M2, 52% of Ctl2, P = 0.0021; n = 3) (Fig. 2a, d). We note that changes in the expression levels of NeuN, DCX, mGluR5 and GluN2B, but not GFAP, were greater in M2, which was exposed to a higher dose of VPA than M1 (300 vs. 200 mg/kg).
In addition to the alterations in the expression of neural markers and a subset of glutamate receptors, we found opposite changes in protein levels of glutamatergic and GABAergic markers in VPA-treated brains. Vesicular glutamate transporter 2 (VGLUT2), a marker for glutamatergic synapses, was significantly lower in the PFC of VPA-treated monkeys (M1, 72.57% of Ctl1, P = 0.0215; M2, 5.63% of Ctl2, P < 0.001; n = 3; Fig. 2a, b). Conversely, vesicular GABA transporter (VGAT), a GABAergic synapse marker, was significantly higher in the PFC of VPA-treated monkeys (M1, 134.63% of Ctl1, P = 0.0087; M2, 223.34% of Ctl2, P = 0.0014; n = 3; Fig. 2a, b). Notably, the embryo exposed to the higher VPA dose (M2) exhibited more prominent changes in the protein level of VGLUT2 and VGAT, similar to the changes of the neuronal markers NeuN and DCX. These results suggest that in utero VPA exposure may alter the balance between excitatory and inhibitory (E/I) synapses in monkey PFC.
Altered transcriptome in VPA-treated monkey brain
While the mechanism of anticonvulsant effect for VPA is not completely understood, one of the known pharmacological actions of VPA is inhibition of HDAC16. Extensive genetic studies of ASD have uncovered the etiological role of epigenetic dysregulation in ASD36,37. We therefore examined a potential effect of VPA on histone acetylation in the monkey brain in two aborted fetuses. In contrast to the increased level of acetylated histone shortly (a few days) after VPA treatment reported in literature38,39, we found a significantly reduced level of acetylation of H3K27 (H3K27ac) in the PFC of M1 (M1, 48.20% of Ctl1; P = 0.0115; n = 5; Fig. 3a, b), but no significant change in acetylation of H3K9 (H3K9ac) in the PFC of both M1 and M2 ~130 days after VPA exposure.

a Representative blots of acetylated histone H3 at K9 and K27 in PFC of VPA-treated and control monkeys. b Statistical analysis shows reduced expression of H3K27ac in M1 PFC. Error bars represent s.e.m.; *P < 0.05 by Student’s t-test. c Heat map representation of differentially expressed genes (DEGs) in VPA-treated monkeys compared to controls. DEGs were distinguished by false discovery rate (FDR) ≤ 0.01 and fold change (FD) ≥ 2 or ≤ 0.5. The columns indicate different samples with three replicates and the rows represent the 1722 DEGs identified between VPA-treated and control monkeys clustered by similar expression patterns. The color denotes the expression levels of different genes as indicated by log2 (FPKM + 1). FPKM, expected fragments per kilobase of transcript per million fragments mapped. d qRT-PCR validation of the RNA-seq results in a VPA-treated male (M1) and a female (M2) monkey. GAPDH was used as the reference gene. The values in both axes denote log2 (FC) in expression. Pearson's correlation coefficient, r = 0.9509, P < 0.0001 for M1 and r = 0.9242, P = 0.0004 for M2. e KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis of the DEGs in response to VPA exposure. Among 1,722 DEGs, 882 are functionally mapped into different KEGG pathways. The vertical line indicates P = 0.05 (−log10 P = 1.301). P value represents Benjamini–Hotchberg correction for multiple tests
VPA could have widespread effects, direct or indirect, on gene expression by affecting histone acetylation levels16,17. Therefore, we performed RNA-seq analysis in the PFC of two VPA-treated embryos (M1 and M2) and two age- and sex-matched control embryos (Ctl1 and Ctl2). We analyzed three independent samples from each animal for biological repeats. The clean reads were aligned to the genome of crab eating macaque (version: Mfa5.0). In total 15,095 expressed genes were retrieved across all samples. Genes expressed in two VPA-treated monkeys were compared to that of two control monkeys, and we focused on genes with shared changes in two VPA-treated monkeys to exclude the effect of sex difference and dosage difference. Gene expression analysis identified 1722 differentially expressed genes (DEGs) in both VPA-treated monkey brains (FDR ≤ 0.01 and log2 FC ≥ 1 or log2 FC ≤ −1; Fig. 3c). Among these, 1187 were upregulated and 535 were downregulated (see Supplementary Table 6 for gene lists). We selected 9 genes that were either having large fold changes in expression or functioning in neurodevelopmental processes (Supplementary Table 2) from RNA-seq for validation analysis using qRT-PCR. Comparison of gene expression levels obtained from RNA-seq and qRT-PCR showed a highly significant Pearson correlation coefficient (r) (M1: r = 0.9509, P < 0.0001; M2: r = 0.9242, P = 0.0004; Fig. 3d). Neuronal marker DCX was significantly decreased in RNA-seq analysis, consisting with the Western results in Fig. 2c. To identify the functional pathways involving these DEGs, we conducted KEGG enrichment analysis. Among the 1722 DEGs, 882 are functionally annotated by the KEGG database. The top 10 enriched pathways are shown in Fig. 3e. The most significantly disrupted pathway in both VPA-treated brains was axon guidance (ko04360), including guidance cues (e.g., netrins and semaphorins) that regulate actin cytoskeleton, thereby affecting neuronal network formation40. In the calcium signaling pathway (ko04020), CaMKII (calcium/calmodulin-dependent protein kinase II), critically involved in learning and memory, was significantly upregulated (Supplementary Table 6), consistent with a previous finding in a VPA rat model20. In summary, the top KEGG pathways induced by VPA treatment are involved in neuronal development and signaling.
Impaired social interaction and stereotypies in VPA-treated monkeys
We also analyzed social behaviors in VPA-treated monkeys (t1–t5), as defects in social interaction are reported in VPA rodent models19. All the live monkeys (5 VPA-treated and 5 untreated controls, Supplementary Table 4) were habituated in the observation cages before the social test. Behaviors of each VPA-treated monkey were quantitatively compared with individual controls within the same cage. The social behaviors of a tested individual were categorized as active or passive based on 1-h daily observation for 5 days, but a 20-min video in the middle of 1-h recording from each day was statistically analyzed. The average time of VPA-treated monkeys (t1 and t3) actively interacting with other monkeys was significantly lower than that of the controls within the same cage (67.32 ± 10.21 s for t1 vs. 382.5 ± 93.16 s for ctl1 and 240.6 ± 54.29 s for ctl2, P = 0.01 and 0.032, respectively; 35.45 ± 4.68 s for t3 vs. 110.7 ± 24.36 s for ctl3, P = 0.035; Fig. 4a). The VPA-treated monkeys (t3 and t4) also received less passive social interaction from other monkeys within the same cage (43.53 ± 17.99 s for t3 and 19.82 ± 8.16 s for t4 vs. 166.5 ± 34.6 s for ctl4, P = 0.014 and 0.012, respectively), while t5 received more social interactions than the control monkey (5.468 ± 2.76 s for ctl5 vs. 54.4 ± 19.88 s for t5, P = 0.041; Fig. 4b). We note that one control monkey (ctl3) displayed mild but significant stereotypy compared with the other three monkeys within cage 2 (Fig. 4c). Importantly, one VPA-treated monkey (t1) exhibited significantly more repetitive walking compared to controls within cage 1 (92.19 ± 18.2 s for t1 vs. 0.84 ± 0.84 s for ctl1 and 2.55 ± 1.79 s for ctl2, P = 0.007 and 0.008, respectively; Fig. 4c and Supplementary Video). However, the time spent on exploration of the surroundings was comparable between VPA-treated and control monkeys within all three cages (Fig. 4d). Therefore, VPA exposure disturbs social behaviors in some juvenile monkeys, while exploratory behaviors appear unaffected.

a–d Active/passive social interaction durations, defined by the average time a monkey initiated a or received b interaction with any other monkey in the group during a 20-min period. Stereotypical c and exploratory d behaviors depicting the durations of repetitive circling and exploration towards the surroundings, respectively. n = 5 for VPA-treated and control monkeys each. Three monkeys (t1, t2, and t3) were exposed to 200 mg/kg VPA twice on GD 26 and 29. The other two monkeys (t4 and t5) were exposed to 300 mg/kg VPA twice on GD 26 and 29. Each boxplot represents the data of one monkey and each dot depicts the data recorded daily from one monkey. Different colors indicate different cages. e Examples of representative stimuli used in the eye-tracking test: “face” (n = 20 different images) and “social & other” (natural complex scene with monkeys, n = 25 images). f The average time spent on each stimulus of 80 total images including “face”, “social & other”, “nonsocial”, and “simple”. VPA-treated monkeys spent significantly more time on “social & other” and “simple” stimuli. g Fixation duration directed at the region of “eye” or “mouth” of the image, and the ratio of duration on “eye” to that of “mouth”. h Fixation duration directed at the region of “social” or “other” of the image, and the ratio of duration on “social” to that of “other”. Each dot represents the data from one monkey. Error bars represent s.e.m., *P < 0.05 and **P < 0.01 by Student’s t-test
Abnormal visual attention in VPA-treated monkeys
Eye tracking is a well-established method to assess visual preferences in patients with ASD by analyzing the focus of visual attention41. ASD patients often exhibit reduced attention to socially relevant stimuli such as faces41. To test whether VPA treatment similarly disrupts visual attention toward social stimuli in non-human primates, we performed eye-tracking analysis following previously published protocols32.
On average, the VPA-treated group spent significantly more time on looking at the 80 stimuli of the four categories (541.1 ± 105.4 ms per stimulus for control vs. 984.6 ± 137.8 ms per stimulus for VPA-treated, P = 0.036; Fig. 4e, f). When exposed to the “face” stimuli, the two groups of animals showed a similar eye-movement pattern with similar eye-to-mouth ratios (P = 0.629; Fig. 4e, g). However, when exposed to the “social & other” images, VPA-treated animals spent significantly more time looking at the nonsocial portion of images (“other”) (759.02 ± 88.52 ms per stimulus) than the control animals did (380.91 ± 109.74 ms per stimulus, P = 0.028; Fig. 4h), while the time spent on “social” scenes was comparable between the two groups. The ratio of fixation duration in “social” versus “other” region appeared reduced in treated monkeys than controls, but there was no significant difference between the two groups (P = 0.199; Fig. 4e, h). These results were consistent with findings of reduced attention to social cues in autism children41,42,43. Together, the VPA-treated monkeys seemed to pay more attention to nonsocial stimuli than the control did.
Discussion
Environmental factors during sensitive brain developmental periods have long-term consequences on brain function44. In utero exposure to antiepileptic drug VPA is linked to brain disorders in offspring, most notably ASD10,45. However, the etiology of environmental factors on neurodevelopment is far from clear, but pivotal for understanding the etiology of ASD without clear genetic causes. In this study, we obtained two aborted monkeys of different sex and different VPA exposure levels and analyzed VPA-induced abnormalities at the molecular and cellular levels. We found that prenatal VPA exposure could disrupt neurogenesis in non-human primates, resulting in reduced numbers of Ki67-positive neuronal precursors and NeuN-positive mature neurons. In utero VPA exposure also altered the expression levels of multiple proteins associated with glutamatergic and GABAergic neurotransmission. Moreover, the phenotype of VPA-treated monkeys appeared to be dose-dependent, as more severe neurogenesis defects and greater decreases in the expression levels of mGluR5, GluN2B, and DCX were observed in the monkey (M2) exposed to a higher dose of VPA, consistent with the known VPA dose-dependent increase in risk for major congenital malformations in human newborns46.
Multiple effects of VPA on neural progenitor cell (NPC) proliferation and neuronal differentiation have been reported in primary cell cultures, including decreased NPC proliferation, increased neuronal differentiation, and inhibition of glial differentiation47. Long-term VPA exposure from embryonic day 1 until birth in mice increased the number of NPCs during the early-middle neurogenic period as well as postnatal neuronal production48. In contrast, we found thinner Ki67-positive EGL in cerebellum of VPA-exposed monkeys, indicative of fewer neuronal precursors. Our finding is consistent with a report that prenatal VPA exposure at E12.5 to mice causes a decrease in cell proliferation by 7 days and decreased cell density in prefrontal cortex at 8 weeks38. In addition, we found fewer neurons, more astrocytes and decreased cell density in full-term fetal brain after prenatal VPA treatment. Glial cells constitute the predominant proportion of human brain cells and play essential roles in neuronal differentiation and metabolism as well as in the regulation of neuroinflammation49. Elevated GFAP expression and more GFAP-positive glial cells in VPA-treated monkey PFC suggest alterations in neuronal differentiation, reactive gliosis, or both.
Factors controlling the formation and function of excitatory and inhibitory synapses affect the E/I ratio; altered ratio of E/I is believed to be a possible pathological mechanism underlying ASD50. The imbalance of E/I synapses has been reported in VPA rodent models51. In this study, western blotting revealed increased expression of the inhibitory synapse marker VGAT but decreased expression of the excitatory marker VGLUT2 in VPA-treated PFC (Fig. 2a, b). The decreased expression of presynaptic VGLUT2 is consistent with the reduced expression of postsynaptic mGluR5 and GluN2B in VPA-treated monkey brain (Fig. 2a, d). In addition to neurogenic defects, the altered E/I synaptic components may also contribute to behavioral abnormalities associated with VPA treatment.
We investigated the changes in transcriptome of monkey brain ~130 days after VPA exposure. Multiple pathways related to neuronal development were altered in VPA-exposed monkey embryos, indicating a long-lasting effect of epigenetic alterations. Gene expression changes right after VPA treatment are attributed mainly to HDAC inhibition16,17, while the long-lasting effect of VPA exposure is unknown. We showed that H3K27ac, a marker of active enhancers, was significantly decreased in M1 PFC. Epigenetic modification at enhancers contributes to the specific gene expression programs that determine cell proliferation and differentiation52. A VPA rat model displays a disrupted amygdala transcriptome at different time points (P10 and P21), suggesting that VPA disrupts molecular pathways related to autism-like behaviors17. Given the dramatic changes at the cellular level revealed by immunostaining, it is not surprising that a vast number of DEGs (1722) were identified upon VPA treatment.
Clinical studies of ASD patients report a defect in engaging attention to socially relevant stimuli. Specifically, individuals with ASD look less at the social scenes and monitor less the play interactions42,43. In agreement with the clinical studies, our eye-tracking analysis showed that VPA-treated monkey offspring paid significantly more attention to the non-social scene when presented with “social & other” images (Fig. 4h). VPA-exposed rodents display more pervasive autism-like behavioral deficits, including reduced sociability, stereotypies, and sensory anomalies53, while VPA-treated monkeys showed a lower penetrance of autism-like behaviors. The low penetrance of autism-like behaviors in monkeys may attribute to sex effects, as male VPA rats showed substantial social interaction deficits compared to female offspring54,55, and only one out of five VPA-treated monkeys reported in this study was male (t5). Individual difference is another explanation for the severe neurogenic defects found in the aborted fetal brain but low penetrance of autism-like behaviors in the five live offspring. Differences in maternal and/or infant metabolism and genetic susceptibility have been reported to affect the teratogenicity of VPA56. Children exposed to VPA during pregnancy also demonstrate variable symptom severity9,10, supporting the existence of individual difference in drug susceptibility. We suspect that monkeys that are more sensitive to VPA exposure probably failed to develop to term, while the less sensitive individuals survived to adulthood. The apparent variability among VPA exposure animals is consistent with human cases, i.e., a large number of pregnant women were exposed to VPA, but only small percentage of the offspring were abnormal, suggesting different individual susceptibility.
In summary, we found pronounced neurogenesis defects and autism-related behavioral abnormalities in VPA-exposed monkey offspring. VPA-exposed monkeys may offer novel insights into the pathophysiology of VPA-induced developmental defects and behavioral impairments in ASD patients. Studies of VPA treatment in both rodents and non-human primates reveal similarities but also clear and substantial differences in neurological and behavioral outcomes. In utero VPA-treated rodents are a valid model for ASD research, while the value of VPA-exposed monkeys for ASD research requires further testing (such as the prevalence and severity of sex-specific social interaction defects). Nonetheless, this study confirms that VPA has substantial effects on neurogenesis that may account for behavior deficits in primates. Further study on the use of this non-human primate model for ASD is warranted.
References
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th edition (American Psychiatric Association, Washington DC, 2013).
Baio, J. et al. Prevalence of autism spectrum disorder among children aged 8 years-autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill. Summ. 67, 1–23 (2018).
Miles, J. H. Autism spectrum disorders-a genetics review. Genet. Med. 13, 278–294 (2011).
Jeste, S. S. & Geschwind, D. H. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat. Rev. Neurol. 10, 74–81 (2014).
Ornoy, A., Weinstein-Fudim, L. & Ergaz, Z. Genetic syndromes, maternal diseases and antenatal factors associated with autism spectrum disorders (ASD). Front. Neurosci. 10, 316 (2016).
Dean, J. C. et al. Long term health and neurodevelopment in children exposed to antiepileptic drugs before birth. J. Med. Genet. 39, 251–259 (2002).
Meador, K. J. et al. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N. Engl. J. Med. 360, 1597–1605 (2009).
Williams, G. et al. Fetal valproate syndrome and autism: additional evidence of an association. Dev. Med. Child. Neurol. 43, 202–206 (2001).
Bromley, R. L. et al. The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J. Neurol. Neurosurg. Psychiatry 84, 637–643 (2013).
Christensen, J. et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 309, 1696–1703 (2013).
Cohen, M. J. et al. Fetal antiepileptic drug exposure: adaptive and emotional/behavioral functioning at age 6years. Epilepsy Behav. 29, 308–315 (2013).
Moore, S. J. et al. A clinical study of 57 children with fetal anticonvulsant syndromes. J. Med. Genet. 37, 489–497 (2000).
Jentink, J. et al. Valproic acid monotherapy in pregnancy and major congenital malformations. N. Engl. J. Med. 362, 2185–2193 (2010).
Grafodatskaya, D., Chung, B., Szatmari, P. & Weksberg, R. Autism spectrum disorders and epigenetics. J. Am. Acad. Child Adolesc. Psychiatry 49, 794–809 (2010).
Sun, W. et al. Histone acetylome-wide association study of autism spectrum disorder. Cell 167, 1385–1397 (2016).
Phiel, C. J. et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 276, 36734–36741 (2001).
Barrett, C. E. et al. Developmental disruption of amygdala transcriptome and socioemotional behavior in rats exposed to valproic acid prenatally. Mol. Autism 8, 42 (2017).
Rodier, P. M., Ingram, J. L., Tisdale, B., Nelson, S. & Romano, J. Embryological origin for autism: developmental anomalies of the cranial nerve motor nuclei. J. Comp. Neurol. 370, 247–261 (1996).
Schneider, T. & Przewlocki, R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology 30, 80–89 (2005).
Rinaldi, T., Kulangara, K., Antoniello, K. & Markram, H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl Acad. Sci. USA 104, 13501–13506 (2007).
Tyzio, R. et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679 (2014).
Juliandi, B. et al. Reduced adult hippocampal neurogenesis and cognitive impairments following prenatal treatment of the antiepileptic drug valproic acid. Stem Cell Rep. 5, 996–1009 (2015).
Sakai, A. et al. Ectopic neurogenesis induced by prenatal antiepileptic drug exposure augments seizure susceptibility in adult mice. Proc. Natl Acad. Sci. USA 115, 4270–4275 (2018).
Jennings, C. G. et al. Opportunities and challenges in modeling human brain disorders in transgenic primates. Nat. Neurosci. 19, 1123–1130 (2016).
Zhao, H., Jiang, Y. H. & Zhang, Y. Q. Modeling autism in non-human primates: opportunities and challenges. Autism Res. 11, 686–694 (2018).
Watson, K. K. & Platt, M. L. Of mice and monkeys: using non-human primate models to bridge mouse- and human-based investigations of autism spectrum disorders. J. Neurodev. Disord. 4, 21 (2012).
Bluemel, J., Korte, S., Schenck, E. & Weinbauer, G. The Nonhuman Primate in Nonclinical Drug Development and Safety Assessment (1st edition). (Academic Press, New York, 2015).
Mast, T. J., Cukierski, M. A., Nau, H. & Hendrickx, A. G. Predicting the human teratogenic potential of the anticonvulsant, valproic acid, from a non-human primate model. Toxicology 39, 111–119 (1986).
Shepard, T. H., Muffley, L. A. & Smith, L. T. Ultrastructural study of mitochondria and their cristae in embryonic rats and primate (N. nemistrina). Anat. Rec. 252, 383–392 (1998).
Rodier, P. M., Ingram, J. L., Tisdale, B. & Croog, V. J. Linking etiologies in humans and animal models: studies of autism. Reprod. Toxicol. 11, 417–422 (1997).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408 (2001).
Machado, C. J., Whitaker, A. M., Smith, S. E., Patterson, P. H. & Bauman, M. D. Maternal immune activation in nonhuman primates alters social attention in juvenile offspring. Biol. Psychiatry 77, 823–832 (2015).
Makori, N., Rodriguez, C. G., Cukierski, M. A. & Hendrickx, A. G. Development of the brain in staged embryos of the long-tailed monkey (Macaca fascicularis). Primates 37, 351–361 (1996).
Carletti, B. & Rossi, F. Neurogenesis in the cerebellum. Neuroscientist 14, 91–100 (2008).
Iijima, Y. et al. Distinct defects in synaptic differentiation of neocortical neurons in response to prenatal valproate exposure. Sci. Rep. 6, 27400 (2016).
Wong, C. C. et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry 19, 495–503 (2014).
Shulha, H. P. et al. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch. Gen. Psychiatry 69, 314–324 (2012).
Kataoka, S. et al. Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int. J. Neuropsychopharmacol. 16, 91–103 (2013).
Lee, H. J., Dreyfus, C. & DiCicco-Bloom, E. Valproic acid stimulates proliferation of glial precursors during cortical gliogenesis in developing rat. Dev. Neurobiol. 76, 780–798 (2016).
Huber, A. B., Kolodkin, A. L., Ginty, D. D. & Cloutier, J. F. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu. Rev. Neurosci. 26, 509–563 (2003).
Klin, A., Lin, D. J., Gorrindo, P., Ramsay, G. & Jones, W. Two-year-olds with autism orient to non-social contingencies rather than biological motion. Nature 459, 257–261 (2009).
Chawarska, K., Macari, S. & Shic, F. Decreased spontaneous attention to social scenes in 6-month-old infants later diagnosed with autism spectrum disorders. Biol. Psychiatry 74, 195–203 (2013).
Shic, F., Bradshaw, J., Klin, A., Scassellati, B. & Chawarska, K. Limited activity monitoring in toddlers with autism spectrum disorder. Brain Res. 1380, 246–254 (2011).
Parras, A. et al. Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 560, 441–446 (2018).
Croen, L. A., Grether, J. K., Yoshida, C. K., Odouli, R. & Hendrick, V. Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch. Gen. Psychiatry 68, 1104–1112 (2011).
Tomson, T. et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 10, 609–617 (2011).
Hsieh, J., Nakashima, K., Kuwabara, T., Mejia, E. & Gage, F. H. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc. Natl Acad. Sci. USA 101, 16659–16664 (2004).
Fujimura, K., Mitsuhashi, T., Shibata, S., Shimozato, S. & Takahashi, T. In utero exposure to valproic acid induces neocortical dysgenesis via dysregulation of neural progenitor cell proliferation/differentiation. J. Neurosci. 36, 10908–10919 (2016).
Herculano-Houzel, S. The glia/neuron ratio: how it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 62, 1377–1391 (2014).
Kim, K. C. et al. Pax6-dependent cortical glutamatergic neuronal differentiation regulates autism-like behavior in prenatally valproic acid-exposed rat offspring. Mol. Neurobiol. 49, 512–528 (2014).
Uzunova, G., Pallanti, S. & Hollander, E. Excitatory/inhibitory imbalance in autism spectrum disorders: implications for interventions and therapeutics. World J. Biol. Psychiatry 17, 174–186 (2016).
Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010).
Nicolini, C. & Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 299, 217–227 (2018).
Kim, K. C. et al. Male-specific alteration in excitatory post-synaptic development and social interaction in prenatal valproic acid exposure model of autism spectrum disorder. J. Neurochem. 124, 832–843 (2013).
de Theije, C. G. et al. Intestinal inflammation in a murine model of autism spectrum disorders. Brain Behav. Immun. 37, 240–247 (2014).
Alsdorf, R. & Wyszynski, D. F. Teratogenicity of sodium valproate. Expert Opin. Drug Saf. 4, 345–353 (2005).
Acknowledgements
This work was supported by the Ministry of Science and Technology of China (2016YFA0501000), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB02020400), the National Natural Science Foundation of China (31110103907, 91332206, 31490592, 31830036, 31671108 and 31800900), the International Partnership Program of Chinese Academy of Sciences (172644KYSB20160026, 172644KYSB20160175), Beijing Municipal Science & Technology Commission (Z181100001518001), Guangdong Province Science and Technology Project (2017B020231001), Guangdong Innovative and Entrepreneurial Research Team Program (No. 2014ZT05S020), Shenzhen Municipal Funding (KQJSCX20170731164702657, JCYJ20170413165053031, JCYJ20151030140325151, and KQTD20140630180249366), and Guangdong Province Science and Technology Project (2015A020212027). We thank Guangdong Blooming-Spring Biological Technology Development Co., Ltd for providing monkeys and animal care. We thank Baoyong Gong and Hang Li for assistance in drug administration.
Author information
Authors and Affiliations
Contributions
H.Z., Y.-H.J. and Y.-Q.Z. designed and supervised the project. Q.-Q.W., H.Z., H.-J.X. and H.-P.Y. performed western blotting, histological staining, and RNA-seq analysis. Q.-Q.W., H.Z., T.Y. and H.-H.Z. performed animal behavior studies and analyzed data. Y.Z., Z.-C.T. and X.-Y.G. provided animal facilities and animal care. Y.-H.J. and X.-J.L. provided advice on results interpretation and discussion. H.Z., Q.-Q.W. and Y.-Q.Z. wrote the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Supplement Information Supplementary Information includes two figures, six tables, and one video which can be found with this article online. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number CRA001719, that are publicly accessible at http://bigd.big.ac.cn/gsa.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, H., Wang, Q., Yan, T. et al. Maternal valproic acid exposure leads to neurogenesis defects and autism-like behaviors in non-human primates. Transl Psychiatry 9, 267 (2019). https://doi.org/10.1038/s41398-019-0608-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0608-1
This article is cited by
-
Reduced excitatory activity in the developing mPFC mediates a PVH-to-PVL transition and impaired social cognition in autism spectrum disorders
Translational Psychiatry (2024)
-
Gestational Fisetin Exerts Neuroprotection by Regulating Mitochondria-Directed Canonical Wnt Signaling, BBB Integrity, and Apoptosis in Prenatal VPA-Induced Rodent Model of Autism
Molecular Neurobiology (2024)
-
Transcriptome Analysis and Epigenetics Regulation in the Hippocampus and the Prefrontal Cortex of VPA-Induced Rat Model
Molecular Neurobiology (2024)
-
Neurobiological Factors of Executive Dysfunction in Autism Spectrum Disorders
Neuroscience and Behavioral Physiology (2023)
-
Focus on your locus with a massively parallel reporter assay
Journal of Neurodevelopmental Disorders (2022)
