
Abstract
Psychosis (delusions or hallucinations) in Alzheimer’s disease (AD + P) occurs in up to 50% of individuals and is associated with significantly worse clinical outcomes. Atypical antipsychotics, first developed for schizophrenia, are commonly used in AD + P, suggesting shared mechanisms. Despite this implication, little empirical research has been conducted to examine whether there are mechanistic similarities between AD + P and schizophrenia. In this study, we tested whether polygenic risk score (PRS) for schizophrenia was associated with AD + P. Schizophrenia PRS was calculated using Psychiatric Genomics Consortium data at ten GWAS p value thresholds (PT) in 3111 AD cases from 11 cohort studies characterized for psychosis using validated, standardized tools. Association between PRS and AD + P status was tested by logistic regression in each cohort individually and the results meta-analyzed. The schizophrenia PRS was associated with AD + P at an optimum PT of 0.01. The strongest association was for delusions where a one standard deviation increase in PRS was associated with a 1.18-fold increased risk (95% CI: 1.06–1.3; p = 0.001). These new findings point towards psychosis in AD—and particularly delusions—sharing some genetic liability with schizophrenia and support a transdiagnostic view of psychotic symptoms across the lifespan.
Similar content being viewed by others


Introduction
Psychosis in Alzheimer’s disease (AD + P)—broadly comprising delusions and hallucinations—is experienced by up to 50% of people over the course of the illness, with prevalence peaking in the later stages1. AD + P is associated with accelerated cognitive decline (independent of disease duration), higher mortality rates and distress to both people with the disease and their carers2,3,4. Moreover, there are wider societal implications with long-term follow-up studies indicating that AD + P is associated with a shorter time to nursing home care5. Despite these compelling reasons for effective management, there is a critical treatment gap, with no licensed treatments available in many jurisdictions. Atypical antipsychotics—developed first for schizophrenia—are frequently used to treat AD + P (in many countries off label) and, while they have some modest benefits, are associated with considerable harms, including a 1.5- to 1.8-fold increase in mortality and a threefold increase in stroke6.
Clinically useful alternatives to antipsychotics are scarce. There are only two new antipsychotic compounds in phase II or later stages of development (pimavanserin and MP-101) but both are refinements of existing mechanisms of action of atypical antipsychotics targeting mechanisms relevant to schizophrenia (e.g. 5HT2A, mGluR2/3) and side effects remain a concern7. The limited understanding of the biological mechanisms underpinning AD + P represents a major challenge to the effective targeting of existing treatments and the identification of novel treatment targets.
One key question is whether some or all of the psychotic symptoms experienced by people with AD have a similar basis to schizophrenia. Phenomenologically the psychotic symptoms in each are different; in AD visual hallucinations are more common than auditory hallucinations, delusions are usually simple, and the first rank symptoms of schizophrenia are very rare. In addition, schizophrenia is characterized by both positive and negative symptoms. While negative symptoms can also accompany psychosis in AD, consensus is yet to be reached on whether these other neuropsychiatric symptoms form part of the AD + P clinical syndrome. Despite the different phenomenology, atypical antipsychotics confer some treatment benefits in some cases of AD + P8, and similar neuropsychological deficits in processing speed and executive function have been observed in individuals with very-late-onset schizophrenia-like psychosis and AD + P9, suggesting some overlap.
A transdiagnostic hypothesis, proposing a mechanistic overlap between AD + P and schizophrenia, is gaining some traction10 and is supported by genetic studies of psychosis in adolescence, the general adult population and Huntington’s disease all showing overlap with schizophrenia11,12,13. In view of these findings and the high heritability of schizophrenia14 and of AD + P (estimated at 81% and 61% respectively)15, it is logical to look for common genetic underpinnings of the two disorders. Comparative studies examining common mechanisms between AD and schizophrenia point towards synaptic elimination and disruption, and telomere length16,17,18, but studies examining AD + P specifically and schizophrenia are less common. It is of note that a recent major GWAS reported a nominally significant genetic correlation between schizophrenia and AD19. It is possible that the presence of psychosis in the AD sample (which was unknown in this study) contributed to part of the association, underscoring the need for dissection of the AD phenotype by psychosis status. In a small study, a copy number variant (CNV) with significant overlap of a duplicated region implicated in schizophrenia and autism (16p11.2) was found in two of 440 AD + P cases but not in AD without psychosis, or in those with more occasional symptoms20. Linkage studies have also implicated regions of the genome in AD + P that have been identified in schizophrenia21,22. Another approach is to examine whether polygenic risk for schizophrenia, summarized in a score (the weighted sum of risk associated alleles) with better discrimination properties than single markers23, is associated with AD + P. Work in this area is limited to only one recent study which, surprisingly, reported that a genetic risk score comprising 94 SNPs reaching genome-wide significance for association with schizophrenia was lower in AD + P compared with AD without psychosis24. While this study represents an important preliminary step in AD + P research, a full genome-wide polygenic risk score (PRS) approach is imperative to address this key question25,26.
Another largely unexplored avenue in AD + P genetic research relates to the split of delusions and hallucinations. Although the two symptoms frequently co-occur in AD, there is evidence from longitudinal cohort studies indicating that 10–20% of people experience hallucinations without delusions and that the two symptoms are associated with different clinical outcomes2,27, suggesting the presence of two distinct clinical phenotypes. While it is commonplace to separate out composite psychotic symptoms in neuroimaging studies of AD + P28,29, their separate genetic associations have not yet been examined in any large-scale AD studies leveraging GWAS data30. This is a particularly relevant issue when assessing genetic overlap with schizophrenia where the emerging evidence from neuroimaging and the clinical similarity supports the hypothesis that shared etiology would be specific to delusions.
We conducted an analysis of the relationship between genetic liability for schizophrenia and AD + P with two principal objectives; firstly, we tested whether PRS for schizophrenia was associated with AD + P and secondly, we examined the association between the PRS and AD with delusions.
Methods
Ethical approval for this analysis protocol was obtained from University of Exeter Medical School Research Ethics Committee (Nov17/D/143).
Cohorts
AD + P target data consisted of 3111 AD cases from 11 cohort studies in Europe and the USA: AddNeuroMed31 (Europe, longitudinal: assessment every 3 months for maximum 1 year), Alzheimer’s Disease Neuroimaging Initiative32 (ADNI; USA, longitudinal: assessment at baseline, 6, 12, 24 and 36 months for maximum 3 years), Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS 1; Italy, cross-sectional), Health and Memory Study in Nord-Trøndelag33 (HMS; Norway, cross-sectional), Resource Use and Disease Couse in Dementia34 (REDIC; Norway, longitudinal: assessment every 6 months for maximum 2.5 years), Norwegian registry of persons assessed for cognitive symptoms35 (NorCog; Norway, cross-sectional), Samhandling mellom avdeling for alderspsykiatri og kommunale sykehjem (SAM-AKS; Norway, cross-sectional), The Dementia Study in Northern Norway36 (NordNorge, Norway, longitudinal: assessment at baseline and 1 year), Progression of Alzheimer’s Disease and Resource Use37 (PADR; Norway, longitudinal: assessment at baseline and 1 year), The Dementia Study in Western Norway38 (DemVest; Norway, longitudinal: assessment every 12 months maximum 6 years); and data from the National Alzheimer’s Coordinating Center (NACC; USA, longitudinal: assessment approximately every 12 months) and the National Institute on Aging Genetics Data Storage Site (NIAGADS), Table 1). Full cohort details are contained in the supplementary material and the Norwegian cohorts are also described in the latest GWAS of Alzheimer’s disease19. Informed consent was obtained by each study for all participants.
Some data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public−private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD). For up-to-date information, see www.adni-info.org.
AD clinical assessments
Diagnosis of AD was performed according to ICD-10 etiological diagnosis, NINCDS-ADRDA criteria or clinical diagnosis by psychiatrist or geriatrician. Longitudinal data were available for seven cohorts (ADNI, AddNeuroMed, DemVest, NordNorge, PADR, REDIC, NACC) and psychotic symptom classification was based on the maximum amount of follow-up data available. Any cases with a history of bipolar disorder or schizophrenia were excluded. For NorCog, PADR, REDIC, SAM-AKS, NACC and ADNI the necessary information on psychiatric history was extracted from source study data resulting in 3, 1, 2, 1, 31 and 1 exclusions, respectively. For AddNeuroMed, DemVest, IRCCS 1 and NordNorge this was an exclusion criterion applied at entry to those individual studies. No information about psychiatric history was available for the HMS study. Dementia severity was assessed in all cohorts by Mini Mental State Examination (MMSE) and psychotic symptoms were assessed by the Neuropsychiatric Inventory (NPI) or its short version, the Neuropsychiatric Inventory Questionnaire (NPI-Q), they are among the most widely used validated instruments to assess psychosis39. Psychotic symptoms are rated on the basis of items A (delusions) and B (hallucinations) of the NPI and NPI-Q. These are two different versions of the same scale, which are strongly correlated and have good between-rater and test−retest reliability, particularly for the psychosis items31,40. Ratings were carried about by trained research staff in all cohorts. In the full NPI, neuropsychiatric symptoms are coded as present or absent first. If rated present they are further scored according to their frequency (1–4) and severity (1–3) with the resulting scores multiplied to give an overall rating (i.e. possible scores are 1, 2, 3, 4, 6, 8, 9 and 12 with 0 indicating no symptoms). The NPI-Q is rated only on a scale of 0−3 according to the severity of the symptom. Both scales have been designed to be completed by verbal interview with a proxy informant who knows the person with AD well. Several diagnostic criteria for AD + P have been proposed but none have been adopted clinically, meaning that where in other psychiatric disorders medical records can be screened, in AD + P this would be unreliable and ratings on specific validated assessment scales must be used. Using such scales, we thus undertook examination of three related but progressively more homogenous psychotic phenotypes:
-
1.
Psychosis wide: Psychosis present: the presence of delusions or hallucinations (NPI/NPI-Q item A or B > 0) at any point; No psychosis: no evidence of delusions or hallucinations (NPI/NPI-Q item A or B = 0) at any point in follow up.
-
2.
Psychosis narrow: Psychosis present: the presence of delusions or hallucinations (NPI/NPI-Q item A or B > 0) at any point; No psychosis: here, an additional level of screening was applied to those rated as having no delusions or hallucinations. In these cases, if an individual was psychosis-free based on criteria for psychosis wide but had not yet reached a moderately severe dementia stage based on available data (defined as MMSE < 20) they were excluded from the analysis. This is a similar approach to that used in most previous AD + P genetic research24,41.
-
3.
Delusions narrow: Delusions present: the presence of delusions (NPI/NPI-Q item A > 0) at any point during follow-up. Thus, the delusion group was the psychosis narrow group above with any individuals rated as having hallucinations only removed. No delusions: as per psychosis narrow.
Genotyping and QC
The genotyping chips used are detailed in Table 1. Raw genotype data for individual cohorts underwent appropriate QC steps (implemented in PLINK). SNPs with a minor allele frequency ≤5% and a Hardy Weinberg equilibrium p < 10−5 were excluded. The SNP and individual genotype failure threshold was set at 5% and individuals with mean heterozygosity ±3 standard deviations were excluded. The analysis was restricted to individuals of European ancestry using genetic principal components computed by EIGENSTRAT. Related (pi-hat > 0.2) or duplicate individuals both within and between cohorts were excluded. Phasing (EAGLE2) and imputation (PBWT) was done via the Sanger Imputation Service using the Haplotype Reference Consortium (r1.1) reference panel on all cohorts. After imputation only SNPs with an imputation quality (INFO) score > 0.4 and MAF > 0.05 were retained. This resulted in 4,895,913 SNPs common across all 11 cohorts available to compute polygenic risk scores.
The most recently published schizophrenia GWAS data from the Psychiatric Genomics Consortium (PGC) was used as base data to generate PRS in the target AD sample26. SNPs with MAF < 0.1, INFO < 0.9 and indels were excluded from the base dataset to leave only the most informative SNPs and only one SNP from the extended MHC region was included42. As a positive control and to evaluate the specificity of the association we then generated PRS of height and depression using the latest GIANT consortium and PGC GWAS results40,43.
Analysis
PRS for schizophrenia were generated in PRSice44 at the following ten GWAS p value thresholds (PT): 5 × 10−8, 1 × 10−5, 1 × 10−4, 1 × 10−3, 0.01, 0.05, 0.1, 0.2, 0.5 and 1. Clumping was performed (250 kb, r2 > 0.1) to retain only the SNP with the strongest association in each window. The resulting PRS were standardized (centering by mean, scaling by standard deviation) for the analysis.
Power was calculated using AVENGEME45, with schizophrenia parameters as set out in Palla and Dudbridge45, number of markers genotyped in both datasets was 76,213 (see section “Schizophrenia PRS is associated with AD psychosis status”), a prevalence of 40%1 and of 36%1 was used for psychosis and delusions, and case-control sample fractions as per Table 2. There are no data available for estimated covariance between AD + P and schizophrenia but if this value is assumed to be 0.08 (less than the 0.13 and 0.17 for schizophrenia and major depressive disorder and bipolar disorder estimated by AVENGEME45), this study has ≥80% power for each PT ≥ 0.01 for psychosis and delusions respectively but <80% power below this value. All statistical analysis was implemented in R. For each cohort ten logistic regression models (one per PT) were run with each of the previously defined psychosis phenotypes as the binary outcome and the first ten ancestry principal components included as covariates. Disease severity is accounted for in our “narrow” phenotype definitions and as there is no strong evidence that age and gender are associated with AD + P1 so these were not included as covariates. Logistic regression assumptions were confirmed using the R “car” package. Proportion of variance explained (R2) by PRS, on the observed scale, was determined by subtracting the Nagelkerke’s pseudo-R2 of the null model from that of the full model. Regression coefficients for each PT across all cohorts were then included in random effects meta-analyses to account for between-study variation in data collection protocols, frequency of psychosis and dementia severity46,47,48. Meta-analysis was undertaken using the “rma” function in the “metafor” package using the REML method49. Because the PRS calculated were correlated, a Bonferroni correction for multiple testing was considered too stringent. Using a correlation matrix of the ten PRS and the matSpD tool (https://gump.qimr.edu.au/general/daleN/matSpD/), the effective number of independent tests was determined to be 5 and the experiment-wide significance threshold for type I error rate of 5% determined to be p = 0.01. All tests reported are two-sided.
Results
On average across all 11 cohorts, individuals were in the mild-moderate stages of dementia at first assessment (mean MMSE of 19). Mean MMSE by cohort ranged from an MMSE of 12 (IRCCS 1) to 24 (ADNI) and this was a correlate of the prevalence of psychosis in each cohort (note the denominator would be the overall cohort N in Table 1), with cohorts that contained individuals with more severe dementia typically having a higher proportion of people with psychosis. Between cohorts, mean age at baseline ranged from 75 to 87 years and the proportion of male participants ranged from 26 to 59%. There was little difference in age between the psychosis and no psychosis groups across all studies but gender distributions did differ.
Frequency of the three psychosis phenotypes by cohort is shown in Table 2. Of the 3111 individuals screened, 1116 (36%) had psychosis (wide definition group). Of the 1995 who were rated as having no psychosis based on their assessment scale result alone, 879 had not yet reached the moderate stages of disease and so were excluded; 1116 AD + P cases and 1116 AD no psychosis “controls” were included in the analysis of the narrow phenotype of psychosis. In all, 936 cases met the criteria for having delusions narrow.
Schizophrenia PRS is associated with AD psychosis status
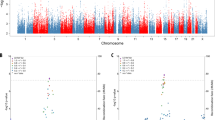
After clumping, 76,213 independent variants were available for computing PRS. Random effects meta-analysis across the 11 cohorts showed the largest OR for the schizophrenia PRS at PT = 0.01 and this was significantly associated with symptom status across the psychosis wide, psychosis narrow and delusions narrow phenotypes despite the progressively smaller sample size in each of these groups (OR: 1.14, 95% CI: 1.05–1.23, p = 0.003; OR: 1.16, 95% CI: 1.06–1.28, p = 0.004; OR: 1.18, 95% CI:1.06–1.30, p = 0.001 respectively) (see Fig. 1 and Table 3). PRS was also significantly associated with both the psychosis narrow and delusions narrow phenotypes at every PT > 0.01. The largest effect size was observed in the delusions narrow group. Overall, there was no evidence of significant heterogeneity; I2 statistics were close to 0% for PT = 0.01 across the three phenotypes.

Each bar represents PRS composed of markers at ten different schizophrenia GWAS P value thresholds (PT). P values shown above each bar
In the individual cohort analysis, we observed that the effect estimates of association between schizophrenia PRS and AD + P in nine of the 11 studies were in the same direction (OR > 1), albeit not statistically significantly (Supplementary Table 1). A forest plot of individual study estimates for delusions narrow at PT = 0.01, the strongest association found in the above meta-analysis, is shown in Fig. 2. A similar plot at PT = 1 for comparison is shown in the Supplementary material along with plots for psychosis wide and psychosis narrow phenotypes. The highest Nagelkerke’s R2 estimate was 2.9% (AddNeuroMed) and the lowest was <0.1% (IRCCS 1). An overall variance explained (Nagelkerke’s R2) in AD + P by schizophrenia PRS of 0.08% was estimated by calculating the weighted average R2 across the 11 studies. To determine the specificity of the signal, PRS for major depression (using the PGC GWAS43) and height (GIANT consortium GWAS40) were generated post-hoc at PT = 1 and tested for association with delusions using the same procedure as described in the section “Analysis”. Neither PRS showed any evidence of association (major depression: OR: 1.03, 95% CI: 0.91–1.18, p = 0.61; height: OR: 0.99, 95% CI: 0.85–1.17, p = 0.99).

Overall estimate from random effects model is represented by the diamond below the individual study estimates
Discussion
We set out to examine whether genetic risk for psychotic symptoms in AD (AD + P) is attributable to common schizophrenia variants. Using polygenic scoring, we found that schizophrenia PRS was associated with AD + P in a collection of over 3000 well-characterized cases and the association persisted as the AD + P phenotype was more precisely defined, despite the progressively smaller N. The largest effect size was observed at PT = 0.01 which was associated with a 1.14-, 1.16- and 1.18-fold (per standard deviation increase in PRS) increased risk of psychosis (wide), psychosis (narrow) and delusions (narrow), respectively. In the individual cohort analysis, the odds ratios of 9 of the 11 studies were in the same direction (OR > 1). In all, these new findings suggest that AD + P is part of a spectrum of neuropsychiatric conditions characterized by psychosis across the lifespan. However, in common with other studies in psychiatric genomics PRS are yet not appropriate for symptom or disease course prediction in AD + P. Although the variance explained by schizophrenia PRS in AD + P is only modest, with the R2 estimates being less than 1%, this should be seen in the context of the same PRS explaining around 2.5% of the variance in bipolar disorder and 1% in MDD in a cross-disorder analysis of the Psychiatric Genomics Consortium with significantly larger target sample sizes50.
In line with our findings, a recent study in UK Biobank found psychotic experiences in the general population to be associated with PRS for schizophrenia, with the strongest association observed for delusions12. Several possible conclusions can be drawn from the finding that the association was still observed in the delusions phenotype in this study, despite a considerably smaller N compared with the broader psychosis phenotypes. This finding may point towards a subset of AD + P patients that have a more schizophrenia-like phenotype. More work is needed to investigate whether further diagnostic refinements to AD + P syndrome definitions are necessary, which may provide a more robust approach for pharmacological intervention trials. Related to this, from a methodological point of view, we show that there is a need for future studies in AD to consider delusions and hallucinations separately. We cannot rule out a genetic association between hallucinations in AD and schizophrenia in these cohorts but the evidence at present suggests a weaker association than for delusions. One might speculate that this is due to visual hallucinations in AD being more often the result of a broader range of causes (e.g. due to medication or delirium) than delusions, thus introducing more noise into the phenotype. The final wider implication is related to the schizophrenia PRS being associated with a broad spectrum of psychotic disorders and personality traits11,12,13,50,51,52. Our findings support a transdiagnostic explanation of delusions, which reaches into neurodegenerative disease and is underpinned by a degree of common genetic liability.
A key strength of our study is the detailed phenotyping with longitudinal data being available in 7 of the 11 cohorts. Rather than relying on medical record screens, which would be highly unreliable for AD + P given the lack of universally accepted and used diagnostic criteria, every individual in our analysis was assessed using specific, reliable assessment tools. We then used this data to dissect AD + P phenotype by focusing on delusions as well as the broader syndrome. We also followed previous research by taking extra measures to screen the “control” groups. This removed any cases in the mild stages of disease who had not yet developed symptoms (i.e. those still at risk1). This approach has been used in most previous genetic research but our extension to focus on delusions in AD + P is novel. Our finding that the association persisted with this more precision definition is consistent with genetic studies of other polygenic traits, like depression43.
For one study (HMS) data on history of major psychiatric conditions were not available. It is possible that some individuals with schizophrenia were present in this cohort; however, HMS is a cohort with a mean age of 87 so it is highly unlikely that the number would be more than one or two out of 178 people in the HMS cohort (this is also supported by the very small numbers we found among the other studies we screened). With over 3000 samples, this is, to our knowledge, the largest analysis of AD + P to exploit GWAS data41. We acknowledge that using different cohorts has led to some variability due to sampling but it is important to acknowledge that there are no single cohorts which are large enough to conduct an analysis of this kind and because of potential sampling and protocol variations across the individual studies we ensured an appropriate analysis was implemented to account for this variability; the same approach as used in other studies examining PRS in complex phenotypes46,47,48. We had access to raw individual-level clinical and genotype data, allowing us to run the same regression models in each study. This included undertaking the same QC across cohorts, imputing all chip data to the same reference panel and analyzing only SNPs present across all cohorts. After ensuring this standardized process was followed for each cohort we ran a random effects meta-analysis, allowing for the effect of the PRS on AD + P to vary across studies. In all, and in the absence of a single large enough study, these measures provide the most robust estimates, as reflected in the low heterogeneity statistics of the meta-analysis and the narrow range of effect estimates and overlapping confidence intervals across the 11 studies included (Fig. 2 and Supplementary Figs. 1–3). Finally, as with all similar studies, these results are not generalizable to individuals with non-European ancestry; there is an equal imperative to extend the genomics of AD + P to other populations as in AD itself.
A previous study that examined a genetic risk score at a more conservative PT comprised of only 94 genome-wide significant schizophrenia SNPs found it to be lower in AD + P cases24. Our study is a similar size to this previous study, and the NACC data were used in both. Given that a PRS with only 94 SNPs will be a less powerful predictor than a full genome-wide score, it is possible larger studies will be needed to confirm associations at this more conservative PT. Nevertheless, schizophrenia is highly polygenic; tens of thousands of markers explain only 7% of the variance on the liability scale, while for optimum cross-trait case-control (e.g. schizophrenia and bipolar) prediction many thousands more SNPs are required50. In addition, cases of schizophrenia in the PGC study (used as base sample to estimate PRS) include patients with both a positive and negative syndrome. There is evidence that negative and disorganized symptoms are more heritable than positive, which—although we report a positive association—may reduce the power of schizophrenia PRS at more conservative PT to discriminate AD cases with or without psychotic symptoms53,54. Accordingly, a full account of association between schizophrenia and AD + P should exploit the full polygenic nature of schizophrenia; our study is the first to do this and the findings represent an important further step towards a complete account of the relationship between common schizophrenia variants and AD + P. Another important milestone will be an appropriately powered discovery GWAS of AD + P and all of these points underscore the need for increasing samples sizes in this field.
In summary, these findings support shared genetic liability between schizophrenia and psychosis in AD. This provides a strong rationale for further work to build a clearer clinical and biological understanding of the psychosis syndrome in AD, an urgently needed step for better management and treatment development.
References
Ropacki, S. A. & Jeste, D. V. Epidemiology of and risk factors for psychosis of Alzheimer’s disease: a review of 55 studies published from 1990 to 2003. Am. J. Psychiatry 162, 2022–2030 (2005).
Connors, M. H., Ames, D., Woodward, M. & Brodaty, H. Psychosis and clinical outcomes in Alzheimer disease: a longitudinal study. Am. J. Geriatr. Psychiatry 26, 304–313 (2018).
Weamer, E. A. et al. The relationship of excess cognitive impairment in MCI and early Alzheimer disease to the subsequent emergence of psychosis. Int. Psychogeriatr. 21, 78–85 (2009).
Savva, G. M. et al. Prevalence, correlates and course of behavioural and psychological symptoms of dementia in the population. Br. J. Psychiatry 194, 212 (2009).
Wergeland, J. N., Selbæk, G., Bergh, S., Soederhamn, U. & Kirkevold, Ø. Predictors for nursing home admission and death among community-dwelling people 70 years and older who receive domiciliary care. Dement. Geriatr. Cogn. Disord. Extra 5, 320–329 (2015).
Corbett, A., Smith, J., Creese, B. & Ballard, C. Treatment of behavioral and psychological symptoms of Alzheimer’s disease. Curr. Treat. Options Neurol. 14, 113–125 (2012).
Creese, B., Da Silva, M. V., Johar, I. & Ballard, C. The modern role of antipsychotics for the treatment of agitation and psychosis in Alzheimer’s disease. Expert Rev. Neurotherapeut. 18, 461–467 (2018).
Ballard, C. & Howard, R. Neuroleptic drugs in dementia: benefits and harm. Nat. Rev. Neurosci. 7, 492–500 (2006).
Van Assche, L. et al. The neuropsychological profile and phenomenology of late onset psychosis: a cross-sectional study on the differential diagnosis of very-late-onset schizophrenia-like psychosis, dementia with Lewy bodies and Alzheimer’s type dementia with psychosis. Arch. Clin. Neuropsychol. 34, 183–199 (2019).
Bebbington, P. & Freeman, D. Transdiagnostic extension of delusions: schizophrenia and beyond. Schizophrenia Bull. 43, 273–282 (2017).
Pain, O. et al. Genome-wide analysis of adolescent psychotic-like experiences shows genetic overlap with psychiatric disorders. Am. J. Med. Genet. Part B: Neuropsychiatr. Genet. 177, 416–425 (2018).
Legge, S. E. et al. Association of Genetic Liability to Psychotic Experiences With Neuropsychotic Disorders and Traits. JAMA Psychiatry bioRxiv 583468 (2019). https://doi.org/10.1001/jamapsychiatry.2019.2508.
Ellis, N. et al. Genetic risk underlying psychiatric and cognitive symptoms in Huntington’s disease. bioRxiv 639658 (2019). https://doi.org/10.1101/639658.
Sullivan, P. F., Kendler, K. S. & Neale, M. C. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60, 1187–1192 (2003).
Bacanu, S. A. et al. Heritability of psychosis in Alzheimer disease. Am. J. Geriatr. Psychiatry. 13, 624–627 (2005).
Alexandrov, P. N., Zhao, Y., Jaber, V., Cong, L. & Lukiw, W. J. Deficits in the proline-rich synapse-associated Shank3 protein in multiple neuropsychiatric disorders. Front. Neurol. 8, 670 (2017).
Cardozo, P. L. et al. Synaptic elimination in neurological disorders. Curr. Neuropharmacol. 17, 1071–1095 (2019).
Anitha, A., Thanseem, I., Vasu, M. M., Viswambharan, V. & Poovathinal, S. A. Telomeres in neurological disorders. Adv. Clin. Chem. 90, 81–132 (2019).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Zheng, X. et al. A rare duplication on chromosome 16p11.2 is identified in patients with psychosis in Alzheimer’s disease. PLoS ONE 9, e111462 (2014).
Hollingworth, P. et al. Increased familial risk and genomewide significant linkage for Alzheimer’s disease with psychosis. Am. J. Med. Genet. Part B, Neuropsychiatr. Genet. 144b, 841–848 (2007).
Shah, C., DeMichele-Sweet, M. A. A. & Sweet, R. A. Genetics of psychosis of Alzheimer disease. Am. J. Med. Genet. Part B: Neuropsychiatr. Genet. 174, 27–35 (2017).
Dudbridge, F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 9, e1003348 (2013).
DeMichele-Sweet, M. A. A. et al. Genetic risk for schizophrenia and psychosis in Alzheimer disease. Mol. Psychiatry 23, 963–972 (2018).
International Schizophrenia C. Common polygenic variation contributes to risk of schizophrenia that overlaps with bipolar disorder. Nature 460, 748–752 (2009).
Ripke, S. et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Qian, W., Fischer, C. E., Schweizer, T. A. & Munoz, D. G. Association between psychosis phenotype and APOE genotype on the clinical profiles of Alzheimer’s disease. Curr. Alzheimer Res. 15, 187–194 (2018).
McLachlan, E., Bousfield, J., Howard, R. & Reeves, S. Reduced parahippocampal volume and psychosis symptoms in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 33, 389–395 (2018).
Darby, R. R., Laganiere, S., Pascual-Leone, A., Prasad, S. & Fox, M. D. Finding the imposter: brain connectivity of lesions causing delusional misidentifications. Brain: J. Neurol. 140, 497–507 (2017).
Fischer, C. E. & Sweet, R. A. Psychosis in Alzheimer’s disease: a review of recent research findings. Curr. Behav. Neurosci. Rep. 3, 308–317 (2016).
Lovestone, S. et al. AddNeuroMed—the European collaboration for the discovery of novel biomarkers for Alzheimer’s disease. Ann. N.Y. Acad. Sci. 1180, 36–46 (2009).
Mueller, S. G. et al. Ways toward an early diagnosis in Alzheimer’s disease: the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Alzheimer’s Dement. 1, 55–66 (2005).
Bergh, S. et al. Cohort profile: The Health and Memory Study (HMS): a dementia cohort linked to the HUNT study in Norway. Int. J. Epidemiol. 43, 1759–1768 (2014).
Roen, I. et al. Resourse use and disease couse in dementia—nursing home (REDIC-NH), a longitudinal cohort study; design and patient characteristics at admission to norwegian nursing homes. BMC Health Serv. Res. 17, 365 (2017).
Helvik, A. S., Engedal, K., Šaltytė Benth, J. & Selbæk, G. Time from symptom debut to dementia assessment by the specialist healthcare service in Norway. Dement. Geriatr. Cogn. Disord. Extra 8, 117–127 (2018).
Andersen, F. et al. Recruitment methods in Alzheimer’s disease research: general practice versus population based screening by mail. BMC Med. Res. Methodol. 10, 35 (2010).
Eldholm, R. S. et al. Progression of Alzheimer’s disease: a longitudinal study in Norwegian memory clinics. J. Alzheimers Dis. 61, 1221–1232 (2018).
Vik-Mo, A. O., Bencze, J., Ballard, C., Hortobágyi, T. & Aarsland D. Advanced cerebral amyloid angiopathy and small vessel disease are associated with psychosis in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 90, 728–730 (2018).
Kaufer, D. I. et al. Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J. Neuropsychiatry Clin. Neurosci. 12, 233–239 (2000).
Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649 (2018).
Hollingworth, P. et al. Genome-wide association study of Alzheimer’s disease with psychotic symptoms. Mol. Psychiatry 17, 1316–1327 (2012).
Vassos, E. et al. An examination of polygenic score risk prediction in individuals with first-episode psychosis. Biol. Psychiatry 81, 470–477 (2017).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
Euesden, J., Lewis, C. M. & O’Reilly, P. F. PRSice: polygenic risk score software. Bioinformatics 31, 1466–1468 (2015).
Palla, L. & Dudbridge, F. A fast method that uses polygenic scores to estimate the variance explained by genome-wide marker panels and the proportion of variants affecting a trait. Am. J. Hum. Genet. 97, 250–259 (2015).
Peyrot, W. J. et al. Does childhood trauma moderate polygenic risk for depression? A meta-analysis of 5765 subjects from the psychiatric genomics consortium. Biol. Psychiatry 84, 138–147 (2018).
Ward, J. et al. Polygenic risk scores for major depressive disorder and neuroticism as predictors of antidepressant response: meta-analysis of three treatment cohorts. PLoS ONE 13, e0203896 (2018).
Zhang, J.-P. et al. Schizophrenia polygenic risk score as a predictor of antipsychotic efficacy in first-episode psychosis. Am. J. Psychiatry 176, 21–28 (2018).
Viechtbauer W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 36, 1–48 (2010).
Consortium C-DGotPG. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet (Lond., Engl.) 381, 1371–1379 (2013).
Smeland, O. B. et al. Identification of genetic loci shared between schizophrenia and the Big Five personality traits. Sci. Rep. 7, 2222 (2017).
Power, R. A. et al. Polygenic risk scores for schizophrenia and bipolar disorder predict creativity. Nat. Neurosci. 18, 953 (2015).
Fanous, A. H. et al. Genome-wide association study of clinical dimensions of schizophrenia: polygenic effect on disorganized symptoms. Am. J. Psychiatry 169, 1309–1317 (2012).
Cardno, A. G., Sham, P. C., Murray, R. M. & McGuffin, P. Twin study of symptom dimensions in psychoses. Br. J. Psychiatry 179, 39–45 (2001).
Acknowledgements
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research provides funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24-AG041689), funded by the National Institute on Aging. The Alzheimer’s Disease Genetics Consortium (ADGC) supported the collection of samples used in this study through National Institute on Aging (NIA) grants U01AG032984 and RC2AG036528. Samples from the National Cell Repository for Alzheimer’s Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Steven Ferris, PhD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016570 (PI Marie-Francoise Chesselet, MD, PhD), P50 AG005131 (PI Douglas Galasko, MD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P50 AG005136 (PI Thomas Montine, MD, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), and P50 AG047270 (PI Stephen Strittmatter, MD, PhD). NIAGADS datasets NG00022, NG00023, and NG00024 contain ADC samples. This paper represents independent research part funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. Funding for genotyping the IRCCS 1 samples was provided by the University of Exeter. IRCCS data were fully supported by “Ministero della Salute”, I.R.C.C.S. Research Program, Ricerca Corrente 2018−2020, Linea n. 2 “Meccanismi genetici, predizione e terapie innovative delle malattie complesse” and by the “5 × 1000” voluntary contribution to the Fondazione I.R.C.C.S. Ospedale “Casa Sollievo della Sofferenza”. The authors are grateful to the investigators of the AddNeuroMed consortium for providing clinical data. AddNeuroMed is funded through the EU FP6 program as part of InnoMed. In addition, we are grateful for additional support from Alzheimer’s Research UK. The authors’ work has been supported in part by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. The authors wish to thank the Norwegian registry of persons assessed for cognitive symptoms (NorCog) for providing access to patient data and biological material. NorCog is financed by South-Eastern Norway Regional Health Authority and Norwegian National Advisory Unit on Ageing and Health. The organization and the data collection of HMS cohort has been funded by the Norwegian Institute of Public Health, the Norwegian University of Science and Technology (NTNU), Nord-Trøndelag Hospital Trust and Innlandet Hospital Trust. The REDIC-NH study was administrated by the research centre for Age-Related Functional Decline and Disease, Innlandet Hospital Trust, and was initiated by the Norwegian Health Directorate, which also provides funding for the data collection. The data collection of the SAM-AKS cohort has been funded by the research centre for Age-Related Functional Decline and Disease, Innlandet Hospital Trust. The authors are grateful to deCODE Genetics for performing the genotyping in the Norwegian cohorts.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
C.B. has received contract grant funding from Lundbeck, Takeda, and Axovant pharmaceutical companies and honoraria from Lundbeck, Lilly, Otusaka, and Orion pharmaceutical companies. D.A. has received research support and/or honoraria from Astra-Zeneca, H. Lundbeck, Novartis Pharmaceuticals, and GE Health, and serves as a paid consultant for H. Lundbeck and Axovant. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Creese, B., Vassos, E., Bergh, S. et al. Examining the association between genetic liability for schizophrenia and psychotic symptoms in Alzheimer’s disease. Transl Psychiatry 9, 273 (2019). https://doi.org/10.1038/s41398-019-0592-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0592-5
This article is cited by
-
The role of neurotransmitter receptors in antipsychotic medication efficacy for Alzheimer’s-related psychosis
The Egyptian Journal of Neurology, Psychiatry and Neurosurgery (2024)
-
Decrease levels of bone morphogenetic protein 6 and noggin in chronic schizophrenia elderly
Cognitive Neurodynamics (2023)
-
The neural signatures of psychoses in Alzheimer’s disease: a neuroimaging genetics approach
European Archives of Psychiatry and Clinical Neuroscience (2023)
-
Genome-wide association of polygenic risk extremes for Alzheimer's disease in the UK Biobank
Scientific Reports (2022)
-
Neuropsychiatric symptoms in AD: the search for mechanisms
Nature Reviews Neurology (2022)
