
Abstract
Obesity and depression are major public health concerns that are both associated with substantial morbidity and mortality. There is a considerable body of literature linking obesity to the development of depression. Recent studies using Mendelian randomization indicate that this relationship is causal. Most studies of the obesity–depression association have used body mass index as a measure of obesity. Body mass index is defined as weight (measured in kilograms) divided by the square of height (meters) and therefore does not distinguish between the contributions of fat and nonfat to body weight. To better understand the obesity–depression association, we conduct a Mendelian randomization study of the relationship between fat mass, nonfat mass, height, and depression, using genome-wide association study results from the UK Biobank (n = 332,000) and the Psychiatric Genomics Consortium (n = 480,000). Our findings suggest that both fat mass and height (short stature) are causal risk factors for depression, while nonfat mass is not. These results represent important new knowledge on the role of anthropometric measures in the etiology of depression. They also suggest that reducing fat mass will decrease the risk of depression, which lends further support to public health measures aimed at reducing the obesity epidemic.
Similar content being viewed by others
Introduction
Obesity, defined as abnormal or excessive accumulation of body fat, is a major public health concern1 as an established risk factor for cardiovascular disease, type II diabetes, certain cancers, and overall decreased life expectancy2. Furthermore, observational studies have shown an association between obesity and depression. For example, Luppino et al.3 found that obese individuals were 55% more likely to develop depression, while depressed individuals were 58% more likely to become obese.
To investigate the observed association between obesity and depression, prior studies have performed Mendelian randomization (MR), a method from genetic epidemiology which uses data from genome-wide association studies (GWAS) to determine whether a risk factor is causal for an outcome4. MR studies have indicated that there is a causal relationship going from obesity to depression, but not vice versa5,6,7,8. These MR studies measured obesity using body mass index (BMI), which is calculated as weight (measured in kilograms) divided by the square of height (meters).
Although BMI is the most common measure of obesity (for example, an obese individual is generally defined as someone with BMI ≥ 30), its use has been repeatedly criticized9,10,11. Most notably, BMI does not distinguish between fat mass and nonfat mass. This distinction is very important from a physiological perspective, due to the fact that adipose tissue has very different properties compared to muscle and bone, the other major tissues contributing to body weight. In particular, adipose tissue is an endocrine organ that produces a range of adipokines and inflammatory proteins, which have been associated with negative systemic effects that may also affect the brain12. Furthermore, BMI does not capture body fat location, and while upper-body and visceral fat, which are more common in men, contribute to the development of an unhealthy cardiometabolic profile13, lower body fat, which is more common in women, seems to be protective of this14. In addition, how much of an individual’s BMI is due to fat, and where this fat is located, might also be of importance, as these factors can influence body satisfaction and social stigma, with psychological consequences15.
The aim of this study was to increase the understanding of the obesity–depression association by assessing the relationship between specific and biologically informative components of BMI (fat mass and nonfat mass, stratified on limbs and trunk) and depression via a MR study using results from large GWAS.
Materials and methods
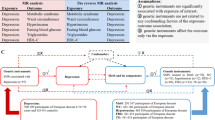
In total, we consider 21 anthropometric measures: the first six are BMI, weight, height, whole-body fat percentage, whole-body fat mass, and whole-body nonfat mass; the remaining 15 are fat percentage, fat mass and nonfat mass for each of trunk, right arm, left arm, right leg, and left leg. Supplementary Fig. 1 reports the genetic correlations between the 21 measures. We use MR to test whether each measure is a causal risk factor for depression, then to test whether depression is a causal risk factor for each measure.
In order to use MR to test whether one phenotype is a causal risk factor for another, we require GWAS summary statistics for both phenotypes. For the anthropometric traits, we use genome-wide summary statistics provided by the Neale Lab (http://www.nealelab.is/uk-biobank/), who performed association analyses for over 2000 phenotypes from the UK Biobank (http://www.ukbiobank.ac.uk/). The UK Biobank is a population-based cohort of approximately 500,000 individuals; 54% are female, the average age is 57 (range 37–73), while 94% report as being White British. For the anthropometric GWAS, the average sample size is 331,910 (see Table 1 for exact numbers); the analysis was performed using linear regression including ten principal components and sex as covariates.
For depression, we use summary statistics from the most recent GWAS of major depressive disorder (MDD) by the Psychiatric Genomics Consortium (PGC)6, available at (https://www.med.unc.edu/pgc/results-and-downloads). This study recruited individuals from seven cohorts; they used a relatively liberal definition of MDD, based on self-reporting, clinical assessment, and examination of medical records, with an estimated population prevalence of 15%. The association analysis was performed using logistic regression, including significantly-associated principal components as covariates. The PGC provides two sets of summary statistics: from their main GWAS of 480,359 samples (135,458 cases, 344,901 controls) they report results for 10,000 of the most strongly-associated SNPs; whereas from a “sub-GWAS” of 173,005 samples (59,851 cases and 113,154 controls), they report results for all SNPs. Therefore, when testing whether depression is a causal risk factor for one of the anthropometric measures (which requires only results for significantly-associated SNPs), we use summary statistics from the main GWAS, whereas when testing whether an anthropometric measure is a causal risk factor for depression (which requires genome-wide results), we use summary statistics from the sub-GWAS.
In total, there are 6,568,396 SNPs common to the UK Biobank and PGC GWAS (we excluded SNPs with alleles A & T or C & G, or with info score < 0.9). We test whether an anthropometric measure is causally associated with depression using inverse-weighted regression, for which we use the R package Mendelian randomization16. To decide which SNPs to use in this regression, we first identify which have P < 5e−8 for the measure, then thin these until no pair remains within 3 cM with correlation squared >0.05. We use the same strategy to test whether depression is a causal risk factor for an anthropometric measure, except now we identify a set of independent, genome-wide significant SNPs for depression.
A key assumption of MR is no pleiotropy17. For example, the SNPs we use when testing the relationship between BMI and depression should be causal for BMI, but not depression. This is difficult to test directly, so instead we perform three sensitivity analyses. Firstly, we repeat the inverse-weighted regression excluding SNPs showing evidence for pleiotropy (P < 0.05/N, where N is the number of independent, genome-wide significant SNPs). Secondly, we instead assess causality using weighted-median regression, which gives unbiased estimates provided at least 50% of the information comes from non-pleiotropic SNPs. Thirdly, we estimate the intercept from Egger Regression; an intercept significantly different to zero (P < 0.05) is an indication of directional pleiotropy.
Our primary analysis focuses on BMI, weight, height, whole-body fat mass, whole-body nonfat mass, and whole-body fat percentage; our secondary analysis considers the 15 location-specific measures of fat mass, nonfat mass and fat percentage. As we are performing a total of 42 tests, we set the (conservative) Bonferroni corrected significance threshold at P < 0.05/42 (this is satisfied when the regression slope is ≥3.0 SDs from zero).
Results
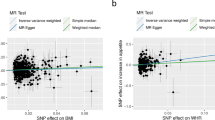
Table 1 reports the number of independent, genome-wide significant SNPs for each of the 21 anthropometric measures, and how much of the phenotypic variation each set of SNPs explains. The results of our primary analysis are displayed in Figs. 1 and 2 and Table 2. Our main conclusions are based on the results from inverse-variance regression using all SNPs (the red lines in each plot), while the remaining three regressions (the orange, green and blue lines) are sensitivity analyses. We note that each time the slope from inverse-variance regression using all SNPs is statistically significant, this finding is supported by the three sensitivity analyses (i.e., the slope remains significant when SNPs showing evidence for pleiotropy are excluded or when we use weighted-median regression, and Egger regression does not find significant directional pleiotropy).

The panels plot per-allele effect sizes for a BMI, b weight, c height, d body fat percentage, e body fat mass and f body fat-free mass (x-axes) against per-allele effect size for depression (y-axis) For the anthropometric measures, effect sizes are measured in SDs, for depression the effect size is log-odds ratio. For each plot we estimate the slope using inverse-variance regression (red solid line), inverse-variance regression after excluding SNPs showing evidence for pleiotropy (orange solid line), weighted-median regression (green solid line) and Egger regression (blue solid line). The corresponding colored dashed lines represent the 95% confidence intervals for the slopes, while the vertical blue segment marks a 95% confidence interval for the intercept from Egger regression. The horizontal black solid line indicates no effect

The panels plot per-allele effect size for depression (x-axis) against per-allele effect sizes for a BMI, b weight, c height, d body fat percentage, e body fat mass and f body fat-free mass (y-axes). For the anthropometric measures, effect sizes are measured in SDs, for depression the effect size is log-odds ratio. For each plot we estimate the slope using inverse-variance regression (red solid line), inverse-variance regression after excluding SNPs showing evidence for pleiotropy (orange solid line), weighted-median regression (green solid line) and Egger regression (blue solid line). The corresponding colored dashed lines represent the 95% confidence intervals for the slopes, while the vertical blue segment marks a 95% confidence interval for the intercept from Egger regression. The horizontal black solid line indicates no effect
Figure 1a confirms that BMI is a causal risk factor for depression. The estimated slope from inverse-weighted regression is 0.17 (SD 0.03), which is significantly greater than zero (P = 1e−7) and indicates that a 1-SD increase in BMI corresponds to a 0.17 increase in the log-odds ratio (OR) for depression. Figure 1b indicates there is a causal relationship between weight and depression; however, the estimated slope (0.13, SD 0.03) is less than that for BMI, which supports the current preference to measure obesity using BMI rather than weight. Figure 1c suggests that height (short stature) is a causal risk factor for depression; the estimated slope is −0.06 (SD 0.02), indicating that a 1-SD increase in height corresponds to a 0.06 decrease in the log-odds ratio (OR) for depression.
Figure 1d and e shows that whole-body fat percentage and whole-body fat mass are both causal risk factors for depression; the estimated slopes are very similar (0.20, SD 0.04 and 0.19, SD 0.03, respectively) indicating that there is no advantage to normalizing (i.e., measuring fat percentage instead of fat mass). The estimated slope in Fig. 1f is 0.06 (SD 0.03), indicating that MR finds no evidence that whole-body nonfat is a causal risk factor for depression.
Figure 2 shows that, consistent with previous studies, there is no significant evidence that depression is a causal risk factor for any of the six anthropometric measures investigated here.
Table 3 reports results from our secondary analysis. For each of the 15 location-specific anthropometric measures, the results are consistent with those from the whole-body version; i.e., regardless of whether we consider trunk, right arm, left arm, right leg, or left arm, we again find that fat percentage and fat mass are causally associated with depression, but that nonfat mass is not. For fat percentage and fat mass, we are interested in comparing slope estimates, as significant differences would indicate that risk of developing depression depends on fat location. The largest difference is observed for fat percentage, where the estimated slope for left leg (0.34, SD 0.06) is approximately twice that for trunk (0.16, SD 0.04); however, although this difference is nominally significance (P = 0.01), it is not significant after correction for multiple comparisons.
Discussion
We have used MR to investigate the causal relationship between different anthropometric measures and depression. We first confirmed that BMI is a causal risk factor for depression, but found no significant evidence that depression causes increased BMI. These results are in line with recent MR studies reporting evidence that higher BMI causally increases the risk of depression, but not the reverse5,6,7,8.
Our main finding is that body fat mass is a causal risk factor for depression, but that body nonfat mass is not, therefore indicating that the BMI-depression causality is driven by fat. We note from Table 1 that the number of genome-wide associated SNPs for whole-body nonfat (n = 667) and the proportion of variance they explain (11.9%) are higher than the corresponding values for fat percentage (n = 336 and 4.9%) and for fat mass (n = 387 and 5.8%), indicating that MR finding no evidence for a causal relationship was not simply a power issue.
We also investigated whether the strength of the causal relationship between body fat and depression depended on the location, but these results were inconclusive; while the point estimates suggested that leg fat more strongly affects risk of depression than either trunk fat or arm fat, the difference was not significant after correcting for multiple comparisons.
The causal relationship going from fat mass to depression is likely to have both psychological and biological components. Psychologically, perceived weight discrimination, stigmatization, and body image dissatisfaction may mediate the causality;18,19,20,21 biologically, obesity is associated with several endocrine and metabolic changes that have been linked to depression, including altered glucocorticoid, adipokine, insulin, leptin, and inflammatory signaling22. Although our study was not aimed at providing insight into how fat increases the risk of depression, the finding that trunk fat mass was not more strongly associated with depression risk than fat mass on the limbs (rather we found a tendency towards the opposite), seems to be in favor of a psychological mechanism—since trunk fat is considered the more metabolically adverse13.
Observational studies have attempted to separate the psychological and physiological components in the relationship between obesity and depression. In support of a physiological component, Jokela et al.23 found that the risk of depressive symptoms associated with obesity increased almost linearly with the number of metabolic risk factors; in particular, obese individuals who were metabolically unhealthy had a higher depression risk than obese individuals who were metabolically healthy (OR = 1.23; 95% CI: 1.05, 1.45). However, the same study also found that metabolically-healthy individuals who were obese had a higher depression risk than metabolically-healthy individuals who were not obese (OR = 1.29; 95% CI: 1.12, 1.50), indicating that metabolic factors only partially explain the increase in depression risk associated with obesity. Similar results were found by the longitudinal study of Hamer et al.24.
A recent study by Tyrrell et al.8 also sought to separate the psychological component of obesity from its adverse metabolic consequences—and employed MR to do so. Specifically, Tyrrell et al. used two genetic instruments, that both represented BMI, but one with and one without its adverse metabolic consequences. They found that both instruments were associated with increased risk of depression - suggesting that the causal association between BMI and depression is primarily driven by psychological consequences of adiposity and not by its adverse metabolic effects8. This conclusion resonates well with the results of our study.
In addition to the results on the impact of body fat on the risk of depression, we also found evidence suggesting that height (short stature) is a causal risk factor for depression. Several large observational studies have found short stature to be associated with poorer mental health25, lower health-related quality of life26, depressive symptoms in adolescents27 and adults28,29, and suicide in men30. Other studies have found no association between height and depression or suicide31 or negligible effects of height on mental health32. The association between short stature and poor mental health may be explained by confounding factors, such as socioeconomic status, prenatal development, or childhood factors, or by a causal effect of height on depression risk33,34. Our results indicate that at least part of the association between short stature and depression is indeed due to a direct causal effect.
Finally, we note some limitations of our work. Firstly, the MR estimates rely on three key assumptions: (i) the SNPs used as genetic predictors for a trait are causal for that trait; (ii) these SNPs are not associated with confounders of the trait-outcome association; (iii) these SNPs only affect the outcome through the causal relationship (i.e., there is no pleiotropy). We can be confident of (i) because our genetic predictors only used SNPs robustly-associated (P < 5e−8) with the trait, while (ii) should be true due to the fact an individual’s genotypes are randomly allocated during gamete formation. It is hard to explicitly test (iii), however, our sensitivity analyses indicate that our conclusions are not the consequence of pleiotropy.
Secondly, the UK Biobank measured fat and nonfat mass via bioelectrical impedance analysis (using a Tanita BC418MA body composition analyzer), which is considered less accurate than techniques such as dual-energy x-ray absorptiometry. However, we would expect measurement error to cause the estimates for fat and nonfat to become more similar, so the fact that we observed a significant difference indicates that the UK Biobank measurements were sufficiently accurate for our purpose.
Thirdly, we note that some samples were common to both the anthropometric and MDD GWAS (the PGC GWAS used approximately 30,000 individuals from the UK Biobank). Although this overlap is likely to have only a small impact35, ideally, our analysis would be repeated using summary statistics from completely independent GWAS.
Fourthly, while we found only suggestive evidence that location of fat affects risk of depression, we recognize that the high correlations between measurements taken in the trunk, arms, and legs (Supplementary Fig. 1) would have reduced our power to detect significant differences.
Fifthly, there may be sex-specific psychological and physiological factors affecting the obesity–depression relationship. While we were not able to test sex differences, because the PGC did not release results from male-only and female-only GWAS of depression, this is worthy of further study.
In conclusion, the present study provides evidence that the causal relationship between BMI and depression is driven by fat mass and height, and not by nonfat mass. These results represent important new knowledge on the role of anthropometric measures in the etiology of depression. They also suggest that reducing fat mass will decrease the risk of depression, which lends further support to public health measures aimed at reducing the obesity epidemic.
References
Swinburn, B. A. et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet 378, 804–814 (2011).
Haslam, D. W. & James, W. P. Obesity. Lancet 366, 1197–1209 (2005).
Luppino, F. S. et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch. Gen. Psychiatry 67, 220–229 (2010).
Lawlor, D. A., Harbord, R. M., Sterne, J. A., Timpson, N. & Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
Hartwig, F. P. et al. Body mass index and psychiatric disorders: a Mendelian randomization study. Sci. Rep. 6, 32730 (2016).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
van den Broek, N. et al. Causal associations between body mass index and mental health: a Mendelian randomisation study. J. Epidemiol. Community Health 72, 708–710 (2018).
Tyrrell, J. et al. Using genetics to understand the causal influence of higher BMI on depression. Int. J. Epidemiol 48, 834–848 (2019).
Kopelman, P. G. Obesity as a medical problem. Nature 404, 635–643 (2000).
Nuttall, F. Q. Body mass index: obesity, BMI, and health: a critical review. Nutr. Today 50, 117–128 (2015).
Wellens, R. I. et al. Relationships between the Body Mass Index and body composition. Obes. Res. 4, 35–44 (1996).
Ouchi, N., Parker, J. L., Lugus, J. J. & Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 11, 85–97 (2011).
Tchernof, A. & Despres, J. P. Pathophysiology of human visceral obesity: an update. Physiol. Rev. 93, 359–404 (2013).
Karpe, F. & Pinnick, K. E. Biology of upper-body and lower-body adipose tissue-link to whole-body phenotypes. Nat. Rev. Endocrinol. 11, 90–100 (2015).
Berner, L. A., Arigo, D., Mayer, L. E., Sarwer, D. B. & Lowe, M. R. Examination of central body fat deposition as a risk factor for loss-of-control eating. Am. J. Clin. Nutr. 102, 736–744 (2015).
Yavorska, O. O. & Burgess, S. Mendelian Randomization: an R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 46, 1734–1739 (2017).
Zheng, J. et al. Recent developments in Mendelian Randomization studies. Curr. Epidemiol. Rep. 4, 330–345 (2017).
Levy, B. R. & Pilver, C. E. Residual stigma: psychological distress among the formerly overweight. Soc. Sci. Med. (1982) 75, 297–299 (2012).
Hunger, J. M. & Major, B. Weight stigma mediates the association between BMI and self-reported health. Health Psychol. 34, 172–175 (2015).
Robinson, E., Sutin, A. & Daly, M. Perceived weight discrimination mediates the prospective relation between obesity and depressive symptoms in U.S. and U.K. adults. Health Psychology 36, 112–121 (2017).
Stevens, S. D., Herbozo, S., Morrell, H. E., Schaefer, L. M. & Thompson, J. K. Adult and childhood weight influence body image and depression through weight stigmatization. J. Health Psychol. 22, 1084–1093 (2017).
Hryhorczuk, C., Sharma, S. & Fulton, S. E. Metabolic disturbances connecting obesity and depression. Front. Neurosci. 7, 177 (2013).
Jokela, M., Hamer, M., Singh-Manoux, A., Batty, G. D. & Kivimaki, M. Association of metabolically healthy obesity with depressive symptoms: pooled analysis of eight studies. Mol. Psychiatry 19, 910–914 (2014).
Hamer, M., Batty, G. D. & Kivimaki, M. Risk of future depression in people who are obese but metabolically healthy: the English longitudinal study of ageing. Mol. Psychiatry 17, 940–945 (2012).
Cheung, Y. B. et al. Height and mental health and health utility among ethnic Chinese in a polyclinic sample in Singapore. Ann. Acad. Med. 42, 73–79 (2013).
Christensen, T. L., Djurhuus, C. B., Clayton, P. & Christiansen, J. S. An evaluation of the relationship between adult height and health-related quality of life in the general UK population. Clin. Endocrinol. 67, 407–412 (2007).
Rees, D. I., Sabia, J. J. & Argys, L. M. A head above the rest: height and adolescent psychological well-being. Econ. Hum. Biol. 7, 217–228 (2009).
Montgomery, S. M., Netuveli, G., Hildon, Z. & Blane, D. Does financial disadvantage at older ages eliminate the potential for better health? J. Epidemiol. Community Health 61, 891–895 (2007).
Osika, W. & Montgomery, S. M. Economic disadvantage modifies the association of height with low mood in the US, 2004: the disappointment paradox. Econ. Hum. Biol. 6, 95–107 (2008).
Magnusson, P. K., Gunnell, D., Tynelius, P., Davey Smith, G. & Rasmussen, F. Strong inverse association between height and suicide in a large cohort of Swedish men: evidence of early life origins of suicidal behavior? Am. J. Psychiatry 162, 1373–1375 (2005).
Bjerkeset, O., Romundstad, P., Evans, J. & Gunnell, D. Association of adult body mass index and height with anxiety, depression, and suicide in the general population: the HUNT study. Am. J. Epidemiol. 167, 193–202 (2008).
Coste, J., Pouchot, J. & Carel, J. C. Height and health-related quality of life: a nationwide population study. J. Clin. Endocrinol. Metab. 97, 3231–3239 (2012).
Batty, G. D. et al. Height, wealth, and health: an overview with new data from three longitudinal studies. Econ. Hum. Biol. 7, 137–152 (2009).
Perkins, J. M., Subramanian, S. V., Davey Smith, G. & Ozaltin, E. Adult height, nutrition, and population health. Nutr. Rev. 74, 149–165 (2016).
Burgess, S., Davies, N. M. & Thompson, S. G. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 40, 597–608 (2016).
Acknowledgements
D.S. is supported by the European Unions Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement number 754513, by Aarhus University Research Foundation (AUFF) and the Independent Research Fund Denmark (7025-00094B). A.D.B. is supported by grants from The Lundbeck Foundation (R102-A9118 and R155-2014-1724). Data handling and analysis on the GenomeDK HPC facility was supported by NIMH (1U01MH109514-01 to Michael O’Donovan and A.D.B.). HPC capacity at GenomeDK was provided by the iSEQ center and Center for Genomics and Personalized Medicine, Aarhus, Denmark (grants to A.D.B.). S.D.Ø. is supported by the Independent Research Fund Denmark (7016-00048).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Speed, M.S., Jefsen, O.H., Børglum, A.D. et al. Investigating the association between body fat and depression via Mendelian randomization. Transl Psychiatry 9, 184 (2019). https://doi.org/10.1038/s41398-019-0516-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0516-4
This article is cited by
-
Body composition, lifestyle, and depression: a prospective study in the UK biobank
BMC Public Health (2024)
-
Brain-derived neurotrophic factor and C-reactive protein (CRP) biomarkers in suicide attempter and non-attempter major depression disorder (MDD) patients
Annals of General Psychiatry (2024)
-
Familial confounding of internalising symptoms and obesity in adolescents and young adults; a co-twin analysis
International Journal of Obesity (2024)
-
Composite healthy lifestyle, socioeconomic deprivation, and mental well-being during the COVID-19 pandemic: a prospective analysis
Molecular Psychiatry (2024)
-
Childhood obesity and adolescent follow-up depressive symptoms: exploring a moderated mediation model of body esteem and gender
European Child & Adolescent Psychiatry (2024)
