
Abstract
Genetic, epidemiological, and biomarker studies suggest that the immune system is involved in the pathogenesis of bipolar disorder (BD). It has therefore been hypothesized that immune activation of microglia, the resident immune cells of the brain, is associated with the disease. Only a few studies have addressed the involvement of microglia in BD so far and a more detailed immune profiling of microglial activation is lacking. Here, we applied a multi-level approach to determine the activation state of microglia in BD post-mortem brain tissue. We did not find differences in microglial density, and mRNA expression of microglial markers in the medial frontal gyrus (MFG) of patients with BD. Furthermore, we performed in-depth characterization of human primary microglia isolated from fresh brain tissue of the MFG, superior temporal gyrus (STG), and thalamus (THA). Similarly, these ex vivo isolated microglia did not show elevated expression of inflammatory markers. Finally, challenging the isolated microglia with LPS did not result in an increased immune response in patients with BD compared to controls. In conclusion, our study shows that microglia in post-mortem brain tissue of patients with BD are not immune activated.
Similar content being viewed by others


Introduction
The aetiology of bipolar disorder (BD) is still largely unknown. Several lines of evidence suggest that the immune system is involved in the pathogenesis of BD. Epidemiological studies on large birth cohorts have shown an association between BD and autoimmune diseases, atopic disorders, and severe childhood infections1,2,3. Additionally, genetic studies found that several genes associated with BD cluster in immune pathways4. Furthermore, many research groups have reported alterations in peripheral immune markers in BD, with increased levels of C-reactive protein (CRP) and several cytokines, such as IL1β, IL6, and TNFα, in blood and cerebrospinal fluid of patients with BD5,6,7. Besides, a decreased percentage of T-regulatory cells8,9,10 and an altered gene expression profile of circulating monocytes in the blood have been described8,11.
It is not yet understood how these immune associations are involved in the pathogenesis of BD, but activation of microglia, the immune cells that reside in the brain parenchyma, has been hypothesized to play a central role12,13,14,15. Microglia are part of the innate immune system and have an important function in initiating and controlling neuroinflammation in the CNS via the secretion of pro- and anti-inflammatory cytokines. In addition, these cells are involved in neurodevelopment and neuronal functioning in adulthood by promoting synaptogenesis and synaptic pruning16,17. Besides, microglia are involved in the regulation and production of serotonin of which an imbalance has been implicated in BD13. In homeostatic conditions, microglia have a ramified morphology with highly motile protrusions that scan the environment for possible danger signals, such as pathogens or cell debris. Upon detection, microglia become immune activated. They migrate to the site of injury, and change their expression profile and shape to a more rounded or so-called ‘amoeboid’ morphology. This process is necessary to adequately respond to the insult via the secretion of cytokines and chemokines, antigen presentation of the microglia, and apoptosis of the injured cell18,19,20. Although microglial immune activation is crucial to minimize the effects of the injury, prolonged activation will lead to the release of reactive oxygen species, which are toxic to neurons and other neighbouring cells21. In BD it has been hypothesized that microglia are immune activated, possibly induced by circulating peripheral immune factors, and result in CNS damage and abnormalities22,23.
However, evidence for immune activation of microglia in BD is still lacking. Five post-mortem studies investigated the density of microglia or the expression of microglia markers in BD. The samples sizes ranged from 9 to 20 subjects, different microglial markers were used, and multiple brain regions were studied. None of the studies found an increased microglial density or elevated expression of microglial markers24,25,26,27. One study even found decreased mRNA expression of the microglial markers ITGAM (CD11b) and CD68 in the anterior cingulate cortex and prefrontal cortex28. Moreover, in a positron emission tomography study with the [(11)C]-(R)-PK11195 tracer, there was no sign for microglia activation in most regions of the brain in BD, except for the hippocampus29. A drawback of these studies was that most used one methodological approach (i.e., number of microglia, microglial mRNA expression or protein level), non-automated analysis and focussed on immunostainings that only give limited information about the diversity of microglial phenotype and function. To conclude, an in-depth microglial immune profile examining more specific signs of microglial activation is still missing in BD.
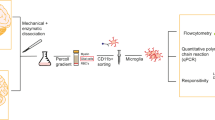
Therefore, the aim of this study was to determine whether microglia in post-mortem brain tissue of patients with BD are immune activated. We applied a multi-level approach and characterized microglial density, and inflammatory functions in the medial frontal gyrus (MFG) of patients with BD. To further profile the microglia and examine their phenotype and functions, we isolated human primary microglia from fresh post-mortem brain tissue of the MFG, superior temporal gyrus and thalamus. This allows us to characterize microglia outside the brain and assess their functions more extensively30,31,32,33. This exclusive approach has been used to profile microglia in neurologic disorders, such as multiple sclerosis and Alzheimer’s disease34,35,36, but has not yet been applied to psychiatric disorders. To our knowledge, the present study is the first investigating microglial immune activation in ex vivo isolated human microglia in combination with immunohistochemistry and mRNA profiling.
Materials and methods
Donors
Paraffin (control N = 12; BD N = 16), snap frozen (control N = 16; BD N = 15), and fresh post-mortem brain tissue (control N = 19; BD N = 12) of patients with BD and controls was obtained from the Netherlands Brain Bank (www.brainbank.nl). The permission to collect human brain material was obtained from the Ethical Committee of the VU University medical center (VUmc, Amsterdam, The Netherlands). An overview of the clinical characteristics and donors used per experiment is summarized in Table 1 and Supplementary Table 1. We selected the medial frontal gyrus (MFG) as the main region of interest (ROI). For fresh post-mortem brain tissue, we also selected the superior temporal gyrus (STG) and thalamus (THA). All three regions have been associated with BD20,37,38. Permission for brain autopsy and the usage of brain tissue and accompanied clinical information for research purposes was obtained per donor ante-mortem. Due to the insufficient quality of the tissue or limited number of viable microglia after isolation, we could not include every donor for all downstream analyses. Control donors are defined as donors without a history of major psychiatric diseases, confirmed by retrospective medical chart review. There was no significant difference between controls and patients with BD in age, post-mortem delay (PMD), and pH (Table 1). Only in frozen tissue, sex was significantly different in both grey (p = 0.004) and white matter (p = 0.008) between patients with BD and controls.
Immunostaining and image analysis
Paraffin-embedded tissue of the MFG of patients with BD and controls was sectioned at 7 μM. The sections were deparaffinised using a standard xylene and alcohol series, followed by blocking of endogenous peroxidase with PBS, 1% H2O2 (Merck, Germany). For antigen retrieval, sections were heated in 0.01 mM citrate buffer (Merck, Darmstadt, Germany), 0.05% Tween-20 (Merck, Darmstadt, Germany), pH = 6.0 for 15 min. Subsequently, aspecific binding was blocked in PBS with 1% normal horse serum (NHS, Thermo Fisher Scientific, Massachusetts, USA), 0.1% bovine serum albumin (BSA, Merck, Darmstadt, Germany), 0.2% Triton X (Merck, Darmstadt, Germany). Sections were incubated with a rabbit polyclonal anti-Iba1 antibody (Wako Pure Chemical Industries, Ltd., 1:1000) at 4 °C. Next day, secondary goat-anti-rabbit biotin (Jackson ImmunoResearch Laboratories, Inc., 1:400) was added, followed by avidin-biotin-peroxidase (AB) complex (Vector Laboratories, USA). To visualize the microglia, the sections were incubated with a 3,3′ diaminobenzidine (DAB) substrate (DAKO, USA). Finally, tissue sections were dehydrated using an alcohol and xylene series and embedded in Entellan (Merck, Darmstadt, Germany). Per tissue section, six pictures of 89,44 um × 119,37 um were taken randomly and blinded for diagnosis from grey and white matter. Two investigators (MS and GS) separately counted the number of microglia in each picture manually and averaged the total number of counted microglia per donor. Microscopic images were also analysed automatically using open source software ImageJ. Microglial cell density was calculated with a particle analysis macro-script, by dividing the cell numbers by the measured area (microglia/mm2) (for further details see Supplementary Methods). There was a high level of correlation between manual cell counts and automated cell counts (Rs = 0.780, p < 0.05) and Cronbachs alpha was greater than >0.80 (CB alpha 0.879). Immunofluorescent staining was performed additionally to determine the number of microglia relative to total cell number. The first part of the staining procedure was similar as for DAB-immunostained microglia, with the usage of donkey anti-rabbit Alexa 488 (1:700) and Hoechst (1:1000) as secondary antibody. An automated macro script was used to quantify the number of microglia (Iba1+Hoechst+) or total cell number (Hoechst+) (See Supplementary Fig. 1a and Supplementary Methods).
Human primary microglia isolation
Human primary microglia (pMG) were isolated from fresh post-mortem brain tissue of the different ROIs based on an earlier established protocol30. Brain tissue was first mechanically dissociated through a metal sieve in a glucose- potassium-sodium buffer (GKN-BSA; 8.0 g/L NaCl, 0.4 g/L KCl, 1.77 g/L Na2HPO4.2H2O, 0.69 g/L NaH2PO4.H2O, 2.0 g/L D-(1)-glucose, 0.3% bovine serum albumin (BSA, Merck, Darmstadt, Germany); pH 7.4) and supplemented with collagenase Type I (3700 units/mL; Worthington, USA) and DNase I (200 µg/mL; Roche, Switzerland) at 37 °C for 30 min (THA) or 60 min (MFG, STG) while shaking. The suspension was put over a 100 µM cell strainer and washed with GKN-BSA buffer in the centrifuge (1800 rpm, slow brake, 4 °C, 10 min) before the pellet was resuspended in 20 mL GKN-BSA buffer. 10 mL of Percoll (Merck, Darmstadt, Germany) was added dropwise and the tissue homogenate was centrifuged at 4000 rpm (fast acceleration, slow brake at 4 °C, 30 min). The middle layer was collected and washed with GKN-BSA buffer, followed by resuspension and centrifuging in a magnetic-activated cell sorting (MACS) buffer (PBS, 1% heat-inactivated fetal cow serum (FCS), 2 mM EDTA; 1500 rpm, 10 °C, 10 min). Microglia were positively selected with CD11b conjugated magnetic microbeads (Miltenyi Biotec, Germany) according to the manufacturer’s protocol. This method relies on the membrane expression of CD11b, which is also present on perivascular and infiltrated macrophages in the central nervous system (CNS)30. Mass cytometry analysis has shown that the percentage of perivascular macrophages (CD206high) was low (<2%) in microglia isolated from control donors and individuals with psychiatric disorders, suggesting that majority of cells that we analyze are indeed microglia39. Microglia were lysed in TRIzol reagent (Invitrogen, USA), stained for flow cytometry or cultured in a poly-l-lysine (PLL; Merck, Germany) coated 96-wells flat bottom plate (Greiner Bio-One, Austria) at a density of 1.0 × 105 cells in a total volume of 200 μL Rosswell-Park-Memorial-Institute medium (RPMI; Gibco Life Technologies, USA) supplemented with 10% FCS, 2 mM l-glutamine (Gibco Life Technologies, USA), 1% penicillin–streptomycin (Gibco Life Technologies, USA) and 100 ng/ml IL-34 (Miltenyi Biotech, Germany). After overnight incubation, pMG were stimulated with 100 ng/mL lipopolysaccharide (LPS) from Escherichia coli 0111:B4 (Merck, Germany) for 6 h. We selected 6 h LPS stimulations for its robust ‘inflammatory’ effect on microglia30. The concentrations and incubation times are based on dose-response curves31. The cells were harvested with TRIzol reagent and stored at −80 °C for further analysis of mRNA expression.
Gene expression analysis
Grey and white matter were manually separated from a 50 µM section of frozen brain tissue at the cryostat and lysed with TRIzol reagent. RNA extraction, cDNA synthesis, and qPCR on these brain tissue samples and pMG were performed as described before30. Considering the low yield of RNA and the consistency when comparing single-well experiments with triplicates, we ran single-well experiments for each reaction. Primer sequences are listed in Supplementary Table 2. Absolute expression was calculated using the ΔΔCT method40. For the selection of reference genes we used the method described by Van den Sompele in 200241. Expression Suite software 1.0.4 was used to select the most stable genes across ROIs, diagnosis, grey/white matter tissue and different donors for normalization. 18S ribosomal RNA (18S) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were most stable for whole brain tissue and β-Actin (ACTB) and GAPDH for directly isolated pMG. LPS stimulated pMG were normalized to GAPDH. Samples with Cycle Threshold (CT) values higher than two times standard deviation above the average of each reference gene were excluded for further analysis. The average mean CT values of total brain tissue samples were between 25 and 31, of pMG between 23 and 30, and LPS stimulated pMG between 19 and 25. Median levels of relative expression between BD and controls were compared using non-parametric tests.
Flow cytometry of human primary microglia
12–20 × 104 human primary microglia were stained in a v-bottom 96-wells plate (Greiner Bio-One, Austria) directly after isolation. pMG were washed with 100 µL PBS with 0.5% BSA (PBA) and incubated in 25 µL PBA with specific monoclonal or isotype control antibodies (Supplementary Table 3). After staining, the cells were washed with PBA, fixated with 4% paraformaldehyde (Riedel-de Haën, Germany) in PBS at room temperature and stored in the fridge for flow cytometry analysis the next day. All microglia samples were processed on a FACS Canto (BD Bioscience) with calibrated settings and similar voltage for the fluorescent channels. The geomean fluorescence intensity (MFI) was determined by subtracting the MFI of the isotype control from the MFI of the positive antigen staining.
Statistical analysis
Statistical analysis was done with GraphPad Prism software (version 7) and SPSS IBM 23. All variables were tested for homogeneity of variances and normality of distribution by means of the Levene and Kolmogorov–Smirnov tests, respectively. Differences between groups were analysed using chi-square (χ2) tests, independent t-tests or the Mann–Whitney U when appropriate. χ2 were used to test for sex difference and, independent t-test for age, PMD and pH differences. Non-parametric Mann–Whitney U test was performed to analyse differences in microglia density, gene expression, and protein levels between BD and controls, since data were non-normally distributed. Results were considered significant if the two-sided p-value was ≤0.05. Bonferroni correction for multiple testing was used for the qPCR and flow cytometry experiments, since more than one marker was analysed per region in the same experiment. For all outcome measures in this study, a spearman ranks correlation was performed to assess whether sex, age, PMD or pH were associated with the outcome. In case we found a significant association (Supplementary Table 4), an ANCOVA was applied to correct for possible confounding effects.
Results
Microglial density and morphology, and expression of microglial markers in total brain tissue
We analysed microglial density with immunohistochemistry using microglial marker ionized calcium-binding adaptor molecule 1 (Iba1). Figure 1a shows four representative pictures of DAB-Iba1+ microglia in the grey (I, III) and white matter (II, IV) in the MFG of a control donor (I–II) and patient with BD (III–IV). Manual and automated counting with ImageJ resulted in a similar microglial density between patients with BD and controls (Fig. 1b, c). Results were validated with immunofluorescent staining, showing no differences in microglial number relative to total cell number in BD compared to controls (Supplementary Fig. 1a, b). Furthermore, we assessed whether the expression of a panel of microglial-related or immune activated genes is changed in both grey and white matter of the MFG in BD. We defined three gene categories: (1) microglial-associated (AIF1, P2Y12, and TMEM119), (2) expressed on all myeloid cells (CD68, CX3CR1 and ITGAM), and (3) (microglial)-immune activation (HLA-DRA, IL1B and IL6) (Table 2 and Fig. 1d–h). We did not find differences in expression between patients and controls for any of these genes.

a–c The medial frontal gyrus of patients with bipolar disorder (BD) and controls was stained for microglial marker ionized calcium-binding adaptor molecule (Iba1) to analyse microglial density and morphology. a Representative pictures are shown of microglia in grey (I, III) and white (II, IV) matter of controls (I–II) and patients with BD (III–IV). b The number of microglia (microglia/mm2) was manually quantified in grey and white matter in controls (N = 12, blue dots) and patients with BD (N = 12, red circles). c The number of microglia (microglia/mm2) was automatically quantified in grey and white matter in controls (N = 12, blue dots) and patients with BD (N = 16, red circles). d–h mRNA expression of microglial markers AIF1 and TMEM119, myeloid marker ITGAM and CX3CR1 and HLA-DRA in patients with BD (N = 15) and controls (N = 16) was determined using qPCR. Gene expression was normalized to 18S ribosomal RNA (18S) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the ΔΔCT method. Graphs show median expression levels and non-parametric testing with Bonferroni correction for multiple testing was performed. ND = number of non-detected samples
Phenotype and function of isolated microglia
Isolation of human primary microglia provided us with the opportunity to profile and characterize microglia as a pure cell population more extensively. We found no differences in the expression of genes involved in pro-inflammatory signalling (IL1B and IL6; Fig. 2a, b), genes related to anti-inflammatory functioning (CD163 and MRC1; Fig. 2c, d), TMEM119 (Fig. 2e), a microglial specific gene known for its homeostatic properties and CX3CR1, a gene related to microglial migration and neuron-glia interaction (Fig. 2f)33,42 in the MFG, STG, and THA (Supplementary Fig. 2). We analysed the expression of a panel of proteins by flow cytometry, including more general markers for microglia and myeloid cells (CD11b, CD11c, CD45, CD14, CD16, CD32, CD40, CD64, CX3CR1) and markers related to immune activation of the cells (CD163, CD172α, CD200R, CD206, HLA-DR, CD83, CD86) (Fig. 2g–j, Supplementary Table 5). Remarkably, the fluorescent mean intensity was quite variable between different donors. We found a significantly decreased expression of CD16 and CX3CR1 in the STG (Supplementary Table 5 and Supplementary Fig. 3d), however, this difference did not survive correction for multiple testing. We did not find differences in protein expression, including CD16 and CX3CR1, between controls and BD in the MGF and THA (Fig. 2j, Supplementary Fig. 3h and Supplementary Table 5).

Human microglia were isolated from the medial frontal gyrus of controls (N = 13, blue dots) and patients with bipolar disorder (BD, N = 10, red circles). a–f After isolation mRNA expression of pro-inflammatory genes (IL1B (a) and IL6 (b)), anti-inflammatory genes (CD163 (c) and MRC1 (d)), the microglial specific gene TMEM119 (e) and the myeloid marker CX3CR1 (f) was determined in patients with BD and controls using qPCR. Gene expression was normalized to β-Actin (ACTB) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the ΔΔCT method. ND = number of non-detected samples. g–j Protein expression of isolated microglia from controls (N = 17) and patients with BD (N = 8) was characterized by flow cytometry. The mean fluorescent intensity (MFI) is determined for CD11b (g), CD45 (h) HLA-DR (i) CX3CR1 j Inflammatory response of isolated microglia from controls (N = 18 and patients with BD (N = 7) after 6 h of stimulation with lipopolysaccharide (LPS) was determined by measuring mRNA expression of IL1B, IL6 and TNF. The fold change was calculated by dividing the mRNA expression of the LPS stimulated sample by mRNA expression of the non-stimulated sample of the same subject. The dotted line represents the baseline mRNA expression of non-stimulated cells. Graphs show median expression levels and non-parametric testing with Bonferroni correction for multiple testing was performed
To determine inflammatory responsiveness, we challenged the isolated human microglia with LPS and determined the mRNA expression levels of cytokines. Elevated expression of these markers in BD relative to controls would suggest increased (primed) immune activity of the cells. In contrast, less expression could indicate ‘silenced’ microglia in which sustained immune activation results in a blunted inflammatory response. We found that microglia from BD and controls both responded to LPS with increased expression of IL1B, IL6, and TNF (Fig. 2k). The effect was not significantly different between microglia of patients with BD and controls. The same was observed in microglia from the STG and THA (Supplementary Fig. 4).
Discussion
The aim of this study was to elucidate whether microglia are immune activated in patients with BD, as is hypothesised by many studies12,13,14,15,22,23. We performed the most extensive, multi-level study to date in characterizing microglial inflammation in BD. We did not find any support for microglial immune activation, since we did not find differences in microglial density, and the expression of mRNA levels genes related to microglial function in fixed and frozen brain tissue. The absence of an immune activated phenotype of microglia in BD was confirmed by the lack of upregulation of inflammatory markers in ex vivo isolated human microglia on mRNA and protein level. Moreover, challenging human microglia of patients with BD with LPS did not result in an altered immune phenotype compared to control microglia. Altogether, the results of post-mortem studies, including ours, are consistent and do not support the hypothesis that microglia are immune activated in BD.
Our findings are in line with previous post-mortem studies that investigated microglial immune activation in BD by quantifying microglial cell numbers24,25,26,27. The results are also in agreement with recent transcriptomic studies on cortex lysates of BD patients and controls43,44,45,46,47,48. None of the activation markers that were examined in our present study were significantly different between BD and controls in any of these studies. For the two largest studies47,48 we extracted the results for these genes from their summary statistic files and depicted those in Supplementary Table 6. Interestingly, a downregulation of CX3CR1, P2RY12, TMEM119, AIF1 and ITGAM was seen in these large studies. In addition, two smaller transcriptomic studies also found a downregulation of several microglial immune activation markers (ITGAM, CX3CR1, AIF1, CD163 and HLA-DR) albeit not significant after correction for multiple testing44,45. In our data, we also found a trend towards downregulation of CX3CR1. A decreased expression of CX3CR1 in grey (p = 0.10) and white (p = 0.15) matter MFG brain tissue was observed on a trend level (see Table 2 and Fig. 1h). Furthermore, we found a significantly decreased protein expression of CX3CR1 in isolated primary human microglia cells in the STG using flow cytometry (see Supplementary Table 5 and Supplementary Fig. 3d). Altogether, these studies indicate that although microglia are not immune activated, the phenotype and function of these cells might be changed. A downregulation of CX3CR1, TMEM119 and P2RY12 has also been described for stage 1 disease-associated microglia in neurodegenerative diseases49. Further studies are therefore warranted to analyse these changes in microglia phenotype in BD.
The hypothesis of immune activation as driving force in BD was mainly based on studies that have described immune alterations in the periphery5,6,7. This is in contrast to our results showing no microglial activation in the CNS, but could be explained in several ways. An important point is the difference in age between the patients in post-mortem studies and peripheral studies, with lower mean age in the latter. Therefore, we are only able to draw conclusions on microglial immune activation in the late phases of the disease. To further understand the relationship between peripheral immune alterations in BD and microglial activation an interesting follow-up study would be to analyse peripheral markers in serum and relate this to markers for microglial activation in the same post-mortem brain donors. At the same time, it is important to look at other explanations for the observed peripheral immune alterations in BD. Immune biomarkers in blood are very sensitive to changes in lifestyle, such as smoking, BMI, diet, stress and sleep problems50,51. We should, therefore, include the possibility that the peripheral changes actually reflect behavioural consequences and not the cause of the disease. On the other hand, this does not explain the increased prevalence of immune-related disorders in BD. Other immune-mediated mechanisms should, therefore, be investigated, such as autoimmunity and the role of non-inflammatory immune pathways in the brain that are for instance involved in glia-neuron communication52. It is becoming more evident that microglia have many more functions than controlling inflammation. Microglia are for example actively participating in modifying synapses in the brain53,54.
The multi-level approach used in this study contributed to a more detailed understanding of microglial activation in BD. Microglial heterogeneity was taken into account by making a distinction between microglia in grey and white matter brain tissue and by analysing isolated microglia from three brain regions. We applied different techniques to frozen, paraffin, and acutely isolated microglia to elucidate whether microglial immune activation is present. Additionally, we validated qualitative assessments of microglia density using scripted and automated analyses. A drawback of this study is the limited sample size per experiment (minimal six donors per condition). We performed a power analysis based on findings of earlier research55. We estimated that 34 subjects in each study arm would be sufficient to obtain a power of 80% at an α-level of 0.05. This number was not reached due to the rarity of the material. We included the maximum availability of brain tissue samples. Although we looked at microglia in three regions, it is also possible that microglial changes are restricted to specific areas or cortical layers of the brain. For the LPS stimulation study, we a priori selected three genes (TNF, IL1B and IL6) to determine the responsiveness of the microglia after 6 h. Quantification of genes other than these ‘inflammatory’ cytokines and long-term effects of LPS stimulations (>24 h) could be explored in future investigations. The availability of fresh post-mortem brain tissue is sparse, especially for psychiatric disorders. Post-mortem studies are subject to many confounders (e.g., chronicity of illness, use of psychotropic medication, somatic comorbidities such as neurologic, infection/autoimmune diseases), smoking, peri-mortem factors, and cause of death (euthanasia, suicide or somatic problems). Many of these factors have been shown to have a potential impact on microglia phenotype, such as mood stabilizers and antipsychotics56,57, neurological diagnoses32,58, aging59, environmental influences and lifestyles, including physical and mental activities, drugs, comorbidities and infections60, and suicide61.This makes primary changes intrinsic to the illness hard to distinguish from secondary, compensatory, and epiphenomenal effects. The absence of microglial activation might be concealed due to the effect of these confounding factors, however, confounder analyses for all these factors is not possible in our cohort due to small sample sizes. For some of our outcome measures, we found a significant correlation with one of the confounders age, sex, pH or PMD. However, correction for these factors resulted in similar findings. Controlling for medication was very difficult, due to the high variability of medication received by the donors. We did study the effect of corticosteroids on mRNA and protein expression and could not find a correlation (data not shown). We matched BD subjects with HC subjects on the presence of neurodegenerative disorders (based on amyloid/braak scores), but this was not always possible due to the rarity of the material. Additionally, we excluded donors with CNS autoimmune diseases, such as multiple sclerosis. No information was available about state of the disease ((hypo)mania or depression) during time of death of the donor. This might be an important factor, since monocyte-induced-microglia generated from patients with BD during a manic or depressed stated have been shown to express a different immune profile62.
In summary, the present study provides an overview of the immune state of microglia in BD. By using a multi-level approach, microglia were extensively profiled in post-mortem brain tissue of the MFG. Microglia in BD did not display any characteristics indicating immune activation. The data on CX3CR1 expression suggest that other microglial functions important for BD pathogenesis, such as the response to stress and their role in synaptic plasticity, could be affected.
References
Benros, M. E. et al. Autoimmune diseases and severe infections as risk factors for mood disorders: a nationwide study. JAMA Psychiatry 70, 812–20 (2013).
Cremaschi, L. et al. Prevalences of autoimmune diseases in schizophrenia, bipolar I and II disorder, and controls. Psychiatry Res. 258, 9–14 (2017).
Wang, L.-Y., Chiang, J.-H., Chen, S.-F. & Shen, Y.-C. Systemic autoimmune diseases are associated with an increased risk of bipolar disorder: A nationwide population-based cohort study. J. Affect Disord. 227, 31–37 (2018).
Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 18, 199–209 (2015).
Fernandes, B. S. et al. C-reactive protein concentrations across the mood spectrum in bipolar disorder: a systematic review and meta-analysis. Lancet Psychiatry 3, 1147–1156 (2016).
Goldsmith, D. R., Rapaport, M. H. & Miller, B. J. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 21, 1696–1709 (2016).
Munkholm, K., Braüner, J. V., Kessing, L. V. & Vinberg, M. Cytokines in bipolar disorder vs. healthy control subjects: a systematic review and meta-analysis. J. Psychiatr. Res. 47, 1119–1133 (2013).
Vogels, R. J., Koenders, M. A., van Rossum, E. F. C., Spijker, A. T. & Drexhage, H. A. T. Cell deficits and overexpression of hepatocyte growth factor in anti-inflammatory circulating monocytes of middle-aged patients with bipolar disorder characterized by a high prevalence of the metabolic syndrome. Front Psychiatry 8, 34 (2017).
Snijders, G. et al. A dynamic course of T cell defects in individuals at risk for mood disorders. Brain Behav. Immun. 58, 11–17 (2016).
do Prado, C. H. et al. Reduced regulatory T cells are associated with higher levels of Th1/TH17 cytokines and activated MAPK in type 1 bipolar disorder. Psychoneuroendocrinology 38, 667–76 (2013).
Becking, K. et al. Inflammatory monocyte gene expression: trait or state marker in bipolar disorder? Int J. Bipolar Disord. 3, 20 (2015).
Keshavarz, M. Glial cells as key elements in the pathophysiology and treatment of bipolar disorder. Acta Neuropsychiatr. 29, 140–152 (2017).
Watkins, C. C., Sawa, A. & Pomper, M. G. Glia and immune cell signaling in bipolar disorder: insights from neuropharmacology and molecular imaging to clinical application. Transl. Psychiatry 4, e350–e350 (2014).
Réus, G. Z. Z. et al. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 300, 141–154 (2015).
Naaldijk, Y. M., Bittencourt, M. C., Sack, U. & Ulrich, H. Kinins and microglial responses in bipolar disorder: a neuroinflammation hypothesis. Biol. Chem. 397, 283–96 (2016).
Kettenmann, H., Kirchhoff, F. & Verkhratsky, A. Microglia: new roles for the synaptic stripper. Neuron 77, 10–18 (2013).
Schafer, D. P. & Stevens, B. Microglia function in central nervous system development and plasticity. Cold Spring Harb. Perspect. Biol. 7, a020545 (2015).
Sklar, P. et al. Whole-genome association study of bipolar disorder. Mol. Psychiatry 13, 558–69 (2008).
Kettenmann, H., Hanisch, U.-K., Noda, M. & Verkhratsky, A. Physiology of microglia. Physiol. Rev. 91, 461–553 (2011).
Maletic, V. & Raison, C. Integrated neurobiology of bipolar disorder. Front Psychiatry 5, 98 (2014).
Takeuchi, H. Neurotoxicity by microglia: mechanisms and potential therapeutic strategy. Clin. Exp. Neuroimmunol. 1, 12–21 (2010).
Rosenblat J. D., McIntyre R. S. Bipolar disorder and immune dysfunction: epidemiological findings, proposed pathophysiology and clinical implications. Brain Sci. 7, 2017. https://doi.org/10.3390/brainsci7110144.
Beumer, W. et al. The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J. Leukoc. Biol. 92, 959–75 (2012).
Hercher, C., Chopra, V. & Beasley, C. L. Evidence for morphological alterations in prefrontal white matter glia in schizophrenia and bipolar disorder. J. Psychiatry Neurosci. 39, 376–385 (2014).
Brisch, R. et al. Microglia in the dorsal raphe nucleus plays a potential role in both suicide facilitation and prevention in affective disorders. Eur. Arch. Psychiatry Clin. Neurosci. 267, 403–415 (2017).
Hamidi, M., Drevets, W. C. & Price, J. L. Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol. Psychiatry 55, 563–569 (2004).
Dean, B. et al. Different changes in cortical tumor necrosis factor-α-related pathways in schizophrenia and mood disorders. Mol. Psychiatry 18, 767–773 (2013).
Seredenina, T. et al. Decreased NOX2 expression in the brain of patients with bipolar disorder: association with valproic acid prescription and substance abuse. Transl. Psychiatry 7, e1206 (2017).
BCMB, Haarman et al. Neuroinflammation in bipolar disorder - A [11C]-(R)-PK11195 positron emission tomography study. Brain Behav. Immun. 40, 219–225 (2014).
Melief, J. et al. Characterizing primary human microglia: a comparative study with myeloid subsets and culture models. Glia 64, 1857–1868 (2016).
Melief, J. et al. Phenotyping primary human microglia: Tight regulation of LPS responsiveness. Glia 60, 1506–1517 (2012).
Mizee, M. R. et al. Isolation of primary microglia from the human post-mortem brain: effects of ante- and post-mortem variables. Acta. Neuropathol. Commun. 5, 16 (2017).
Olah M., et al. A transcriptomic atlas of aged human microglia. Nat. Commun. 2018; 9. https://doi.org/10.1038/s41467-018-02926-5.
de Haas, A. H., Boddeke, H. W. G. M., Brouwer, N. & Biber, K. Optimized isolation enables ex vivo analysis of microglia from various central nervous system regions. Glia 55, 1374–84 (2007).
Moore, C. S. et al. P2Y12 expression and function in alternatively activated human microglia. Neurol. Neuroimmunol. Neuroinflamm. 2, e80 (2015).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017).
Emsell, L. & McDonald, C. The structural neuroimaging of bipolar disorder. Int Rev. Psychiatry 21, 297–313 (2009).
Price, J. L. & Drevets, W. C. Neurocircuitry of mood disorders. Neuropsychopharmacology 35, 192–216 (2010).
Böttcher, C. et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat. Neurosci. 22, 78–90 (2019).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Vandensompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometricaveraging of multiple internal control genes. Genome Biol. 18, 7 (2002).
Satoh, Jichi et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology 36, 39–49 (2016).
Darby, M. M., Yolken, R. H. & Sabunciyan, S. Consistently altered expression of gene sets in postmortem brains of individuals with major psychiatric disorders. Transl. Psychiatry 6, e890 (2016).
Akula, N. et al. RNA-sequencing of the brain transcriptome implicates dysregulation of neuroplasticity, circadian rhythms and GTPase binding in bipolar disorder. Mol. Psychiatry 19, 1179–85 (2014).
Cruceanu, C. et al. Transcriptome sequencing of the anterior cingulate in bipolar disorder: dysregulation of g protein-coupled receptors. Am. J. Psychiatry 172, 1131–1140 (2015).
Pacifico, R. & Davis, R. L. Transcriptome sequencing implicates dorsal striatum-specific gene network, immune response and energy metabolism pathways in bipolar disorder. Mol. Psychiatry 22, 441–449 (2017).
Gandal M. J., et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science (80-) 362, 2018. https://doi.org/10.1126/science.aat8127.
Gandal, M. J. et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science (80-) 359, 693–697 (2018).
Keren-shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’ s disease article a unique microglia type associated with restricting development of Alzheimer’ s disease. Cell 169, 1–15 (2017).
Duivis, H. E., Vogelzangs, N., Kupper, N., de Jonge, P. & Penninx, B. W. J. H. Differential association of somatic and cognitive symptoms of depression and anxiety with inflammation: findings from the Netherlands Study of Depression and Anxiety (NESDA). Psychoneuroendocrinology 38, 1573–85 (2013).
Lamers, F., Milaneschi, Y., de Jonge, P., Giltay, E. J. & Penninx, B. W. J. H. Metabolic and inflammatory markers: associations with individual depressive symptoms. Psychol. Med. 48, 1102–1110 (2018).
Suzumura, A. Neuron-microglia interaction in neuroinflammation. Curr. Protein Pept. Sci. 14, 16–20 (2013).
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S. & Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–80 (2009).
Tremblay, M.-È., Lowery, R. L. & Majewska, A. K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8, e1000527 (2010).
Van Kesteren et al. Immune involvement in the pathogenesis of schizophrenia: A meta-analysis on postmortem brain studies. Transl. Psychiatry 2017; 7. https://doi.org/10.1038/tp.2017.4.
Bloomfield, P. S. et al. The effects of haloperidol on microglial morphology and translocator protein levels: An in vivo study in rats using an automated cell evaluation pipeline. J. Psychopharmacol. 32, 1264–1272 (2018).
Watkins C. C., Sawa A., Pomper M. G. Glia and immune cell signaling in bipolar disorder: Insights from neuropharmacology and molecular imaging to clinical application. Transl. Psychiatry. 4, 2014. https://doi.org/10.1038/tp.2013.119.
Hansen, D. V., Hanson, J. E. & Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472 (2018).
von Bernhardi R., Eugenín-von Bernhardi L., Eugenín J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015; 7.https://doi.org/10.3389/fnagi.2015.00124.
Streit W. J., Xue Q. S., Tischer J., Bechmann I. Microglial pathology. Acta Neuropathol Commun 2, 2014. https://doi.org/10.1186/s40478-014-0142-6.
Steiner, J. et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J. Psychiatr. Res. 42, 151–157 (2008).
Ohgidani, M. et al. Microglial CD206 gene has potential as a state marker of bipolar disorder. Front Immunol. 7, 676 (2016).
Acknowledgements
This project is financially supported by a 2014 NARSAD Young Investigator Grant provided by the Brain & Behaviour Research Foundation. This study was further supported by the psychiatric donor program of the Netherlands Brain Bank (NBB-Psy), which is supported by the Netherlands Organization for Scientific Research (NWO). Funding was also provided by the a/Mental Illness, Education, and Clinical Center (MIRECC) of the JJPVA Medical Center in the Bronx, NY. The authors thank the team of the Netherlands Brain Bank for their services and P.R. Ormel and Y. He for their help with the microglial isolations and LPS response experiments.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sneeboer, M.A.M., Snijders, G.J.L.J., Berdowski, W.M. et al. Microglia in post-mortem brain tissue of patients with bipolar disorder are not immune activated. Transl Psychiatry 9, 153 (2019). https://doi.org/10.1038/s41398-019-0490-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0490-x
This article is cited by
-
Biomarkers of Bipolar Disorder in Late Life: An Evidence-Based Systematic Review
Current Psychiatry Reports (2024)
-
The role of microglia in neuropsychiatric disorders and suicide
European Archives of Psychiatry and Clinical Neuroscience (2022)
-
Glial Cell Abnormalities in Major Psychiatric Diseases: A Systematic Review of Postmortem Brain Studies
Molecular Neurobiology (2022)
-
Human microglial models to study HIV infection and neuropathogenesis: a literature overview and comparative analyses
Journal of NeuroVirology (2022)
-
Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies
Nature Genetics (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}