
Abstract
Individuals affected with different neuropsychiatric disorders such as autism (AUT), schizophrenia (SCZ) and bipolar disorder (BPD), may share similar clinical manifestations, suggesting shared genetic influences and common biological mechanisms underlying these disorders. Using brain transcriptome data gathered from postmortem donors affected with AUT, SCZ and BPD, it is now possible to identify shared dysregulated gene sets, i.e., those abnormally expressed in brains of neuropsychiatric patients, compared to non-psychiatric controls. Here, we apply a novel aberrant gene expression analysis method, coupled with consensus co-expression network analysis, to identify gene sets with shared dysregulated expression in cortical brains of individuals affected with AUT, SCZ and BPD. We identify eight gene sets with dysregulated expression shared by AUT, SCZ and BPD, 23 by AUT and SCZ, four by AUT and BPD, and two by SCZ and BPD. The identified genes are enriched with functions relevant to amino acid transport, synapse, neurotransmitter release, oxidative stress, nitric oxide synthase biosynthesis, immune response, protein folding, lysophosphatidic acid-mediated signaling and glycolysis. Our method has been proven to be effective in discovering and revealing multigene sets with dysregulated expression shared by different neuropsychiatric disorders. Our findings provide new insights into the common molecular mechanisms underlying the pathogenesis and progression of AUT, SCZ and BPD, contributing to the study of etiological overlap between these neuropsychiatric disorders.
Similar content being viewed by others


Introduction
Autism (AUT), schizophrenia (SCZ) and bipolar disorder (BPD) are three major neuropsychiatric disorders. AUT patients present with impairments in social interaction and communication, and repetitive and restricted behaviors. SCZ is characterized by delusions, hallucinations, disordered thoughts and blunted affect. The symptoms of BPD include recurrent mania and depression, frequently with delusions. Patients with these severe neuropsychiatric disorders share similar behavioral, social, cognitive, and perceptual impairments. Up to 30% of individuals diagnosed with AUT during childhood will develop SCZ in adulthood1. The presence of SCZ or BPD in first-degree relatives is a consistent and significant risk factor for AUT2. Similarity in clinic symptoms, as well as shared genetic influences, between AUT, SCZ and BPD, have been the focus of several recent studies3,4,5,6,7.
Genetic studies have identified genetic variants that contribute to the risk of developing AUT8,9,10,11,12,13,14, SCZ15,16,17,18 and BPD19,20,21,22. However, for any of these disorders, it remains elusive how these reported variants lead to the pathogenesis of disorder. One possibility is that these variants cause gene expression alternations in brain (during a certain stage or across several stages of brain development) and eventually lead to the neuropsychiatric disorders. Measuring and analyzing gene expression information in postmortem brains is thus valuable for understanding the pathogenesis of neuropsychiatric disorders. The availability of samples from brains of diseased cases and healthy controls makes it possible to measure the gene expression from this primarily affected organ for understanding AUT23,24, SCZ25,26 and BPD27,28. To study the effect of genetic correlation in these disorders, some researchers focused on studying the overlap of gene expression alterations between diseases29,30,31,32. For example, a transcriptome analysis of anterior cingulate cortex samples from SCZ, BPD and controls was conducted by Zhao et al.31. They showed the gene expression of SCZ and BPD are correlated, and dysregulation of cytoskeleton remodeling and lysosomal function underlies the common causes of SCZ and BPD. In a more recent study, Ellis et al. integrated and analyzed the transcriptomic RNA-sequencing data of cortex samples of AUT, SCZ, BPD and controls combined from two previous studies24,31. Ellis et al. used univariate analysis method, i.e., single gene-based differential expression analysis, to first identify differentially expressed genes associated with each disease, and then obtained a list of differentially expressed genes shared by those disorders. They reported the transcriptomes of AUT and SCZ are correlated, and altered neurotransmission and synapse regulation are shared between these two disorders32.
To reveal shared dysregulated gene sets between AUT, SCZ and BPD, herein we re-analyze the gene expression data of cortical brain tissue samples from AUT, SCZ, BPD and healthy controls32 but applying multivariate analysis methods. We construct gene co-expression networks to find consensus modules shared across disorders. To consider the gene expression dispersion, we apply aberrant gene expression analysis to identify shared gene sets tend to be aberrantly expressed across diseases. By analyzing the shared dysregulated gene sets, we evaluate the extent of similarity between gene expression of AUT, SCZ and BPD and gain better understanding the downstream impact of genetic overlap in these neuropsychiatric disorders.
Materials and methods
Gene expression data
The gene expression data of 104 cortical brain tissue samples (47 AUT and 57 controls), which was normalized using conditional quantile normalization (CQN)33 to remove technical variability, was obtained from the study of Ellis et al.32. We denoted it as data I. These 104 samples were from three brain regions, of which 62, 14 and 28 were from cerebral cortex (BA 19), anterior prefrontal cortex (BA 10), and a part of the frontal cortex (BA 44) respectively, involving 40 healthy and 32 autistic individuals. The transcriptomes of the 104 samples were originally sequenced by Gupta et al.24. The CQN normalized gene expression data of 82 anterior cingulate cortex (BA24) samples (involving 31 SCZ patients, 25 BPD patients, and 26 controls) was also obtained from the study of Ellis et al.32, which we denoted as data II. These samples were originally from Stanley Medical Research Institute (SMRI) and the whole transcriptome sequencing was performed by Zhao et al.31. For these two datasets (data I and data II), the sequencing reads were subjected to a common pre-processing pipeline for obtaining the gene expression data32.
In this study, we first combined data I and data II, and processed the combined data using the algorithm of ComBat34 for removing the effect caused by different data sources and regressing out the covariates (age, gender and brain region). Next, we applied the algorithm of probabilistic estimation of expression residuals (PEER)35 to discover up to 20 possible hidden determinants of expression variation and then regressed out the hidden factors that were uncorrelated with disease status. The lowly expressed genes with expression median < 2 were excluded. The final data matrix contains the expression level of 8485 protein-coding genes in 186 samples (47 AUT, 31 SCZ, 25 BPD and 83 controls).
Curated gene sets
The gene sets (n = 17,786) used in the Gene Set Enrichment Analysis (GSEA) were obtained from the molecular signatures database (MSigDB v6.1)36. The GSEA gene sets include 50 hallmark gene sets, 326 positional gene sets, 4738 curated gene sets, 836 motif gene sets, 858 computational gene sets, 5917 GO gene sets, 189 oncogenic signatures and 4872 immunologic signatures. A total of 1007 AUT-candidate genes were downloaded from gene scoring module in Simons Foundation Autism Research Initiative (SFARI), which includes 68, 25, 59, 176, 406, 157, and 21 genes from categories S (syndromic), 1 (high confidence), 2 (strong candidate), 3 (suggestive evidence), 4 (minimal evidence), 5 (hypothesized but untested) and 6 (evidence does not support a role). We downloaded 2752 genes associated with SCZ from SZDB37, database for schizophrenia genetic research, and these genes were identified by different kinds of studies including convergent functional genomics, CNV, differentially expression, GWAS, genetic linkage and association studies, Sherlock integrative analysis and Pascal gene-based test. We also downloaded 599 BPD-associated genes from BDgene database38, and each gene is positively supported by at least one kind of studies.
Aberrant gene expression analysis
The aberrant gene expression analysis39 is a multivariate method, which adopts Mahalanobis distance (MD)40 to quantify the dissimilarity in multigene expression patterns between diseased samples and control group. Here we firstly applied aberrant gene expression analysis to identify gene sets that may be expressed aberrantly in each of the three disorders (AUT, SCZ and BPD). Specifically, based on the final data matrix including 186 samples (47 AUT, 31 SCZ, 25 BPD and 83 controls), we first calculated, for each disorder and each given gene set, the MD from each diseased sample i to the robust multivariate centroid of control group (including 83 controls), denoted as MDi. MD measures the number of standard deviations from case sample to the robust mean of controls. To reduce the influence of possible outliers in control group, we adopted the algorithm of Minimum Covariance Determinant (MCD)41 to obtain the robust location estimator (expression mean) and scattering estimator (covariance matrix) of the controls. The MCD algorithm subsamples h observations (set h = 0.8n, where n is the number of controls) whose covariance matrix had the smallest covariance determinant, and the MCD robust estimates of location and scattering were imputed from these h controls. Then MDi was calculated as:
where xi∙ is the vector of gene expression levels for diseased sample i, yc is the vector of expression means of genes across h control samples, and ψc is the covariance matrix estimated from the h controls.
Next, the sum of squared MDi, denoted as SSMD:
was calculated to measure the overall dispersion of m cases to the robust centroid of control group. To assess the significance of SSMD of a given gene set, we performed permutation tests using N randomly reconstructed gene sets with the same size. As SSMD measures the overall dispersion of cases relative to the control group, even if the robust centroid of controls may change when we performed permutation tests, but it would not affect the calculation of a relative measure, i.e., SSMD. The P-value of permutations, Pperm, was determined by M/N, where M is the number of random gene sets whose SSMD values are greater than that of the given gene set, N is the total number of random gene sets. The correction for multiple testing was performed by controlling the false discovery rate (FDR) with the Benjamini–Hochberg method42.
To assess the relative contribution of each gene in a significant gene set to the total SSMD, we calculated the difference between the total SSMD value and the SSMD value calculated after the gene was excluded from the gene set, which we denoted as ΔSSMD. The ΔSSMD of gene j was calculated as:
Then we sorted the genes by their ΔSSMD values for a significant gene set.
Clustering of aberrantly expressed gene sets
For any pair of significant aberrantly expressed gene sets a and b, we adopted Jaccard distance43 to measure their dissimilarity, which was calculated as:
where J(a, b) is Jaccard similarity coefficient and is defined as the number of genes in the intersection of a and b divided by the number of genes in the union of a and b. Based on the Jaccard distance, we generated a hierarchical cluster tree to group the gene sets whose distances are less than 0.3.
Consensus gene co-expression network analysis
In addition to aberrant gene expression analysis, we also performed gene co-expression network analysis. We first split the final data matrix into three sub-datasets, denoted as datasets 1, 2 and 3, each containing the data for a disorder and their respective controls. Then weighted gene co-expression network analysis (WGCNA)44 was applied to find consensus modules between datasets 1, 2 and 3. The signed consensus modules were built using the function of blockwiseConsensusModules in WGCNA package45. Modules were defined using biweight midcorrelation (bicor) which is more robust to outliers compared to Pearson correlation46, along with the soft-threshold power of 5 for all datasets achieving approximate scale-free topology (R2 > 0.8), minimum module size of 20 and FALSE pamStage. The module eigengene (the first principal component) was used to represent the expression level of each consensus module and was associated with the traits (age, gender, and disease status) to compute the correlation coefficients and P-values. The P-values were then corrected by controlling the FDR with the Benjamini–Hochberg method42. Gene ontology analysis was performed using the database for annotation, visualization and integrated discovery (DAVID)47,48. Gene lists of modules were uploaded, and the GO terms whose Bonferroni-adjusted P-value < 0.05 were reported as significant in the ‘Functional Annotation Chart’ generated using the minimum number of genes involved in the term of 10.
Results
Analysis workflow
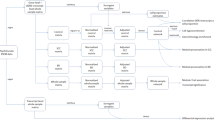
Figure 1 describes the analysis workflow. The analyses of this study were conducted with the gene expression datasets, normalized using conditional quantile normalization (CQN)33 to remove technical variability, from the study of Ellis et al.32, including (1) the gene expression data of 104 cortical brain tissue samples (47 AUT and 57 controls), here denoted as data I, and (2) the gene expression data of 82 anterior cingulate cortex samples (31 SCZ, 25 BPD, and 26 controls), denoted as data II. Firstly, we combined data I and data II and processed the combined data with the algorithm of ComBat34 to remove batch effect. Then the data was subjected to the algorithm of probabilistic estimation of expression residuals (PEER)35 to regress out up to 20 possible hidden factors that were uncorrelated with disease status (Materials and Methods). The final data matrix contains the expression level of 8,485 protein-coding genes in 186 samples (47 AUT, 31 SCZ, 25 BPD and 83 controls) (Supplementary Data 1).

The analysis workflow used in parallel, including aberrant gene expression analysis and consensus gene co-expression analysis
Next, we applied the aberrant gene expression analysis39 to identify significant gene sets for each disorder and then obtained shared ones across disorders by generating a hierarchical cluster tree. In addition, we used weighted gene co-expression network analysis (WGCNA)44 to find consensus modules shared across disorders. We then compared the results from these two kinds of analysis methods, aberrant gene expression analysis and WGCNA. By analyzing the identified shared sets of dysregulated genes, we assessed the commonality between AUT, SCZ, and BPD.
Shared aberrantly expressed gene sets across disorders
Dysregulated gene expression is characterized by increased level of expression dispersion between individuals23,39. To assess the gene expression dispersion in diseased individuals, we used aberrant gene expression analysis method39 to identify gene sets tend to be aberrantly expressed in AUT, SCZ and BPD separately based on the final data matrix including 47 AUT, 31 SCZ, 25 BPD samples and 83 controls (Materials and Methods). From 17,786 GSEA (Gene Set Enrichment Analysis) gene sets36, 156, 102, 51 gene sets were identified to be associated with AUT, SCZ and BPD respectively (P-value ≤ 0.01) (Fig. 2). To consider the effect of different case sample sizes for different diseases, we re-run the aberrant gene expression analysis by using a same diseased sample size (i.e., the minimum sample size among AUT, SCZ and BPD cases, which is equal to 25). In specific, we randomly picked 25 case samples from all AUT cases, and used their gene expression to perform aberrant gene expression analysis. This process was repeated for 100 times, and we found that the average number of identified gene sets associated with AUT is 137. For SCZ, we also performed aberrant gene expression analysis for 100 times by using the gene expression of 25 randomly selected SCZ cases in each time. We found that the average number of identified gene sets associated with SCZ is 97. It can be seen that after we controlled the effect of different case sample sizes, the differences in the number of identified gene sets associated with different disorders still exist, especially between AUT and BPD, SCZ and BPD. It may because for our analyzed gene expression data AUT cases tend to show disruption in more gene sets or more biological functions relative to SCZ, and especially BPD.

The Venn diagram shows the number of identified aberrantly expressed gene sets associated with AUT, SCZ, and BPD
Then we clustered the significant gene sets (156, 102 and 51 gene sets associated with AUT, SCZ and BPD respectively) to obtain shared ones by more than one disorders (Materials and Methods). As a result, there are 21 gene sets shared by AUT and SCZ, four shared by AUT and BPD, one shared by SCZ and BPD, four shared by all three disorders (Fig. 2). The shared GSEA gene sets are listed in Table 1. When the cutoff of P-value was set to 0.05, 119 gene sets are shared by AUT and SCZ, 44 shared by AUT and BPD, 25 shared by SCZ and BPD, 31 shared by all three disorders (Supplementary Table 1). To assess the overlap between genes in identified gene sets and disease-associated genes, we downloaded 1,007 AUT-associated genes from Simons Foundation Autism Research Initiative (SFARI), 2752 SCZ candidate genes from SZDB37 and 599 BPD candidate genes from BDgene38 (Materials and Methods). Many dysregulated gene sets are enriched with disease-associated genes (Table 1). To measure the relative contribution of each gene in a significant gene set, we calculated ΔSSMD value of each gene (Materials and Methods) and sorted the genes by their ΔSSMD values. For each disease, the genes with top three ΔSSMD values are listed for each significant gene set, and the ones overlapping with disease-associated genes are bold (Table 1).
Shared gene sets involved in amino acid transport
In Table 1, five gene sets, associated with AUT and SCZ, are related to amino acid transport including GO_AMINO_ACID_TRANSMEMBRANE_TRANSPORTER_ACTIVITY, GO_NEUTRAL_AMINO_ACID_TRANSPORT, GO_L_AMINO_ACID_TRANSMEMBRANE_TRANSPORTER_ACTIVITY, GO_L_AMINO_ACID_TRANSPORT and REACTOME_AMINO_ACID_TRANSPORT_ACROSS_THE_PLASMA_MEMBRANE. This is in line with the known link between amino acid transport and AUT or SCZ—amino acids are essential in cognitive functioning and brain development49. For example, increased transport capacity of alanine across the cell membrane and decreased affinity for transport sites of tyrosine may be associated with the development of AUT in children, demonstrating the disturbances in transport mechanisms for amino acids at the membrane level may influence the transport of amino acids across the blood brain barrier49. The solute carrier transporter 7a5 (SLC7A5), a large neutral amino acid transporter localized at the blood brain barrier, is important in maintaining normal levels of brain branched chain amino acids; the abnormalities in branched-chain amino acid catabolic pathway may cause AUT50. The aberrant amino acid transport activities, such as the aberrant tyrosine transport across the cell membrane51 and excitatory amino acid transport52, were also found in patients with SCZ.
In these five shared gene sets, genes CLN8, KCNJ10, SLC1A1, SLC1A2, SLC25A12, SLC38A10, SLC6A1, SLC6A8, SLC7A3, SLC7A5, and SLC7A7 are overlapped with SFARI AUT-associated genes. Nine of them belong to SLC family, of which SLC1A2 and SLC6A1 belong to category S (syndromic), and SLC6A1 is a strong candidate (category 2) for AUT. Among these genes, SLC1A1 and SLC1A2 are also associated with SCZ. In particular, gene SLC1A2 have been linked to SCZ by studies of convergent functional genomics, linkage and association, and differential expression analysis according to the explanation in SZDB37. There are 15 more genes associated with SCZ including ARL6IP5, OCA2, SLC16A12, SLC1A3, SLC1A4, SLC32A1, SLC38A5, SLC38A7, SLC3A1, SLC43A2, SLC6A11, SLC6A17, SLC7A1, SLC7A4, and SLC7A6. Thirteen of these gene belong to SLC family, of which SLC1A3, SLC32A1 and SLC38A7 are strong candidates for SCZ. Most disease-associated genes in the gene sets belong to SLC family, implying the important role of SLC in AUT and SCZ.
Shared gene sets involved in synapse and neurotransmitter release
Identified gene sets shared by AUT and SCZ also include REACTOME_NEUROTRANSMITTER_RELEASE_CYCLE, GO_SYNAPTIC_VESICLE_RECYCLING, GO_SYNAPTIC_VESICLE_ ENDOCYTOSIS, GO_GLUTAMATE_SECRETION, GO_NEGATIVE_REGULATION_OF_ CATECHOLAMINE_SECRETION, and GO_INHIBITORY_SYNAPSE. The function of brains is dependent on neurotransmission and its alteration is linked to neuropsychiatric disorders. Neurotransmitter release requires the rapid recycling of synaptic vesicles by endocytosis. Evidence has shown synaptic vesicle recycling and endocytosis are related to AUT and SCZ53,54,55,56. In a study57, the identified pathways related to SCZ involved neuronal systems and one hub centered around the neurotransmitter release cycle including the release cycles for dopamine, serotonin, norepinephrine and glutamate neurotransmitters. Glutamate is the main excitatory neurotransmitter of brains and may be a key neurotransmitter involved in neuropsychiatric disorders58. Glutamatergic dysfunction is a possible mechanism of AUT59,60 and SCZ61,62. Catecholamines are monoamine neurotransmitters, including epinephrine, norepinephrine and dopamine. Studies in human neural stem cell models indicated that the dysregulation in catecholamine secretion may contribute to the pathogenesis of AUT63,64. It has also been demonstrated that SCZ cases show increased secreted catecholamine and SCZ neuronal cultures show a higher percentage of tyrosine hydroxylase-positive neurons, the first enzymatic step for catecholamine biosynthesis65. There also have been studies demonstrating the presence of excitatory and inhibitory imbalance in AUT and SCZ66,67, and SCZ-associated cortical inhibitory neurons68, thus GO_INHIBITORY_SYNAPSE is also likely a related gene set.
In these six shared gene sets, 26 genes are associated with AUT, of which ALDH5A1, NF1, NTRK2, SLC1A2, SLC6A1 and STXBP1 belong to category S (syndromic), NLGN3, RIMS1 and SLC6A1 belong to category 2 (strong candidate), CNR1, OPHN1 and STXBP1 belong to category 3 (suggestive evidence) in SFARI. There are 26 genes associated with SCZ, of which DRD2, CNR1 and SLC1A2 are the top three strongest candidates, and genes GABBR1, GAD1, IGSF9B, MAOA, SLC1A3, SLC32A1 and SYN2 have been also linked to SCZ by two kinds of studies in SZDB. Thirteen genes are associated with both AUT and SCZ, including CNR1, DRD2, GAD1, MAOA, NTRK2, OPHN1, PPFIA1, RIMS1, SLC1A1, SLC1A2, SNAP25, STX1A and SYN2.
Shared gene sets involved in oxidative stress and nitric oxide synthase biosynthesis
Two gene sets involved in oxidative stress induced apoptosis were identified to be associated with AUT and SCZ, including GO_REGULATION_OF_OXIDATIVE_STRESS_INDUCED_INTRINSIC_APOPTOTIC_SIGNALING_PATHWAY and GO_NEGATIVE_REGULATION_OF_OXIDATIVE_STRESS_INDUCED_INTRINSIC_APOPTOTIC_SIGNALING_PATHWAY. The intrinsic apoptotic pathway can be activated following oxidative and peroxidative damage driven by excessive levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS)69. When the levels of ROS exceed the antioxidant capacities of a cell, oxidative stress occurs which often leads to the death of a cell70. There have been studies showing that oxidative stress is higher in AUT70 and SCZ71. It also has been shown that oxidative stress can affect the apoptosis of neurons as a mediator in neuropsychiatric disorders, such as SCZ72.
Gene sets, associated with nitric oxide synthase biosynthesis, GO_POSITIVE_REGULATION_OF_ NITRIC_OXIDE_SYNTHASE_BIOSYNTHETIC_PROCESS and GO_REGULATION_OF_NITRIC_ OXIDE_SYNTHASE_BIOSYNTHETIC_PROCESS, are also related to AUT and SCZ. There have been some clues showing the link between nitric oxide synthase biosynthesis and neuropsychiatric disorders. The unbalance between antioxidant capacity and oxidative stress leads to an excess of RNS, such as nitric oxide (NO)73. NO, produced by NO synthase (NOS), modulates short-term and long-term synaptic plasticity and is essential in the regulation of many physiological processes such as neurotransmitter release, neuronal excitability, long-term potentiation and neurovascular coupling74. NO affects the function of ROS in the local cellular environment, in which biological antioxidants are present. NO shows both neuroprotective and neurotoxic effects. As the disease progresses, NO can depend on the adaptive functions of the antioxidant capacity and oxidative stress-related ROS/RNS73. Given the roles of NO, its alteration may lead to the neurodevelopmental changes associated with neuropsychiatric diseases. Indeed, several studies have indicated that NO or NOS is involved in the pathogenesis of many neuropsychiatric disorders including AUT74,75,76,77 and SCZ71.
In these four gene sets, there are two genes associated with AUT, including GPX1 (glutathione peroxidase 1) and SOD1 (superoxide dismutase 1), both of which belong to category 4 (minimal evidence). There are 12 genes associated with SCZ, of which BAG5 and CPEB1 have been linked to SCZ by two kinds of studies in SZDB. Checking the function of these genes, we found some of them are indeed involved in oxidative stress. The protein encoded by gene GPX1 catalyzes the reduction of organic hydroperoxides and hydrogen peroxide by glutathione, protecting cells against oxidative damage. The protein encoded by gene SOD1 is an isozyme responsible for destroying free superoxide radicals in the body, converting naturally generating but harmful superoxide radicals to molecular oxygen and hydrogen peroxide. The protein encoded by gene BAG5 belongs to BAG1-related protein family, and BAG1 is an anti-apoptotic protein interacting with a variety of proteins involved in cell apoptosis and growth.
Shared gene sets involved in immune response
Gene sets GO_CELL_ACTIVATION_INVOLVED_IN_IMMUNE_RESPONSE and GO_LYMPHOCYTE_ACTIVATION_INVOLVED_IN_IMMUNE_RESPONSE were identified associated with AUT and BPD. Abnormal immunological phenomena have been noted in AUT, involving cytokines, immunoglobulins, inflammation and cellular activation. The increased pro-inflammatory cytokines in brains and the activation of resident immune cells known as microglia in AUT individuals, may interfering with the development and function of normal brains, potentially lead to AUT78. In a study79, the finding suggested immune activation, including activation of T-lymphocyte subsets, may be essential in modulating and potentially improving behaviors in some AUT patients. It was also found that AUT individuals often have alterations in immune cells, immunoglobulins and autoantibodies. For BPD, the immunological dysfunction was also described, including distinct immune cell profile, release of/altered cytokines by stimulated mononuclear cells, elevated levels of circulating immune markers, inflammatory changes in the central nervous system80 and the expansion of activated T cells81.
These two shared gene sets contain 10 SFARI AUT candidate genes, and four genes associated with BPD. The 10 genes are ADA, CX3CR1, FOXP1, IFNG, IL6, KIT, LAT, PIK3CG, RORA and TSC1. TSC1 belong to category S, and FOXP1 is also a strong candidate for AUT. There are two genes associated with both AUT and BPD, which are IFNG and RORA. Both belong to category 5 (hypothesized but untested) in SFARI; nevertheless, the link between these genes and AUT, especially for RORA, is well supported82,83,84. Among the four genes associated with BPD, RORA and IFNG also appear as the strongest candidates for BPD85,86,87.
Shared gene sets involved in protein folding
Two gene sets, GO_DE_NOVO_POSTTRANSLATIONAL_PROTEIN_FOLDING and GO_DE_NOVO_PROTEIN_FOLDING, were identified to be associated with AUT, SCZ and BPD. There have been some clues showing the link between protein folding and neuropsychiatric disorders. Neuroligins (NLs), postsynaptic cell-adhesion molecules, are essential for the normal function of synapse. Mutations in neuroligin-4 (NL4) (gene symbol: NLGN4) have been associated with AUT, for instance R87W substitution (a single amino-acid substitution in NL4)88. R87W substitution, a loss-of-function mutation, impairs the normal folding of NL4, completely traps NL4 in the endoplasmic reticulum and blocks NL4 transport to the cell surface. As a result, the synapse formation activity of NL4 will be inactivated and the functional effect of NL4 on synapse strength will be blocked88.
In the cellular environment, molecular chaperones are required to ensure the correct folding of many other proteins89. Heat shock protein (HSP) is a kind of molecular chaperone targeting misfolded proteins that accumulate in response to cellular stress, facilitating protein refolding and targeting damaged proteins for degradation in proteasomes90. HSPs play an essential role in the development of neuropsychiatric disorder, such as HSP70 (heat shock protein-70). HSP70 participates in many cellular processes including protein folding, transport across membranes, prevention of protein aggregation and degradation. Genetic variations of HSP70 have been associated with the presence of SCZ91. Looking at the genes contained in these two gene sets, we found seven genes are associated with SCZ, including CHCHD4, DNAJB1, HSPA8, HSPD1, HSPE1, HSPH1, TOR1A. Over half of these genes belong to HSP family, implying the important role of HSP in SCZ.
Shared gene sets involved in miscellaneous functions
Gene set PID_LPA4_PATHWAY was identified associated with all three diseases. Lysophospholipids (LPs) are an important family of lipid signaling molecules, and lysophosphatidic acid (LPA) is a major member of this family within the nervous system. LPA effects are now known to act through cognate, cell-surface G protein-coupled receptors termed LPA receptors (LPARs). There are currently six LPARs: protein names LPA1-6, gene names LPAR1-6. Large evidence has been shown that LPA1 signaling is essential in normal cognition. For instance, LPAR1 null mice display a variety of negative behavioral signs and cognitive deficits including the traits commonly seen in AUT and SCZ patients. LPA is also known to reduce glutamate uptake involving an LPA1-independent mechanism while glutamatergic signaling alterations are implicated in AUT, SCZ and other related neuropsychiatric disorders92. LPA1 has been particularly associated with neuropsychiatric disorders, while other LPA receptor subtypes may also have disease relevance93, which may provide new insights into complex neuropsychiatric disorders. In the gene set, genes ADCY3 and ADCY5 are associated with AUT; ADCY8 and RPS6KA5 are associated with SCZ; ADCY2, ADCY8, ADCY9, and CREB1 are associated with BPD.
Gene set REACTOME_GLYCOLYSIS was identified associated with AUT and SCZ. The majority of glucose metabolites are significantly disturbed in SCZ patients, suggesting the disturbance of glucose metabolism may be implicated in SCZ94,95. Additionally, the findings of a recent study in 2018 have also suggested an elevation of glycolysis through the phenomenon of aerobic glycolysis in AUT, while the dysregulation of aerobic glycolysis had been proposed as a candidate cause of AUT96. In the gene set, genes PPP2R1B and PPP2R5D belong to category 4 (minimal evidence) in SFARI, and PPP2R5D also belongs to category S. Genes ALDOA, ALDOB, PFKFB1, PGK1 and PPP2CA are linked to SCZ.
Gene set GO_CAMP_BIOSYNTHETIC_PROCESS was associated with AUT and BPD. cAMP (cyclic adenosine monophosphate, or cyclic AMP) is a second messenger, which is important in many biological processes. There have been studies showing the link between cAMP and AUT, such as the study showing reduced cAMP induction may be a cause of fragile X and AUT97, and the study pointing out the role of cAMP pathology in AUT98. Furthermore, a genetic association study of cAMP signaling genes with BPD found several statistically significant single-SNP associations and SNP-SNP associations with BPD, suggesting that variants in several cAMP signaling pathway genes increase the risk of BPD99. In the gene set, genes ADCY3 belongs to category 3 (suggestive evidence), and ADCY5 and ADORA2A belong to category 4 (minimal evidence) in SFARI. Genes ADCY2, ADCY8, ADCY9, and ADM are associated with BPD.
Gene set ST_PAC1_RECEPTOR_PATHWAY is associated with AUT and SCZ. PAC1 is a receptor of PACAP (pituitary adenylate cyclase-activating polypeptide, a neuropeptide with neurotransmission modulating activity). PACAP and its receptor PAC1 are important for the development and function of brains, psychiatric conditions and stress response100. Studies raised the potential relation between PACAP signaling dysfunctions and neuropsychiatric disorders characterized by social reciprocity impairments such as AUT101. PAC1 gene contains many putative splicing factor recognition sites which might be activated at different stages of neuronal activation. PAC1 signaling controls many cellular and physiological responses, such as proliferation, differentiation, cell cycle regulation, neurotransmitter, and hormone release and adaptation to stressful challenges102. The regulation of PAC1 splicing and its outcomes might be relevant to the etiology of some neurological and psychiatric disorders102. A genetic study103 showed that the variants of the genes encoding PACAP and PAC1 receptor are associated with SCZ. The transcriptome sequencing of the cortex of SCZ patients revealed significant differences in the alternative splicing of PAC1 receptor104. The evidences above show the link of PAC1 receptor pathway to neuropsychiatric disorders.
Results obtained using consensus co-expression analysis
In addition to aberrant gene expression analysis, we performed gene co-expression analysis using WGCNA44. Specifically, after obtaining final data matrix containing the expression level of 8,485 protein-coding genes in 186 samples (47 AUT, 31 SCZ, 25 BPD and 83 controls), we firstly split the data into three sub-datasets, denoted as datasets 1, 2, and 3, each of which contains respective cases and controls (Fig. 1). Dataset 1 contains the gene expression level of 47 AUT and 57 controls from data I; dataset 2 contains 31 SCZ and 26 controls from data II; dataset 3 contains 25 BPD and 26 controls from data II. Note that, SCZ and BPD cases are originally from data II, so the respective controls are also from data II. Next, we applied WGCNA to find consensus modules between datasets 1, 2 and 3, and 22 modules were identified (Materials and Methods, Fig. 3a). As the 26 controls included in datasets 2 and 3 are shared, we used a strategy similar to that in the study of Ellis et al.32, where these controls were split randomly into two halves. Specifically, we divided the 26 controls randomly into two halves, and one half was assigned to a new dataset 2 along with all SCZ cases, and another half was assigned to a new dataset 3 along with all BPD cases. Then we reconstructed networks to find consensus modules with the same parameters between dataset 1, new datasets 2 and 3. The procedure above was repeated for 100 times. To demonstrate the robustness of the consensus modules built in full data with datasets 2 and 3 containing 26 controls respectively, we used a similar method in a previous study46 showing the clustering tree (dendrogram) of genes together with consensus modules built from the full data and the 100 ones built from the resampled data with new datasets 2 and 3 containing 13 controls respectively (Supplementary Figure 1). Most of the 22 identified consensus modules built in full data are robust and can be identified in most or all resampled data sets, so we used these 22 consensus modules as the final module assignments.

a The clustering tree (dendrogram) of genes and the identified consensus modules. The mentioned modules (M1, M2, M3, M4, M8, M11 and M22) in the main text are marked. For these modules, b shows the correlation between module eigengenes and disease status. FDR-corrected P < 0.001(***); <0.01 (**); <0.05 (*); <0.1 (#). c The top three significant GO terms enriched in responding modules are shown with Bonferroni adjusted P-values
We associated each consensus module with traits (age, gender and disease status) (Materials and Methods) and found 17 modules are shared by more than one diseases (FDR-corrected P-value < 0.1) (Supplementary Table 2). We used the database for annotation, visualization and integrated discovery (DAVID)47,48 to perform gene ontology analysis and seven modules left were enriched in GO categories. Modules M1, M2, M4 and M11 are associated with all disorders; M3 and M22 are shared by AUT and SCZ; M8 is shared by SCZ and BPD (Fig. 3b). The top three significant GO terms enriched in these modules include neuron part, synapse, neuron projection, adherens junction, biological adhesion, anchoring junction, poly(A) RNA binding, RNA binding, nuclear part, nervous system development, mitochondrion, macromolecular complex, ncRNA metabolic process, and negative regulation of biosynthetic and nitrogen compound metabolic processes (Fig. 3c).
Comparing aberrant gene expression analysis with WGCNA, we found functions of gene sets identified by these two methods are similar and largely overlapped (Supplementary Table 3). There are 301 unique genes included in the 30 shared aberrantly expressed gene sets and 736 genes included in the seven shared WGCNA gene modules. Among them, 52 genes were overlapped and their functions include many of those related to synapse, neuron, cell projection, localization, cell-cell signaling and so on (Supplementary Table 3). Thus, aberrant gene expression analysis and WGCNA produce consistent results and are complementary to each other to guarantee the identification of comprehensive gene sets associated with neuropsychiatric disorders.
Discussion
Between AUT, SCZ and BPD, similar clinic symptoms and shared genetic etiology have been reported3,4,5,6,7. With the availability of human brain transcriptome data from postmortem donors affected with different neuropsychiatric disorders, gene expression anlaysis has been widely used to identify shared genetic component underlying different disorders; nevertheless, depending on the analytic methods adopted, the results of analysis may vary, each capturing different features of underlying mechanisms of dysregulation. For example, Ellis et al. analyzed the transcriptomic RNA-sequencing data of cortex samples of AUT, SCZ, BPD and controls from two previous study24,31 using single gene-based differential expression analysis. They found the transcriptomes of AUT and SCZ are correlated, and altered neurotransmission and synapse regulation are shared between these two disorders32. Their analysis method focused on obtaining single differentially expressed genes for each disease and then finding the shared ones across disorders. Different from that, here we used multivariate analysis methods to re-analyze the gene expression data for identifying shared dysregulated gene sets across AUT, SCZ and BPD.
The most common gene expression analysis method for identifying shared genes across disorders is based on differential gene expression analysis. Differential gene expression analysis is used to detect the genes with significant difference in gene expression means between diseased and control samples and then the shared differentially expressed genes across diseases are identified. Except from the difference of gene expression means between groups, the difference of gene expression variability also need to be captured. In addition, the major assumption underlying differential expression analysis is: diseased cases have the same or similar gene expression change phenotypes, which makes them as a separate group have significantly higher or lower gene expression than the controls. This assumption contradicts the fact that neuropsychiatric disorders have substantial genetic and phenotypic heterogeneity. Therefore, we need to capture the gene expression variability in diseased samples affected by complex neuropsychiatric disorders. For this, we applied aberrant gene expression analysis method39, quantitatively measuring the departure of multigene expression dispersion between groups, to identify shared dysregulated gene sets across AUT, SCZ and BPD. The identified shared gene sets include the ones associated with amino acid transport activity, neurotransmitter release, synaptic vesicle, excitatory and inhibitory synapse, oxidative stress, nitric oxide synthase biosynthesis, immune response, protein folding, lysophosphatidic acid-mediated signaling and glycolysis. Our method is not dependent on the prior knowledge about gene function or mutations in genes. Thus, it can be used for discovering and identifying genes or gene sets previously unknown to be involved in the progression of AUT, SCZ and BPD. Aberrant gene expression analysis is effective in discovering and revealing shared dysregulated gene sets across disorders, contributing to the study of gene expression overlap between AUT, SCZ and BPD.
In addition to aberrant gene expression analysis, we also applied weighted gene co-expression network analysis to identify shared gene modules across disorders. The functions of several identified modules have been associated with neuropsychiatric disorders. For instance, the modules shared by AUT, SCZ and BPD are enriched for GO terms related to neuron, synapse, adherens junction, RNA binding and nervous system development. For adherens junction, there has been literature documenting the links with AUT, SCZ and BPD105,106,107,108. Adherens junctions are cadherin-based intercellular adhesions109 and the cadherin genes implicated in psychiatric disorders were overrepresented in cell-cell adhesion and adherens junction organization108. There also have been many studies documenting the contribution of dozens of RNA binding proteins to neurodegenerative and neurodevelopmental disorders such as AUT and SCZ110. For example, the loss of function of FMRP, a polyribosome-associated neuronal RNA-binding protein, causes Fragile X syndrome and autistic features. FMRP interacts with the coding region of transcripts which encode pre- and postsynaptic proteins and transcripts implicated in AUT. FMRP target genes significantly overlap with AUT candidate genes in SFARI111. Recently, a paper has studied the link between poly(A) binding protein nuclear I and synaptic plasticity112, implying the relation between poly(A) RNA binding and neuropsychiatric disorders. Another WGCNA gene module, shared by AUT and SCZ, is enriched for GO terms related to mitochondrion. Large literature reported the mitochondrial dysfunction in AUT and SCZ, including evidences of decreased activity of mitochondrial respiratory chain complexes, the presence of biomarkers of oxidative stress and mitochondrial dysfunction and an indication of mtDNA mutations113,114,115,116. Comparing aberrant gene expression analysis and WGCNA, we found the functions of gene sets identified by these two kinds of methods are similar and overlapped. These two kinds of analysis methods complement to each other and promote to identify more comprehensive gene sets associated with neuropsychiatric disorders.
To identify genetic overlap between AUT, SCZ, and BPD, we applied multigene aberrant expression analysis, along with consensus co-expression network analysis, to identify shared dysregulated gene sets in cortical brains of individuals affected with different diseases. Our findings provide new insights into the common molecular mechanisms underlying the pathogenesis and progression of AUT, SCZ and BPD, contributing to the study of etiological overlap between these neuropsychiatric disorders. We show that the aberrant gene expression analysis reveals the variability in gene expression among diseased samples. This method, complementing with the network-based method, is effective in detecting dysregulated gene sets shared across neuropsychiatric disorders, and is applicable in genetic overlap analysis for other complex diseases.
Code availability
Computer codes used throughout for data pre-processing and data analyses are available from corresponding author.
References
Burbach, J. P. H. & van der Zwaag, B. Contact in the genetics of autism and schizophrenia. Trends Neurosci. 32, 69–72 (2009).
Sullivan, P. F. et al. Family history of schizophrenia and bipolar disorder as risk factors for autism. Arch. Gen. psychiatry 69, 1099–1103 (2012).
Cross-Disorder Group of the Psychiatric Genomics C. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 45, 984–994 (2013).
Carroll, L. S. & Owen, M. J. Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med. 1, 102–102 (2009).
Ruderfer, D. M. et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol. psychiatry 19, 1017–1024 (2014).
McCarthy, S. E. et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. psychiatry 19, 652–658 (2014).
Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014).
Turner, T. N. et al. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am. J. Hum. Genet 98, 58–74 (2016).
O’Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012).
Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012).
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012).
Iossifov, I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299 (2012).
Gaugler, T. et al. Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885 (2014).
Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Ripke, S. et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45, 1150–1159 (2013).
Consortium SPG-WAS. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43, 969–976 (2011).
O’donovan, M. C. et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 40, 1053–1055 (2008).
Hou, L. et al. Genetic variants associated with response to lithium treatment in bipolar disorder: a genome-wide association study. Lancet 387, 1085–1093 (2016).
Ikeda, M. et al. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol. Psychiatry 23, 639–647 (2018).
Mühleisen, T. W. et al. Genome-wide association study reveals two new risk loci for bipolar disorder. Nat. Commun. 5, 3339 (2014).
Group PGCBDW. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43, 977–983 (2011).
Voineagu, I. et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 (2011).
Gupta, S. et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 5, 5748 (2014).
Fromer, M. et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016).
Hwang, Y. et al. Gene expression profiling by mRNA sequencing reveals increased expression of immune/inflammation-related genes in the hippocampus of individuals with schizophrenia. Transl. Psychiatry 3, e321 (2013).
Cruceanu, C. et al. Transcriptome sequencing of the anterior cingulate in bipolar disorder: dysregulation of G protein-coupled receptors. Am. J. Psychiatry 172, 1131–1140 (2015).
Seifuddin, F. et al. Systematic review of genome-wide gene expression studies of bipolar disorder. BMC psychiatry 13, 213 (2013).
Kim, S., Hwang, Y., Webster, M. & Lee, D. Differential activation of immune/inflammatory response-related co-expression modules in the hippocampus across the major psychiatric disorders. Mol. psychiatry 21, 376–385 (2016).
Darby, M., Yolken, R. & Sabunciyan, S. Consistently altered expression of gene sets in postmortem brains of individuals with major psychiatric disorders. Transl. Psychiatry 6, e890 (2016).
Zhao, Z. et al. Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol. psychiatry 20, 563–572 (2015).
Ellis, S. E., Panitch, R., West, A. B. & Arking, D. E. Transcriptome analysis of cortical tissue reveals shared sets of downregulated genes in autism and schizophrenia. Transl. psychiatry 6, e817 (2016).
Hansen, K. D., Irizarry, R. A. & Wu, Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216 (2012).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Stegle, O., Parts, L., Durbin, R. & Winn, J. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol. 6, e1000770 (2010).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Wu, Y., Yao, Y.-G. & Luo, X.-J. SZDB: a database for schizophrenia genetic research. Schizophr. Bull. 43, 459–471 (2017).
Chang, S.-H. et al. BDgene: a genetic database for bipolar disorder and its overlap with schizophrenia and major depressive disorder. Biol. psychiatry 74, 727–733 (2013).
Guan, J. et al. Exploiting aberrant mRNA expression in autism for gene discovery and diagnosis. Hum. Genet. 135, 797–811 (2016).
Mahalanobis, P. C. On the generalised distance in statistics. Proc. Natl. Inst. Sci., India 2, 49–55 (1936).
Rousseeuw, P. J. & Van Driessen, K. A fast algorithm for the minimum covariance determinant estimator. Technometrics 41, 212–223 (1999).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met 57, 289–300 (1995).
Jaccard, P. Distribution de la flore alpine dans le bassin des Dranses et dans quelques régions voisines. Bull. de. la Société Vaud. des. Sci. Nat. 37, 241–272 (1901).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Langfelder, P. & Horvath, S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 1, 54 (2007).
Langfelder, P. & Horvath, S. Fast R functions for robust correlations and hierarchical clustering. J. Stat. Softw. 46, 1–17 (2012).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Fernell, E. et al. Aberrant amino acid transport in fibroblasts from children with autism. Neurosci. Lett. 418, 82–86 (2007).
Tarlungeanu, D. C. et al. Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell 167, 1481–1494.e1418 (2016).
Flyckt, L. et al. Aberrant tyrosine transport across the cell membrane in patients with schizophrenia. Arch. Gen. psychiatry 58, 953–958 (2001).
Smith, R. E., Haroutunian, V., Davis, K. L. & Meador-Woodruff, J. H. Expression of excitatory amino acid transporter transcripts in the thalamus of subjects with schizophrenia. Am. J. Psychiatry 158, 1393–1399 (2001).
Zhao, H. et al. SCAMP5 plays a critical role in synaptic vesicle endocytosis during high neuronal activity. J. Neurosci. 34, 10085–10095 (2014).
Kononenko, N. L. et al. Compromised fidelity of endocytic synaptic vesicle protein sorting in the absence of stonin 2. Proc. Natl. Acad. Sci. 110, E526–E535 (2013).
Egbujo, C. N., Sinclair, D. & Hahn, C.-G. Dysregulations of synaptic vesicle trafficking in schizophrenia. Curr. psychiatry Rep. 18, 77–77 (2016).
Kavalali, E. T. Synaptic vesicle reuse and its implications. Neuroscientist 12, 57–66 (2006).
Narla, S. et al. Common developmental genome deprogramming in schizophrenia—role of integrative nuclear FGFR1 signaling (INFS). Schizophr. Res. 185, 17–32 (2017).
Rojas, D. C. The role of glutamate and its receptors in autism and the use of glutamate receptor antagonists in treatment. J. Neural Transm. 121, 891–905 (2014).
El-Ansary, A. & Al-Ayadhi, L. GABAergic/glutamatergic imbalance relative to excessive neuroinflammation in autism spectrum disorders. J. Neuroinflamm. 11, 189 (2014).
Blaylock, R. & Strunecka, A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr. Med. Chem. 16, 157–170 (2009).
Howes, O., McCutcheon, R. & Stone, J. Glutamate and dopamine in schizophrenia: an update for the 21(st) century. J. Psychopharmacol. 29, 97–115 (2015).
Marsman, A. et al. Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophr. Bull. 39, 120–129 (2013).
de la Torre-Ubieta, L., Won, H., Stein, J. L. & Geschwind, D. H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 22, 345–361 (2016).
Paşca, S. P. et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 17, 1657 (2011).
Hook, V. et al. Human iPSC neurons display activity-dependent neurotransmitter secretion: aberrant catecholamine levels in schizophrenia neurons. Stem Cell Rep. 3, 531–538 (2014).
Gao, R. & Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 15, 146–167 (2015).
Nelson, S. B. & Valakh, V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698 (2015).
Lewis, D. A., Hashimoto, T. & Volk, D. W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312 (2005).
Morris, G., Walker, A. J., Berk, M., Maes, M. & Puri, B. K. Cell death pathways: a novel therapeutic approach for neuroscientists. Mol. Neurobiol. 55, 5767–5786 (2018).
Pardo, C. A. & Eberhart, C. G. The neurobiology of autism. Brain Pathol. 17, 434–447 (2007).
Nasyrova, R. F., Ivashchenko, D. V., Ivanov, M. V. & Neznanov, N. G. Role of nitric oxide and related molecules in schizophrenia pathogenesis: biochemical, genetic and clinical aspects. Front. Physiol. 6, 1–16 (2015).
Loh, K. P., Huang, S. H., De Silva, R., Tan, B. K. & Zhu, Y. Z. Oxidative stress: apoptosis in neuronal injury. Curr. Alzheimer Res. 3, 327–337 (2006).
Kunio, Y., Yohei, K., Hiroshi, Y. & Shintaro, O. Oxidative stress and nitric oxide in autism spectrum disorder and other neuropsychiatric disorders. CNS Neurol. Disord. Drug Targets 15, 587–596 (2016).
Frye, R. E. et al. Metabolic effects of sapropterin treatment in autism spectrum disorder: a preliminary study. Transl. Psychiatry 3, e237 (2013).
Kim, H.-W. et al. Family-based association study between NOS-I and IIA polymorphisms and autism spectrum disorders in Korean trios. Am. J. Med. Genet. Part B: Neuropsychiatr. Genet. 150B, 300–306 (2009).
Delorme, R. et al. Mutation screening of NOS1AP gene in a large sample of psychiatric patients and controls. BMC Med. Genet. 11, 108–108 (2010).
Frye, R. E. & Rossignol, D. A. Treatments for biomedical abnormalities associated with autism spectrum disorder. Front. Pediatr. 2, 66–66 (2014).
Goines, P., Van & de Water, J. The Immune System’s Role in the Biology of Autism. Curr. Opin. Neurol. 23, 111–117 (2010).
Ashwood, P. et al. Altered T cell responses in children with autism. Brain, Behav., Immun. 25, 840–849 (2011).
Barbosa, I. G., Machado-Vieira, R., Soares, J. C. & Teixeira, A. L. The immunology of bipolar disorder. Neuroimmunomodulation 21, 117–122 (2014).
Barbosa, I. G. et al. Monocyte and lymphocyte activation in bipolar disorder: a new piece in the puzzle of immune dysfunction in mood disorders. Int. J. Neuropsychopharmacol. 18, 1–7 (2015).
Nguyen, A., Rauch, T. A., Pfeifer, G. P. & Hu, V. W. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 24, 3036–3051 (2010).
Sarachana, T. & Hu, V. W. Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Molecular. Autism 4, 14–14 (2013).
Sayad, A., Noroozi, R., Omrani, M. D., Taheri, M. & Ghafouri-Fard, S. Retinoic acid-related orphan receptor alpha (RORA) variants are associated with autism spectrum disorder. Metab. Brain Dis. 32, 1595–1601 (2017).
Lai, Y.-C. et al. Investigation of associations between NR1D1, RORA and RORB genes and bipolar disorder. PloS ONE 10, e0121245 (2015).
Soria, V. et al. Differential association of circadian genes with mood disorders: CRY1 and NPAS2 are associated with unipolar major depression and CLOCK and VIP with bipolar disorder. Neuropsychopharmacology 35, 1279–1289 (2010).
Clerici, M. et al. Cytokine polymorphisms in the pathophysiology of mood disorders. CNS Spectr. 14, 419–425 (2009).
Zhang, C. et al. A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and ER export. J. Neurosci. 29, 10843–10854 (2009).
Hartl, F. U., Bracher, A. & Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 475, 324 (2011).
Lin, M. et al. Heat shock alters the expression of schizophrenia and autism candidate genes in an induced pluripotent stem cell model of the human telencephalon. PloS ONE 9, e94968 (2014).
Kim, J. J. et al. Association analysis of heat shock protein 70 gene polymorphisms in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 258, 239–244 (2008).
Yung, Y. C., Stoddard, N. C., Mirendil, H. & Chun, J. Lysophosphatidic acid (LPA) signaling in the nervous system. Neuron 85, 669–682 (2015).
Choi, J. W. & Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. et. Biophys. acta 1831, 20–32 (2013).
Liu, M.-L. et al. Severe disturbance of glucose metabolism in peripheral blood mononuclear cells of schizophrenia patients: a targeted metabolomic study. J. Transl. Med. 13, 226 (2015).
Martins-de-Souza, D. et al. Proteome analysis of the thalamus and cerebrospinal fluid reveals glycolysis dysfunction and potential biomarkers candidates for schizophrenia. J. Psychiatr. Res. 44, 1176–1189 (2010).
Vallée, A. & Vallée, J.-N. Warburg effect hypothesis in autism Spectrum disorders. Mol Brain 11, 1 (2018).
Kelley, D. J. et al. The cyclic AMP cascade is altered in the fragile X nervous system. PloS ONE 2, e931–e931 (2007).
Kelley, D. J. et al. The cyclic AMP phenotype of fragile X and autism. Neurosci. Biobehav. Rev. 32, 1533–1543 (2008).
McDonald, M. L., MacMullen, C., Liu, D. J., Leal, S. M. & Davis, R. L. Genetic association of cyclic AMP signaling genes with bipolar disorder. Transl. Psychiatry 2, e169–e169 (2012).
Shen, S., Gehlert, D. R. & Collier, D. A. PACAP and PAC1 receptor in brain development and behavior. Neuropeptides 47, 421–430 (2013).
Nicot, A., Otto, T., Brabet, P. & DiCicco-Bloom, E. M. Altered social behavior in pituitary adenylate cyclase-activating polypeptide type I receptor-deficient mice. J. Neurosci. 24, 8786–8795 (2004).
Blechman, J. & Levkowitz, G. Alternative splicing of the pituitary adenylate cyclase-activating polypeptide receptor PAC1: mechanisms of fine tuning of brain activity. Front. Endocrinol. 4, 55–55 (2013).
Hashimoto, R. et al. Pituitary adenylate cyclase-activating polypeptide is associated with schizophrenia. Mol. psychiatry 12, 1026 (2007).
Wu, J. Q. et al. Transcriptome sequencing revealed significant alteration of cortical promoter usage and splicing in schizophrenia. PloS ONE 7, e36351–e36351 (2012).
Yoon, K.-J. et al. Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell 15, 79–91 (2014).
Folmsbee, S. S. et al. αT-catenin in restricted brain cell types and its potential connection to autism. J. Mol. Psychiatry 4, 2 (2016).
Carter, C. J. Susceptibility genes are enriched in those of the herpes simplex virus 1/host interactome in psychiatric and neurological disorders. Pathog. Dis. 69, 240–261 (2013).
Hawi, Z. et al. The role of cadherin genes in five major psychiatric disorders: a literature update. Am. J. Med. Genet. Part B: Neuropsychiatr. Genet. 177, 168–180 (2018).
Efimova, N. & Svitkina, T. M. Branched actin networks push against each other at adherens junctions to maintain cell–cell adhesion. J. Cell Biol. 217, 1827–1845 (2018).
Bryant, C. D. & Yazdani, N. RNA binding proteins, neural development and the addictions. Genes, brain, Behav. 15, 169–186 (2016).
Darnell Jennifer, C. et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011).
Vlatkovic I. et al. Poly(A) Binding protein nuclear 1 regulates the polyadenylation of key synaptic plasticity genes and plays a role in homeostatic plasticity. bioRxiv (2017).
Siddiqui, M. F., Elwell, C. & Johnson, M. H. Mitochondrial dysfunction in autism spectrum disorders. Autism 6, 1000190 (2016).
Rossignol, D. A. & Frye, R. E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 5, 150 (2014).
Prabakaran, S. et al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol. psychiatry 9, 684 (2004).
Rajasekaran, A., Venkatasubramanian, G., Berk, M. & Debnath, M. Mitochondrial dysfunction in schizophrenia: pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 48, 10–21 (2015).
Acknowledgements
This work has been supported by the National Natural Science Foundation of China (Nos. 61803320, 61573296, 61871463 and 61802323), and the Fundamental Research Funds for the Central Universities in China (Xiamen University: 20720190022).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guan, J., Cai, J.J., Ji, G. et al. Commonality in dysregulated expression of gene sets in cortical brains of individuals with autism, schizophrenia, and bipolar disorder. Transl Psychiatry 9, 152 (2019). https://doi.org/10.1038/s41398-019-0488-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0488-4
This article is cited by
-
Genetic regulation of human brain proteome reveals proteins implicated in psychiatric disorders
Molecular Psychiatry (2024)
-
Molecular profiling of the hippocampus of children with autism spectrum disorder
Molecular Psychiatry (2024)
-
Analysis of human neuronal cells carrying ASTN2 deletion associated with psychiatric disorders
Translational Psychiatry (2024)
-
Immunoglobulin genes expressed in lymphoblastoid cell lines discern and predict lithium response in bipolar disorder patients
Molecular Psychiatry (2023)
-
Identification of schizophrenia symptom-related gene modules by postmortem brain transcriptome analysis
Translational Psychiatry (2023)
