
Abstract
Alzheimer’s disease (AD) is a public health priority for the 21st century. Risk reduction currently revolves around lifestyle changes with much research trying to elucidate the biological underpinnings. We show that self-report of parental history of Alzheimer’s dementia for case ascertainment in a genome-wide association study of 314,278 participants from UK Biobank (27,696 maternal cases, 14,338 paternal cases) is a valid proxy for an AD genetic study. After meta-analysing with published consortium data (n = 74,046 with 25,580 cases across the discovery and replication analyses), three new AD-associated loci (P < 5 × 10−8) are identified. These contain genes relevant for AD and neurodegeneration: ADAM10, BCKDK/KAT8 and ACE. Novel gene-based loci include drug targets such as VKORC1 (warfarin dose). We report evidence that the association of SNPs in the TOMM40 gene with AD is potentially mediated by both gene expression and DNA methylation in the prefrontal cortex. However, it is likely that multiple variants are affecting the trait and gene methylation/expression. Our discovered loci may help to elucidate the biological mechanisms underlying AD and, as they contain genes that are drug targets for other diseases and disorders, warrant further exploration for potential precision medicine applications.
Similar content being viewed by others

Introduction
The genetic epidemiology of late-onset Alzheimer’s disease (LOAD) has advanced over the last decade1, with >20 independent loci associated with the disease in addition to APOE2. Presently, the largest meta-analytic genome-wide association study (GWAS) for LOAD employed a two-stage study design. First, 17,008 cases were compared to 37,154 controls. A total of 11,632 single-nucleotide polymorphisms (SNPs) with P < 1 × 10−3 from this meta-analysis were included in the second stage that compared 8,572 cases to 11,312 controls. A meta-analysis of the SNPs included in stages 1 and 2 was also performed3.
One difficulty in traditional studies of AD is case ascertainment4—either directly for prevalent cases or indirectly through prospective cohort studies for incident cases. A recent GWAS study on a subset of the UK Biobank cohort used information from family history (parent or first-degree relative with AD or dementia) as a proxy-phenotype for the participants5. When meta-analysed with the GWAS summary data highlighted above3, four new loci were identified.
The UK Biobank proxy-phenotype AD question, which is used here, does not incorporate biomarker data that are required for a clinical diagnosis. However, it is easy to administer at scale and we show that it has a near-unit genetic correlation with the AD results from the LOAD meta-analysis3, where many of the samples also lacked a confirmed diagnosis by biomarker levels or autopsy.
In the present study, we related proxy-phenotype information on dementia (i.e., reporting a parent with Alzheimer’s dementia or dementia) to genetic data from 314,278 individuals from the UK Biobank cohort to identify new AD-associated loci. This sample includes individuals from the previous proxy-phenotype AD study by Liu et al.5 GWA studies were conducted separately for maternal and paternal AD due to a 1.7-fold difference in disease prevalence—9.6% and 5.5%, respectively. The summary statistics from these models were meta-analysed with those from the largest publicly available case–control study3. Sensitivity analyses showed that an overlap of controls in the maternal and paternal GWAS did not bias the results. Genetic correlation analysis showed the self-reported measure of parental AD to be an accurate proxy for clinical diagnosis, validating the global meta-analysis. In addition, we tested for causal evidence of our SNP–AD associations being mediated through gene expression and DNA methylation in the prefrontal cortex.
Subjects and methods
UK Biobank cohort
UK Biobank data6 (http://www.ukbiobank.ac.uk) were collected on over 500,000 individuals aged between 37 and 73 years from across Great Britain (England, Wales and Scotland) at the study baseline (2006–2010), including health, cognitive and genetic data.
The Research Ethics Committee (REC) granted ethical approval for the study—reference 11/NW/0382—and the current analysis was conducted under data application 10,279.
Genotyping
Genotyping details for the UK Biobank cohort have been reported previously7,8. Briefly, two custom genotyping arrays were utilised with 49,950 participants typed using the UK BiLEVE Axiom Array and 438,427 participants typed using the UK Biobank Axiom Array7,8. The released genotyped data contained 805,426 markers on 488,377 individuals. Imputed genotypes were supplied with the UK Biobank data with the Haplotype Reference Consortium (HRC) used as the imputation reference panel7.
Downstream quality control steps conducted for the current analysis included removing (1) those with non-British ancestry based on both self-report and a principal components analysis, (2) outliers based on heterozygosity and missingness, (3) individuals with sex chromosome configurations that were neither XX nor XY, (4) individuals whose reported sex did not match inferred sex from their genetic data and (5) individuals with >10 putative third-degree relatives from the kinship table. This left a sample of 408,095 individuals. To remove the possibility of double contributions from sibs, whose parents will have the same AD status, we first considered a list of all participants with a relative (N = 131,790). A genetic relationship matrix was built for these individuals using GCTA-GRM9 and a relationship threshold of 0.025 was applied to exclude related individuals. After removing one person from each pair of related individuals, the sample size was 332,050. Quality control thresholds applied to the GWAS included: minor allele frequency >0.01, imputation quality score >0.3 and restriction to HRC-imputed SNPs, leaving a total of 7,795,605 SNPs for the GWAS.
Phenotypes
Family history of Alzheimer’s disease was ascertained via self-report. Participants were asked “Has/did your father ever suffer from Alzheimer’s disease/dementia?” and “Has/did your mother ever suffer from Alzheimer’s disease/dementia?” Self-report data from the initial assessment visit (2006–2010), the first repeat assessment visit (2012–2013) and the imaging visit (2014+) were aggregated with exclusions made for participants whose parents were: aged under 60 years; dead before reaching age 60 years; without age information. After merging with the genetic data, this left 27,696 cases of maternal AD with 260,980 controls, and 14,338 cases of paternal AD with 245,941 controls. There were 314,278 instances where AD information was available on at least one parent. Given the expected difference in disease prevalence due to sex differences in longevity—AD prevalence was 1.7-fold higher in mothers compared to fathers—GWA studies were performed separately for maternal and paternal AD.
Genome-wide association study
The GWA studies were conducted using BGENIE7. The outcome variable was the residuals from a linear regression model of maternal or paternal AD on age of parent at death or at time of the offspring’s self-report, assessment centre, genotype batch, array and 40 genetic principal components. The predictor variable was the autosomal SNP and an additive model was considered.
The GWAS linear regression coefficients were converted to odds ratios using observed sample prevalences of 0.096 and 0.055 for maternal and paternal AD, respectively10. Subsequently, the log-odds were multiplied by two so that the effect sizes are reported on the same scale as a traditional case–control design5. Briefly, the conversion to odds ratios uses the following equation, derived in Lloyd-Jones et al.10, where k = disease prevalence, p = population allele frequency and β = the estimated SNP regression coefficient on the binary disease scale from the GWAS: \({\mathrm{OR}} = \frac{{\left( {{{k}} + {{\beta }}\left( {1 - {{p}}} \right)} \right) \times \left( {1 - {{k}} + {{\beta p}}} \right)}}{{\left( {{{k}} - {{\beta p}}} \right) \times \left( {1 - {{k}} - {{\beta }}\left( {1 - {{p}}} \right)} \right)}}\). SEs for the log-odds were then calculated based on the adjusted OR and the P-value from the initial GWAS (Supplementary Note 1). The ORs and SEs were then carried forward to a SE-weighted meta-analysis in METAL11, first to create a UK Biobank parental meta-analysis, and then with the stage 2 summary output from the International Genomics of Alzheimer’s Project (IGAP) study3 and the stage 1 output for the SNPs that did not contribute to stage 2. Linkage disequilibrium score (LDSC) regression was used to estimate the genetic correlation between the maternal and paternal AD GWAS results and to test for residual confounding in the meta-analysis by examining the LDSC intercept12,13.
The number of independent loci from the meta-analysis was determined by using the default settings in FUMA14. Independent lead SNPs had P < 5 × 10−8 and were independent at r2 < 0.6. Within this pool of independent SNPs, lead SNPs were defined as those in LD at r2 < 0.1. Loci were defined by combining lead SNPs within a 250 kb window and all SNPs in LD of at least r2 = 0.6 with one of the independent SNPs. A gene-based analysis was carried out on all SNP output using the MAGMA software15 with default settings (SNP-wise (mean) model for each gene), and assuming a constant sample size for all genes. A Bonferroni-adjusted P-value of 0.05/18,251 = 2.7 × 10−6 was used to identify significant genes. The 1000 genomes phase 3 data16 were used to map LD in both the independent locus and MAGMA analyses.
Summary data-based Mendelian randomisation
To test for pleiotropic associations between SNPs and AD and gene expression/DNA methylation in the brain, summary data-based Mendelian randomisation (SMR) was performed17. GWAS summary output from the meta-analysis of UK Biobank and IGAP (sample size specified as 314,278 + 74,046 = 388,324) were included along with expression Quantitative Trait Loci (QTL) summary output from the Common Mind Consortium, which contains data on >600 dorsolateral prefrontal cortex samples, and DNA methylation QTL summary output on 258 prefrontal cortex samples (age >13)18. The reference genotypes were based on the Health and Retirement Study, imputed to the 1000 Genomes phase 1 reference panel. SNP exclusions included: imputation score <0.3, Hardy–Weinberg P-value < 1 × 10−6 and a minor allele frequency <0.01. Related individuals, based on a genomic-relationship matrix cutoff of 0.05, were removed. Two sets of eQTL summary data were considered (1) after adjustment for diagnosis, institution, sex, age of death, post-mortem interval, RNA integrity number (RIN), RIN2, and clustered library batch (2) with additional adjustments for 20 surrogate variables to account for additional possible confounders. Five ancestry vectors were included as covariates in the eQTL analyses. Further details are available at: https://www.synapse.org/#!Synapse:syn4622659. Default parameters for the SMR analysis were used and cis eQTLs/methQTLs were considered for analysis. Bonferroni-corrected P-value thresholds were applied (P < 0.05/2011 = 2.5 × 10−5 for eQTL data set 1, P < 0.05/4380 = 1.1 × 10−5 for eQTL data set 2 and P < 0.05/54,624 = 9.2 × 10−7 for the methQTL data set). The SMR P-value highlights candidate transcripts or methylation sites through which a cis SNP may be acting on the outcome, AD. The heterogeneity in dependent instruments (HEIDI) P-value indicates evidence for a single causal SNP (effect on AD is mediated through the transcript/methylation site if P > 0.05) or different SNPs affecting AD and the transcript/methylation site if P < 0.05.
Results
UK Biobank GWAS
There were 27,696 cases of maternal AD (260,980 controls, prevalence of 9.6%) and 14,338 cases of paternal AD (245,941 controls, prevalence of 5.5%) in the UK Biobank. The genetic correlation between maternal and paternal AD was not significantly different from unity (rg = 0.65, SE 0.59), although the SE is large. Both traits had a high genetic correlation with the case–control summary output from the IGAP study: rg with maternal and paternal AD was 0.91 (SE 0.24) and 0.67 (0.40), respectively, both not significantly different from unity but with large SEs.
Prior to meta-analysing the UK Biobank parental summary statistics with the IGAP output, we investigated the influence of overlapping proxy controls in UK Biobank. The P-values from a single GWAS of parental AD status (0, 1 or 2 parents with AD) correlated 0.99 with those from a meta-analysis of separate maternal AD and paternal AD; the regression of −log10 P-values on each other gave an intercept of 0 and a slope of 1. A meta-analysis of the summary statistics from the maternal and paternal results is therefore essentially equivalent to the analysis of parental AD status. The linear regression effect sizes from the GWAS were converted to odds ratios prior to the meta-analysis10.
UK Biobank paternal and maternal meta-analysis
Six genome-wide significant loci were identified in the UK Biobank paternal and maternal GWAS meta-analysis (Supplementary Table 1, GWAS summary statistics available at www.ccace.ed.ac.uk/node/335). All were located in established AD loci. The top lead SNPs were rs679515 (P = 5.2 × 10−9, chr1, CR1 locus), rs6733839 (P = 1.1 × 10−27, chr2, BIN1 locus), rs7384878 (P = 1.3 × 10−10, chr7, PILRA locus), rs3851179 (P = 1.8 × 10−12, chr11, PICALM locus), rs3845261 (P = 4.0 × 10−8, chr17, ZNF232 locus) and rs10119 (P = 3.5 × 10−308, chr19, APOE/TOMM40 locus). The chromosome 17 locus is ~100 kb from the SCIMP gene-variant identified by Liu et al.5
UK Biobank parental and IGAP meta-analysis
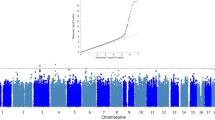
The meta-analysis of the maternal and paternal AD history in UK Biobank with the IGAP data identified 83 lead SNPs and 264 independent significant SNPs with P < 5 × 10−8 from 26 genomic risk loci. The majority (n = 45) of the lead SNPs were located in the gene-dense APOE/TOMM40 locus on chromosome 19 (Fig. 1 and Supplementary Tables 2 and 3; GWAS summary statistics are available for the out-meta-analysed SNPs online: www.ccace.ed.ac.uk/node/335). The LDSC regression intercept term from the meta-analysis summary output was 1.029 (SE 0.0097), and around 81% of the genomic inflation (λ = 1.11) was due to the polygenic signal, rather than sources of confounding including population stratification and cryptic relatedness.

The red line indicates P = 5 × 10−8 and the blue line indicates P = 1 × 10−5. P-values truncated at 1 × 10−20
Novel genome-wide significant loci
Of the 26 significant risk loci, seven were novel. These include the three loci highlighted in the previous version of the manuscript (Table 1 and Supplementary Figures 1–3). The top lead SNPs were: rs593742 (ADAM10, locus chr15:58873555–59132827); rs889555 210 (BCKDK/KAT8, locus chr16:30916129–31155458); and rs138190086 (ACE, locus 211 chr17:61001497–61559625).
We now report four additional genome-wide significant loci: rs9381040 (TREML2, locus chr6: 41031273-41164788); rs59685680 (SPPL2A, locus chr15:50716633-51060127); rs4985556 (IL-34, locus chr16:70638035-70694991); rs12444183 (PLCG2, locus chr16:81773003-81773816).
Replication of IGAP SNPs
Seventeen of the 21 previously reported SNPs3 associated with AD were genome-wide significant (P < 5 × 10−8) in the current meta-analysis, with two other SNPs (rs2718058 and, rs10838725) having P < 1 × 10−7 (Supplementary Table 4). The MEF2C variant, rs190982, had a meta-analysis P-value of 3.7 × 10−4 and rs8093731 (a DSG2 variant), which was genome-wide significant in stage 1 but not stage 2 of IGAP, had a meta-analysis P-value of 7.2 × 10−4. There was complete sign-concordance between UK Biobank and IGAP with the exception of rs190982 (Supplementary Table 4). The odds ratios between the maternal and paternal analysis for the top 21 IGAP SNPs were correlated r = 0.76. Both also correlated highly with the effect sizes reported in the IGAP analysis (r = 0.89 and 0.85, respectively).
Gene-based analysis
A total of 87 genes were significant at a Bonferroni threshold of P < 2.7 × 10−6 (Supplementary Table 5). Gene ontology analysis, using a Fisher exact test to compare the number of significant genes in each gene set with the expected number, showed significant enrichment for 51 gene sets, including those linked to the regulation of amyloid-beta, neurofibrillary tangle assembly, immune response, and cholesterol and lipid transport (Supplementary Table 6).
Summary data-based Mendelian randomisation
Pleiotropic associations between AD and gene expression in the brain were tested using SMR17. GWAS summary data for AD were taken from the UK Biobank and IGAP meta-analysis. eQTL summary data came from the Common Mind Consortium (n > 600 dorsolateral prefrontal cortex samples: data set 1 adjusted for age at death, sex and institution; data set 2 made additional adjustments for 20 surrogate variables). MethQTL data came from 258 dorsolateral prefrontal cortex samples (participants aged 13 years and older—adjustments were made for the first 5 genetic MDS components and first 11 methylation PCs)18. We found evidence of brain expression and DNA methylation associated with AD in the TOMM40 gene (part of the APOE/TOMM40 cluster on chromosome 19) in both the second eQTL model and also in the methQTL model (Supplementary Tables 7–9). However, the HEIDI P-values were <0.05 for both analyses, indicating that the associations were unlikely to be driven by a single causal variant affecting both expression/methylation and AD.
Different top QTL SNPs in TOMM40 were identified in the two analyses: rs760136 and rs7259620 although they were in perfect LD in European samples19,20 (R2 = 1.00). The SNPs have differing LD patterns with the APOE allele defining SNPs, rs7412 (R2 = 0.001, D′ = 0.03) and rs429358 (R2 = 0.15, D′ = 1.0). A significant SMR association with HEIDI P-value >0.05, indicating pleiotropy, was observed for KAT8 and CR1 in the second eQTL analysis (Supplementary Table 8 and Supplementary Figures 4 and 5) and for STAG3 (chr7, PILRA locus), CD2AP (chr6), HLA-DRB1 (chr6, locus not reported in Lambert et al.3), and PSMC3 (chr11, CELF locus) in the methQTL analysis (Supplementary Table 9 and Supplementary Figures 6–9).
Discussion
Using recently established proxy-phenotype methods for case ascertainment, we show this approach to be valid in GWAS of AD. Meta-analysing the proxy-phenotype summary statistics from UK Biobank with those from a case–control study (IGAP), we identified three new genome-wide significant loci that contain several relevant genes.
ACE determines levels of angiotensin II, which has trophic actions within the brain and contributes to the regulation of cerebral blood flow21. Previous meta-analyses of candidate gene studies identified variants within ACE to be associated with AD, though not at genome-wide significance22,23. ACE variants have also been linked to atrophy of the hippocampus and amygdala24, and CSF-ACE protein levels correlate with CSF tau and phosphorylated tau25,26.
ADAM10 is involved in the cleavage of amyloid-beta precursor protein27, which is involved in the deposition of amyloid beta, a major neurological hallmark of AD. ADAM10 has been proposed as potential therapeutic agent in AD therapy27,28. Rare variants in ADAM10 have also been linked to LOAD29.
The BCKDK/KAT8 locus contains the VKORC1 gene, which was a genome-wide significant gene-based finding (P = 5.1 × 10−8). The VKORC1 variant, rs9923231, whose T allele was suggestively associated with an increased risk of AD (P = 1.8 × 10−7), is strongly associated with the need for a reduced dose of warfarin anticoagulation30,31. KAT8 is regulated by KANSL1, which has been linked to AD as a genome-wide significant finding in APOE e4-negative individuals32.
Our cutoff for relatedness of 0.025 in UK Biobank was very stringent, in particular since disease status was on parents of genotyped individuals, so that correlations in proxy-phenotypes from relatives (e.g., cousins) are likely to be very small. When we used a more relaxed threshold of 0.4 (excluding first-degree relatives only), we increased our sample size to 385,869 and found three additional loci and top lead SNPs: a gene desert on chr4 (rs6448453, P = 2.8 × 10−9, chr4:11014822–11041549) that is ~400 kb from the CLNK gene; a region on chromosome 17 that includes the benzodiazepine receptor-associated protein 1 (BZRAP1) gene (rs2526378, P = 2.0 × 10−8, chr17:56398006-56410041) and a locus including variants in the amyloid precursor protein (APP) gene on chromosome 21 (rs4817090, P = 4.8 × 10−8, chr21:27454995-27534261). The corresponding P-values for these three lead SNPs in the GWAS meta-analysis with the more stringent relatedness cutoff were P = 9.6 × 10−8, 7.5 × 10−7, and 1.3 × 10−7, respectively. Two additional loci were reported as significant using the less stringent relatedness threshold in the previous version of the manuscript. Both were just below genome-wide significance in the updated analysis: rs4575098, P = 8.8 × 10−8, ADAMTS4 and rs1171812, P = 6.4 × 10−8, CCDC6. Moreover, the novel KAT8/BCKDK locus, which is reported in the main results section, is not genome-wide significant in the analysis using the less stringent relatedness threshold (rs889555, P = 1.7 × 10−7).
The gene-set enrichment analysis implicated pathways involved in AD neuropathology (amyloid and neurofibrillary tangles) in addition to immune response, lipid and cholesterol metabolism, as previously reported in a gene-set analysis on the IGAP data33. A separate gene-set analysis on the meta-analysed paternal and maternal summary statistics from UK Biobank showed a highly overlapping set of enriched pathways (Supplementary Table 10).
An integrative analysis of eQTL and methQTL with the GWAS summary data identified one previously identified AD gene, TOMM40, as having its gene expression and methylation levels associated with AD. The most parsimonious explanation of these results is the existence of multiple causal variants, some affecting AD and others affecting expression or methylation. A previous SMR analysis of AD and LDL cholesterol identified evidence of 16 pleiotropic SNPs, 12 of which were located in the APOE region34. Given the involvement of immunity in the aetiology of AD, as highlighted by the pathway analysis, it may be relevant to consider immune cells, such as monocytes, for the SMR analysis.
The main strength of the study is the proxy-phenotype approach, which resulted in over 42,000 proxy cases for the GWAS analysis. However, the question used to determine parental AD status may have resulted in some responders being unable to discriminate Alzheimer’s disease and dementia from other dementia sub-types, which have different presentations and genetic architectures35,36. This method of proxy-case ascertainment may have influenced the loci uncovered. Parental dementia status is partly dependent on longevity, with age being the biggest risk factor for AD. We partially controlled for this by excluding participants whose parents were younger than or died prior to reaching the age of 60 years when AD incidence is extremely low. The misclassification of case status via incorrect informant reporting will have reduced the power to detect true effects.
Conclusion
We identified three new AD-associated loci that have known and putative biological processes associated with Alzheimer’s disease. These findings help to elucidate the biological mechanisms underlying AD and, given that some loci (VKORC1, ACE) are existing drug targets for other diseases and disorders, warrant further exploration for potential precision medicine and clinical trial applications.
Disclaimer
The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Change history
06 June 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Huang, Y. & Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222 (2012).
Van Cauwenberghe, C., Van Broeckhoven, C. & Sleegers, K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430 (2016).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Sibbett, R. A., Russ, T. C., Deary, I. J. & Starr, J. M. Dementia ascertainment using existing data in UK longitudinal and cohort studies: a systematic review of methodology. BMC Psychiatry 17, 239 (2017).
Liu, J. Z., Erlich, Y. & Pickrell, J. K. Case-control association mapping by proxy using family history of 363 disease. Nat. Genet. 49, 325–331 (2017).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Bycroft, C. et al. Genome-wide genetic data on ~500,000 UK Biobank participants. (2017) Preprint at https://www.biorxiv.org/content/early/2017/07/20/166298.
Wain, L. V. et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir. Med. 3, 769–781 (2015).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Lloyd-Jones, L. R., Robinson, M. R., Yang, J. & Visscher, P. M. Transformation of summary statistics from linear mixed model association on all-or-none traits to odds ratio. Genetics 208, 1397–1408 (2018).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Watanabe, K., Taskesen, E., Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
1000 Genomes Project Consortium et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Jaffe, A. E. et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci. 19, 40–47 (2016).
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Machiela, M. J. & Chanock, S. J. LDassoc: an online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics 34, 887–889 (2017).
Starr, J. M. & Whalley, L. J. ACE Inhibitors: Central Actions (Raven Press, New York, 1994).
Elkins, J. S., Douglas, V. C. & Johnston, S. C. Alzheimer disease risk and genetic variation in ACE: a meta-analysis. Neurology 62, 363–368 (2004).
Lehmann, D. J. et al. Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of Alzheimer’s disease. Am. J. Epidemiol. 162, 305–317 (2005).
Sleegers, K. et al. ACE gene is associated with Alzheimer’s disease and atrophy of hippocampus and amygdala. Neurobiol. Aging 26, 1153–1159 (2005).
Jochemsen, H. M. et al. The association of angiotensin-converting enzyme with biomarkers for Alzheimer’s disease. Alzheimers Res. Ther. 6, 27 (2014).
Kauwe, J. S. K. et al. Genome-wide association study of CSF levels of 59 alzheimer’s disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet. 10, e1004758 (2014).
Suh, J. et al. ADAM10 missense mutations potentiate β-amyloid accumulation by impairing prodomain chaperone function. Neuron 80, 385–401 (2013).
Yuan, X.-Z., Sun, S., Tan, C.-C., Yu, J.-T. & Tan, L. The role of ADAM10 in Alzheimer’s disease. J. Alzheimers Dis. 58, 303–322 (2017).
Kim, M. et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum. Mol. Genet. 18, 3987–3996 (2009).
Takeuchi, F. et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 5, e1000433 (2009).
Wadelius, M. et al. Common VKORC1 and GGCX polymorphisms associated with warfarin dose. Pharm. J. 5, 262–270 (2005).
Jun, G. et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 21, 108 (2016).
International Genomics of Alzheimer’s Disease Consortium (IGAP). Convergent genetic and expression data implicate immunity in Alzheimer’s disease. Alzheimers Dement. 11, 658–671 (2015).
Zhu, Z. et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun 9, 224 (2018).
Ferrari, R. et al. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatry 88, 152–164 (2017).
Ikram, M. A. et al. Genetics of vascular dementia—review from the ICVD working group. BMC Med. 15, 48 (2017).
Acknowledgements
R.E.M. is supported by Alzheimer’s Research UK major project grant number ARUK-PG2017B-10. The research was conducted, using the UK Biobank Resource, in The University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology (CCACE), part of the cross-council Lifelong Health and Wellbeing Initiative (MR/K026992/1); funding from the Biotechnology and Biological Sciences Research Council (BBSRC) and Medical Research Council (MRC) are gratefully acknowledged. CCACE supports I.J.D., with some additional support from Dementias Platform UK (MR/L015382/1). This report represents independent research part-funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The University of Queensland authors acknowledge funding from the Australian National Health and Medical Research Council (grants 1113400, 1078901 and 1078037) and the Charles & Sylvia Viertel Fellowship (J.Y.). W.D.H. is supported by a grant from Age UK (Disconnected Mind Project). A.G. is supported by the National Institutes of Health (AG049508, AG052411 and RF1AG054014) and the JPB Foundation. A.G. is on the scientific advisory board for Denali Therapeutics and has served as a consultant for AbbVie and Cognition Therapeutics.
Author information
Authors and Affiliations
Contributions
Conception and design of the work, drafting the manuscript: R.E.M. Interpretation: R.E.M., P.M.V., A.F.M. and Q.Z. Analysis: R.E.M., W.D.H. and Q.Z. Critical revision of the manuscript: all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marioni, R.E., Harris, S.E., Zhang, Q. et al. GWAS on family history of Alzheimer’s disease. Transl Psychiatry 8, 99 (2018). https://doi.org/10.1038/s41398-018-0150-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-018-0150-6
