
Abstract
Major depressive disorder (MDD) is a heterogeneous disease. Efforts to identify biomarkers for sub-classifying MDD and antidepressant therapy by genome-wide association studies (GWAS) alone have generally yielded disappointing results. We applied a metabolomics-informed genomic research strategy to study the contribution of genetic variation to MDD pathophysiology by assaying 31 metabolites, including compounds from the tryptophan, tyrosine, and purine pathways, in plasma samples from 290 MDD patients. Associations of metabolite concentrations with depressive symptoms were determined, followed by GWAS for selected metabolites and functional validation studies of the genes identified. Kynurenine (KYN), the baseline plasma metabolite that was most highly associated with depressive symptoms, was negatively correlated with severity of those symptoms. GWAS for baseline plasma KYN concentrations identified SNPs across the beta-defensin 1 (DEFB1) and aryl hydrocarbon receptor (AHR) genes that were cis-expression quantitative trait loci (eQTLs) for DEFB1 and AHR mRNA expression, respectively. Furthermore, the DEFB1 locus was associated with severity of MDD symptoms in a larger cohort of 803 MDD patients. Functional studies demonstrated that DEFB1 could neutralize lipopolysaccharide-stimulated expression of KYN-biosynthesizing enzymes in monocytic cells, resulting in altered KYN concentrations in the culture media. In addition, we demonstrated that AHR was involved in regulating the expression of enzymes in the KYN pathway and altered KYN biosynthesis in cell lines of hepatocyte and astrocyte origin. In conclusion, these studies identified SNPs that were cis-eQTLs for DEFB1 and AHR and, which were associated with variation in plasma KYN concentrations that were related to severity of MDD symptoms.
Similar content being viewed by others
Introduction
Major depressive disorder (MDD) is a common, life-threatening psychiatric disease worldwide1,2. However, the pathophysiology of MDD is not fully understood. Deficiency of the neurotransmitter serotonin (5-HT) appears to play a role in the pathophysiology of MDD and, as a result, drugs, such as selective serotonin reuptake inhibitors (SSRIs) that enhance serotonergic neurotransmission are used to treat MDD. SSRIs are the standard of care drug therapy for MDD. However, many MDD patients fail to respond to SSRI therapy and response may be delayed for weeks or months3. Several genome-wide association studies (GWAS) for both MDD risk4,5,6 and for SSRI treatment outcomes have been performed7,8,9,10,11,12, but few of the top signals from those GWAS have been replicated or functionally validated. This may result, in part, from phenotypic heterogeneity for this disease and/or the lack of biologically based phenotypes13.
To advance our understanding of MDD pathophysiology, the Mayo Clinic Pharmacogenomics Research Network-Antidepressant Medication Pharmacogenomics Study (PGRN-AMPS) performed an SSRI trial in which MDD patients were treated with citalopram or escitalopram, with clinical evaluation and blood sampling at baseline and after 4 and 8 weeks of SSRI therapy14. GWAS for response10 and for plasma drug and drug metabolite levels15 in this trial have been published. In an effort to move beyond genomics alone, we have now applied a metabolomics-informed genomic research strategy in which plasma samples from MDD patients were assayed using a “targeted” metabolomic platform that focused on metabolites that might potentially be related to MDD risk and/or response to SSRI therapy. Those metabolites were often related to monoamine neurotransmitters or their metabolites. We then used the metabolomics data to “inform” subsequent genomic analyses, with the goal of discovering mechanisms related to variation in disease risk and/or drug response16,17. Previous metabolomic studies in depression and SSRI treatment response have revealed novel signatures18,19,20, and by using a metabolomic-informed genomic research strategy, we had previously identified genome-wide significant single-nucleotide polymorphism (SNP) signals that were associated with plasma 5-HT concentrations and SSRI treatment outcomes, with replication in other SSRI GWAS21. However, whether any metabolite(s) or SNP genotype(s) might be associated with the baseline severity of MDD symptoms has not been determined.
The pathophysiology of MDD is almost certainly heterogeneous. Stratification of MDD patients may help identify novel mechanisms that contribute to this disease and could potentially lead to new therapeutic interventions. Extensive data support the hypothesis that variation in monoamine neurotransmission contributes to MDD pathophysiology. In addition, inflammation may also play an important role in MDD pathophysiology22. Concentrations of pro-inflammatory cytokines have been shown to be increased in MDD patients as compared with non-depressed patients23,24 and have been positively correlated with depressive symptom severity25,26. In addition, infection, exposure to endotoxin, therapeutic use of cytokines and psychological stress can all trigger inflammation and lead to depressive symptoms22,27. Gut microbe-associated release of inflammatory cytokines may also be a mechanism that can result in depressive symptoms through the “microbiota–gut–brain” axis28,29,30. Importantly, MDD patients with high levels of inflammation have demonstrated poor response to conventional antidepressant therapies, such as SSRIs, but they may respond to cytokine antagonism31,32,33.
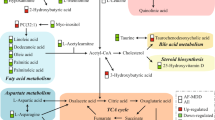
In addition, the “neurotransmission” and the “inflammation” hypotheses are not mutually exclusive and might be linked by tryptophan (TRP) metabolism. TRP is the precursor for both 5-HT and KYN (Fig. 1). Given the importance of 5-HT neurotransmission, the shunting of TRP metabolism from 5-HT to the KYN biosynthetic pathway might contribute to the pathophysiology of depression34,35. The first step in the “KYN Pathway” (Fig. 1) is catalyzed by indoleamine 2,3-dioxygenase 1 (IDO1) and IDO2 and/or tryptophan 2,3-dioxygenase 2 (TDO2)36. Inflammatory cytokines can increase IDO expression, which leads to activation of the “KYN Pathway” and results in depressive-like behavior in mice37,38. However, underlying mechanism(s) remain unclear39,40. KYN, unlike 5-HT, can cross the blood–brain barrier, and the majority of KYN in the brain originates from peripheral blood36. KYN is then further metabolized by either kynurenine aminotransferases (KATs) or kynurenine 3-monooxygenase (KMO) and kynureninase (KYNU), resulting in the generation of “downstream” metabolites, such as kynurenic acid (KYNA), which is neuroprotective, and quinolinic acid (QUIN), which is neurotoxic36. These “downstream” metabolites could play a role in disease pathophysiology through their effects on the N-methyl-D-aspartate (NMDA) receptor36 (Fig. 1). Activation of microgila by immune stimuli accelerates the biosynthesis of neurotoxic QUIN and raises the QUIN/KYNA ratio, which has been suggested as a mechanism that might contribute to depression symptoms36. Increased QUIN concentrations in cerebrospinal fluid (CSF) have been associated with the development of depressive symptoms in patients after interferon treatment, but no differences in QUIN/KYNA ratios were found39. Therefore, even though activation of the KYN pathway has been associated with depression and depressive-like behavior, the cause and effect relationship between the KYN pathway and depression requires clarification, and underlying molecular mechanisms remain unclear.

Tryptophan (TRP) is metabolized by two major pathways: the “kynurenine (KYN) pathway” and the “serotonin (5-HT)” pathways. 5-HT cannot pass the blood–brain barrier (BBB). The majority of TRP is metabolized to form KYN in the liver and is released into peripheral blood. The initial and rate-limiting step in the KYN pathway is catalyzed by indoleamine 2,3-dioxygenase 1 (IDO1), IDO2 and/or tryptophan 2,3-dioxygenase (TDO2)—depending on the tissue involved—to form N-formyl-kynurenine. N-formyl-kynurenine is metabolized by formamidase to produce KYN, which can cross the BBB. Approximately 60% of KYN in the CNS originates from the liver. In the periphery and in the CNS, KYN can be further metabolized by either kynurenine aminotransferases (KATs) or by kynurenine 3-monooxygenase (KMO) and kynureninase (KYNU), leading to the generation of the neuroactive metabolites kynurenic acid (KYNA) or quinolinic acid (QUIN), respectively. KYNA, which is neuroprotective, and QUIN, which is neuroactive in the CNS, have opposite effects on the NMDA receptor. Four KATs, encoded by AADAT, CCBL1, CCBL2, and GOT2, have been shown to catalyze the conversion of KYN to KYNA. The other branches of the KYN pathway involve KMO and KYNU, which catalyze the metabolism of kynurenine to form 3-hydroxykynurenine and 3-hydroxyanthranilic acid, respectively. KYNU can also metabolize KYN to form anthranilic acid, which can then be coverted to 3-hydroxyanthranilic acid by nonspecific hydroxylation. 3-Hydroxyanthranilic acid is metabolized further by 3-hydroxyanthranilic acid 3,4-dioxygenase (HAAO) to form either QUIN or, after a series of reactions, picolinic acid
In the present study, we have applied a metabolomics-informed genomic research strategy to study the severity of MDD symptoms. This approach began with the association of metabolite concentrations with disease severity, followed by GWAS using those metabolites as phenotypes to identify genetic loci associated with variation in concentrations of the metabolites. Of the 31 metabolites that we assayed, plasma KYN concentration was most significantly associated with disease severity in MDD patients prior to antidepressant therapy. GWAS for plasma KYN concentrations identified SNP signals across the beta-defensin 1 (DEFB1) and aryl hydrocarbon receptor (AHR) genes. DEFB1 is an antimicrobial peptide that is secreted from epithelial cells at the surface of multiple tissues, such as the gastrointestinal (GI) and urinary tracts, and which plays an important role in host defense against microbial infections41,42,43. AHR, a ligand-activated transcription factor that is best known for its role in response to environmental toxins44,45, has been reported to regulate the expression of IDO1/2 46,47,48, and KYN is an endogenous AHR ligand49,50. We then performed DEFB1 and AHR functional validation to determine how variation in the expression of those genes might influence phenotypes—e.g. biochemical pathways—related to KYN in cell lines, including cell lines of immune system, liver and central nervous system (CNS) origin.
Materials and methods
Subjects, samples and metabolomic profiling
Patient selection, symptomatic evaluation, and blood sample collection for the PGRN-AMPS clinical trial, which recruited a total of 803 MDD patients, have been described elsewhere10,14,15,21. Patients were required to have a baseline Hamilton Depression Rating Scale (HAMD-17) score >1414. Plasma samples from 306 randomly selected patients for whom samples at all three time points were available were used to perform metabolomic assays with a quantitative, targeted liquid chromatography electrochemical coulometric array (LCECA) platform. We previously used this dataset to perform our studies of plasma 5-HT in these same patients21. See Supplementary Methods for details.
Correlation of metabolites with disease symptoms
Associations of baseline plasma metabolite concentrations with HAMD-17 scores were assessed using Spearman (partial) correlations. All associations with baseline metabolite levels were adjusted for age and sex. Of the 306 subjects for whom metabolite concentrations were measured, six non-Caucasian and 10 non-compliant patients (based on blood drug assays) were removed from the analysis. Therefore, 290 subjects who had plasma metabolite data were included in the present correlation and GWAS analyses.
GWAS analyses
GWAS genotyping and imputation using “1000 Genomes” data as reference have been described previously10,15. GWAS for baseline plasma KYN concentrations and KYN/TRP (K/T) ratio were performed using linear regression assuming additive allelic effects. See Supplementary Methods for details.
Expression quantitative trait loci (eQTL) analyses for DEFB1 and AHR SNPs
DEFB1 and AHR expression by genotype were determined using the GTEx dataset51. We also used eQTL data from the “BRAINEAC” dataset52 to check the DEFB1 and AHR expression in brain tissue. Differences in DEFB1 and AHR expression levels were considered significant when P-values were ≤0.05.
DEFB1–lipopolysaccharide (LPS) neutralization and KYN biosynthesis in monocytic cells
LPS (Sigma, St. Louis, MO) was pre-incubated with or without recombinant human DEFB1 protein (Creative Biolabs, Shirley, NY) in water with 1 mM of dithiothreitol at 37 °C for 30 min. The DEFB1–LPS mixture was then added to serum-free culture media for THP-1 cells (ATCC, Manassas, VA). Cells were pelleted by centrifugation at 4 °C at 100 × g for 5 min for extraction of total RNA or protein lysate preparation. The culture media was collected for quantification of KYN and TRP using high-performance liquid chromatography (HPLC) with UV detection. See Supplementary Methods for details.
AHR and KYN pathways in hepatocyte and CNS-derived cells
HepaRG cells (Biopredict, Rennes, France) were cultured and differentiated into hepatocyte-like cells using the manufacture’s protocol. Differentiated HepaRG cells and human U-87 MG glioblastoma cells (ATCC, Manassas, VA) were transfected with pooled mRNA-specific siRNAs for knockdown (KD) studies. Total RNA and whole cell lysates were then prepared for mRNA quantification and Western blot analysis. Culture media was used for HPLC analyses of KYN and TRP. See Supplementary Methods for details.
Statistical analyses
Data analyses were performed using JMP (SAS Institute, Cary, NC), and graphs were plotted using GraphPad Prism (GraphPad Software, La Jolla, CA). Statistical comparisons for the functional studies were made using Student’s t-test or one-way ANOVA.
Results
Association of plasma metabolite concentrations with depressive symptoms
Baseline plasma KYN was the metabolite that was most significantly associated with the baseline severity of depressive symptoms, as measured by the HAMD-17 (P = 0.008) (Supplementary Table S1). The “nominal” P-values listed in the table have not been corrected for multiple comparisons because our purpose was to identify metabolites for use in GWAS to discover genetic variation associated with variation in metabolite concentrations. The negative correlation coefficient (r = −0.157) indicated that lower plasma KYN concentrations were associated with more severe depressive symptoms. To pursue these observations, we performed a discovery GWAS using baseline plasma KYN concentration as the phenotype.
Plasma KYN GWAS
The Manhattan plot of the GWAS for baseline plasma KYN concentrations showed a cluster of SNPs across the DEFB1 gene (top SNP rs5743467, P = 8.18E−07) and another cluster of SNPs across the AHR gene (top SNP rs17137566, P = 6.22E−06) (Fig. 2), neither of which were genome-wide significant. Other SNPs with P-values lower than those for the SNPs across DEFB1 and AHR were either located in “gene deserts” or had very low (≤0.02) minor allele frequencies (MAFs), which increased the possibility that they might be false positive associations (Supplementary Table S2). Plasma KYN concentrations were higher in patients homozygous for the DEFB1 variant SNP (rs5743467) genotype (G/G), but lower in patients homozygous for the AHR variant SNP (rs17137566) genotype (C/C) as compared with the major alleles (Supplementary Fig. S1). A combination of both AHR and DEFB1 SNP genotypes resulted in the most significant differences in KYN concentrations in which patients who were homozygous wild type for the DEFB1 SNPs (C/C) and homozygous variant for the AHR SNPs (C/C) had the lowest KYN concentrations, and vice versa (Supplementary Fig. S1C). DEFB1 and AHR GWAS SNPs with P-values < 5.0E−04 are listed in Supplementary Tables S3 and S4, respectively. MAFs for DEFB1 SNPs ranged from 0.18 to 0.49 and between 0.17 and 0.23 for AHR SNPs in the GWAS subjects, all of whom were Caucasian. These MAFs are consistent with those reported by the 1000 Genomes Project.

a Manhattan plot for baseline plasma KYN concentrations. SNPs across the DEFB1 and AHR genes have been highlighted, with rs5743467 as the “top” DEFB1 SNP (P-value = 8.18E−07) and, rs17137566 as the “top” AHR SNP (P-value = 6.22E−06). Regional association plots (Locus Zooms) for the DEFB1 gene B and the AHR gene C are also shown. Circles and diamonds represent observed and imputed SNPs, respectively. The color of each SNP represents its’ linkage disequilibrium (LD) with the “top SNP”, which is colored purple
Since KYN is a metabolite of TRP and since the plasma K/T ratio is commonly used as a marker for inflammation, we asked whether the K/T ratio might also be associated with severity of MDD symptoms. We found that, like baseline KYN, the K/T ratio was also negatively associated with severity of MDD symptoms as measured by the HAMD-17 (r = −0.168, P = 0.004), indicating that a decrease in K/T ratio was associated with more severe MDD symptoms. Therefore, we also performed a discovery GWAS for baseline plasma K/T ratio (Supplementary Fig. S2). The same SNP signal across the DEFB1 gene that was associated with plasma KYN concentrations, was associated with plasma K/T ratio in these MDD patients (top SNP rs5743467, P = 2.15E−07) (Supplementary Table S5). Other SNPs that were associated with variation in the K/T ratio had very low MAFs (≤0.04) (Supplementary Table S6). No other SNP signals were present in both the KYN and K/T ratio GWAS with P-values less than 1E−04.
As mentioned previously, the goal of this study was to use metabolomics-informed genomics to understand the biology underlying MDD pathophysiology. DEFB1 is an antimicrobial peptide associated with innate immunity41,42, and bacterial infection-induced inflammation has been associated with depression22. AHR has been shown to regulate KYN biosynthesis46,47,48, and KYN is an AHR ligand49,50. Therefore, we pursued both the DEFB1 and AHR SNP signals functionally even though the P-values for these signals were not genome-wide significant.
DEFB1 and AHR SNPs are cis-eQTLs
We next determined the relationship of the SNPs identified in the GWAS with DEFB1 and AHR expression using a number of databases. We began with the GTEx dataset51 and found that the variant genotype for the DEFB1 “top SNP”, rs5743467, was associated with decreased DEFB1 expression, and that the variant genotype for the AHR “top SNP”, rs17137566, was associated with decreased AHR expression in many tissues, including brain, colon, and esophagus (Fig. 3). We also performed eQTL analyses using the BRAINEAC dataset52. The “top SNPs” for DEFB1 and AHR were also cis-eQTLs in human cerebellar cortex, thalamus, and other brain tissues (Supplementary Table S7). The variant alleles were once again associated with decreased DEFB1 and AHR expression. We also used RNA-seq data from 48 lymphoblastoid cell lines (LCLs) generated from European-American subjects to verify these obsersvations53,54. DEFB1 was not expressed in this cell line but AHR mRNA levels were significantly decreased in LCLs homozygous for variant genotypes for the AHR “top SNP” (rs17137566, P = 0.042).

eQTL analysis for the top DEFB1 A and AHR B SNPs based on the GTEx dataset51. A DEFB1 mRNA expression was significantly decreased in human cerebellum (left), transverse colon (middle) and esophageal mucosa (right) from individuals with rs5743467 variant genotypes (G) when compared with WT genotype (C) (P < 0.05). B AHR mRNA expression was significantly decreased in human cerebellum (left), sigmoid colon (middle) and esophageal mucosa (right) from subjects with rs17137566 variant genotypes (C) when compared with those for subjects homozygous for the WT genotype (T) (P < 0.05). mRNA levels were determined by RNA sequencing data available in GTEx
DEFB1 and KYN pathway functional genomics in monocytic cells
DEFB1 is an antimicrobial peptide that plays an important role in gut microbiome homeostasis and is constitutively secreted by epithelial cells41. Like other antimicrobial peptides, DEFB1 kills bacteria by cell wall permeabilization41 and it neutralizes LPS55—immune-stimulating molecules on the surface of Gram-negative bacteria—thus protecting the host from bacterial infection and subsequent immune response. Exposure to bacteria or bacterial products such as LPS can activate circulating monocytes, which release pro-inflammatory cytokines that can then activate the KYN pathway by upregulating IDO1 expression22. Since infection and inflammation have been associated with increased KYN concentrations and the development of depressive symptoms and since the “microbiota–gut–brain” axis has been hypothesized to play a role in MDD pathophysiology, we investigated the possible influence of DEFB1 on KYN concentrations.
Specifically, we used THP-1 cells, a widely used human monocytic cell line56,57,58, exposed to LPS to mimic the “innate immune response” triggered by bacterial infection or exposure to bacterially derived compounds, such as LPS. The mRNA expression of the KYN biosynthesizing enzyme IDO1 was significantly increased after LPS treatment (see Figs. 1 and 4A). Tumor necrosis factor alpha (TNF) mRNA was also highly induced after LPS exposure. We used TNF expression as an indicator of LPS stimulation. The level of IDO1 protein was also significantly induced after LPS treatment (Fig. 4B). The induction of IDO1 was associated with an increase in KYN concentration, a decrease in TRP concentration and—as expected—an increased K/T ratio in the cell culture media (Fig. 4C). We also observed that the expression of TDO2 and IDO2 mRNA was induced after LPS treatment (data not shown). However, TDO2 protein was undetectable, perhaps because the level of TDO2 expression in this cell line is very low, suggesting a limited contribution to KYN synthesis in this cell line. IDO2 mRNA was also much less highly induced than that of IDO1. Therefore, immune stimulation, as modeled by LPS treatment, altered KYN pathway metabolism in THP-1 monocytic cells primarily by the induction of IDO1 expression.

A mRNA expression was determined by qRT-PCR after THP-1 cells were exposed to 10 and 100 ng/ml of LPS at different time points. Compared to vehicle treated cells, IDO1 mRNA levels were significantly increased after LPS treatment. mRNA levels for TNF, a pro-inflammatory cytokine used as a positive control for LPS effect, were also significantly increased after LPS treatment. B IDO1 protein expression was increased as analyzed by Western blot after LPS treatment. C KYN concentrations (left) in cell culture media were undetectable after 24 and 48 h of vehicle treatments, but were significantly increased after 10 ng/ml LPS treatment. At the same time, TRP concentrations (middle) were significantly decreased in cell culture media after LPS treatment and the K/T ratio was increased (right). After 3, 6, or 12 h of LPS treatment, KYN concentrations were undetectable. D mRNA levels for IDO1 and TNF were significantly increased after 10 ng/ml LPS treatment, but recombinant human DEFB1 co-incubation with LPS significantly decreased mRNA levels for IDO1 and TNF when compared with LPS treatment alone. E When DEFB1 was co-incubated with LPS as compared to LPS alone, KYN concentrations (left) in cell culture media were significantly decreased, TRP concentrations were increased (middle) and K/T ratios (right) were decreased after DEFB1 was pre-incubated with LPS when compared with results for cells treated with LPS alone. N ≥ 3 for all the experiments. Data = mean ± SEM, with statistical significance determined by two-tailed t-test denoted as *P < 0.05, **P < 0.01, and ***P < 0.001
We then used THP-1 cells to investigate the effect of DEFB1 on LPS-stimulated KYN metabolism. Addition of recombinant DEFB1 to THP-1 cell culture media decreased LPS response as measured by TNF expression. In addition, IDO1 expression was decreased after the addition of DEFB1, with a parallel decrease in KYN concentration, an increase in TRP concentration and a decrease in the K/T ratio as compared to LPS treatment alone (Fig. 4D, E). Therefore, DEFB1 could alter the response of THP-1 cells to LPS treatment, as previously described55, and it could alter KYN biosynthesis. These results were consistent with our GWAS data in which the variant genotype (G/G) for the DEFB1 “top SNP” was associated with decreased DEFB1 expression (Fig. 3A), higher plasma KYN concentrations (Supplementary Fig. S1A and S1C) and a higher K/T ratio in MDD patients (Supplementary Table S5).
AHR and KYN pathway functional genomics in hepatic-derived and astrocytic-derived cells
The majority of plasma KYN is synthesized in the liver where TDO2 is highly expressed, and peripheral KYN is the source of ∼60% of KYN in the brain36. Therefore, we also performed functional studies with HepaRG cells—liver progenitor cells that can be differentiated into hepatocyte-like cells that retain many of the characteristics of human hepatocytes59. To mimic the effect of the variant AHR SNPs that were associated with decreased AHR expression (Fig. 3B), we knocked down AHR in HepaRG cells and assayed the mRNA expression of enzymes, such as TDO2, IDO1, IDO2, KMO, KYNU, CCBL1, AADAT, CCBL2, GOT2, and HAAO that regulate KYN pathway metabolism (see Fig. 1). We also determined the expression of CYP1A1 as a prototypic gene that is regulated by AHR60. As anticipated, KD of AHR dramatically decreased CYP1A1 expression (Fig. 5A). AHR KD also resulted in significantly increased mRNA expression for TDO2, the gene that encodes the rate-limiting enzyme for KYN biosynthesis in liver36, as well as KMO and KYNU, genes encoding “downstream” KYN pathway enzymes that catalyze reactions leading to the generation of QUIN—a neurotoxic NMDA receptor agonist (Fig. 1). IDO1 is not expressed in this cell line, and mRNA levels for other KYN pathway genes that we tested were unchanged after AHR KD. The increase in TDO2, KMO, and KYNU mRNA expression was also reflected in protein levels as measured by Western blot analysis (Fig. 5B). After treatment with a prototypic AHR ligand, 3-methylcholanthrene (3MC)61, expression of TDO2, KMO, and KYNU was significantly decreased (Fig. 5C), while the expression of CYP1A1, as anticipated, increased.

A mRNA expression determined by qRT-PCR after AHR KD in HepaRG cells. TDO2, KMO, and KYNU expression was significantly increased and AHRR and CYP1A1 expression was significantly decreased following AHR KD. B Protein expression analysis for AHR, TDO2, KMO, and KYNU by Western blot analysis after AHR KD in HepaRG cells. C mRNA expression in HepaRG cells determined by qRT-PCR after 24-h treatment with 1 µM 3-MC, an AHR agonist. TDO2, KMO, and KYNU expression was significantly decreased. AHRR, CYP1A1 (in HepaRG), was induced by 3-MC, indicating that AHR was activated by the treatment. D KYN concentrations in HepaRG cell culture media after AHR and AHR plus KMO or KYNU KD. E TDO2, KMO, and KYNU expression was significantly increased while AHRR and CYP1B1 expression was significantly decreased following AHR KD. F AHR, TDO2, KMO, and KYNU protein concentrations were significantly altered in U-87 MG cells after AHR KD. G mRNA expression in U-87 MG cells after 1 µM 3-MC treatment showed significant decreases in TDO2, KMO, and KYNU expression and significant increases in AHRR and CYP1B1 expression. H KYN concentrations in U-87 MG cell culture media were significantly decreased after AHR KD but were less decreased after AHR KD together with KMO or KYNU KD. N ≥ 3 for all experiments. Data = mean ± SEM, with statistical significance determined by two-tailed t-test denoted as *P < 0.05, **P < 0.01 and ***P < 0.001 when compared with control. ns = not significant
To determine the relationship between these results and KYN biosynthesis, concentrations of KYN and TRP were assayed in the HepaRG culture media before and after AHR KD. KYN concentrations in the cell culture media decreased significantly after AHR KD when compared with cells exposed to control siRNA (Fig. 5D). This occurred in spite of induction of the expression of TDO2, the enzyme that catalyzes the rate-limiting step in the biosynthesis of KYN in liver, probably as a result of increased expression of KMO and KYNU—“downstream” enzymes that utilize KYN as a substrate. In support of that hypothesis, KD of AHR together with KD of either KMO or KYNU resulted in increased KYN culture media concentrations when compared with AHR KD alone (Fig. 5D). Meanwhile, media TRP concentrations decreased both after AHR KD and AHR KD together with KMO or KYNU KD (Supplementary Fig. S3B). Although KYN concentration did not display a statistically significant change after AHR + KMO KD vs. AHR KD alone, there was a trend toward KYN concentration increase after AHR + KMO KD. Finally, after AHR KD combined with exposure to a potent KMO inhibitor (Ro 61-8048, IC50 = 0.037 µM), KYN concentrations in the cell culture media increased although TRP concentrations remained decreased, further supporting the conclusion that increased KMO expression after AHR KD contributed to the observed decrease in KYN media concentrations (Supplementary Fig. S3C).
These results showed that AHR KD, a model that we had used to mimic the effect of variant AHR SNP genotypes on AHR expression, resulted in decreased KYN concentrations in cell culture media. This result was consistent with our GWAS, which showed that variant AHR SNP genotypes (C/C) were associated with lower AHR mRNA (Fig. 3B) and with lower levels of plasma KYN in MDD patients (Supplementary Fig. S1B).
KYN biosynthesis in brain primarily occurs in astrocytes36. To determine whether the regulation of KYN pathway genes by AHR might also occur in CNS-derived cells, we used an astrocyte-derived cell line, the U-87 MG glioblastoma cells, to perform experiments parallel to those performed with HepaRG cells. U-87 MG glioblastoma cells express TDO2 as well as KYNU while expressing KMO at a relatively low level. We found that AHR KD significantly increased TDO2, KMO, and KYNU expression at both the mRNA and protein levels (Figs. 5E, F) and that 3-MC treatment down-regulated TDO2, KMO, and KYNU expression (Fig. 5G), similar to the HepaRG results (Fig. 5A-D). We also measured KYN and TRP concentrations in U-87 MG culture media. Consistent with the HepaRG results, KYN concentrations in U-87 MG cell culture media were significantly decreased following AHR KD but rose when KMO or KYNU were knocked-down together with AHR (Fig. 5H), indicating that increased expression of KMO and KYNU after AHR KD plays a role in the decrease in KYN concentration after AHR KD alone. We observed that the increase in KYN concentration after AHR KD together with KYNU KD was more significant than that for KMO KD (Fig. 5H), which might indicate that KYNU KD affects two KYN downstream metabolizing pathways (Fig. 1). We also observed that culture media TRP concentrations decreased after AHR KD but that treatment with the KMO inhibitor increased KYN media concentrations after AHR KD, with continued decrease in TRP concentrations, supporting the conclusion that increased KMO expression after AHR KD is one cause of the decrease in KYN concentrations (Supplementary Fig. S3E–H). Taken together, these results strongly suggest that the decrease in KYN concentration after AHR KD resulted from enhanced KYN downstream metabolism as a result of increased expression of KMO and KYNU.
Finally, although TRP catabolism in the brain occurs mainly in glial cells, we also tested the effect of AHR KD using neurons differentiated from human-induced pluripotent stem cells (iPSCs). TDO2, KMO, and KYNU mRNA levels also increased after AHR KD in these neuronal cells (Supplementary Fig. S4). Baseline levels of expression for these genes were low in the iPSC-derived neurons, which resulted in low KYN concentrations (0.23 + 0.02 µM) in the culture media, perhaps explaining why we did not observe significant changes in these concentrations after AHR KD (data not shown).
Association of DEFB1 SNP signals with severity of MDD symptoms
To determine whether the DEFB1 and AHR SNPs that were identified during the GWAS for plasma KYN concentrations might be directly associated with severity of MDD symptoms, we determined associations of these SNPs with the severity of symptoms measured by both HAMD-17 and QIDS-C16 scores in the larger cohort of all 803 patients who were recruited to the Mayo-PGRN AMPS study14. One of the DEFB1 SNPs, rs2702877, was significantly associated with the severity of MDD symptom as measured by both HAMD-17 (P = 1.74E−04) and QIDS-C16 scores (P = 1.25E−05) for all 803 patients (Supplementary Table S8). The variant allele for this SNP was associated with more severe symptoms at baseline. This SNP was associated with increased DEFB1 expression and lower KYN concentrations. None of the AHR SNPs was associated with HAMD-17 or QIDS-C16 scores in these 803 patients.
This rs2702877 SNP was not in LD with the rs5743467 “top hit” SNP that was most highly associated with plasma KYN concentrations (r 2 = 0.09 in Caucasian, 1K Genomes data). However, rs2702877 was a stronger eQTL (Supplementary Fig. S5) and had a higher MAF (0.31) than the rs5743467 SNP.
Discussion
We have applied a metabolomics-informed genomic research strategy to study the severity of MDD symptoms in an attempt to address the molecular basis for these symptoms. We began by assaying 31 metabolites that might be relevant to MDD pathophysiology, including compounds from the tryptophan, tyrosine, and purine pathways, in plasma samples from 290 MDD patients. Plasma KYN was the metabolite for which concentrations were most highly associated with baseline depressive symptoms as measured by HAMD-17 scores. KYN concentrations were negatively correlated with severity of depressive symptoms (Supplementary Table S1). This result was consistent with a recently published study that reported plasma KYN concentrations were negatively associated with suicidal ideation, one of the most severe symptoms of MDD62.
KYN can cross the blood–brain barrier and peripheral KYN, primarily generated in the liver, is the source of ∼60% of CNS KYN36. However, the relationship of plasma KYN concentration to MDD symptom severity remains unclear. Previous studies have not consistently determined whether increased or decreased plasma KYN concentrations were associated with symptoms of MDD, perhaps because of the metabolomic assays used, MDD phenotypic heterogeneity, sample size and/or cohort composition39,63. In an attempt to further understand possible mechanisms underlying the association of plasma KYN concentrations with severity of MDD symptoms, we performed a GWAS with plasma KYN concentration as the phenotype to identify genetic factors that might contribute to inter-individual variation in plasma KYN concentrations. The GWAS identified SNPs across the DEFB1 and AHR genes that were associated with variation in baseline plasma KYN concentrations in these MDD patients (Fig. 2). The same SNP signal for DEFB1 was also associated with variation in the plasma K/T ratio (Supplementary Fig. S2). The SNP signals across DEFB1 and AHR were cis-eQTLs for DEFB1 and AHR expression, respectively (Fig. 3).
Depression has been associated with inflammation, which can be influenced by the interaction between the microbiome and the host. DEFB1 is an antimicrobial peptide that plays a role in host defense against bacterial infection. Therefore, we performed functional studies to investigate the possible relationship of DEFB1 to KYN biosynthesis in innate-immune-related cell lines. DEFB1 inhibited LPS-stimulated transcription of KYN pathway genes, which resulted in smaller increases in KYN concentrations in cell culture media after LPS exposure. These results were consistent with our GWAS data indicating that WT genotype for the DEFB1 “top SNP”, which was associated with increased DEFB1 expression (Fig. 3), was associated with lower plasma KYN concentrations in MDD patients (Supplementary Fig. S1A).
DEFB1 kills bacteria by the same mechanism as other antimicrobial peptides but, unlike other antimicrobial peptides that are induced by immune stimuli, DEFB1 is constitutively expressed and secreted from epithelial cells41. Furthermore, the expression of DEFB1 is not affected by pro-inflammatory or bacterial molecules43, such as LPS64. DEFB1 is also known to play a role in maintaining gut–microbiome homeostasis43.Therefore, intrinsic variation in DEFB1 expression, such as that resulting from genetic polymorphisms, could be related to inter-individual variation in disease phenotypes. In fact, genetic polymorphisms in DEFB1 have been associated with inflammatory bowel disease65 and other immune-related diseases66,67,68. The SNPs across DEFB1 identified in our GWAS for baseline plasma KYN concentrations and the GWAS for plasma K/T ratio in our 290 MDD patients were strong cis-eQTLs for DEFB1 expression in many tissues, including gut (Fig. 3). One of the DEFB1 SNPs was also directly associated with the severity of MDD symptoms in all 803 patients who had been recruited to the Mayo-PGRN AMPS trial (Supplementary Table S8). Although broadly expressed in many tissues, DEFB1 appears to be functionally important at mucosal membranes, since its antimicrobial activity can be increased in an anaerobic environment which changes its structure to a more active form by disulfide bond reduction42. The DEFB1 SNPs are not only associated with plasma KYN concentrations but also with plasma KYN/TRP ratios. Maintenance of homeostatic balance among commensal microbes is critical for health69,70, and evidence suggests that gut dysbiosis could alter brain function and mental illness, including MDD28,29. Therefore, our data suggest that SNPs across DEFB1 may be associated with individual variation in DEFB1 levels and variation in host–microbiome interaction, which could result in altered KYN biosynthesis and MDD symptom severity. Future studies that assay DEFB1 and inflammatory cytokines in patient samples, as well as studies of MDD patient microbiota will be required to test the hypothesis that DEFB1 may play a role in MMD pathophysiology through the “microbiota–gut–brain” axis.
In addition to the DEFB1 signal, SNPs across AHR were also associated with plasma KYN concentrations in our MDD patients. Since the majority of KYN in the brain originates from the liver, and since AHR has been shown to play a role in KYN biosynthesis by influencing the expression of IDO1, we also investigated the impact of AHR expression on KYN biosynthesis in both hepatocytes and CNS-derived cell lines. We demonstrated that AHR KD resulted in increased expression of TDO2, KMO, and KYNU, enzymes that catalyze KYN biosynthesis and its downstream metabolism (Fig. 1). TDO2 is the major enzyme responsible for the biosynthesis of KYN in the liver71. KMO catalyzed biotransformation ultimately results in the conversion of KYN to the neurotoxic metabolite, QUIN, an NMDA receptor agonist, which has been reported to be associated with depressive symptoms39 and other neuropsychiatric diseases72,73,74. The increase in KMO and KYNU expression after AHR KD may help explain why variant AHR SNP genotypes, which were associated with lower AHR expression, were also associated with lower plasma KYN concentrations. Since the potential actions of KYN metabolites on the glutamatergic system75,76 are thought to contribute to MDD risk, our results suggest that AHR may play a role in regulating KYN biosynthesis and, as a result, may contribute to MDD pathophysiology.
In summary, our results raise the possibility of the involvement of DEFB1 and AHR genetic polymorphisms in MDD pathophysiology—at least in part through an effect on the biosynthesis of KYN.
References
Martinowich, K., Jimenez, D. V., Zarate, C. A. Jr. & Manji, H. K. Rapid antidepressant effects: moving right along. Mol Psychiatry 18, 856–863 (2013).
Kessler, R. C. et al. Prevalence and effects of mood disorders on work performance in a nationally representative sample of U.S. workers. Am J Psychiatry 163, 1561–1568 (2006).
Trivedi, M. H. et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 163, 28–40 (2006).
Sullivan, P. F. et al. Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol Psychiatry 14, 359–375 (2009).
Wray, N. R. et al. Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry 17, 36–48 (2012).
Ripke, S. et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 18, 497–511 (2013).
Ising, M. et al. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch. Gen. Psychiatry 66, 966–975 (2009).
Garriock, H. A. et al. A genomewide association study of citalopram response in major depressive disorder. Biol. Psychiatry 67, 133–138 (2010).
Uher, R. et al. Genome-wide pharmacogenetics of antidepressant response in the GENDEP project. Am. J. Psychiatry 167, 555–564 (2010).
Ji, Y. et al. Pharmacogenomics of selective serotonin reuptake inhibitor treatment for major depressive disorder: genome-wide associations and functional genomics. Pharmacogenom. J. 13, 456–463 (2013).
Biernacka, J. M. et al. The International SSRI Pharmacogenomics Consortium (ISPC): a genome-wide association study of antidepressant treatment response. Transl. Psychiatry 5, e553 (2015).
Tansey, K. E. et al. Contribution of common genetic variants to antidepressant response. Biol. Psychiatry 73, 679–682 (2013).
Krishnan, V. & Nestler, E. J. The molecular neurobiology of depression. Nature 455, 894–902 (2008).
Mrazek, D. A. et al. Treatment outcomes of depression: the pharmacogenomic research network antidepressant medication pharmacogenomic study. J. Clin. Psychopharmacol. 34, 313–317 (2014).
Ji, Y. et al. Citalopram and escitalopram plasma drug and metabolite concentrations: genome-wide associations. Br. J. Clin. Pharmacol. 78, 373–383 (2014).
Kaddurah-Daouk, R. & Weinshilboum, R. M. Pharmacometabolomics: implications for clinical pharmacology and systems pharmacology. Clin. Pharmacol. Ther. 95, 154–167 (2014).
Neavin, D., Kaddurah-Daouk, R. & Weinshilboum, R. Pharmacometabolomics informs pharmacogenomics. Metabolomics 12, 121 (2016).
Kaddurah-Daouk, R. et al. Pretreatment metabotype as a predictor of response to sertraline or placebo in depressed outpatients: a proof of concept. Transl. Psychiatry 1, e26 (2011).
Kaddurah-Daouk, R. et al. Pharmacometabolomic mapping of early biochemical changes induced by sertraline and placebo. Transl. Psychiatry 3, e223 (2013).
Zhu, H. et al. Pharmacometabolomics of response to sertraline and to placebo in major depressive disorder—possible role for methoxyindole pathway. PLoS One 8, e68283 (2013).
Gupta, M. et al. TSPAN5, ERICH3 and selective serotonin reuptake inhibitors in major depressive disorder: pharmacometabolomics-informed pharmacogenomics. Mol. Psychiatry 21, 1717–1725 (2016).
Miller, A. H. & Raison, C. L. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 16, 22–34 (2016).
Raison, C. L., Capuron, L. & Miller, A. H. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 27, 24–31 (2006).
Miller, A. H., Maletic, V. & Raison, C. L. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 65, 732–741 (2009).
Thomas, A. J. et al. Increase in interleukin-1beta in late-life depression. Am. J. Psychiatry 162, 175–177 (2005).
Alesci, S. et al. Major depression is associated with significant diurnal elevations in plasma interleukin-6 levels, a shift of its circadian rhythm, and loss of physiological complexity in its secretion: clinical implications. J. Clin. Endocrinol. Metab. 90, 2522–2530 (2005).
Iwata, M. et al. Psychological stress activates the inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 receptor. Biol. Psychiatry 80, 12–22 (2016).
Rogers, G. B. et al. From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol. Psychiatry 21, 738–748 (2016).
Foster, J. A. & McVey Neufeld, K. A. Gut-brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. 36, 305–312 (2013).
Maes, M., Kubera, M. & Leunis, J. C. The gut-brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol. Lett. 29, 117–124 (2008).
Raison, C. L. et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70, 31–41 (2013).
Kohler, O. et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 71, 1381–1391 (2014).
Kappelmann, N., Lewis, G., Dantzer, R., Jones, P. B., Khandaker, G. M. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol. Psychiatry advance online publication, 18 October 2016; doi:10.1038/mp.2016 . 167 (2016).
Lapin, I. P. & Oxenkrug, G. F. Intensification of the central serotoninergic processes as a possible determinant of the thymoleptic effect. Lancet 1, 132–136 (1969).
Oxenkrug, G. F. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: the serotonin hypothesis revisited 40 years later. Isr. J. Psychiatry Relat. Sci. 47, 56–63 (2010).
Schwarcz, R., Bruno, J. P., Muchowski, P. J. & Wu, H. Q. Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477 (2012).
O’Connor, J. C. et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 14, 511–522 (2009).
O’Connor, J. C. et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J. Neurosci. 29, 4200–4209 (2009).
Raison, C. L. et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol. Psychiatry 15, 393–403 (2010).
Agudelo, L. Z. et al. Skeletal muscle PGC-1alpha1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 159, 33–45 (2014).
Ganz, T. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3, 710–720 (2003).
Schroeder, B. O. et al. Reduction of disulphide bonds unmasks potent antimicrobial activity of human beta-defensin 1. Nature 469, 419–423 (2011).
Ostaff, M. J., Stange, E. F. & Wehkamp, J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol. Med. 5, 1465–1483 (2013).
Murray, I. A., Patterson, A. D. & Perdew, G. H. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat. Rev. Cancer 14, 801–814 (2014).
Denison, M. S. & Nagy, S. R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43, 309–334 (2003).
Vogel, C. F., Goth, S. R., Dong, B., Pessah, I. N. & Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 375, 331–335 (2008).
Jux, B., Kadow, S. & Esser, C. Langerhans cell maturation and contact hypersensitivity are impaired in aryl hydrocarbon receptor-null mice. J. Immunol. 182, 6709–6717 (2009).
Nguyen, N. T. et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 107, 19961–19966 (2010).
Opitz, C. A. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 (2011).
Bessede, A. et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 511, 184–190 (2014).
GTExConsortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
Niu, N. et al. Radiation pharmacogenomics: a genome-wide association approach to identify radiation response biomarkers using human lymphoblastoid cell lines. Genome Res. 20, 1482–1492 (2010).
Liu, D. et al. Breast cancer chemoprevention pharmacogenomics: Deep sequencing and functional genomics of the ZNF423 and CTSO genes. npj Breast Cancer 3, 30 (2017).
Lee, S. H., Jun, H. K., Lee, H. R., Chung, C. P. & Choi, B. K. Antibacterial and lipopolysaccharide (LPS)-neutralising activity of human cationic antimicrobial peptides against periodontopathogens. Int. J. Antimicrob. Agents 35, 138–145 (2010).
Takashiba, S. et al. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kappaB. Infect. Immun. 67, 5573–5578 (1999).
Park, E. K. et al. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm. Res. 56, 45–50 (2007).
Daigneault, M., Preston, J. A., Marriott, H. M., Whyte, M. K. & Dockrell, D. H. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5, e8668 (2010).
Cerec, V. et al. Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology 45, 957–967 (2007).
Denison, M. S. & Whitlock, J. P. Jr. Xenobiotic-inducible transcription of cytochrome P450 genes. J. Biol. Chem. 270, 18175–18178 (1995).
Bersten, D. C., Sullivan, A. E., Peet, D. J. & Whitelaw, M. L. bHLH-PAS proteins in cancer. Nat. Rev. Cancer 13, 827–841 (2013).
Setoyama, D. et al. Plasma metabolites predict severity of depression and suicidal ideation in psychiatric patients—a multicenter pilot analysis. PLoS One 11, e0165267 (2016).
Orlikov, A. B., Prakhye, I. B. & Ryzov, I. V. Kynurenine in blood plasma and DST in patients with endogenous anxiety and endogenous depression. Biol. Psychiatry 36, 97–102 (1994).
O’Neil, D. A. et al. Expression and regulation of the human beta-defensins hBD-1 and hBD-2 in intestinal epithelium. J. Immunol. 163, 6718–6724 (1999).
Kocsis, A. K. et al. Association of beta-defensin 1 single nucleotide polymorphisms with Crohn’s disease. Scand. J. Gastroenterol. 43, 299–307 (2008).
Chen, Q. X. et al. Genomic variations within DEFB1 are associated with the susceptibility to and the fatal outcome of severe sepsis in Chinese Han population. Genes Immun. 8, 439–443 (2007).
Jurevic, R. J., Bai, M., Chadwick, R. B., White, T. C. & Dale, B. A. Single-nucleotide polymorphisms (SNPs) in human beta-defensin 1: high-throughput SNP assays and association with Candida carriage in type I diabetics and nondiabetic controls. J. Clin. Microbiol. 41, 90–96 (2003).
Kalus, A. A. et al. Association of a genetic polymorphism (-44 C/G SNP) in the human DEFB1 gene with expression and inducibility of multiple beta-defensins in gingival keratinocytes. BMC Oral Health 9, 21 (2009).
Rooks, M. G. & Garrett, W. S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352 (2016).
Sekirov, I., Russell, S. L., Antunes, L. C. & Finlay, B. B. Gut microbiota in health and disease. Physiol. Rev. 90, 859–904 (2010).
Zhang, Y. et al. Crystal structure and mechanism of tryptophan 2,3-dioxygenase, a heme enzyme involved in tryptophan catabolism and in quinolinate biosynthesis. Biochemistry 46, 145–155 (2007).
Wonodi, I. et al. Downregulated kynurenine 3-monooxygenase gene expression and enzyme activity in schizophrenia and genetic association with schizophrenia endophenotypes. Arch. Gen. Psychiatry 68, 665–674 (2011).
Lavebratt, C. et al. The KMO allele encoding Arg452 is associated with psychotic features in bipolar disorder type 1, and with increased CSF KYNA level and reduced KMO expression. Mol. Psychiatry 19, 334–341 (2014).
Zwilling, D. et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 145, 863–874 (2011).
Myint, A. M. et al. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J. Affect. Disord. 98, 143–151 (2007).
Muller, N. & Schwarz, M. J. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol. Psychiatry 12, 988–1000 (2007).
Acknowledgements
This work was supported, in part, by NIH grants U19 GM61388 (The Pharmacogenomics Research Network), RO1 GM28157, R24 GM078233 (The Metabolomics Research Network for Drug Response Phenotype), RO1 MH108348, and by RC2 GM092729 (The Metabolomics Network for Drug Response Phenotypes). A.P.A. was supported by an NCSA/CompGen fellowship from the University of Illinois at Urbana-Champaign and a Summer Research Fellowship from the Mayo Clinic Center for Individualized Medicine. R.K.I. was supported by an IBM Faculty Award, NSF MRI grant CNS 13-37732, and NSF grants, CNS 16-24790 and CNS 16-24615, for the Center for Computational Biology and Genomic Medicine. We also want to acknowledge the critical contributions of the late Dr. David Mrazek, our colleague who designed and initiated the Mayo PGRN SSRI trial. Trial name: Pharmacodynamics and Pharmacokinetics of Citalopram and Escitalopram (PGRN-SSRI). URL: https://clinicaltrials.gov/show/NCT00613470. Registration number: NCT00613470.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
Dr. Balmiki Ray is a salaried employee of Assurex Health Inc. Dr. Wayne Matxon and Dr. Swati Bhasin are salaried employees of Ixcela. Dr. Mark Frye receives grant support from AssureRx, Janssen Research & Development, Myriad, and Pfizer. He is also a consultant for Janssen Research & Development, LLC, Mitsubishi Tanabe Pharma Corporation, Myriad Genetics, Sunovion, Supernus Pharmaceuticals, Teva Pharmaceuticals, and Neuralstem, Inc. and he receives CME/Travel Support from the American Physician Institute and CME Outfitters. Drs. Wang and Weinshilboum are founders and stockholders in OneOme LLC.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, D., Ray, B., Neavin, D.R. et al. Beta-defensin 1, aryl hydrocarbon receptor and plasma kynurenine in major depressive disorder: metabolomics-informed genomics. Transl Psychiatry 8, 10 (2018). https://doi.org/10.1038/s41398-017-0056-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-017-0056-8
This article is cited by
-
Stress-induced despair behavior develops independently of the Ahr-RORγt axis in CD4 + cells
Scientific Reports (2022)
-
Detection of the role of intestinal flora and tryptophan metabolism involved in antidepressant-like actions of crocetin based on a multi-omics approach
Psychopharmacology (2022)
-
TSPAN5 influences serotonin and kynurenine: pharmacogenomic mechanisms related to alcohol use disorder and acamprosate treatment response
Molecular Psychiatry (2021)
-
A metabolome-wide association study in the general population reveals decreased levels of serum laurylcarnitine in people with depression
Molecular Psychiatry (2021)
-
ERICH3: vesicular association and antidepressant treatment response
Molecular Psychiatry (2021)
