
Abstract
Angioedema in the mouth or upper airways is a feared adverse reaction to angiotensin-converting enzyme inhibitor (ACEi) and angiotensin receptor blocker (ARB) treatment, which is used for hypertension, heart failure and diabetes complications. This candidate gene and genome-wide association study aimed to identify genetic variants predisposing to angioedema induced by these drugs. The discovery cohort consisted of 173 cases and 4890 controls recruited in Sweden. In the candidate gene analysis, ETV6, BDKRB2, MME, and PRKCQ were nominally associated with angioedema (p < 0.05), but did not pass Bonferroni correction for multiple testing (p < 2.89 × 10−5). In the genome-wide analysis, intronic variants in the calcium-activated potassium channel subunit alpha-1 (KCNMA1) gene on chromosome 10 were significantly associated with angioedema (p < 5 × 10−8). Whilst the top KCNMA1 hit was not significant in the replication cohort (413 cases and 599 ACEi-exposed controls from the US and Northern Europe), a meta-analysis of the replication and discovery cohorts (in total 586 cases and 1944 ACEi-exposed controls) revealed that each variant allele increased the odds of experiencing angioedema 1.62 times (95% confidence interval 1.05–2.50, p = 0.030). Associated KCNMA1 variants are not known to be functional, but are in linkage disequilibrium with variants in transcription factor binding sites active in relevant tissues. In summary, our data suggest that common variation in KCNMA1 is associated with risk of angioedema induced by ACEi or ARB treatment. Future whole exome or genome sequencing studies will show whether rare variants in KCNMA1 or other genes contribute to the risk of ACEi- and ARB-induced angioedema.
Similar content being viewed by others

Introduction
Angiotensin-converting enzyme inhibitor (ACEi) and angiotensin receptor blocker (ARB) containing medications are often used for hypertension, heart failure and diabetes complications, and currently 14% of the population in Sweden is treated with these drugs [1]. Although being mostly effective and safe, around 0.2–0.7% of patients starting an ACEi, and 0.1–0.2% starting an ARB experience the adverse drug reaction (ADR) angioedema [2,3,4,5,6]. Compared with other populations, the risk is higher in African Americans, suggesting a genetic effect modification [6,7,8]. The reaction is not dose-related and displays large discrepancies in time to onset, ranging from hours to years [9, 10].
ACEi- and ARB-induced angioedema is characterised by swelling of the reticular dermis and subcutaneous skin layers or mucosa [9, 11, 12]. It is usually localised to the head and neck region, and may become life threatening when breathing is impaired due to swelling in the oral cavity, tongue, pharynx or larynx [12]. In rare instances the intestines or genitals may be involved [13]. The pathophysiological mechanism is believed to involve the accumulation of the vasoactive peptide bradykinin [11, 12]. ACEis directly inhibit the inactivation of bradykinin while blocking the conversion of angiotensin I to angiotensin II [14]. ARBs have no direct effect on bradykinin [15], but may increase bradykinin levels through indirect inhibition of ACE and metallo-endopeptidase [16]. Drugs conventionally used in immune-mediated angioedema such as antihistamines, glucocorticoids and adrenaline have limited effects on ACEi- and ARB-induced angioedema [12, 17]. Alternative treatments are the bradykinin receptor antagonist icatibant, complement C1-esterase inhibitors, ecallantide, and fresh frozen plasma, but definite proof of their benefit is still lacking [12, 18, 19].
Two recent reviews describe nine studies with possible genetic associations to ACEi-induced angioedema [20, 21]. The majority are case–control studies that focus on candidate genes in bradykinin pathways. Three studies found significant associations with variation in the bradykinin-degrading enzyme aminopeptidase P gene XPNPEP2 [22,23,24], and one found an association with a variant of the type 2 bradykinin receptor gene (BDKRB2) [25]. Mutations in serpin super family and clotting factor genes causing the rare condition hereditary angioedema are also of interest [26]. It is recommended that these patients avoid ACEis [27], but a mutation in the Serpin Family E Member 1 gene (SERPINE1) has been described in at least one patient with apparent ACEi-induced angioedema [28], and a mutation in the Coagulation Factor XII gene (F12) in another [29]. None of the mentioned candidate genes was significantly associated in the only published genome-wide association study (GWAS) on ACEi-induced angioedema [30]. The main findings were a non-significant association between risk of angioedema and the ETS Variant Transcription Factor 6 gene (ETV6) in African Americans, while a variant of the Protein Kinase C Theta gene (PRKCQ) tended to be protective in European Americans [30]. Furthermore, in a candidate gene analysis of the same study, a nominal association with the bradykinin degrading enzyme Neprilysin gene (Membrane Metalloendopeptidase, MME) was detected in African Americans [30]. Neprilysin-inhibitors are a recent addition to the arsenal of agents used in cardiovascular disease, and are known to augment the risk of angioedema in ACEi-treated patients [31, 32].
The objective of this genome-wide association and candidate gene study was to investigate whether any genetic variants predispose to angioedema associated with ACEi or ARB treatment in a Northern European population, and if so to assess the functionality of detected associations.
Subjects and methods
Ethics statement
Research was carried out in accordance with the latest update of the Declaration of Helsinki. Written informed consent was obtained from all participants in the discovery and replication cohorts. The study protocols were approved by Ethics Committees or Institutional Review Boards in each country. Please see the Supplementary Methods for details.
Discovery cohort description
Angioedema cases associated with ACEi or ARB treatment reported to the Swedish national ADR registry at the Medical Products Agency or referred from collaborating clinicians were recruited according to SWEDEGENE’s standardised procedure (www.swedegene.se). Please see the Supplementary Methods for details. Inclusion and exclusion was according to published phenotype criteria [11]. All cases were reviewed and adjudicated by a clinical expert in allergology. Patients provided an EDTA blood sample that was stored at −70 °C until DNA extraction. Unrelated population controls were obtained from the Swedish Twin registry. The subgroup treated controls had collected at least two prescriptions of an ACEi at a Swedish pharmacy according to the Swedish Prescribed Drug Register [1, 33]. Indications for treatment were searched for in the Swedish National Patient Register, but were only available for 71% of the controls since the register has a low sensitivity for primary care diagnosis [34]. Controls who had been given the diagnoses angioedema or larynx-oedema were excluded [35].
Discovery genotyping, imputation and data analysis
Cases were genotyped on the Illumina HumanOmni2.5 BeadChip array and controls had previously been genotyped on the Illumina HumanOmniExpress 700 K BeadChip (Illumina, CA, USA). Quality control and data management was done with PLINK v1.9 [36]. Sex chromosomes were filtered out at an early stage. The resulting merged data set included 622992 single nucleotide polymorphisms (SNPs). Imputation of genotypes was performed with PhaseIT [37] and Impute v2 using 1000 genomes as reference [38]. Post-imputation, variants with MAF below 1% or a missing rate above 2%, and individuals with more than 2% missing markers were filtered out. SNPs deviating from the Hardy-Weinberg equilibrium (p < 10−6) among controls were removed. The final dataset contained 7585599 SNPs. The quantile–quantile (Q-Q) plot for analysis of cases vs all controls after imputation is shown in Supplementary Fig. S1. Principal component analysis was performed on non-imputed data in order to account for possible population stratification. Logistic regression was used to estimate univariate and multiple models. SNP effects were modelled as additive. The conventional genome-wide significance threshold p < 5 × 10−8 was used to correct for multiple testing [39]. PLINK v1.9 was used for genome-wide analyses, and R 3.2.2 (R Foundation for Statistical Computing, Vienna, Austria) for individual SNP analyses.
In addition, we performed candidate gene analyses for BDKRB2, ETV6, F12, MME, PRKCQ, and SERPINE1 in the imputed data set [20, 21]. All variants within the chromosomal positions (Table 1) ±10,000 bases upstream and downstream were extracted. XPNPEP2, which is located on the X chromosome, could not be evaluated since sex chromosomes were filtered out pre-imputation. SNP effects were modelled as additive. The number of tested genetic variants was 1730, and the Bonferroni cut-off for correction for multiple testing was p < 2.89 × 10−5 (= 0.05/1730 SNPs). All 173 cases were tested vs 4890 unmatched controls, and vs the 1345 matched controls.
Power calculation
Our sample size was powered to detect common genetic variants with effect sizes of clinical utility [40]. Power calculations were based on an ADR prevalence of 1%, and a minor allele frequency (MAF) of 10%, 173 cases, 4890 unmatched and 1345 matched controls, and an additive genetic model. Given a genome-wide significance level of 5 × 10−8, we had 80% power to detect an odds ratio (OR) of 3.0 when using unmatched controls, and 80% power to detect an OR of 3.1 when using matched controls (Supplementary Fig. S2a, b). Given a candidate gene significance level of 2.89 × 10−5, we had 80% power to detect an OR of 2.4 when using unmatched controls, and 80% power to detect an OR of 2.5 when using matched controls (Supplementary Fig. S3a, b).
Replication cohort description
Data from the published ACEi-angiodema GWAS cohort from Vanderbilt and Marshfield, USA, was obtained through collaboration [30]. Cases were defined as patients who developed swelling of the lips, pharynx or face for the first time while taking an ACE inhibitor. Controls had been treated with an ACEi for at least 6 months without developing angioedema.
The Northern European cohort was recruited in four countries, and was part of the PREDICTION-ADR study. Please see the Supplementary Methods for details. In Sweden, cases were recruited according to SWEDEGENE’s standardised procedure. In Denmark, cases were referred by physicians at participating hospitals and a general practitioner. In the Netherlands, cases were referred by physicians at participating hospitals or identified by screening electronic medical records from all hospital admissions for angioedema. In the UK, cases were identified by referral from consultant immunologists, and by review of archived clinical letters. The expert who assessed the discovery cases also assessed all replication cases and used identical criteria [11]. Treated controls from Sweden were recruited by collaborating clinicians. Danish treated controls were selected from existing databases [41]. Dutch treated controls were participants of Utrecht Cardiovascular Pharmacogenomics (UCP) studies [42]. British treated controls were patients in the Genetics of Diabetes Audit and Research Tayside Study (GoDARTS) [43]. Cases and controls provided a blood or saliva sample (2 ml Oragene® OG-500 collection kit, DNA Genotek, Canada). Blood samples were kept at −70 °C and saliva samples at room temperature until DNA extraction according to standard procedures.
Replication genotyping and meta-analysis
The American cohort was genotyped using a 610Quadv1.B Illumina Bead Chip. Genotyping of the Northern European replication cohort was performed on the Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to standard procedures. The TaqMan SNP Genotyping Assay kit for rs2253202 (C__15924034_20) was used.
Meta-analysis was performed using one genotyped SNP in the Northern European replication cohort, and summary data for this SNP in the published American GWAS [30] and the discovery GWAS. Logistic regression was used to evaluate the association between the SNP and angioedema. The SNP effect was modelled as additive. Meta-analysis was performed using a fixed-effect model in the metafor R-package [44]. The cut-off for a statistically significant association was set to p < 0.05.
Functional analysis
Functional annotations were obtained by intersecting top GWAS SNPs and SNPs in high linkage disequilibrium (LD) with transcription factor binding sites reported by the ENCODE project, and with chromatin state models from the Roadmap Epigenomics project [45,46,47]. Chromatin state was based on deoxyribonuclease (DNAse) I hypersensitive clusters and histone modifications that indicate active regulatory regions (H3K4me3 and H3K27ac). We used annotations in skin, mucosa-derived tissues, oesophagus, aorta, smooth muscle and the umbilical vein endothelial cell line HUVEC. Motif analysis was based on a position weight matrix library and enhancer tissue-specific motif clustering [47,48,49].
Results
Discovery cohort characteristics
One hundred and seventy-three cases of angioedema associated with ACEi and/or ARB treatment passed adjudication. Almost all were of Swedish origin (93.1%), and there were more males than females (Table 2). One outlier among the cases was detected with principal component analysis, but not removed (Supplementary Fig. S4a, b). Most patients had a history of cardiovascular disease, including hypertension, angina pectoris, myocardial infarction or stroke, and approximately one in four suffered from diabetes. The angioedema event was in 144 patients associated with ACEi treatment, most frequently enalapril and ramipril. In 31 patients, the event was associated with an ARB, most frequently candesartan and losartan. Two patients developed angioedema during treatment with both an ACEi and an ARB (enalapril/candesartan and ramipril/losartan respectively). Frequent co-medications during the three months preceding the onset of angioedema were diuretics (47.4%), lipid-modifying agents (44.5%), beta-blockers (42.8%), antiplatelet drugs (42.2%), and selective calcium channel blockers (31.8%).
Data from 4890 unrelated population controls were obtained from the Swedish Twin Registry [33]. The subgroup of treated controls (n = 1345) had collected at least two prescriptions of an ACEi at a Swedish Pharmacy without being diagnosed with angioedema or larynx-oedema (Table 2). Sex distribution among treated controls was similar to that of the cases with more males than females. Hypertension, diabetes and heart failure were common among the treated controls that had data on diagnoses (n = 956). The most frequently prescribed ACEis were enalapril and ramipril.
Discovery cohort genome-wide analysis data analysis
The calcium-activated potassium channel subunit alpha-1 (KCNMA1) gene on chromosome 10 was significantly associated with angioedema on a genome-wide level. The top hit in intron 1 of KCNMA1 was rs2253201 (OR = 2.47, 95% CI = 1.79–3.42, p = 4.17 × 10−8), and several nearby SNPs had p values around 10−7 (Table 3a, Supplementary Table S1). After imputation, five additional SNPs in intron 1 of KCNMA1 were associated on a genome-wide level: rs2253202 (OR = 2.47, 95% CI = 1.79–3.42, p = 4.31 × 10−8), rs2673471 (OR = 2.47, 95% CI = 1.79–3.41, p = 4.35 × 10−8) and rs2619635, rs2670121 and rs2673455 (all OR = 2.47, 95% CI = 1.79–3.41, p = 4.59 × 10−8) (Fig. 1a, Table 3b, Supplementary Table S2). Comparisons with the 1345 treated controls supported that the association with KCNMA1 was not due to confounding by indication (OR = 2.34, 95% CI = 1.67–3.29, p = 8.91 × 10−7) (Figs. 1b and 2, Table 3c).

Analysis of 7585599 genome-wide single nucleotide polymorphisms in 173 cases of angioedema associated with angiotensin-converting enzyme inhibitors and/or angiotensin receptor blockers adjusted by sex and genetic principal components 1–4. The threshold for genome-wide significance (p < 5 × 10−8) is indicated by the red line on the x-axis where the scale is –log10(p). a Intronic SNPs in the Calcium-activated potassium channel subunit alpha-1 (KCNMA1) gene on chromosome 10 were significantly associated on a genome-wide level when cases were compared with all 4890 controls. b Associations were similar, but did not reach genome-wide significance, when cases were compared with 1345 controls matched for treatment.

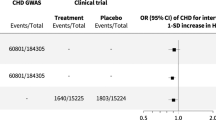
A forest plot of the association with rs2253202 in the 173 discovery cases compared with all 4890 controls and with 1345 treated controls. Estimated odds ratios (OR) with 95% confidence intervals (CI), p value and numbers (N) included are shown. The effect is adjusted for sex and genetic principal components 1–4.
Discovery cohort candidate gene analysis
ETV6, BDKRB2, MME, and PRKCQ were nominally associated with angioedema (p < 0.05), but none of the candidate genes was significantly associated after correction with multiple testing (p < 2.89 × 10−5) (Fig. 3a, Table 4a, Supplementary Table S3). Results were consistent when cases were compared with matched controls (Fig. 3b, Table 4b). The only published candidate SNPs present in our data were the presumed protective PRKCQ rs500766 and deleterious MME rs989692 [30]. Odds ratios for these SNPs were close to 1 in our study (Table 5).

Analysis of 1730 candidate gene single nucleotide polymorphisms in 173 cases of angioedema associated with angiotensin-converting enzyme inhibitors and/or angiotensin receptor blockers adjusted by sex and genetic principal components 1–4. The threshold for candidate gene-wide significance (p < 2.89 × 10−5) is indicated by the red line on the x-axis where the scale is –log10(p). a None of the candidate genes BDKRB2, ETV6, F12, MME, PRKCQ or SERPINE1 was significantly associated when cases were compared with all 4890 controls. b Results were consistent when cases were compared with 1345 controls matched for treatment.
Replication cohort characteristics
The American cohort included 175 angioedema cases and 489 ACEi-exposed controls [30]. Sixty-six of the cases were of African, and 109 of European descent. Ethnic origin of the controls was 157 African, 330 European, and two unknown. The Northern European cohort included 238 angioedema cases and 110 treated controls. Specifically, there were 96 cases and 38 controls from Sweden, 60 cases and 20 controls from Denmark, 64 cases and 52 controls from the Netherlands, and 18 cases from the UK. All cases had been treated with an ACEi, and one also with an ARB. All controls were treated with an ACEi, and eight also with an ARB.
Replication and meta-analysis
We sought replication for the top hit rs2253202 in the American cohort [30] and the Northern European cohort divided into Swedish-Danish and Dutch-British (Fig. 3). All individual cohorts had odds ratios above 1, but the results did not reach significance. Meta-analysis of the 413 cases and 599 ACEi-exposed controls included in the replication likewise gave non-significant results (OR = 1.25, 95% CI = 0.85−1.83, p = 0.255) (Fig. 3). The discovery cohort was then added to the meta-analysis, making a grand total of 586 cases and 1944 ACEi-exposed controls, which rendered a statistically significant association between rs2253202 and angioedema (OR = 1.62, 95% CI = 1.05–2.50, p = 0.030) (Fig. 4). This means that the odds of experiencing angioedema increased 1.62 times per minor allele, i.e. that the risk would be (1.62)2 = 2.62 times increased in homozygous carriers of rs2253202.

Meta-analysis of the top single nucleotide polymorphism rs2253202 in the replication cohorts from the US [30] and Northern Europe. Meta-analysis was performed with and without the Swedish discovery cohort using a fixed-effect model in the metafor R-package. Logistic regression was used to evaluate the association between the SNP and angioedema. SNP effects were modelled as additive. Estimated odds ratios with 95% confidence intervals (CI), p value and numbers (N) included are shown. The cut-off for a statistically significant association was set to p < 0.05.
Functional analysis
The six significantly associated SNPs in the first intron of the KCNMA1 gene are in a haplotype block spanning ∼0.1 Mb, and lack evidence of functional activity. They are in high LD (r2 > 0.8) with 15 non-coding SNPs located in enhancer elements defined in several angioedema relevant tissues such as skin, mucosa, oesophagus, aorta, visceral smooth muscle and endothelial cells (Supplementary Tables S4a, b, and Supplementary Fig. S5). According to motif analyses, eight of the SNPs alter binding sites for transcription factors enriched in skin, mucosa or smooth muscle (Supplementary Table S3b) [47]. Previous results from chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-seq) point to the SNP rs816827 G > A that overlaps binding sites of 22 transcription factors in different cell lines (Supplementary Fig. S6) [46, 47, 49]. Tissue-specific analyses show that rs816827 alters binding motifs for transcription factors enriched in enhancer elements in skin (orange), mucosa (pink) and smooth muscle (brown) (Supplementary Table S4b). The TRAP tool [49] indicates that the transcription factor MAF, which is expressed in for example skin, oral mucosa, oesophagus, aorta and smooth muscle, would bind significantly stronger to the minor than the major allele (difference log(p) for two alleles = −0.812 or A/p value = 0.0253 vs G/p value = 0.164).
Discussion
This study aimed to identify common genetic risk factors for angioedema induced by ACEi or ARB treatment. Genetic variants in the calcium-activated potassium channel subunit alpha-1 (KCNMA1) gene were significantly associated with angioedema on a genome-wide level (p < 5 × 10−8). Calcium-activated potassium channel genes are expressed in vascular smooth muscle and endothelium, and encode effector proteins crucial for control of vascular tone and blood pressure [50]. We ascertained that our finding was not due to confounding by indication for treatment by redoing the analysis using only ACEi-treated controls. Furthermore, meta-analysis of four cohorts of cases and treated controls supported the association between KCNMA1 and ACEi- or ARB-induced angioedema (Fig. 3).
The KCNMA1 intron 1 variants significantly associated with angioedema in our study lack evidence of functional activity, but are in linkage disequilibrium with several genetic variants located in regulatory elements. Motif analyses indicate that one variant in particular alters binding sites for tissue-specific transcription factors, which could lead to deregulation of a target gene. The target gene could be distant, but it is more plausible that it is KCNMA1 itself. KCNMA1 encodes the pore-forming alpha subunit of the large-conductance calcium-dependent potassium big K (BK) channel, which is important for the repolarization of the cell membrane [50]. A study using an alpha subunit BK channel knockout mouse model suggested that this gene modulates the renin-angiotensin-aldosterone system [51]. KCNMA1 is one among several genes considered to be involved in control of vascular tone and hypertension [50, 52]. A previous GWAS detected an association between a KCNMA1 intron 1 SNP (rs603788, GRCh37 chr10: 79211262) and diastolic blood pressure in a subgroup of 3014 elderly patients (p = 9.37 × 10−7) [53]. In a candidate gene study, a KCNMA1 intron 17 SNP (rs16934182, GRCh37 chr10: 78778734) was nominally associated with severe systolic hypertension (p = 0.03), and severe general hypertension (p = 0.04) [54]. These two intronic SNPs are not in LD with the six intron 1 variants significantly associated in our study (GRCh37 chr10:79356393-79359111) [55].
Genetic variants of calcium/potassium channel genes have previously been implicated in studies on ACEi-induced ADRs. A GWAS on ACEi-induced cough detected an association with the calcium-activated potassium channel gene KCNMB2 [56], while another GWAS found an association with the calcium-binding potassium channel-interacting protein gene KCNIP4 [57]. A clinical study showed that patients who reacted with angioedema to an ACEi were more often treated with a calcium channel blocker than those who reacted with cough [58]. Furthermore, angioedema is in rare cases triggered by a calcium channel blocker (e.g. amlodipine) or potassium channel blocker (e.g. amiodarone) alone [59, 60]. It is thus reasonable to believe that deficient transport of calcium and/or potassium across the cell membrane plays a role in the development of angioedema.
In addition, we analysed data for the candidate genes BDKRB2, ETV6, F12, MME, PRKCQ, and SERPINE1. There was no significant association with single nucleotide polymorphisms in the candidate genes after correcting for multiple testing. ETV6 showed promising results with relatively high odds ratios and p values around 0.001. A larger sample size would be needed to determine whether any candidate gene is truly associated with angioedema induced by ACEi or ARB treatment.
A strength of this study is that the cases were well-characterised and the diagnostic accuracy was high. There are also limitations. First, matching of controls was done using ACEi exposure as a proxy for the indication for treatment. We were thus unable to identify whether treated controls were prescribed ACEis for hypertension, which was the most common diagnosis among cases. Secondly, although this is the largest GWAS on ACEi- or ARB-induced angioedema in people of European ancestry to date, the low number of cases does not provide enough power to detect association with rare variants. Nor could we show whether the trait in question is affected by multiple weakly associated variants that interact to increase the risk.
Our GWAS suggests that common genetic variants of KCNMA1 are associated with risk of ACEi- or ARB-induced angioedema. Comparisons between cases and treated controls confirm that the finding is not due to confounding by indication. Meta-analysis of replication cohorts from the US [30] and Northern Europe together with the discovery cohort further supports the association. This is a step forward in the understanding of mechanisms behind ACEi- or ABR-induced angioedema. It is, however, reasonable to believe that intron 1 of KCNMA1 is only one of several susceptibility loci. Future studies based on whole exome or genome sequencing will show whether rare coding variants contribute to the risk of ACEi- or ABR-induced angioedema to a clinically important degree. When the complete picture is known, and patients have DNA sequence data available in the medical record before treatment, personalised medical decisions can be used to prevent this potentially fatal ADR.
References
The National Board of Health and Welfare. Swedish Prescribed Drug Register. https://www.socialstyrelsen.se/statistik-och-data/register/alla-register/lakemedelsregistret/.
Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Kober L, Maggioni AP, et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N. Engl J Med. 2003;349:1893–906.
Piller LB, Ford CE, Davis BR, Nwachuku C, Black HR, Oparil S, et al. Incidence and predictors of angioedema in elderly hypertensive patients at high risk for cardiovascular disease: a report from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). J Clin Hypertens (Greenwich). 2006;8:649–56. quiz 57–8.
Kostis JB, Kim HJ, Rusnak J, Casale T, Kaplan A, Corren J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165:1637–42.
Banerji A, Blumenthal KG, Lai KH, Zhou L. Epidemiology of ACE inhibitor angioedema utilizing a large electronic health record. J Allergy Clin Immunol Pr. 2017;5:744–9.
Miller DR, Oliveria SA, Berlowitz DR, Fincke BG, Stang P, Lillienfeld DE. Angioedema incidence in US veterans initiating angiotensin-converting enzyme inhibitors. Hypertension. 2008;51:1624–30.
Mahoney EJ, Devaiah AK. Angioedema and angiotensin-converting enzyme inhibitors: are demographics a risk? Otolaryngol Head Neck Surg. 2008;139:105–8.
Brown NJ, Ray WA, Snowden M, Griffin MR. Black Americans have an increased rate of angiotensin converting enzyme inhibitor-associated angioedema. Clin Pharm Ther. 1996;60:8–13.
Rasmussen ER, Mey K, Bygum A. Angiotensin-converting enzyme inhibitor-induced angioedema–a dangerous new epidemic. Acta Derm Venereol. 2014;94:260–4.
Norman JL, Holmes WL, Bell WA, Finks SW. Life-threatening ACE inhibitor-induced angioedema after eleven years on lisinopril. J Pharm Pr. 2013;26:382–8.
Wadelius M, Marshall SE, Islander G, Nordang L, Karawajczyk M, Yue QY, et al. Phenotype standardization of angioedema in the head and neck region caused by agents acting on the angiotensin system. Clin Pharm Ther. 2014;96:477–81.
Bas M. The angiotensin-converting-enzyme-induced angioedema. Immunol Allergy Clin North Am. 2017;37:183–200.
Korniyenko A, Alviar CL, Cordova JP, Messerli FH. Visceral angioedema due to angiotensin-converting enzyme inhibitor therapy. Cleve Clin J Med. 2011;78:297–304.
Bas M, Adams V, Suvorava T, Niehues T, Hoffmann TK, Kojda G. Nonallergic angioedema: role of bradykinin. Allergy. 2007;62:842–56.
Gohlke P, Pees C, Unger T. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension. 1998;31:349–55.
Campbell DJ, Krum H, Esler MD. Losartan increases bradykinin levels in hypertensive humans. Circulation. 2005;111:315–20.
Winters ME, Rosenbaum S, Vilke GM, Almazroua FY. Emergency department management of patients with ACE-inhibitor angioedema. J Emerg Med. 2013;45:775–80.
Lawlor CM, Ananth A, Barton BM, Flowers TC, McCoul ED. Pharmacotherapy for angiotensin-converting enzyme inhibitor-induced angioedema: a systematic review. Otolaryngol Head Neck Surg. 2018;158:232–9.
Riha HM, Summers BB, Rivera JV, Van Berkel MA. Novel therapies for angiotensin-converting enzyme inhibitor-induced angioedema: a systematic review of current evidence. J Emerg Med. 2017;53:662–79.
Liau Y, Chua I, Kennedy MA, Maggo S. Pharmacogenetics of angiotensin-converting enzyme inhibitor-induced angioedema. Clin Exp Allergy. 2019;49:142–54.
Ali HA, Lomholt AF, Hamidreza Mahmoudpour S, Hermanrud T, Bygum A, von Buchwald C, et al. Genetic susceptibility to angiotensin-converting enzyme-inhibitor induced angioedema: a systematic review and evaluation of methodological approaches. PLoS ONE. 2019;14:e0224858.
Duan QL, Nikpoor B, Dube MP, Molinaro G, Meijer IA, Dion P, et al. A variant in XPNPEP2 is associated with angioedema induced by angiotensin I-converting enzyme inhibitors. Am J Hum Genet. 2005;77:617–26.
Woodard-Grice AV, Lucisano AC, Byrd JB, Stone ER, Simmons WH, Brown NJ. Sex-dependent and race-dependent association of XPNPEP2 C-2399A polymorphism with angiotensin-converting enzyme inhibitor-associated angioedema. Pharmacogenet Genom. 2010;20:532–6.
Cilia La Corte AL, Carter AM, Rice GI, Duan QL, Rouleau GA, Adam A, et al. A functional XPNPEP2 promoter haplotype leads to reduced plasma aminopeptidase P and increased risk of ACE inhibitor-induced angioedema. Hum Mutat. 2011;32:1326–31.
Moholisa RR, Rayner BR, Patricia Owen E, Schwager SL, Stark JS, Badri M, et al. Association of B2 receptor polymorphisms and ACE activity with ACE inhibitor-induced angioedema in black and mixed-race South Africans. J Clin Hypertens (Greenwich). 2013;15:413–9.
Zuraw BL, Bernstein JA, Lang DM, Craig T, Dreyfus D, Hsieh F, et al. A focused parameter update: hereditary angioedema, acquired C1 inhibitor deficiency, and angiotensin-converting enzyme inhibitor-associated angioedema. J Allergy Clin Immunol. 2013;131:1491–3.
Maurer M, Magerl M, Ansotegui I, Aygoren-Pursun E, Betschel S, Bork K, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2017 revision and update. Allergy. 2018;73:1575–96.
Levy J, Rivard GE, Wagner E, Beezhold D, Berlin N, Fan L, et al. Examination of genetic variants involved in generation and biodisposition of kinins in patients with angioedema. Allergy Asthma Clin Immunol. 2014;10:60.
Veronez CL, Serpa FS, Pesquero JB. A rare mutation in the F12 gene in a patient with ACE inhibitor-induced angioedema. Ann Allergy Asthma Immunol. 2017;118:743–5.
Pare G, Kubo M, Byrd JB, McCarty CA, Woodard-Grice A, Teo KK, et al. Genetic variants associated with angiotensin-converting enzyme inhibitor-associated angioedema. Pharmacogenet Genom. 2013;23:470–8.
Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E. Omapatrilat and enalapril in patients with hypertension: the omapatrilat cardiovascular treatment vs. enalapril (OCTAVE) trial. Am J Hypertens. 2004;17:103–11.
Bas M, Greve J, Strassen U, Khosravani F, Hoffmann TK, Kojda G. Angioedema induced by cardiovascular drugs: new players join old friends. Allergy. 2015;70:1196–200.
Magnusson PK, Almqvist C, Rahman I, Ganna A, Viktorin A, Walum H, et al. The Swedish twin registry: establishment of a biobank and other recent developments. Twin Res Hum Genet. 2013;16:317–29.
Ludvigsson JF, Andersson E, Ekbom A, Feychting M, Kim JL, Reuterwall C, et al. External review and validation of the Swedish national inpatient register. BMC Public Health. 2011;11:450.
The National Board of Health and Welfare. Swedish Patient Register. https://www.socialstyrelsen.se/statistik-och-data/register/alla-register/patientregistret/.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10:5–6.
Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529.
Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet. 2014;15:335–46.
Purcell S, Cherny SS, Sham PC. Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003;19:149–50.
Poulsen FR, Munthe S, Soe M, Halle B. Perindopril and residual chronic subdural hematoma volumes six weeks after burr hole surgery: a randomized trial. Clin Neurol Neurosurg. 2014;123:4–8.
Peters BJ, Rodin AS, Klungel OH, van Duijn CM, Stricker BH, van’t Slot R, et al. Pharmacogenetic interactions between ABCB1 and SLCO1B1 tagging SNPs and the effectiveness of statins in the prevention of myocardial infarction. Pharmacogenomics. 2010;11:1065–76.
Dujic T, Zhou K, Tavendale R, Palmer CN, Pearson ER. Effect of serotonin transporter 5-HTTLPR polymorphism on gastrointestinal intolerance to metformin: a GoDARTS study. Diabetes Care. 2016;39:1896–901.
Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw. 2010;36:1–48.
Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–7.
Bernstein BE, Birney E, Dunham I, Green ED, Gunter C. ENCODE Project Consortium. et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A. Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30.
Kheradpour P, Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014;42:2976–87.
Manke T, Heinig M, Vingron M. Quantifying the effect of sequence variation on regulatory interactions. Hum Mutat. 2010;31:477–83.
Kohler R. Single-nucleotide polymorphisms in vascular Ca2+-activated K+-channel genes and cardiovascular disease. Pflug Arch. 2010;460:343–51.
Sausbier M, Arntz C, Bucurenciu I, Zhao H, Zhou XB, Sausbier U, et al. Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation. 2005;112:60–8.
Jordan J. Sluggish genes and hypertension. J Hypertens. 2008;26:2093–5.
Simino J, Shi G, Bis JC, Chasman DI, Ehret GB, Gu X, et al. Gene-age interactions in blood pressure regulation: a large-scale investigation with the CHARGE, Global BPgen, and ICBP Consortia. Am J Hum Genet. 2014;95:24–38.
Tomas M, Vazquez E, Fernandez-Fernandez JM, Subirana I, Plata C, Heras M, et al. Genetic variation in the KCNMA1 potassium channel alpha subunit as risk factor for severe essential hypertension and myocardial infarction. J Hypertens. 2008;26:2147–53.
Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4.
Hallberg P, Persson M, Axelsson T, Cavalli M, Norling P, Johansson HE, et al. Genetic variants associated with angiotensin-converting enzyme inhibitor-induced cough: a genome-wide association study in a Swedish population. Pharmacogenomics. 2017;18:201–13.
Mosley JD, Shaffer CM, Van Driest SL, Weeke PE, Wells QS, Karnes JH, et al. A genome-wide association study identifies variants in KCNIP4 associated with ACE inhibitor-induced cough. Pharmacogenomics J. 2016;16:231–7.
Hallberg P, Nagy J, Karawajczyk M, Nordang L, Islander G, Norling P, et al. Comparison of clinical factors between patients with angiotensin-converting enzyme inhibitor-induced angioedema and cough. Ann Pharmacother. 2017;51:293–300.
Kuruvilla ME, Sanan N. Amlodipine-induced angioedema: an unusual complication of a common medication. Allergy Rhinol (Providence). 2018;9:2152656718764139.
Lahiri K, Malakar S, Sarma N. Amiodarone-induced angioedema: report of two cases. Indian J Dermatol Venereol Leprol. 2005;71:46–7.
Acknowledgements
We are grateful to all physicians, research nurses and supporting staff, who assisted in recruiting patients and controls or administering phenotype databases. In particular, Dr Leif Nordang, Dr Nina Bylund, and assistant Eva Prado, Uppsala University Hospital, RN Ulrica Ramqvist, RN Charlotta Haglund, and assistant Sofie Collin, Uppsala University, Sweden; Prof. Frantz Rom Poulsen (Open Patient Data Explorative Network), Odense University Hospital, Odense, Denmark; Dr Alexander SF Doney (GoDARTS bio resource) and Prof. Sara Marshall, Ninewells Hospital, Dundee, RN Diane van Ecker and RN Ruth Westhead, University of Liverpool, Liverpool, UK; Dr L.J.J.M. Bauwens, Westfriesgasthuis, Hoorn, Dr M. van der Torn, Westfriesgasthuis, Hoorn, Dr Chris Nieuwhof, UMC Maastricht, Dr Heike Röckmann, UMC Utrecht, Mr M.Q.N.G. Ruigrok, VUMC Amsterdam, Ms H.D.M. Smidt-Huizinga, UMC Groningen, and Ms M.H. Bugter, UMC Groningen, Netherlands. Lastly, we are grateful to Prof. Nancy J Brown at Vanderbilt University Medical Center for generously sharing GWAS data from the American study. This research was supported by the Swedish Research Council (Medicine 521–2011–2440, 521–2014–3370 and 2018-03307), Swedish Heart and Lung Foundation (20120557, 20140291 and 20170711), Medical Products Agency, Selander’s Foundation, Thureus’ Foundation, Clinical Research Support Avtal om Läkarutbildning och Forskning at Uppsala University, and Swedish Diabetes foundation (DIA2017-269). PREDICTION-ADR received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under Grant Agreement no. 602108. UCP studies were funded by the Netherlands Heart Foundation and the Dutch Top Institute Pharma Mondriaan Project. Folkert Asselbergs is supported by UCL Hospitals NIHR Biomedical Research Centre. We acknowledge the Swedish Twin Registry for access to data from controls. The Swedish Twin Registry is managed by Karolinska Institutet, and receives funding through the Swedish Research Council under grant no. 2017-00641. Genotyping of the discovery cohort was performed by the SNP&SEQ Technology Platform at Uppsala University (www.genotyping.se). The facility is part of the National Genomics Infrastructure supported by the Swedish Research Council for Infrastructures and Science for Life Laboratory, Sweden. The SNP&SEQ Technology Platform is also supported by the Knut and Alice Wallenberg Foundation. Computations were done on resources provided by the Swedish National Infrastructure for Computing through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX).
Author information
Authors and Affiliations
Contributions
Study design: MW, PH, and QYY. Case, control and data collection: PH, MW, ERR, EVB, GI, SH, IT, FWA, PN, HEJ, MKS, CCL, PKEM, CvB, AB, AA, AHMZ, and CNAP. Adjudication of cases: MK. Genotyping: ACS, HK, CM, and CNAP. Data analysis: NE, CJ, AV, and CM. Functional analysis: MC and CW. Data interpretation: MW, PH, NE, CJ, ERR, CM, AV, MKS, and CNAP. Drafting manuscript: MW, ERR, PH, NE, MC, HK, and CW. Revising manuscript content: MW, ERR, PH, NE, IT, FWA, HEJ, MC, CW, AB, AA, and AHMZ. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
No funder took part in the study design, recruitment, analysis, interpretation of data, writing of the report or in the decision to submit the paper for publication.The study was part of Dr Eva Rye Rasmussen’s PhD thesis, which was financially supported by CSL Behring and by Shire with an investigator initiated research grant (grant no. IIR-DNK-001219). No other conflicts of interest exist.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rasmussen, E.R., Hallberg, P., Baranova, E.V. et al. Genome-wide association study of angioedema induced by angiotensin-converting enzyme inhibitor and angiotensin receptor blocker treatment. Pharmacogenomics J 20, 770–783 (2020). https://doi.org/10.1038/s41397-020-0165-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41397-020-0165-2
This article is cited by
-
Prediction and prevention of ACE-inhibitor-induced angioedema—an unmet clinical need in management of hypertension
Hypertension Research (2024)
-
Leveraging Genetics for Hereditary Angioedema: A Road Map to Precision Medicine
Clinical Reviews in Allergy & Immunology (2021)
-
ACE inhibitors/angiotensin receptor blockers
Reactions Weekly (2021)
