
Abstract
Thaumarchaeota (now the class Nitrososphaeria in the phylum Thermoproteota in GTDB taxonomy) are abundant across marine and soil habitats; however, their genomic diversity and evolutionary history in freshwater environments remain elusive. Here, we reconstructed 17 high-quality metagenome-assembled genomes of Nitrososphaeria from a deep lake and two great rivers, and compared all available genomes between freshwater and marine habitats regarding their phylogenetic positions, relative abundance, and genomic content. We found that freshwater Nitrososphaeria were dominated by the family Nitrosopumilaceae and could be grouped into three distinct clades closely related to the genera Nitrosopumilus, Nitrosoarchaeum, and Nitrosotenuis. The Nitrosopumilus-like clade was exclusively from deep lakes, while the Nitrosoarchaeum-like clade was dominated by species from deep lakes and rivers, and the Nitrosotenuis-like clade was mainly from rivers, deep lakes, and estuaries. Interestingly, there was vertical niche separation between two clades in deep lakes, showing that the Nitrosopumilus-like species dominated shallow layers, whereas the relative abundance of the Nitrosoarchaeum-like clade increased toward deep waters. Phylogenetic clustering patterns in the Nitrosopumilaceae supported at least one freshwater-to-marine and two marine-to-freshwater transitions, the former of which refined the potential terrestrial-to-marine evolutionary path as previously proposed. The occurrence of the two marine-to-freshwater transitions were accompanied by horizontal transfer of the genes involved in nutrition regulation, osmoregulation, and cell motility during their colonization to freshwater habitats. Specifically, the Nitrosopumilus-like clade showed losses of genes encoding flagella assembly and ion transport, whereas the Nitrosoarchaeum-like clade had losses of intact genes involved in urea uptake and utilization and gains of genes encoding osmolarity-mediated mechanosensitive channels. Collectively, our results reveal for the first time the high genomic diversity of the class Nitrososphaeria across freshwater ecosystems and provide novel insights into their adaptive mechanisms and evolutionary histories.
Similar content being viewed by others

Introduction
Ammonia-oxidizing archaea (AOA) of the phylum Thaumarchaeota (now the class Nitrososphaeria in the phylum Thermoproteota [1]) are widespread across a great variety of ecosystems, including oceans [2], soils [3], and freshwater [4], and play a significant role in global nitrogen and carbon cycling [5, 6]. Since the first report of the archaea in the marine water column [7, 8], our understanding of the diversity, ecology, and evolutionary history of the class Nitrososphaeria has been advanced through culture or enrichment approaches combined with culture-independent molecular technologies [9]. Representative examples include the heterotrophic lifestyle of the non-AOA lineages in dark ocean water [10] and coastal marine water [11], the metabolic adaptation of novel AOA clades to the hadal zone [12, 13], habitat expansion associated with the extensive lateral transfer of the genes encoding ATPase [14], genome expansion of the Nitrososphaerales-like lineage driven by lateral gene transfer and extensive gene duplication [15], the evolutionary transition from terrestrial to deep and shallow waters of the ocean driven by oxygen [16] and linked to major geological events of glaciation and oxygenation in Earth’s history [17]. All these studies, largely depending on reconstruction of metagenome-assembled genomes (MAGs), show the critical role of metagenome technology in expanding and elucidating the genomic diversity, metabolic potential and genomic evolution of the class Nitrososphaeria. Nevertheless, compared with marine and soil environments, less information regarding the archaea is available for freshwater habitats [18].
Freshwater members of the class Nitrososphaeria are ubiquitous and abundant in lakes [19], rivers [20], wastewater treatment plants [21], groundwaters [22], and estuaries [23], as revealed by archaeal 16 S rRNA and ammonia monooxygenase subunit genes, and they constitute up to 58% of the archaeal community in deep oligotrophic lakes [24]. AOA of the class Nitrososphaeria are proposed to be dominant nitrifiers in freshwater habitats, especially in oligotrophic waters. For example, the abundance of AOA population is positively correlated with in situ nitrification rates along the depth in oligotrophic Lake Superior [25]. A single AOA species, accounting for 13–21% of bacterioplankton, could involve in ammonia oxidation in the hypolimnion of deep oligotrophic Lake Constance based on the negative relationships between its population abundance and total ammonia and the measured active nitrification by 15N-isotope dilution [26]. In addition to their global distribution, distinct phylogenetic lineages of the Nitrososphaeria have been observed across freshwater habitats. For example, the Nitrososphaeria-associated population in Lake Redon mainly comprises two clades closely related to the Nitrosoarchaeum and Nitrosotalea genera, with the latter clade showing higher abundance at the surface and lower at depth [27], whereas members of the Nitrososphaeria in the hypolimnion of Lake Constance is dominated by a single Nitrosopumilus species [26], and a cluster of Nitrosoarchaeum-like species is identified from the Great Lakes [28]. Their phylogenetic divergence may have resulted from the limited resolution using the single marker gene-based approach [29], or ecological adaptation to local environments [30]. Although these results show an enormous diversity of freshwater Nitrososphaeria, few studies have comprehensively investigated their genomic diversities and explored the mechanisms underlying genomic differentiation. Due to the limited number of genome sequences from pure or enriched cultures in the class Nitrososphaeria [9], metagenome-assembled genomes (MAGs) using a genome-resolved metagenomics approach provide the opportunity to study the diversification and adaptation mechanisms of the archaea such as in freshwater environments.
Here, we reconstructed the MAGs of freshwater Nitrososphaeria from a deep lake and two great rivers, and performed large-scale phylogenomic analyses by inclusion of other Nitrososphaeria-associated genomes from freshwater and non-freshwater habitats around the world (Supplementary Table S1). We aimed to elucidate the phylogenetic differentiation of freshwater clades, their evolutionary history, and potential genetic determinants associated with their adaptation to freshwater habitats. Briefly, we obtained 41 surface sediments along a 94-m water depth gradient in Lake Lugu, China (Supplementary Table S2–4). The diversity of Nitrososphaeria along the water depth and the associated environmental explanatory factors were examined with archaeal 16 S rRNA gene amplicon. We recovered the Nitrososphaeria-associated MAGs from metagenomic sequence of Lake Lugu and the Yangtze River [31] and Amazon River (Supplementary Table S5, [32]), and further integrated these obtained MAGs with other available genomes in public databases to build phylogenomic trees of 102 Nitrososphaeria species (Supplementary Table S1). The relative abundance of these archaeal species across five aquatic biomes, that is, deep lakes, rivers, estuaries, coasts, and marine, was estimated with 126 metagenomic samples (Supplementary Table S2). Genomic differentiation of the class Nitrososphaeria between freshwater and marine habitats were revealed with the comparative genomic analyses, including genomic signature, ANI, and average amino acid identity. The habitat transition history for the family Nitrosopumilaceae was evaluated according to phylogenetic structure and ancestral genomic content estimation. Further details of field sampling, sequence collection, metagenome analyses, and phylogenomic analyses were provided in the supplementary material.
Results and discussion
Divergence of Nitrososphaeria in freshwater habitats
We reconstructed 17 high-quality Nitrososphaeria MAGs with an average completeness of 95.43% (±3.92%) and contamination of 2.34% (±1.44%, Supplementary Table S1), including four from Lake Lugu, seven from the Yangtze River, and six from the Amazon River. To determine the phylogenetic positions of these MAGs and infer the evolutionary history of freshwater members of the class Nitrososphaeria, 102 genomes from diverse habitats, especially freshwater habitats (Fig. 1A), were selected and used to build the phylogeny. All major lineages in the phylogenomic trees generated by both Bayesian inference and maximum likelihood approaches have high bootstrap values or posterior probabilities (Fig. 1B and Supplementary Fig. S1), especially the freshwater and marine clades within the family Nitrosopumilaceae, showing good congruent with the previous results [9, 10, 16, 33]. The phylogenomic trees supported that freshwater Nitrososphaeria are mainly partitioned into three clades (Fig. 1B and Supplementary Fig. S1, Supplementary Table S6), which are closely related to the genera Nitrosoarchaeum (17 genomes, n = 17), Nitrosopumilus (n = 5), and Nitrosotenuis (n = 25). The three clades hereafter are referred to as the Fresh-I, Fresh-II, and Fresh-III clades (Fig. 1A, B). The partitioning of the three clades was also confirmed by dendrograms based on either the distance of genome-wide average nucleotide identity (ANI) or amino acid (AAI) values among these archaeal genomes (Supplementary Fig. S2).

A The geographic distribution of freshwater Nitrososphaeria-associated genomes, including 17 MAGs reconstructed in this study (blank stars) and 40 accessed from public databases. Genomes are colored according to their phylogenetic clustering, as shown below. Detailed habitat information about these genomes was shown in Supplementary Table S1. Three species that are enriched non-freshwater habitats but phylogenetically clustered with most freshwater members were underlined. B The phylogenomic tree of 102 Nitrososphaeria-associated genomes was constructed by using a Bayesian inference approach implemented in MrBayes using 48 concatenated single copy gene families (Supplementary Table S6). The phylogeny was rooted by seven Aigarchaeota species, and only the species belonging to the family Nitrosopumilaceae (based on the rank-normalized GTDB taxonomy) were shown. Different phylogenetic groups are highlighted with distinct colors, as shown in the legend. C The relative abundance of freshwater Nitrososphaeria across aquatic biomes was estimated using a metagenomic read-recruiting approach. The biomes include lakes (63 metagenomic samples, n = 63), rivers (n = 79), estuaries (n = 7), coasts (n = 9) and marine environments (n = 9). Details regarding the genomes, selection of single copy genes, model parameters for phylogenetic construction, and abundance estimation are provided in the supplementary material.
Among the three main freshwater clades, however, it should be noted that there were few exceptions regarding their sampling habitats (that is, the species with underlined names in Fig. 1A). For example, the Fresh-I clade contained the arch.aeon Candidatus (Ca.) Nitrosoarchaeum koreensis MY1 enriched from soils [34]. Among the Fresh-III clade (n = 25), two archaeal genomes were enriched from non-freshwater habitats, including Ca. Nitrosotenuis chungbukensis MY2 from agricultural soils [35] and Ca. Nitrosotenuis uzonensis N4 from thermal spring sediments [36]. These results show that these non-freshwater strains were phylogenetically nested with the freshwater clades, which indicates that these archaea may have expanded to new niches after colonizing freshwater habitat or have adapted to these habitats in parallel with freshwater Nitrososphaeria. Furthermore, we noted that members of Nitrososphaeria in wastewater are phylogenetically diverse with only some lineages closely related to freshwater clades. This is indicated by the fact that Ca. Nitrosotenuis cloacae SAT1 from municipal wastewater [37] are assigned to the Fresh-III clade, whereas another wastewater-enrichment archaeon Ca. Nitrosocosmicus hydrocola G61 [38] belongs to the Nitrosocosmicus-like clade that exclusively comprises soil members (Supplementary Fig. S1). The scattered pattern of wastewater Nitrososphaeria could be alternatively explained by source contamination. For example, the source of Nitrososphaeria in wastewater phylogenetically related to marine Nitrosopumilus is associated with the seawater used for toilet flushing [39].
These three freshwater clades not only had niche separation, but also showed a magnitude of niche overlap, as revealed by their relative abundance across different aquatic biomes (Fig. 1C). Briefly, the Fresh-I clade contained the species from the water column and sediments of deep lakes, great rivers, and groundwater, whereas the Fresh-II clade was exclusively dominated by members inhabiting the water column of deep lakes. Members within the Fresh-III clade were mainly from great rivers, lakes, and estuaries (Supplementary Table S1). The niche preferences of these freshwater clades were further supported by their relative abundance across different aquatic biomes (Fig. 1C), which were estimated using a metagenomic read-recruiting approach. Specifically, the Fresh-I clade had a relatively higher abundance in deep lakes and rivers, with the genome LGS11MAG012 recovered from Lake Lugu showing a peak in deep lake samples, and the Fresh-II clade exhibited a relatively higher abundance in deep lakes of Lake Fuxian (the second-deepest freshwater lake in China) and Lake Baikal (the deepest lake on Earth). Within the Fresh-III clade, the abundance of the members in rivers and estuaries was higher than that in other aquatic biomes, with the genome AM_0615 from the upstream section of the Amazon showing a peak in estuarine samples (Fig. 1C).
The divergence of freshwater Nitrososphaeria was consistently observed in different layers of deep lakes around the world. For example, four Nitrososphaeria-associated MAGs from Lake Fuxian [40] fell into two separate clades, with one in the Fresh-I clade and three in the Fresh-II clade (Fig. 1B). This was also true for the MAGs from Lake Baikal [41, 42]. The Fresh-I and Fresh-II clades further showed contrasting water-depth patterns in their abundance in deep lakes based on metagenomic and amplicon sequencing data, with one dominating the shallow layers and the other increasing toward deep waters (Supplementary Fig. S3).
Community analyses using the archaeal 16 S rRNA amplicon data from Lake Lugu consolidates niche separation among freshwater Nitrososphaeria. Specifically, the Nitrososphaeria diversity and abundance showed hump-shaped patterns along the water depth, with a peak at the depths ranging from 20 to 50 m around the thermocline zone of Lugu lake (Fig. 2A, Supplementary Fig. S3A, B). Further, the Nitrososphaeria-associated communities were well separated by water depth showing less variations toward deep layers below 50 m (Fig. 2B). The peak pattern and relatively high diversity of the archaea around the thermocline zone are likely associated with the occurrence of a niche suitable for archaeal growth, such as low temperature (10–15 °C) and oxygen (169–215 µM). For example, marine Nitrososphaeria have been demonstrated to exhibit cold tolerance [43, 44] and high oxygen affinity [45]. Such a niche separation hypothesis was supported by the random forest model results showing that water temperature and oxygen were among the most important environmental factors in shaping species richness and composition of the Nitrososphaeria population (Fig. 2C, D). In addition to vertical divergence of the archaeal population in Lake Redon based on archaeal 16 S rRNA [27], our results collectively support that niche separation of the Nitrososphaeria could be a general phenomenon within deep lakes.

To explore their distribution along a water depth gradient in deep lakes and potential environmental factors, archaeal amplicon sequencing data were analyzed in parallel with metagenomic data (see the supplementary material). A Species richness of thaumarchaeotal communities along a water depth gradient. B Nonmetric multidimensional scaling (nMDS) plot of Nitrososphaeria communities. Each point in (A, B) represents a sample, which is colored by water depth, scaling from light to deep blue. Random forest analysis is used to identify the important environmental factors driving species richness (C) and community composition of Thaumarchaeota in Lake Lugu (D). Environmental factors suffixed with “bottom” represent the physiochemical parameters collected at the water-sediment interface of each sampling sites, whereas the factors suffixed with “water” represent the ones collected from surface water.
Niche separation is often observed among prokaryotes and driven by different biotic or abiotic factors. For instance, the growth and emissions of N2O from AOA differentiate them from those of ammonia-oxidizing bacteria in marine and soil environments owing to their difference in ammonium affinity [46, 47]. Two distinct clades of aerobic methane-oxidizing bacteria are responsible for methane emissions in lakes. Specifically, one clade dominates the methanotrophy throughout the water column, and the other thrives in oxygenated bottom waters, which has been proposed to be driven by oxygen concentration [48, 49]. Expanding to a relatively large spatial scale, we found that members of the Nitrososphaeria from great rivers were also partitioned in the phylogeny (Fig. 1B). For instance, MAGs from the Yangtze River belonged to the Fresh-I and III clades (Fig. 1A, B), whereas all MAGs from the Amazon River were assigned to the Fresh-III clade. The Amazon River-derived MAGs fell into two separate groups: one group is exclusively from this river, while the other group is mixed with another two MAGs originating from Lake Tanganyika [50] and an archaeon enriched out of a freshwater aquarium biofilter [51]. These results indicate that members of the Nitrososphaeria in freshwater show varying magnitude of niche separation across habitats.
In addition to these three main clades mentioned, five freshwater Nitrososphaeria-associated MAGs were scattered in some lineages that had not been previously reported to harbor freshwater species (Supplementary Fig. S1). The first three MAGs, which were recovered from Lake Tanganyika (the second-largest freshwater lake on Earth) [50], the Yangtze River, and an aquifer adjacent to the Colorado River [52], deeply branched at the base of the most recent ancestral lineage leading to the genera Nitrosoarchaeum, Nitrosopumilus, and Nitrosopelagicus (Fig. 1B). The paraphyletic group here was named as the Fresh-IV clade. The genome-wide ANI between the Fresh-IV clade and other freshwater and marine clades ranged from 70.82% to 72.79%, a value lower than the similarity threshold of genus demarcation recently proposed [53], indicating at least the existence of two novel genera within this diverse family Nitrosopumilaceae. To date, no pure or enrichment cultures have been successfully obtained within the clade. The fourth MAG from a meromictic deep lake of Lake Powell in Canada [54] is phylogenetically related to the Nitrosotalea genus, which contains obligately acidophilic ammonia oxidizers in soils [55]. The Nitrosotalea-like clade has been identified in the surface layers of alpine deep lakes based on archaeal 16 S rRNA genes [27], however, we failed to recruit any Nitrosotalea-like sequences from the surface water (0–5 cm) of Lake Lugu and Fuxian in our previous study [56] or from the water column at different depths (20–140 m) in Lake Fuxian. The fifth and last MAG from Lake Tanganyika was clustered with three genomes from deep ocean waters [10, 11], which are assigned to the basal groups of the Nitrososphaeria, a clade incapable of oxidizing ammonia (the pSL12 group). Due to the limited number of these distantly related MAGs, these genomes were not included in downstream analyses, but could still shed light for future genomic diversity study of freshwater Nitrososphaeria.
Significant differences in genomic and proteomic features were observed among the three main clades of freshwater Nitrososphaeria and their marine relatives (Supplementary Figs. S4–6). Marine AOA within the family Nitrosopumilaceae were classified into two groups for operational purposes, namely Marine-shallow and Marine-deep groups. In regard to genomic features (Supplementary Fig. S4), the Fresh-III clade showed the largest GC content among these clades (39.87 ± 1.77%), which was significantly higher than that of the two marine groups (33.97 ± 1.11%), the Fresh-I (33.09 ± 0.63%) and Fresh-II clades (30.30 ± 0.80%). The genome size of the Marine-shallow group (1.65 ± 0.23 Mb) and Fresh-III clade (1.50 ± 0.20 Mb) was significantly larger than that of other clades, especially the Marine-deep group (1.14 ± 0.09 Mb). Low GC content and genome size in free-living bacterioplankton have been considered signatures of genome streamlining [57, 58] and may be associated with lower inorganic nitrogen supply in the surface ocean [59]. The relatively lower GC contents in the Fresh-I, Fresh-II, and marine groups are consistent with the oligotrophic status of deep lakes (Supplementary Table S3), whereas the higher GC content in the Fresh-III may reflect a substantial nitrogen supply in estuaries and great rivers. For the proteomic features (Supplementary Fig. S5), the Fresh-III clade exhibited the highest nitrogen contents but lowest carbon contents, which were represented by the average number of nitrogen and carbon atoms per amino acid residual side chain (C-ARSC and N-ARSC). Interestingly, we found that GC content across these freshwater and marine clades showed a decreasing trend toward high protein carbon content but an opposite trend for nitrogen content (Supplementary Fig. S5), consistent with previous findings [60,61,62]. Higher nitrogen content in their encoded proteins (N-ARSC) indicates an adaptation of freshwater Nitrososphaeria to the higher nitrogen availabilities in estuarine and great rivers where the Fresh-III clade inhabits. We further observed significant differences in their amino acid composition and codon usage among the archaea (Supplementary Fig. S6), suggesting their functional difference and evolutionary divergence. Collectively, the differences in genomic and proteomic features among freshwater Nitrososphaeria likely reflect their distinct adaptation strategies to nutrition availability in their surroundings during evolutionary processes, therefore promoting the inference of this evolutionary history for the freshwater clades.
Habitat transitions between freshwater and marine within the family Nitrosopumilaceae
The phylogenetic relationships among the three main freshwater clades and few freshwater MAGs scattered along the phylogeny suggest a complex historical evolution of freshwater Nitrososphaeria within the phylum, which could be explained by the independent evolution or repeated transitions from non-freshwater to freshwater environments, or both. The inference of evolutionary history for the family Nitrosopumilaceae (based on the rank-normalized GTDB taxonomy recently proposed [1]) could be enabled mainly due to the clear phylogenetic separation among two marine groups (Marine-shallow and Marine-deep), three major (Fresh-I, II, and III) and one minor (Fresh-IV) freshwater clades (Fig. 3). The phylogenetic structure of the family Nitrosopumilaceae further showed that there were at least three major habitat transition events: one freshwater-to-marine and two subsequent marine-to-freshwater transitions.

The tree was modified from the phylogeny in Supplementary Fig. S1b with the main clades collapsed and the basal groups suppressed. The color scheme for these collapsed clades is the same as that in Fig. 1. The rank-normalized archaeal taxonomy proposed in the GTDB database is used to define the family Nitrosopumilaceae here. The ancestral habitat transition events are marked with gray arrows and labeled (N1, N2, and N3) at the corresponding nodes. The posterior probability of ancestral habitat and the estimated diverging time of the transition events are shown in the bottom table. The bar plots in the left panel show the distribution of archaeal COG (arCOG) functional categories for the gene families gained/lost at the ancestral nodes. The annotations for arCOG categories are listed as follows: C, energy production and conversion; D, cell cycle control, cell division, and chromosome partitioning; E, amino acid transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure and biogenesis; K, transcription; L, replication, recombination and repair; M, cell wall, membrane, and envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, and chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolite biosynthesis, transport and catabolism; R, general function prediction only; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; and V, defense mechanisms. Detailed analyses of ancestral habitat estimation and molecular dating are provided in the supplementary material.
Firstly, both Marine-shallow and Marine-deep groups within the family were embedded within two freshwater clades (Fig. 3), namely, the monophyletic Fresh-III clade (genus Nitrosotenuis) and the paraphyletic Fresh-IV clade, indicating that their most recent common ancestor had been expanded from freshwater Nitrososphaeria through at least one transition event (the label “N1” in Fig. 3). This hypothesis was supported by a high posterior probability of 0.972 that the ancestral lineage involved in the transition was a marine inhabitant through an ancestral state reconstruction approach (Supplementary Fig. S7), and consistent with the transition from terrestrial to marine environments proposed in recent studies [16, 17]. The availability of genomes of distinct freshwater Nitrososphaeria here allows us to provide a relatively higher resolution for the evolutionary path, that is, the transition expansion from freshwater (including river, estuary, and groundwater) to marine environments. Secondly, one marine-to-freshwater transition was further inferred based on the observation that the monophyletic Fresh-I clade was nested within two marine lineages on the phylogeny (Fig. 3). One is the Marine-deep group containing the Nitrosopelagicus-like clade from marine-shallow waters [63] and a clade from marine-deep layers [5, 16, 64, 65], and the other is the Cenarchaeum-like clade containing a sponge symbiont Cenarchaeum symbiosum [66] and a marine free-living archaeon C. sp. HMK20 [9]. This nested pattern for the Fresh-I clade is consistent with an evolutionary scenario where the ancestor of the Fresh-I clade may have originated from marine habitats and later colonized freshwater environments through at least one transition (the label “N2” in Fig. 3). Finally, a second marine-to-freshwater transition could have occurred within the Marine-shallow group, resulting in the formation of the Fresh-II clade (the label “N3” in Fig. 3). These two marine-to-freshwater transition events were further supported by the high posterior probabilities of 0.996 and 0.995, respectively (Supplementary Fig. S7).
The Bayesian relaxed clock model showed that the occurrences of the three transition events of N1, N2, and N3 were dated at 1222 million years ago (Mya, posterior 95% confidence interval (CI): 1370-1067 Mya), 288 Mya (95% CI: 363-216 Mya) and 47 Mya (95% CI: 93-10 Mya), respectively (Fig. 3 and Supplementary Fig. S8, Supplementary Table S7–9). The time of first freshwater-to-marine transition N1 is largely consistent with the origin of marine AOA within the family Nitrosopumilaceae reported previously [16, 17]. However, the time of the latter two transitions expanding to freshwater habitats are much older than the formation time of any deep lakes ranging from 4 to 30 Mya (Supplementary Table S3), indicating there may be intermediate habitats for the ancestor of these freshwater clades before colonizing deep lakes.
Habitat transition between freshwater and marine involves many eco-physiological challenges for the prokaryotes in many aspects, including environment changes, nutrient availability, and the adaptation to salinity change. Salinity preference is considered a conserved trait involving in many genes and complex cellular processes [67] and transitions between marine and freshwater ecosystems are difficult and infrequent [68, 69]. However, marine-to-freshwater transition events are proposed to have occurred across distinct microbial lineages. For example, SAR11, representing up to 25–50% of total planktonic cells in marine environments, has a well-known freshwater clade LD12 (or subclade IIIb), which may have evolved from a genome-streamlined marine ancestor [70,71,72]. Moreover, some archaeal lineages, such as Bathyarchaeia (formerly Bathyarchaeota phylum or the Miscellaneous Crenarchaeotic Group), have undergone salinity-freshwater transition events, leading to the adaptations specific to marine and freshwater sediments [73]. Our findings of these marine-to-freshwater transitions within the family Nitrosopumilaceae provide novel evidence for habitat transitions across salinity barrier and further insights into the evolutionary history and ecological adaptation for freshwater counterparts.
Genomic content changes involved in the transition to freshwater environments
Evolutionary transitions between marine and freshwater environments are often accompanied by changes in the genome repertoire in response to distinct physiochemical and ecological conditions, such as salinity changes and biotic interactions [68]. To pinpoint the relevant genes associated with habitat transitions, we used two complementary approaches: the ancestral genome reconstruction analysis using the asymmetric Wagner parsimony and the gene enrichment analysis with Fisher’s exact test (Supplementary Fig. S9). The former approach reconstructs ancestral genome contents and infers the gains and losses of lineage-specific gene families along the phylogeny, and the latter one identifies the genes that are significantly under- or overrepresented among three main freshwater clades and two marine groups of the family Nitrosopumilaceae. Among the candidate gene families screened by these two approaches, we manually selected a set of functional genes associated with osmoregulation and nutrient acquisition (Supplementary Table S10), which likely facilitate habitat transitions of these freshwater clades within Nitrosopumilaceae and contribute to specialization to the local environment during their diversification.
-
(1)
Urea utilization
Urea in aquatic habitats is often utilized as a nitrogen source because of its properties of direct assimilation and fast intracellular hydrolysis, which provides two atoms of nitrogen in reduced form [74]. Most members of the Fresh-II and Fresh-III clades were found to harbor the genes encoding the intact components of urea utilization, including the core urease operon (ureABC) and four additional urease accessory genes (ureEFGH) (Fig. 4), whereas these genes were rarely dispersed in members of the Fresh-I clade and two marine groups. Urea utilization in the class Nitrososphaeria was firstly reported in a marine sponge symbiont Cenarchaeum symbiosum A [66] and then demonstrated in the laboratory using pure cultures [75] and in marine environments [76]. Two types of urea transporters, the sodium:solute symporter (SSS) family and urea transporter (UT) family, have been identified in terrestrial Nitrososphaeria [75, 77], while only the former was detected in the Fresh-II clade and nearly absent in the other two freshwater clades (Fig. 4). The expression level of the SSS family transporter could be two orders of magnitude higher than that of the urease genes in a marine archaeon Ca. Nitrosopelagicus brevis U25 when growing with urea [78], indicating that this transporter is involved in taking up urea from the surroundings. As one of the most important components of the dissolved organic nitrogen pool, urea provides 10–50% of bioavailable nitrogen in lake and river surface waters [79]. Urea usually accumulates from external sources and internal processes, with the former dominated by agricultural fertilizer [80] and the latter dominated by phytoplankton decomposition and microbial metabolism of nitrogenous substrates [74]. The nearly exclusive presence of these urea utilization genes in most members of the Fresh-II and Fresh-III clades indicates that these clades might be able to use urea instead of ammonia as the sole energy source in lakes and rivers, as have been shown in some marine lineages [81, 82].
Fig. 4: The distribution of functional genes enriched in three freshwater clades of the class Nitrososphaeria. 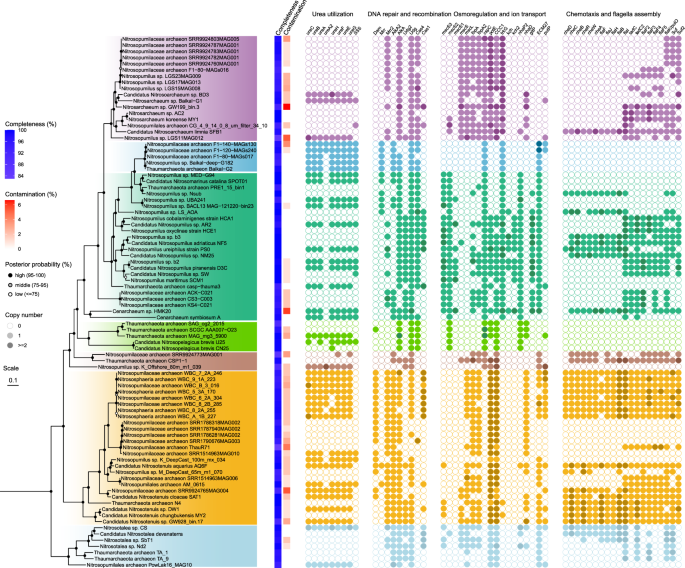
The enriched genes among three freshwater clades and two marine groups of Nitrososphaeria were identified using Fisher’s exact test (p < 0.05), followed by manual inspection. An empty cycle in the right panel represents the absence of a gene family in an archaeon, whereas the filled cycles with light and deep colors represent the single copy form and the multiple copy of a gene family, respectively. The phylogeny in the left panel and color scheme for these clades are the same as the phylogeny in Fig. 1B. Details of these functional genes are provided in the main text and Supplementary Table S10.
-
(2)
Osmoregulation
To cope with osmotic pressure variations in freshwater environments, a variety of strategies are exploited by prokaryotes, including the regulation of mechanosensitive channels, biosynthesis and uptake of compatible solutes and ion transport mechanisms [83, 84]. The Fresh-I and III clades exclusively harbored the gene encoding the large conductance of mechanosensitive (MS) channels (MscL, Fig. 4), which protects the cells from osmotic downshock by allowing fast efflux of intracellular nonspecific solutes [85, 86]. In contrast, all members in the Fresh-II clade and two marine groups of the family Nitrosopumilaceae lacked the mscL gene, suggesting that the gene may have been lost from these lineages or their ancestors (Supplementary Fig. S10). In addition, six separate gene families encoding the homolog of the small conductance of the MS channel (MscS) were identified in members of the class Nitrososphaeria (Supplementary Fig. S11), including the one exclusively present in the Fresh-I clade and two mscS genes in both marine groups. In the typical strain of Nitrosopumilus maritimus SCM1, one mscS gene (Nmar_1342) is adjacent to the ectABCD gene cluster, which encodes the biosynthesis of ectoine/hydroxyectoine as a compatible solute. The mscS gene in the marine archaeon is reported to be coexpressed with the ectABCD cluster under high salinity conditions [87], indicating that the gene may be involved in osmotic stress protection using the MscS-type mechanosensitive channel. The Fresh-III clade also enriched a putP gene encoding the high-affinity Na+:proline symporter, which catalyzes the sodium ion-dependent uptake of proline as compatible solute during adaptation to osmotic stress [88]. Despite the ectABCD cluster missing in all freshwater clades, the presence of mscL and mscS genes and the additional genes responsible for osmolyte uptake likely facilitate their adaptation to osmotic shocks resulting from physical water mixing in lakes and rivers.
Ion transport is also a necessary strategy adopted by prokaryotes to cope with osmotic stress, in which inorganic ions accumulate in the cytoplasm driven by an electrochemical gradient to maintain cellular homeostasis. Three clades of freshwater Nitrososphaeria have different sets of gene families involved in ion transport when compared with their marine relatives. For example, the trkG gene encoding the membrane component of the Trk-type potassium transport system is present in the Fresh-I and II clades and Marine-shallow group, but absent in all members of the Fresh-III clade and Marine-deep group (Fig. 4). In contrast, the Marine-shallow group exclusively contained two separate kch genes encoding the voltage-gated potassium channel. The nhaP2 gene encoding the K+:H+ antiporter was only detected in the Fresh-II clade and two marine groups, whereas the Fresh-III clade exclusively possessed the nhaD gene encoding the Na+:H+ antiporter NhaD. Together, the identification of these genes involved in the regulation of mechanosensitive channels and cellular homeostasis shed light on the processes that confer osmoadaptation in freshwater Nitrososphaeria.
-
(3)
Stress regulation
In addition to osmoregulation, the genes involved in the regulation of other stresses, including cold and nutrient limitation, were also enriched in aquatic Nitrososphaeria. For example, two separate gene families (ibpA) encoding the small heat shock protein of the Hsp20 family were exclusively present in the Fresh-I and Fresh-III clades and Marine-shallow group (Fig. 4). The Hsp20 family protein is a molecular chaperone protein that binds to unfolded proteins to prevent them from irreversible aggregation [89] and enhances the thermotolerance of a hyperthermophilic archaeon at temperatures lower than the optimum [90]. The Hsp20 gene has been found to be differentially expressed in marine AOA, such as in response to ammonia limitation in Ca. N. brevis CN25 [78] and under Cu limitation or toxicity conditions in N. maritimus SCM1 [91], indicating a potential role of the ibpA genes for mediating nutrient limitation stress in freshwater environments. Moreover, all aquatic AOA except for the Fresh-III clade harbored the gene cspC, which encodes a cold shock protein in the CspA family and is involved in the regulation of the global stress response regulator RpoS and numerous universal stress protein family A UspA [92].
-
(4)
Motility and chemotaxis
Archaeal flagella consist of motor components, flagellar filaments, and accessory proteins, providing motility in liquids and on the surface for archaea [93]. A list of genes involved in flagella/pili assemblies and chemotaxis was present in freshwater clades of the class Nitrososphaeria (Fig. 4). The members from lakes, estuaries, and groundwater within the Fresh-III clade contained genes flaF, flaI, flaJ, flaB, flaH, and flaK, whereas a subcluster of MAGs from the Amazon River within the same clade lacked these genes. The majority of the archaeal genomes with flagella/pili-related genes possessed the gene responsible for the chemotaxis signal transduction system (cheA, cheB, cheC, cheD, cheR, cheW, cheY, MCP, and methyl-accepting chemotaxis proteins). These genes were absent in the Fresh-I and Fresh-II clades except for the archaeon Nitrosoarchaeum limnia SFB1 within the Fresh-I clade, which encodes all genes required to assemble archaea flagellum that have been confirmed using microscopy [94]. These flagella-related genes were present in MAGs from groundwater, estuary, and surface sediments (Fresh-I and III clades) but absent from the water column in deep layers (Fresh-II), pointing to a possible scenario in which motility is required for these particle-attached AOA to adapt to varying nutrients in the sediments and estuaries rather than as free-living populations in the water column. Interestingly, motility-related genes were also harbored by most members in the Marine-shallow group, including several pure cultures, such as Nitrosopumilus sp. b3, N. ureiphilus PS0, and Cenarchaeum sp. HMK20, whereas all members in the Marine-deep group lacked these genes. It is worth noting that the presence of flagella or pili genes does not indicate the capability for motility. For example, the flagella apparatus was not observed in strain PS0 in the laboratory using electron microscopy despite the existence of these genes [95].
-
(5)
DNA repair and defense systems
The genes responsible for DNA repair and defense systems were significantly enriched in different freshwater clades, and the discrepancies in the metabolic category probably played a key role in their genomic evolution. Additional DNA repair-related gene families were often detected in different clades. For example, two alkA genes encoding 3-methyladenine/8-oxoguanine DNA glycosylase that remove alkylation damage from duplexes and single-stranded DNA were identified, with one conserved in all members and the other exclusively present in the Fresh-II and Fresh-III clades (Fig. 4 and Supplementary Fig. S12). In contrast, the Marine-shallow group possesses an additional udg gene, which encodes uracil-DNA glycosylase, a DNA repair enzyme that initiates base-excision repair and removes uracil from damaged DNA. In addition to the DNA repair genes, the genes encoding the components of microbial immune systems [96], including restriction-modification (RM) and clustered regularly interspaced short palindromic repeats (CRISPR-Cas), were also enriched in these freshwater clades. For example, the Fresh-II clade contained the mrr gene encoding endonuclease, whereas the mrr2 gene encoding restriction endonuclease was present in all aquatic AOA except in the Fresh-II clade. The Fresh-III clade exclusively had a gene (dam) encoding DNA adenine methylase or site-specific DNA methylase. The genes encoding CRISPR-associated protein Cas4 was absent in the Fresh-III clade but present in all other groups, while the gene encoding the Cas1 protein was present in part of the Marine-shallow group and the Fresh-III clade. The pattern of CRISPR-related genes was consistent with previous analyses of mobile genetic elements in members of the class Nitrososphaeria [97], indicating distinct immune mechanisms in these freshwater clades. We hypothesized that the enrichment of these genes involved in DNA repair and defense processes was likely associated with different magnitudes of selection pressures in freshwater and marine environments.

Conclusions
Our 17 MAGs reconstructed from deep lakes and great rivers present a great opportunity to study the diversification and adaptation mechanisms of the class Nitrososphaeria in freshwater environments. Based on phylogenomic analyses of these new MAGs and the other Nitrososphaeria-associated genomes from diverse habitats, we concluded that freshwater Nitrososphaeria mainly belong to the family Nitrosopumilaceae and consist of three distinct clades namely Nitrosopumilus-, Nitrosoarchaeum- and Nitrosotenuis-like clades. To the best of our knowledge, this is the first study to comprehensively evaluate the genomic diversity of the class Nitrososphaeria in freshwater habitats and their phylogenetic divergence. The separation of the three clades mainly by habitats and distinct abundance patterns across different aquatic biomes consistently supports niche separation among these freshwater archaea. We further provide genomic evidence that one freshwater-to-marine and two subsequent marine-to-freshwater transition events have occurred in the evolutionary history of the family Nitrosopumilaceae, the former of which provides a highly resolved evolutionary path compared to the terrestrial-to-marine evolutionary path proposed previously. The latter two marine-to-freshwater habitat transitions were accompanied by horizontal transfer of the genes involved in osmoregulation, cell motility, and nutrition regulation for their adaptations and recolonization to freshwater habitats. Specifically, the first transition was characterized by the presence of urea utilization genes in the Fresh-II and Fresh-III clades, whereas the second one was characterized by the exclusive presence of the genes encoding mechanosensitive channels and cell motility in the Fresh-I and Fresh-III clades. Our findings are an indispensable complement to the current understanding of the class Nitrososphaeria by providing novel insights into distinct mechanisms underlying their freshwater adaptations.
References
Rinke C, Chuvochina M, Mussig AJ, Chaumeil P-A, Davín AA, Waite DW, et al. A standardized archaeal taxonomy for the Genome Taxonomy Database. Nat Microbiol. 2021;6:946–59.
Karner MB, DeLong EF, Karl DM. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature. 2001;409:507–10.
Buckley DH, Graber JR, Schmidt TM. Phylogenetic analysis of nonthermophilic members of the kingdom Crenarchaeota and their diversity and abundance in soils. Appl Environ Microbiol. 1998;64:4333–9.
Casamayor EO, Schäfer H, Bañeras L, Pedrós-Alió C, Muyzer G. Identification of and spatio-temporal differences between microbial assemblages from two neighboring sulfurous lakes: Comparison by microscopy and denaturing gradient gel electrophoresis. Appl Environ Microbiol. 2000;66:499–508.
Francis CA, Roberts KJ, Beman JM, Santoro AE, Oakley BB. Ubiquity and diversity of ammonia-oxidizing Archaea in water columns and sediments of the ocean. Proc Natl Acad Sci USA. 2005;102:14683–8.
Stahl DA, de la Torre JR. Physiology and diversity of ammonia-oxidizing archaea. Annu Rev Microbiol. 2012;66:83–101.
DeLong EF. Archaea in coastal marine environments. Proc Natl Acad Sci USA. 1992;89:5685–9.
Fuhrman JA, McCallum K, Davis AA. Novel major archaebacterial group from marine plankton. Nature. 1992;356:148–9.
Qin W, Zheng Y, Zhao F, Wang Y, Urakawa H, Martens-Habbena W, et al. Alternative strategies of nutrient acquisition and energy conservation map to the biogeography of marine ammonia-oxidizing archaea. ISME J. 2020;14:2596–609.
Aylward FO, Santoro AE. Heterotrophic Thaumarchaea with small genomes are widespread in the dark ocean. mSystems. 2020;5:e00415–00420.
Reji L, Francis CA. Metagenome-assembled genomes reveal unique metabolic adaptations of a basal marine Thaumarchaeota lineage. ISME J. 2020;14:2105–15.
Wang Y, Huang J-M, Cui G-J, Nunoura T, Takaki Y, Li W-L, et al. Genomics insights into ecotype formation of ammonia-oxidizing archaea in the deep ocean. Environ Microbiol. 2019b;21:716–29.
Zhong H, Lehtovirta-Morley L, Liu J, Zheng Y, Lin H, Song D, et al. Novel insights into the Thaumarchaeota in the deepest oceans: their metabolism and potential adaptation mechanisms. Microbiome. 2020;8:78.
Wang B, Qin W, Ren Y, Zhou X, Jung M-Y, Han P, et al. Expansion of Thaumarchaeota habitat range is correlated with horizontal transfer of ATPase operons. ISME J. 2019a;13:3067–79.
Sheridan PO, Raguideau S, Quince C, Holden J, Zhang L, Gaze WH, et al. Gene duplication drives genome expansion in a major lineage of Thaumarchaeota. Nat Commun. 2020;11:5494.
Ren M, Feng X, Huang Y, Wang H, Hu Z, Clingenpeel S, et al. Phylogenomics suggests oxygen availability as a driving force in Thaumarchaeota evolution. ISME J. 2019;13:2150–61.
Yang Y, Zhang C, Lenton TM, Yan X, Zhu M, Zhou M, et al. The evolution pathway of ammonia-oxidizing archaea shaped by major geological events. Mol Biol Evol. 2021;38:3637–48.
Alves RJE, Minh BQ, Urich T, von Haeseler A, Schleper C. Unifying the global phylogeny and environmental distribution of ammonia-oxidising archaea based on amoA genes. Nat Commun. 2018;9:1517.
Llirós M, Casamayor EO, Borrego C. High archaeal richness in the water column of a freshwater sulfurous karstic lake along an interannual study. FEMS Microbiol Ecol. 2008;66:331–42.
Wang Z, Wang Z, Huang C, Pei Y. Vertical distribution of ammonia-oxidizing archaea (AOA) in the hyporheic zone of a eutrophic river in North China. World J Microbiol Biotechnol. 2014;30:1335–46.
Mußmann M, Brito I, Pitcher A, Sinninghe Damsté JS, Hatzenpichler R, Richter A, et al. Thaumarchaeotes abundant in refinery nitrifying sludges express amoA but are not obligate autotrophic ammonia oxidizers. Proc Natl Acad Sci USA. 2011;108:16771–6.
Biller S, Mosier A, Wells G, Francis C. Global biodiversity of aquatic ammonia-oxidizing archaea is partitioned by habitat. Front Microbiol. 2012;3:252.
Beman JM, Francis CA. Diversity of ammonia-oxidizing archaea and bacteria in the sediments of a hypernutrified subtropical estuary: Bahia del Tobari, Mexico. Appl Environ Microbiol. 2006;72:7767–77.
Auguet J-C, Nomokonova N, Camarero L, Casamayor EO. Seasonal changes of freshwater ammonia-oxidizing archaeal assemblages and nitrogen species in oligotrophic alpine lakes. Appl Environ Microbiol. 2011;77:1937–45.
Small GE, Bullerjahn GS, Sterner RW, Beall BFN, Brovold S, Finlay JC, et al. Rates and controls of nitrification in a large oligotrophic lake. Limnol Oceanogr. 2013;58:276–86.
Herber J, Klotz F, Frommeyer B, Weis S, Straile D, Kolar A, et al. A single Thaumarchaeon drives nitrification in deep oligotrophic Lake Constance. Environ Microbiol. 2020;22:212–28.
Auguet J-C, Triadó-Margarit X, Nomokonova N, Camarero L, Casamayor EO. Vertical segregation and phylogenetic characterization of ammonia-oxidizing Archaea in a deep oligotrophic lake. ISME J. 2012;6:1786–97.
Podowski JC, Paver SF, Newton RJ, Coleman ML. Genome streamlining, proteorhodopsin, and organic nitrogen metabolism in freshwater nitrifiers. bioRxiv. 2021;2021.2001.2019.427344.
Gohl DM, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotech. 2016;34:942–9.
Restrepo-Ortiz CX, Auguet J-C, Casamayor EO. Targeting spatiotemporal dynamics of planktonic SAGMGC-1 and segregation of ammonia-oxidizing thaumarchaeota ecotypes by newly designed primers and quantitative polymerase chain reaction. Environ Microbiol. 2014;16:689–700.
Liu S, Wang H, Chen L, Wang J, Zheng M, Liu S, et al. Comammox Nitrospira within the Yangtze River continuum: community, biogeography, and ecological drivers. ISME J. 2020;14:2488–504.
Santos-Júnior CD, Sarmento H, de Miranda FP, Henrique-Silva F, Logares R. Uncovering the genomic potential of the Amazon River microbiome to degrade rainforest organic matter. Microbiome. 2020;8:151.
Jung M-Y, Sedlacek CJ, Kits KD, Mueller AJ, Rhee S-K, Hink L, et al. Ammonia-oxidizing archaea possess a wide range of cellular ammonia affinities. ISME J. 2022;16:272–83.
Kim BK, Jung MY, Yu DS, Park SJ, Oh TK, Rhee SK, et al. Genome sequence of an ammonia-oxidizing soil archaeon, “Candidatus Nitrosoarchaeum koreensis” MY1. J Bacteriol. 2011;193:5539–40.
Jung MY, Park SJ, Kim SJ, Kim JG, Sinninghe Damste JS, Jeon CO, et al. A mesophilic, autotrophic, ammonia-oxidizing archaeon of thaumarchaeal group I.1a cultivated from a deep oligotrophic soil horizon. Appl Environ Microbiol. 2014;80:3645–55.
Lebedeva EV, Hatzenpichler R, Pelletier E, Schuster N, Hauzmayer S, Bulaev A, et al. Enrichment and genome sequence of the group i.1a ammonia-oxidizing archaeon “Ca. Nitrosotenuis uzonensis” representing a clade globally distributed in thermal habitats. PLoS One. 2013;8:e80835.
Li Y, Ding K, Wen X, Zhang B, Shen B, Yang Y. A novel ammonia-oxidizing archaeon from wastewater treatment plant: Its enrichment, physiological and genomic characteristics. Sci Rep. 2016;6:23747.
Sauder LA, Albertsen M, Engel K, Schwarz J, Nielsen PH, Wagner M, et al. Cultivation and characterization of Candidatus Nitrosocosmicus exaquare, an ammonia-oxidizing archaeon from a municipal wastewater treatment system. ISME J. 2017;11:1142–57.
Wang Y, Qin W, Jiang X, Ju F, Mao Y, Zhang A, et al. Seasonal prevalence of ammonia-oxidizing archaea in a full-scale municipal wastewater treatment plant treating saline wastewater revealed by a 6-year time-series analysis. Environ Sci Technol. 2021;55:2662–73.
Xing P, Tao Y, Luo J, Wang L, Li B, Li H, et al. Stratification of microbiomes during the holomictic period of Lake Fuxian, an alpine monomictic lake. Limnol Oceanogr. 2020;65:S134–S148.
Cabello-Yeves PJ, Zemskaya TI, Rosselli R, Coutinho FH, Zakharenko AS, Blinov VV, et al. Genomes of novel microbial lineages assembled from the sub-ice waters of Lake Baikal. Appl Environ Microbiol. 2017;84:e02132–02117.
Cabello-Yeves PJ, Zemskaya TI, Zakharenko AS, Sakirko MV, Ivanov VG, Ghai R et al. Microbiome of the deep Lake Baikal, a unique oxic bathypelagic habitat. Limnol Oceanogr. 2020;65:1471–88.
Qin W, Amin SA, Martens-Habbena W, Walker CB, Urakawa H, Devol AH, et al. Marine ammonia-oxidizing archaeal isolates display obligate mixotrophy and wide ecotypic variation. Proc Natl Acad Sci USA. 2014;111:12504–9.
Bayer B, Vojvoda J, Offre P, Alves RJ, Elisabeth NH, Garcia JA, et al. Physiological and genomic characterization of two novel marine thaumarchaeal strains indicates niche differentiation. ISME J. 2016;10:1051–63.
Bristow LA, Dalsgaard T, Tiano L, Mills DB, Bertagnolli AD, Wright JJ, et al. Ammonium and nitrite oxidation at nanomolar oxygen concentrations in oxygen minimum zone waters. Proc Natl Acad Sci USA. 2016;113:10601–6.
Hink L, Gubry-Rangin C, Nicol GW, Prosser JI. The consequences of niche and physiological differentiation of archaeal and bacterial ammonia oxidisers for nitrous oxide emissions. ISME J. 2018;12:1084–93.
Martens-Habbena W, Berube PM, Urakawa H, de la Torre JR, Stahl DA. Ammonia oxidation kinetics determine niche separation of nitrifying Archaea and Bacteria. Nature. 2009;461:976–9.
Mayr MJ, Zimmermann M, Guggenheim C, Brand A, Bürgmann H. Niche partitioning of methane-oxidizing bacteria along the oxygen–methane counter gradient of stratified lakes. ISME J. 2020;14:274–87.
Reis PCJ, Thottathil SD, Ruiz-González C, Prairie YT. Niche separation within aerobic methanotrophic bacteria across lakes and its link to methane oxidation rates. Environ Microbiol. 2020;22:738–51.
Tran PQ, Bachand SC, McIntyre PB, Kraemer BM, Vadeboncoeur Y, Kimirei IA, et al. Depth-discrete metagenomics reveals the roles of microbes in biogeochemical cycling in the tropical freshwater Lake Tanganyika. ISME J 2021;15:1971–86.
Sauder LA, Engel K, Lo C-C, Chain P, Neufeld JD. Candidatus Nitrosotenuis aquarius, an ammonia-oxidizing archaeon from a freshwater aquarium biofilter. Appl Environ Microbiol. 2018;84:e01430-18.
Hug LA, Thomas BC, Brown CT, Frischkorn KR, Williams KH, Tringe SG, et al. Aquifer environment selects for microbial species cohorts in sediment and groundwater. ISME J. 2015;9:1846–56.
Barco RA, Garrity GM, Scott JJ, Amend JP, Nealson KH, Emerson D. A genus definition for Bacteria and Archaea based on a standard genome relatedness index. MBio. 2020;11:e02475–02419.
Haas S, Desai DK, LaRoche J, Pawlowicz R, Wallace DWR. Geomicrobiology of the carbon, nitrogen and sulphur cycles in Powell Lake: a permanently stratified water column containing ancient seawater. Environ Microbiol. 2019;21:3927–52.
Herbold CW, Lehtovirta-Morley LE, Jung M-Y, Jehmlich N, Hausmann B, Han P, et al. Ammonia-oxidising archaea living at low pH: Insights from comparative genomics. Environ Microbiol. 2017;19:4939–52.
Shen M, Li Q, Ren M, Lin Y, Wang J, Chen L, et al. Trophic status is associated with community structure and metabolic potential of planktonic microbiota in plateau lakes. Front Microbiol. 2019;10:2560–2560.
Giovannoni SJ, Cameron Thrash J, Temperton B. Implications of streamlining theory for microbial ecology. ISME J. 2014;8:1553–65.
Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, González JM, et al. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci USA. 2013;110:11463–8.
Grzymski JJ, Dussaq AM. The significance of nitrogen cost minimization in proteomes of marine microorganisms. ISME J. 2012;6:71–80.
Bragg JG, Hyder CL. Nitrogen versus carbon use in prokaryotic genomes and proteomes. Proc R Soc Lond B Biol Sci. 2004;271:S374–7.
Mende DR, Bryant JA, Aylward FO, Eppley JM, Nielsen T, Karl DM, et al. Environmental drivers of a microbial genomic transition zone in the ocean’s interior. Nat Microbiol. 2017;2:1367–73.
Baudouin-Cornu P, Schuerer K, Marlière P, Thomas D. Intimate evolution of proteins: Proteome atomic content correlates with genome base composition. J Biol Chem. 2004;279:5421–8.
Santoro AE, Dupont CL, Richter RA, Craig MT, Carini P, McIlvin MR, et al. Genomic and proteomic characterization of Candidatus Nitrosopelagicus brevis: An ammonia-oxidizing archaeon from the open ocean. Proc Natl Acad Sci USA. 2015;112:1173–8.
Luo H, Tolar BB, Swan BK, Zhang CL, Stepanauskas R, Ann Moran M, et al. Single-cell genomics shedding light on marine Thaumarchaeota diversification. ISME J. 2014;8:732–6.
Reji L, Tolar BB, Smith JM, Chavez FP, Francis CA. Depth distributions of nitrite reductase (nirK) gene variants reveal spatial dynamics of thaumarchaeal ecotype populations in coastal Monterey Bay. Environ Microbiol. 2019;21:4032–45.
Hallam SJ, Konstantinidis KT, Putnam N, Schleper C, Watanabe Y, Sugahara J, et al. Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc Natl Acad Sci USA. 2006;103:18296–301.
Martiny JBH, Jones SE, Lennon JT, Martiny AC. Microbiomes in light of traits: A phylogenetic perspective. Science. 2015;350:aac9323.
Logares R, Bråte J, Bertilsson S, Clasen JL, Shalchian-Tabrizi K, Rengefors K. Infrequent marine–freshwater transitions in the microbial world. Trends Microbiol. 2009;17:414–22.
Paver SF, Muratore D, Newton RJ, Coleman ML, Flynn TM. Reevaluating the salty divide: Phylogenetic specificity of transitions between marine and freshwater systems. mSystems. 2018;3:e00232–00218.
Henson MW, Lanclos VC, Faircloth BC, Thrash JC. Cultivation and genomics of the first freshwater SAR11 (LD12) isolate. ISME J. 2018;12:1846–60.
Luo H, Csűros M, Hughes AL, Moran MA, Azam F, Zehr J. Evolution of divergent life history strategies in marine Alphaproteobacteria. MBio. 2013;4:e00373–00313.
Zaremba-Niedzwiedzka K, Viklund J, Zhao W, Ast J, Sczyrba A, Woyke T, et al. Single-cell genomics reveal low recombination frequencies in freshwater bacteria of the SAR11 clade. Genome Biol. 2013;14:R130.
Fillol M, Auguet J-C, Casamayor EO, Borrego CM. Insights in the ecology and evolutionary history of the Miscellaneous Crenarchaeotic Group lineage. ISME J. 2016;10:665–77.
Siuda W, Kiersztyn B. Urea in lake ecosystem: The origin, concentration and distribution in relation to trophic state of the Great Mazurian Lakes (Poland). Pol J Ecol. 2015;63:110–23. 114
Spang A, Poehlein A, Offre P, Zumbragel S, Haider S, Rychlik N, et al. The genome of the ammonia-oxidizing Candidatus Nitrososphaera gargensis: insights into metabolic versatility and environmental adaptations. Environ Microbiol. 2012;14:3122–45.
Kitzinger K, Padilla CC, Marchant HK, Hach PF, Herbold CW, Kidane AT, et al. Cyanate and urea are substrates for nitrification by Thaumarchaeota in the marine environment. Nat Microbiol. 2019;4:234–43.
Kerou M, Offre P, Valledor L, Abby SS, Melcher M, Nagler M, et al. Proteomics and comparative genomics of Nitrososphaera viennensis reveal the core genome and adaptations of archaeal ammonia oxidizers. Proc Natl Acad Sci USA. 2016;113:E7937–E7946.
Carini P, Dupont Christopher L, Santoro, Alyson E. Patterns of thaumarchaeal gene expression in culture and diverse marine environments. Environ Microbiol. 2018;20:2112–24.
Bogard MJ, Donald DB, Finlay K, Leavitt PR. Distribution and regulation of urea in lakes of central North America. Freshw Biol. 2012;57:1277–92.
Glibert PM, Harrison J, Heil C, Seitzinger S. Escalating worldwide use of urea – a global change contributing to coastal eutrophication. Biogeochemistry. 2006;77:441–63.
Alonso-Sáez L, Waller AS, Mende DR, Bakker K, Farnelid H, Yager PL, et al. Role for urea in nitrification by polar marine Archaea. Proc Natl Acad Sci USA. 2012;109:17989–94.
Tolar BB, Wallsgrove NJ, Popp BN, Hollibaugh JT. Oxidation of urea-derived nitrogen by thaumarchaeota-dominated marine nitrifying communities. Environ Microbiol. 2017;19:4838–50.
Gunde-Cimerman N, Plemenitaš A, Oren A. Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol Rev. 2018;42:353–75.
Hagemann M. Molecular biology of cyanobacterial salt acclimation. FEMS Microbiol Rev. 2011;35:87–123.
Blount P, Iscla I. Life with bacterial mechanosensitive channels, from discovery to physiology to pharmacological target. Microbiol Mol Biol Rev. 2020;84:e00055–00019.
Martinac B, Bavi N, Ridone P, Nikolaev YA, Martinac AD, Nakayama Y, et al. Tuning ion channel mechanosensitivity by asymmetry of the transbilayer pressure profile. Biophys Rev. 2018;10:1377–84.
Widderich N, Czech L, Elling FJ, Konneke M, Stoveken N, Pittelkow M, et al. Strangers in the archaeal world: osmostress-responsive biosynthesis of ectoine and hydroxyectoine by the marine thaumarchaeon Nitrosopumilus maritimus. Environ Microbiol. 2016;18:1227–48.
Jung H, Hilger D, Raba M. The Na+/L-proline transporter PutP. Front Biosci-Landmark. 2012;17:745–59.
Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–88.
Li D-C, Yang F, Lu B, Chen D-F, Yang W-J. Thermotolerance and molecular chaperone function of the small heat shock protein HSP20 from hyperthermophilic archaeon, Sulfolobus solfataricus P2. Cell Stress Chaperones. 2012;17:103–8.
Qin W, Amin SA, Lundeen RA, Heal KR, Martens-Habbena W, Turkarslan S, et al. Stress response of a marine ammonia-oxidizing archaeon informs physiological status of environmental populations. ISME J. 2017a;12:508–19.
Phadtare S, Inouye M. Role of CspC and CspE in regulation of expression of RpoS and UspA, the stress response proteins in Escherichia coli. J Bacteriol. 2001;183:1205–14.
Albers S-V, Jarrell KF. The archaellum: An update on the unique archaeal motility structure. Trends Microbiol. 2018;26:351–62.
Mosier AC, Lund MB, Francis CA. Ecophysiology of an ammonia-oxidizing archaeon adapted to low-salinity habitats. Micro Ecol. 2012;64:955–63.
Qin W, Heal KR, Ramdasi R, Kobelt JN, Martens-Habbena W, Bertagnolli AD, et al. Nitrosopumilus maritimus gen. nov., sp. nov., Nitrosopumilus cobalaminigenes sp. nov., Nitrosopumilus oxyclinae sp. nov., and Nitrosopumilus ureiphilus sp. nov., four marine ammonia-oxidizing archaea of the phylum Thaumarchaeota. Int J Syst Evol Microbiol. 2017b;67:5067–79.
Dupuis M-È, Villion M, Magadán AH, Moineau S. CRISPR-Cas and restriction–modification systems are compatible and increase phage resistance. Nat Commun. 2013;4:2087.
Krupovic M, Makarova KS, Wolf YI, Medvedeva S, Prangishvili D, Forterre P, et al. Integrated mobile genetic elements in Thaumarchaeota. Environ Microbiol. 2019;21:2056–78.
Acknowledgements
We thank Dr. Jindong Zhao from Peking University, Dr. Tao Li from Institute of Hydrobiology, Chinese Academy of Sciences and Dr. Fengping Wang from Shanghai Jiao Tong University for valuable discussions, Dr. Michael Wagner and Dr. Craig Herbold from University of Vienna for helpful comments about the early version of the manuscript, Dr. Hao Zhang from the Chinese University of Hong Kong for the discussion of molecular dating analyses. This study was supported by National Natural Science Foundation of China (42002304, 91851117), Research Program of Sino-Africa Joint Research Center, Chinese Academy of Sciences (151542KYSB20210007, 151542KYSB20200015, SAJC202106), the National International Science and Technology Cooperation Project (KY201901006), National Natural Science Foundation of China (41571058, 41871048), and CAS Key Research Program of Frontier Sciences (QYZDB-SSW-DQC043). We also thank the anonymous reviewers for providing critical comments to improve the manuscript.
Author information
Authors and Affiliations
Contributions
The research was conceived by MLR and JJW. Data analyses and drafting of the manuscript were undertaken by MLR with the contribution of JJW. All authors were involved in the discussion and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ren, M., Wang, J. Phylogenetic divergence and adaptation of Nitrososphaeria across lake depths and freshwater ecosystems. ISME J 16, 1491–1501 (2022). https://doi.org/10.1038/s41396-022-01199-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-022-01199-7
This article is cited by
-
Extreme summers impact cropland and grassland soil microbiomes
The ISME Journal (2023)
-
Pan-kinome of Legionella expanded by a bioinformatics survey
Scientific Reports (2022)
