
Abstract
The formation and fate of pyrite (FeS2) modulates global iron, sulfur, carbon, and oxygen biogeochemical cycles and has done so since early in Earth’s geological history. A longstanding paradigm is that FeS2 is stable at low temperature and is unavailable to microorganisms in the absence of oxygen and oxidative weathering. Here, we show that methanogens can catalyze the reductive dissolution of FeS2 at low temperature (≤38 °C) and utilize dissolution products to meet cellular iron and sulfur demands associated with the biosynthesis of simple and complex co-factors. Direct access to FeS2 is required to catalyze its reduction and/or to assimilate iron monosulfide that likely forms through coupled reductive dissolution and precipitation, consistent with close associations observed between cells and FeS2. These findings demonstrate that FeS2 is bioavailable to anaerobic methanogens and can be mobilized in low temperature anoxic environments. Given that methanogens evolved at least 3.46 Gya, these data indicate that the microbial contribution to the iron and sulfur cycles in ancient and contemporary anoxic environments may be more complex and robust than previously recognized, with impacts on the sources and sinks of iron and sulfur and other bio-essential and thiophilic elements such as nickel and cobalt.
Similar content being viewed by others


Introduction
Pyrite (FeS2) is the most abundant sulfide mineral in Earth’s crust and its formation and fate are key controls on the biogeochemical cycles of iron (Fe), sulfur (S), carbon, and oxygen (O2) [1]. However, the specific role of sulfide minerals, such as FeS2, in these cycles has changed drastically over geological time [2, 3]. Isotopic evidence suggests that FeS2 has formed in sedimentary or volcanic environments since at least 3.81 Gya [4]. Yet, prior to the advent of oxygenic photosynthesis and the gradual accumulation of O2 ~2.4 Gya (as reviewed in [5, 6]), biotic and abiotic oxidative weathering of FeS2 with O2, the primary driver of sulfate (SO42−) input into oceans [7], was of minimal importance [3, 6, 8, 9]. Furthermore, reduction of FeS2 has only been demonstrated at high temperature (>90 °C) in abiotic laboratory reactors containing artificially high (>15 bar) concentrations of hydrogen (H2) [10, 11]. Together, these data imply that once iron sulfide minerals such as FeS2 form in low temperature sulfidic and anoxic environments, they remain largely unavailable to biology since they cannot be extensively mobilized through oxidative or reductive weathering [3]. This raises key questions as to how anaerobic microorganisms acquire Fe (and other thiophilic metals such as nickel and cobalt) and S to meet biosynthetic demands in contemporary and past anoxic environments that favor formation of iron sulfide minerals, such as sulfidic freshwater or marine sediments [1, 12,13,14].
All cells require Fe and S, which are key components of amino acids, vitamins, and a variety of co-enzymes and co-factors. This includes both simple and complex biological iron–sulfur ([Fe-S]) clusters that function in electron transfer, catalysis, substrate binding and activation, and other purposes [15, 16]. As a consequence of their functional versatility, [Fe-S] clusters are ubiquitous in all life forms [15, 17] and have central roles in photosynthesis, respiration, and fermentation [18].
Methanogenic archaea are argued to be the most primitive of extant organisms [19, 20], and isotopic evidence preserved in the rock record indicates their presence as early as ~3.46 Gya [21]. Despite their early emergence during the anoxic Archean, when free Fe and/or S may have been limited due to precipitation as sulfide minerals [2, 3], methanogens have been shown to utilize [Fe-S] clusters more extensively than other organisms. Specifically, Methanococcus maripaludis cells were shown to contain ~15 fold more [Fe-S] clusters per mg protein than Escherichia coli cells [22] and methanogen genomes code for a higher number of putative [Fe-S] cluster binding proteins than those of facultative anaerobes and obligate aerobes [23].
The types of S sources that support methanogens vary based on their evolutionary history. Methanogens that belong to more primitive (Class I) lineages (Methanopyrales, Methanobacteriales, and Methanococcales) tend to be grown with sulfide (HS−/H2S) as their S source whereas more recently evolved (Class II) lineages (Methanocellales, Methanomicrobiales, and Methanosarcinales) can also be grown with cysteine as their sulfur source [22, 24]. Sulfide is toxic to methanogens [25], but toxicity can be alleviated by precipitation with metals, such as ferrous iron (Fe(II)), nickel (II), or cobalt (II) [26]. Sulfide readily reacts with Fe(II), resulting in the formation of transient aqueous iron monosulfide clusters (FeS(aq)) via Eq. (1) [27] that can condense to form metastable and less soluble nanoparticulate iron sulfide mineral phases such as mackinawite (FeS(mack)) via Eq. (2) [27,28,29]. Depending on the type and abundance of aqueous S species in an environment (e.g., H2S or polysulfide (Sx2−)), dissolution and/or solid-state transformation of FeS(mack) can occur and can result in the formation of FeS2 via Eqs. (3) and (4) [13, 27, 29]:
The anoxic, sulfidic environments that anaerobic methanogens inhabit can have high concentrations of Fe(II), which would tend to favor the formation of iron sulfide minerals including FeS(mack) and FeS2 [27, 29]. However, FeS2 is reportedly stable under anoxic, low temperature conditions [10, 11] and, to date, has not been shown to be bioavailable to microorganisms in the absence of oxygen or alternative oxidants (e.g., manganese oxides, ferric iron ions) that themselves require O2 to generate [30].
Here, we characterized the growth and methanogenesis activity of the model Class I and II methanogens, Methanococcus voltae strain A3 and Methanosarcina barkeri strain MS, respectively, in defined medium (Supplementary Online Materials (SOM) Tables S1 and S2). M. voltae A3 was originally isolated from a saltmarsh [24], whereas M. barkeri MS was originally isolated from an anaerobic sewage digestor [31]. M. voltae and M. barkeri were provided with canonical Fe and S sources (Fe(NH4)2(SO4)2 • 6H2O and Na2S • 9H2O or L-cysteine HCl, respectively), laboratory synthesized nanoparticulate FeS2 or FeS(mack), or powdered specimen FeS2 (63–125 µm diameter) as their sole Fe and S sources. All minerals used in our experiments were subjected to multiple washing steps to remove trace Fe and S sources that may have formed during synthesis or mineral crushing. X-ray diffraction spectra of minerals used in growth experiments confirm that they correspond to previously characterized FeS2 or FeS(mack) standards (SOM Fig. S1). The size distribution of laboratory synthesized FeS2, determined via field emission scanning electron microscopy (FE-SEM), ranged from 0.22 to 4.49 µm with small framboids commonly aggregated into larger particles (SOM Fig. S2). Growth was monitored via direct cell counts (M. voltae) or DNA yield due to cell aggregation (M. barkeri), and methanogenesis activity was monitored by methane (CH4) production via gas chromatography.
Materials and methods
Preparation of synthetic pyrite
All chemicals used in mineral synthesis were American Chemical Society grade or higher. All glassware was first washed in 10% nitric acid and rinsed three times with 18.2 Ω MilliQ H2O (MQ H2O). Pyrite (FeS2) was synthesized in the lab according to Berner [32]. Briefly, within an anaerobic chamber (Coy Labs, Grass Lake, MI), 14.4 g Na2S • 9H2O and 16.7 g FeSO4 • 7H2O were separately dissolved in 50 ml of anoxic MQ H2O. These two solutions were then combined into a 500 ml bottle and stirred vigorously for 15 min, at which point 2.1 g of elemental sulfur was added. The bottle was then sealed with a butyl rubber stopper, removed from the chamber, and bubbled with N2 passed over heated (200 °C) and H2-reduced copper shavings for 45 min. Following purging, the solution was incubated anoxically for 4 days at 65 °C and then for another 4 days at 85 °C. After incubation, the FeS2 was washed (via centrifugation and decanting) in the following series to remove unreacted HS−, Fe(II), FeS, and S0: four rinses with 1 N HCl, one rinse with boiling 6 N HCl, two rinses with MQ H2O, three rinses with >99.5% acetone, and, finally, three rinses with 0.2 µm filter-sterilized MQ H2O. After washing, the FeS2 was pelleted via centrifugation, brought into an anaerobic chamber, the aqueous phase decanted, and the pellet resuspended in sterile, anoxic MQ H2O in a sterile serum bottle and was stoppered. Finally, the headspace of the bottle was purged with 0.2 µm filtered N2 gas. The % w/v of the slurry was determined by drying 1 ml of slurry in triplicate under N2 in pre-weighed glass serum bottles.
Mackinawite (FeS(mack)) was synthesized similarly to FeS2. Within an anaerobic chamber 1.14 g Na2S • 9H2O and 1.67 g FeSO4 • 7H2O were separately dissolved in 10 ml of anoxic MQ H2O. The solutions were then combined and vigorously stirred in a 50 ml glass flask for 15 min. The FeS(mack) solution was washed three times with anoxic MQ H2O, as described above. Inside an anaerobic chamber, the final mineral pellet was resuspended in 15 ml of 0.2 µm filter-sterilized, anoxic MQ H2O and then transferred to a sterile serum bottle and stoppered. Finally, the headspace of the bottle was purged with 0.2 µm filtered N2 gas. The % w/v of the slurry was determined as described above.
Specimen FeS2, obtained from Zacatecas, Mexico (Ward’s Science, Rochester, NY), was crushed with an ethanol sterilized jaw crusher (Gilson, Lewis Center, OH) in a laminar flow hood. Crushed mineral was applied to a sterile sieve stack (U.S. Standard #10/2000 μm, U.S. Standard #35/500 μm, U.S. Standard #60/250 μm, U.S. Standard #120/125 μm, U.S. Standard #230/63 μm, and catch pan; all 8″ diameter) in a laminar flow hood. The sieve stack was mechanically shaken (~275 oscillations and ~150 taps per minute) for 10 min using a Ro-Tap® RX-29 mechanical sieve shaker (W.S. Tyler, Mentor, OH). Following sieving, the sieve stack was returned to the laminar flow hood and the 63–125 µm fraction was collected, washed as described above for laboratory synthesized FeS2, and dried at room under N2 in a sterile serum bottle sealed with a butyl rubber stopper.
Minerals were characterized using a SCINTAG X-1 system X-ray powder diffraction (XRD) spectrometer (XRD Eigenmann GmbH, Mannheim, Germany), a JEOL JSM-6100 scanning electron microscope equipped with an energy dispersive X-ray spectrometer (JEOL USA INC., Peabody, MA), and a Zeiss SUPRA 55VP FE-SEM (Zeiss, Oberkochen, Germany). Samples for mineral characterization were prepared by drying ~10 ml of the mineral slurry or reacted mineral in a sealed serum bottle under a stream of N2.
Strains and cultivation media
Methanococcus voltae strain A3 was obtained from the American Type Culture Collection (ATTC-BAA-1334). M. voltae was grown in Fe- and S-free basal medium that contained (g l−1): NaCl, 21.98; MgCl2 • 6H2O, 5.10; NaHCO3, 5.00; NH4Cl, 0.50; K2HPO4, 0.14; KCl, 0.33; CaCl2 • 2H2O, 0.10 (SOM Table S2). The basal medium was amended with 0.01 g l−1 Fe(NH4)2(SO4)2 • 6H2O and 0.48 g l−1 Na2S • 9H2O for Fe (II)/HS− grown cells. Sulfide was added from an anoxic, sterile stock solution 30 min prior to inoculation. Basal medium was amended (each 1% v/v) with trace element, vitamin, and organic solutions. The trace element solution used was based on Whitman et al. [33] and was amended to omit Fe and replace sulfate salts with chloride salts at the same molar concentrations. The trace element solution contained (g l−1): nitriloacetic acid, 1.500; MnCl2 • 4H2O, 0.085; CoCl2 • H2O, 0.100; ZnCl2, 0.047; CuCl2 • 2H2O, 0.0683; NiCl2 • 6H2O, 0.0683; Na2SeO3, 0.200; Na2MoO4 • 2H2O, 0.100; Na2WO4 • 2H2O, 0.100. The vitamin solution contained (g l−1): pyridodoxine HCl, 0.01; thiamine HCl, 0.005; riboflavin, 0.005 g; nicotinic acid, 0.005; calcium D(+) pantothenate, 0.005; biotin, 0.002; folic acid, 0.002; cobalamin, 0.0001. The organics solution consisted of 1 M sodium acetate • 3H2O, 75 mM L-leucine HCl, and 75 mM L-isoleucine HCl. Cultures of M. voltae grown with N2–CO2 were supplemented with a 40% (wt/v) sodium formate stock solution added to a final concentration of 0.4% (v/v) prior to inoculation.
Methanosarcina barkeri strain MS was purchased from the ATCC (BAA-2329) and grown on Fe- and S-free basal medium that contained (g L−1): NaCl, 1.0; MgCl2 • 6H2O, 0.4, NaHCO3, 2.00; NH4Cl, 0.54; KCl, 0.5; K2HPO4, 0.35; KH2PO4, 0.23; CaCl2 • 2H2O, 0.10 (SOM Table S3). The basal medium was amended with 0.01 g l−1 Fe(NH4)2(SO4)2 • 6H2O and 0.35 g l−1 L-cysteine HCl for Fe(II)/cysteine grown cells. Sterile, anoxic stock trace metals and vitamins solutions were prepared and added to the basal medium (1% v/v final concentration) prior to inoculation. The iron-free SL-10 trace metals solution contained (g l−1): ZnCl2, 0.070; MnCl2 • 4H2O, 0.100; H3BO3, 0.006; CoCl2 • 6H2O, 0.190; CuCl2 • 2H2O, 0.002; NiCl2 • 6H2O, 0.024; Na2MoO4 • 2H2O, 0.036. The vitamin solution used was as described for M. voltae above. Sterile, anoxic stock solutions of methanol and acetate were added to final concentrations of 0.5% (v/v) and 40 mM, respectively.
All medium components, with the exception of NaHCO3, were dissolved in MQ H2O and then boiled for 15 min. After boiling, the bottle containing the medium was sealed with a butyl rubber stopper and sparged with N2 passed over a heated copper column for 1 h L−1 of medium. After degassing, the medium was sealed, brought into an anaerobic chamber, and NaHCO3 was added as specified above. After adding these components, the pH of the medium was adjusted to 7.00 using anoxic 2 M HCl or 1 M NaOH. Seventy-five ml of media were dispensed into 165 ml serum bottles (DWK Life Sciences, Milville, NJ) that had been acid-washed in 10% nitric acid and sealed with black rubber stoppers (Glasgerätebau Ochs GmbH, Bovenden, Germany) and aluminum crimp caps. The serum bottles were then brought out of the chamber and the headspace was purged with N2 for 30 min. The bottles were then autoclaved for 20 min at 123 °C.
Synthesized FeS2 was added to microcosms from the prepared slurry at a final concentration of 2 mM of Fe, and specimen FeS2 powder was added to microcosms at 1.5 g per 75 ml of medium (167 mM). This amount was chosen to normalize for differences in the estimated surface area between laboratory synthesized FeS2 and powdered specimen FeS2. Surface area of laboratory synthesized FeS2 was estimated by calculating the average particle size of synthesized FeS2 framboids (0.74 µm diameter) then calculating surface area of the particles assuming framboids were octahedral. Briefly, ten fields of view were randomly collected using back-scattered secondary electron imaging on a Zeiss SUPRA 55VP FE-SEM. The images were imported into ImageJ software (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997–2018.) and particle sizes were calculated using the “Analyze Particles” function. The surface area of specimen FeS2 was estimated assuming an average particle diameter of 94 µm, the midpoint of the 63–125 µm size fraction, and octahedral form.
Cultivation procedures
Cultures of M. voltae and M. barkeri were maintained by weekly transfers (10% v/v) into fresh medium with Fe(II) as Fe source, HS− or L-cysteine as S source, and formate or methanol as methanogenesis substrate, respectively. Cells were washed prior to inoculation by pelleting them in sealed 50 ml centrifuge tubes (Globe Scientific, Mahwah, NJ) at 4700 × g for 20 min at 4 °C in a swing-out bucket rotor. Spent medium was decanted in an anaerobic chamber and the cell pellet was resuspended in sterile and anoxic Fe/S-free basal medium. All experiments used washed M. voltae and M. barkeri cells grown with 26 µM Fe(II) and either 2 mM HS− or 2 mM L-cysteine as inoculum (10% v/v), respectively. After inoculation, the headspaces of microcosms were flushed with an 80:20 (v/v) mixture of N2–CO2 gas that had been passed through a 0.2 µm filter for at least 15 min before being pressurized (final pressure of 3.14 atm). M. voltae and M. barkeri were incubated at 38 °C. All cultures were incubated statically on their sides to minimize disruption of microbe-mineral interactions while maximizing gas diffusion.
Measurement of activity and growth
Headspace gas from microcosms was sampled with a N2 flushed syringe and stopcock and diluted with ultra-high purity N2 into CaliBond bags (Calibrated Instruments Inc., Manhasset, NY) prior to CH4 determination. CH4 was determined by gas chromatography by injecting 5 ml of sample into an injector valve set at 55 °C into an SRI 8610C gas chromatograph (SRI instruments, Torrance, CA) equipped with a 4.5 m × 0.125″ OD Hayesep DB 100/120 packed column with the oven set to 44 °C (Valco Instrument Company Inc., Houston, TX). CH4 was detected by a flame-ionization detector set at 156 °C with He as carrier gas. CH4 peak area values were converted to ppm using a standard CH4 curve (EGAS Depot, Nampa, ID). Dissolved HS− was determined via colorimetry (670 nm) using the methylene blue assay [34] and converted to total HS− (dissolved and gas phase) using Henry’s law. Absorbance was measured using a Genesys 10S Vis Spectrophotometer (Thermo Scientific, Waltham, MA).
Growth in cultures of M. voltae was determined by direct counting of cells using a Petroff-Hausser counting chamber on a Nikon YS100 light microscope with a ×100 oil objective (Nikon, Tokyo, Japan). Prior to counting, cells were concentrated by centrifugation at 15,000 × g for 15 min in a fixed-angle rotor. Despite efforts to minimize cell aggregation in cultures of M. barkeri, cells still aggregated necessitating an alternative approach to quantify growth. Quantification of DNA from M. barkeri was used as a proxy for cell growth. DNA was extracted using the FastDNA SPIN kit for soils (MP Biomedicals, Santa Ana, CA) with a modified protocol. First, 2 ml of culture were pelleted via centrifugation at 20,000 × g for 30 min at 20 °C. The supernatant was carefully decanted and 489 µl of sodium phosphate buffer (MP Biomedicals, Santa Ana, CA) and 61 µl of MT lysis buffer (MP Biomedicals) were used to resuspend the cell pellet. The resuspended pellet and buffer mixture were subjected to three rounds of freezing at −80 °C and thawing at 70 °C. The cell and lysis buffer mixture were transferred to a Lysis E tube (MP Biomedicals), homogenized on a bead beater for 40 s (Biospec Products, Bartlesville, OK), then centrifuged 14,000 × g for 15 min at 4 °C to pellet cell debris. DNA in the supernatant was quantified using a Qubit HS dsDNA assay (Invitrogen, Carlsbad, CA).
Dialysis membrane experiments
Dialysis membranes of varying pore sizes were used to examine the requirement for direct cell contact with FeS2 or the size of extracellular proteins involved in catalyzing its reduction. A 12 cm length of Spectra/Por (Spectrum Labs Inc., Rancho Dominguez, CA) 3 dialysis tubing with either 3.5, 25, 50, or 100 kDa molecular weight cutoff was used for each bottle. The dialysis membranes were first washed by three cycles of soaking precut tubing in MQ H2O for 30 min at 65 °C, followed by decanting and rinsing with fresh MQ H2O. The tubing was then transferred to a bottle containing 50% ethanol, and the submerged membranes were then incubated overnight at 65 °C. The tubes were then handled in a UV-sterilized molecular hood with ethanol sterilized gloves and forceps where they were tied at one end with ethanol-soaked monofilament line (Berkley, Columbia, SC). To seal the bags in preparation for addition of FeS2, the end of the tubing was first folded over twice, then crimped with a dialysis clip (Spectrum Labs Inc., Rancho Dominguez, CA). The clip was then removed, leaving a fold that was amenable to tying with monofilament. The tubing was then returned to the 50% ethanol bottle, sparged with N2 gas for 1 h, and brought into an anaerobic chamber. Monofilament line rather than dialysis clips was used to facilitate the entry of tubing into narrow mouthed (20 mm diameter) serum bottles for cultivation experiments.
Inside the anaerobic chamber, the dialysis bags were rinsed in sterile and anoxic MQ H2O to remove residual ethanol. Then, laboratory -synthesized FeS2 was added to dialysis bags as a slurry (2 mM final concentration Fe once in medium bottles), and the bags were resealed as described above with monofilament line. Finally, the outsides of all bags were rinsed with anoxic and sterile MQ H2O and were transferred to pre-sterilized 165 ml serum bottles containing 75 ml of basal medium amended with vitamins, trace metals, and organics solution. The bottles were sealed with sterilized butyl rubber stoppers and purged with an 80:20 mixture of 0.2 µm filtered N2:CO2 for 45 min to remove gas originating from the anaerobic chamber and were then pressurized (final pressure of 3.14 atm). The bottles were then inoculated with washed, Fe(II)/HS−-grown cells (10% v/v inoculum). Positive controls contained 3.5 kDa dialysis membranes and FeS2 that was not enclosed in the membrane. Unamended controls contained 3.5 kDa dialysis membranes without any added Fe or S source.
Electron microscopy
Subsamples of cultures of M. voltae for use in FE-SEM imaging analyses were collected and incubated in 10 ml of freshly prepared 2% glutaraldehyde solution in 0.1 M phosphate buffer for 2 h at room temperature. Glutaraldehyde-fixed samples were applied to a Au-sputtered 0.2 µm black Isopore polycarbonate filter (MilliporeSigma, Burlington, MA). The filter was subjected to an ethanol series (25, 50, 70, 85, 95, and 100%) for complete dehydration, then stored dry at 4 °C until imaging in the Imaging and Chemical Analysis Laboratory at Montana State University. Images were taken using a high-resolution FE-SEM (Supra 55VP, Zeiss, Thornwood, NY) with a primary electron beam energy of 1 keV at different magnifications. Samples were mounted on the FE-SEM holder using double-sided carbon tape and sputtered with a thin film of iridium for conductivity before loading to the FE-SEM chamber. Elemental mapping was acquired using energy dispersive X-ray spectroscopy with a Scanning Auger Electron Nanoprobe-Physical Electronics 710 and 10 and 20 kV energy beam.
Results and discussion
Reductive dissolution of FeS2 at low temperature
Cultivation of M. voltae and M. barkeri, when provided with formate or methanol and acetate as the methanogenesis substrates, respectively, resulted in significant cell and CH4 production in cultures containing nanoparticulate FeS2 or canonical forms of Fe and S but were not observed in cultures unamended with Fe or S (Fig. 1a–d and Extended Data File 1). Growth of both M. voltae and M. barkeri was also observed with powdered specimen FeS2 as the sole Fe and S source (SOM Fig. S3). These observations indicate that both synthetic nanoparticulate FeS2 and specimen FeS2, the latter of which should be reflective of FeS2 commonly encountered by methanogens in natural environments, can serve as the sole Fe and S source for these cells.

Production of biomass, methane, and sulfide in cultures of Methanococcus voltae provided with formate (a, c, e, respectively) or Methanosarcina barkeri provided with methanol and acetate (c, d, f, respectively) as the methanogenesis substrate when grown with different iron and sulfur sources in defined medium. Legends in a and c and in b and d are the same. Ferrous iron and sulfide or cysteine were added to final concentrations of 26 µM and 2 mM, respectively. Laboratory synthesized pyrite was added to a final concentration of 2 mM Fe. CH4, total methane; HS−, total sulfide; Fe(II), ferrous iron; Cys, cysteine; FeS2, pyrite. Data shown are the mean values for each condition with error bars reflecting the standard deviation of three replicate biological reactors; a single reactor for abiotic controls was monitored and thus error bars are not presented. Total HS− was monitored only in the FeS2 growth conditions. See Extended Data File 1 for CH4 and HS− data.
Sulfide (referred to as HS− from here on) was detected in cultures of M. voltae and M. barkeri provided with nanoparticulate FeS2 but not in abiotic controls; concentrations of HS− increased with increased production of biomass and CH4 (Fig. 1e, f and Supplementary Data File 1). This is consistent with cells catalyzing the reductive dissolution of FeS2 to produce HS−. The accumulation of HS− suggests that the rate of FeS2 reduction by methanogens exceeds the rate of cellular S uptake. To the extent that cells are not taking up HS− (discussed below), the amount of HS− that accumulated in the growth medium can be combined with the predicted cellular S requirement per cell to provide a conservative estimate of the amount of FeS2 reduction in laboratory grown cultures. The estimated S content of log phase Methanococcus maripaludis, a close relative of M. voltae, is 349 nmol per mg protein [22]. The amount of protein in log phase M. voltae is 8.88 × 10−10 mg cell−1 (E.S. Boyd, unpublished data). As such, an individual M. voltae cell during log phase is estimated to contain 0.31 fmol S. When this is combined with the amount of HS- that accumulates during FeS2 reduction, as normalized to an individual cell (0.15 fmol cell−1, see Extended Data File 1), it is conservatively estimated that M. voltae catalyzes the reduction of 0.23 fmol FeS2 cell−1 during log phase growth.
Importantly, acid labile Fe(II) was not detected in spent medium via the ferrozine assay (~11 µM lower detection limit) at any point during the growth of M. voltae or M. barkeri (data not shown). This could indicate that the cells maintain aqueous Fe(II) below the limits of detection of our assay or that the Fe(II) phase that forms via FeS2 reduction (reverse of Eqs. (2)–(4)) undergoes rapid precipitation on the mineral surface. A coupled reductive dissolution and precipitation reaction has been proposed for abiotic reduction of FeS2 at high temperature, with iron monosulfide as mackinawite or pyrrhotite forming as the predominant Fe(II) phase(s) on the surface of FeS2 [10]. Together, these data indicate that representatives of both Class I and II methanogens can catalyze the reduction of FeS2 at low temperatures (≤38 °C) and raise the question of the form(s) of Fe and S that is assimilated to meet biosynthetic demands.
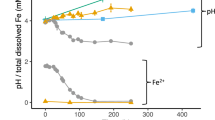
One source of Fe and S that could potentially support growth of methanogens during the reductive dissolution of FeS2 is FeS(mack), which would be expected to be produced if biological FeS2 reduction is taking place via the reverse reaction of Eqs. (3) or (4). Further, FeS(mack) is a commonly detected form of iron monosulfide in low temperature settings [35] and is a possible precursor to the formation of FeS2 in these environments [29, 36]. Equilibrium dissolution of FeS(mack) in non-acidic (pH > 6.0) aqueous solutions results in the formation of FeS(aq) as the dominant form of Fe(II) (reverse of Eq. (2)) when HS– is at a concentration exceeding 1 µM [28]. Thus, to further evaluate the potential form of Fe and S supporting growth of cells, M. voltae was provided with nanoparticulate, laboratory -synthesized FeS2 or FeS(mack) in the presence or absence of 1 mM added HS–. This concentration of HS− was chosen such that any aqueous Fe(II) either dissociated from FeS(mack) or from a trace laboratory contaminant, if present, would predominantly be in the form of FeS(aq) [28, 29]. M. voltae cultures showed no apparent difference in growth or activity with FeS(mack) in the presence or absence of added HS− and these did not differ from the Fe(II)/HS− positive control (Fig. 2). M. barkeri also demonstrated growth on FeS(mack) as the sole Fe and S source (SOM Fig. S4). We also tested the growth of M. voltae on FeS2 in the presence or absence of 1 mM HS− and observed growth and surprisingly also observed HS− production from FeS2 even in the presence of added HS− (SOM Fig. S5). Sulfide concentrations could not be accurately monitored in growth experiments with FeS(mack) given that this mineral is partially soluble, acid labile, and forms nanoparticles (~2 nm in size) [29] that cannot be readily size fractionated; these features interfere with the sulfuric acid-based methylene blue assay that was used to quantify HS−. Collectively, these results suggest that cells can use mineral forms of Fe and S (as FeS2 and FeS(mack)) to meet both Fe and S demands and that they may even prefer these forms over canonical forms (e.g., Fe(II) or HS−/cysteine).

Production of cells (a) and methane (b) for M. voltae cultures. Cells were grown with synthetic mackinawite (FeS) added to a final concentration of 2 mM Fe with or without added 1 mM added HS−. Cells provided with Fe(II) and HS− served as the positive control. CH4, methane; Fe(II), ferrous iron; HS−, sulfide; FeS, mackinawite. Data shown are the mean values for each condition with error bars reflecting the standard deviation of three replicate biological reactors. See Extended Data File 1 for CH4 and HS− data.
Comparing growth on FeS2 to growth under Fe or S limiting conditions
To determine whether M. voltae can directly assimilate aqueous HS- and to verify that trace Fe or S in the cultivation medium was not sufficient to meet cellular demands, we compared the growth and activity of M. voltae cultures provided with only FeS2 to those provided with only Fe(II) or only HS− (SOM Fig. S6). When cells were provided with only Fe(II), there was no production of cells and CH4 production was minimal. However, production of cells and CH4 was observed in cultures provided with only HS−, albeit at 40% of the cell yield for FeS2-grown cells. These results indicate that trace Fe in the medium can partially fulfill biological demands in cultures provided with HS− but that trace S cannot, consistent with a higher demand for S than Fe in cells [22]. Our findings agree with the apparent ability of Class I and Class II methanogens to assimilate HS– during growth [22, 37]. However, given that HS− readily reacts with Fe(II) to form FeS(aq) and FeS(mack) and the fact that both HS− and Fe(II) tend to be provided in the growth medium of methanogens, it is likely that these and other Class I and II methanogens can assimilate FeS(aq) or iron monosulfide precipitates and may supplement S demands by assimilating free HS−.
Interactions between methanogens and FeS2
The requirement for cells to associate with the surface of FeS2 for mineral-dependent growth was investigated using dialysis membrane experiments. M. voltae was grown with formate as the electron donor and with FeS2 free in solution or sequestered in dialysis membranes with 3.5 kDa pore size to limit direct access to the mineral surface. Growth of M. voltae and the reduction of FeS2 was assessed by monitoring production of cells, CH4, and HS− (Fig. 3 and SOM Table S3). Cells, CH4, and HS− were produced when FeS2 was provided free in solution but not when FeS2 was sequestered in dialysis tubing with a 3.5 kDa pore size (Fig. 3). Cells, CH4, and HS− were also not detected when FeS2 was sequestered in dialysis tubing with 25, 50, or 100 kDa pore sizes (data not shown), which would allow for the passage of small extracellular proteins or metabolites across the dialysis membrane that may be involved in FeS2 reduction or acquisition of reductive dissolution products. Collectively, these results indicate that cells require direct contact with FeS2 to catalyze its reduction and/or that cells require direct access to FeS2 to acquire Fe and S following the coupled reductive dissolution-precipitation reaction. Alternatively, these results could point to high molecular weight (>100 kDa) extracellular proteins, metabolites, or other cellular assemblages (such as enzyme containing vesicles) as being involved in catalyzing FeS2 reduction or in acquiring mineral-associated Fe and S. The involvement of vesicles in mineral sulfide oxidation has been demonstrated in the crenarchaeote Metallosphaera sedula, where they were shown to adhere to and promote the oxidation and solubilization of chalcopyrite (CuFeS2) [38].

FeS2 was added to a final concentration of 2 mM Fe. The cell density was determined after 5 days. Data shown are the mean values for each condition with error bars reflecting the standard deviation of three replicate biological reactors. CH4, methane; HS−, sulfide. See Supplementary Table 3 for data used to generate the plot.
We therefore examined associations between M. voltae (and possible vesicles) and both synthetic and specimen forms of FeS2 using FE-SEM imaging (Fig. 4). We did not observe any vesicles, but this is potentially attributable to the 0.4 µm pore size used for the EM grid which may not have allowed vesicles to be visualized. Rather, formate-grown M. voltae cells provided with FeS2 were observed to be attached to the mineral during growth, perhaps mediated by extracellular appendages (Fig. 4a, b and SOM Figs. S7 and S8). Archaeal flagella, or archaella, are well characterized in M. voltae and other Archaea where they function in motility, attachment to surfaces, and in biofilm formation [39]. The apparent affinity for cells to attach to the mineral surfaces and form biofilms is further supported by observations that when cells are grown with FeS2, CH4 production (seen as bubble generation) and cellular biomass (biofilms) are co-localized with settled synthetic (Supplementary Video 1) or specimen (Supplementary Video 2) mineral grains. While one would expect bubble generation to occur more extensively on nucleation points under highly saturated conditions, this phenomenon is also observed early in growth when CH4 levels are low. These observations provide further evidence indicating that methanogens likely attach to mineral sources to meet Fe and S biosynthetic demands through bio-catalyzed reductive dissolution.

a Cells of M. voltae grown with synthetic pyrite (FeS2) or b specimen FeS2. c Synthetic FeS2 or d specimen FeS2 in the absence of M. voltae. Samples were fixed with 2% glutaraldehyde before FE-SEM imaging on gold-sputtered 0.2 µm polycarbonate filters. Scale bar equals 0.5 µm in a–c and 1.0 µm in d.
Conclusions
Data presented here reveal that representatives of both Class I and II methanogens, including strains from both freshwater (M. barkeri) and marine (M. voltae) environments, can catalyze the reductive dissolution of FeS2. This activity results in the liberation of Fe and S, likely as precipitated secondary minerals such as FeS(mack) and/or pyrrhotite, that are used to meet biosynthetic demands. These observations raise intriguing questions related to the mechanism(s) of biological FeS2 reduction, the mechanism(s) of iron monosulfide acquisition and assimilation, and intracellular processing and trafficking of iron monosulfide for [Fe-S] cluster, amino acid, cofactor, and vitamin biosynthesis. Further, these observations highlight the ability of methanogens to transform Earth abundant and inert iron sulfide minerals, such as FeS2, into co-factors for enzymes capable of interconverting substrates (e.g., CO2/CH4 and H+/H2) of ecological and biotechnological relevance. These enzymes include simple and complex [Fe-S] clusters associated with [NiFe]-hydrogenases, carbon monoxide dehydrogenases, and numerous other Fe-S metalloproteins and co-factors synthesized by methanogens. The ability of anaerobic methanogens to acquire Fe and S from abundant mineral sources long considered to be non-bioavailable necessitates a re-evaluation of the Fe and S cycles in anoxic ecosystems of today and in the geological past.
References
Berner RA. Sedimentary pyrite formation: an update. Geochim Cosmochim Acta. 1984;48:605–15.
Fike DA, Bradley AS, Rose CV. Rethinking the ancient sulfur cycle. Ann Rev Earth Planet Sci. 2015;43:593–622.
Canfield DE, Habicht KS, Thamdrup B. The archean sulfur cycle and the early history of atmospheric oxygen. Science. 2000;288:658–61.
Smith PE, Evensen NM, York D, Moorbath S. Oldest reliable terrestrial 40Ar-39Ar age from pyrite crystals at Isua west Greenland. Geophys Res Lett. 2005;32:L21318.
Lyons TW, Reinhard CT, Planavsky NJ. The rise of oxygen in Earth’s early ocean and atmosphere. Nature 2014;506:307–15.
Canfield DE. The early history of atmospheric oxygen: homage to Robert M. Garrels. Annu Rev Earth Planet Sci. 2005;33:1–36.
Langmuir D. Aqueous environmental geochemistry. Vol. 8. Upper Saddle River, New Jersey: Prentice Hall; 1997.
Farquhar J, Bao H, Thiemens M. Atmospheric influence of Earth’s earliest sulfur cycle. Science. 2000;289:756–8.
Anbar AD, Duan Y, Lyons TW, Arnold GL, Kendall B, Creaser RA, et al. A whiff of oxygen before the great oxidation event? Science. 2007;317:1903–6.
Truche L, Berger G, Destrigneville C, Guillaume D, Giffaut E. Kinetics of pyrite to pyrrhotite reduction by hydrogen in calcite buffered solutions between 90 and 180°C: implications for nuclear waste disposal. Geochim Cosmochim Acta. 2010;74:2894–914.
Hall AJ. Pyrite-pyrrhotine redox reactions in nature. Miner Mag. 1986;50:223–9.
Thiel J, Byrne JM, Kappler A, Schink B, Pester M. Pyrite formation from FeS and H2S is mediated through microbial redox activity. Proc Nat Acad Sci USA. 2019;116:6897–902.
Rickard D. Kinetics of pyrite formation by the H2S oxidation of iron (II) monosulfide in aqueous solutions between 25 and 125 °C: the rate equation. Geochim Cosmochim Acta. 1997;61:115–34.
Schoonen MAA. Mechanisms of sedimentary pyrite formation. In: Sulfur biogeochemistry—past and present. Amend JP, Edwards KJ, Lyons TW, editors. Geological Society of America; 2004. p. 379.
Beinert H, Holm RH, Munck E. Iron-sulfur clusters: nature’s modular, multipurpose structures. Science. 1997;277:653–9.
Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 2005;74:247–81.
Imlay JA. Iron-sulphur clusters and the problem with oxygen. Mol Microbiol. 2006;59:1073–82.
Ayala-Castro C, Saini A, Outten FW. Fe-S cluster assembly pathways in bacteria. Microbiol Mol Biol Rev. 2008;72:110–25.
Russell MJ, Martin W. The rocky roots of the acetyl-CoA pathway. Trends Biochem Sci. 2004;29:358–63.
Weiss MC, Sousa FL, Mrnjavac N, Neukirchen S, Roettger M, Nelson-Sathi S, et al. The physiology and habitat of the last universal common ancestor. Nat Microbiol. 2016;1:16116.
Ueno Y, Yamada K, Yoshida N, Maruyama S, Isozaki Y. Evidence from fluid inclusions for microbial methanogenesis in the early Archaean era. Nature. 2006;440:516.
Liu Y, Sieprawska-Lupa M, Whitman WB, White RH. Cysteine is not the sulfur source for iron-sulfur cluster and methionine biosynthesis in the methanogenic archaeon Methanococcus maripaludis. J Biol Chem. 2010;285:31923–9.
Major TA, Burd H, Whitman WB. Abundance of 4Fe–4S motifs in the genomes of methanogens and other prokaryotes. FEMS Microbiol Lett. 2004;239:117–23.
Whitman WB, Shieh J, Sohn S, Caras DS, Premachandran U. Isolation and characterization of 22 mesophilic methanococci. Syst Appl Microbiol. 1986;7:235–40.
Koster IW, Rinzema A, de Vegt AL, Lettinga G. Sulfide inhibition of the methanogenic activity of granular sludge at various pH-levels. Water Res. 1986;20:1561–7.
Paulo LM, Stams AJM, Sousa DZ. Methanogens, sulphate and heavy metals: a complex system. Rev Environ Sci Bio. 2015;14:537–53.
Luther GW, Rickard DT. Metal sulfide cluster complexes and their biogeochemical importance in the environment. J Nanopart Res. 2005;7:389–407.
Rickard D. The solubility of FeS. Geochim Cosmochim Acta. 2006;70:5779–89.
Rickard D, Luther GW. Chemistry of iron sulfides. Chem Rev. 2007;107:514–62.
Schippers A, Jørgensen BB. Biogeochemistry of pyrite and iron sulfide oxidation in marine sediments. Geochim Cosmochim Acta. 2002;66:85–92.
Bryant MP, Boone DR. Emended description of strain MST(DSM 800T), the type strain of Methanosarcina barkeri. Int J Syst Evol. 1987;37:169–70.
Berner RA. The synthesis of framboidal pyrite. Econ Geol. 1969;64:383–84.
Whitman WB, Ankwanda E, Wolfe RS. Nutrition and carbon metabolism of Methanococcus voltae. J Bacteriol. 1982;149:852–63.
Fogo JK, Popowsky M. Spectrophotometric determination of hydrogen sulfide. Anal Chem. 1949;21:732–4.
Alistair RL, Katharine ERE, David JV. Transformation of synthetic mackinawite to hexagonal pyrrhotite: a kinetic study. Am Miner. 1995;80:960–7.
Benning LG, Wilkin RT, Barnes HL. Reaction pathways in the Fe–S system below 100 °C. Chem Geol. 2000;167:25–51.
Rauch BJ, Perona JJ. Efficient sulfide assimilation in Methanosarcina acetivorans is mediated by the MA1715 protein. J Bacteriol. 2016;198:1974.
Johnson TB, Mach C, Grove R, Kelly R, Van Cott K, Blum P. Secretion and fusion of biogeochemically active archaeal membrane vesicles. Geobiology. 2018;16:659–73.
Jarrell KF, Ding Y, Nair DB, Siu S. Surface appendages of archaea: structure, function, genetics and assembly. Life. 2013;3:86–117.
Acknowledgements
This work was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through Grant DE-SC0020246 to ESB. DP was partially supported by a fellowship from the Montana Space Grant Consortium. We thank Dr. David Mogk and Elif Roehm for their assistance with electron microscopy at the Montana Nanotechnology Facility, a National Nanotechnology Infrastructure Center member supported by National Science Foundation Grant ECCS-1542210.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Payne, D., Spietz, R.L. & Boyd, E.S. Reductive dissolution of pyrite by methanogenic archaea. ISME J 15, 3498–3507 (2021). https://doi.org/10.1038/s41396-021-01028-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-01028-3
This article is cited by
-
Micrometric pyrite catalyzes abiotic sulfidogenesis from elemental sulfur and hydrogen
Scientific Reports (2024)
-
Acute stress reduces population-level metabolic and proteomic variation
BMC Bioinformatics (2023)
-
Assimilatory sulfate reduction in the marine methanogen Methanothermococcus thermolithotrophicus
Nature Microbiology (2023)
-
Influence of sulfide on diazotrophic growth of the methanogen Methanococcus maripaludis and its implications for the origin of nitrogenase
Communications Biology (2023)
-
The minimal SUF system is not required for Fe–S cluster biogenesis in the methanogenic archaeon Methanosarcina acetivorans
Scientific Reports (2023)
