
Abstract
Hydrothermal sediments contain large numbers of uncultured heterotrophic microbial lineages. Here, we amended Guaymas Basin sediments with proteins, polysaccharides, nucleic acids or lipids under different redox conditions and cultivated heterotrophic thermophiles with the genomic potential for macromolecule degradation. We reconstructed 20 metagenome-assembled genomes (MAGs) of uncultured lineages affiliating with known archaeal and bacterial phyla, including endospore-forming Bacilli and candidate phylum Marinisomatota. One Marinisomatota MAG had 35 different glycoside hydrolases often in multiple copies, seven extracellular CAZymes, six polysaccharide lyases, and multiple sugar transporters. This population has the potential to degrade a broad spectrum of polysaccharides including chitin, cellulose, pectin, alginate, chondroitin, and carrageenan. We also describe thermophiles affiliating with the genera Thermosyntropha, Thermovirga, and Kosmotoga with the capability to make a living on nucleic acids, lipids, or multiple macromolecule classes, respectively. Several populations seemed to lack extracellular enzyme machinery and thus likely scavenged oligo- or monomers (e.g., MAGs affiliating with Archaeoglobus) or metabolic products like hydrogen (e.g., MAGs affiliating with Thermodesulfobacterium or Desulforudaceae). The growth of methanogens or the production of methane was not observed in any condition, indicating that the tested macromolecules are not degraded into substrates for methanogenesis in hydrothermal sediments. We provide new insights into the niches, and genomes of microorganisms that actively degrade abundant necromass macromolecules under oxic, sulfate-reducing, and fermentative thermophilic conditions. These findings improve our understanding of the carbon flow across trophic levels and indicate how primary produced biomass sustains complex and productive ecosystems.
Similar content being viewed by others


Introduction
Hydrothermally influenced marine sediments are characterized by fluctuating temperatures, and inorganic and organic carbon substrates that can support a diverse range of microbial metabolic activities [1,2,3]. The focus of research at hydrothermal systems often lies on the microbial transformation of inorganic compounds and hydrocarbons that originate in hydrothermal fluids and in the subsurface [4, 5]. However, a substantial proportion of the diversity and community function at hydrothermal systems is attributed to heterotrophic microorganisms [6, 7]. This heterotrophic community comprises many uncultured and understudied lineages which likely meet their carbon and energy demands using biological macromolecules such as proteins [8], nucleic acids [9], lipids [10], and polysaccharides [11]. Whether supplied as detritus from the overlying water column [12], as necromass from cell lysis, or as exudates, biological macromolecules are considered the most abundant class of organic compounds available for catabolism in marine sediments [13]. Identifying the microorganisms and enzymes involved in the degradation of macromolecules in hydrothermal sediments is thus relevant for our understanding of biogeochemical cycling and ecosystem functioning in deep-sea environments.
Hydrothermally influenced environments have been shown to host archaeal and bacterial heterotrophs, including Thermotogales, Thermococcales, and Archaeoglobales [14] that are able to grow on biological macromolecules. The use of macromolecules for growth requires a wide range of enzymes involved in extracellular cleavage, transmembrane import, and intracellular degradation. Generally, these polymeric substrates first need to be hydrolyzed by enzymes that are attached to cells or are released to the environment [15]. Subsequently, membrane receptors transport the depolymerized oligo- or monomers into the cell [16], which then enter catabolic pathways. The substrates produced by carbohydrate-active enzymes (CAZymes) and peptidases can be metabolized directly by glycolysis and subsequent oxidation via respiration or fermentation, while nucleosides and fatty acids require further hydrolysis and oxidation before entering the central carbon metabolic pathways [17].
Here we discovered and characterized heterotrophic thermophiles capable of degrading biological macromolecules in hydrothermal sediments. We targeted key metabolisms through amending organic-rich marine sediments from Guaymas Basin, Gulf of California, Mexico, with either proteins, polysaccharides, nucleic acids or lipids as sole added carbon and energy source (Fig. 1A, B). We investigated whether these macromolecule-degrading communities comprise uncultured microbial lineages that have been detected previously in metagenomic surveys of hydrothermal seafloor habitats, providing insights into so far overlooked ecological niches and the vast diversity of heterotrophs in these hydrothermal sediments.

A Core 4862-8 was recovered from hot hydrothermal sediments covered by an orange Beggiatoa mat and characterized by a steep thermal gradient that reached up to 146 °C in 30 cm below seafloor (cmbsf). B Core 4871-20 was recovered from temperate sediments characterized by a light-gray patch of sulfur deposits and infaunal worms and a moderate thermal gradient that reached ca. 14 °C in 50 cmbsf. C Enrichment cultures from slurries of cores 4862-8 and 4871-20 were supplemented with four types of carbon sources and incubated under oxic, sulfate-amended and fermentative conditions. The resulting 24 cultures were duplicated, yielding 48 enrichments. D Timeline of data collection during the period of incubation.
Methods
Enrichment cultures
Sediments were collected from two Alvin dives (4862 and 4871) during R/V Atlantis research cruise AT37-06 (December 2016) in the Guaymas Basin, Gulf of California, Mexico. Core 4862-8 site was collected from a hydrothermal hot spot near the Mat Mound area (27˚00.43′/111˚24.56′) in the southern Guaymas axial valley [18], and was characterized by an orange Beggiatoa mat and a very steep thermal gradient that reached up to 146 °C in 30 cm below seafloor (cmbsf) (Fig. 1A) [19]. Core 4871-20 was collected from a temperate site, termed “Site 2” (27˚02.77′/111˚23.09′) in the “Northern Towers” area, ca. 3 miles to the northeast of the frequently sampled hydrothermal locations [18]; the sediment was characterized by a light-gray patch of sulfur deposits and infaunal worms and a moderate thermal gradient that reached ca. 14 °C in 50 cmbsf (Fig. 1B) [19]. Both cores were strongly sulfidic and gas-rich, as observed immediately after shipboard recovery; oxygen penetrates Guaymas Basin hydrothermal sediments only by 1–2 mm, if at all [18]. Sediment (~150–200 ml) including porewater was sampled from three horizons (0–2, 3–4 and 5–10 cm) and stored without headspace in wide-mouth glass bottles at 4 °C (for details see Supplementary Information). Determination of optimal sample volume for inoculation is provided (see Supplementary Information). Slurries (1:10 sediment:anoxic artificial seawater) of 4862-8 (“hot”) and 4871-20 (“temperate”) sediments served as inocula for enrichment cultures incubated with either proteins, polysaccharides, nucleic acids, or lipids as carbon and energy source, and either oxygen (from air) or sulfate as an electron acceptor, or without an added electron acceptor (fermentative condition), in duplicates (n = 48). 50 ml anoxic cultures were grown in 100 ml serum bottles under argon headspace and 50 ml oxic cultures were grown in 250 ml baffled flasks in a shaking incubator (Thermo MaxQ6000). We used basal saltwater medium with sulfate for sulfate-reducing and oxic cultures and basal saltwater medium lacking sulfate (which was replaced by equimolar chloride) for fermentative cultures [20]. Exact concentrations of the media constituents are provided (Table S1). All enrichment cultures were incubated at 60 °C for 4 weeks.
Choice of carbon sources
Microbial cell lysate contains macromolecular organic matter, including proteins, lipids, nucleic acids, and polysaccharides (e.g., [21]). To test which microorganisms have specialized in the degradation of each of these compound classes, we chose analogues, i.e., model substrates, of defined origin whose structural key linkages (peptide, phosphodiester, and lipid glycerol ester bonds) are targeted consistently by the hydrolytic enzymes for each substrate class [22]. We used bovine serum albumin (BSA) as an analogue for water-soluble protein, because BSA is in the same size range (~600 amino acids, ~60 kDa) as many soluble microbial proteins (e.g., [23, 24]). We chose bacterial DNA of typical gram-negative Gammaproteobacteria (genus Salmonella), because we can produce it in sufficient quantity and high purity. The phosphatidylcholine lipid that we chose is derived from soy but is widespread in the domain Bacteria as well [25]. Although microbial cells harbor many more different polysaccharides, we included seven polysaccharides (alginate, amylopectin (starch), carrageenan, chitosan, chondroitin, dextran, and xanthan) that represent a variety of monomeric building blocks and glycosyl bonds found in microbial cells and marine algae [26,27,28,29,30].
Gas measurements
Hydrogen and methane concentrations of the anoxic enrichments were measured weekly using an SRI 310C gas chromatograph with thermal conductivity detector and molecular sieve column (see also Supplementary Information). After each measurement the headspace was purged with Argon to exclude product inhibition by the produced hydrogen. Thus, an increase in hydrogen from one week to another does not only represent an increase in the amount of hydrogen, but also in the rate of its production.
Cell counts
Cells were enumerated by direct microscopic counts from the cultures during the first and third week of incubation. In brief, the paraformaldehyde fixed cells were sonicated, filtered onto a polycarbonate filter (0.2 µm pore size), stained with DAPI, embedded in Citifluor:Vectashield (4:1) and counted using 20 grids (100 × 100 µm) per sample (see also Supplementary Information).
Statistical analyses
To determine differential activity (H2 concentrations and microbial cell numbers) among culture conditions, mixed-effects repeated measures analysis of variance (ANOVA) was employed using the lme function within the nlme package [31]. When significant effects were found, multiple comparisons between culture conditions were performed using the glht function within the multcomp package [32].
DNA extraction
Total nucleic acids were extracted from all 48 enrichments with a modified protocol based on Zhou et al. [33]. In brief, we treated 5 ml of sample using chemical (extraction buffer without CTAB), physical (3× freeze thaw) and enzymatic steps (overnight at 37 °C in lysozyme solution) for extraction of the DNA. Extraction blanks were included to assess potential laboratory contamination during extraction. DNA concentrations were measured fluorometrically using a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Canada).
16S rRNA gene amplicon sequencing and processing
The 16S rRNA gene V4-V5 variable regions were amplified using the bacterial primers 518F and 926R [34] and the archaeal primers 517F and 958R [35] (for details see Supplementary Information). Amplicons were sequenced using Illumina’s v3 600-cycle (paired-end) reagent kit on a MiSeq (Illumina Inc., San Diego, CA, USA). Reads were demultiplexed based on the combination of index (CASAVA 1.8) and barcode (custom python scripts). Raw sequences were processed and amplicon sequence variants (ASVs) were generated following the DADA2 [36] Pipeline Tutorial v1 (https://benjjneb.github.io/dada2/tutorial.html), for details see Supplementary Information.
16S rRNA-based community analyses
Community composition, diversity indices, and dissimilarity matrices were obtained using ASV relative abundances. Data analysis was performed using VisuaR (https://github.com/EmilRuff/VisuaR), a workflow based on custom scripts and R packages, including vegan v2.5-6 [37] and ggplot2 v3.3.2 [38] (for details see Supplementary Information).
Metagenomic sequencing, assembly, binning and analyses
Total DNA from 11 samples was used to prepare libraries with an Ovation Ultralow V2 DNA-Seq Kit (TECAN Group Ltd., Mannedorf, Switzerland). Sequencing was performed on an Illumina NextSeq 550 using the paired-end 2 × 150 bp run-type mode. Quality control was performed using PRINSEQ [39] (for details see Supplementary Information). Reads were co-assembled using SPAdes with the --meta option [40] and binned using MetaWRAP processing modules with initial extraction using MaxBin2, metaBAT2, and CONCOCT, bin refinement, and reassembly [41] (for details see Supplementary Information). Assembled genomes with more than 90% completeness, less than 3% contamination, as assessed using CheckM [42], and less than 100 contigs were further analyzed (Table 1). Taxonomy and closest phylogenetic relatives were assigned using GTDB-tk (v1.3.0) [43], average nucleotide identity (ANI), average amino acid identity (AAI) (Supplementary Data 1), and single-copy genes. A maximum likelihood phylogenetic tree of genomes was generated using PATRIC RaxML service [44] and visualized using the Interactive Tree of Life webtool.
Genomes were annotated using Distilled and Refined Annotation of Metabolism (DRAM) [45], with default parameters. DRAM searches amino acid sequences against multiple databases such as KEGG, MEROPS and dbCAN/CAZYmes, and provides all database hits in a single output file. We searched for key genes in the raw (Supplementary Data 2) and metabolism summary files (Supplementary Data 3) provided by DRAM.py annotate and DRAM.py distill, respectively. Localization analyses for genes of interest were done using PSORTb [46]. We classified the metagenome-assembled genomes (MAGs) as potential degraders of the carbon substrates based on their presence in enrichment cultures amended with the particular macromolecule and the presence of enzymes involved in extracellular cleavage, transmembrane import, and intracellular degradation of the particular macromolecule in their genome (Supplementary Information).
Abundance of MAGs
The abundances of MAGs across enrichment cultures and in environmental metagenomes were estimated using the metaWRAP Quant bins module [41]. Quant bins uses Salmon [47] to align reads from each sample to the assembly contigs producing coverage values for each contig. These coverage values (CPM) are standardized by library size (for every 1,000,000 metagenomic reads) and by contig length, similar to transcripts per million (TPM) in RNAseq analysis. Then, Quant bins estimates the abundance of MAGs in each sample by computing a length-weighted average of the MAG’s contigs CPMs. We analyzed published metagenomes of hydrothermal ecosystems in Guaymas Basin [7, 48, 49] (16 datasets), of the Mid-Atlantic Ridge (4 datasets) [50], of Okinawa Trough (4 datasets) [51] and Brothers submarine volcano (6 datasets) [52], and of hadal sediments from the Yap Trench (3 datasets) [53] (Supplementary Data 4).
Results
Experimental design summary
Hot and temperate hydrothermal sediments from the southern spreading center of Guaymas Basin (Fig. 1A, B), were amended with either proteins, nucleic acids, lipids or polysaccharides as sole added carbon and energy source at 60 °C under oxic, sulfate-reducing, or fermentative (no added electron acceptor) conditions (Fig. 1C). Enriched bacterial and archaeal lineages and their catabolic potential were analyzed by 16S rRNA gene and metagenomic analyses (Fig. 1D), methane and hydrogen production was measured using gas chromatography and growth was estimated using cell counts.
Hydrogen, not methane, is a product of anoxic macromolecule degradation
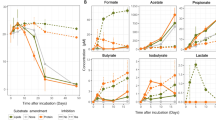
To monitor the enrichment of anaerobic, heterotrophic thermophiles, we followed the production of methane and molecular hydrogen, key products of methanogenesis and fermentation respectively, in the headspace of anoxic cultures weekly (1–4 weeks) (Fig. 2). We did not detect methane in any of the enrichments, despite a detection limit of 200 ppm (8 µM methane). In addition, we did not find evidence of methanogens in the 16S rRNA gene and metagenomic sequencing datasets. Thus, enrichments under anoxic conditions that were expected to support methanogenesis apparently did not contain a syntrophic network including methanogens. However, we observed an increase in the concentration of H2 in the headspace with time, indicating a successful enrichment of fermentative microbes (Fig. 2). Fermentative enrichments had significantly more hydrogen than the sulfate-containing cultures (p < 0.0001, Table S2), while H2 concentrations were highest in cultures supplemented with proteins or DNA and lowest in cultures supplemented with lipids (Fig. 2, Table S3).

Hydrogen was measured weekly in sulfate-reducing and fermentative anoxic cultures that were supplemented with proteins (PRO), nucleic acids (DNA), lipids (LIP), or polysaccharides (POL) having a temperate or a hot sediment as inoculum. The headspace was exchanged every week, hence a straight line represents constant production, and increasing line means increasing production of hydrogen per time.
The rate of H2 production was associated with microbial growth over time, as the production of H2 and the cell counts positively correlated (R2: 0.35, p = 0.048). At week 3, the microbial cell numbers were highest in cultures supplemented with polysaccharides or proteins and lowest in cultures supplemented with lipids (Fig. S1a), indicating that proteins and polysaccharides were turned over faster or were more labile than DNA and lipids (Fig. S1b). Although we did not constrain how much of the added carbon was consumed, we can estimate how much carbon was used to produce new cells (Supplementary Information, Supplementary Data 5). Based on our estimates using the increase in cell numbers, about 6.6% of the added polysaccharide-carbon was used for the production of cells. For proteins (4.5%), DNA (4.2%), and lipids (2.2%), this percentage was lower (Supplementary Data 5).
Macromolecule type and redox conditions selectively enriched for distinct communities
The differences in H2 production and cell counts coincided with differences in microbial community structure. Protein-amended cultures (with high H2 production) displayed a decreased bacterial diversity, indicating selective enrichment of specific taxa, while lipid-amended cultures (low H2 production) retained much of the original diversity (Fig. 3A, B). In the hot and temperate sediment, Proteobacteria and Bacteroidetes accounted for ~40 and ~20% of the 16S rRNA gene amplicons, respectively. After 4 weeks of incubation, the relative sequence abundance of Proteobacteria and Bacteroidetes decreased to an average of 16% and 1%, respectively (Fig. 3C). Firmicutes increased from 1% in both inocula, to an average of 41% across all enrichment cultures (Fig. 3C). The PerMANOVA results based on genus-level bacterial relative sequence abundance detected significant differences in community structure between redox conditions (p = 0.008, R2 = 0.15) as well as between carbon sources (p = 0.010, R2 = 0.20). However, significant post-hoc comparisons were only found between oxic and anoxic conditions (Fig. S2, P values are reported in Table S4). Lineages found in more than one culture condition, low number of samples, and variation between replicates potentially limited our statistical power (Fig. S3). Differences in archaeal community composition also reflected a selective enrichment of microbes affiliated with specific taxa. In the hot sediment inoculum, anaerobic methane-oxidizing archaea of the ANME-1 lineage accounted for 75% of the archaeal 16S rRNA gene amplicons. After 4 weeks of incubation, the relative sequence abundance of ANME-1 decreased in most of the enrichment cultures, while Thermococcus increased from 3% in the hot sediment to an average of 51% across enrichment cultures (Fig. S4).

A Shannon’s entropy, B non-metric multidimensional scaling (NMDS) analyses (95% confidence ellipse) and C relative sequence abundance of the most abundant bacterial clades in the temperate and hot sediment used as inocula and in enrichment cultures supplemented with proteins (PRO), nucleic acids (DNA), lipids (LIP) or polysaccharides (POL) under oxic, sulfate-reducing or fermentative (no added electron acceptor) conditions after four weeks of incubation. The used inoculum is indicated at the top of the graph. Relative sequence abundances are based on 16S rRNA V4/V5 gene amplicons.
Uncharacterized bacterial lineages are involved in the degradation of biological macromolecules
Metagenomic sequencing, assembly and binning of DNA extracted from 11 cultures revealed 3 archaeal and 17 bacterial medium-to-high-quality MAGs [54]. These MAGs were assigned to seven phyla, with a majority affiliating with Firmicutes (Table 1). The taxonomic analyses from GTDB-tk, ANI, AAI, and single-copy genes confirmed that all MAGs represented populations of new species (Supplementary Data 1). Four MAGs were affiliated with the uncultivated lineages UBA2242, UBA5301, and ZCTH02-B6 (Fig. 4). The class-level UBA2242 lineage falls within the candidate phylum Marinisomatota/“Candidatus Marinimicrobia” (previously Marine Group A or candidate phylum SAR406) and the class-level lineage UBA5301 falls within the phylum Firmicutes [55]. The ZCTH02-B6 lineage is an uncultured family-level lineage within the phylum Firmicutes and affiliated with the class Limnochordia [56]. Three MAGs that were unclassified at the genus-level were assigned to the families Brevibacillaceae, Calderihabitantaceae, and Desulforudaceae (Supplementary Data 1) within the phylum Firmicutes (Fig. 4). The 13 remaining MAGs could be assigned to known genera (Supplementary Data 1). All Firmicutes MAGs encoded genes for sporulation (Supplementary Data 6). MAG abundance estimates in each set of culture conditions revealed that several MAGs were in high abundance only under one enrichment regime, while others were found in several conditions (Fig. 5), suggesting specialist and generalist lifestyles. Moreover, the abundance estimates revealed that the MAGs represent lineages with widespread occurrence in other hydrothermal and sedimentary environments. Particularly, high coverages of UBA2242, Thermotogae, Thermodesulfobacteria, and Synergistia populations were found in environmental samples of Guaymas Basin hydrothermal sediments (Fig. 5). Brevibacillus, ZCTH02-B6, and UBA5301 lineages were abundant in hydrothermal sediments from the Mid-Atlantic Ridge and Okinawa Trough (Iheya Ridge).

Culture conditions enriching MAGs are shown. Frequently, different enrichment conditions (carbon sources and redox conditions) selected for the same MAG, as determined by mapping the sequences of each MAG to the metagenomes of each cultivation regime.

(Left) Guaymas Basin enrichment cultures supplemented with proteins (blue), nucleic acids (pink), lipids (purple) or polysaccharides (orange) under oxic, sulfate-reducing or fermentative conditions. (Right) Environmental metagenomes from Guaymas Basin, Mid-Atlantic Ridge, Iheya Ridge and Yap Trench. The abundance estimates are expressed as contigs per million reads (CPM) calculated with metaWRAP’s Quant bins module. Quant bins uses Salmon to align reads from each sample to the bins producing coverage values for each contig in the same manner as transcripts per million (TPM). The abundance of each MAG in each sample is calculated by taking the length-weighted average of the MAG’s contig coverage.
Thermophilic protein degrading populations
Protein degradation under oxic conditions was apparently carried out by Bacilli and Limnochordia (ZCTH02-B6), whose MAGs were enriched in the oxic protein-fed cultures (Fig. 5) and contained the highest number of genes for extracellular and cellular peptidases (Figs. 6, 7, Supplementary Data 7), as well as peptide and amino acid transporters (Fig. 7). The communities that anaerobically degraded proteins were more diverse. These cultures included populations that had extracellular peptidases and the means for peptide uptake (UBA5301, Calderihabitantaceae, Thermotogae, Thermococcus), as well as populations that were only able to uptake peptides or amino acids (Desulfotomaculum, Thermovirga, Thermodesulfobacterium, UBA2242, Archaeoglobus) (Figs. 7 and 8).

Main culture conditions enriching MAGs are shown: proteins (blue), nucleic acids (pink), lipids (purple) or polysaccharides (orange) under oxic (triangle), sulfate-reducing (square) or fermentative (circle) conditions. A complete list of the annotations is reported in Supplementary Data 2 (peptidases) and 3 (CAZymes).

Culture conditions enriching MAGs are shown: protein (blue), nucleic acid (pink), lipid (purple), polysaccharide (orange), oxic (triangle), sulfate-reducing (square), or fermentative (circle). Culture conditions for which a given MAG encoded the necessary extracellular enzymes are shown with symbols having a red outline. Different capabilities belonging to the same category are given as numbers, e.g., if a MAG has three different extracellular peptidases the blue square in column 1 will show the number 3, if the MAG has only one extracellular peptidase only the blue square is shown. A complete list of the annotations is reported in Supplementary Data 2, 3.

Shown are each MAGs major extracellular enzymes and transporters necessary to make a living on protein (blue), nucleic acid (pink), lipid (purple), and polysaccharides (orange). Extracellular enzymes are depicted close to the MAG in which they occur. Arrows represent processes that each MAG is capable of, while dotted arrows show potential flow of monomers/oligomers provided by other MAGs. As an example, GB067 has extracellular peptidases, transporters, and enzymes to grow on proteins. Despite lacking the extracellular nucleases and CAZymes GB067 was also abundant in the nucleic acid and polysaccharide enrichments, likely because it contained the necessary transporters and catabolic pathways to scavenge monomers. Transporters that occur in only one of the population genomes are shown in colored font, e.g. Aap, ABC-SP and Agl were only present in MAG GB067.
Thermophilic polysaccharide degrading populations
Polysaccharide degradation under oxic conditions was largely carried out by a population affiliating with the genus Bacillus (Table S5). Like other Bacilli, this population did not occur in other conditions although it had the capability to process all provided substrates. Analogous to anaerobic protein degradation, the anaerobic polysaccharide degrading communities contained populations with all necessary enzymes (Thermosipho, Kosmotoga, and UBA2242), populations that can uptake oligo- /monomers (Thermococcus, UBA5301), and populations that apparently lacked the enzymes for extracellular cleavage and uptake (Desulforudaceae, Thermovirga) (Figs. 7 and 8). Thermotogae populations had very broad polysaccharide-degrading capabilities including several extracellular CAZymes (Supplementary Data 7), numerous sugar transport systems, as well as CAZymes belonging to glycoside hydrolase families which include enzymes to process almost all the polysaccharides we provided (Fig. 7). Kosmotoga MAG GB055, for example, encoded two extracellular CAZymes (beta-N-acetylglucosaminidase and 1,4-beta-cellobiosidase) that may be involved in the degradation of chitosan (chitin) and xanthan. This population also had the necessary transporters and CAZymes to make a living on these substrates (Fig. 7). The capabilities of the Thermotogae were only rivaled by the two Marinisomatota MAGs GB43 (and GB103). These populations were apparently able to degrade a large number of different polysaccharides and had 6 (3) extracellular CAZymes (e.g., chitin deacetylase, endo-beta-1,3(4)-glucanase and 1,4-beta-cellobiosidase) likely involved in the degradation of chitosan and xanthan (Figs. 8 and S5). The MAGs also encoded 6 (1) polysaccharide lyases and CAZymes belonging to 35 (21) different glycoside hydrolase families, which comprise enzymes for the degradation of all seven provided polysaccharides including alginate, chondroitin, and carrageenan. The Marinisomatota MAGs lacked most of the common sugar transporters that were found in the other populations, yet contained many genes annotated as TonB proteins (K03832) and TonB-dependent receptors.
Thermophilic nucleic acid or lipid degrading populations
Although the majority of the MAGs enriched in this study were derived from cultures supplemented with protein and polysaccharides, several bacterial lineages were reconstructed from cultures supplemented with nucleic acids or lipids. MAGs affiliating with the genera Thermosyntropha, Kosmotoga, Desulfohalotomaculum, the family Calderihabitantaceae, and the uncultured classes UBA5301 and UBA2242 were found in cultures supplemented with DNA (Fig. 5). Thermosyntropha, Kosmotoga, and Desulfohalotomaculum MAGs featured an extracellular nuclease (EC 3.1.31.1, Fig. 7, Supplementary Data 7). Kosmotoga, Desulfohalotomaculum, and UBA5301 also contained nucleoside transporters in addition to an extracellular nuclease (Fig. 8). Known genes for purine and pyrimidine degradation were found in UBA2242 and Kosmotoga (Fig. 7). Thermovirga, Thermosipho, and Thermodesulfobacterium MAGs were derived from cultures supplemented with lipids. Thermovirga featured a membrane phospholipase, phospholipid transporters and long-chain acyl-CoA synthetase (Fig. 8), while Thermodesulfobacterium encoded phospholipid transporters (Fig. 7).
Central carbon and energy metabolisms
The use of biopolymers and lipids as an energy source was also reflected by the presence of genes encoding pathways for the degradation of monomeric substrates and smaller molecules produced by the activity of peptidases, CAZymes, lipases, and nucleases in the MAGs. All Bacilli MAGs that were enriched in oxic cultures contained a complete glycolysis pathway, citrate (TCA/Krebs cycle) and glyoxylate cycles, and cytochrome c oxidases (Fig. 9). Pathways for sulfate reduction and fermentation were detected in MAGs under these respective anoxic conditions. Desulfohalotomaculum, Desulforudaceae, Calderihabitantaceae, Thermodesulfobacterium, and Archaeoglobus featured genes for dissimilatory sulfate reduction (sat, aprAB, and dsrAB) (Fig. 9). These populations likely couple the oxidation of macromolecule-derived carbon to the reduction of sulfate. In Thermotogae we found FeFe hydrogenases (hydABC) which have been shown to be involved in fermentative hydrogen evolution (Fig. 9). These hydrogenases likely contributed to the increased hydrogen production we observed in the fermentative enrichments. Genes that could support fermentative metabolism, phosphate acetyltranferase (pta) and acetate kinase (ackA), were found in Thermosyntropha and UBA2242 MAGs (Fig. 9).

A complete list of the annotations is reported in Supplementary Data 2.
Discussion
Uncultured heterotrophs contribute to macromolecule degradation in hydrothermal sediments
We combined cultivation with metagenomics to gain insights into heterotrophic Bacteria and Archaea with the potential for protein, polysaccharide, nucleic acid, and lipid degradation in hydrothermally influenced sediments. Each of the tested cultivation conditions enriched a microbial community that was capable of metabolizing the supplemented carbon sources. In previous Guaymas Basin sequence-based surveys [1, 6, 7, 57,58,59,60,61,62] and in the sediment cores analyzed here, Firmicutes were infrequently detected. Yet, we found Firmicutes at high relative sequence abundances in many of the enrichment cultures, and half of the MAGs (10 of 20) were affiliated with uncultured Firmicutes lineages. The cultivation conditions apparently caused the germination of bacterial thermospores present in Guaymas Basin sediments. Cultivated Firmicutes contained sporulation proteins and were closely related to spore formers [63,64,65,66]. Spore-forming lineages were found in oxic (Bacilli) and anoxic conditions (Moorellia and Desulfotomaculia).
The presence of sulfate-reducing Firmicutes also indicates a potential role of these lineages in thermophilic sulfate reduction in Guaymas Basin [67]. Previous studies have shown that high temperature incubation of cold sediments activates and supports the growth of endospore-forming bacteria via sulfate reduction and fermentation [68]. Under suitable environmental conditions these spores germinate and may become major contributors to community functions in the warm hydrothermal sediments, which potentially represent a source of thermospores widely distributed in marine sediments [69, 70]. For example, we detected the Firmicutes uncultured lineages UBA5301 and ZCTH02-B6 in hydrothermal sediments from Mid-Atlantic and Okinawa Trough. ZCTH02-B6 protein- and UBA5301 protein- and polysaccharide- degradation capabilities suggest that sediments in these environments provide the particular substrates and can sustain heterotrophic communities. Interestingly, the closest relatives of our unclassified Bacillaceae MAGs such as Brevibacillus brevis are not associated with hot environments [66], expanding the habitat range of this group. Further research should address the question whether these organisms originate from deeper sediment layers and reach the surface via geofluids [69, 71] or whether they are deposited by ocean currents [72].
The enrichment of uncultured lineages such as UBA2242 within the candidate phylum Marinisomatota underlines the successful cultivation of uncultured diversity of Guaymas Basin. The candidate phylum Marinisomatota is considered a highly diverse group, participating in marine sulfur and nitrogen cycles [73,74,75]. Marinisomatota (formerly SAR406, Marine Group A, Marinimicrobia) was reported to comprise lineages that degrade complex carbohydrates in marine oxygen minimum zones [76] featuring numerous glycoside hydrolase families with enzymes involved in the degradation of chitin, cellulose, starch, sulfated galactans among others (Fig. S6). Our finding of two Marinisomatota MAGs with similar capabilities suggests that the ability to degrade complex polysaccharides may be more widespread among Marinisomatota. Besides Guaymas Basin [7], UBA2242 have been previously detected in cold-water geyser fluids in Utah, USA [77].
The microbial utilization of environmental DNA, either to salvage nucleosides or as a carbon and energy source, was shown to occur in marine sediments [78,79,80,81]. At least nine of the enriched thermophiles had the capability to extracellularly cleave DNA, take up the nucleosides, remove the ribose and use it as carbon and energy source via the pentose phosphate pathway (Figs. 7 and 9). GB116 affiliating with Bacillus additionally had the enzymes to further catabolize pyrimidines (Fig. 7). However, despite having extracellular nucleases, nucleoside transporters, and nucleoside phosphorylases not all of these populations were enriched in the DNA-amended cultures (Figs. 7 and 9). The widespread metabolic capabilities and the general abundance of DNA in marine sediments [9] suggest that nucleic acid degradation is a common process and ecological niche in hydrothermal sediments.
Lipids have also been shown to be microbially degraded in marine sediments [82, 83] and serve as carbon and energy source for microorganisms [10, 84]. Lipids were apparently the most difficult substrate for the enriched thermophiles promoting the lowest increase in cell numbers and the least community shifts. Surprisingly, the populations that were most abundant in the lipid-amended microcosms seemed to lack most (GB013—Thermovirga, GB111—Thermodesulfobacterium) or all genes (GB058—Thermosipho) that are needed to import and metabolize the lipids or fatty acids (Figs. 7 and 9). These organisms thus may not use lipids after all, but other organic compounds released from the perished cells of the inoculum. Unknown genes, pathways, or processes, e.g., non-classical secretion of extracellular enzymes [85], or novel metabolisms could also explain this gap in knowledge and merit further investigation.
Niches of thermophilic heterotrophs involved in macromolecule degradation
The findings of our metabolic analyses suggest that Guaymas Basin sediments harbored specialists that were adapted to the utilization of a particular carbon polymer (e.g., Brevibacillales) as well as generalists that were able to degrade a wide range of biopolymers under different redox conditions (e.g., Thermotogae) (Fig. 8). Most of the cultivated lineages were capable of secreting enzymes, suggesting the ability to hydrolyze the polymers into oligomers extracellularly. The lineages that lacked exoenzymes (e.g., Archaeoglobus) contained oligomer transporters instead. These populations potentially have opportunistic trophic interactions with exoenzyme-producing decomposers and feed on the products of extracellular hydrolysis (Fig. 8) carried out by other microbes [86]. We found that protein and polysaccharide degradation is likely carried out by multiple bacterial and archaeal lineages. Consistent with the literature, Archaeoglobus species were capable of coupling the oxidation of carbon compounds to the reduction of sulfate [87, 88], Thermococcus were thriving on peptides and carbohydrates and likely produced hydrogen [89], and Thermotogae and Marinisomatota were able to degrade a wide range of polysaccharides [90, 91].
Nucleic acids and lipids were found to be less labile or turned over slower than proteins and polysaccharides, especially the slow growth on lipids in anoxic conditions is consistent with previous findings [10]. Thermosyntropha and Thermovirga however thrived in cultures supplemented with nucleic acids and lipids, respectively. Although DNA and lipids are energy-rich and abundant in marine sediments [9], the identity and distribution of nucleases and lipases across genomes, organisms and databases is not well characterized, in contrast to the enzymes involved in the breakdown of proteins [92, 93] and polysaccharides [94, 95]. Few studies have identified bacterial extracellular nucleases [96, 97] and lipases [98] associated with catabolic activities. Furthermore, nucleases and lipases can also participate in synthesis and salvage pathways, and are not necessarily involved in catabolism [99, 100]. Interestingly, although the energy yield of all provided substrates is highest using oxygen as electron acceptor [101], we did not find populations that were specialized on the aerobic degradation of lipids and nucleic acids. Overall, despite the environmental pressure of high temperature and minimal media the enriched food webs apparently contained primary degraders, secondary degraders and potentially even hydrogen-oxidizing sulfate reducers revealing insights into heterotrophic food webs and the ecophysiology of uncultured lineages [102].
Implications of macromolecule degradation for marine carbon cycling
We show that Guaymas Basin hydrothermal sediments host a variety of thermophilic heterotrophs with the potential to hydrolyze the tested cellular macromolecules and transport their oligomers. The oligomers can be further oxidized via glycolysis fed respiration and fermentation. Although we provided conditions that are considered suitable for hydrogenotrophic methanogenesis, methane was not detected in the headspace of any culture and we were not able to enrich hydrogenotrophic, acetoclastic, or methylotrophic methanogens. This indicates that thermophilic macromolecule degradation does not support methanogenesis or syntrophic interactions required to cross-feed methanogens under the tested conditions. Stable carbon isotopic evidence for methanogenesis (δ13C for porewater methane near −70‰) in Guaymas Basin sediments was limited to cool sediments, possibly due to overprinting by microbial methane oxidation [1]. Previous anoxic high temperature (50 °C) incubations of Guaymas sediments over 5–9 days have resulted in hydrogen accumulation [59], suggesting that hydrogenotrophic methanogens were not enriched under these conditions either. Hyperthermophilic hydrogenotrophic methanogens have been isolated from Guaymas Basin sediments [103], and the key genes of methanogenesis, methyl coenzyme M reductase (mcrA), including those of hyperthermophilic lineages, are found in Guaymas Basin sediments [104]. Hence, these methanogenic populations may respond to hyperthermophilic enrichments (80 °C). Thermophilic Methermicoccaceae were found in Guaymas sediments [59], yet these organisms are obligately methylotrophic and thus may have not found suitable substrate in our enrichments. Hydrogenotrophic and acetoclastic methanogens, on the other hand, may have been too low in abundance in the inoculum, due to being inhibited by methane or outcompeted by sulfate reducers [105]. However, most evidence for the presence of hydrogenotrophic, acetoclastic, and methylotrophic methanogens in Guaymas Basin sediments is based on gene sequencing, which may underrepresent active but infrequently occurring community members [106]. In cold and temperate anoxic sediments, it was shown that methane was produced from e.g., lipid degradation [10, 83]. The finding that methane was not produced during thermophilic degradation of biomass constituents thus merits further investigation and if confirmed has implications for the deep-sea methane cycle. Biomass carbon that was assimilated using subsurface-derived methane at hydrothermal sediments may not be converted back into methane, but into carbon dioxide and organic acids.
Conclusion
The heterotrophic populations enriched in our study showed the potential to consume energy-rich complex carbon compounds including proteins, polysaccharides, nucleic acids, and lipids. These populations can mineralize the organic matter from primary production in overlying waters and from local chemosynthesis in hydrothermal sediments. Beyond Guaymas Basin, macromolecule-degrading thermophiles apparently occur more broadly in hydrothermal systems worldwide and are not limited to sediments but could thrive in hydrothermal chimneys or in surface-attached biofilms. The degradation of primary produced biomass or of necromass by uncultured heterotrophs may be a widespread process relevant for our understanding of the diversity and carbon cycling in marine ecosystems. Future studies are needed to characterize the abundance and activity of uncultured macromolecule degraders at cold seeps and hydrothermal vents to further improve our understanding of carbon exchanges between the lithosphere and biosphere.
Data availability
All sequence data is available at NCBI under BioProject ID PRJNA635695. Accession numbers for individual metagenomes can be found in Supplementary Information (Table S6).
References
McKay L, Klokman VW, Mendlovitz HP, LaRowe DE, Hoer DR, Albert D, et al. Thermal and geochemical influences on microbial biogeography in the hydrothermal sediments of Guaymas Basin, Gulf of California. Environ Microbiol Rep. 2016;8:150–61.
Price RE, Lesniewski R, Nitzsche KS, Meyerdierks A, Saltikov C, Pichler T, et al. Archaeal and bacterial diversity in an arsenic-rich shallow-Sea hydrothermal system undergoing phase separation. Front Microbiol. 2013;4:158.
Gugliandolo C, Maugeri TL. Phylogenetic diversity of Archaea in shallow hydrothermal vents of Eolian Islands. Italy Diversity. 2019;11:156.
Wankel SD, Germanovich LN, Lilley MD, Genc G, Diperna CJ, Bradley AS, et al. Influence of subsurface biosphere on geochemical fluxes from diffuse hydrothermal fluids. Nat Geosci. 2011;4:461–8.
McCollom TM, Shock EL. Geochemical constraints on chemolithoautotrophic metabolism by microorganisms in seafloor hydrothermal systems. Geochim Cosmochim Acta. 1997;61:4375–91.
Ruff SE, Biddle JF, Teske AP, Knittel K, Boetius A, Ramette A. Global dispersion and local diversification of the methane seep microbiome. Proc Natl Acad Sci. 2015;112:4015–20.
Dombrowski N, Teske AP, Baker BJ. Expansive microbial metabolic versatility and biodiversity in dynamic Guaymas Basin hydrothermal sediments. Nat Commun. 2018;9:4999.
Orsi WD, Richards TA, Francis WR. Predicted microbial secretomes and their target substrates in marine sediment. Nat Microbiol. 2018;3:32–37.
Dell’Anno A, Danovaro R. Extracellular DNA plays a key role in deep-sea ecosystem functioning. Science. 2005;309:2179.
Harvey HR, Fallon RD, Patton JS. The effect of organic matter and oxygen on the degradation of bacterial membrane lipids in marine sediments. Geochim Cosmochim Acta. 1986;50:795–804.
Arnosti C, Repeta DJ, Blough NV. Rapid bacterial degradation of polysaccharides in anoxic marine systems. Geochim Cosmochim Acta. 1994;58:2639–52.
Ruff SE, Probandt D, Zinkann AC, Iversen MH, Klaas C, Würzberg L, et al. Indications for algae-degrading benthic microbial communities in deep-sea sediments along the Antarctic Polar Front. Deep Sea Res Part II Top Stud Oceanogr. 2014;108:6–16.
Burdige DJ. Preservation of organic matter in marine sediments: Controls, mechanisms, and an imbalance in sediment organic carbon budgets? Chem Rev. 2007;107:467–85.
Andrade CMMC, Pereira NJ, Antranikian G. Extremely thermophilic microorganisms and their polymerhydrolytic enzymes. Rev Microbiol. 1999;30:287–98.
Arnosti C, Bell C, Moorhead DL, Sinsabaugh RL, Steen AD, Stromberger M, et al. Extracellular enzymes in terrestrial, freshwater, and marine environments: perspectives on system variability and common research needs. Biogeochemistry. 2014;117:5–21.
Davidson AL, Dassa E, Orelle C, Chen J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev. 2008;72:317–64.
Li M, Baker BJ, Anantharaman K, Jain S, Breier JA, Dick GJ. Genomic and transcriptomic evidence for scavenging of diverse organic compounds by widespread deep-sea archaea. Nat Commun. 2015;6:8933.
Teske A, de Beer D, McKay LJ, Tivey MK, Biddle JF, Hoer D, et al. The Guaymas Basin hiking guide to hydrothermal mounds, chimneys, and microbial mats: complex seafloor expressions of subsurface hydrothermal circulation. Front Microbiol. 2016;7:75.
Teske A, Wegener G, Chanton JP, White D, MacGregor B, Hoer D, et al. Microbial communities under distinct thermal and geochemical regimes in axial and off-axis sediments of Guaymas Basin. Front Microbiol. 2021;12:633649.
Widdel F, Bak F. Gram-negative mesophilic sulfate-reducing bacteria. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H, (eds). The Prokaryotes. 2nd ed. New York, N.Y.: Springer-Verlag; 1992. p. 3352–78.
Zheng Q, Lin W, Wang Y, Li Y, He C, Shen Y, et al. Highly enriched N-containing organic molecules of Synechococcus lysates and their rapid transformation by heterotrophic bacteria. Limnol Oceanogr. 2021;66:335–48.
Schada Von Borzyskowski L, Bernhardsgrütter I, Erb TJ. Biochemical unity revisited: microbial central carbon metabolism holds new discoveries, multi-tasking pathways, and redundancies with a reason. Biol Chem. 2020;401:1429–41.
Balti R, Zayoud N, Hubert F, Beaulieu L, Massé A. Fractionation of Arthrospira platensis (Spirulina) water soluble proteins by membrane diafiltration. Sep Purif Technol. 2021;256:117756.
Cain TC, Lubman DM, Weber WJ, Vertes A. Differentiation of bacteria using protein profiles from matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry. Rapid Commun Mass Spectrom. 1994;8:1026–30.
Geiger O, López-Lara IM, Sohlenkamp C. Phosphatidylcholine biosynthesis and function in bacteria. Biochim Biophys Acta Mol Cell Biol Lipids. 2013;1831:503–13.
Arnosti C, Wietz M, Brinkhoff T, Hehemann JH, Probandt D, Zeugner L, et al. The biogeochemistry of marine polysaccharides: sources, inventories, and bacterial drivers of the carbohydrate cycle. Ann Rev Mar Sci. 2021;13:9.1–8.
Hettle AG, Hobbs JK, Pluvinage B, Vickers C, Abe KT, Salama-Alber O, et al. Insights into the κ/ι-carrageenan metabolism pathway of some marine Pseudoalteromonas species. Commun Biol. 2019;2:474.
Mühlenbruch M, Grossart HP, Eigemann F, Voss M. Mini-review: Phytoplankton-derived polysaccharides in the marine environment and their interactions with heterotrophic bacteria. Environ Microbiol. 2018;20:2671–85.
Zhang Z, Chen Y, Wang R, Cai R, Fu Y, Jiao N. The fate of marine bacterial exopolysaccharide in natural marine microbial communities. PLoS One. 2015;10:e0142690.
Grondin JM, Tamura K, Déjean G, Abbott DW, Brumer H. Polysaccharide utilization loci: fueling microbial communities. J Bacteriol. 2017;199:e00860–16.
Pinheiro J, Bates D, DebRoy S, Sarkar D, Heisterkamp S, Van Willigen B, et al. Linear and nonlinear mixed effects models. 2020; R package version 3.1-148.
Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biometrical J. 2008;50:346–63.
Zhou J, Bruns MA, Tiedje JM. DNA recovery from soils of diverse composition. Appl Environ Microbiol. 1996;62:316–22.
Nelson MC, Morrison HG, Benjamino J, Grim SL, Graf J. Analysis, optimization and verification of Illumina-generated 16S rRNA gene amplicon surveys. PLoS One. 2014;9:e94249.
Topçuoğlu BD, Stewart LC, Morrison HG, Butterfield DA, Huber JA, Holden JF. Hydrogen limitation and syntrophic growth among natural assemblages of thermophilic methanogens at deep-sea hydrothermal vents. Front Microbiol. 2016;7:1240.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, Hara RBO, et al. vegan: Community Ecology Package. 2017; R package version 2.4-4.
Wickham H. ggplot2: elegant graphics for data analysis. New York, NY: Springer-Verlag; 2016.
Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Uritskiy GV, Diruggiero J, Taylor J. MetaWRAP - A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:158.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2020;36:1925–7.
Wattam AR, Davis JJ, Assaf R, Boisvert S, Brettin T, Bun C, et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017;45:D535–42.
Shaffer M, Borton MA, Mcgivern BB, Zayed AA, Leanti S, Rosa L, et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020;48:8883–900.
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–15.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9.
Li M, Jain S, Dick GJ. Genomic and transcriptomic resolution of organic matter utilization among deep-sea bacteria in guaymas basin hydrothermal plumes. Front Microbiol. 2016;7:1125.
Dombrowski N, Seitz KW, Teske AP, Baker BJ. Genomic insights into potential interdependencies in microbial hydrocarbon and nutrient cycling in hydrothermal sediments. Microbiome. 2017;5:106.
Zhou Z, Liu Y, Xu W, Pan J, Luo ZH, Li M. Genome and community-Level interaction insights on wide carbon utilizing and element cycling function of Hydrothermarchaeota from hydrothermal sediment. mSystems. 2020;5:e00795–19.
Wang HL, Sun L. Comparative metagenomics reveals insights into the deep-sea adaptation mechanism of the microorganisms in Iheya hydrothermal fields. World J Microbiol Biotechnol. 2017;33:86.
Reysenbach AL, St John E, Meneghin J, Flores GE, Podar M, Dombrowski N, et al. Complex subsurface hydrothermal fluid mixing at a submarine arc volcano supports distinct and highly diverse microbial communities. Proc Natl Acad Sci USA. 2020;117:32627–38.
Zhang X, Xu W, Liu Y, Cai M, Luo Z, Li M. Metagenomics reveals microbial diversity and metabolic potentials of seawater and surface sediment from a hadal biosphere at the Yap trench. Front Microbiol. 2018;9:2402.
Bowers RM, Kyrpides N, Stepanauskas R, Harmon-Smith M, Doud D, Reddy T, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–31.
Parks DH, Rinke C, Chuvochina M, Chaumeil PA, Woodcroft BJ, Evans PN, et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2017;2:1533–42.
Lemos LN, Pereira RV, Quaggio RB, Martins LF, Moura LMS, da Silva AR, et al. Genome-centric analysis of a thermophilic and cellulolytic bacterial consortium derived from composting. Front Microbiol. 2017;8:644.
Edgcomb VP, Kysela DT, Teske AP, de Vera Gomez A, Sogin ML. Benthic eukaryotic diversity in the Guaymas Basin hydrothermal vent environment. PNAS. 2002;99:7658–62.
Biddle JF, Cardman Z, Mendlovitz H, Albert DB, Lloyd KG, Boetius A, et al. Anaerobic oxidation of methane at different temperature regimes in Guaymas Basin hydrothermal sediments. ISME J. 2012;6:1018–31.
Dowell F, Cardman Z, Dasarathy S, Kellermann MY, Lipp JS, Ruff SE, et al. Microbial communities in methane- and short chain alkane-rich hydrothermal sediments of Guaymas Basin. Front Microbiol. 2016;7:17.
Vigneron A, Cruaud P, Roussel EG, Pignet P, Caprais J-C, Callac N, et al. Phylogenetic and functional diversity of microbial communities associated with subsurface sediments of the Sonora Margin, Guaymas Basin. PLoS One. 2014;9:e104427.
Cruaud P, Vigneron A, Pignet P, Caprais JC, Lesongeur F, Toffin L, et al. Comparative study of Guaymas Basin microbiomes: cold seeps vs. hydrothermal vents sediments. Front Mar Sci. 2017;4:417.
Teske A, Hinrichs K, Edgcomb V, Gomez ADV, Kysela D, Sylva SP, et al. Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Am Soc Microbiol. 2002;68:1994–2007.
Ko KS, Oh WS, Lee MY, Lee JH, Lee H, Peck KR, et al. Bacillus infantis sp. nov. and Bacillus idriensis sp. nov., isolated from a patient with neonatal sepsis. Int J Syst Evol Microbiol. 2006;56:2541–4.
Jeon CO, Lim JM, Lee JM, Xu LH, Jiang CL, Kim CJ. Reclassification of Bacillus haloalkaliphilus Fritze 1996 as Alkalibacillus haloalkaliphilus gen. nov., comb. nov. and the description of Alkalibacillus salilacus sp. nov., a novel halophilic bacterium isolated from a salt lake in Chi. Int J Syst Evol Microbiol. 2005;55:1891–6.
Hugon P, Mishra AK, Lagier JC, Nguyen TT, Couderc C, Raoult D, et al. Non-contiguous finished genome sequence and description of Brevibacillus massiliensis sp. nov. Stand Genom Sci. 2013;8:1–14.
Panda AK, Bisht SS, DeMondal S, Senthil Kumar N, Gurusubramanian G, Panigrahi AK. Brevibacillus as a biological tool: a short review. Antonie van Leeuwenhoek Int J Gen Mol Microbiol. 2014;105:623–39.
Elsgaard L, Isaksen MF, Jørgensen BB, Alayse AM, Jannasch HW. Microbial sulfate reduction in deep-sea sediments at the Guaymas Basin hydrothermal vent area: Influence of temperature and substrates. Geochim Cosmochim Acta. 1994;58:3335–43.
Bell E, Sherry A, Pilloni G, Suárez-Suárez A, Cramm MA, Cueto G, et al. Sediment cooling triggers germination and sulfate reduction by heat-resistant thermophilic spore-forming bacteria. Environ Microbiol. 2020;22:456–65.
Chakraborty A, Ellefson E, Li C, Gittins D, Brooks JM, Bernard BB, et al. Thermophilic endospores associated with migrated thermogenic hydrocarbons in deep Gulf of Mexico marine sediments. ISME J. 2018;12:1895–906.
Müller AL, De Rezende JR, Hubert CRJ, Kjeldsen KU, Lagkouvardos I, Berry D, et al. Endospores of thermophilic bacteria as tracers of microbial dispersal by ocean currents. ISME J. 2014;8:1153–65.
Ruff SE, Felden J, Gruber-Vodicka HR, Marcon Y, Knittel K, Ramette A, et al. In situ development of a methanotrophic microbiome in deep-sea sediments. ISME J. 2019;13:197–213.
Hubert C, Loy A, Nickel M, Arnosti C, Baranyi C, Brüchert V, et al. A constant flux of diverse thermophilic bacteria into the cold arctic seabed. Science. 2009;325:1541–4.
Lewis WH, Tahon G, Geesink P, Sousa DZ, Ettema TJG. Innovations to culturing the uncultured microbial majority. Nat Rev Microbiol. 2021;19:225–40.
Bertagnolli AD, Padilla CC, Glass JB, Thamdrup B, Stewart FJ. Metabolic potential and in situ activity of marine Marinimicrobia bacteria in an anoxic water column. Environ Microbiol. 2017;19:4392–416.
Hawley AK, Nobu MK, Wright JJ, Durno WE, Morgan-Lang C, Sage B, et al. Diverse Marinimicrobia bacteria may mediate coupled biogeochemical cycles along eco-thermodynamic gradients. Nat Commun. 2017;8:1507.
Thrash JC, Seitz KW, Baker BJ, Temperton B, Gillies LE, Rabalais NN, et al. Metabolic roles of uncultivated bacterioplankton lineages in the northern gulf of Mexico “dead zone”. MBio. 2017;8:e01017–17.
Probst AJ, Ladd B, Jarett JK, Geller-Mcgrath DE, Sieber CMK, Emerson JB, et al. Differential depth distribution of microbial function and putative symbionts through sediment-hosted aquifers in the deep terrestrial subsurface. Nat Microbiol. 2018;3:328–36.
Wasmund K, Pelikan C, Watzka M, Richter A, Noel A, Hubert CRJ, et al. DNA-foraging bacteria in the seafloor. bioRxiv. 2019:528695.
Paul JH, DeFlaun MF, Jeffrey WH. Mechanisms of DNA utilization by estuarine microbial populations. Appl Environ Microbiol. 1988;54:1682–8.
Lennon JT. Diversity and metabolism of marine bacteria cultivated on dissolved DNA. Appl Environ Microbiol. 2007;73:2799–805.
Dell’Anno A, Corinaldesi C. Degradation and turnover of extracellular DNA in marine sediments: ecological and methodological considerations. Appl Environ Microbiol. 2004;70:4384–6.
Sun MY, Aller RC, Lee C, Wakeham SG. Effects of oxygen and redox oscillation on degradation of cel-associated lipids in surficial marine sediments. Geochim Cosmochim Acta. 2002;66:2003–12.
Logemann J, Graue J, Köster J, Engelen B, Rullkötter J, Cypionka H. A laboratory experiment of intact polar lipid degradation in sandy sediments. Biogeosciences. 2011;8:2547–60.
Pelikan C, Wasmund K, Glombitza C, Hausmann B, Herbold CW, Flieder M, et al. Anaerobic bacterial degradation of protein and lipid macromolecules in subarctic marine sediment. ISME J. 2021;15:833–47.
Bendtsen JD, Kiemer L, Fausbøll A, Brunak S. Non-classical protein secretion in bacteria. BMC Microbiol. 2005;5:58.
Schimel JP, Schaeffer SM. Microbial control over carbon cycling in soil. Front Microbiol. 2012;3:348.
Hocking WP, Stokke R, Roalkvam I, Steen IH. Identification of key components in the energy metabolism of the hyperthermophilic sulfate-reducing archaeon Archaeoglobus fulgidus by transcriptome analyses. Front Microbiol. 2014;5:95.
Birkeland NK, Schönheit P, Poghosyan L, Fiebig A, Klenk HP. Complete genome sequence analysis of Archaeoglobus fulgidus strain 7324 (DSM 8774), a hyperthermophilic archaeal sulfate reducer from a North Sea oil field. Stand Genom Sci. 2017;12:79.
Hensley SA, Jung JH, Park CS, Holden JF. Thermococcus paralvinellae sp. nov. and Thermococcus cleftensis sp. nov. of hyperthermophilic heterotrophs from deep-sea hydrothermal vents. Int J Syst Evol Microbiol. 2014;64:3655–9.
Frock AD, Notey JS, Kelly RM. The genus Thermotoga: Recent developments. Environ Technol. 2010;31:1169–81.
Urios L, Cueff-Gauchard V, Pignet P, Postec A, Fardeau ML, Ollivier B, et al. Thermosipho atlanticus sp. nov., a novel member of the Thermotogales isolated from a Mid-Atlantic Ridge hydrothermal vent. Int J Syst Evol Microbiol. 2004;54:1953–7.
Nguyen TTH, Myrold DD, Mueller RS. Distributions of extracellular peptidases across prokaryotic genomes reflect phylogeny and habitat. Front Microbiol. 2019;10:413.
Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018;46:D624–32.
Martinez-Garcia M, Brazel DM, Swan BK, Arnosti C, Chain PSG, Reitenga KG, et al. Capturing single cell genomes of active polysaccharide degraders: an unexpected contribution of Verrucomicrobia. PLoS One. 2012;7:e35314.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:490–5.
Binnenkade L, Kreienbaum M, Thormann KM. Characterization of ExeM, an extracellular nuclease of Shewanella oneidensis MR-1. Front Microbiol. 2018;9:1761.
Mulcahy H, Charron-Mazenod L, Lewenza S. Pseudomonas aeruginosa produces an extracellular deoxyribonuclease that is required for utilization of DNA as a nutrient source. Environ Microbiol. 2010;12:1621–9.
Ishibashi Y, Aoki K, Okino N, Hayashi M, Ito M. A thraustochytrid-specific lipase/phospholipase with unique positional specificity contributes to microbial competition and fatty acid acquisition from the environment. Sci Rep. 2019;9:16357.
Borkar PS, Bodade RG, Rao SR, Khobragade CN. Purification and characterization of extracellular lipase from a new strain: Pseudomonas aeruginosa SRT 9. Brazilian. J Microbiol. 2009;40:358–66.
Zrenner R, Riegler H, Marquard CR, Lange PR, Geserick C, Bartosz CE, et al. A functional analysis of the pyrimidine catabolic pathway in Arabidopsis. N. Phytol. 2009;183:117–32.
LaRowe DE, Van, Cappellen P. Degradation of natural organic matter: a thermodynamic analysis. Geochim Cosmochim Acta. 2011;75:2030–42.
Saad S, Bhatnagar S, Tegetmeyer HE, Geelhoed JS, Strous M, Ruff SE. Transient exposure to oxygen or nitrate reveals ecophysiology of fermentative and sulfate-reducing benthic microbial populations. Environ Microbiol. 2017;19:4866–81.
Kurr M, Huber R, König H, Jannasch HW, Fricke H, Trincone A, et al. Methanopyrus kandleri, gen. and_sp. nov. represents a novel group of hyperthermophilic methanogens, growing at 110◦C. Arch Microbiol. 1991;156:239–47.
Lever MA, Teske AP. Diversity of methane-cycling archaea in hydrothermal sediment investigated by general and group-specific PCR primers. Appl Environ Microbiol. 2015;81:1426–41.
Zhuang GC, Montgomery A, Sibert RJ, Rogener MK, Samarkin VA, Joye SB. Effects of pressure, methane concentration, sulfate reduction activity, and temperature on methane production in surface sediments of the Gulf of Mexico. Limnol Oceanogr. 2018;63:2080–92.
Dhillon A, Lever M, Lloyd KG, Albert DB, Sogin ML, Teske A. Methanogen diversity evidenced by molecular characterization of methyl coenzyme M reductase A (mcrA) genes in hydrothermal sediments of the Guaymas Basin. Appl Environ Microbiol. 2005;71:4592–601.
Yoon S-H, Ha SM, Lim J. Kwon S, Chun J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek 2017;110:1281–86.
Kim M, Oh SK, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. International Journal of Systematic and Evolutionary Microbiology 2014;64(Pt_2):346–51
Luis M, Rodriguez-R, Konstantinos T, Konstantinidis. Bypassing Cultivation To Identify Bacterial Species. Microbe Magazine 2014:9:111–118
Acknowledgements
We are grateful to the captain and crew of the R/V Atlantis AT37-06 as well as the crew of the human occupied vehicle Alvin for their tireless support. Sampling at Guaymas Basin was supported by NSF (OCE-1357238). We thank Gunter Wegener and Barbara McGregor for help with onboard research activities. We thank Michael Nightingale and Bernhard Meyer for advice concerning geochemical measurements. We also thank Aleksey Morozov and Nicole Robichaud at the W. M. Keck Ecological and Evolutionary Genetics Facility of the Marine Biological Laboratory (MBL) for sequence processing. We thank Richard Fox and Anna Shipunova for computing resources assistance provided by the MBL Bay Paul Center and Bhavya Papudeshi and Miguel Desmarais for bioinformatics analyses assistance. We appreciate the laboratories that collected and analyzed the data that allow our MAG comparisons.
Author information
Authors and Affiliations
Contributions
SPC processed the metagenomic data, analyzed the data, and wrote the manuscript with input from all co-authors. MAB processed metagenomes metabolic annotations (DRAM). KR performed cell counts. IHdA processed 16S rRNA data. KCW processed metagenomes metabolic annotations (DRAM). APT, cruise chief scientist, and collected samples. MS provided chemicals and helped design the study. SER designed the study, collected samples, performed headspace measurements and analyzed data.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pérez Castro, S., Borton, M.A., Regan, K. et al. Degradation of biological macromolecules supports uncultured microbial populations in Guaymas Basin hydrothermal sediments. ISME J 15, 3480–3497 (2021). https://doi.org/10.1038/s41396-021-01026-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-01026-5
This article is cited by
-
Anaerobic degradation of organic carbon supports uncultured microbial populations in estuarine sediments
Microbiome (2023)
-
The majority of microorganisms in gas hydrate-bearing subseafloor sediments ferment macromolecules
Microbiome (2023)
-
Global patterns of diversity and metabolism of microbial communities in deep-sea hydrothermal vent deposits
Microbiome (2022)
-
Microbiomes in the Challenger Deep slope and bottom-axis sediments
Nature Communications (2022)
