
Abstract
Exploration of oxygen-depleted marine environments has consistently revealed novel microbial taxa and metabolic capabilities that expand our understanding of microbial evolution and ecology. Marine blue holes are shallow karst formations characterized by low oxygen and high organic matter content. They are logistically challenging to sample, and thus our understanding of their biogeochemistry and microbial ecology is limited. We present a metagenomic and geochemical characterization of Amberjack Hole on the Florida continental shelf (Gulf of Mexico). Dissolved oxygen became depleted at the hole’s rim (32 m water depth), remained low but detectable in an intermediate hypoxic zone (40–75 m), and then increased to a secondary peak before falling below detection in the bottom layer (80–110 m), concomitant with increases in nutrients, dissolved iron, and a series of sequentially more reduced sulfur species. Microbial communities in the bottom layer contained heretofore undocumented levels of the recently discovered phylum Woesearchaeota (up to 58% of the community), along with lineages in the bacterial Candidate Phyla Radiation (CPR). Thirty-one high-quality metagenome-assembled genomes (MAGs) showed extensive biochemical capabilities for sulfur and nitrogen cycling, as well as for resisting and respiring arsenic. One uncharacterized gene associated with a CPR lineage differentiated hypoxic from anoxic zone communities. Overall, microbial communities and geochemical profiles were stable across two sampling dates in the spring and fall of 2019. The blue hole habitat is a natural marine laboratory that provides opportunities for sampling taxa with under-characterized but potentially important roles in redox-stratified microbial processes.
Similar content being viewed by others


Introduction
Oxygen-depleted marine water columns have been rich targets for exploring novel microbial processes. Within the past five years alone, studies in these systems have shed new light on microbial processes of arsenic respiration [1], cryptic sulfur [2] and oxygen cycling [3], low oxygen adapted nitrification [4] and denitrification [5], and anaerobic methane oxidation [6]. Moreover, oxygen-depleted waters are expanding worldwide [7], making it critical to understand how oxygen concentration impacts microbially regulated nutrient and energy budgets, and how these impacts vary among sites. Most microbes in these systems are uncultivated and phylogenetically divergent from better-studied relatives. Many of these taxa remain unclassified beyond the phylum or class level and likely represent lineages uniquely adapted to low oxygen conditions. The importance of understanding microbial ecology in marine low oxygen environments is underscored by the recent discovery of new bacterial and archaeal “superphyla,” the Candidate Phyla Radiation and DPANN groups, respectively [8,9,10]. Discovery of these groups has massively expanded the tree of life and provided an important opportunity to further our understanding of microbial ecology and evolution.
Blue holes are subsurface caverns found in karst bedrock environments. They formed during climatic periods when low sea levels exposed the bedrock to weathering, and subsequently became submerged as sea levels rose [11]. Marine blue holes differ from anchialine blue holes, such as those found in the Bahamas and the Yucatán peninsula, as they do not have freshwater layers and are not exposed to the atmosphere. Anchialine blue holes can be highly stratified, with anoxic and sulfidic bottom waters [12,13,14] and microbial communities distinct from other marine and freshwater systems [15,16,17,18]. However, data on true marine blue holes are limited. A recent study on the Sansha Yongle blue hole, the deepest known marine blue hole with a bottom depth of 300 m, found the water column became anoxic around 100 m with increases in hydrogen sulfide, methane, and dissolved inorganic carbon below that depth [19]. This and two additional studies also found that microbial communities in Yongle were notably different from those of the surrounding pelagic water column, with anoxic layers in the hole dominated by taxa linked to sulfur oxidation and nitrate reduction [19,20,21]. Further, deep blue hole waters may be resistant to mixing with waters outside the hole, especially in regions with limited seasonal variation or water mass intrusion. These observations suggest the potential for blue holes to harbor novel microbial lineages as a consequence of both unique geochemistry and environmental isolation.
Locations of 18 blue holes have been recorded in offshore waters on the west Florida shelf; many more may exist but remain undiscovered due to a lack of systematic survey [22] (J. Culter, Mote Marine Laboratory, unpublished data, December 7, 2017). According to anecdotal reports from recreational divers and fishers, the rims of these holes feature dense communities of corals, sponges, and other invertebrates, in contrast with the more barren sandy bottom of the surrounding shelf. Commercially and recreationally valuable fishes also congregate at the rims. High biomass and elevated nutrient levels at these sites have broad relevance for coastal ecology in the Gulf of Mexico, particularly as they may fuel phytoplankton blooms [23, 24]. Indeed, the west coast of Florida experiences frequent and intense harmful algal blooms (HABs). While numerous nutrient sources have been identified as potential HAB triggers, the relative importance of these sources remains geographically unconstrained and our knowledge of what drives HABs is by no means complete [25,26,27]. The prevalence of HAB-causing or other phytoplankton species at these holes would be fueled by microbially driven nutrient cycles, underscoring the importance of characterizing their microbial communities and associated biogeochemistry.
Gulf of Mexico blue holes may be chemically stratified and devoid of oxygen. In one of the only biological studies of these features, Garman et al. [16] explored Jewfish Sink, a coastal blue hole near Hudson, Florida. Oxygen concentrations fell from near saturation at the rim (~2 m water depth) to zero around 20 m water depth and remained below detection to the bottom of the hole (~64 m water depth), with the anoxic layer further characterized by pronounced sulfide accumulation with depth. A clone library of 16S rRNA genes from microbial mats in the hole revealed a taxonomically rich community (338 operational taxonomic units, including 150 bacterial and 188 archaeal taxa), with sequences closely related to those from low oxygen habitats including deep-sea sediments, salt marshes, cold seeps, and whale falls [16]. These sequences represent taxa linked to a range of metabolisms, including dissimilatory sulfur, methane, and nitrogen cycling (both oxidative and reductive). This study also reported dense and chemically variable clouds of particulates within the hole; these included iron-sulfide minerals, suggesting the potential for microbial-metal interactions in the water column. This work, alongside evidence from the Yongle blue hole [19], suggests marine blue holes are biogeochemically complex features and potential hotspots for microbial diversity.
Here, we used metagenomics to describe the microbial ecosystem in a marine blue hole on the west Florida shelf. To help interpret microbial processes, we also present a comprehensive electrochemistry-based analysis of redox chemical speciation. Amberjack (AJ) Hole lies ~50 km west of Sarasota, Florida at a water depth of 32 m. Like Jewfish Sink, AJ is conical in shape, with a narrow rim (25 m diameter) and a wider floor (~100 m diameter, as determined by initial reconnaissance dives for this study). Water depth at the floor ranges from 110 m at the edge to 90 m at the center, where a debris pile of fine-grained sediment has accumulated. Our knowledge of AJ, like that of other marine blue holes, is limited and based primarily on exploration by a small number of technical divers. The hole’s shape and depth present a challenge for SCUBA, as well as for instrumentation or submersibles. The goals of our study were to determine what microbial taxa drive the physical and chemical processes shaping the AJ Hole water column and to link microbial taxonomic and biochemical diversity to low oxygen processes in an unexplored marine environment. During two expeditions in May and September 2019, we used a combination of technical divers and Niskin bottles to sample water column microbial communities. These collections spanned oxygenated waters at the rim, an anoxic but non-sulfidic intermediate layer, and a bottom layer rich in reduced sulfur compounds. Our results reveal a system that is highly stratified, apparently stable between timepoints, and phylogenetically diverse, with high representation by uncultivated and poorly understood taxa. The results suggest marine blue holes may serve as unique natural laboratories for exploring redox-stratified microbial ecosystems, while highlighting a need for future biogeochemical exploration of other blue holes and similar habitats.
Results
Sampling scheme
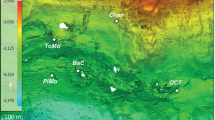
We sampled physical, chemical, and biological features of Amberjack Hole in May and September 2019. The May data set included one conductivity, temperature, and depth (CTD) profile with a coupled dissolved oxygen sensor, along with water column samples for chemical analyses (5 depths) and microbial community sequencing (11 depths, including 5 from within the hole). These samples were collected via a combination of diver bottle water exchange, hand-cast Niskin bottles, and automated Niskin sampling on a rosette (Supplementary Table S1). The CTD profile was acquired by attaching a combined CTD and optical dissolved oxygen sensor to an autonomous lander deployed for 24 h to measure sediment respiratory processes and fluxes (data not included in this study). The lander was positioned at 106 m water depth on the slope of the debris pile; the deepest May water sample was acquired by a diver at this depth (Fig. 1).

The benthic lander positions are shown for the May 2019 and September 2019 sampling expeditions, and those depths also correspond to the deepest collected water samples for microbial community analysis (106 m in May, 95 m in September).
In September, improved sampling design resulted in higher spatial resolution for all water column parameters. We obtained two CTD profiles and eight samples from within the hole for microbiome analysis. All September water samples were acquired by hand-cast Niskin bottles, with the deepest sample from 95 m, the presumed peak of the debris pile. In contrast to the May sampling, all water sampling in September was performed on the day of the CTD casts, ensuring that chemical and biological measurements were temporally coupled. We therefore focus primarily on September results (below), with exceptions where noted.
Water column physical and chemical profiles
Figure 1 provides a schematic of Amberjack Hole, the benthic lander positions (and corresponding deepest water samples) in May and September, and the general water column features.
Amberjack Hole was highly stratified (Figs. 1, 2 and Supplementary Fig. S1). Salinity increased sharply by one PSU between the overlying water column (35.2) and the blue hole water mass (~30 m, 36.2). Within the hole, salinity decreased slightly with depth (maximum difference 0.4 PSU) until 75 m and then increased to ~36 PSU at the bottom (Fig. 2A). Dissolved oxygen decreased sharply upon entry into the hole, from 100% saturation at the rim (32 m) to <5% saturation by 40 m (Fig. 2B). Oxygen then increased gradually to a secondary maximum of 40% at 75 m, before dropping to near 0% (anoxia) below 80 m.

A Compared to the overlying water, salinity was slightly higher and pH was slightly lower inside the hole (i.e., below 30 m). A coincident dip in salinity and rise in pH was present at 75 m. B Dissolved oxygen concentrations varied widely, with both a primary and secondary oxycline. At 80 m, the onset of anoxia immediately below the secondary oxycline coincided with a spike in turbidity. Water density is represented by σT, defined as ρ(S,T)-1000 kg m−3 where ρ(S,T) is the density of a sample of seawater at temperature T and salinity S, measured in kg m−3, at standard atmospheric pressure. C Dissolved inorganic carbon (DIC) increased slightly from 20 to 50 m but more intensely between 70 and 90 m, from ~2.2 to 2.5 mM. A sharp increase in NOx (NO2− + NO3−) between 40 and 50 m was followed by a return to near 0 between 60 and 90 m. Phosphate (PO43−) and ammonium (NH4+) remained below 1 µm before increasing to 5–6 µm between 70 and 80 m, respectively. D Dissolved ferrous iron (Fe(II)d) and total dissolved iron (Fed) increased with the transition to anoxia. Sulfur species are presented as follows: S2O32− (thiosulfate, S in the +II oxidation state). S(0) represents combined dissolved and colloidal elemental sulfur measured after sample acidification; however, the dissolved fraction may also include a small amount of S(0) derived from the acid-dissociations of polysulfide species (i.e., Sx2−). Finally, ∑S(−II) represents primarily hydrogen sulfide (HS−) removed by acidification but could also be minorly redundant with HS− released by acidification of Sx2−. Thiosulfate peaked between 80 and 90 m, and all iron and sulfur species increased sharply by 70–85 m, with S(0) representing the largest component of the reduced sulfur pool.
Dissolved inorganic nitrogen (nitrate (NO3−) + nitrite (NO2−), or NOx) spiked between 40 and 70 m from 0 to ~12 µM in May (data not shown) and nearly 17 µM in September (Fig. 2C). Ammonium (NH4+) and phosphate (PO43−) increased sharply below 80 m in September, with NH4+ reaching nearly 50 µM and PO43− reaching nearly 6 µM (Fig. 2C). Particulate nutrients (N, P, and carbon (C)) and chlorophyll a concentrations were seemingly less variable throughout the water column in May compared to September (Supplementary Fig. S2), although this pattern may be an artifact of the lower sampling resolution. In September, particulate nutrients and chlorophyll a spiked at the hole opening (between 30 and 40 m) with P at 0.1 µM, N at 1.2 µM, C at 11 µM, and chlorophyll a at nearly 1.5 µg L−1. Particulate nutrients remained low deeper in the water column with the exception of P, which spiked again to 0.14 µM at 80 m. Particulate C and N also increased slightly below 80 m in both May and September (Supplementary Fig. S2).
Both iron and sulfur species increased below the second oxycline at 80 m. Coinciding with anoxia, ferrous iron (Fe(II)d) increased from 16 nM (0.015 µM) to 187 nM (0.187 µM) between 70 and 80 m, respectively. With increasing depth, sequentially more reduced sulfur compounds were observed (Fig. 2D):
-
a.
At 85 m, S2O32− (thiosulfate, sulfur (S) in the +II oxidation state), reached a maximum of 264 µM;
-
b.
at 90 m, S(0), representing either S8 (i.e., elemental sulfur, S in the 0 oxidation state) or most S within polysulfide (Sx2−, e.g., S42− or S82−), peaked at 725 µM and was comprised of both dissolved (<0.7 µm; 126 µM) and particulate (599 µM) fractions (fractional data not shown); and
-
c.
at 95 m (deepest sample), combined ΣS(−II) (i.e., either as hydrogen sulfide (HS−), polysulfide (Sx2−), or both) reached a maximum of 61 µM.
Due to analytical uncertainties associated with the voltammetric quantification scheme, the ΣS(−II) as presented may contribute redundantly to both (b) and (c) from a sulfur mass balance perspective. Only 6% of the S(−II) signal in the unfiltered 95 m sample was lost upon filtering through 0.7 μm GFF filters (not shown), compared to 93–100% losses in the 85 and 80 m samples, respectively. The analytical conditions employed during the cathodic Hg/Au voltammetric analyses are unable to resolve separately the speciation of truly dissolved hydrogen sulfide (HS−), elemental sulfur (S8), or polysulfides (Sx2−), all of which react at similar potentials. These species are therefore represented collectively as ΣS(−II). The filtration results, however, indicate that the deepest waters (i.e., the 95 m sample) are probably enriched in truly dissolved hydrogen sulfide (HS−), whereas the shallower waters are enriched in colloidal or particulate S8 unable to pass through the filter. Indeed, this suggests significant HS− is fluxing from sediments, confirmed by benthic flux and pore water measurements (data not shown).
Notably, the zone of S(0) (i.e., elemental sulfur) accumulation between 80 and 90 m was marked by a spike in turbidity (Fig. 2B). In addition, particulate P (Supplementary Fig. S2B) was elevated concomitantly with Fed and Fe(II)d between 70 and 80 m (Fig. 2D). The latter suggests the presence of particulate or colloidal minerals and associated adsorption sites (e.g., Fe oxide colloids).
The May sampling resulted in a single data point from a depth of 85 m, which showed levels of 19.8 ± 2.3 µM S2O32−, 93 µM ΣS(−II), and no S(0), similar to the September measurements from that depth.
Microscopy and cell counts
Microscopy-based cell counts (prokaryotes) ranged from 5 × 106 to 7 × 106 cells mL−1 within the hole (data available only from September), almost an order of magnitude lower than counts above the hole (30 m sample; Supplementary Fig. S3). Counts were lowest at 80 m, the depth of the observed turbidity spike.
Microbial community composition
Taxonomic composition was assessed using both 16S rRNA gene amplicon and metagenome-assembled genome sequences. The taxonomy databases for these methods (SILVA [28] for amplicons, the Genome Taxonomy Database (GTDB; [29]) for metagenome-assembled genomes (MAGs)) differ in their naming conventions; as GTDB is more up-to-date with recent literature (for example, the Woesearchaeotal amplicon sequence variants (SVs) were classified as “Nanoarchaeota” using SILVA), we have used its annotations where possible, with notes when older taxonomic labels may be useful.
16S rRNA gene amplicon sequencing yielded 12,692–118,397 reads per sample after filtering for quality (Supplementary Table S1). Water column microbial communities differed significantly based on depth grouping (PERMANOVA p = 0.016), partitioning into shallow (0–32 m, oxic zone above the hole), middle (40–70 m, hypoxic zone), and deep (80–106 m, anoxic zone) groups (Fig. 3A). The shallow group was characterized by the ubiquitous cyanobacteria Synechococcus and Prochlorococcus, as well as several clades of the heterotrophic alphaproteobacterium SAR11 (particularly clade Ia) (Fig. 3B). Cyanobacteria were absent below 32 m in both May and September. The middle water column featured high frequencies of Nitrosopumilus sp., a member of the ammonia-oxidizing Thaumarchaeota, as well as members of the sulfur-oxidizing family Thioglobaceae (Fig. 3B). Other groups, composing 1–10% of the community in this depth zone, included Marine Groups II and III Thermoplasmatota (formerly MGII/III Euryarchaeota) and members of the family Gimesiaceae (phylum Planctomycetes).

A Principal components analysis shows communities were highly similar within each depth grouping regardless of the sampling date. B Community composition of representative samples from each of the three depth groupings show both middle and deep water column layers feature high levels (~40% frequency) of a single taxon, the ammonia-oxidizing Nitrosopumilus sp. in the middle water column and the Woesearchaeota in the deepest layers. Samples shown here are from September 2019 at 30 m, 40 m, and 85 m. A taxon is defined as the sum of all SVs classified at the indicated level. Taxa comprising the “Other” category (<5% relative abundance) included Ca. Actinomarina, Rhodospirillales AEGEAN-169 marine group, and SAR202 in the 10 m sample; the alphaproteobacterial family Rhodobacteraceae in the 40 m sample; and the Patescibacterial phylum ABY1 in the 85 m sample.
The anoxic and sulfidic deep water column was dominated by Woesearchaeota of the DPANN superphylum. Woesearchaeota represented 33–56% of all sequences below 75 m in both May and September and were comprised of 74 sequence variants (SVs), with two SVs representing between 84% (May 85 m) and 99% (September 90 m) of the total Woesearchaeotal fraction. Other well-represented groups included Nitrosopumilus sp, Thioglobaceae (SUP05 clade), and a member of the phylum Bacteroidota (formerly Bacteroidetes) associated with hydrothermal vents (“Bacteroidetes VC2.1 Bac22” in the SILVA database) (Fig. 3B). While Shannon diversity was similar among depth groups, Simpson diversity was ~50% higher in the deep compared to the middle and shallow groups (Supplementary Fig. S4).
Depth patterns in community composition were highly similar between May and September. The most abundant taxa showed only minor differences in frequency between these months, with a few exceptions (Fig. 4). Marine Group II Thermoplasmatota reached 12% relative abundance in the middle hypoxic zone in September while in May this taxon never exceeded 1%; in contrast, Marine Group III peaked at around 6% in May and in September only reached about half that level. In May, Thioglobaceae frequency increased steadily below 30 m to peak at >10% of the community in the deep anoxic zone. In contrast, Thioglobaceae was not detected in the anoxic zone in September. Rather, Arcobacter sp. (represented by a single SV) spiked at 80 m in September to nearly 10% of the community. In all other samples, this taxon never exceeded 0.5% at any sampled depth. The 80 m depth was not sampled in May.

A May 2019, major taxa (10–50% of the community), B May 2019, minor taxa (1–12% of the community), C September 2019, major taxa, D September 2019, minor taxa. Sampled depths are represented by points on each line. Overall patterns were consistent between the 2 months, reflecting the separation of microbial communities by depth grouping. Notably, Nitrosopumilus sp. and Woesearchaeota dominated the middle and deep water column, respectively. The sulfur-oxidizing SUP05 clade increased continuously with depth in May but decreased in relative abundance below 70 m in September, while Arcobacter sp. spiked sharply at 80 m in September.
Metagenomes and MAG taxonomy
We obtained four metagenomes from two depths in both May and September. These represent communities at 60 m (both months) in the hypoxic (~15% O2 saturation) and Nitrosopumilus-dominated zone, and at 106 m (May) and 95 m (September) in the anoxic, sulfidic, and Woesearchaeota-dominated zone. Sequencing and assembly results are provided in Supplementary Table S2. Analysis with Nonpareil [30], a tool to assess the fraction of the total extracted DNA that was sequenced based on the level of redundancy among sequenced reads, showed estimated metagenome coverage values between 50% and 80%, with the 60 m samples showing lower diversity and higher estimated coverage than deep samples (Supplementary Fig. S5). Between 82 and 93% of all reads were unclassified by Kraken 2 against the RefSeq database (Supplementary Table S2). Genome equivalent values were higher in the 60 m metagenomes than the deep metagenomes for both months, although the values for both September samples were higher than both May samples (Supplementary Table S2), despite sub-sampling metagenomes at even depths. This discrepancy may be related to overall sequencing quality; however, it is unlikely to have affected functional annotation or binning results. Metagenome binning involving data from all samples yielded 31 high-quality, non-redundant MAGs representing a diverse array of microbial taxa (Table 1). These included eight archaeal MAGs, seven of which were generated from the 60 m samples, including six belonging to the phylum Thermoplasmatota and one belonging to Nitrosopumilus (Thaumarchaeota). The deep metagenomes yielded two MAGs classified as members of the phylum ABY1 (superphylum Patescibacteria/CPR), one of which contained a 16S rRNA gene SV classified as Ca. Uhrbacteria (phylum ABY1). The only archaeal MAG from the deep samples was classified as a member of the order Woesearchaeota. This MAG contained a 16S rRNA gene with 100% nucleotide identity to the most abundant SV recovered in amplicon sequencing of the bottom water communities (Table 1 and Supplementary Table S3). Queries against the Genome Taxonomy Database (GTDB; using MAG single-copy core genes) and SILVA database (using the 16S rRNA gene SV) classified this MAG (BH21) as belonging to the order “Woesearchaeia” in the phylum Nanoarchaeota. However, this classification is outdated, as “Woesearchaeia” has recently been given the phylum-level designation Woesearchaeota [31]. Only nine MAGs had a reference genome in GTDB that exceeded the minimum alignment fraction (65%); the remainder had no known close relative (Table 1).
Three MAGs representing the highest amplicon frequencies (from all recovered MAGs) were placed in phylogenies with closely related taxa (Supplementary Table S4), confirming their taxonomic assignments (based on GTDB and SILVA) as members of the Woesearchaeota (BH21; Fig. 5), Nitrosopumilus (BH19; Supplementary Fig. S6), and Thioglobaceae (SUP05 clade) (BH20; Supplementary Fig. S7). The two taxa most closely related to the Woesarchaeotal MAG are both from oxygen minimum zones in the Arabian Sea, with the next most closely related taxa from an iron-rich terrestrial hot spring and a freshwater lake (Lake Baikal). The phylogeny shows Woesearchaeotal MAGs from a wide range of environments interspersed across branches, providing weak support for clustering according to habitat type (Fig. 5). The AJ Nitrosopumilus MAG was most closely related to a MAG from a cold seep sponge, and relatively distantly removed from the nearest cultured Nitrosopumilus isolate (Supplementary Fig. S6). The AJ Thioglobaceae MAG (SUP05 clade) was most closely related to a group of eight Thioglobaceae MAGs from deep-sea hydrothermal vents. Other closely related genomes were associated with deep-sea invertebrates (Supplementary Fig. S7).

The phylogeny was constructed using an alignment of 44 single-copy gene amino acid sequences in a maximum likelihood analysis using a GAMMA model of rate heterogeneity, a BLOSUM62 protein substitution model, and 999 bootstraps.
MAG gene content
Gene content analysis of MAGs from the deep metagenome assemblies showed the three DPANN and CPR MAGs (BH21, BH22, and BH28) devoted a higher proportion of their genome to genetic information processing, while genes for metabolism represented a lower proportion of the genomes (Fig. 6A). This pattern was most pronounced for BH21, the Woesearchaeotal genome. Within the “Metabolism” category, genes for metabolism of amino acids, nucleotides, and other complex molecules were roughly equal across all MAGs, but genes for metabolism of amino acids, carbohydrates, energy, lipids, and cofactors/vitamins were underrepresented in the same three MAGs relative to all others (Fig. 6B). BH21 contained a higher proportion of genes across all sub-categories in “Genetic Information Processing,” with the CPR MAGs having similarly low levels of transcription-related genes to all other deep MAGs (Fig. 6C).

The bars represent the number of genes in each category or subcategory divided by the total number of open reading frames of the genome.
Functional annotation
Based on the analysis of all metagenome contigs (prior to MAG binning), broad functional categories (KEGG “subgroup2” level) separated the 60 m samples from the deep samples (Supplementary Fig. S8). The most differentially enriched genes between the depth groups included many with hypothetical functions as well as some involved in metabolism. Genes involved in genetic information processing were only enriched in the deep samples, and one in the “Environmental Information Processing” category was annotated by KEGG as K23573, a gene for the eukaryotic protein dentin sialophosphoprotein. This gene was nearly 5000× more abundant in the deep metagenomes relative to the 60 m metagenomes. BLASTX queries against the NCBI-nr database linked this sequence to hypothetical proteins from genomes of the candidate phylum Uhrbacteria (hereafter Patescibacteria ABY1), although these proteins shared only 30% amino acid identity with the gene in our data. This gene was identified in one of the two MAGs (BH22) classified as Patescibacteria ABY1. The longest contig containing a gene with this annotation (out of 14 total contigs in the deep co-assembly) also contained genes for signal transduction, membrane transport, and DNA replication/repair (Supplementary Table S5).
Although they were not among the most differentiating categories, sulfur and nitrogen metabolism categories also differed in frequency between the depths (Fig. 7). These included genes for both assimilatory and dissimilatory metabolism. Some of the most pronounced differences in sulfur metabolism involved the dissimilatory thiosulfate (phsA/psrA) and sulfite (dsrAB) reductases, both of which were enriched in deep metagenomes. Sulfur metabolism genes enriched in the 60 m metagenomes also included dmdABCD, which encode enzymes for the catabolism of dimethylsulfoniopropionate (DMSP).

Color bars show the depth group of the metagenome (60 m or “Deep,” 95 or 106 m). A Genes involved in sulfur metabolism. B Genes involved in nitrogen metabolism. Some dissimilatory genes, such as dsrAB and napAB, are enriched in the deep water layer. Others, including nitrate, nitrite, and nitrous oxide reductases, are enriched in the 60 m layer. Samples clustered by depth grouping in both categories.
Blue hole MAGs encoded a diverse suite of metabolisms (Table 2). We detected genes involved in both reductive and oxidative pathways of dissimilatory sulfur and nitrogen cycling, arsenic respiration, methylotrophy, and carbon monoxide metabolism, among others. One MAG (BH25) is one of only two reported members of the Bacteroidota (formerly Bacteroidetes) phylum potentially capable of dissimilatory sulfur metabolism, with the other represented by a MAG from a hot spring [32]. Most MAGs (23 out of 31) contained genes involved in arsenic resistance. Genes for arsenic respiration were detected in three MAGs, with two occurrences of arsenite oxidase gene aioA (BH16, Actinobacterial order Microtrichales, and BH24, alphaproteobacterial order Rhodospirillales) and one of arsenate reductase gene arrA (BH30, Desulfobacterota taxon NaphS2) (Supplementary Fig. S9). The latter had 82% amino acid identity with a gene from another NaphS2 strain isolated from anoxic sediment in the North Sea (DSM:14454); this gene is annotated in the FunGene database as arrA. Other MAG-affiliated proteins included nitrous oxide reductase (nosZ, in four MAGs representing three phyla), carbon monoxide dehydrogenase (cooFS, in BH30 (NaphS2, phylum Desulfobacterota)), and the ammonia and methane monooxygenases amoAB (in BH19, Nitrosopumilus) and pmoAB (in BH9 (family Methylomonadaceae, Gammaproteobacteria) (Table 2)). BH9 also contained several other genes involved in C1 metabolism including mxaD, mch, mtdAB, and fae.
The Woesearchaeotal MAG (BH21) was notable for its small size (679 Kbp, with 67% (CheckM) and 73% (anvi’o) completion values). As with other Woesearchaeotal genomes, genes for several core biosynthetic pathways were missing, including those for glycolysis/gluconeogenesis, the citric acid cycle, and the pentose phosphate pathway. Genes linked to glyoxylate and glycarboxylate metabolism and fructose and mannose metabolism were detected, along with the carbamate kinase arcC; however, no full energetic pathway could be reconstructed.
Only one MAG (BH24, order Rhodospirillales) contained genes for the full denitrification pathway (NO3 → N2). Other MAGs had the potential to perform individual steps:
NO3 → NO2, seven MAGs; NO2 → NO, six MAGs; NO → N2O, six MAGs; and N2O → N2, three MAGs (Table 2).
Discussion
Oxygen-deficient marine water columns are crucial habitats for understanding ecosystem function under oxygen limitation and across gradients of redox substrates, representing conditions that are predicted to expand substantially in the future [7]. Moreover, these systems have been a critical resource for discoveries of novel microbial diversity and unforeseen linkages between chemical cycles [1,2,3,4,5,6]. While in recent years these discoveries have been facilitated by community DNA, RNA, and protein sequencing, most oxygen-depleted waters have not yet been characterized, either from an -omics perspective or via cultivation-dependent methods. This is due partly to the fact that these systems are challenging to sample and span a gradient of environmental conditions. For example, the Pacific oxygen minimum zones (OMZs) are anoxic through several hundreds of meters of the water column, cover hundreds of square kilometers of open ocean, and are relatively unaffected by processes in the underlying sediment [33]. Microbial communities in these systems, especially those along the peripheries, presumably have periodic exchange with microbial communities outside the OMZ, for example via eddy intrusion, storms, or offshore transport [2]. In contrast, we describe a very different oxygen-deficient zone with intense but apparently stable stratification. A similar formation in the South China Sea, the Sansha Yongle blue hole, was recently investigated and provides an opportunity to identify features potentially common to blue hole environments worldwide [20, 21, 34]. The Yongle formation also featured an anoxic, sulfidic bottom water layer, but the geochemical profile differed from Amberjack in notable ways, including the absence of a second oxycline. Microbial amplicon and metagenomic data show some microbial taxa and functional genes are common to both holes, including the taxa SAR406 and Marine Group II Thermoplasmata in the intermediate layers and sulfate-reducing Deltaproteobacteria and Arcobacter spp. in the bottom anoxic layers [21], as well as the dissimilatory sulfate reductase gene dsrB in the bottom layer [20]. Importantly, however, the frequency and potential roles of DPANN and CPR lineages in Yongle were not discussed [20, 21]. We show here that the semi-enclosed Amberjack blue hole environment exhibits a physiochemical profile and microbial community both similar to but also remarkably distinct from that of other oxygen-deficient water columns.
Amberjack Hole is characterized by unusual profiles of oxygen, NOx, and reduced sulfur species (Figs. 1 and 2). The oxygen profile is unlike that of most OMZs, with a secondary oxygen peak around 75 m where dissolved oxygen rose to 43% saturation before dropping back to zero (Fig. 2B). In open ocean OMZs, oxygen profiles are typically unimodal, with concentrations falling along an upper oxycline, staying hypoxic or anoxic through a core layer, and then gradually increasing below the core as organic substrates are depleted with depth and microbial respiration slows [35, 36]. The Yongle blue hole has a stratification scheme fundamentally different from that of AJ, with only one oxycline and the bottom anoxic layer comprising two-thirds of the water column [34]. In AJ, the secondary oxygen peak at 75 m represents either a decline in net oxygen consumption driven by decreased microbial respiration, a transport-related phenomenon that affects oxygen supply, or both. AJ Hole is also characterized by a subsurface maximum in NO3− (nitrate) and/or NO2− (nitrite) below the main chemocline (Supplementary Fig. S2), suggesting that dissolved oxygen is also consumed by nitrification. The decline in NOx coincides with the second sharp drop in oxygen concentration around 80 m. This drop is followed by the detection of reduced inorganic compounds, consistent with decreasing redox free energy expectations as a function of depth. The presence of ΣS(−II) below 80 m suggests active SO42− reduction. However, preliminary evidence from pore water profiles and sediment flux measurements indicates that ΣS(−II) was generated at millimolar concentrations in the sediments (data not shown). Thus, it is likely that the reduced sulfur in the deep water column largely originates from benthic, rather than water column, sulfate reduction.
Reduced sulfur is clearly a key energy source in the deepest layers of the blue hole. We detected high concentrations of sulfur in intermediate redox states, notably S(II) (i.e., thiosulfate, S2O32−) and S(0) (elemental sulfur), overlying a pronounced zone of S(−II) (hydrogen sulfide) below 80 m. Generally, oxidative sulfur metabolism proceeds with H2S/HS− being oxidized to sulfite and sulfate (SO32−/SO42−) with S(0) and S2O32− produced as intermediates [37, 38], with the relative completion depending on the pH or the microbial community. In some species, for example, pH affects the relative rates of different steps of S2O32− oxidation via the sox pathway, notably favoring elemental sulfur production compared to consumption (S(0) oxidation to sulfate) under low-pH conditions [39]. Elemental sulfur accumulation—either intra- or extra-cellularly—is also controlled by the gene content of the sox pathway itself; in organisms lacking soxCD, sulfur remains bound to the SoxY enzyme rather than completely oxidized to sulfite/sulfate [40, 41]. The intermediate S2O32− can be produced enzymatically via sulfide oxidation, notably via sulfide:quinone oxidoreductase (SQR), or chemically by a reaction of elemental sulfur with sulfite [42]. Here, certain MAGs contained genes involved only in one step of S(0)/S2O32− production or consumption (e.g., MAG BH25 (Bacteroidota) from the deep samples possesses only the sqr pathway), highlighting the potential modular nature by which sulfur may be cycled in this zone. Interestingly, the sulfide-oxidizing taxon Arcobacter is not present in the deepest samples but is enriched at 80 m. One possible explanation is that this species has an advantageous niche given that it is capable of coupling the oxidation of sulfide to the reduction of nitrate, an alternative oxidant, which is in elevated concentration at this depth. In AJ Hole, the peak concentration of S2O32− was observed at a depth above that of the peak concentration of S(0), suggesting that different steps in the overall oxidation of ΣS(−II) may be performed by various vertically-distributed niches within a complex cycle. We argue that if a single microbial population was performing complete H2S/HS− oxidation to SO42− (with S(0) or S(+II) as intermediates), these compounds should exhibit similar depth distributions, not stratification.
The peak sulfur zone (peak sulfur concentrations of all measured dissolved sulfur compounds) occurred below 80 m (Fig. 1D) and appeared vertically decoupled from detectable dissolved oxygen, which fell below detection between 74 and 79 m (Fig. 1B). This decoupling suggests the anaerobic sulfur oxidation may also proceed with NOx as a terminal oxidant, as NOx remained detectable above 90 m and decreased in concentration with depth as reduced sulfur concentrations increased (Fig. 1). Although representing only the deepest, most sulfidic layers of the blue hole, metagenomes contained diverse genes for oxidizing sequentially reduced sulfur compounds, as well as high abundances of genes for each step of the denitrification pathway (Fig. 5). Sulfur-driven denitrification is common in marine OMZs (e.g., ref. [43]), notably in conjunction with gammaproteobacteria of the SUP05 lineage (e.g., [26]). In some cases, we detected both sulfur oxidation and denitrification genes in the same MAG, including in the SUP05 (Thioglobaceae) MAG (BH20). We also recovered MAGs encoding only sulfur oxidation proteins or incomplete denitrification pathways (Table 2), a pattern observed in other low oxygen water columns and suggesting the likely cross-feeding of sulfur and nitrogen cycle intermediates between taxa (e.g., refs. [44, 45]). However, the sulfur intermediate stratification zone, roughly 70–90 m, overlaps with the zone of oxygen availability (down to 80 m) as measured with the fluorometric sensor, which has a detection limit only in the low micromolar range only.
Thus, it is likely that the community in this depth range consumes both oxygen and oxidized nitrogen species (and potentially other oxidants) for use in sulfur oxidation, with the relative concentrations of oxidants and sulfur intermediates potentially driving the vertical separation of microbial niches. Versatility in oxidant and reductant use is common in pelagic low oxygen communities [44], as well as in lineages within these communities. The gammaproteobacterial sulfur oxidizer (GSO) group, for example, comprises multiple lineages across and within OMZs, hydrothermal vent plumes, and other low oxygen environments. These lineages differ in their genomic potential for autotrophy versus heterotrophy, low versus high oxygen affinity during aerobic growth, partial versus complete denitrification during anaerobic growth, or reduced sulfur versus hydrogen oxidation [46,47,48]. Such metabolic diversity—including that in AJ—invites basic questions about diversification in marine microbes. Notably, is high phenotypic versatility driven by temporal versus spatial separation of niches? Or both? Presumably, in Amberjack and other waters sheltered from physical disturbance, functional diversification is driven by an atypically high level of vertical stability. The implications of this hypothesis for questions about microbial “endemism” and rates of vertical chemical exchange remain to be explored.
Interestingly, the oxic-anoxic transition at 80 m coincided with a sharp spike in turbidity. This spike was not due to an increase in microbial cell counts (Supplementary Fig. S3). However, at 80 m, we recorded a sharp increase (to 10% of total) in the frequency of 16S rRNA genes assigned to Arcobacter, a sulfide-oxidizing bacterium known to produce filamentous sulfur at oxic-anoxic interfaces [49]. Between 80 and 90 m, 70–83% of the combined elemental sulfur (S(0)) and polysulfide (ΣS(−II)) signal was lost after 0.7 μm filtration, suggesting that this lost fraction was in fact elemental sulfur, as polysulfide is soluble [50, 51]. Arcobacter sulfidicus is motile and thrives at oxic-anoxic interfaces under high-sulfide conditions; [52] a discrete layer containing high levels of this taxon is therefore further evidence for a stratified water column. The dominant Arcobacter SV in our study had two nucleotide mismatches relative to Arcobacter sulfidicus. Based on the environments in which different Arcobacter strains have been detected, it is likely that members of this genus have similar metabolisms. For example, mat-forming Arcobacter spp. have been observed at deep-sea hydrothermal vents with high levels of elemental sulfur [53]. We did not sample at 80 m in May and therefore cannot confirm whether the sharp turbidity and Arcobacter spikes are stable over time. However, the production of filamentous or other particulate forms of sulfur under microaerophilic conditions is consistent with the observed sulfide gradient in September 2019 (Fig. 2) and oxygen gradients in both months (Fig. 2 and Supplementary Fig. S1). Alternatively, Arcobacter may be using NOx to oxidize S(−II), as has been shown in denitrifying members of the genus [54].
Other oxidants may also have a role in AJ. Dissolved ferrous iron (Fe(II)) reached a maximum concentration near the turbidity spike at ~80 m, below the zone of peak O2/NOx and above the zone of reduced sulfur (Fig. 2D). Further, total dissolved iron (Fed), which includes dissolved organically-stabilized Fe(III) and FeS colloids, remained <300 nM, but increased below 40 m, peaking just above the dynamic reduced sulfur zone (Fig. 2D). These patterns hint at a possible cryptic iron cycle, in which ΣS(−II) diffusing upwards is rapidly oxidized by dissolved Fe(III) [55] to form Fe(II), S(0), and ΣSx2−. Dissolved Fe(III) may then be recycled by reoxidation of Fe(II) by O2, and the S(0) and Sx2− simultaneously oxidized by O2 or dissolved Fe(III) to form thiosulfate, S2O32−. Alternatively, ΣS(−II) may first react with Fe(II) to form the detected FeS colloids, which subsequently oxidize to form Fe(II), S(0), and Sx2−. Metabolic pathways for iron oxidization (or reduction) are not easy to detect with metagenomic data, as iron metabolism genes have roles in other cellular processes and are therefore suggestive but not diagnostic of iron metabolism [56, 57]. Nonetheless, additional metagenomic sampling at finer spatial resolution may help identify a role for microbial activity in blue hole iron cycling. The oxidation of iron, however, is rapid even abiotically, and the vertical distribution of iron in this system can partially explain the apparent vertical disconnect between the ΣS(−II) and O2. Indeed, this phenomenon has been observed in a nearshore Florida hole [16], in which FeS colloids formed at the interface between 30 and 50 m. AJ appears to be enriched instead in colloidal S(0), as FeS colloids remained undetected.
The pronounced stratification of blue hole chemistry is consistent with the differentiation of microbial communities with depth. In both May and September, microbiome composition varied distinctly among oxic, hypoxic, and anoxic, sulfidic layers (Fig. 3). Shannon diversity was similar among all communities, but Simpson diversity was notably higher in the anoxic depth group (Supplementary Fig. S4), reflecting the lower evenness of these microbiomes. Alpha diversity was not assessed in the Yongle blue hole; however, as with AJ Hole, the microbial communities reflected the water column layers characterized by varying dissolved oxygen levels [21, 34]. In both blue hole environments, metagenomic data from the anoxic zone confirm the potential for sulfur oxidation linked to denitrification. In the AJ water column, sulfur speciation suggests that most ΣS(−II) originates from sediment sulfate reduction and upward ΣS(−II) diffusion. Nonetheless, patterns in our molecular data raise the possibility of water column sulfate reduction and ΣS(−II) production. First, the gene dsrA, which encodes the dissimilatory sulfite reductase necessary for sulfate reduction, was enriched in the bottom water of AJ Hole, similar to the pattern seen in Yongle (Fig. 7). This enzyme, however, can also operate in reverse and we, therefore, cannot rule out its involvement in sulfur oxidation. Second, known sulfate-reducing deltaproteobacteria, including the genera Desulfobacula, Desulfatiglans, and Desulfobacter, were detected in the deep water column samples in both May and September, collectively comprising 6–11% of the community (Fig. 3B). These levels are comparable to the abundances (11–17%) of known sulfate reducers reported in Yongle [21]. Experimental measurements of sulfur reduction and oxidation rates (e.g., ref. [58]) will help clarify the connections between sediment and water column sulfur cycling in blue holes.
In contrast to Yongle, however, the deepest AJ communities were dominated by a recently described archaeal lineage, the Woesearchaeota [9]. Woesearchaeota have been detected in a wide variety of biomes, including groundwater, terrestrial and marine sediments, wetlands, deep-sea hydrothermal vents, and hypersaline lakes [59,60,61,62]. However, their relative abundance is consistently low, at most ~5% of the total microbial community in any given environment, with the highest proportions observed in freshwater sediments [61] and high-altitude lakes [62]. In contrast, Woesearchaeota comprised at least one-third of the blue hole microbiome between 75 and 106 m, reaching a maximum of nearly 60% in the anoxic sulfidic layer in September (Figs. 3 and Fig. 4). Remarkably, one 16S rRNA sequence variant represented up to 97% (and only two SVs represented up to 99%) of all Woesearchaeotal amplicons in each sample, suggesting low intra-population diversity. Dominance (>50% of the community) by a single strain variant is relatively uncommon in pelagic marine microbiomes; it is even rarer that such occurrences involve members of the Archaea. The AJ Woesearchaeotal MAG, BH21, which contained the dominant 16S rRNA Woesearchaeota amplicon sequence, was 679 Kbp and similar in size to other Woesearchaeotal genomes, which rarely exceed 1 Mbp [9, 61]. BH21 was most closely related to two of the few available marine Woesarchaeotal MAGs, both of which originated from an OMZ in the Arabian Sea (Fig. 5). Furthermore, a recent study in the Black Sea showed Woesearchaeota were highly enriched in protein-amended water samples from an anoxic, sulfidic environment similar to AJ Hole [63]. Based on the apparent ubiquity of Woesearchoaeota across a wide range of biomes, intra-phylum diversity is likely high and the limited number of MAGs are unlikely to span all sub-clades, precluding any conclusions about biome-specific Woesearchaeotal adaptations. Nevertheless, their prevalence at hydrothermal vents [60], OMZs, and the Black Sea suggest certain members of this phylum have traits that make it particularly suited to a marine, low oxygen niche.
Two groups of recently discovered microbial lineages, the archaeal DPANN superphylum and the bacterial superphylum Patescibacteria (also known as Candidate Phyla Radiation, or CPR), have garnered considerable attention for encompassing large fractions of total microbial diversity and for their apparent ubiquity among diverse environments including terrestrial groundwater, estuaries, deep-sea hydrothermal vents, and hypersaline lakes [60, 61, 64,65,66,67]. While members of both superphyla have been observed in marine environments, they are more typically found in freshwater systems; Patescibacteria in particular seem to be associated with subsurface aquifers [66]. Sequences from both groups were detected in the Yongle blue hole at low levels (<5% of the total microbial community) [20, 21], which is a typical level of representation among studied biomes. Woesearchaeota belongs to the DPANN, while two other MAGs from the AJ deep water column samples are members of the ABY1 phylum within Patescibacteria, providing an opportunity to compare their biochemical and metabolic potential with related lineages. Both Patescibacteria and DPANN are characterized by small genomes and cell sizes, low per-cell ribosome counts, and predicted minimal metabolic capabilities [8, 64, 68, 69], including reliance on fermentation rather than respiration [68]. These observations have led some to propose that they rely on syntrophic partners or host organisms for their energy needs and some cellular components like amino acids [68, 70]. Three AJ MAGs (one Woesearchaeota and two Patescibacteria) support the growing body of evidence for streamlined genomes and a likely reliance on co-occurring microbes for basic cellular components. None of these MAGs contained genes for metabolic processes common to low oxygen marine systems, notably dissimilatory sulfur or nitrogen cycling or autotrophy. A comparison of gene content among all deep water column MAGs showed that the Woesearchaeotal (BH19) and Patescibacterial (BH22, BH28) MAGs devote a much larger fraction of their genomes to genetic processes like DNA replication and translation, and a much smaller fraction to metabolism, than other members of the microbial community (Fig. 6). This pattern was most pronounced in the Woesearchaeotal MAG, which had virtually no genes involved in energy metabolism, even compared to the Patescibacteria (Fig. 6B). The single gene in this category was the carbamate kinase arcC, which is also involved in arginine biosynthesis and purine metabolism. A relationship between genome size and preferential accumulation of genes for particular cellular processes has been previously shown [71] and is well-characterized in symbiotic organisms [72]. In the case of DPANN and CPR microbes, their small genomes may reflect ancestral traits retained in a nutrient-limited, highly stable environment, rather than a loss of more recent metabolic strategies as is typically assumed for symbionts [72, 73]. The apparent high level of clonality of this population also points to a highly conserved genetic repertoire with little gene flow. However, certain genes in these MAGs may reflect adaptations for microbe-microbe interactions. Notably, the Woesearchaeotal MAG contained pilA encoding an archaeal type IV pilus assembly protein. Pilus-like structures have been observed in CPR cells and proposed to facilitate diverse cell-cell interactions or nutrient scavenging [69]; these structures have also been associated with biofilm formation and DNA exchange in other archaea [74, 75]. These combined observations suggest AJ Woesearchaeota and Patescibacteria rely on other microbes capable of harvesting energy from reduced sulfur species, such as Thioglobaceae or Rhodospirillales (see below; Table 2), in a type of syntrophy which has been previously proposed for DPANN and CPR lineages [70]. Physical attachment to larger cells may even be the reason Woesearchaeota were so prevalent in our samples, as their predicted tiny cell sizes (<0.1 µm) [64, 67, 69] would have led to free-living cells escaping our 0.22 µm filters.
The DPANN and CPR MAGs were three of several novel lineages in the AJ water column. From the deepest samples (95 and 106 m), recovered MAGs included members of the sulfur-oxidizing SUP05 clade of Thioglobaceae and three members of the phylum Marinimicrobia (SAR406), among others (Table 1). The middle water column (60 m) was also populated by underdescribed taxa, including several lineages of marine Thermoplasmatota and members of the bacterial phyla Planctomycetota and Myxococcota. Only eight out of nineteen MAGs from 60 m had identifiable close representatives in the GTDB, suggesting a high level of taxonomic novelty. Surprisingly, automatic binning did not recover a Thaumarchaeal MAG from the 60 m assembly despite the large fraction of amplicon SVs belonging to the ammonia-oxidizing genus Nitrosopumilus. However, a manually binned MAG could be phylogenomically placed in this lineage (Supplementary Fig. S6) and contained the dominant SV from the amplicon data set, which comprised up to 86% of all Nitrosopumilus spp. amplicons. As with the Woesearchaeota, this low level of SV diversity implies population homogeneity. It remains to be determined if this homogeneity is driven by a potential dearth of ecological niches. Indeed, the AJ Nitrosopumilus MAG, at 725 Kbp, was estimated to be only 53–63% complete (Table 1 and Supplementary Table S3); its full biochemical potential, therefore, remains to be characterized. However, in contrast to recently described and putatively heterotrophic Thaumarchaeota [76, 77], blue hole Nitrosopumilus, like all known members of this genus, contain amoCAB encoding ammonia monooxygenase and therefore likely contribute to nitrification in the blue hole.
The 60 m samples also contained six MAGs belonging to the archaeal phylum Thermoplasmatota. One of these MAGs (BH15) could only be placed in the family Thalassoarchaeaceae. The others included four (BH4, BH11, BH12, and BH13) from the Marine Group II (MGII) lineage, which is one of the four major planktonic archaeal groups [78], and one from the less well-characterized Marine Group III (MGIII) lineage (BH3). The Marine Group II have recently been proposed as an order-level lineage, Candidatus Poseidoniales, containing two families delineating the current MGIIa and MGIIb clades [79]. Most MGIIa members have been identified from the photic zone, while MGIIb sequences are largely limited to depths below 200 m, although there are exceptions [80]. Notably, all AJ MGII MAGs belonged to the MGIIb lineage (Table 1). Several studies have identified genes related to low oxygen metabolism including reduction of sulfate [81,82,83] and nitrate [79], suggesting these taxa are more likely to be adapted to microaerophilic or anaerobic conditions. We found a gene for thiosulfate reduction to sulfite (phsA) in two of the four MGIIb MAGs (Table 2), but no complete pathways for any forms of anaerobic respiration. Nevertheless, the MGII lineage comprised over 25% (five out of nineteen) of the MAGs recovered from 60 m, suggesting this group is an important component of the hypoxic zone community.
Metabolic potential differentiated the middle and deep water column communities (Fig. 7 and Supplementary Fig. S8). Many of these functions could be linked to MAGs and represented a range of anaerobic metabolisms. Genes for dissimilatory sulfide and sulfite oxidation (or potentially reduction; dsrAB, aprAB), denitrification (nar, nir, nor, nos), and dissimilatory nitrate reduction to ammonia (DNRA, nrf) were enriched in deep anoxic samples, but also common in the hypoxic zone (60 m) (Table 2, Fig. 7). Other genes linked to MAGs included aioAB and arrA, encoding arsenite oxidase and arsenate reductase, respectively (Supplementary Fig. S9). The reduction of arsenate to arsenite is thought to be an ancient metabolic pathway, originating before oxygenation of the Earth’s atmosphere and oceans [84]. It has recently been proposed as an important microbial metabolic strategy in oxygen-deficient marine environments, potentially providing arsenite for use as an energy source by other microbes [1]. Arsenic concentrations in Amberjack were not measured. However, the detection of aioAB and arrA, of which the latter was also detected in unbinned contigs from the deep co-assembly (Supplementary Fig. S9), as well as genes for arsenic detoxification (arsCM) in diverse MAGs (including in the Nitrosopumilus MAG described above), suggest a role for arsenic cycling in AJ. Remarkably, one MAG (BH24, Rhodospirillales) contained genes for several of these pathways, representing an unusual repertoire of respiration strategies (Supplementary Table S4). Finally, one of the genes with the largest differences in frequency between 60 m and deep samples belonged to BH22 (Patescibacteria) and encodes an unknown protein previously observed in other genomes of this group (previously called Uhrbacteria). This gene (K23573) has no known prokaryotic homologs, and other genes on this contig did not provide further insight into its function (Supplementary Table S5). This likely reflects the massive uncharacterized protein family diversity of CPR bacteria, which has been estimated to exceed that of all other bacterial groups [85]. Indeed, approximately 90% of metagenome reads from both deep samples could not be taxonomically classified by the RefSeq database, possibly reflecting a high representation of uncharacterized lineages. It is beyond the scope of our study to fully assess the evolutionary relationships of all genes of interest; however, the AJ microbiome and particularly the prevalence of DPANN and CPR taxa, which are rarely found at high levels in marine environments, provide an important new context for understanding the diversity and function of uncharacterized genes.
Conclusions
The Amberjack Hole water column is potentially highly stable, with complete turnover (driven either by wind or the sinking of cold water masses) unlikely. Thus, AJ microbiomes, particularly those in the deepest layers, potentially have limited connectivity to other marine communities. The extent to which such isolation explains the observed unique community composition or drives divergence of individual microbial lineages remains uncertain. Undoubtedly, the vertical transport, transformation, and stratification of redox-active elements—notably sulfur, nitrogen, and iron—also have a significant role in structuring this unusual microbiome. The cause of the unusual oxygen profile, with a secondary oxygen peak and two hypoxic zones, remains unexplained and may be influenced by subsurface freshwater flow, a phenomenon that would undoubtedly affect the microbial ecology. The presumed stability of this community, its high metabolic diversity, and its dominance by understudied microbial taxa highlight AJ as a model for detecting novel biogeochemical processes under low oxygen. Marine blue holes in general are potentially valuable sites for the study of microbial diversification and linked elemental cycling.
Methods
Sampling scheme
Sampling was conducted May 15–17, 2019 and September 19, 2019 using the vessels R/V William R. Mote and R/V Eugenie Clark. Amberjack Hole is located ~50 km west of Sarasota, Florida, at 27.28748 N, −83.16139 W. One component of the research involved the deployment of an autonomous benthic lander to the bottom of the hole for 24–48 h at a time for benthic electrochemistry measurements; these data are not included in this paper. Technical SCUBA divers who guided the lander into and out of the hole also collected water samples (see details below).
In both May and September, all water samples used for nutrient measurements and microbial DNA preservation were filtered immediately on board. Microbial DNA samples were stored on ice until they return to Mote Marine Laboratory, where they were stored at −20 °C. Subsequent nutrient analyses described below were conducted at Mote except where noted.
Water column physical parameters and dissolved oxygen
During the May sampling, an EXO2 sonde (YSI Inc/Xylem Inc, Yellow Springs, Ohio) was attached to the autonomous lander deployed for 24 h to the bottom of the hole. The sensor provided extensive data from the surface (1–2 m) and the bottom position (106 m) but low-resolution readings from the water column due to rapid ascent and descent velocities. Dissolved oxygen, temperature, and salinity data were retrieved from the instrument and plotted in R.
During the fall sampling, an integrated SBE-19plus V2 CTD with an SBE43 DO sensor, WET Labs FLNTUrt chlorophyll and turbidity sensor, Satlantic cosine PAR sensor, and an SBE18 pH sensor (Sea-Bird Electronics Inc) was lowered twice by hand at a rate that provided high-resolution dissolved oxygen, turbidity, salinity, and density data on September 19, 2019. CTD casts were performed the same day as water was sampled for microbial DNA and nutrients. Data were retrieved and plotted in R as above.
SBE data were processed using SBE Data Processing software after compensating for sensor thermal mass, sensor alignment, timing offset (the delay associated with pumped sensors, i.e., conductivity), and changes in instrument velocity (due to ship heave). Data were binned (~0.2 m increments) to smooth data variability. Processed and derived data included temperature (°C), potential temperature (°C), salinity (PSU), water density (σT in kg m−3), chlorophyll a (µg L−1 based on relative fluorescence), turbidity (NTU), dissolved oxygen (mg L−1), O2 saturation (%), pH, PAR, and Brunt–Väisälä frequency (N = stratification index). Manufacturer recommendations for calibration and service for all sensors were followed.
Water column electrochemical measurements
Water samples (~10 m vertical resolution) were subject to solid-state Hg/Au voltammetric analyses [86] for measurement of the redox environment, i.e., O2, Mn(+II), organic-Fe(III) complexes, Fe(II), S(+II) (in the form of S2O3−), and ΣH2S (S2−, HS−, H2S, S(0), ΣSx2−). Briefly, samples collected from the water column at 10 m intervals were carefully transferred via a Tygon (formula 2375) transfer tube line into LDPE bottles while filling from bottom to top to minimize atmospheric oxygen contaminations. Sample bottles remained sealed until analyses and were stored at 4 °C. Upon return to the lab (within 4–6 h of the collection), 20 mL of each sample was carefully pipetted into an electrochemical cell (Analytical Instrument Systems, Inc.) holding a custom fabricated Hg/Au amalgam microelectrode working electrode, a Pt counter electrode, and a fritted Ag/AgCl reference electrode (with 3 M KCl electrolyte solution). N2 was gently blown over the top of the solution to minimize mixing with the atmosphere. Within 5 min, samples were subject to a series of anodic square wave voltammograms (ASWVs), cathodic square wave voltammograms (CSWVs), and linear sweep voltammograms (LSVs) to quantify the above redox analytes [86, 87]. To analytically distinguish between S(0) and ΣS(−II) (S2−, HS−, H2S), which both react at the same potential at a Hg electrode [50], CSWV measurements were repeated after acidification with HCl (final pH < 3) and N2 sparging of the solution for 2 min to remove free ΣS(−II). This same electrochemical speciation experiment was conducted a second time in separate aliquots that were filtered through 0.7 µm GFF filters directly in the N2-degassed electrochemical cell (this filter size was selected for future organic carbon analyses).
Nutrient, iron, and DIC measurements
Water samples were collected for nutrients and included chlorophyll a, dissolved ammonium (NH4+), dissolved NOx (nitrate (NO3) + nitrite (NO2)), dissolved orthophosphates (PO43−), dissolved total and ferrous iron (Fe), dissolved inorganic carbon (DIC) and particulate carbon (C), nitrogen (N), and phosphorous (P). All samples were collected and immediately filtered once shipboard.
Chlorophyll a was measured according to EPA method 445.0 [88]. Briefly, samples were filtered through a glass fiber filter until clogging, then stored in the dark until analyses. Prior to analyses, filters were sonicated in 90% acetone to extract chlorophyll from algal cells and then centrifuged for clarification. The fluorescence of the clarified extract was then measured using a fluorometer (Turner 10-AU Fluorometer) with special narrow bandpass filters at an excitation wavelength of 436 nm and an emission wavelength of 680 nm. Analytical quality assurance followed standard laboratory practices assessing precision and accuracy using sample replicates, container blanks, duplicate, and spiked analyses with results meeting acceptable levels of precision and accuracy.
Samples for dissolved NH4+ and NOx were filtered through Pall Supor [46] 450 47 mm (0.45 µm pore size) membrane filters while samples for particulates were filtered through pre-combusted (450 °C, 3 h) 47 mm (0.7 µm pore size) Whatman GF/F filters. For particulate carbon and nitrogen, 200 mL was filtered per sample, and filters were rinsed with acidified (10% HCl) filtered seawater to remove inorganic carbon. For particulate P, 500 mL was filtered. Samples were then stored on ice until their return to the lab and analyzed within 48 h (dissolved NH4+ and NOx) or within 28 days (particulates). Analyses for dissolved nutrients followed colorimetric, segmented flow, autoanalyzer techniques on an AA3 with method reference and method detection limits (MDL) as follows: dissolved NH4+ ([89]; 0.07 µM), dissolved NOx ([90]; 0.07 µM). Particulate phosphorus was analyzed according to [91, 92] with in-house modifications for analysis on a segmented flow analyzer and MDLs of 0.03 µM. For particulate carbon and nitrogen, samples were analyzed on a Thermo FlashEA1 1112 Elemental Analyzer with MDLs of 0.2 and 0.1 µM, respectively.
To measure DIC, samples were immediately poisoned with HgCl2 and stored until analyses. DIC was analyzed (Apollo AS-C6 DIC Analyzer) following methods by [93]. The accuracy and precision of the instrument were regularly monitored using Certified Reference Materials for Seawater CO2 Measurements (Dickson Laboratory, Scripps Institution of Oceanography, San Diego, CA; Batch #181 and 186).
Soluble orthophosphates (PO43−) were measured spectrophotometrically using the molybdate-blue technique [94].
Finally, the speciation of iron was obtained by measuring Fe(II) by the ferrozine assay [95] in filtered samples before (dissolved Fe(II)) and after (total dissolved Fe) reduction by hydroxylamine (0.2 M) using a long waveguide spectrophotometric flow cell. The analytical quality assurance follows standard laboratory practices assessing precision and accuracy using sample replicates, container blanks, duplicate, and spiked analyses with results meeting acceptable levels of precision and accuracy.
Microbial sampling and preservation
Water column samples were collected on May 15 and 17, 2019 and September 19, 2019. In May, 800 mL water was collected by divers from depths of 46, 61, and 106 m inside the hole. Sterilized Nalgene bottles filled with deionized water were taken down by the divers, opened at the appropriate depth, and DI water was replaced with ambient seawater. Niskin bottles deployed on the CTD rosette also collected 1.9 L samples from depths of 8, 15, and 23 m above the hole. In May and September, hand-cast Niskin bottles collected 1 L water from 0 m (surface), 30, 60, and 85 m (May), and from 0 m (surface) to 90 m at 10 m intervals, as well as 85 m and 95 m (September). Water samples were stored at 4 °C until processing, with time to processing ranging from immediate to 2 h following collection. Water was filtered onto 0.22 µm Sterivex (MilliporeSigma) cartridges (PES filter) with a peristaltic pump and preserved with ~3 mL DNA/RNA stabilization buffer (25 mM sodium citrate, 10 mM EDTA, 5.3 M ammonium sulfate (pH 5.2)) at −20 °C during transport back to the lab (1-3 h) and then at −80 °C or on dry ice until further processing at Georgia Tech. In September, unfiltered water from the following depths was also preserved (1.4 mL water, 150 µL PBS-buffered formaldehyde) for nucleic acid staining and microscopy and kept frozen at −20 °C until processing: 30 m, 50 m, 60 m, 70 m, 80 m, 85 m, 90 m, 95 m.
Cell staining and microscopy
Duplicate samples from the following depths taken in May 2019 were processed for microscopy: 30, 50, 70, 80, and 95 m. Samples preserved in PBS-buffered formaldehyde (1.55 mL total) were combined with 2.0 mL milliQ water and filtered onto 0.2 µm GTBP filters (Millipore) using vacuum filtration. Filters were dried for ~20 min and incubated in the dark on ice with 50 µL DAPI (0.2 µg mL−1). Filters were then rinsed in milliQ water and 100% ethanol, dried for ~10 min, and placed on a microscope slide with one drop of Citifluor (Electron Microscopy Sciences). Slides were visualized with a Zeiss Axio Observer D1 confocal epifluorescence microscope using a DAPI filter (Zeiss filter set 49). Between 20 and 25 photographs were taken in a grid pattern from each slide and counted in ImageJ [96] using a custom script. Cells were assumed to be uniformly distributed on the filter, and manual validation of count values was performed for a subset of photographs. Numbers of cells mL−1 were calculated using a membrane conversion factor (filtration area/area of micrometer field) multiplied by N*D, where N is the total number of cells counted/number of micrometer fields counted and D = total volume of a sample stained/volume of original sample available. The average value of cell counts from all photos for each sample was plotted in Microsoft Excel.
Sequencing library preparation
DNA was extracted from each Sterivex cartridge using a custom protocol as described in ref. [97], except for the September surface (0 m) sample which was lost during sample transit to Georgia Tech. Briefly, cells were lysed by flushing out RNA stabilizing buffer and replacing it with lysis buffer (50 mM Tris-HCl, 40 mM EDTA, 0.73 M sucrose) and lysozyme (2 mg in 40 mL of lysis buffer per cartridge), then incubating cartridges for 45 min at 37 °C. Proteinase K was added and cartridges were resealed and incubated for 2 h at 55 °C. The lysate was removed, and the DNA was extracted once with phenol:chloroform:isoamyl alcohol (25:24:1) and once with chloroform:isoamyl alcohol (24:1). Finally, DNA was concentrated by spin dialysis using Ultra-4 (100 kDA, Amicon) centrifugal filters. The yield was assessed using a Qubit 2.0 dsDNA high-sensitivity assay (Invitrogen, Carlsbad, CA).
Illumina MiSeq libraries were prepared by amplifying the V4 region of the 16S rRNA gene using the environmental DNA protocol adapted from [98]. Briefly, amplicons were generated using Platinum® PCR SuperMix (Life Technologies, Carlsbad, CA) with Earth Microbiome Project primers 515FB and 806RB appended with Illumina-specific adapters. Template DNA of all samples was diluted to approximately 5 ng µL−1 and PCRs were performed in 25-µL reactions using Platinum® PCR SuperMix (Life Technologies) (22 µL), BSA (Invitrogen) (1 µL), and 0.5 µL each of forward and reverse primer (10 ng L−1 stock concentration) with 1 µL template DNA. The thermal cycling protocol consisted of 26 cycles with the following steps: denaturation at 98 °C (30 s), followed by 30 cycles of denaturation at 98 °C (5 s), primer annealing at 55 °C (5 s), and primer extension at 72 °C (8 s), followed by extension at 72 °C for 1 min. Negative control reactions were run using 1 µL milliQ water in place of the DNA template. Amplicons were analyzed by gel electrophoresis to verify size (~400 bp, including barcodes and adaptor sequences) and purified using Diffinity RapidTip2 PCR purification tips (Diffinity Genomics, West Chester, PA). Amplicons from different samples were pooled at equimolar concentrations and sequenced using a paired-end Illumina MiSeq 500 cycle kit (2 × 250 bp) with 5% PhiX to increase read diversity.
Metagenomes were generated from four water column samples: two from May (60 m and 106 m) and two from September (60 m and 95 m). Libraries were prepared using the Illumina Nextera XT DNA library preparation kit (Illumina Inc., San Diego, CA) according to the manufacturer’s instruction and run on a Bioanalyzer 2100 instrument (Agilent) using a high-sensitivity DNA chip to determine library insert sizes. An equimolar mixture of the libraries (final loading concentration of 12 pM) was sequenced on an Illumina MiSeq instrument (School of Biological Sciences, Georgia Institute of Technology), using a MiSeq reagent v2 kit for 600 cycles (2 × 300 bp paired-end protocol).
Amplicon sequence data processing and analysis
Demultiplexed amplicon sequences are available in the NCBI Sequence Read Archive (BioProject ID PRJNA689047) and in the Patin FigShare account: https://figshare.com/projects/Amberjack_Blue_Hole/85013. Raw sequences were run through the DADA2 algorithm [99] in QIIME2 [100] to assess sequences at sequence variant resolution, using the following command parameters: --p-trim-left-f 70 --p-trim-left-r 70 --p-trunc-len-f 150 --p-trunc-len-r 150. Resulting SVs were assigned taxonomy using the naïve Bayes classifier trained on the Silva 132 database (99% OTUs, 515F/805R sequence region).
The SV table with raw read counts of all water samples was exported from QIIME2 and transformed in R using the variance stabilizing transformation (vst) in the DESeq2 package [101]. Tables were not rarefied to preserve the maximum available information. Metadata including depth and SV taxonomy were imported and combined into a Phyloseq object [102, 103]. Alpha and beta diversity analyses were run in DivNet [104] at the SV level. Shannon and Simpson diversity results were extracted and plotted by depth grouping using ggplot2 [105]. Beta diversity was assessed using the resulting Bray-Curtis distance matrix in a principal component analysis using the prcomp() function in R and visualized in a PCA using ggplot2. The distance matrix was also used to test for significant difference among samples by depth grouping and month using the adonis() function in the R vegan package [106] with the following command: adonis(formula = bc ~ Depth + Month, data = metadata, permutations = 999).
Metagenomic sequence data processing and analysis
Metagenome sequences are available in the NCBI Sequence Read Archive (BioProject ID PRJNA689047) and in the Patin FigShare account: https://figshare.com/projects/Amberjack_Blue_Hole/85013. Raw sequences from the four metagenomes (May 60 m, May 106 m, September 60 m, September 95 m) were trimmed and checked for quality and adapter contamination as described for the amplicons above. The Nd value, a metric based on sequence diversity, was calculated for each metagenome using Nonpareil 3.0 [30] and used to generate curves of estimated average coverage as a function of sequencing depth. Quality controlled reads were run through MicrobeCensus [107] to generate genome equivalent (GE) values for each metagenome. Normalization of read counts by GE values corrects for variation in coverage across samples and generates more accurate relative abundance estimates than normalizing by single-copy housekeeping genes [108, 109]. To account for the different sequencing depths of the metagenomes, we subsampled each metagenome to 3,000,000 reads for the estimation. To generate more even coverage distribution across the metagenomes, quality-controlled reads were normalized using BBNorm, part of the open source BBMap package (https://sourceforge.net/projects/bbmap/).
Six assemblies were generated from the four water column metagenomes. Individual assemblies of each sample were generated using metaSPAdes (v3.14.0) [110]. Co-assemblies for each depth pair (May 60 m with September 60 m, and May 106 m with September 95 m) were also generated using MEGAHIT (v1.2.9) [111] using a minimum contig length of 2500 bp. All assemblies were assessed for quality using metaQUAST [112] and annotated using Prokka [113] and the KEGG database.
For the latter, open reading frames were generated using Prodigal [114] and clustered using MeShClust [115] at 90% nucleotide identity. The longest sequence from each cluster was extracted using a custom Python script and these representative sequences were run against the KEGG ortholog profile HMM models (KOfams) using KofamScan with the “prokaryote” database [116]. The parameter “-f mapper” was applied to provide only the most confident annotations (those assigned an individual KO). Orthologies were matched to their corresponding functions using a parsed version of the “ko00001.keg” database text file (https://github.com/edgraham/GhostKoalaParser), which provides a three-tiered hierarchical categorization of each gene, referred to here as “Group”, “Subgroup1”, and “Subgroup2”. Sequence coverage of each gene was generated by mapping metagenomic short reads against each one using Magic-BLAST [117]. The Magic-BLAST output was filtered to include only the best hit for each read and the read counts were normalized by the GE value of the corresponding metagenome.
The normalized counts were used to run hierarchical clustering analyses in Python using the Seaborn “clustermap” function. KofamScan outputs were grouped by the three hierarchical categories to generate heat maps at different levels of categorization. At the highest categorical level (Group) the categories “Human Diseases,” “Organismal Systems,” “Cellular community—eukaryotes,” and “Brite Hierarchies” were removed before performing the cluster analysis. The first three groups are not relevant to microbial gene functions and the fourth provides a different hierarchical categorization scheme for the same annotations and was thus redundant. One heat map was generated for all “Subgroup2” categories, with values square-root transformed for better visualization. A separate heat map was made for all genes (KOs) with an average normalized read count above 0.2 and a difference between the means of 60 m and deep samples >20×. Values were fourth-root transformed for better visualization. All cluster analyses were run using ward linkage and Euclidean distance methods.
Open reading frames generated from the SPAdes individual assemblies were run through the Hidden Markov Model search tool described in ref. [118] and available at https://github.com/ShadeLab/PAPER_Dunivin_meta_arsenic/tree/master/HMM_search to query for genes involved in arsenic metabolism. HMM outputs were run through the data_preparation.R script (also available in the GitHub repository) to generate plots showing the quality distributions of hits to the aioA and arrA genes. The amino acid sequence of a potential sequence for the arrA gene extracted from BH30 (alignment fraction 89%, bitscore 237) was also queried against the arrA BLAST database from the same toolkit.
A read-based assessment of taxonomic composition was performed using Kraken 2 [119] followed by Bracken [120] to estimate relative abundances of taxa from each metagenome without the PCR bias incurred by amplicon sequencing. We used the “Standard” pre-built Kraken 2 database (https://benlangmead.github.io/aws-indexes/k2), which includes classification schemes for archaea, bacteria, viruses, plasmids, human DNA, and vectors (“UniVec_Core”).
Metagenome-assembled genomes
Genomic bins were generated from all individual assemblies as well as co-assemblies. For each co-assembly, short reads from both metagenomes were submitted to MaxBin to leverage contig co-variation patterns across samples. All bins were assessed for quality using CheckM [121] and anvi’o (v6.1) [122]. Bins with a quality score greater 40 were retained for dereplication, with quality score calculated as completion—5 × contamination (CheckM values). Dereplication was performed with dRep [123] with an ANI cutoff of 95% for secondary clustering and a 20% minimum pairwise overlap between genomes. For bins belonging to the same secondary cluster (i.e., likely representing the same microbial population), the highest-quality was chosen as a representative. In cases where the qualities of two MAGs were within 1%, the completeness and redundancy values from anvi’o were applied to determine the higher quality bin. In only one case (BH31) was a MAG from the single assembly of higher quality than a highly similar MAG from the co-assembly; in all other cases, the co-assemblies produced higher quality bins, likely due to the longer contigs representing more sequence space. Further analyses were performed on these representative bins.
All dereplicated high-quality MAGs were assessed for taxonomy with anvi’o and the Genome Taxonomy Database toolkit (GTDB-Tk) [29, 124]. Each MAG was checked for the presence of rRNA genes using the anvi’o HMM models and one MAG from the 60 m assembly was found to contain a eukaryotic 18S rRNA gene with an 80% match to a copepod sequence (Calanus sp.). This contig was removed from the co-assembly of origin and the process of binning, quality assessment, and dereplication was repeated.
All MAGs were queried for the presence of 16S rRNA gene amplicons by using the SVs as a query for a BLASTn analysis against the two co-assembly contigs. MAG contigs containing 100% identity matches across the entire SV length were identified, and the SV SILVA-based taxonomy was compared with the whole-genome taxonomy assignment from GTDB.
All MAGs were functionally annotated using both Prokka and KofamScan as described above, without the read mapping step. The following genes were verified by running amino acid sequences against the NCBI-nr database using blastp: pmoA-amoA (K10944; KEGG), pmoB-amoB (K10955; KEGG), pmoC-amoC (K10956; KEGG), K23573 (DSPP, dentin sialophosphoprotein; KEGG), pmoA (Prokka), pmoB (Prokka), aioB (Prokka), arsC2 (Prokka), arsM (Prokka). To confirm the annotations of arsenic respiration and resistance genes, and to query other genes involved in arsenic metabolism, including the arsenate reductase gene arrA, all MAGs were run through the Hidden Markov Model search tool described in ref. [118] and available at https://github.com/ShadeLab/PAPER_Dunivin_meta_arsenic/tree/master/HMM_search.
Gene content of all deepwater column MAGs (BH20-32) was compared using the KofamScan annotations. The number of genes belonging to each KEGG category and subcategory was calculated and divided by the total number of open reading frames in the MAG to assess the fraction of the genome devoted to each category. The results for the most low-resolution KEGG categories as well as the “Metabolism” and “Genetic Information Processing” sub-categories were plotted using the BlueHole_MAGs_gene_content.ipynb script.
Manual MAG refinement and generation
Six MAGs contained more than one 16S rRNA gene SV. In cases where the SVs within a MAG were distantly related to each other, any contigs containing a copy that was divergent from the genome-wide GTDB-TK taxonomic assessment were removed from the bin. Quality assessments were repeated on the edited MAG using CheckM and anvi’o, and if the quality score (as defined above) was over 5 points lower than the original MAG, the edited MAG was discarded. This happened in only one case (BH22); all other MAGs were kept in their edited form.
One of these edited MAGs was BH28, classified by GTDB as Patescibacteria but containing an SV classified as Magnetospiraceae (Alphaproteobacteria, Rhodospirillales) by SILVA. Conversely, BH24, which was classified by GTDB as Rhodospirillales, contained no 16S rRNA genes. The contig containing the Magnetospiraceae SV was removed from the Patescibacterial bin and added to BH24 based on its tetranucleotide clustering proximity to other contigs in that bin (as seen in the anvi’o interactive interface) and the matching taxonomy between the MAG and the SV.
Finally, BH22 (Patescibacteria ABY1) was edited by the addition of a contig clustering very closely to other MAG contigs in the anvi’o interface dendrogram. This contig contained the hypothetical K23573 gene, which has previously been identified in other ABY1 MAGs.
The dominant Nitrosopumilus amplicon SV was not present in any of the MAGs generated from the 60 m samples. However, it was found on an unbinned contig in the 60 m co-assembly. The anvi’o interactive interface was used to manually generate a bin containing this sequence, which also contained an archaeal 23S rRNA gene sequence with a top BLAST hit in NCBI to that of Nitrosomarinus catalina (99% identity; the full 16S rRNA gene sequence identities of N. catalina and this MAG were also >99%), and other contigs of similar tetranucleotide frequency, GC content, and coverage (BH19). This MAG was assessed for quality using CheckM and anvi’o.
All MAGs were run through GTDB using the “classify_wf” (for taxonomic classification) and “ani_rep” (for a closest relative by ANI) commands in GTDB-Tk [124], and through Prokka [113] and KofamScan [116] for functional annotation. Closest relatives were determined using a minimum 65% alignment fraction; if no reference genome exceeded this minimum there was no result.
All MAG sequences are available in the NCBI Whole Genome Sequence repository (BioProject ID PRJNA689047) and in the Patin FigShare account: https://figshare.com/projects/Amberjack_Blue_Hole/85013.
Phylogenomic analyses
Phylogenomic trees were generated for the MAGs BH19 (Nitrosopumilus), BH20 (SUP05 clade, Thioglobaceae), and BH21 (Woesearchaeota). In each case, genomes with the same or similar taxonomy as the MAG were downloaded from the NCBI Assembly database and run through anvi’o for the quality assessment using anvi-estimate-genome-completeness. High-quality MAGs (quality score > 40 as defined above) were retained (Supplementary Table S4) and single-copy genes were extracted using the anvi’o script anvi-get-sequences-for-hmm-hits. The two archaeal MAGs (BH19 and BH21) were run against the “Archaea_76” HMM database and the bacterial MAG (BH20) was run against the “Bacteria_71” HMM database. Genes occurring in most or all MAGs were concatenated and aligned (see Supplementary Table S4 for taxon-specific minimum gene occurrences). The alignment was run through RAxML [125] with optimization of substitution rates and a GAMMA model of rate heterogeneity (PROTGAMMA) with 999 bootstraps for a maximum likelihood phylogeny. The resulting.tre files were uploaded to the interactive Tree of Life website [126] (itol.embl.de) and labeled with their strain or sample of origin.
Hypothetical protein
The gene with the biggest difference in relative abundance between the 60 m and deep co-assemblies was annotated as K23573 in KofamScan. All contigs in the deep assembly containing this gene were identified and the longest of these was chosen for further investigation. All KofamScan annotations for ORFs located on this contig are provided in Supplementary Table S5.
Code availability
Python scripts used for calculating gene relative abundances, comparing these abundances across metagenomes, and calculating and comparing gene content in MAGs are available in the corresponding author’s GitHub repository: https://github.com/nvpatin/BlueHole_manuscript.
Change history
25 May 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41396-021-00996-w
References
Saunders JK, Fuchsman CA, McKay C, Rocap G. Complete arsenic-based respiratory cycle in the marine microbial communities of pelagic oxygen-deficient zones. Proc Natl Acad Sci USA. 2019;116:9925–30.
Callbeck CM, Lavik G, Ferdelman TG, Fuchs B, Gruber-Vodicka HR, Hach PF, et al. Oxygen minimum zone cryptic sulfur cycling sustained by offshore transport of key sulfur oxidizing bacteria. Nat Commun. 2018;9:1729.
Garcia-Robledo E, Padilla CC, Aldunate M, Stewart FJ, Ulloa O, Paulmier A, et al. Cryptic oxygen cycling in anoxic marine zones. Proc Natl Acad Sci USA. 2017;114:8319–24.
Sun X, Kop LFM, Lau MCY, Frank J, Jayakumar A, Lücker S, et al. Uncultured Nitrospina-like species are major nitrite oxidizing bacteria in oxygen minimum zones. ISME J. 2019;13:2391–402.
Tsementzi D, Wu J, Deutsch S, Nath S, Rodriguez-R LM, Burns AS, et al. SAR11 bacteria linked to ocean anoxia and nitrogen loss. Nature. 2016;536:179–83.
Thamdrup B, Steinsdóttir HGR, Bertagnolli AD, Padilla CC, Patin NV, Garcia-Robledo E, et al. Anaerobic methane oxidation is an important sink for methane in the ocean’s largest oxygen minimum zone. Limnol Oceanogr. 2019;64:2569–85.
Breitburg D, Levin LA, Oschlies A, Grégoire M, Chavez FP, Conley DJ, et al. Declining oxygen in the global ocean and coastal waters. Science. 2018;359:eaam7240.
Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature. 2015;523:208–U173.
Castelle CJ, Wrighton KC, Thomas BC, Hug LA, Brown CT, Wilkins MJ, et al. Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol. 2015;25:690–701.
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, et al. A new view of the tree of life. Nat Microbiol. 2016;1:16048.
Mylroie JE, Carew JL, Moore AI. Blue holes: definition and genesis. Carbonates Evaporates. 1995;10:225–33.
Canganella F, Bianconi G, Kato C, Gonzalez J. Microbial ecology of submerged marine caves and holes characterized by high levels of hydrogen sulphide. Rev Environ Sci Biotechnol. 2007;6:61–70.
Gischler E, Shinn EA, Oschmann W, Fiebig J, Buster NA. A 1500-year holocene caribbean climate archive from the blue hole, Lighthouse Reef, Belize. J Coast Res. 2008;246:1495–505.
Pohlman JW. The biogeochemistry of anchialine caves: progress and possibilities. Hydrobiologia. 2011;677:33–51.
Davis MC, Garey JR. Microbial function and hydrochemistry within a stratified anchialine sinkhole: A window into coastal aquifer interactions. Water. 2018;10:972–972.
Garman KM, Rubelmann H, Karlen DJ, Wu T, Garey JR. Comparison of an inactive submarine spring with an active nearshore anchialine spring in Florida. Hydrobiologia. 2011;677:65–87.
Gonzalez BC, Iliffe TM, Macalady JL, Schaperdoth I, Kakuk B. Microbial hotspots in anchialine blue holes: Initial discoveries from the Bahamas. Hydrobiologia. 2011;677:149–56.
Seymour JR, Humphreys WF, Mitchell JG. Stratification of the microbial community inhabiting an anchialine sinkhole. Aquat Microb Ecol. 2007;50:11–24.
Yao P, Wang XC, Bianchi TS, Yang ZS, Fu L, Zhang XH, et al. Carbon cycling in the world’s deepest blue hole. J Geophys Res. 2020;125:e2019JG005307.
He H, Fu L, Liu Q, Fu L, Bi N, Yang Z, et al. Community Structure, abundance and potential functions of bacteria and archaea in the Sansha Yongle blue hole, Xisha, South China Sea. Front Microbiol. 2019;10:2404–2404.
He P, Xie L, Zhang X, Li J, Lin X, Pu X, et al. Microbial diversity and metabolic potential in the stratified Sansha Yongle Blue Hole in the South China Sea. Sci Rep. 2020;10:5949–5949.
DeWitt D. Submarine springs and other Karst features in offshore waters of the Gulf of Mexico and Tampa Bay, Southwest Florida Water Management District. 2003.
Hu C, Muller-Karger FE, Swarzenski PW. Hurricanes, submarine groundwater discharge, and Florida’s red tides. Geophys Res Lett. 2006;33:L11601.
Smith CG, Swarzenski PW. An investigation of submarine groundwater-borne nutrient fluxes to the west Florida shelf and recurrent harmful algal blooms. Limnol Oceanogr. 2012;57:471–85.
Vargo GA, Heil CA, Fanning KA, Dixon LK, Neely MB, Lester K, et al. Nutrient availability in support of Karenia brevis blooms on the central West Florida Shelf: What keeps Karenia blooming? Continental Shelf Res. 2008;28:73–98.
Walsh DA, Zaikova E, Howes CG, Song YC, Wright JJ, Tringe SG, et al. Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones. Science. 2009;326:578–82.
Weisberg RH, Liu YG, Lembke C, Hu CM, Hubbard K, Garrett M. The coastal ocean circulation influence on the 2018 West Florida Shelf K. brevis Red Tide Bloom. J Geophys Res Oceans. 2019;124:2501–12.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36:996–1004.
Rodriguez RLM, Gunturu S, Tiedje JM, Cole JR, Konstantinidis KT. Nonpareil 3: fast estimation of metagenomic coverage and sequence diversity. Msystems. 2018;3: e00039-18.
Baker BJ, De Anda V, Seitz KW, Dombrowski N, Santoro AE, Lloyd KG. Diversity, ecology and evolution of Archaea. Nat Microbiol. 2020;5:887–900.
Thiel V, Costas AMG, Fortney NW, Martinez JN, Tank M, Roden EE, et al. “Candidatus Thermonerobacter thiotrophicus,” a non-phototrophic member of the Bacteroidetes/Chlorobi with dissimilatory sulfur metabolism in hot spring mat communities. Front Microbiol. 2019;9:3159–3159.
Helly JJ, Levin LA. Global distribution of naturally occurring marine hypoxia on continental margins. Deep-Sea Res Part I. 2004;51:1159–68.
Xie LP, Wang BD, Pu XM, Xin M, He PQ, Li CX, et al. Hydrochemical properties and chemocline of the Sansha Yongle blue hole in the South China Sea. Sci Total Environ. 2019;649:1281–92.
Thamdrup B, Dalsgaard T, Revsbech NP. Widespread functional anoxia in the oxygen minimum zone of the Eastern South Pacific. Deep-Sea Res Part I. 2012;65:36–45.
Wyrtki K. The oxygen minima in relation to ocean circulation. Deep-Sea Res Oceanographic Abstr. 1962;9:11–23.
Ghosh W, Dam B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol Rev. 2009;33:999–1043.
Luther GW, Findlay AJ, MacDonald DJ, Owings SM, Hanson TE, Beinart RA, et al. Thermodynamics and kinetics of sulfide oxidation by oxygen: a look at inorganically controlled reactions and biologically mediated processes in the environment. Front Microbiol. 2011;2:1–9.
Houghton JL, Foustoukos DI, Flynn TM, Vetriani C, Bradley AS, Fike DA. Thiosulfate oxidation by Thiomicrospira thermophila: metabolic flexibility in response to ambient geochemistry. Environ Microbiol. 2016;18:3057–72.
Kelly DP, Shergill JK, Lu WP, Wood AP. Oxidative metabolism of inorganic sulfur compounds by bacteria. Antonie Van Leeuwenhoek Int J Gen Mol Microbiol. 1997;71:95–107.
Grimm F, Franz B, Dahl C. Thiosulfate and sulfur oxidation in purple sulfur bacteria. In: Dahl C, Friedrich C, editors. Microbial sulfur metabolism. Springer: Heidelberg, Germany; 2008. p. 101–16.
Zopfi J, Ferdelman TG, Fossing H. Distribution and fate of sulfur intermediates - sulfite, tetrathionate, thiosulfate, and elemental sulfur - in marine sediments. In: Amend JP, Edwards KJ, Lyons TW, editors. Sulfur biogeochemistry: past and present. The Geological Society of America: Boulder, Colorado; 2004. p. 97–116.
Wright JJ, Konwar KM, Hallam SJ. Microbial ecology of expanding oxygen minimum zones. Nat Rev Microbiol. 2012;10:381–94.
Bertagnolli AD, Stewart FJ. Microbial niches in marine oxygen minimum zones. Nat Rev Microbiol. 2018;16:723–9.
Hawley AK, Brewer HM, Norbeck AD, Pasǎ-Tolić L, Hallam SJ. Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc Natl Acad Sci USA. 2014;111:11395–400.
Anantharaman K, Hausmann B, Jungbluth SP, Kantor RS, Lavy A, Warren LA, et al. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 2018;12:1715–28.
Murillo AA, Ramírez-Flandes S, DeLong EF, Ulloa O. Enhanced metabolic versatility of planktonic sulfur-oxidizing gamma-proteobacteria in an oxygen-deficient coastal ecosystem. Front Mar Sci. 2014;1:1–13.
Shah V, Chang BX, Morris RM. Cultivation of a chemoautotroph from the SUP05 clade of marine bacteria that produces nitrite and consumes ammonium. ISME J. 2017;11:263–71.
Wirsen CO, Sievert SM, Cavanaugh CM, Molyneaux SJ, Ahmad A, Taylor LT, et al. Characterization of an autotrophic sulfide-oxidizing marine Arcobacter sp. that produces filamentous sulfur. Appl Environ Microbiol. 2002;68:316–25.
Luther GW, Glazer BT, Hohmann L, Popp JI, Tailefert M, Rozan TF, et al. Sulfur speciation monitored in situ with solid state gold amalgam voltammetric microelectrodes: polysulfides as a special case in sediments, microbial mats and hydrothermal vent waters. J Environ Monit. 2001;3:61–6.
Rozan TF, Theberge SM, Luther G. Quantifying elemental sulfur (S0), bisulfide (HS-) and polysulfides (S(x)2-) using a voltammetric method. Analyt Chim Acta. 2000;415:175–84.
Sievert SM, Wieringa EBA, Wirsen CO, Taylor CD. Growth and mechanism of filamentous-sulfur formation by Candidatus Arcobacter sulfidicus in opposing oxygen-sulfide gradients. Environ Microbiol. 2007;9:271–6.
Moussard H, Corre E, Cambon-Bonavita MA, Fouquet Y, Jeanthon C. Novel uncultured Epsilonproteobacteria dominate a filamentous sulphur mat from the 13 degrees N hydrothermal vent field, East Pacific Rise. FEMS Microbiol Ecol. 2006;58:449–63.
Heylen K, Vanparys B, Wittebolle L, Verstraete W, Boon N, De PV. Cultivation of denitrifying bacteria: optimization of isolation conditions and diversity study. Appl Environ Microbiol. 2006;72:2637–43.
Taillefert M, Bono AB, Luther GW. Reactivity of freshly formed Fe(III) in synthetic solutions and (pore)waters: voltammetric evidence of an aging process. Environ Sci Technol. 2000;34:2169–77.
Barco RA, Emerson D, Sylvan JB, Orcutt BN, Jacobson Meyers ME, Ramírez GA, et al. New insight into microbial iron oxidation as revealed by the proteomic profile of an obligate iron-oxidizing chemolithoautotroph. Appl Environ Microbiol. 2015;81:5927–37.
Garber AI, Nealson KH, Okamoto A, McAllister SM, Chan CS, Barco RA, et al. FeGenie: a comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Front Microbiol. 2020;11:37.
Canfield DE, Stewart FJ, Thamdrup B, De Brabandere L, Dalsgaard T, Delong EF, et al. A cryptic sulfur cycle in oxygen-minimum-zone waters off the Chilean Coast. Science. 2010;330:1375–8.
Ding J, Zhang Y, Wang H, Jian H, Leng H, Xiao X. Microbial community structure of deep-sea hydrothermal vents on the ultraslow spreading Southwest Indian Ridge. Front Microbiol. 2017;8:1012.
Leon-Zayas R, Peoples L, Biddle JF, Podell S, Novotny M, Cameron J, et al. The metabolic potential of the single cell genomes obtained from the Challenger Deep, Mariana Trench within the candidate superphylum Parcubacteria (OD1). Environ Microbiol. 2017;19:2769–84.
Liu X, Li M, Castelle CJ, Probst AJ, Zhou Z, Pan J, et al. Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome. 2018;6:102–102.
Ortiz-Alvarez R, Casamayor EO. High occurrence of Pacearchaeota and Woesearchaeota (Archaea superphylum DPANN) in the surface waters of oligotrophic high-altitude lakes. Environ Microbiol Rep. 2016;8:210–7.
Suominen S, Dombrowski N, Damste JSS, Villanueva L. A diverse uncultivated microbial community is responsible for organic matter degradation in the Black Sea sulphidic zone. Environ Microbiol. 2021. https://doi.org/10.1111/1462-2920.14902.
Castelle CJ, Banfield JF. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell. 2018;172:1181–97.
Dombrowski N, Lee JH, Williams TA, Offre P, Spang A. Genomic diversity, lifestyles and evolutionary origins of DPANN archaea. FEMS Microbiol Lett. 2019;366:fnz008.
Tian RM, Ning DL, He ZL, Zhang P, Spencer SJ, Gao SH, et al. Small and mighty: adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome. 2020;8:51.
Vigneron A, Cruaud P, Langlois V, Lovejoy C, Culley AI, Vincent WF. Ultra-small and abundant: Candidate phyla radiation bacteria are potential catalysts of carbon transformation in a thermokarst lake ecosystem. Limnol Oceanogr Lett. 2020;5:212–20.
Beam JP, Becraft ED, Brown JM, Schulz F, Jarett JK, Bezuidt O, et al. Ancestral absence of electron transport chains in Patescibacteria and DPANN. Front Microbiol. 2020;11:1848.
Luef B, Frischkorn KR, Wrighton KC, Holman HYN, Birarda G, Thomas BC, et al. Diverse uncultivated ultra-small bacterial cells in groundwater. Nat Commun. 2015;6:6372.
Wrighton KC, Castelle CJ, Wilkins MJ, Hug LA, Sharon I, Thomas BC, et al. Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer. ISME J. 2014;8:1452–63.
Konstantinidis KT, Tiedje JM. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc Natl Acad Sci USA. 2004;101:3160–5.
Moya A, Pereto J, Gil R, Latorre A. Learning how to live together: genomic insights into prokaryote-animal symbioses. Nat Rev Genet. 2008;9:218–29.
Moran NA, Plague GR. Genomic changes following host restriction in bacteria. Curr Opin Genet Dev. 2004;14:627–33.
Chaudhury P, Quax TEF, Albers SV. Versatile cell surface structures of archaea. Mol Microbiol. 2018;107:298–311.
Pohlschroder M, Esquivel RN. Archaeal type IV pili and their involvement in biofilm formation. Front Microbiol. 2015;6:190.
Aylward FO, Santoro AE. Heterotrophic Thaumarchaea with small genomes are widespread in the dark ocean. mSystems. 2020;5:e00415–20.
Reji L, Francis CA. Metagenome-assembled genomes reveal unique metabolic adaptations of a basal marine Thaumarchaeota lineage. ISME J. 2020;14:2105–15.
Santoro AE, Richter RA, Dupont CL. Planktonic marine Archaea. Annu Rev Mar Sci. 2019;11:131–58.
Rinke C, Rubino F, Messer LF, Youssef N, Parks DH, Chuvochina M, et al. A phylogenomic and ecological analysis of the globally abundant Marine Group II archaea (Ca. Poseidoniales ord. nov.). ISME J. 2019;13:663–75.
Pereira O, Hochart C, Auguet JC, Debroas D, Galand PE. Genomic ecology of Marine Group II, the most common marine planktonic Archaea across the surface ocean. Microbiol Open. 2019;8:e00852.
Martin-Cuadrado AB, Garcia-Heredia I, Moltó AG, López-Úbeda R, Kimes N, López-García P, et al. A new class of marine Euryarchaeota group II from the mediterranean deep chlorophyll maximum. ISME J. 2015;9:1619–34.
Martin-Cuadrado AB, Rodriguez-Valera F, Moreira D, Alba JC, Ivars-Martínez E, Henn MR, et al. Hindsight in the relative abundance, metabolic potential and genome dynamics of uncultivated marine archaea from comparative metagenomic analyses of bathypelagic plankton of different oceanic regions. ISME J. 2008;2:865–86.
Moreira D, Rodríguez-Valera F, López-García P. Analysis of a genome fragment of a deep-sea uncultivated Group II euryarchaeote containing 16S rDNA, a spectinomycin-like operon and several energy metabolism genes. Environ Microbiol. 2004;6:959–69.
Sforna MC, Philippot P, Somogyi A, Van Zuilen MA, Medjoubi K, Schoepp-Cothenet B, et al. Evidence for arsenic metabolism and cycling by microorganisms 2.7 billion years ago. Nat Geosci. 2014;7:811–5.
Meheust R, Burstein D, Castelle CJ, Banfield JF. The distinction of CPR bacteria from other bacteria based on protein family content. Nat Commun. 2019;10:4173.
Luther GW, Glazer BT, Ma S, Trouwborst RE, Moore TS, Metzger E, et al. Use of voltammetric solid-state (micro)electrodes for studying biogeochemical processes: laboratory measurements to real time measurements with an in situ electrochemical analyzer (ISEA). Mar Chem. 2008;108:221–35.
Brendel PJ, Luther GW. Development of a gold amalgam voltammetric microelectrode for the determination of dissolved Fe, Mn, O2, and S(-II) in porewaters of marine and freshwater sediments. Environ Sci Technol. 1995;29:751–61.
Arar EJ, Collins GB. Method 445.0 in vitro determination of chlorophyll a and pheophytin a in marine and freshwater algae by fluorescence. Washington, DC: U.S. Environmental Protection Agency; 1997.
Bran+Luebbe/Seal. Ammonia in water and seawater, in Method No G-171-96. 2005. Norderstedt, Germany.
Bran+Luebbe/Seal. Nitrate and nitrite in water and seawater; total nitrogen in persulfate digests, in Metho No G-172-96. 2010. Norderstedt, Germany.
Solórzano L, Sharp JH. Determination of total dissolved nitrogen in natural waters. Limnol Oceanogr. 1980;25:751–4.
Solórzano L, Sharp JH. Determination of total dissolved phosphorus and particulate phosphorus in natural waters. Limnol Oceanogr. 1980;25:754–8.
Dickson AG, Sabine CL, Christian JR. Guide to best practices for ocean CO2 measurements. PICES Special Publication 3. 2007.
Murphy J, Riley JP. A modified single solution method for the determination of phosphate in natural waters. Analy Chim Acta. 1962;27:31–6.
Stookey LL. Ferrozine—a new spectrophotometric reagent for iron. Anal Chem. 1970;42:779–81.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Padilla CC, Bertagnolli AD, Bristow LA, Sarode N, Glass JB, Thamdrup B, et al. Metagenomic binning recovers a transcriptionally active gammaproteobacterium linking methanotrophy to partial denitrification in an anoxic oxygen minimum zone. Front Mar Sci. 2017;4:23–23.
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–20.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550–550.
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217–e61217.
McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10:e1003531.
Willis AD, Martin BD. DivNet: estimating diversity in networked communities. bioRxiv. 2018. Available from https://www.biorxiv.org/content/10.1101/305045v1.
Wickham H. Elegant graphics for data analysis. New York: Springer-Verlag; 2016.
Oksanen J, Blanchet F, Friendly M, Kindt R, Legendre P, Mcglinn D, et al. vegan: Community Ecology package, in R package version 2.5-5. 2019. https://cran.r-project.org/package=vegan.
Nayfach S, Pollard KS. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 2015;16:51–51.
Nayfach S, Bradley PH, Wyman SK, Laurent TJ, Williams A, Eisen JA, et al. Automated and accurate estimation of gene family abundance from shotgun metagenomes. PLoS Comput Biol. 2015;11:e1004573.
Nayfach S, Pollard KS. Toward accurate and quantitative comparative metagenomics. Cell. 2016;166:1103–16.
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. MetaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–34.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6.
Mikheenko A, Saveliev V, Gurevich A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics. 2016;32:1088–90.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119–119.
James BT, Luczak BB, Girgis HZ. MeShClust: an intelligent tool for clustering DNA sequences. Nucleic Acids Res. 2018;46:e83.
Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2019;36:2251–2.
Boratyn GM, Thierry-Mieg J, Thierry-Mieg D, Busby B, Madden TL. Magic-BLAST, an accurate RNA-seq aligner for long and short reads. BMC Bioinformatics. 2019;20:405–405.
Dunivin TK, Yeh SY, Shade A. A global survey of arsenic-related genes in soil microbiomes. BMC Biol. 2019;17:45–45.
Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257.
Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. PeerJ Comput Sci. 2017;3:e104.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Eren A. M., Kiefel E., Shaiber A., et al. 2021. Community-led, integrated, reproducible multi-omics with anvi’o. Nature Microbiology volume 6, pages 3–6. https://doi.org/10.1038/s41564-020-00834-3.
Olm MR, Brown CT, Brooks B, Banfield JF. DRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–8.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–7.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Letunic I, Bork P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019;47:p. W256–9.
Acknowledgements
We thank the captains of the R/V William R. Mote and R/V Eugenie Clark for their support and professionalism, and staff at Mote Marine Lab for their work in sample collection and processing. We are also indebted to all of the volunteer technical divers who made this mission possible. We are grateful to Dr. Navid Constantinou for his help modeling the wind speeds required for the turnover of the water column. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Funding
This work was funded by the National Oceanic and Atmospheric Administration Office of Exploration and Research [7NA18OAR0110291].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: one reference was added and the subsequent references were re-numbered accordingly.
Rights and permissions
About this article

Cite this article
Patin, N.V., Dietrich, Z.A., Stancil, A. et al. Gulf of Mexico blue hole harbors high levels of novel microbial lineages. ISME J 15, 2206–2232 (2021). https://doi.org/10.1038/s41396-021-00917-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-00917-x
This article is cited by
-
DNA metabarcoding reveals ecological patterns and driving mechanisms of archaeal, bacterial, and eukaryotic communities in sediments of the Sansha Yongle Blue Hole
Scientific Reports (2024)
-
Trait biases in microbial reference genomes
Scientific Data (2023)
-
Phylogenetically and metabolically diverse autotrophs in the world’s deepest blue hole
ISME Communications (2023)
-
MeShClust v3.0: high-quality clustering of DNA sequences using the mean shift algorithm and alignment-free identity scores
BMC Genomics (2022)
-
Genomic evidence of functional diversity in DPANN archaea, from oxic species to anoxic vampiristic consortia
ISME Communications (2022)
