
Abstract
Dominant coral-associated Endozoicomonas bacteria species are hypothesized to play a role in the coral sulfur cycle by metabolizing dimethylsulfoniopropionate (DMSP) into dimethylsulfide (DMS); however, no sequenced genome to date harbors genes for this process. In this study, we assembled high-quality (>95% complete) draft genomes of strains of the recently added species Endozoicomonas acroporae (Acr-14T, Acr-1, and Acr-5) isolated from the coral Acropora sp. and performed a comparative genomic analysis on the genus Endozoicomonas. We identified DMSP CoA-transferase/lyase—a dddD gene homolog in all sequenced genomes of E. acroporae strains—and functionally characterized bacteria capable of metabolizing DMSP into DMS via the DddD cleavage pathway using RT-qPCR and gas chromatography (GC). Furthermore, we demonstrated that E. acroporae strains can use DMSP as a carbon source and have genes arranged in an operon-like manner to link DMSP metabolism to the central carbon cycle. This study confirms the role of Endozoicomonas in the coral sulfur cycle.
Similar content being viewed by others


Introduction
Coral reefs are one of the most diverse ecosystems on Earth, with over 800 different coral species known to date. Of these, corals in the genus Acropora are some of the most abundant reef-building corals across the Indo-Pacific region [1]. They are also a significant producer of dimethylsulfoniopropionate (DMSP) [2, 3], an organosulfur compound abundant in animals that harbor symbiotic algae such as scleractinian corals and giant clams [2]. DMSP is present in coral tissues, mucus, and endosymbiotic dinoflagellates (Symbiodiniaceae) [4, 5]. In marine algae, DMSP protects against various stresses, such as oxidative and osmotic stress [6]. Moreover, DMSP also acts as an attractant for specific bacterial groups that have been reported to be part of coral-associated bacterial communities and underpin coral health [7].
Once released from marine planktonic dinoflagellates, most DMSP emanates to surrounding water where it is readily available for microbial catabolic conversion as a source of reduced carbon and sulfur [8, 9]. DMSP is a central molecule in the marine sulfur cycle, and is degraded by bacteria via two pathways: a cleavage pathway and a demethylation pathway [9, 10]. The majority (~75%) of the DMSP is metabolized via the demethylation/demethiolation pathway, producing methylmercaptopropionate [11,12,13]. The cleavage pathway, which accounts for the remaining ~25% of DMSP, produces dimethylsulfide (DMS)—a climate-active gas and acrylic acid [13,14,15]. Several DMSP lyases have been identified to date—including DddP, DddY, DddQ, DddW, DddK, and DddL—in organisms like Rhodobacterales, Roseobacters, Sulfitobacter, and Pseudomonadales [9, 13, 16]. Another gene, dddD, was identified in Marinomonas sp., which cleaves DMSP to produce DMS and 3-hydroxypropionate (3-HP)—not acrylate, which is produced by other genes [13]. Moreover, DMSP metabolism yielding DMS and acrylate was also recently identified in coccolithophore algae [17]. The DMS produced after DMSP cleavage is then released into the surrounding water [12].
Coral-associated bacterial communities are highly diverse and dynamic [18,19,20,21]. These bacterial communities can be found in various niches associated with corals like coral mucus [22], spaces in the skeleton [23,24,25], and coral tissues [26,27,28]. Raina et al. [8] confirmed that coral-associated bacteria have the potential to metabolize organic sulfur compounds present in the coral tissues. They inferred that the majority of the DMSP-degrading bacteria belong to class Gammaproteobacteria, including Alteromonas-, Arhodomonas-, Idiomarina-, Pseudomonas-, and Spongiobacter (Endozoicomonas)-related organisms. Of these, Arhodomonas-, Pseudomonas-, and Roseobacter-related species harbor DMSP-metabolizing genes.
Of all the organisms in the tremendously diverse coral holobionts, genomes of Endozoicomonas species have been widely studied for their functional and ecological roles [29, 30]. Endozoicomonas species were found to be abundant in the coral holobionts across the globe [31] and are hypothesized to be potential indicators of coral health [32]—they have high abundance in healthy corals and a relatively low abundance in diseased or stressed corals [33,34,35]. Furthermore, nearly complete and draft genomes of Endozoicomonas species isolated from corals and other marine invertebrates [29, 36, 37] have recently been assembled. First, a near-complete genome of the Endozoicomonas isolate Endozoicomonas montiporae CL-33T from the encrusting pore coral Montiporae aequituberculata has provided insights into how this bacterium interacts with its host both outside and inside the coral cell with the help of potential effector proteins [30]. A recent comparative genomics analysis also identified a high number of Type III secretion system (T3SS)-related genes and suggested that (1) most gene ontology terms are associated with the generic transport of molecules and (2) genomes of Endozoicomonas species show high plasticity [30]. Endozoicomonas species have been hypothesized to play a role in the coral sulfur cycle by effectively metabolizing DMSP into DMS [38, 39]. However, no study has confirmed the genus’ role and no sequenced genome has been found to harbor genes related to this process [30]. Hence, the role of this coral endosymbiont in the coral sulfur cycle remains elusive.
In this study, we assembled high-quality draft genomes of newly added Endozoicomonas acroporae strains and profiled their abundances in different coral species in the Indo-pacific region. We identified and functionally characterized a DMSP Co-A transferase/lyase, encoded by a dddD homolog, in all sequenced E. acroporae genomes. This gene was not present in any other Endozoicomonas genomes used in this study. Furthermore, we provide conclusive evidence that E. acroporae has a role in the coral sulfur cycle by effectively metabolizing DMSP into DMS and can use DMSP as a carbon source for growth and survival.
Materials and methods
Culturing and whole-genome sequencing of E. acroporae Acr-1, Acr-5, and Acr-14T
Strains of E. acroporae Acr-1, Acr-5, and Acr-14T were isolated from the coral Acropora sp. from Kenting, off the southern coast of Taiwan, and cultured using a method described previously [40]. Genomic DNA was isolated using the cetyltrimethylammonium bromide method [41]; the quality of the isolated DNA was assessed using NanoDrop 1000 (Thermo Scientific, USA). High-quality DNA was sent to the core sequencing facility at Biodiversity Research Center, Academia Sinica, Taiwan for whole-genome sequencing on the Illumina Miseq platform, with a TRUSeq DNA paired-end library generated to achieve an insert size of 500 bp.
Genome assembly and annotation
Reads obtained from Illumina MiSeq were quality-filtered and trimmed (Phred score ≥ 30) using NGS QC toolkit v2.3.3 [42]. Quality-filtered and trimmed reads were de novo assembled using CLC Genomics Workbench version 1.10.1 (Qiagen) with a bubble size of 40 and automatic word size enabled. Minimum contig length was set to ≥500 bp (no scaffolding was performed). Assembled genomes were quality checked for completeness, contamination, and heterogeneity using CheckM [43]. Other Endozoicomonas species genomes were downloaded from the NCBI genomes database (last accessed January 2018). Gene prediction on all genomes used in this study was performed with Prodigal [44] wrapped in Prokka [45] with default settings to make GFF files compatible for down-stream pan-genome analysis with Roary [46]. Furthermore, genome annotations and up-to-date higher level-functional categories at subsystem levels were obtained from rapid annotation using the subsystem technology (RAST) server [47] with predicted gene calls preserved (February 2018).
Identification of genomic characteristics
In silico genome–genome distances (GGDs) among the genomes of genus Endozoicomonas were calculated using the GGD calculator from the DSMZ server [48], and the average nucleotide identity (ANI) (https://enve-omics.ce.gatech.edu/ani/) calculator was used to calculate the ANI values. Furthermore, amino acid identity (AAI) among the genomes was calculated with CompareM (https://github.com/dparks1134/CompareM). All plots were generated with R (R Core Team, 2016) [49] using the ggplot2 [50] package. Clustered regularly interspaced short palindromic repeat (CRISPR) structures in all genomes were identified using Prokka; prophages and phages within the genomes were identified using the PHAge Search Tool (PHAST) [51], which classified the phages as intact, incomplete, or questionable. T3SS proteins were identified by EffectiveT3 [52] using EffectiveDB with the animal classification module and selective (0.9999) restriction value method enabled. Type IV secretion system (T4SS) proteins were predicted using the web-based T4SEpre (beta) (https://effectors.csb.univie.ac.at/effective) with a minimum score of 0.05. Insertion sequence (IS) elements in the genomes of E. acroporae strains and E. montiporae were identified using the ISfinder database (https://www.is.biotoul.fr) with blastn and an e-value threshold of 1e−5. Furthermore, proteins with eukaryotic repeat domains (Ankyrin repeats (ARPs) and WD40 domain) were identified in all E. acroporae strains using the web-based Batch Conserved Domain-Search (CD-search) tool [53] with the CDD-52910 PSSMs database and an e-value threshold of 1e-5 (October 2019).
16S rRNA gene phylogenetic analysis
To determine robust phylogenetic relationships within the genus Endozoicomonas, all available 16S rRNA sequences (68 total) were downloaded from the NCBI taxonomy database, for which host information was available to understand the distribution of Endozoicomonas species in different marine invertebrates and identify the position of E. acroporae strains within the genus Endozoicomonas. Sequences were aligned using cmalign from the infernal package [54], which performs a covariance model (CM)-guided SSU rRNA alignment. The CM model for domain bacteria was obtained from the rfam database [55]. A maximum likelihood (ML) phylogeny was computed using IQ-TREE v1.6.11 [56] with the TIM3+F+I+G4 model (Bayesian Information Criterion values, Supplementary Table S1) and 1000 bootstraps. ModelFinder [57] was invoked to automatically select the best model in IQ-TREE [56]. A consensus tree was visualized in iTOL v4 [58].
E. acroporae distribution and abundance in different coral species from the Indo-Pacific region
We analyzed microbial community data publically available from three different studies—1) our laboratory’s previous study [34]; (2) a study of coral-associated bacterial communities in the Red Sea [59]; and (3) coral-associated bacterial community from reefs in the east and west coast of Australia, including Ningaloo Reef, Lizard Island, reefs from the northern sector of the Great Barrier Reef, and Lorde Howe Island [60]—to profile the abundance of E. acroporae strains in different coral species from Penghu Archipelago, Taiwan; the Red Sea, Saudi Arabia; and east and west Australia. All three studies used the same operational taxonomic unit (OTU) clustering threshold of 97% and the Greengenes database (v99) [61] for taxonomic assignment. OTU abundance profiles and their representative sequences were downloaded from supplementary materials in Shiu et al. [34], Ziegler et al. [59], and Pollock et al. [60]. We performed similarity searches on all OTU sequences from the three studies against the in-house blast [62] database of all available Endozoicomonas 16S rRNA gene sequences with standalone blastn [62] and profiled the relative abundance of E. acroporae strains at the three locations with e-value < 1e–5 and identity threshold ≥ 99%. A map was drawn using the Generic Mapping Tool [63].
Comparative genomics: pan-genome analysis and core genome phylogeny
Pan-genome analysis was performed with Roary [46] using GFF files of all the genomes, including Parendozoicomonas haliclonae S-B4-1UT (outgroup), obtained from Prokka [45]. Core genome identification and alignment were performed on all genomes using the parameters -i 80, -e, –n –cd 90. A Bayesian inference phylogenetic tree was constructed with MrBayes [64] run for one-million generations using the GTR+F+I+G4 (Supplementary Table S1) model, pruning the initial 25% of trees. The model was selected automatically by ModelFinder [57] wrapped in IQ-TREE [56]. An alternate ML tree was also constructed with 1000 bootstraps using IQ-TREE [56]. Trees were visualized with iTOL v4 [58].
Identification of stress response genes, dddD CoA-transferase/lyase, and DMSP metabolism-related operon
The Stress Response Subsystem was analyzed for the distribution of different categories of stress-responsive genes present in the genomes of Endozoicomonas. The Sulfur Metabolism Subsystem in the RAST analysis annotated a dddD gene capable of metabolizing DMSP into DMS within the “sulfur metabolism—no subcategory”. Furthermore, the presence of domains in the dddD gene and DMSP metabolism-related operon genes was determined with a web-based CD-Search [53], NCBI, with default parameters. In addition, we performed a similarity search to annotate DMSP demethylation pathway (sulfur metabolism) genes using the web-based blastkoala [65]. Other DMSP lyases were similarity searched against full-length seed sequences obtained from the pfam database (PF16867) [66] using blastp [62] for all Endozoicomonas genomes with e-value < 1e–5 and percent identity ≥ 50%. We also performed a phylogenetic analysis on proteins involved in the DMSP metabolism-related operon (see Supplementary Data: Materials and Methods, Supplementary Fig. S8 and Supplementary Table S9).
DMSP degradation by strain Acr-14T
Endozoicomonas acroporae Acr-14T was cultivated on Modified Marine Broth Version 4 (MMBV4) medium [29] with several modifications (Supplementary Table S2) at 25 °C for 48 h. Carbon sources, ~3 mM maltose, and 0.5 mM DMSP were added to the enrichment culture of strain Acr-14T and kept at 25 °C for 24 h (OD600 of ∼1.0 after incubation). The enrichment culture was centrifuged at 2000 × g for 10 min, and the supernatant was discarded. The enrichment culture pellet was washed with 1 ml fresh minimal medium (Supplementary Table S3) twice to remove the MMBV4 medium containing DMSP. Two experimental groups were made for the test: (1) 1 ml of washed bacteria was resuspended in a 40 ml minimal medium containing 0.2% casamino acid and 1 mM DMSP; (2) 1 ml of washed bacteria was resuspended in a 40 ml minimal medium containing 0.2% casamino acid without DMSP. The cultures were sampled at 0, 16, 24, and 48 h for RT-qPCR.
Gene expression of dddD by RT-qPCR
Total RNA was extracted using a TRI-reagent solution (Invitrogen, Carlsbad, CA, USA). The cultured samples (1–2 ml) were centrifuged for 1 min at 12000 × g and 4 °C following the manufacturer’s guidelines. The RNA pellet was air-dried and resuspended in nuclease-free water. Residual DNA was removed using a TURBO DNA-free Kit (Invitrogen). The RNA quality was determined using NanoDrop ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies). RNA integrity was assessed by electrophoresis on a 1% agarose-guanidine thiocyanate gel. Complementary DNA (cDNA) was synthesized from purified RNA using the SuperScript IV First-Strand Synthesis System for RT-qPCR (Invitrogen) following the manufacturer’s guidelines. For cDNA, RT and non-RT samples were screened for residual DNA contamination using the hypervariable V6V8 region of the bacterial 16S rRNA gene (U968F and U1391R).
A primer pair was made for the dddD gene based on the genome data of strain Acr-14T for Quantitative PCR using DNASTAR Lasergene [67] and Primer-BLAST tool on BLAST search (NCBI): dddD-F (5′-ACCGCATCGCACCACTCAGG-3′) and dddD-R (5′-GGCCCCGGTTGTTTCATCAT-3′). It is important to note that the dddD gene sequence is 99.24% identical among E. acroporae strains. The endogenous control was performed with the rpoD gene using rpoD-F (5′-AAGGCGGTGGACAAGTTCG-3′) and rpoD-R (5′-GATGGTGCGGGCCTGGTCTG-3′). RT-qPCR assays were carried out using the Applied Biosystems QuantStudioTM 5 Real-Time PCR System. The standard cycling program consisted of cycles of UDG activation at 50 °C for 2 min, initial denaturation activation at 95 °C for 2 min and 40 cycles of denaturation at 95 °C for 10 s and annealing at 60 °C for 40 s using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, USA). A dissociation step was performed to confirm the specificity of the product and avoid the production of primer dimers. For all reactions, 10 ng of template DNA was added to a reaction of 10 μl. The 10 μl reactions contained 5 μl of PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, USA), 3.4 μl sterilized nuclease-free water, 0.3 μl each of the forward and reverse primers (final conc. 0.3 µM), and 1 μl of DNA template. Each sample was performed in duplicate. The relative quantification of the expression ratio was calculated by the comparative 2−ΔΔCт method. Differences between treatments were statistically tested using the t-test.
Quantification of released DMS
To assay the functional activity of the dddD protein in Endozoicomonas acroporae strain Acr-14T, cultures were first grown in MMB medium with 0.5 mM DMSP for 24 h. After that, 3 ml culture was collected, spun down, and washed twice with minimal medium, and then resuspended with 1 ml minimal medium. This 1 ml culture was then injected into sterile 60 ml vials sealed with a rubber stoppers containing designated medium Treatment (a): 20 ml minimal medium with 0.2% casamino acid and 1 mM DMSP; Treatment (c): 20 ml minimal medium with 0.2% casamino acid, acting as the negative control; and a culture-free Treatment (b): 20 ml minimal medium with 0.2% casamino acid and 1 mM DMSP, acting as the control. Vials were then incubated at 25 °C, 200 rpm, in the dark. After incubation for 0, 24, and 48 h, 1 ml of the headspace air sample was collected and injected into a gas chromatograph (Shimadzu, GC-14B) fitted with a flame ionization detector (150 °C) and column (SGE, 60 m × 0.53 mm ID BP624 × 3.0 µm) to determine the DMS concentration.
E. acroporae grown with DMSP as the carbon source
E. acroporae Acr-1, Acr-5, and Acr-14T and E. montiporae CL-33T strains were cultivated on MMB medium (along with 0.1% maltose for E. montiporae CL-33T) at 25 °C for 72 h. The carbon source, 0.1 mM DMSP, was added to the enrichment culture of E. acroporae strains and kept at 25 °C for 24 h (OD600 of ~0.3 after incubation). 0.1 mM DMSP and 3 mM maltose were added to the enrichment culture of E. montiporae and kept at 25 °C for 24 h (OD600 of ~0.8 after incubation). The enrichment culture was centrifuged at 2000 × g for 10 min, and the supernatant was discarded. The enrichment culture pellet was washed twice by 1 ml fresh minimal medium. After resuspending with minimal medium, the enrichment cultures were added to the treatments and adjusted to OD600 of 0.06. All the treatments were kept at 25 °C, 200 rpm, and the OD600 of treatment was recorded after incubating for 24, 48, and 72 h.
E. acroporae Acr-14T, Acr-1, and Acr-5 were cultivated on a minimal medium with 0.2% casamino acid and 0.1, 1, or 3 mM DMSP to test their ability to use DMSP as a carbon source (three replicates each). Treatments with 0.1, 1, or 3 mM DMSP with 0.2% casamino acid were also used to test whether E. montiporae CL-33T can use DMSP. In these treatments, E. montiporae CL-33T and E. acroporae were also grown on 3 mM maltose.
Results
16S rRNA gene phylogeny and E. acroporae abundance profiling in different coral species from the Indo-Pacific region
All three strains of E. acroporae had only one copy of the 16S rRNA gene, compared with seven copies in the E. montiporae CL-33T genome (Supplementary Table S4) [26]. 16S rRNA gene-based phylogeny clustered sequences reflect the host phylogeny in most cases (Supplementary Fig. S1). We identified separate clades for E. montiporae and E. acroporae (Supplementary Fig. S1). The closest relative of E. acroporae strains was a new species within the same clade, Endozoicomonas coralli, whose genome has not been sequenced yet. It is worth noting that the E. acroporae and E. coralli 16S rRNA gene share high (98.69%) sequence identity.
Relative abundance profiles of E. acroporae strains were determined (at >99% identity) to obtain an overview of the distribution of this new species in different coral species from three distinct geographical regions (Fig. 1). We determined that E. acroporae strains were abundant in corals from the Red Sea, Saudi Arabia (~10–22% of coral bacterial communities); Penghu Archipelago, Taiwan (~20%); and coral reefs in Eastern (~4–22%) and Western (~2–20%) Australia. The presence and relative abundances of E. acroporae strains provide evidence that this species is abundant in coral holobionts spread across distinct geographic locations, similar to the distribution of other Endozoicomonas species.

The green color in pie charts represents relative abundance of E. acroporae strains (identified at ≥99% identity and e-value < 1e–5) in coral microbial communities as assessed by 16S rRNA amplicon sequencing.
Genome assembly features
Whole-genome sequencing of E. acroporae isolates produced genome assemblies with 98.56, 98.99, and 95.25% completeness and contamination levels of 1.39, 2.25, and 2.25% in E. acroporae Acr-14T, E. acroporae Acr-5, and E. acroporae Acr-1, respectively (Supplementary Fig. S2). Contigs were checked for similarity against all sequenced Symbiodiniaceae and coral Acropora genomes (obtained from www.reefgenomics.org) for contamination, but no significant hit (e-value < 1e−5, percent identity ≥ 90, alignment coverage ≥ 50%) was obtained. Thus, our genomes are currently high-quality drafts based on MIMAG standards of completeness and contamination [68]. The E. acroporae Acr-1 genome was assembled into 299 contigs with a total of 6.024 Mb, and we predicted 77 tRNAs, 5,144 genes (avg. length: 1002 bp), a gene density of 839 genes per Mb, and 5,059 coding sequences (CDS). E. acroporae Acr-5 genome was 6.034 Mb with 295 contigs coding for 80 tRNAs, 5190 genes (avg. length: 904 bp), a gene density of 898 genes per Mb, and 5,101 CDS. The assembled genome of E. acroporae Acr-14T was 6.048 Mb with 309 contigs coding for 79 tRNAs, 5104 genes (avg. length: 1014 bp) [37], a gene density of 829 genes per Mb, and 5018 CDS. A list of all the genomes used in this study—along with their sizes (4.049–6.69 Mb) and hosts (coral, sea slug, comb pen shell, and sponge)—is shown in Supplementary Table S5. All E. acroporae genomes (Acr-1, Acr-5, and Acr-14T) had similar genome sizes (6.024–6.048 Mb) and gas chromatography (GC) contents (49.2, 49.3, and 49.3%, respectively).
Genomic characteristics of genus Endozoicomonas
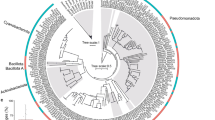
Endozoicomonas species have large genomes ranging from 4.049 Mb (Endozoicomonas sp. AB1) to 6.69 Mb (E. elysicola DSM22380). The average genome has ~5100 protein-coding genes and a GC content of 47.6%. The average GGD, ANI, and AAI values among the Endozoicomonas species were ~46, ~73, and ~75%, respectively, which are at the lower end of the 62–100% range of interspecies variation within a genus [69], indicating high genomic diversity (Fig. 2a–c).

Heatmaps based on (a) average amino acid identity (AAI), (b) average nucleotide identity (ANI), and (c) genome–genome distance (GGD) for all the sequenced genomes from genus Endozoicomonas. (d) Core genome (313)-based Bayesian inference phylogenetic tree generated from MrBayes run for one-million generations using the GTR+F+G4 model; posterior probabilities are mentioned at the branch-points. Parendozoicomonas haliclonae S-B4-1UT was used as the outgroup.
A diverse array of IS elements were identified in genomes of E. acroporae and E. montiporae. For example, of the total 44 IS elements identified, 22 were only present in E. acroporae genomes, while 18 were only present in E. montiporae genomes. Relatively few IS elements (ISEc46, ISEcret1, ISEc42, and ISPa18) were present in both species. Each genome also harbored some unique IS elements (Supplementary Fig. S3).
Three E. acroporae genomes (Acr-14T, Acr-5, and Acr-1) encoded more T3SS effector proteins (523, 499, and 499, respectively) —than other Endozoicomonas species (Supplementary Table S6). Moreover, all Endozoicomonas species also harbored a large number of T4SS effector proteins within the range of 106 (Endozoicomonas sp. AB1) to 226 (E. ascidiicola KASP37) (Supplementary Table S6). Interestingly E. acroporae genomes also had a high count of proteins containing ARPs and WD40 domains, though most of these were annotated as hypothetical proteins (Supplementary Table S7). We further identified different phage insertions in all Endozoicomonas genomes (Supplementary Table S8) and different CRISPR counts in the genomes of E. acroporae Acr-14T, Acr-5, and Acr-1 (Supplementary Table S4).
Core genome phylogeny
Core gene (n = 313)-based phylogenetic analysis reflected the host phylogeny in some cases, and also hinted towards a high genomic divergence within the species of genus Endozoicomonas. Endozoicomonas genomes isolated from the same host clustered very tightly together, e.g. E. acroporae and E. montiporae with the coral host, and E. ascidiicola and E. arenosclerae with a sponge host (Fig. 2d and Supplementary Fig. S4). Moreover, E. numazuensis and E. arenosclerae genomes shared a branch, and were both isolated from the sponge. E. acroporae Acr-1 and Acr-5 clustered tightly while sharing a branch with E. acroporae Acr-14T, which confirms that Acr-1 and Acr-5 are closer to each other (ANI: 100%) than to Acr-14T (ANI: 98.10% and 99.02%, respectively).
Analyzing RAST subsystems
RAST subsystems—carbohydrates, protein metabolism, and amino acids and derivatives—had the highest number of genes, representing an average of 11.69%, 10.85%, and 13.29% of all the annotated genes, respectively (Supplementary Fig. S5), in all Endozoicomonas genomes. We focused our analysis on two other important subsystems: stress response and sulfur metabolism. The highest number of genes were annotated for oxidative stress (39.16% of stress genes) in the stress response subsystem, followed by unclassified (16%) and detoxification stresses (12.53%) (Supplementary Fig. S6). Interestingly, we identified a dddD gene homolog involved in sulfur metabolism in the E. acroporae Acr-14T, Acr-5, and Acr-1 genomes. This gene was not present in other Endozoicomonas genomes (Supplementary Table S5). Furthermore, no other genes involved in DMSP metabolic pathways were identified in any Endozoicomonas genomes. The dddD gene present in E. acroporae genomes has two identical CaiB domains (positions: 1–416, 439–821) belonging to the coenzyme-A transferase superfamily (Supplementary Fig. S7).
DMSP metabolism operon and links to the central carbon cycle
We identified homologs of the DMSP transcriptional regulator (LysR family), a sulfur transporter belonging to the betaine/carnitine/choline transporter family hypothesized to transport DMSP and genes involved in producing acetate from DMSP (Fig. 3a). We discerned a complete pathway with genes arranged in a consecutive manner to form an operon that can yield acetate from DMSP metabolism via three-step enzymatic reactions mediated by DddD, 3-HP dehydrogenase (EC 1.1.1.59) and malonate-semialdehyde dehydrogenase (EC 1.2.1.18) (Fig. 3b) in all the E. acroporae genomes, with significant gene and protein identity for all the three genes (>97%) and proteins (>97%) (Fig. 3c). Identification of the DMSP cleavage pathway leading to central carbon metabolism suggested that E. acroporae species can use DMSP as a carbon source. Furthermore, DMSP metabolism-related proteins of E. acroporae formed distinct clades in phylogenetic analysis (Supplementary Fig. S8) suggesting presence of high diversity in these genes across different bacteria.

(a) DMSP metabolism operon, with genes arranged based on the layout of the operon within all E. acroporae strains, with a similar arrangement in Marinomonas sp. MWYL1 and different putative DMSP transporters (BetS and BetL). (b) The metabolic pathway represented by the genes, linking DMSP metabolism to the central carbon cycle, producing acetate from DMSP when DMS is released in E. acroporae. (c) Heatmaps based on gene/protein identity for the three enzymes (from all E. acroporae strains) used in the metabolic cycle: DddD, 3-hydroxypropionate dehydrogenase (EC 1.1.1.59), and malonate-semialdehyde dehydrogenase (EC 1.2.1.18).
dddD gene activity and DMS quantification
RT-qPCR was used to examine the expression of the dddD gene in E. acroporae Acr-14T. dddD gene expression increased with sampling time in the condition with 1 mM DMSP (Fig. 4a). dddD gene expression in this condition was 42.77, 56.52, and 91.37 times higher than samples without DMSP at 16, 24, and 48 h, respectively (t-test, p < 0.05), confirming that dddD was active in E. acroporae. After confirming the dddD gene expression, we quantified the amount of DMS released by E. acroporae when incubated in a DMSP-rich environment with a time series detection (0, 24, and 48 h). The DMS signal could only be detected in Treatment (a), and there was no DMS signal in the control groups (Treatments (b) and (c)) (Fig. 4b). A temporal increase in released DMS concentration in Treatment (a) confirmed the ability of E. acroporae to metabolize DMSP into DMS.

(a) RT-qPCR of the dddD gene from E. acroporae Acr-14T to measure temporal expression change in the presence of 1 mM DMSP (Student’s t test: *p < 0.05, ***p < 0.01). (b) GC-based quantification of DMS released from E. acroporae Acr-14T cultures in the presence of DMSP (1 mM). (c) Growth on 0.1 mM DMSP; E. acroporae strains can use DMSP as its carbon source, whereas E. montiporae CL-33T did not show signs of using DMSP as its carbon source.
DMSP as the carbon source
E. acroporae strains grew on 0.1 and 1 mM DMSP, but showed no signs of growth on 3 mM DMSP (Fig. 4c and Supplementary Fig. S9), suggesting that it is able to use only a certain range of DMSP as its carbon source. In 0.1 mM DMSP, Acr-14T, Acr-1, and Acr-5 had a mean OD600 value of ~0.67, ~0.45, and ~0.31, respectively, after 72-h of incubation (Fig. 4c). In contrast, E. montiporae CL-33T cells appeared to form aggregates in all DMSP concentrations after 24 h (Supplementary Fig. S10) and were visualized with viability staining (details in Supplementary Data: Material and Methods). These result showed that E. montiporae CL-33T formed aggregates, making OD600 measurements unreliable and suggesting that E. montiporae CL-33T can use maltose, but not DMSP, as its carbon source (Supplementary Fig. S10). E. acroporae Acr-14T also used maltose for growth (Supplementary Fig. S10).
Discussion
In this study, we performed genomic (and comparative genomic) analyses, functional assays, and abundance profiling of strains from a new species E. acroporae in the marine bacterial genus Endozoicomonas. We assembled high-quality draft genomes of E. acroporae strains and identified the dddD gene involved in DMSP metabolism in all E. acroporae strains. Furthermore, we characterized the functional activity of the gene and the ability of E. acroporae strains to grow on DMSP as a carbon source. This is the first study to establish genomic and functional evidence that Endozoicomonas species play a role in the coral sulfur cycle.
DMSP breakdown by E. acroporae and its utilization for growth and survival
Endozoicomonas species have been suggested to play a role in the coral sulfur cycle via DMSP metabolism [8, 70]. However, no gene related to DMSP metabolism was found in previously sequenced Endozoicomonas genomes (Supplementary Table S5) [30]. Furthermore, using enrichment cultures from coral mucus, tissue, and skeleton, certain bacterial species have been reported to degrade DMSP, including Spongiobacter (Endozoicomonas) nickelotolerans, but no gene related to this function was identified, and it was hypothesized to use a different pathway for DMSP degradation [8]. In this study, we investigated the sulfur metabolism pathway in high-quality draft genomes (Supplementary Fig. S2) of E. acroporae strains and identified a dddD gene that codes for a bi-functional enzyme with Co-A transferase/lyase activity that cleaves DMSP into volatile DMS [9, 71]. Among the eight identified DMSP lyases in different bacterial species, only dddD produces 3-HP and DMS, whereas others form acrylate and DMS [72]. 3-HP is not the usual product of other DMSP lyases, which directly produce acrylate after the cleavage due to their unusual organization of class III Co-A transferase domains [73]. Co-A transferase domains present in the DddD protein are similar to CaiB (Supplementary Fig. S7), which is a homodimer capable of adding a Co-A to carnitine [13], a biochemical analysis showed that DddD is unlikely to be a carnitine CoA-transferase [71]. RT-qPCR-based temporal analysis of E. acroporae strains in a DMSP-rich environment showed that dddD activity increased significantly (Fig. 4a); moreover, the GC analysis also showed that high amounts of DMS were released, which is evidence that dddD is functionally active (Fig. 4b).
Genomic analysis also identified genes arranged in consecutive order to form an operon with DMSP transport, metabolism, and transcriptional regulator genes (Fig. 3a) in all the strains of E. acroporae. dddT is a transporter that imports molecules like DMSP [13, 74], and thus has been suggested to import DMSP. dddR is a transcription regulator able to activate the expression of the dddD gene in response to DMSP [13]. The functions of dddB and dddC have not been characterized biochemically, but they are hypothesized to be involved in oxido-reductive functions based on their sequence similarities to other oxidoreductases, which modify DMSP either before or after the addition of the acyl CoA moiety by dddD [13]. Interestingly, the arrangement of genes around dddD is of a ‘pick ‘n’ mix’ form where, in some cases, corresponding genes have very similar sequence identity and the same function, but in others, overall corresponding gene sequences are very different but may have the same function and mechanism [74]. This arrangement is present in several DddD+ bacteria, like Marinomonas sp. MWYL1, Sagittus sp. E37, and B. cepacia AMMD [74]; although a similar gene arrangement was observed between Marinomonas sp. MWYL1 and E. acroporae strains (Fig. 3a).
The ability of E. acroporae strains to use DMSP as their carbon source (Fig. 4c, Supplementary Fig. S9)—unlike E. montiporae, which is unable to utilize DMSP, even at higher concentrations (Supplementary Fig. S10)—is further evidence that E. acroporae strains can metabolize DMSP and use it for growth and survival. This finding indicates that DMSP metabolism is linked to the central carbon cycle in these species, as we predicted in the genomic analysis (Fig. 3b). Furthermore, it also confirmed the hypothesis that marine bacteria that harbor dddD have the ability to use DMSP as the carbon source [75].
Genome architecture of Endozoicomonas species and putative relationship with the host
Bacterial genome size can reflect its evolutionary dependency on the host when they are engaged in obligate symbiosis [76]. A striking feature of obligate symbionts is that they have smaller genome sizes than facultative symbionts, and lower gene densities than the bacterial average of 85–90% [75, 76]. However, genomes of Endozoicomonas species are relatively large, ranging from 4.09 to 6.69 Mb; this includes E. acroporae strains, which have an average genome length of ~6.03 Mb, suggesting that they might have a free-living stage. The possibility that Endozoicomonas species might have a free-living stage is further supported by their large number of coding genes (avg. ~5000 proteins) and average gene density of ~855 genes per Mb (Supplementary Table S4) [77, 78]. These features suggest that genome streamlining [79], a notable feature of symbiotic bacteria, is not prominent in genus Endozoicomonas. Moreover, a diverse array of phages (Supplementary Table S8) and IS elements (Supplementary Fig. S3) provides clues about the infection and colonization histories of different marine hosts, aided by frequent divergence events.
A recent study by Ding et al. [29] reported a high proportion of repeat sequences and a variety of IS elements in the genome of E. montiporae, which may help the bacterium adapt to the host and also identified the N-deglycosylation enzyme that helps penetrate the mucus layer of the host. These findings suggest that Endozoicomonas can transition between different symbiotic lifestyles. We identified a high number of eukaryotic repeat proteins in E. acroporae genomes (Supplementary Table S7); our results are similar to a recent study on Porites lutea microbial symbionts, which identified >50 copies of eukaryotic repeat proteins in a metagenome-derived Endozoicomonas genome [80], suggesting a symbiotic relationship. Furthermore, we also identified a high count of secretory (T3SS and T4SS) proteins in the genomes of E. acroporae strains (Supplementary Table S6) that may help transport organic macromolecules and effector proteins between the host and symbiont. Both T3SS and T4SS proteins help bacteria interact with their host [81, 82], and our study identified a complete gene set for the assembly of these vital secretory systems. E. acroporae has secretory genes that regulate host mechanisms, similar to those of E. montiporae, and can increase the chances that bacteria survive in the host while improving the host fitness [29]. In addition, other genes found in E. acroporae may also provide clues about the survival strategy of E. acroporae in hosts. For example, identification of a catalase gene in E. acroporae strains—along with phosphoenolpyruvate synthase and 7,8-dihydro-8-oxoguanine triphosphatase as secretory genes from T3SS—can help the bacterium survive by scavenging H2O2, modulating the gluconeogenesis, and confer resistance from oxidative stresses to the host [29, 83, 84]. Upregulating genes involved in gluconeogenesis have been proposed as a response to stress-induced starvation in corals [84].
Phylogenetic analysis based on a core genome (n = 313) clustered the species to reflect their host phylogeny (Fig. 2d), similar to results obtained in earlier studies [30, 31, 60]. However, even when the host type was the same (i.e. stony coral), E. montiporae and E. acroporae did not share any branch, and their strains clustered tightly within their clade, which suggests that the co-diversification between host and symbiont is complex in nature (Fig. 2d and Supplementary Fig. S4). Moreover, a highly reduced core genome and similar ANI, GGD, and AAI at the lower end of interspecies range give clues about the diversity in genus Endozoicomonas (Fig. 2a–c).
Oxidative stress response, E. acroporae distribution, and potential implications for DMSP cycling in corals
Reef-building corals, along with soft corals, are prolific producers of DMSP in both temperate and tropical reefs, making them important in DMSP cycling [4, 85]. Furthermore, there is evidence that DMSP helps coral mitigate intracellular oxidative stress and the process considered to initiate bleaching induced mortality [86, 87]. Endozoicomonas species have been found to dominate the microbiomes of diverse marine hosts residing in shallow depths or intertidal zones, such as corals in tropical and temperate reefs [88, 89]. These hosts experience changes in environmental conditions such as thermal stress, exposure to ultraviolet radiation, and tidal heights, which lead to oxidative stress and has been shown to influence DMSP upregulation [86, 90]. Interestingly, genomes of Endozoicomonas species, including E. acroporae, harbor a high percentage of oxidative stress-responsive genes (Supplementary Fig. S6) which provide clues for their potential to mitigate oxidative stress. Furthermore, the conclusion that E. acroporae, which is widely distributed in diverse coral genera in the Indo-pacific region (Fig. 1), can metabolize DMSP to DMS suggests that it might play a role in the coral sulfur cycle. Interestingly, this bacterium may also protect its host from the coral pathogen Vibrio coralliilyticus, which uses DMSP as a cue to find physiologically stressed corals, but shows no chemoattraction towards DMS [91]. The presence of E. acroporae in different coral species also supports the idea that this new species has a broad host range similar to other members of Endozoicomonas. Moreover, it also highlights the importance of studying Endozoicomonas in greater detail. With only very few cultured isolates known to date, identification of new culturable isolates will enhance our understanding of this diverse marine genus and help establish its functional and ecological roles in a diversity of hosts.
Conclusion
Endozoicomonas species have been long hypothesized to play a role in the coral sulfur cycle, and this study provides the first genomic and functional evidence to support this hypothesis. E. acroporae strains cannot only metabolize DMSP to produce DMS, but also use DMSP as a carbon source for growth and survival. In our study, we also identified the first DMSP-related operon in E. acroporae, which links DMSP metabolism to the central carbon cycle. Furthermore, the presence of stress-responsive genes at higher proportions gives clues about how this genus adapted in marine environments. Since very few species in this diverse marine genus have been cultured to date, the possibility that there are other genes or mechanisms of DMSP metabolism, along with other functional roles in coral reefs, cannot be ignored. More focus on this genus in regards to coral reef health can provide better insights in the near future.
Data availability
Draft genomes of E. acroporae Acr-1 and E. acroporae Acr-5 have been deposited into GenBank under accession IDs SAUT00000000 and SAUU00000000, respectively. The E. acroporae Acr-14T genome was previously made public under accession ID PJPV00000000.
References
Veron JEN. Corals of the World. In: Stafford-Smith M, editor. Townsville, Australia: Australian Institute of Marine Sciences. Vol. 1–3; 2000.
Broadent AD, Jones GB, Jones RJ. DMSP in Corals and Benthic Algae from the Great Barrier Reef. Estuar Coast Shelf Sci. 2002;55:547–55.
Raina JB, Tapiolas DM, Foret S, Lutz A, Abrego D, Ceh J, et al. DMSP biosynthesis by an animal and its role in coral thermal stress response. Nature. 2013;502:677–80.
Broadent AD, Jones GB. DMS and DMSP in mucus ropes, coral mucus, surface films and sediment pore waters from coral reefs in the Great Barrier Reef. Mar Freshw Res. 2004;55:849–55.
LaJeunesse TC, Parkinson JE, Gabrielson PW, Jeong HJ, Reimer JD, Voolstra CR, et al. Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr Bio. 2018;28:2570–80.
Sunda W, Kieber DJ, Kiene RP, Huntsman S. An antioxidant function for DMSP and DMS in marine algae. Nature. 2002;418:317–20.
Raina JB, Dinsdale EA, Willis BL, Bourne DG. Do the organic sulfur compounds DMSP and DMS drive coral microbial associations? Trends Microbiol. 2010;18:101–8.
Raina JB, Tapiolas D, Willis D, Bourne DG. Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl Environ Microbiol. 2009;75:3492.
Reisch CR, Moran MA, Whitman WB. Bacterial catabolism of Dimethylsulfoniopropionate (DMSP). Front Microbiol. 2011;2:172.
Curson ARJ, Todd JB, Sullivan MJ, Johnston AWB. Catabolism of dimethylsulphoniopropionate: microorganisms, enzymes and genes. Nat Rev Microbiol. 2011;9:849–59.
Howard EC, Henriksen JR, Buchan A, Reisch CR, Burgmann H, Welsh R, et al. Bacterial taxa that limit sulfur flux from the ocean. Science. 2006;314:649–52.
Kiene RP, Linn LJ, Burton JA. New and important roles for DMSP in marine microbial communities. J Sea Res. 2000;43:209–24.
Todd JD, Rogers R, Li YG, Wexler M, Bond PL, Sun L. Structural and regulatory genes required to make the gas dimethylsulfide in bacteria. Science. 2007;315:666–9.
Moran MA, Buchan A, Gonzalez JM, Heidelberg JF, Whitman WB, Kiene RP, et al. Genome sequence of Silibacter pomeroyi reveals adaptation to the marine environment. Nature. 2004;432:910–3.
Yoch DC. Dimethylsulfoniopropionate: its source, role in the marine food web, and biological degradation of dimethylsulfide. Appl Environ Microbiol. 2002;68:5804–15.
Curson ARJ, Rogers R, Todd JD, Brearley CA, Johnston AWB. Molecular genetic analysis of a dimethylsulfoniopropionate lyase that liberates the climate-changing gas dimethylsulfide in several marine α-proteobacteria and Rhodobacter spharoides. Environ Microbiol. 2008;10:757–67.
Alcolombri U, Ben-Dor S, Feldmesser E, Levin Y, Tawfik DS, Vardi A. Identification of the algal dimethyl sulfide releasing enzyme: a missing link in the marine sulfur cycle. Science. 2015;348:1466–9.
Kooperman N, Ben-Dov E, Kramarsky-Winter E, Barak Z, Kushmaro A. Coral mucus-associated bacterial communities from natural and aquarium environments. FEMS Microbiol Lett. 2007;276:106–13.
Rohwer F, Seguritan V, Azam F, Knowlton N. Diversity and distribution of coral-associated bacteria. Mar Ecol Prog Ser. 2002;243:1–10.
Yang SH, Tseng CH, Huang CR, Chen CP, Tandon K, Lee STM, et al. Long-term survey is necessary to reveal various shifts of microbial compositions in corals. Front Microbiol. 2017;8:1094.
Zanevald JR, Burkepile DE, Shantz AA, Pritchard CE, McMinds R, Payet JP, et al. Overfishing and nutrient pollution interact with temperature to disrupt coral reefs down to microbial scales. Nat Commun. 2016;7:11833.
Lee STM, Davy S, Tang SL, Kench PS. Mucus sugar content shapes the bacterial community structure in thermally stressed Acropora muricata. Front Microbiol. 2016;7:1371.
Ferrer LM, Szmant AM. Nutrient regeneration by the endolithic community in coral skeletons. In: Choat JH, Barnes D, Borowitzka MA, Coll JC, Davies PJ, Flood P, et al. editors. Proceedings of the 6th International Coral Reef Symposium Townsville, Australia: Contributed Papers. Vol. 1;1988. p. 1–4.
Yang SH, Lee STM, Huang CR, Tseng CH, Chiang PW, Chen CP, et al. Prevalence of potential nitrogen-fixing, green sulfur bacteria in the skeleton of reef-building coral Isopora palifera. Limnol Oceanogr. 2016;61:1078–86.
Yang SH, Tandon K, Lu CY, Wada N, Shih CJ, Hsiao SSY, et al. Metagenomic, phylogenetic, and functional characterization of predominant endolithic green sulfur bacteria in the coral Isopora palifera. Microbiome. 2019;7:3.
Reshef L, Koren O, Loya Y, Zilber-Rosenberg I, Rosenberg E. The coral probiotic hypothesis. Environ Microbiol. 2006;8:2068–73.
Ritchie KB. Regulation of microbial populations by coral surface mucus and mucus-associated bacteria. Mar Ecol Prog Ser. 2006;322:1–14.
Rosenberg E, Koren O, Reshef L, Efrony R, Zilber-Rosenberg I. The role of microorganisms in coral health, disease and evolution. Nat Rev Microbiol. 2007;5:355–62.
Ding JY, Shiu JH, Chen WM, Chiang YR, Tang SL. Genomic insight into the host-endosymbiont relationship of Endozoicomonas montiporae CL-33(T) with its coral host. Front Microbiol. 2016;7:251.
Neave MJ, Michell CT, Apprill A, Voolstra CR. Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci Rep. 2017;7:40579.
Neave MJ, Apprill A, Ferrier-Pagès C, Voolstra CR. Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl Microbiol Biotechnol. 2016;100:8315–24.
Bourne DG, Iida Y, Uthicke S, Smith-Keune C. Changes in coral-associated microbial communities during a bleaching event. ISME J. 2008;2:350–63.
Vezzulli L, Pezzati E, Huete-Stauffer C, Pruzzo C, Cerrano C. 16S rDNA pyrosequencing of the Mediterranean Gorgonian Paramuricea clavata reveals a link among alterations in bacterial holobiont members, anthropogenic influence and disease outbreaks. PLoS One. 2013;8:6745.
Shiu JH, Keshavmurthy S, Chiang PW, Chen HJ, Lou SP, Tseng CH, et al. Dynamics of coral-associated bacterial communities acclimated to temperature stress based on recent thermal history. Sci Rep. 2017;7:14933.
O’Brien P, Smith HA, Fallon S, Fabricus K, Willis BT, Morrow KM, et al. Elevated CO2 has little influence on the bacterial communities associated with the pH-tolerant coral, massive Porites spp. Front Microbiol. 2018;9:2621.
Neave MJ, Michell CT, Apprill A, Voolstra CR. Whole-genome sequences of three symbiotic Endozoicomonas strains. Genome Announc. 2014;2:e00802–14.
Tandon K, Chiang PW, Chen WM, Tang SL. Draft genome sequence of Endozoicomonas acroporae strain Acr-14T, isolated from Acropora Coral. Genome Announc. 2018;6:e0156–17.
Ransome E, Rowley SJ, Thomas S, Tait K, Munn CB. Disturbance to conserved bacterial communities in the coral water gorgonian coral Eunicella verrucosa. FEMS Micorbiol Ecol. 2014;90:404–16.
Pike RE, Haltli R, Kerr RG. Description of Endozoicomonas euniceicola sp. nov. and Endozoicomonas gorgoniicola sp. nov., bacteria isolated from the octocoral Eunicea fusca and Plexaura sp., and an emended description of the genus Endozoicomonas. Int J Syst Evol Microbiol. 2014;63:4294–302.
Sheu SY, Lin KR, Hsu MY, Sheu DS, Tang SL, et al. Endozoicomonas acroporae sp. nov. isolated from Acropora coral. Int J Syst Evol Microbiol. 2017;67:3791–7.
Wilson K. Preparation of genomic DNA from bacteria. Curr Protoc Mol Biol. 2001;56:241–5.
Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE. 2012;7:e30619.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 2010;11:119.
Seeman T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;20:2068–9.
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3.
Aziz RK, Bartels D, Best DD, DeJogh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Meier-Kolthof JP, Klenk HP, Goker M. Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int J Syst Microbiol. 2014;64:325–56.
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2016. https://www.R-project.org/.
Wickham H. ggplo2: elegant graphics for data analysis. New York: Springer-Verlag; 2009.
Zhou Y, Liang Y, Lynch KH, Denis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;37:W347–W352.
Eichinger V, Nussbaumer T, Platzer A, Jehl MA, Arnold R, Rattei T, et al. EffectiveDB- updates and novel feature annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 2016;44:D669–74.
Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2005;32:W327–W331.
Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–5.
Nawrocki EP. Annotating functional RNAs in genomes using Infernal. Methods Mol Biol. 2014;1097:163–7.
Nguyen LT, Schmidt HA, Haeseler von A, Minh BQ. IQ-Tree: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Bio Evol. 2015;32:268–74.
Kalyaanamoorthy S, Minh BQ, Wong TKF, Haeseler von A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat methods. 2017;14:587–9.
Letunic I, Bork P. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47:256–9.
Ziegler M, Roik A, Porter A, Zubier K, Mudarris SM, Ormond R, et al. Coral microbial community dynamics in response to anthropogenic impacts near a major city in the central Red Sea. Mar Pollut Bull. 2016;105:629–40.
Pollock FJ, McMinds R, Smith S, Bourne DG, Willis BL, Medina M, et al. Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat Commun. 2018;9:4921.
DeSantiz TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Wessel P, Smith WH, Scharroo R, Luis J, Wobbe F. Generic mapping tools: improved version released. Eos Trans Am Geophys Union. 2013;94:409–10.
Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;8:754–5.
Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–31.
El-Gebali S, Misry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:427–32.
Burland TG. DNASTAR’s Lasergene sequence analysis software. Methods Mol Biol. 2000;132:71–91.
Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–31.
Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64:346–51.
Bourne DG, Morrow KM, Webster NS. Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu Rev Microbiol. 2016;70:317–40.
Alcolombri U, Laurino P, Lara-Astiaso P, Vardi A, Tawfik DS. DddD is a CoA-transferase/lyase producing dimethyl sulfide in the marine environment. Biochemistry. 2014;53:5473–5.
Bullock HA, Luo HW, Whitman WB. Evolution of dimethylsulfoniopropionate metabolism in marine phytoplankton and bacteria. Front Microbiol. 2017;8:637.
Yoch DC. Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide. Appl Environ Microbiol. 2002;68:5804–15.
Johnston AWB, Todd JD, Sun L, Nikolaidou-Katsaridou MN, Curson ARJ, Rogers R. Molecular diversity of bacterial production of the climate active gas, dimethylsulphide, a molecule that impinges on local and global symbioses. J Exp Bot. 2008;59:1059–67.
Todd JD, Curson ARJ, Nikolaidou-Katsaridou N, Brealey CA, Watmough NJ, Chan Y, et al. Molecular dissection of bacterial acrylate catabolism- unexpected links with dimethylsulfoniopropionate catabolism and dimethylsulfide production. Environ Microbiol. 2010;12:327–43.
Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–90.
Kuo CH, Moran AN, Ochman H. The consequences of genetic drift for bacterial genome complexity. Genome Res. 2009;19:1450–4.
McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2011;10:13–36.
Kwan JC, Donia MS, Han AW, Hirose E, Haygood MG, Schmidt EW. Genome streamlining and chemical defense in a coral reef symbiosis. Proc Natl Acad Sci USA. 2012;109:20655–60.
Robbins SJ, Singleton CM, Chan CX, Messer LF, Geers AU, Ying H, et al. A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat. Microbiol. 2019. https://doi.org/10.1038/s41564-019-532-4.
Preston GM. Metropolitan microbes: type III secretion in multihost symbionts. Cell Host Microbe. 2007;2:291–4.
Grohmann E, Christie PJ, Waksman G, Backert S. Type IV secretion in Gram-negative and Gram-positive bacteria. Mol Microbiol. 2018;107:455–71.
Kondrashov FA, Koonin EV, Morgunov IG, Finogenova TV, Kondrashova MN. Evolution of glyoxylate cycle enzymes in Metazoa: evidence of multiple horizontal transfer events and pseudogene formation. Biol Direct. 2006;1:31.
Kenkel C, Meyer E, Matz M. Gene expression under chronic heat stress in populations of the mustard hill coral (Porites asteroids) from different thermal environments. Mol Ecol. 2013;22:4322–4.
Haydon TD, Seymour JR, Suggett DJ. Soft corals are significant DMSP producers in tropical and temperate reefs. Mar Biol. 2018;165:109.
Deschaseaux ESM, Jones GB, Deseo MA, Shepherd KM, Kiene RP, Swan HB. Effects of environmental factors on dimethylated sulfur compounds and their potential role in the antioxidant system of the coral holobiont. Limnol Oceanogr. 2014;59:758–68.
Downs CA, McDougall KE, Woodley CM, Fauth JE, Richmond RH, Kushmaro A, et al. Heat-stress and light-stress induce different cellular pathologies in the symbiotic dinoflagellate during coral bleaching. PLoS ONE. 2013;8:e77173.
Neave MJ, Rachmawati R, Xun L, Michell CT, Bourne DG, Apprill A, et al. Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 2017;11:186–200.
Van de Water JA, Melkonan A, Voolstra CR, Junca H, Beraud E, Allemand D, et al. Comparative assessment of mediterranean gorgonian-associated microbial communities reveals conserved core and locally vibrant bacteria. Microb Ecol. 2017;73:466–78.
Lesser MP. Oxidative stress in marine environments: biochemistry and physiological ecology. Annu Rev Physiol. 2006;68:253–78.
Garren M, Son K, Raina JB, Rusconi R, Menolascina F, Shapiro OH, et al. A bacterial pathogen uses dimethylsulfoniopropionate as a cue to target heat-stressed corals. ISME J. 2014;8:999–1007.
Acknowledgements
This study was supported by funding from Academia Sinica. KT would like to acknowledge the Taiwan International Graduate Program (TIGP) for its fellowship. The authors would like to acknowledge support from Dr Shu-Fen Chiou during the GC experiment. We would like to thank Noah Last of Third Draft Editing for his English language editing.
Author information
Authors and Affiliations
Contributions
KT and S-LT conceived the idea for this study. KT assembled the genomes, performed the bioinformatics analysis, and wrote and revised the manuscript. C-YL performed the DMS quantification and GC experiment. P-WC cultured the strains and performed the RT-qPCR analysis and E. montiporae experiments. Y-FC helped with GC experiments and analysis. NW and S-HY helped write the manuscript and modify the illustrations. P-YC, H-YC, and M-SC helped with the GC experiments and provided the instruments for conducting the experiment. Y-JC performed the dose-dependent experiment for E. acroporae. W-MC provided the cultures. S-LT supervised the overall study. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tandon, K., Lu, CY., Chiang, PW. et al. Comparative genomics: Dominant coral-bacterium Endozoicomonas acroporae metabolizes dimethylsulfoniopropionate (DMSP). ISME J 14, 1290–1303 (2020). https://doi.org/10.1038/s41396-020-0610-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0610-x
This article is cited by
-
Coral microbiomes are structured by environmental gradients in deep waters
Environmental Microbiome (2024)
-
A genome-centric view of the role of the Acropora kenti microbiome in coral health and resilience
Nature Communications (2024)
-
The coral microbiome in sickness, in health and in a changing world
Nature Reviews Microbiology (2024)
-
DNA from non-viable bacteria biases diversity estimates in the corals Acropora loripes and Pocillopora acuta
Environmental Microbiome (2023)
-
Genomic exploration of coral-associated bacteria: identifying probiotic candidates to increase coral bleaching resilience in Galaxea fascicularis
Microbiome (2023)

{kind=link}