
Abstract
The remineralization of organic material via heterotrophy in the marine environment is performed by a diverse and varied group of microorganisms that can specialize in the type of organic material degraded and the niche they occupy. The marine Dadabacteria are cosmopolitan in the marine environment and belong to a candidate phylum for which there has not been a comprehensive assessment of the available genomic data to date. Here in, we assess the functional potential of the marine pelagic Dadabacteria in comparison to members of the phylum that originate from terrestrial, hydrothermal, and subsurface environments. Our analysis reveals that the marine pelagic Dadabacteria have streamlined genomes, corresponding to smaller genome sizes and lower nitrogen content of their DNA and predicted proteome, relative to their phylogenetic counterparts. Collectively, the Dadabacteria have the potential to degrade microbial dissolved organic matter, specifically peptidoglycan and phospholipids. The marine Dadabacteria belong to two clades with apparent distinct ecological niches in global metagenomic data: a clade with the potential for photoheterotrophy through the use of proteorhodopsin, present predominantly in surface waters up to 100 m depth; and a clade lacking the potential for photoheterotrophy that is more abundant in the deep photic zone.
Similar content being viewed by others


Introduction
Heterotrophy in the marine environment is a complex process with many organisms contributing to the remineralization of organic matter. In the surface ocean, ~50% of new organic carbon is remineralized by heterotrophs within the first 100 m [1, 2]. Despite the importance of this process to the overall ocean carbon budget, the specific contributions of the phylogenetically diverse marine bacterioplankton community remain poorly constrained. The metabolic capacity of the community members directly governs the types of organic material that can be degraded in a particular environment [3]. Heterotrophs occupy a spectrum of metabolic diversity and growth strategies [4]. While copiotrophs exploit multiple organic resources and/or undergo rapid growth in response to nutrient availability, oligotrophs specialize in a limited number of resources and dominate in low nutrient environments [5]. Because of the interplay of heterotrophs on this spectrum of metabolic diversity, it is important to understand the role(s) that specific groups play in the degradation of organic matter in the surface ocean.
An evolutionary feature that has been observed among marine oligotrophs is the reduction and simplification of the genome. This evolutionary trajectory has been posited as the theory of genome streamlining, in which organisms that grow in nutrient limited environments undergo selection to reduce cellular demand for specific compounds and nutrients [6]. While originating in the marine environment [7, 8], genome streamlining has been identified in numerous habitats for a variety of microorganisms [9,10,11,12]. Streamlined genomes will tend to have smaller genome sizes as a result of increased coding density and a decreased number of paralogs/gene duplication events, which overall reduce cellular demand for nutrients [13]. Additionally, in nitrogen-limited environments, streamlined genomes may reduce the contribution of nitrogen to the DNA by decreasing genomic GC content and the proteome through the selection of amino acids with side chains that contain fewer nitrogen atoms [13]. The theory of genome streamlining is an important avenue for understanding microbiology and provides important insights into the evolutionary history and ecological distributions of a microorganism.
Here in, we assess the potential contributions of the Dadabacteria to marine heterotrophy. A phylum level group phylogenetically clustered near the phyla Campylobacteria, Aquificota, and Deferribacteres. The Dadabacteria (formerly SBR1093) lack a cultured representative and have not been extensively assessed for their potential contributions to biogeochemical cycles though they have been detected in numerous environments. The first Dadabacteria genome was reconstructed from industrial activated sludge and reported to possess the capacity for carbon fixation through the 3-hydroxybutyrate/4-hydroxypropionate cycle [14]. Interestingly, multiple Dadabacteria metagenome-assembled genomes (MAGs) were reconstructed from the Tara Oceans global, marine metagenomic samples, though their exact role in the marine environment was unknown [15,16,17]. Our analysis reveals that the marine Dadabacteria are likely heterotrophic oligotrophs that have undergone genome streamlining with the capacity to degrade microbially derived peptidoglycan as a carbon source with further metabolic diversification between shallow and deep photic zone niches.
Materials and methods
Collect, assess and clean genomes, and construct phylogenomic trees
MAGs generated from several studies using the Tara Oceans metagenomics dataset were initially identified as Dadabacteria based on 16S rRNA phylogeny and 16 concatenated ribosomal proteins (ribosomal proteins L2, L3, L4, L5, L6, L14, L16, L18, L22, L24, S3, S8, S10, S17, and S19) [18]. All Dadabacteria metagenome-assembled genomes (MAGs) identified in NCBI (as of August 2019) [19,20,21,22,23] and one Dadabacteria genome (formally Candidate Phylum SBR1093) derived from Wang et al. [14] were also included. Genomes reconstructed from Tully et al. [15] and Tully et al. [24] were subjected to manual assessments for quality using the same methodology as in Graham et al. [25]. Briefly, read coverage and DNA compositional data were utilized to bin additional contigs (>5 kb) from the Tara Oceans Longhurst province where the original Dadabacteria MAG was reconstructed using CONCOCT (v.0.4.1; parameters: -c 800 -I 500) [26]. Bins determined through CONCOCT with overlapping contigs in a Dadabacteria MAG were profiled (anvi-profile default parameters), combined (anvi-merge default parameters) and visualized (anvi-interactive default parameters) in anvi’o [27] (v5.0). MAGs were manually refined by removing contigs with incongruent composition metrics or coverage values. Genomes from Delmont et al. [17] were also visualized in anvi’o and manually curated based on composition metrics only. Bin refinement was conducted to minimize contamination estimates and improve genome completion.
Dadabacteria MAGs were assessed for quality through the PhyloSanity workflow (default parameters) of the tool MetaSanity [28] (beta version; v1). Estimated completeness, contamination, and strain heterogeneity were determined using CheckM (v1.0.18; lineage_wf default parameters) [29]. The estimated completeness and MAG size were used to calculate an approximate genome size for the complete genome. Additionally, the CheckM Reported Statistics subcommand (checkm qa --tab-table) was used to calculate the coding density. Phylogeny was confirmed using GTDB-Tk (v1.0.0; database ver. 89; classify_wf default parameters) [30]. The GTDB-Tk de novo workflow was used to construct a multiple sequence alignment (MSA) of the Dadabacteria MAGs using the bac120 marker set and with f_SZUA-79 set as the outgroup. The full MSA was reduced to include the following lineages related to the Dadabacteria: SZUA-79, Chrysiogenetota, Deferribacterota, Thermosulfidibacterota, Aquificota, Camplyobacterota. The MSA was refined using MUSCLE (v3.8.31, parameter: -refine) [31] and FastTree (v2.1.10, parameters: -lg, -gamma) [32] was used to generate a phylogenetic tree that was visualized using the Interactive Tree of Life (IToL) [33] (Supplementary Data 1).
Functional annotation
For functional annotation and evidence of genomic streamlining, due to the limited number of available MAGs, all genomes were considered during the analysis. Dadabacteria MAGs were assessed for putative metabolic functionality through the FuncSanity workflow of the tool MetaSanity [28] (beta version;v1). All downstream analyses use the putative CDS (coding DNA sequences) as predicted by Prokka (v1.13.3) [34]. Putative CDS were assigned to carbohydrate-active enzyme (CAZy) families based on HMMs (hidden Markov models) from dbCAN (v6) [35] using hmmsearch (v3.1b2; parameter: -T 75) [36]. The output from MetaSanity that combines the CAZy matches for all submitted genomes (MetaSanity output file: combined.cazy) was used to determine the number of CAZy matches per Mbp in each MAG, including a curated selection of glycoside hydrolases (GH) and carbohydrate-binding module (CBM) containing proteins and excluding matches to CAZy subfamily HMMs (e.g., matches to GH13 model were included, while matches to GH13_9 model, etc. were excluded).
CDS were determined to be putative peptidases through hmmsearch (parameter: -T 75) using PFAM [37] HMMs selected to represent the MEROPS families [38]. Putative peptidases were screened for signatures denoting possible extracellular localization using PSORTb (v3.0; parameters: --cutoff 1, --divergent 1, -M 10, -c 70) [39] and SignalP (v4.1; defaults) [40]. First, PSORTb was used to identify all putative peptidases with the localization assignment of “extracellular”, “cellwall”, or “unknown”. For any putative peptidase that had “unknown” localization, if SignalP predicted a transmembrane helix, the peptidase was determined to be putatively extracellular.
Metabolic functions of interest were identified based on the KEGG-Decoder [25] output (v1.0.10) as implemented in MetaSanity (MetaSanity output file: KEGG.final.tsv). As part of this workflow, CDS were assigned to KEGG Ontology (KO) identifiers using KofamScan (v1.2.0) [41] and the accompanying KOfam HMMs. KO annotations were then assigned to a set of manually curated pathways and processes. Additionally, metabolisms of interest, especially those lacking KOfam HMMs, were searched independently and incorporated using KEGG-Expander as implemented in MetaSanity.
Additional databases were used to identify feature of interests within the Dadabacteria MAGs. Putative metabolic functions of interest shared between the four phylogenetic clades were identified using eggNOG-mapper [42] (http://eggnog-mapper.embl.de/; default parameters for “Auto adjust per query”) and precomputed eggNOG clusters (v5.0) [43]. antiSMASH (v5.0.0) [44] was used to detect secondary metabolite biosynthetic gene clusters (parameters: --cb-general --cb-knownclusters --cb-subclusters --asf --pfam2go --smcog-trees). Based on matches to the rhodopsin PFAM HMM model (PF01036) performed as part of the KEGG-Decoder analysis, putative rhodopsin CDS were compared to the MicRhoDE database [45] using BLASTP [46] (http://application.sb-roscoff.fr/micrhode/doblast; default parameters for “All Micrhode” option) and assigned to a previously identified clades of rhodopsins based on the highest scoring result (Supplementary Data 2). Additionally, putative rhodopsins were aligned with MUSCLE (parameter: -iter 8) and the 17 amino acid (aa) region that contains the crucial aa for determining function (aa site 97 & 108) and spectral tuning (aa site 105) were categorized based on known rhodopsin relationships (Supplementary Data 3).
Genomic streamlining
Putative CDS were used to calculate the total number of carbon and nitrogen atoms present in the predicted proteome and the corresponding ratio of each MAG (https://github.com/edgraham/CNratio). For identifying duplicate genes in a MAG, first, all putative CDS in a MAG was compared against each other using DIAMOND BLASTP [47] (parameters: --more-sensitive –max-taget-seqs 300). BLAST matches were filtered using the minbit approach [48], where significant matches were determined based on the relative comparison of bitscore values. Minbit was calculated for protein A compared to protein B, as in Eq. (1),
retaining all BLAST matches ≥0.5. BLAST matches above this threshold were reformatted and clustered using MCL [49] (mcxload parameters: --abc --stream-mirror --stream-neg-log10 -stream-tf ceil(200); mcl default parameters; mcxdump parameter: -icl). All clusters in the mcxdump output were considered to be gene duplication events within the MAG.
Ecological distribution and environmental correlations
For determining the ecological distribution and environmental correlations, a nonredundant set of MAGs was determined using FastANI [50] (v1.3; parameters: --frag-length 1500) with a representative selected from a cluster of genomes with ≥98.5% average nucleotide identity [51]. Metagenomes derived from bioGEOTRACES [52] (bGT) and Tara Oceans [53] were mapped against the nonredundant set of Dadabacteria genomes using bowtie2 [54] (v2.3.4.1, parameters: -q, --no-unal), converted from a SAM to BAM file using samtools [55] (v.1.9; view; sort), and filtered using BamM (v1.7.0, parameters: --percentage_id 0.95, --percentage_aln 0.75). featureCounts [56] (v1.5.3, default parameters) implemented through Binsanity-profile [57] (v0.3.3, default parameters) was used to generate read counts for each contig from the filtered BAM files. Read counts were used to calculate the relative fraction of each genome in the sample (Eq. (2)) and determine the reads per kbp of each genome per Mbp of metagenomic sample (RPKM) (Eq. (3)).
Environmental data were accessed from GEOTRACES Intermediate Data Product 2017 (Version 2) [58] and paired with the corresponding metagenome sample ID. In many cases there were multiple CTD casts associated with a particular station and depth (Supplementary Data 4). The mean value was used in cases where a parameter was measured multiple times at the same depth and station. Environmental data were paired with a metagenome only if the depth was within 1 m of the metagenome. RPKM values for Dadabacteria genomes from all samples with available environmental data were used in a canonical correspondence analysis (CCA) in Past4 [59] (v.4.01). Only environmental data that were measured for ≥90% of the samples were used to perform the CCA. RPKM values were normalized (log(n + 1)) prior to CCA. Transect plots were made in Ocean Data View (v5.2.1; DIVA Gridding; Schlitzer, Reiner, Ocean Data View, https://odv.awi.de, 2020). Bathymetry was pulled from General Bathymetric Chart of the Oceans (GEBCO 2014; https://doi.org/10.1564/PANGAEA.708081).
Results and discussion
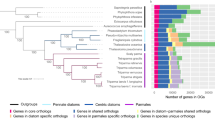
As a candidate phylum, a broad understanding of the ecological role of the Dadabacteria has remained elusive due to the limited amount of metabolic information available for the clade. Based on the phylogenetic reconstruction of 48 MAGs (mean ± s.d. completeness 75.72% ± 17.77% and contamination 1.85 ± 1.48%; Fig. 1a; Supplementary Table 1), the phylum partitions into three distinct clades which share common environmental features: hydrothermal systems (terrestrial hot springs and hydrothermal vents), organic carbon-associated systems (the terrestrial subsurface, oil-polluted marine systems, marine sponges, marine sediment, and hydrothermal vent sediments), and marine pelagic systems. Within the “marine pelagic” clade, there are two distinct subclades, designated as marine pelagic clade I and II. The marine pelagic clades harbor genomic features that differentiate them from the other clades, specifically with regards to genomic evolutionary selection (e.g., streamlining) and putative metabolisms.

a A phylogenomic tree of the bac120 marker set for the Dadabacteria and related phyla and a heatmap displaying functions of interest for each Dadabacteria MAG. Bootstrap (1000 resamples) values are scaled proportionally between 0.75 and 1. Putative extracellular peptidase, secondary metabolite, glycoside hydrolase, and carbohydrate-binding module counts are displayed on a scale from 0 to 5. Functions inferred from eggNOG counts are displayed on a scale from 0–20+. Metabolic processes inferred from KEGG are displayed on a scale for 0–1, as a fraction of a particular metabolism detected. MAGs abbreviations: TOBG from Tully et al. [15]; TMED from Tully et al. [24]; TARA from Delmont et al. [17]; MED from López-Pérez et al. [69]; UBA from Parks et al. [16]. b A scatter plot of percent G + C (%G + C) and approximate complete genome size in megabase pairs (Mbp) for each Dadabacteria MAG. c A scatterplot of putative proteome carbon-to-nitrogen content ratio and percent coding density for each Dadabacteria MAG. d The number of duplicate gene events in each Dadabacteria MAG.
The pelagic marine Dadabacteria have undergone a genome streamlining process in comparison to the organic carbon-associated and hydrothermal lineages. The marine pelagic Dadabacteria exhibit all five traits associated with genome streamlining: reduced genome size, decreased %GC content, increased C/N ratio in the predicted proteome, increased coding density, and limited/no gene duplication events (Fig. 1b–d; Supplementary Table 1) [6, 13]. The estimated complete marine pelagic Dadabacteria genome is ~1.22 Mb (± 0.05 95% CI) with >96% coding density, smaller in size and similar in coding density to the well-studied marine SAR11 clades [8, 60]. The presence of the Dadabacteria MAGs reconstructed from multiple oligotrophic Tara Oceans regions would suggest that these organisms, like other oligotrophs, are adapted to environments with low nutrient concentrations [6] (Supplementary Fig. 1). Modifications in GC content and proteome C/N ratio are associated with lowering the nitrogen demand for organisms in nitrogen-limited environments [6]. While small genomes, devoid of paralogs and with high coding density, are thought to have reduced energy requirements for division and growth. These genomic modifications which confer an advantage in oligotrophic marine environments are the result of changes in selection pressure that occurred at the transition between the marine pelagic and hydrothermal/organic carbon-associated Dadabacteria clades [61, 62]. These results provide further evidence that the theory of genome streamlining is a common evolutionary response to organisms that undergo a transition from nutrient rich to nutrient poor environments [63].
While the SBR1093 MAG was implicated in carbon fixation via the 3-hydroxypriopinate/4-hydroxybutyrate cycle [14], analysis of the Dadabacteria phylum reveals, especially for the marine pelagic clades, a predominantly heterotrophic lifestyle (Fig. 1a). Except for the SBR1093 MAG, none of the publicly available Dadabacteria MAGs have the potential for carbon fixation (Supplementary Table 2). Several MAGs from the hydrothermal and organic carbon-associated clades have the potential to interface with the nitrogen and sulfur cycles with metabolic processes involved in denitrification, dissimilatory nitrate reduction to ammonia (DNRA), sulfate reduction, sulfide oxidation, and the production of dimethylsulfoniopropionate (DMSP) (Fig. 1a). However, while both marine pelagic clades lack these particular metabolic pathways, all four clades share in the potential for the heterotrophic degradation of proteins and complex carbohydrates, including starch/glycogen (β-glucosidase and α-amylase). One consistent target for the extracellular peptidases (LysM) and carbohydrate-active enzymes (CAZymes; peptidoglycan lyase and CBM Family 50) across the Dadabacteria clades is peptidoglycan, the polymer of the microbial cell wall. It may be possible that these predicted proteins are responsible for the internal recycling of the cell wall during cell division or an indication that the Dadabacteria occupy a niche capable of recycling microbially derived dissolved organic matter (DOM).
Interestingly, the number of extracellular peptidases, CAZymes, and ATP-binding cassette-type (ABC-type) transporter components normalized for MAG length across all four clades remains consistent even as the overall diversity within each group of proteins decreases (Fig. 1a; Supplementary Tables 3–5). This may highlight an interplay between heterotrophic metabolic diversity and changes in carbon utilization as Dadabacteria genome size decreases during streamlining. Additionally, there are several other metabolic processes that distinguish the four clades and highlight the divide between the hydrothermal and organic carbon-associated clades and marine pelagic clades. Specifically, for the hydrothermal clade, the prevalence of CRISPR-associated proteins (used as proxy for CRISPR arrays due low recovery in MAGs), motility, and two-component regulatory chemotaxis suggest that both avoidance of viral predation and physical adjustments within the hydrothermal environment are important evolutionary advantages (Supplementary Tables 2 and 5). Distinct for the hydrothermal and organic carbon-associated clades, are the presence of phosphonate and phosphate ABC transporters, the Entner-Doudoroff pathway, an alternative pathway to glycolysis for glucose degradation, and a Type II secretion system (Supplementary Tables 2 and 6). In many marine systems, phosphorous, like nitrogen, can be a limiting resource. All four clades possess ABC-type phospholipid transporters (Supplementary Table 6), so while most of the marine pelagic clades (63%) lack phosphonate and phosphate transporters, the presence of phospholipid transporters suggest these organisms may recover phosphorous for cellular demand from DOM.
The marine pelagic I and II clades have several distinguishing metabolic properties. Potentially most importantly are the mechanisms related to utilizing light energy. Uniquely amongst the Dadabacteria, the marine pelagic I clade possesses rhodopsins and the biosynthetic capacity for retinal synthesis (Fig. 1a). Based on the present amino acids, it is predicted that all of the identified rhodopsins are H+-pumping proteorhodopsins [64] (Supplementary Table 7). For the eight identified proteorhodopsins within the marine pelagic I clade, all but one are predicted to be spectrally tuned to absorb blue light [65, 66] (Supplementary Table 7). The marine pelagic I clade also has the capacity to produce terpene secondary metabolites (Supplementary Table 8). Terpenes are organic hydrocarbons that have been shown to be associated with carotenoid synthesis [67]. These terpenes may be related to the production of β-carotene, a biological precursor to retinal, or to production of other unidentified carotenoids (Supplementary Table 6). The marine pelagic II clade lack proteorhodopsins, retinal biosynthesis, and terpene secondary metabolites (Fig. 1a). Like all other Dadabacteria clades, the marine pelagic clades possess starch/glycogen and peptidoglycan degradation mechanisms may suggest that these heterotrophic processes are the predominant avenues for energy acquisition.
The metabolic division based on the utilization of light via proteorhodopsins between the marine pelagic clades is reflected in the ecological distribution of the clades. Using a nonredundant set of the marine pelagic Dadabacteria MAGs, the large global metagenomic datasets (Tara Oceans and bGT) were mapped against the MAGs and used to assess where the Dadabacteria occurred through the water column (Supplementary Tables 9 and 10). The two datasets have distinct properties that allow for varying perspectives on the ecology of the Dadabacteria. Tara Oceans is globally distributed with multiple size fractions and samples from the mesopelagic, while bGT provides several high-resolution cruise tracks with multiple depths between the surface and ~250 m depth. The results from Tara Oceans demonstrate that, broadly, the marine clades are present in the planktonic size fraction (<3 μm) and almost exclusively found in the epipelagic (Supplementary Fig. 2).
As exemplified by the GA03 cruise track in the North Atlantic, the resolution provided by bGT reveals that the marine pelagic I and II clades tend to be dominant above and below ~100 m depth (~1% light level), respectively, and that this niche transition can be sharp, with the marine pelagic I clade dropping to a negligible component of the microbial community at this partitioning depth (Fig. 2; Supplementary Table 11). This relationship can be observed for the other three cruise tracks, station ALOHA (Hawaii Ocean Time-series), and hydrostation S (Bermuda Atlantic Time-series) with some localized variation, potentially due to surficial mixing and/or downwelling/upwelling events, where the marine pelagic II clade can be found at the surface and the marine pelagic I clade can be found at 250 m. However, for many of the sampling stations there remains a divide between the two clades at the ~1% light depth (Supplementary Figs. 2 and 3). Canonical correspondence analysis (CCA) of the GA03 environmental parameters support this niche transition as a majority of the marine pelagic II clade MAGs correlated with depth and depth-associated parameters (nutrients, temperature, etc.; Fig. 2c). Similar correlations between depth-associated parameters and the marine pelagic clades are observed for the other cruise tracks (Supplementary Fig. 4). As has been shown previously, deep euphotic zone blue-light proteorhodopsins are adapted to low light incidence and capture a limited amount of light at 75 m [68], the apparent depth partitioning linked to encoding proteorhodopsin likely reflects an evolutionary selective pressure against maintaining a light-responsive protein apparatus at depth and manifests as depth-specific niche boundaries between the two marine pelagic clades.

a Ocean Data View plot of percent relative fraction for the Dadabacteria marine pelagic I clade along the GEOTRACES transect GA03. b Ocean Data View plot of percent relative fraction for the Dadabacteria marine pelagic II clade along the GEOTRACES transect GA03. c Canonical correspondence analysis of the nonredundant marine Dadabacteria MAGs. Vectors denote correlations with environmental parameters and have been modified for easier visualization: trioplot amp 1.5, scaling type 2. d Cruise track of GA03. Red circle denotes start of cruise track (0 km).
Conclusion
The Dadabacteria phylum is an understudied clade with a limited number of genomic representatives. The broad analysis of the four major clades represented among publicly available genomes reveals a broad range of heterotrophic organisms, putatively involved in the recycling of microbially derived DOM, such as peptidoglycan and phospholipids. The hydrothermal and organic carbon-associated clades appear to be facultative anaerobes capable of using alternative electron acceptors, while the marine pelagic clades appear to be obligate aerobes. The marine pelagic clades have genomic features indicating extensive genome streamlining evolutionary pressures that mirror their ecological distribution in oligotrophic environments. Genome streamlining theory is an important hypothesis for explaining the prevalence of small genomes among cosmopolitan microorganisms and the Dadabacteria represent a clear example of the theory in action. The two distinct marine pelagic clades are differentiated in metabolic potential by the presence of light-associated adaptations, such as proteorhodopsin, terpenes, and carotenoids, supporting an argument that marine pelagic I clade possess a photoheterotrophic lifestyle. These adaptations are reflected in the ecological distribution of these clades with depth-partitioned niches for marine pelagic I and II clades. The Dadabacteria have multiple transitions that are of interest for understanding evolutionary pressures and adaptations in different environments, including: terrestrial to marine transitions; high to moderate/low temperature transitions; and adaptations from organic rich to organic poor environments. Further studies and the expansion of available genomes for this clade may provide specific insights as to how these transitions occur and manifest in microbial genomes.
Data availability
Several of the MAGs (TOBG-EAC99, TARA-RED-00009, TOBG-IN994, TOBG-MED731, TOBG-MED713, and TOBG-SP357) used in this study and underwent manual curation originated from the Tara Oceans dataset and were never submitted to NCBI to avoid duplication in GenBank. These curated MAGs are noted in Supplementary Table 1 and are available here: https://doi.org/10.6084/m9.figshare.12344207. As noted in Supplementary Table 1, MAGs with corresponding submissions in NCBI GenBank have been updated.
References
Cole JJ, Findlay S, Pace ML. Bacterial production in fresh and saltwater ecosystems: a cross-system overview. Mar Ecol Prog Ser. 1988;43:1–10.
Ducklow HW, Kirchman DL, Quinby HL, Carlson CA, Dam HA. Stocks and dynamics of bacterioplankton carbon during the spring bloom in the eastern North Atlantic Ocean. Deep Sea Res II. 1993;40:245–63.
Arnosti C. Microbial extracellular enzymes and the marine carbon cycle. Annu Rev Mar Sci. 2011;3:401–25.
Malik AA, Martiny JBH, Brodie EL, Martiny AC, Treseder KK, Allison SD. Defining trait-based microbial strategies with consequences for soil carbon cycling under climate change. ISME J. 2019;14:1–9.
Vergin KL, Done B, Carlson CA, Giovannoni SJ. Spatiotemporal distributions of rare bacterioplankton populations indicate adaptive strategies in the oligotrophic ocean. Aquat Microb Ecol. 2013;71:1–13.
Giovannoni SJ, Cameron Thrash J, Temperton B. Implications of streamlining theory for microbial ecology. ISME J. 2014;8:1553–65.
Rocap G, Larimer FW, Lamerdin J, Malfatti S, Chain P, Ahlgren NA, et al. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature. 2003;424:1042–7.
Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, et al. Genome streamlining in a cosmopolitan oceanic bacterium. Science. 2005;309:1242–5.
Luo H, Swan BK, Stepanauskas R, Hughes AL, Moran MA. Evolutionary analysis of a streamlined lineage of surface ocean Roseobacters. ISME J. 2014;8:1428–39.
Castelle CJ, Wrighton KC, Thomas BC, Hug LA, Brown CT, Wilkins MJ, et al. Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol. 2015;25:690–701.
Brewer TE, Handley KM, Carini P, Gilbert JA, Fierer N. Genome reduction in an abundant and ubiquitous soil bacterium ‘Candidatus Udaeobacter copiosus’. Nat Microbiol. 2016;2:16198–7.
Neuenschwander SM, Ghai R, Pernthaler J, Salcher MM. Microdiversification in genome-streamlined ubiquitous freshwater Actinobacteria. ISME J. 2018;12:185–98.
Getz EW, Tithi SS, Zhang L, Aylward FO. Parallel evolution of genome streamlining and cellular bioenergetics across the marine radiation of a bacterial phylum. mBio. 2018;9:1034–14.
Wang Z, Guo F, Liu L, Zhang T. Evidence of carbon fixation pathway in a bacterium from candidate phylum SBR1093 revealed with genomic analysis. PLoS One. 2014;9:e109571–9.
Tully BJ, Graham ED, Heidelberg JF. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci Data. 2018;5:170203.
Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2017;2:1–10.
Delmont TO, Quince C, Shaiber A, Esen ÖC, Lee ST, Rappé MSR, et al. Nitrogen-fixing populations of Planctomycetes and Proteobacteria are abundant in surface ocean metagenomes. Nat Microbiol. 2018;326:1–12.
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, et al. A new view of the tree of life. Nat Microbiol. 2016;1:16048.
Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7:13219.
Hug LA, Thomas BC, Brown CT, Frischkorn KR, Williams KH, Tringe SG, et al. Aquifer environment selects for microbial species cohorts in sediment and groundwater. ISME J. 2019;9:1–11.
Kato S, Sakai S, Hirai M, Tasumi E, Nishizawa M, Suzuki K, et al. Long-term cultivation and metagenomics reveal ecophysiology of previously uncultivated thermophiles involved in biogeochemical nitrogen cycle. Microbes Environ. 2018;33:107–10.
Tully BJ, Sachdeva R, Graham ED, Heidelberg JF. 290 metagenome-assembled genomes from the Mediterranean Sea: a resource for marine microbiology. PeerJ 2017;5:e3558–15. https://doi.org/10.7717/peerj.3558.
Ward LM, Idei A, Nakagawa M, Ueno Y, Fischer WW, McGlynn SE. Geochemical and metagenomic characterization of jinata onsen, a proterozoic-analog hot spring, reveals novel microbial diversity including iron-tolerant phototrophs and thermophilic lithotrophs. Microbes Environ. 2019;34:278–92.
Ward LM, Idei A, Nakagawa M, Ueno Y, Fischer WW, McGlynn SE. Geochemical and metagenomic characterization of jinata onsen, a proterozoic-analog hot spring, reveals novel microbial diversity including iron-tolerant phototrophs and thermophilic lithotrophs. Microbes Environ. 2019;34:278–92.
Graham ED, Heidelberg JF, Tully BJ. Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 2018;350:1–6.
Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al. Binning metagenomic contigs by coverage and composition. Nat Meth. 2014;11:1144–6.
Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, et al. Anvi’o: an advanced analysis and visualization platform for’omics data. PeerJ. 2015;3:e1319.
Neely CJ, Graham ED, Tully BJ. MetaSanity: an integrated microbial genome evaluation and annotation pipeline. Bioinformatics. 2020;10:D233.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;1–3. https://doi.org/10.1093/bioinformatics/btz848.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Price MN, Dehal PS, Arkin AP. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–51.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37.
El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018;47:D427–32.
Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2013;42:D503–9.
Yu NY, Wagner JR, Laird MR, Melli G, Rey SB, Lo R, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–15.
Petersen TN, Brunak S, Heijne, von G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Meth. 2011;8:785–6.
Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2.
Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol. 2017;34:2115–22.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018;47:D309–14.
Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY, et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;79:629–7.
Boeuf D, Audic S, Brillet-Guéguen L, Caron C, Jeanthon C. MicRhoDE: a curated database for the analysis of microbial rhodopsin diversity and evolution. Database. 2015;2015:bav080–8.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinforma. 2009;10:421–9.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Meth. 2014;12:59–60.
Benedict MN, Henriksen JR, Metcalf WW, Whitaker RJ, Price ND. ITEP: an integrated toolkit for exploration of microbial pan-genomes. BMC Genomics. 2014;15:8.
van Dongen S, Abreu-Goodger C. Using MCL to extract clusters from networks. Methods Mol Biol. 2012;804:281–95. https://doi.org/10.1007/978-1-61779-361-5_15.
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:7200–8.
Tully BJ. Metabolic diversity within the globally abundant Marine Group II Euryarchaea offers insight into ecological patterns. Nat Commun. 2019;10:1–12.
Biller SJ, Berube PM, Dooley K, Williams M, Satinsky BM, Hackl T, et al. Data descriptor: marine microbial metagenomes sampled across space and time. Sci Data. 2018;5:1–7.
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Ocean plankton. Structure and function of the global ocean microbiome. Science. 2015;348:1261359.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012;9:357–9.
Li H, Handsaker B, Fennell T, Ruan J, Homer N. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30.
Graham ED, Heidelberg JF, Tully BJ. BinSanity: unsupervised clustering of environmental microbial assemblies using coverage and affinity propagation. PeerJ. 2017;5:e3035–19.
Schlitzer R, Anderson RF, Dodas EM, Lohan M, Geibert W, Tagliabue A, et al. The GEOTRACES intermediate data product 2017. Chem Geol. 2018;493:210–23.
Hammer Ø, Harper D, Ryan PD. PAST: paleontological statistics software package for education and data analysis. Palaeontologia Electron. 2001;4:9.
Grote J, Thrash JC, Huggett MJ, Landry ZC, Carini P, Giovannoni SJ, et al. Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio. 2012;3:e00252–12.
Luo H, Thompson LR, Stingl U, Hughes AL. Selection maintains low genomic GC content in marine SAR11 lineages. Mol Biol Evolution. 2015;32:2738–48.
Martinez-Gutierrez CA, Aylward FO. Strong purifying selection is associated with genome streamlining in epipelagic marinimicrobia. Genome Biol Evolution. 2019;11:2887–94.
Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, González JM, et al. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci. 2013;110:11463–8.
Béjá O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP, et al. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science. 2000;289:1902–6.
Béjá O, Spudich EN, Spudich JL, Leclerc M, DeLong EF. Proteorhodopsin phototrophy in the ocean. Nature. 2001;411:786–9.
Man D, Wang W, Sabehi G, Aravind L, Post AF, Massana R, et al. Diversification and spectral tuning in marine proteorhodopsins. EMBO J. 2003;22:1725–31.
Gershenzon J, Dudareva N. The function of terpene natural products in the natural world. Nat Chem Biol. 2007;3:408–14.
Wang W-W, Sineshchekov OA, Spudich EN, Spudich JL. Spectroscopic and photochemical characterization of a deep ocean proteorhodopsin. J Biol Chem. 2003;278:33985–91.
López-Pérez M, Haro-Moreno JM, Gonzalez-Serrano R, Parras-Moltó M, Rodriguez-Valera F. Genome diversity of marine phages recovered from Mediterranean metagenomes: Size matters. PLoS Genet. 2017;13:e1007018. https://doi.org/10.1371/journal.pgen.1007018.
Acknowledgements
We would like to acknowledge and thank the Tara Oceans consortium and, now, the authors of Biller et al. for providing access to such important global marine metagenomic datasets. Their commitment to the open-access data has proven to be a valuable asset to all who build on the shoulder of giants. And we thank the Center for Dark Energy Biosphere Investigations (C-DEBI) for providing funding to BJT (OCE-0939654). This is C-DEBI contribution number 554.
Author information
Authors and Affiliations
Contributions
Analyses were conducted by EDG and BJT Specifically, EDG performed quality assessments, manual improvement of the MAGs, reconstructed the phylogeny, and recruitment procedure to determine ecological distributions. BJT performed analyses related to functional annotations and genome streamlining. EDG and BJT wrote the manuscript. The study was conceived by BJT.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Graham, E.D., Tully, B.J. Marine Dadabacteria exhibit genome streamlining and phototrophy-driven niche partitioning. ISME J 15, 1248–1256 (2021). https://doi.org/10.1038/s41396-020-00834-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-00834-5
This article is cited by
-
Genome streamlining in Parcubacteria transitioning from soil to groundwater
Environmental Microbiome (2024)
-
Prokaryotic communities of the French Polynesian sponge Dactylospongia metachromia display a site-specific and stable diversity during an aquaculture trial
Antonie van Leeuwenhoek (2024)
-
Sponge-associated microbes in the twilight zone of Curaçao
Symbiosis (2024)
-
Candidatus Nemesobacterales is a sponge-specific clade of the candidate phylum Desulfobacterota adapted to a symbiotic lifestyle
The ISME Journal (2023)
-
Replicated life-history patterns and subsurface origins of the bacterial sister phyla Nitrospirota and Nitrospinota
The ISME Journal (2023)
