
Abstract
Microbes in ecosystems often develop coordinated metabolic interactions. Therefore, understanding metabolic interdependencies between microbes is critical to deciphering ecosystem function. In this study, we sought to deconstruct metabolic interdependencies in organohalide-respiring consortium ACT-3 containing Dehalobacter restrictus using a combination of metabolic modeling and experimental validation. D. restrictus possesses a complete set of genes for amino acid biosynthesis yet when grown in isolation requires amino acid supplementation. We reconciled this discrepancy using flux balance analysis considering cofactor availability, enzyme promiscuity, and shared protein expression patterns for several D. restrictus strains. Experimentally, 13C incorporation assays, growth assays, and metabolite analysis of D. restrictus strain PER-K23 cultures were performed to validate the model predictions. The model resolved that the amino acid dependency of D. restrictus resulted from restricted NADPH regeneration and predicted that malate supplementation would replenish intracellular NADPH. Interestingly, we observed unexpected export of pyruvate and glutamate in parallel to malate consumption in strain PER-K23 cultures. Further experimental analysis using the ACT-3 transfer cultures suggested the occurrence of an interspecies malate–pyruvate shuttle reconciling a redox imbalance, reminiscent of the mitochondrial malate shunt pathway in eukaryotic cells. Altogether, this study suggests that redox imbalance and metabolic complementarity are important driving forces for metabolite exchange in anaerobic microbial communities.
Similar content being viewed by others


Introduction
Microbial function is critical to global element cycling, agriculture, bioremediation, human health, and industrial biotechnology [1,2,3,4]. The interactions among microbes are central topics in microbial ecology, which can only be observed in microbial communities but not in isolated cultures [5]. These interactions have been classified into three main types: syntrophy, cross-feeding, and competition [6]. Several methods have been proposed to elucidate these complex interactions, from simple analysis of co-cultures to complex time-dependent microbial co-association network analysis [7].
Metabolic complementarity is a driving force for microbial mutualism [8, 9]. While mutualism confers robustness to a microbial community, a trade-off is that co-adapted microbes are susceptible to the loss of nonessential functions via genome streamlining, which can lead to auxotrophy in other environments [10]. As a result, isolation of microorganisms from their syntrophic partners or natural habitats is often challenging, as demonstrated by the scarcity of culturable isolates [11]. An alternative approach is metabolic modeling based on microbial genomes [12,13,14]. Genome-scale constraint-based metabolic models have been increasingly used to predict microbial function at the community level [15, 16]. Nevertheless, physiological information and genome annotation verification for organisms in complex communities are often lacking, which results in the inclusion of misannotated genes and nongene associated reactions [17], rendering predictions irrelevant to community function. Integration of laboratory experiments with metabolic modeling can significantly improve the accuracy of function prediction while reduce laboratory efforts [18], as demonstrated in the study of amino acid cross-feedings in synthetic E. coli communities [3].
Mineralization of organic compounds in anaerobic environments requires tightly coupled metabolic coordination between microbes due to redox and thermodynamic constraints [19, 20]. Organohalide-respiring microbial communities are great models to study metabolic interdependency between microbes because they are often co-inhabited by a variety of anaerobic microbes possessing distinct ecological function, including acetogens, fermenting bacteria, methanogens, sulfate-reducing bacteria, and organohalide-respiring bacteria (OHRB) [21, 22]. Dehalobacter restrictus strains, active participants in global halogen cycling, are specialized in respiring a variety of organohalides [23,24,25,26,27,28,29]. D. restrictus strains PER-K23, CF, and DCA require the addition of either amino acids or parent culture supernatants to support growth in isolation [23, 30], indicating unexplored metabolic interdependencies with other microbes in their natural habitats. In CF-dechlorinating consortium ACT-3, D. restrictus strain CF grows in association with a Bacteroides sp. [31], while in another enrichment culture, hexachlorocyclohexane-dechlorinating D. restrictus strain E1 grows with a Sedimentibacter sp. [26, 32]. Only isolated strain UNSWDHB was shown not to required amino acid supplementation [28]. However, comparative genomic analysis and refined metabolic annotation suggested that genomes of the strains CF, DCA, PER-K23, E1, and UNSWDHB all possess a complete set of genes to synthesize all amino acids, including a salvage pathway to obtain serine from threonine [25, 30].
In this study, we elucidated some metabolic interdependencies in D. restrictus-containing consortium ACT-3, which first required resolving the discrepancy between observed amino acid auxotrophy and the genome annotation of D. restrictus. We explored the metabolic potential of D. restrictus using a genome-scale, constraint-based metabolic model built on a highly annotated D. restrictus strain CF genome [33]. The metabolic annotation was systematically curated with: (a) genome analysis [25] and (b) shared protein expression data available for strains DCA, PER-K23, E1, and UNSWDHB [34,35,36,37]. Additionally, the model incorporated refinements of annotations in central metabolism and in amino acid/cofactor biosynthesis that were experimentally supported [30]. The model simulations considered cofactor availability, enzyme promiscuity, physiological redox conditions, and further model constraints based on experimental values obtained in this study. Experimentally, D. restrictus strain PER-K23 isolate cultures were used to validate model predictions because the most complete and tiered datasets in physiology, genomics, transcriptomics and proteomics are available for this particular strain. Using this combined and iterative computational and experimental approach, we determined that the amino acid dependency of D. restrictus resulted from restricted NADPH regeneration, which could be restored with malate supplementation via the function of the NADP+-dependent malic enzyme. Furthermore, strain PER-K23 cultures grown in mineral medium supplemented with malate exhibited an unexpected export of pyruvate and glutamate. Growth experiments and metabolite analysis using the ACT-3 transfer cultures suggested that the specialized malate requirement of D. restrictus arose through co-adaption with a malate-producing, glutamate-auxotrophic Bacteroides sp. in ACT-3.
Materials and methods
Flux balance analysis (FBA)
The experimentally refined genome of D. restrictus strain CF (accession no. NC_018866) [30] was used to reconstruct a draft Dehalobacter genome-scale metabolic model [33]. Flux balance analysis and flux variability analysis simulations were conducted with COBRApy [38]. Constraints were applied to the genome-scale metabolic model based on five considerations to improve the accuracy of flux predictions: (i) cofactor availability for metabolic reactions; (ii) shared expression patterns in available proteomes of D. restrictus strains (Table S1); (iii) cellular redox state under given growth conditions; (iv) potential promiscuous enzyme activity to rescue missing pathways; and (v) integration of experimental values from culture growth assays and metabolite profile analysis. Based on these considerations, constraints were applied to the model as follows: (a) inactivating certain reactions; (b) limiting the direction of specific reactions (making reactions irreversible); (c) applying experimentally relevant metabolite fluxes; and (d) disabling/enabling the export of metabolites (Table S2).
Microbial cultures and growth conditions
All Chemicals were ordered from Sigma-Aldrich (Oakville, ON, Canada) at highest purity available unless specified otherwise. D. restrictus strain PER-K23 was provided by the Löffler Lab at University of Tennessee (Knoxville, USA). Escherichia coli strain BL21(DE3) was purchased from New England Biolabs Ltd. Consortium ACT-3 was originally enriched from 1,1,1-trichloroethane-contaminated groundwater in 2001 from a northeastern United States industrial area [39], and a subculture (1.8 l) was adapted to respire chloroform and has been maintained ever since [40]. E. coli strain BL21 was grown on LB broth except in the 13C incorporation assay. The strain PER-K23 cultures and the consortium ACT-3 and CF subcultures are maintained in a FeS-reduced, bicarbonate-based mineral medium described previously [30, 40]. The growth assays for the strain PER-K23 cultures and consortium ACT-3 were performed following the established protocol reported previously with some modifications (described in SI) [30, 37].
DNA extraction, quantitative PCR, and illumina amplicon sequencing
Culture DNA was extracted from 1 ml samples. Cells were harvested by centrifugation at 16,000×g for 10 min at 4 °C. Since Dehalobacter cell pellets are easily resuspended, in each tube, most of supernatant was remove (0.9 ml), and the cell pellets were resuspended using the remaining supernatant (0.1 ml), and the DNA was extracted using the MO BIO PowerSoil® DNA isolation kit following the manufacturer’s recommendations. Real-time quantitative polymerase chain reaction (qPCR) assays were performed to track the gene copy numbers of D. restrictus using specific 16S rRNA gene primers reported previously (described in SI) [41]. For the 16S rRNA gene-based population analysis, the ACT-3 transfer cultures (2 ml) from each triplicate trials were combined, and the DNA was extracted as described above. The DNA samples were sent to the Genome Quebec Innovation Centre (McGill University, Canada) for Illumina MiSeq amplicon sequencing. After sequencing, the raw data were processed following an established pipeline described previously [42]. The assemblage of pair-end reads, primer removal, quality filtering, chimera and singleton detections, and read number normalization were implemented using the sequence analysis tool USEARCH, and the taxonomic assignment of OTUs was performed against the Silva database (version 128; https://www.arb-silva.de/documentation/release-128/). The taxonomic assignment and abundance of individual OTUs is available in Table S3.
13C incorporation assay
A 13C-pyruvate incorporation assay was performed following a previous study on amino acid biosynthesis of Dehalococcoides with some modifications (described in SI) [43]. The 13C incorporation pattern into the amino acids is available in Table S4.
Phylogenetic analysis
A species tree was created with a modified method recently published [44]. Briefly, 10 ribosome protein subunits in the selected bacterial species were independently aligned with MAFFT v7.245, trimmed to remove unaligned N and C termini residues using default parameters with Gblocks version 0.91b [45], and concatenated to reconstruct a maximum likelihood tree with 100 bootstrap values using PHYML v3.2.0 [46]. Ortholog groups were predicted via OrthoMCL [47] and OrthoDB [48]. Enzyme orthologs were mapped to the species tree with Evolview v2 [49].
Enzyme activity assays
The conditions for cell extract preparation and dehalogenase activity assays (using trichloroethene as substrate) were described previously [30], and o-phosphoserine phosphatase activity assays were conducted following established protocols with some modifications (described in SI) [50]. Enzyme activity is defined as µmol product produced min−1 mg protein−1.
Analytical procedures
Chlorinated hydrocarbons were measured by injecting a 0.3 ml headspace sample into a Hewlett-Packard 5890 Series II GC fitted with a GSQ column (30-m-by-0.53-mm [inner diameter] PLOT column; J&W Scientific, Folsom, CA) as described previously [30]. For the metabolite profile analysis, in an anaerobic chamber (Coy), each culture was sampled (0.2 ml), and gently filtered through a 0.1 µm-pore-size syringe filter (Millipore). The flow-through was collected in a plastic microcentrifuge tube, followed by a centrifugation at 16,000×g for 10 min at 4 °C. The supernatants were stored at −80 °C before analysis. The amount of acetate, malate and pyruvate from the strain PER-K23 cultures was determined by high-performance liquid chromatography (HPLC) using an ICS5000 system (Thermo scientific) equipped with an Aminex HPX-87H column (BioRad) connected to a ultraviolet (UV) detector. Each sample (25 µl) was injected onto the column incubated at 35 °C, using 5 mM H2SO4 eluent at a flow rate of 0.6 ml min−1 with the UV wavelength set to 210 nm. Amino acids and organic acids were detected using liquid chromatography electrospray-coupled high resolution mass spectrometry (LC-ESI-HRMS) with a Dionex UHPLC system and a Q-Exactive mass spectrometer (Thermo Scientific) equipped with a HESI II source (Thermo Scientific) and a Micro-splitter valve (IDEX Health & Science) (described in SI).
Results and discussion
Validation of serine autotrophy in D. restrictus
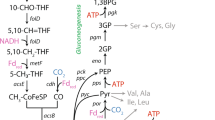
Previously, we refined genome annotations for central metabolism and biosynthesis of amino acids/cofactors comparing D. restrictus strains CF, DCA, PER-K23, E1, and UNSWDHB [25, 30], and observed that genes involved in these metabolic pathways are highly conserved in these five D. restrictus genomes, except completeness of the genes in corrinoid biosynthesis pathway. Common features among these Dehalobacter genomes are gaps in the TCA cycle and serine biosynthesis. We then built a genome-scale constraint-based Dehalobacter metabolic model based on the refined metabolic annotation [33]. To increase accuracy in model reconstruction, we analyzed the presence/absence of enzyme orthologs involved in these metabolic pathways in a range of OHRB species, Firmicutes species, Bacteroides species, and some well-characterized or widely studied model organisms in Bacteria (Fig. 1). Malate dehydrogenase and succinate dehydrogenase/fumarate reductase genes are lost in D. restrictus but not in its close relative Desultofobacterium hafniense. The membrane-bound transhydrogenase PntAB is absent in D. restrictus and in many Firmicutes species, reducing their flexibility in NADH/NADPH metabolism. Interestingly, the orthologs of o-phosphoserine phosphatase (SerB; EC 3.1.3.3), the enzyme catalyzing the final step in the classical serine biosynthesis pathway (Fig. 2a) [51], are notably absent in Firmicutes. Conservation of SerA gene in most Firmicutes likely results from the overlapping biosynthesis pathway between serine and pyridoxine, an essential cofactor involved in central metabolism (Fig. 1) [52].

Presence/absence of genes for enzyme orthologs in the TCA cycle, NADPH regeneration, and serine biosynthesis mapped to a species tree of organohalide-respiring bacteria and selected organisms. Solid green, orange, and purple bars show presence. Open circles represent cases where the enzyme activity is present in the corresponding organisms based on experimental evidence but corresponding genes for the enzyme orthologs were not found; the dashed line boxes highlight missing TCA cycle enzymes in Dehalobacter strains and the Bacteroides sp. in consortium ACT-3. Note that isocitrate dehydrogenase (Icd) in the TCA cycle is also involved in NADPH regeneration (orange star). SDH succinate dehydrogenase/fumarate reductase, Mdh malate dehydrogenase, Cit citrate synthase, Aco aconitase, MAE malic enzyme, MetF 5,10-methelene-tetrahydrofolate reductase/dehydrogenase, HndABCD (or HymABC) NADP+-reducing hydrogenase, NfnAB NADP+:ferredoxin oxidoreductase, PntAB membrane-bound transhydrogenase, SerA 3-phosphoglycerate dehydrogenase, SerB o-phosphoserine phosphatase, SerC o-phosphoserine aminotransferase, Thrald threonine aldolase, Shmt serine hydroxymethyltransferase, PdxB erythrose-4-phosphate dehydrogenase

Serine biosynthesis in Dehalobacter restrictus via threonine. a Schematic of amino acid biosynthesis (alanine, aspartate, glutamate, and serine; in dashed boxes) and carbon incorporation in Dehalobacter restrictus using [3-13C]pyruvate as the precursor. Serine is synthesized via the salvage pathway in D. restrictus due to the lack of SerB (highlighted in orange), the enzyme catalyzing the final step in the classical serine biosynthesis pathway. The 13C-labeled carbon originating from [3-13C]pyruvate is shown in red and the carbons derived from pyruvate are highlighted in cyan. b o-Phosphoserine phosphatase (SerB) activities in cell lysates of the strain PER-K23 and E. coli, respectively. (−) control, cell-lysate-free control. Tetrachloroethene dehalogenase (PceA) activity (trichloroethene as the substrate) was used as a quality control of the strain PER-K23 cell lysates. Data are means ± SE of three replicates in each experiment as shown on figures. c Relative abundance of different mass isotopomers (M1/M0) of serine, aspartate, alanine, and glutamate obtained from the strain PER-K23 cells cultivated on the defined medium supplemented with unlabeled pyruvate or [3-13C]pyruvate. Ac-CoA acetyl-CoA, AKG α-ketoglutarate, CH2 = THF 5,10-methylenetetrahydrofolate, MAL, OAA oxaloacetate, SER-Pi o-phosphoserine, SerA phosphoglycerate dehydrogenase, SerB o-phosphoserine phosphatase, SerC phosphoserine aminotransferase
Previously, we identified a promiscuous Dehalobacter serine hydroxymethyltransferase (Shmt; EC 2.1.2.1) possessing threonine aldolase activity [30], which allows serine salvage from threonine (Fig. 2a). Moreover, this promiscuous serine hydroxymethyltransferase is significantly upregulated in the proteome of the strain UNSWDHB (reaction ID 00692 in Table S1). Nevertheless, it is likely that a phylogenetically distant SerB or a promiscuous phosphatase with SerB-like activity is present in D. restrictus. We used D. restrictus cultures to validate these hypotheses. We first examined the growth (via trichloroethene (TCE) dechlorination activity) of H2-fed D. restrictus strain PER-K23 cultures supplemented with only acetate or with both acetate and serine as carbon sources. TCE dechlorination was only observed in cultures supplemented with both acetate and serine as carbon sources (Figure S1A). We then examined potential promiscuous phosphatase activity in D. restrictus cell lysates. While we observed dechlorinating activity in assays containing the D. restrictus cell lysates, dephosphorylation of o-phosphoserine was only observed in assays containing E. coli cell lysates (positive control) (Fig. 2b). These preliminary results indicate that SerB-dependent classical serine biosynthesis pathway is not functioning in D. restrictus cells.
Subsequently, we followed 13C incorporation to elucidate serine biosynthesis in D. restrictus. [3-13C]pyruvate or unlabeled pyruvate (5 mM), a precursor of serine in the classical pathway (Fig. 2a) [53], was supplemented to a mineral medium [23, 30] containing 1 mM acetate and 0.1 mM each of arginine, histidine, and threonine required to cultivate strain PER-K23 cultures [23]. Therefore, the 3-13C in pyruvate would be incorporated into serine if D. restrictus synthesizes serine via the classical pathway, and MS analysis of isotopomer distribution would reveal an enrichment in relative abundance of the M1 isotopomer of serine (M1/M0). Alternatively, the M1 serine isotopomer would remain at natural abundance if D. restrictus salvages serine from threonine (Fig. 2a). Consistent with this second option, serine obtained from both unlabeled and [3-13C]pyruvate-fed D. restrictus cells revealed a comparable M1/M0 (4.2–4.7%) (Fig. 2c). However, alanine, aspartate, and glutamate obtained from [3-13C]pyruvate-fed D. restrictus cells revealed at least threefold enrichment in M1/M0 (~15%). Altogether, based on genome annotation and multiple lines of experimental evidence, the classical serine biosynthesis pathway is absent in D. restrictus, and serine is synthesized from the salvage pathway (Fig. 2a). Interestingly, this salvage pathway is likely a major route for serine biosynthesis in all SerB-lacking Firmicutes and in Geobacter spp. (Fig. 1). Therefore, our results reinforce the need to consider the presence of peripheral or salvage pathways in bioinformatics-based predictions of microbial phenotypes.
Restricted NADPH regeneration in D. restrictus
The absence of the classical serine biosynthesis pathway in D. restrictus can result in many metabolic defects because serine is the cellular C1 pool donor and a precursor for purines and other amino acids, including glycine, cysteine, methionine, and tryptophan (Fig. 3a) [54]. Recent studies also discovered that serine can support NADPH regeneration via the folate cycle (Fig. 3a) [54, 55]. Furthermore, biosynthesis of serine through the classical pathway is NADPH-independent [53]. In contrast, the biosynthesis of serine from threonine increases the demand for NADPH for amino acid synthesis by approximately 30% (see Table under Fig. 3b), assuming a protein composition similar to that of Bacillus subtilis [56].

Amino acid dependency of Dehalobacter restrictus results from redox cofactor imbalance (NADPH and ferredoxin). a Proposed central carbon metabolism and redox cofactor regeneration system in Dehalobacter restrictus with acetate and malate as the carbon sources. The name of enzymes and the missing cofactors (in square bracket) are shown in blue. The red crosses (X) represent the missing genes or missing cofactors in the D. restrictus genome. NAD(P)+/NAD(P)H are shown in red beside the corresponding metabolic reactions. The black arrows represent the metabolic reactions involved in NADPH and ferredoxin regeneration, while gray arrows represent other reactions. Double-headed arrows indicate reversible reactions, and the bigger arrowhead indicates the direction of the reaction under physiological conditions. b Amino acid profile (serine concentration is shown in Fig. 4b) in the supernatants of strain PER-K23 cultures after the consumption of at Day 45 (Fig. 4a). The amounts of NADPH required to synthesize each amino acid via the classical pathway (SerB (+)) or salvage pathway (SerB (−)) are listed in the table below. b Data are means ± SE of three replicates in each experiment as shown on Figures. AC acetate, AC-CoA acetyl-CoA, AKG α-ketoglutarate, CH2 = THF 5,10-methylenetetrahydrofolate, CH2≡THF 5,10-methenyltetrahydrofolate, CH3-THF 5-methyltetrahydrofolate, CHO-THF 5-formyl-tetrahydrofolate, CIT citrate, FOR formate, FUM fumarate, FRD ferredoxin, ICIT isocitrate, MAL malate, OAA oxaloacetate, PYR pyruvate, SER-Pi o-phosphoserine, SUC succinate, SUC-CoA succinyl-CoA, THF tetrahydrofolate, CODH carbon monoxide dehydrogenase, FDH formate dehydrogenase, FUM/SDH, fumarate reductase/succinate dehydrogenase, MAE malic enzyme, MTHFD 5,10-methylenetetrahydrofolate dehydrogenase, MTHFR 5,10-methylenetetrahydrofolate reductase, MDH malate dehydrogenase, SerB o-phosphoserine phosphatase, WL pathway Wood–Ljungdahl pathway (or folate cycle)
According to the annotation of D. restrictus genomes, seven potential enzyme reactions can contribute to NADPH regeneration (Fig. 3a), including: a putative ferredoxin:NADP+ oxidoreductase (NfnAB; EC 1.6.1.4); a putative NADP+-reducing hydrogenase (HndABCD; EC 1.12.1.3); isocitrate dehydrogenase (EC 1.1.1.42); NADP-dependent malic enzyme (MAE; EC 1.1.1.40); MoCo-dependent NADP+-specific formate dehydrogenase (FdhAB; EC 1.2.1.43); and 5,10-methylenetetrahydrofolate reductase/dehydrogenase (MetF; EC 1.5.1.20; FolD; EC 1.5.1.5). However, D. restrictus cannot synthesize the molybdopterin cofactor [30] to functionalize formate dehydrogenase. Also, given that D. restrictus employs the TCA cycle and Wood–Ljungdahl pathway for anabolism but not respiration, NADPH regenerated from isocitrate dehydrogenase and 5,10-methylenetetrahydrofolate reductase/dehydrogenase is insufficient to support amino acid biosynthesis using acetate as the sole carbon source. Finally, the putative NADP+-reducing hydrogenase was not expressed in the proteomes of strains UNSWDHB and PER-K23, with the putative ferredoxin:NADP+ oxidoreductase found to be up-regulated only in the proteome of strain UNSWDHB (reaction IDs 14159 and 16350 in Table S1). Strain UNSWDHB was reported to grow on acetate as the sole carbon source [28] and was isolated from an acetate/H2-fed enrichment, while strain PER-K23 was isolated from lactate-fed enrichment cultures [39, 57]. Such difference in ferredoxin:NADP+ oxidoreductase expression among D. restrictus strains is consistent with the observed phenotypes.
Among the seven NADPH-regenerating enzymes, MAE can replenish the intracellular NADPH pool with malate supplementation (Fig. 3a). Also, D. restrictus genomes possess malate permease (maeP; Accession K4LDY2) to uptake malate, with MAE found to be expressed in the proteomes of strains UNSWDHB and PER-K23 and partial proteome of strain CF (reaction ID 00161 in Table S1). Thus we examined if malate supplementation can support D. restrictus growth on the acetate-based mineral medium. Consistently, strain PER-K23 cultures supplemented with acetate and malate showed a three-fold faster dechlorination rate than that of cultures supplemented with acetate and serine (Figure S1A). Supplementing acetate, malate and serine to strain PER-K23 cultures resulted in the highest and sustainable dechlorination rate. Therefore, the lack of SerB and a limited ability to regenerate NADPH likely explains the amino acid dependency of D. restrictus.
Unexpected pyruvate export in D. restrictus cultures
To examine the hypothesis of restricted NADPH regeneration in D. restrictus, we performed three consecutive 1% transfers of the strain PER-K23 cultures using the FeS-reduced mineral medium supplemented with both malate and serine but not acetate. Strain PER-K23 cultures were able to deplete 1 mM TCE after each of these three 1% transfers (Figure S1B). We then examined the long-term growth of strain PER-K23 cultures with repeated feedings without transfer. The cultures readily depleted three feedings of 1 mM TCE (Fig. 4a). qPCR analysis revealed that D. restrictus cell density in the cultures increased by 100-fold after the consumption of 3 mM TCE (~2 × 107 cells ml−1) (Fig. 4a). However, after the fourth feeding on Day 20, the cultures displayed a significant drop in dechlorination rate. Before each feed, cultures were purged with N2/CO2 to remove accumulated cis-dichloroethene, to adjust the pH, and to replenish the medium with H2. The metabolite profile of culture supernatants over this time revealed that only a small portion of the added malate (~50 µM) and serine (~0.15 mM) were consumed (Fig. 4b). Thus the slowing of dechlorination rate was not due to a shortage of electron donor/carbon sources, accumulation of cis-dichloroethene, or acidic pH. Unexpectedly, the metabolite profile of strain PER-K23 culture supernatants revealed pyruvate production (~0.1 mM) at Day 20, which can be produced from malate using MAE, from serine using serine deaminase (SdaA; EC 4.3.1.17), or from acetate using pyruvate:ferredoxin oxidoreductase (Fig. 3a). This unexpected pyruvate export suggests an unfavorable sink for pyruvate in D. restrictus cells. Indeed, the lack of malate dehydrogenase and succinate dehydrogenase/fumarate reductase genes prevents D. restrictus from fermenting pyruvate to succinate, and the absence of the classical serine pathway prevents pyruvate from entering the Wood–Ljungdahl pathway via serine (Fig. 3a). We presumed that the loss in dechlorination activity in the PER-K23 cultures after Day 20 (Fig. 4a) was likely related to these defects in pyruvate metabolism. Therefore we turned back to our metabolic flux model to further validate this hypothesis.

Growth assays of Dehalobacter restrictus strain PER-K23. a Cumulative tetrachloroethene (TCE) dechlorination profile (blue circles) and cell growth (brown squares) by the strain PER-K23 cultures cultivated on the defined mineral medium amended with malate and serine. When amended TCE was depleted (*), the cultures were purged with 80% N2/CO2 to remove dechlorination product cis-dichloroethene (cDCE) and re-fed with H2 (5 ml) and TCE (10 µl). b Time-course metabolite profile (malate, circles; serine, squares; pyruvate, triangles) in culture supernatants. The black-colored plots represent the killed controls. The slightly gradual increase in substrate concentration in the killed controls is due to water evaporation during culture purging. The cumulative TCE consumption is also shown. Data are means ± SE of three replicates in each experiment
Reconciling redox imbalance in D. restrictus using complementary FBA and experimental validation
We next simulated D. restrictus metabolism using the model constrained by experimental values for cell growth and substrate consumption (TCE, malate, and serine) in strain PER-K23 cultures at Day 20 (Fig. 4b). In addition to pyruvate, the resulting model also predicted significant CO export via ferredoxin-dependent CO dehydrogenase (CooS; EC 1.2.7.4) (Figure S2A), indicating excess reduced ferredoxin (FRDred) accumulation (Fig. 3a). Given that D. restrictus is grown with excess H2 as the electron donor, the model predicted FRDred production from the FRD-reducing hydrogenases (Ech) (reaction ID 05759 in Table S2). FRDred accumulation will inhibit the conversion of pyruvate to acetyl-CoA by pyruvate-ferredoxin oxidoreductase in central carbon metabolism (Table 1), resulting in acetyl-CoA shortage. Since acetyl-CoA is a substrate for citrate synthase in the oxidative TCA cycle, acetyl-CoA shortage would disable citrate synthesis, preventing pyruvate from entering the TCA cycle for glutamate biosynthesis (Fig. 3a), resulting in the slowing of dechlorination in strain PER-K23 cultures after Day 20 (Fig. 4a). If this is true, then acetate addition to strain PER-K23 cultures would replenish the acetyl-CoA pool in D. restrictus and ameliorate the redox cofactor imbalance. Consistently, acetate supplementation (1 mM) at Day 35 led to an apparent increase in dechlorination rate, and the strain PER-K23 cultures were able to deplete the remaining TCE and a subsequent feeding in 10 days (Fig. 4a), along with a significant pyruvate production (~0.3 mM) in parallel to the consumption of acetate (~0.15 mM), malate (~0.3 mM) and serine (~0.2 mM). In the first three feeding cycles, strain PER-K23 cells likely used the residual acetate in the medium components (≤0.1 mM based on HPLC analysis) to synthesize acetyl-CoA. These experiments suggest that the limited sink for pyruvate and excess FRDred production in D. restrictus resulted in pyruvate export.
After resolving redox cofactor imbalance in D. restrictus driven by FRD, we further constrained the model with experimental values of strain PER-K23 metabolites at Day 45, including pyruvate export (0.3 mM over 45 days; Fig. 4b). However, the resulting model revealed excess NADPH production from the consumed malate (0.3 mM) via the MAE reaction, unless the model allowed (a) export of citrate in the oxidative TCA cycle to prevent NADPH production via isocitrate dehydrogenase (Fig. 3a) or (b) export of glutamate, valine, leucine, and proline to consume NADPH (Figure S2B). However, citrate was not detected in the strain PER-K23 culture supernatants at Day 45 (<0.1 µM). Instead, metabolite profiling of the strain PER-K23 culture supernatants at Day 45 revealed predicted production of glutamate (~35 µM), valine (~20 µM), and iso/leucine (~20 µM) against Day 1 and the killed control (Fig. 3b), and these amino acids were not detected (<0.1 µM) in the culture supernatants of E. coli cultures cultivated on the defined medium at comparable cell density (4 × 107 cells ml−1). Moreover, the concentration of exported glutamate, leucine, and valine was higher than the concentrations of exported glycine and alanine, the most abundant amino acids in the biomass of Firmicutes (Ala, 1.25; Gly, 1.75 mmol per g of protein in Bacillius subtilis) [56]. Therefore, the observed export of glutamate, leucine and valine is unlikely due to cell lysis and protein degradation. Thus, D. restrictus appears to export these amino acids to consume excess NADPH when a NADPH-regenerating source like malate is available in the growth medium. Additionally, some alanine and aspartate were also detected in the culture supernatants. Since these two amino acids are directly downstream of pyruvate (Fig. 2a), the model also predicted that D. restrictus exports them to consume excess FRDred and to prevent excess NADPH production by isocitrate dehydrogenase (Fig. 3a, S2A). Altogether, our data suggested that both acetate and malate are required to sustain minimal growth of D. restrictus, and that serine supplementation can reduce the NADPH requirements of D. restrictus for amino acid biosynthesis.
In summary, our results provide an explanation for the previously identified arginine and threonine requirements of the strain PER-K23 [23]. Due to the lack of SerB, D. restrictus utilizes threonine to synthesize serine and derived amino acids. Also, due to a limited number of options for NADPH regeneration, arginine is degraded to malate via fumarate for NADPH regeneration (Fig. 3a) [58]. When malate (or fumarate) is available, D. restrictus becomes an amino acid producer, exporting amino acids to consume excess NADPH (Fig. 3b), along with the decarboxylated product pyruvate (Fig. 4b). This finding may explain the common co-occurrence of the amino acid- and pyruvate-fermenting Sedimentibacter spp. in D. restrictus-enriched consortia [29, 32, 59, 60]. However, amino/organic acid export also limits the growth of D. restrictus. Indeed, the average cell growth yield ((6.9 ± 0.26) × 1012 cells or ~0.8 g dry weight per mol of Cl− released) (Table S5) is lower than the reported values of its close relative Desulfitobacterium spp. ((3 ± 1) × 1013 cells or ~1.75 g dried weight per mol of Cl− released) [61]. Interestingly, other OHRB possess more flexible NADPH regeneration systems (Fig. 1). For example, the classical serine biosynthesis pathway is present and active in Dehalococcoides based on a 13C incorporation experiment, and an incomplete Wood–Ljungdahl pathway can support NADPH regeneration (Fig. 3a) [43]. Geobacter lovleyi, while lacking SerB, possesses a functional TCA cycle to oxidize acetate completely and generate sufficient NADPH using isocitrate dehydrogenase (Fig. 3a) [62, 63]. Finally, Desulfitobacterium hafniense, in contrast to its close relative D. restrictus, retains malate dehydrogenase and fumarate reductase genes in the TCA cycle, allowing pyruvate fermentation to malate for NADPH regeneration via MAE (Fig. 3a) [64].
Metabolic interdependencies in D. restrictus consortium ACT-3
After identifying D. restrictus’s carbon source requirement (acetate, malate, and serine), we wondered if these nutrients are available in a mixed consortium such as ACT-3. Acetate is a final product of acetogenic and fermenting bacteria that often co-exist in organohalide-respiring microbial communities [65], while the presence of malate or serine has not been reported before. Therefore, we analyzed the metabolites in supernatants of consortium ACT-3 during chloroform dechlorination to dichloromethane. After lactate and chloroform were fed, time-course metabolite profile revealed a time-dependent spike of malate (~0.5 µM) in the ACT-3 supernatants (Figure S3) along with accumulation of dichloromethane. The elevated malate concentration (relative to time zero) lasted until lactate was depleted. Traces of pyruvate were present, but the concentrations remained unchanged throughout dechlorination (Figure S3). We made sure that malate was not detected in the 1 M lactate stock based on LC–MS analysis (<50 nM). Therefore, malate is a natural substrate for D. restrictus in ACT-3. The detection of malate production in ACT-3 supernatant is consistent with: (a) the occurrence of a malate permease gene in D. restrictus genomes but not in its close relative D. hafniense; and (b) observed MAE expression in D. restrictus strains UNSWDHB and PER-K23 and in strain CF from consortium ACT-3. Also, finding available malate in ACT-3 supernatant suggests that D. restrictus has enough NADPH to drive a more NADPH-consuming pathway (SerB-independent pathway) for amino acid synthesis (see Table below Fig. 3b). However, in contrast to strain PER-K23 cultures, no amino acid, including serine, was detected (<0.1 µM) in ACT-3 supernatants. Therefore, the amino acid exchange in ACT-3 remains elusive.
We then managed to identify the malate producer in ACT-3. Based on 16S rRNA pyrotag sequences (accession SRX181448), an unculturable Bacteroides sp. is a dominant population in ACT-3 [39, 40, 60]. Fermentative malate/fumarate production by Bacteroides species has been studied extensively [66,67,68,69]. Bacteroides species lack the ability to synthesize heme, and therefore only ferment glucose to acetate, H2, and malate/fumarate via lactate [69], unless exogenous heme is present to support further malate fermentation to succinate (Fig. 5a). Similarly, the closed genome of the Bacteroides sp. in ACT-3 (accession CP006772) reveals complete pathways for lactate fermentation to acetate and succinate [31] and multiple genes for H2-producing hydrogenases (Hyf; EC 1.12.1.4) [70], but lacks most genes for heme biosynthesis. Moreover, citrate synthase, aconitase, and isocitrate dehydrogenase genes in the TCA cycle are missing from the closed genome of the Bacteroides sp. (Fig. 5b), which hamper glutamate biosynthesis. Therefore, these data, along with the specific malate requirement and unexpected glutamate export observed in D. restrictus cultures, seem to portend a complementary metabolic interdependency between D. restrictus and the Bacteroides sp. in ACT-3.

Proposed interspecies malate-pyruvate shuttle between Dehalobacter restrictus and the Bacteroides sp. in consortium ACT-3. a Proposed metabolic interdependencies between Dehalobacter restrictus and the Bacteroides sp. in ACT-3 including syntrophic lactate fermentation/chloroform dechlorination and exchange of malate, pyruvate, and amino acids. In this simplified ACT-3 model, lactate (LAC) is used as the sole electron donor and carbon source, and chloroform (CF) is used as the final electron acceptor. The red crosses (X) represent the missing genes in D. restrictus genomes; the orange square bracket [heme] represents the lack of heme cofactor due to a missing heme biosynthesis pathway in the closed genome of the Bacteroides sp. b Proposed genome streamlining between NADPH-restricted D. restrictus and the glutamate-auxotrophic Bacteroides sp. demonstrated by complementary genes in the TCA cycle and in malate metabolism (present in D. restrictus, red; present in the Bacteroides sp., blue; present in both, purple). c Dechlorination profile of 4% ACT-3 transfer cultures provided with lactate only (Heme (−)); lactate and heme (Heme (+)); or lactate, heme, and malate (0.25 mM; Heme (+) and MAL). A second addition of lactate (0.5 mM) was given to all transfer cultures on Day 12 after supernatant sampling. d LC–MS analysis of malate (red bar) and pyruvate (orange bar) in the supernatants of Heme (−) and Heme (+) cultures at Day 12. The black asterisks indicate that the metabolites were undetectable. e Microbial community composition (%) in Heme (−) and Heme (+) cultures at Day 18 determined using 16S rRNA gene amplicon sequencing. AC acetate Ac-CoA acetyl-CoA, DCM dichloromethane, FUM fumarate, OAA oxaloacetate, SUC succinate, MAE malic enzyme, MDH malate dehydrogenase. Data are means ± SE of three replicates in each experiment
Based on literature and integrated multi-omics data, we propose a metabolic interdependency in ACT-3 (Fig. 5a). Given that (a) fermentative malate production from lactate is thermodynamically unfavorable under standard conditions (Table 1) and (b) heme is insoluble in water (<2 nM) [71] and never added to the growth medium, lactate is mainly fermented to H2, acetate and CO2 by the Bacteroides sp. Nevertheless, fermentative malate production from lactate would become thermodynamically favorable when (a) the H2 production is coupled to organohalide respiration by D. restrictus (Table 1) and (b) an efficient consumer is present to lower malate concentration. Therefore, we expect that the Bacteroides sp. exports malate to consume excess NADH generated from lactate and to facilitate NADPH regeneration in D. restrictus. In return, D. restrictus exports glutamate and potentially other amino acids to consume excess NADPH and to facilitate growth of the Bacteroides sp. Since pyruvate did not accumulate in ACT-3 (Figure S3), the exported pyruvate from D. restrictus is likely recycled by other community members, including the Bacteroides sp., which shapes an intercellular metabolic cycle, enabling D. restrictus and the Bacteroides sp. to indirectly exchange NADH/NADPH across the cell membrane, resembling the function of a transhydrogenase (Table 1) [72].
Decoupling malate–pyruvate exchange in consortium ACT-3 via heme addition
We sought to validate the hypothesis of proposed malate–pyruvate exchange in ACT-3. Given that Bacteroides species ferment lactate to malate as a result of heme-auxotrophy, heme addition to ACT-3 would enable further malate fermentation to succinate and would reduce H2 production in the Bacteroides sp., thereby limiting growth and dechlorination of D. restrictus (Fig. 5a). On the other hand, if heme is intrinsically available for the Bacteroides sp. in ACT-3, heme addition should not cause an apparent effect on growth and dechlorination of D. restrictus in ACT-3. Therefore, we monitored the effect of heme addition to ACT-3 transfer cultures under limiting electron donor conditions (one time electron equivalents needed for dechlorination). After 12 days of incubation, chloroform dechlorination (50 µM) only occurred in ACT-3 transfer cultures without heme addition (Heme (−) cultures) but not in cultures supplemented with 1 mg l−1 heme (Heme (+) cultures) (Fig. 5c). Moreover, malate and pyruvate were only detected in supernatants of Heme (−) cultures but not in supernatants of Heme (+) cultures at Day 12 (Fig. 5d). In contrast, Heme (+) cultures supplemented with malate (0.25 mM) revealed significant chloroform dechlorination (0.2 mM). These data indicate that chloroform dechlorination in Heme (+) cultures is limited by the shortage of electron donors and/or malate. Consistently, when we provided excess electron donor lactate (three times electron equivalents needed for dechlorination) to the transfer cultures at Day 12, all three cultures depleted chloroform within 6 days. Finally, since fumarate reduction to succinate in Bacteroides species is coupled to the generation of a proton motive force, heme supplementation will enable Bacteroides species to synthesize more ATP via oxidative phosphorylation rather than via substrate-level phosphorylation [66, 73] (Table 1), resulting in higher cell density. Consistently, the relative abundance of the Bacteroides sp. population in Heme (+) cultures at Day 18 is 10 times higher than that in Heme (−) cultures (Fig. 5e).
Previously, we found that D. restrictus can synthesize heme de novo, an essential cofactor in Hup-type hydrogenases used for H2 oxidation during respiration [30]. Therefore, we expected that D. restrictus could cross-feed heme to the Bacteroides sp. in ACT-3. However, our data suggest that heme is not accessible for the Bacteroides sp. in ACT-3. Moreover, the ABC-type heme transporter is expressed in both proteomes of strains PER-K23 (Dehre_1618) and UNSWDHB (unswdhb_2853) that were cultivated on heme-free medium [34, 35]. Perhaps the persistent expression of the heme uptake transporter in D. restrictus prevents heme leakage from its cells, thereby limiting heme for the Bacteroides sp. in ACT-3 for further malate fermentation to succinate. Taken all together, the data reported herein support the occurrence of malate–pyruvate exchange in ACT-3.
Implications for microbial ecology
In this study, the proposed malate–pyruvate exchange in consortium ACT-3 resembles the mitochondrial malate–pyruvate shuttle in eukaryotic cells [74, 75]. In the eukaryotic malate–pyruvate shuttle, cytoplasmic pyruvate is first transported to mitochondria, and is reduced to malate via NADH-dependent malate dehydrogenase in the TCA cycle (Fig. 3a). Malate is then exported to the cytoplasm via an antiporter, and decarboxylated to pyruvate by MAE for NADPH regeneration. Although the paring of metabolic partners in nature can appear random, the complementary gaps in the TCA cycle and malate metabolism between genomes of D. restrictus and the Bacteroides sp. in ACT-3 (complementarity illustrated in Fig. 5b) are likely a consequence of genome streamlining driven by co-adaption. This idea is supported by the presence of malate dehydrogenase and succinate dehydrogenase/fumarate reductase genes in its closed relative D. hafniense but not in D. restrictus (dashed boxes in Fig. 1). Also, the oxidative TCA cycle is present in the genomes of all available Bacteroides isolates but not in the genome of the Bacteroides sp. in ACT-3. Accordingly, this study has found a relevant example of a bacterial interspecies malate–pyruvate shuttle to buttress an endosymbiotic hypothesis proposing that the fusion of an ancestral mitochondrial-like organism and an ancestral eukaryotic-like organism was driven by organic acid exchange [76]. It thus seems that this “malate-pyruvate shuttle” is possibly a common strategy of organisms to circumvent the impermeability of cell membranes to essential reducing equivalents (i.e., NADH/NADPH).
In conclusion, the data present in this study demonstrate that metabolic complementarity and redox imbalance (e.g., NADPH regeneration and FRD accumulation) are important driving forces for metabolite exchange in anaerobic microbial communities. Furthermore, the success of our model predictions further emphasizes the need for careful curation to improve the accuracy of gene and metabolic annotations through consideration of cofactor availability, enzyme promiscuity, and potential salvage pathways. Finally, finding D. restrictus as a potential amino acid producer in parent consortium ACT-3 reinforces the caveat that observed phenotypes of isolate cultures sometimes deviate from their functions in ecosystems. Therefore, integration of informed laboratory experiments with iterative cycles of computational metabolic modeling offers great opportunities to decipher the metabolic interdependencies of fastidious, or currently unculturable, microbes.
References
Dolfing J. Syntrophy in microbial fuel cells. ISME J. 2013;8:4.
Embree M, Liu JK, Al-Bassam MM, Zengler K. Networks of energetic and metabolic interactions define dynamics in microbial communities. Proc Natl Acad Sci. 2015;112:15450–5.
Mee MT, Collins JJ, Church GM, Wang HH. Syntrophic exchange in synthetic microbial communities. Proc Natl Acad Sci. 2014;111:E2149–56.
Zhao M, Xue K, Wang F, Liu S, Bai S, Sun B, et al. Microbial mediation of biogeochemical cycles revealed by simulation of global changes with soil transplant and cropping. ISME J. 2014;8:2045–55.
Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10:538.
Seth EC, Taga ME. Nutrient cross-feeding in the microbial world. Front Microbiol. 2014;5:350.
Cardona C, Weisenhorn P, Henry C, Gilbert JA. Network-based metabolic analysis and microbial community modeling. Curr Opin Microbiol. 2016;31:124–31.
Mori M, Ponce-de-León M, Peretó J, Montero F. Metabolic complementation in bacterial communities: necessary conditions and optimality. Front Microbiol. 2016;7:1553.
Wintermute EH, Silver PA. Emergent cooperation in microbial metabolism. Mol Syst Biol. 2010;6:407–407.
McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10:13.
Wade W. Unculturable bacteria—the uncharacterized organisms that cause oral infections. J R Soc Med. 2002;95:81–83.
Manor O, Levy R, Borenstein E. Mapping the inner workings of the microbiome: genomic- and metagenomic-based study of metabolism and metabolic interactions in the human microbiome. Cell Metab. 2014;20:742–52.
Roling WF, van Bodegom PM. Toward quantitative understanding on microbial community structure and functioning: a modeling-centered approach using degradation of marine oil spills as example. Front Microbiol. 2014;5:125.
Tan J, Zuniga C, Zengler K. Unraveling interactions in microbial communities - from co-cultures to microbiomes. J Microbiol. 2015;53:295–305.
Magnúsdóttir S, Thiele I. Modeling metabolism of the human gut microbiome. Curr Opin Biotechnol. 2018;51:90–96.
Zhuang K, Izallalen M, Mouser P, Richter H, Risso C, Mahadevan R, et al. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 2011;5:305.
Suthers PF, Zomorrodi A, Maranas CD. Genome‐scale gene/reaction essentiality and synthetic lethality analysis. Mol Syst Biol. 2009;5:301.
Amador-Noguez D, Feng X-J, Fan J, Roquet N, Rabitz H, Rabinowitz JD. Systems-level metabolic flux profiling elucidates a complete, bifurcated tricarboxylic acid cycle in Clostridium acetobutylicum. J Bacteriol. 2010;192:4452–61.
McInerney MJ, Sieber JR, Gunsalus RP. Syntrophy in anaerobic global carbon cycles. Curr Opin Biotechnol. 2009;20:623–32.
Sieber JR, McInerney MJ, Gunsalus RP. Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annu Rev Microbiol. 2012;66:429–52.
Adrian L, Loeffler FE. Organohalide-respiring bacteria, vol. 85. Berlin, Heidelberg, Germany: Springer; 2016.
Duhamel M, Edwards EA. Growth and yields of dechlorinators, acetogens, and methanogens during reductive dechlorination of chlorinated ethenes and dihaloelimination of 1,2-dichloroethane. Environ Sci Technol. 2007;41:2303–10.
Holliger C, Hahn D, Harmsen H, Ludwig W, Schumacher W, Tindall B, et al. Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra- and trichloroethene in an anaerobic respiration. Arch Microbiol. 1998;169:313–21.
Justicia-Leon SD, Ritalahti KM, Mack EE, Löffler FE. Dichloromethane fermentation by a Dehalobacter sp. in an enrichment culture derived from pristine river sediment. Appl Environ Microbiol. 2012;78:1288–91.
Tang S, Wang PH, Higgins S, Löffler F, Edwards EA. Sister Dehalobacter genomes reveal specialization in organohalide respiration and recent strain differentiation likely driven by chlorinated substrates. Front Microbiol. 2016 https://doi.org/10.3389/fmicb201600100.
van Doesburg W, van Eekert MH, Middeldorp PJ, Balk M, Schraa G, Stams AJ. Reductive dechlorination of beta-hexachlorocyclohexane (beta-HCH) by a Dehalobacter species in coculture with a Sedimentibacter sp. FEMS Microbiol Ecol. 2005;54:87–95.
Wang S, Zhang W, Yang KL, He J. Isolation and characterization of a novel Dehalobacter species strain TCP1 that reductively dechlorinates 2,4,6-trichlorophenol. Biodegradation. 2014;25:313–23.
Wong YK, Holland SI, Ertan H, Manefield M, Lee M. Isolation and characterization of Dehalobacter sp. strain UNSWDHB capable of chloroform and chlorinated ethane respiration. Environ Microbiol. 2016 https://doi.org/10.1111/1462-292013287.
Yoshida N, Ye L, Baba D, Katayama A. A novel Dehalobacter species is involved in extensive 4,5,6,7-tetrachlorophthalide dechlorination. Appl Environ Microbiol. 2009;75:2400–5.
Wang PH, Tang S, Nemr K, Flick R, Yan J, Mahadevan R, et al. Refined experimental annotation reveals conserved corrinoid autotrophy in chloroform-respiring Dehalobacter isolates. ISME J. 2016;11:626–40.
Tang S, Edwards EA. Complete genome sequence of Bacteroidales strain CF from a chloroform-dechlorinating enrichment culture. Genome Announc. 2013a;1:e01066–13.
Maphosa F, van Passel MW, de Vos WM, Smidt H. Metagenome analysis reveals yet unexplored reductive dechlorinating potential of Dehalobacter sp. E1 growing in co-culture with Sedimentibacter sp. Environ Microbiol Rep. 2012;4:604–16.
Correia K, Ho H, Mahadevan R (2018). Genome-scale metabolic network reconstruction of the chloroform-respiring Dehalobacter restrictusstrain CF. bioRxiv. https://doi.org/10.1101/375063
Jugder BE, Ertan H, Wong YK, Braidy N, Manefield M, Marquis CP, et al. Genomic, transcriptomic and proteomic analyses of Dehalobacter UNSWDHB in response to chloroform. Environ Microbiol Rep. 2016;8:814–24.
Rupakula A, Kruse T, Boeren S, Holliger C, Smidt H, Maillard J. The restricted metabolism of the obligate organohalide respiring bacterium Dehalobacter restrictus: lessons from tiered functional genomics. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120325.
Rupakula A, Lu Y, Kruse T, Boeren S, Holliger C, Smidt H, et al. Functional genomics of corrinoid starvation in the organohalide-respiring bacterium Dehalobacter restrictus strain PER-K23. Front Microbiol. 2015;5:751.
Tang S, Edwards EA. Identification of Dehalobacter reductive dehalogenases that catalyse dechlorination of chloroform, 1,1,1-trichloroethane and 1,1-dichloroethane. Philos Trans R Soc Lond B Biol Sci. 2013b;368:20120318.
Ebrahim A, Lerman JA, Palsson BO, Hyduke DR. COBRApy: constraints-based reconstruction and analysis for python. BMC Syst Biol. 2013;7:74.
Grostern A, Edwards EA. A 1,1,1-trichloroethane-degrading anaerobic mixed microbial culture enhances biotransformation of mixtures of chlorinated ethenes and ethanes. Appl Environ Microbiol. 2006;72:7849–56.
Grostern A, Duhamel M, Dworatzek S, Edwards EA. Chloroform respiration to dichloromethane by a Dehalobacter population. Environ Microbiol. 2010;12:1053–60.
Puentes Jácome LA, Edwards EA. A switch of chlorinated substrate causes emergence of a previously undetected native Dehalobacter population in an established Dehalococcoides-dominated chloroethene-dechlorinating enrichment culture. FEMS Microbiol Ecol. 2017;93:fix141.
Chen Y-L, Fu H-Y, Lee T-H, Shih C-J, Huang L, Wang Y-S, et al. Identification of estrogen degradation pathway and estrogen degraders in an activated sludge. Appl Environ Microbiol. 2018 https://doi.org/10.1128/AEM.00001-18.
Zhuang W-Q, Yi S, Bill M, Brisson VL, Feng X, Men Y, et al. Incomplete Wood–Ljungdahl pathway facilitates one-carbon metabolism in organohalide-respiring Dehalococcoides mccartyi. Proc Natl Acad Sci. 2014;111:6419–24.
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, et al. A new view of the tree of life. Nat Microbiol. 2016;1:16048.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77.
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL‐DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinforma. 2011;6.12:11–16.12. 19.
Zdobnov EM, Tegenfeldt F, Kuznetsov D, Waterhouse RM, Simao FA, Ioannidis P, et al. OrthoDBv9. 1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2016;45:744–9.
He Z, Zhang H, Gao S, Lercher MJ, Chen W-H, Hu S. Evolviewv2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016;44:W236–41.
Kuznetsova E, Proudfoot M, Gonzalez CF, Brown G, Omelchenko MV, Borozan I, et al. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J Biol Chem. 2006;281:36149–61.
Greenberg DM, Ichihara A. Further studies on the pathway of serine formation from carbohydrate. J Biol Chem. 1957;224:331–40.
Lam H, Winkler ME. Metabolic relationships between pyridoxine (vitamin B6) and serine biosynthesis in Escherichia coli K-12. J Bacteriol. 1990;172:6518–28.
Grundy FJ, Henkin TΜ. In: Sonenshein AL, Hoch JA, Losick R (eds.) Chapter 18: Synthesis of serine, glycine, cysteine, and methionine. In Bacillus subtilis and its closest relatives. Washington, DC: ASM Press, 2002; p. 245–54.
Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298.
Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi S, et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013;4:e877.
Dauner M, Sauer U. Stoichiometric growth model for riboflavin‐producing Bacillus subtilis. Biotechnol Bioeng. 2001;76:132–43.
Holliger C, Schraa G, Stams A, Zehnder A. A highly purified enrichment culture couples the reductive dechlorination of tetrachloroethene to growth. Appl Environ Microbiol. 1993;59:2991–7.
Cunin R, Glansdorff N, Pierard A, Stalon V. Biosynthesis and metabolism of arginine in bacteria. Microbiol Rev. 1986;50:314.
Imachi H, Sakai S, Kubota T, Miyazaki M, Saito Y, Takai K. Sedimentibacter acidaminivorans sp. nov., an anaerobic, amino-acid-utilizing bacterium isolated from marine subsurface sediment. Int J Syst Evol Microbiol. 2016;66:1293–300.
Tang S, Gong Y, Edwards EA. Semi-automatic in silico gap closure enabled de novo assembly of two dehalobacter genomes from metagenomic data. PLoS One. 2012;7:e52038.
Ding C, Zhao S, He J. A Desulfitobacterium sp. strain PR reductively dechlorinates both 1,1,1-trichloroethane and chloroform. Environ Microbiol. 2014;16:3387–97.
Galushko AS, Schink B. Oxidation of acetate through reactions of the citric acid cycle by Geobacter sulfurreducens in pure culture and in syntrophic coculture. Arch Microbiol. 2000;174:314–21.
Sung Y, Fletcher KE, Ritalahti KM, Apkarian RP, Ramos-Hernandez N, Sanford RA, et al. Geobacter lovleyi sp. nov. strain SZ, a novel metal-reducing and tetrachloroethene-dechlorinating bacterium. Appl Environ Microbiol. 2006;72:2775–82.
Peng X, Yamamoto S, Vertès AA, Keresztes G, Inatomi K-i, Inui M, et al. Global transcriptome analysis of the tetrachloroethene-dechlorinating bacterium Desulfitobacterium hafniense Y51 in the presence of various electron donors and terminal electron acceptors. J Ind Microbiol Biotechnol. 2012;39:255–68.
Heimann AC, Batstone DJ, Jakobsen R. Methanosarcina spp. drive vinyl chloride dechlorination via interspecies hydrogen transfer. Appl Environ Microbiol. 2006;72:2942–9.
Macy J, Probst I, Gottschalk G. Evidence for cytochrome involvement in fumarate reduction and adenosine 5’-triphosphate synthesis by Bacteroides fragilis grown in the presence of hemin. J Bacteriol. 1975;123:436–42.
Macy JM, Ljungdahl LG, Gottschalk G. Pathway of succinate and propionate formation in Bacteroides fragilis. J Bacteriol. 1978;134:84–91.
Miller T. The pathway of formation of acetate and succinate from pyruvate by Bacteroides succinogenes. Arch Microbiol. 1978;117:145–52.
Chen M, Wolin MJ. Influence of heme and vitamin B12 on growth and fermentations of Bacteroides species. J Bacteriol. 1981;145:466–71.
Trchounian K, Trchounian A. Escherichia coli hydrogenase 4 (hyf) and hydrogenase 2 (hyb) contribution in H2 production during mixed carbon (glucose and glycerol) fermentation at pH 7.5 and pH 5.5. Int J Hydrog Energy. 2013;38:3921–9.
Ketchum MA, Olafson KN, Petrova EV, Rimer JD, Vekilov PG. Hematin crystallization from aqueous and organic solvents. J Chem Phys. 2013;139:09B611_611.
Voordouw G, Vies SM, Themmen AP. Why are two different types of pyridine nucleotide transhydrogenase found in living organisms? FEBS J. 1983;131:527–33.
Madej MG, Nasiri HR, Hilgendorff NS, Schwalbe H, Unden G, Lancaster CRD. Experimental evidence for proton motive force-dependent catalysis by the diheme-containing succinate: menaquinone oxidoreductase from the Gram-positive bacterium Bacillus licheniformis. Biochemistry. 2006;45:15049–55.
Liu YQ, Jetton TL, Leahy JL. β-Cell adaptation to insulin resistance increased pyruvate carboxylase and malate-pyruvate shuttle activity in islets of nondisbetic zucker fatty rats. J Biol Chem. 2002;277:39163–8.
MacDonald MJ. Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets further implication of cytosolic NADPH in insulin secretion. J Biol Chem. 1995;270:20051–8.
Searcy DG. Metabolic integration during the evolutionary origin of mitochondria. Cell Res. 2003;13:229.
Flamholz A, Noor E, Bar-Even A, Milo R. eQuilibrator—the biochemical thermodynamics calculator. Nucleic Acids Res. 2011;40:770–5.
Acknowledgments
Support was provided by the Government of Ontario through Genome Ontario SPARK Research Grant. We also acknowledge the BioZone Mass Spectrometry facility for UPLC-ESI-HRMS analyses. We are grateful to the gift of active Dehalobacter restrictus strain PER-K23 culture from Prof. Frank Löffler in University of Tennessee (Knoxville, USA).
Author contributions
E.A.E., R.M., and P.H.W. conceptualized this study. C.H. and K.C. re-constructed the Dehalobacter model. K.C. performed the Flux Balance Analysis. P.H.W. performed the experiments. K.N. performed the organic acid analysis. R.F. performed the LC–MS analysis. P.H.W. and N.V. proposed the mechanism of interspecies malate–pyruvate shuttle. E.A.E., R.M., and P.H.W. wrote this paper with helps from all the authors. All the authors participated in data analysis and discussion.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Wang, PH., Correia, K., Ho, HC. et al. An interspecies malate–pyruvate shuttle reconciles redox imbalance in an anaerobic microbial community. ISME J 13, 1042–1055 (2019). https://doi.org/10.1038/s41396-018-0333-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0333-4
This article is cited by
-
Common principles and best practices for engineering microbiomes
Nature Reviews Microbiology (2019)
