
Abstract
Lucinidae clams harbor gammaproteobacterial thioautotrophic gill endosymbionts that are environmentally acquired. Thioautotrophic lucinid symbionts are related to metabolically similar symbionts associated with diverse marine host taxa and fall into three distinct phylogenetic clades. Most studies on the lucinid–bacteria chemosymbiosis have been done with seagrass-dwelling hosts, whose symbionts belong to the largest phylogenetic clade. In this study, we examined the taxonomy and functional repertoire of bacterial endosymbionts at an unprecedented resolution from Phacoides pectinatus retrieved from mangrove-lined coastal sediments, which are underrepresented in chemosymbiosis studies. The P. pectinatus thioautotrophic endosymbiont expressed metabolic gene variants for thioautotrophy, respiration, and nitrogen assimilation distinct from previously characterized lucinid thioautotrophic symbionts and other marine symbionts. At least two other bacterial species with different metabolisms were also consistently identified in the P. pectinatus gill microbiome, including a Kistimonas-like species and a Spirochaeta-like species. Bacterial transcripts involved in adhesion, growth, and virulence and mixotrophy were highly expressed, as were host-related hemoglobin and lysozyme transcripts indicative of sulfide/oxygen/CO2 transport and bactericidal activity. This study suggests the potential roles of P. pectinatus and its gill microbiome species in mangrove sediment biogeochemistry and offers insights into host and microbe metabolisms in the habitat.
Similar content being viewed by others

Introduction
Chemosymbiosis is widespread in marine habitats, where endosymbiotic or episymbiotic chemolithoautotrophs use inorganic chemical energy for the synthesis of organic compounds that benefit their hosts [1, 2]. One of the most ancient examples of marine chemosymbiosis is found in the bivalve family Lucinidae [3], which has a fossil record arguably dating back to the Silurian period [4]. Despite being capable of suspension feeding, all living lucinids studied to date fulfill a considerable proportion of their nutritional needs through obligate chemosymbiotic associations with gammaproteobacterial endosymbionts occupying bacteriocytes in their gills [3]. Lucinid species examined so far acquire their thioautotrophic endosymbionts from free-living environmental bacterial populations [5,6,7,8,9]. Enzymatic assays, stable isotope analyses, and clone-based amplicon sequencing methods demonstrate that lucinid endosymbionts mainly use energy derived from the oxidation of reduced sulfur compounds to fix inorganic carbon for their hosts [10]. Other reported functions of lucinid endosymbionts included mixotrophy, denitrification, assimilation of nitrogenous compounds, and diazotrophy [11,12,13,14].
Because of the widespread distribution of lucinids in marine habitats, ranges in host and endosymbiont phylogenetic diversity, as well as the possibility that lucinids may harbor non-thioautotrophic symbionts [15,16,17], the lucinid–bacteria chemosymbiotic system has the potential to address fundamental cellular to ecological questions about host–symbiont interactions, cues, and communication across individual hosts, among species, and within populations. However, there is still relatively poor understanding of lucinid and gill microbiome diversity and metabolic functions. For instance, although 16S rRNA gene sequences of thioautotrophic lucinid endosymbionts form a paraphyletic group consisting of three distinct clades [10, 18], only the genomes, transcriptomes, and proteomes of two lucinid endosymbiont species from clade A have been sequenced [13, 14]. Clade A symbionts are mainly associated with diverse seagrass-dwelling lucinids, but symbiont clades B and C are from predominately mangrove-dwelling Anodontia spp. and Phacoides pectinatus, respectively [18]. Almost no diversity or functional diversity study has centered on either of these bacterial clades.
To begin to fill these gaps, our study characterizes the metabolic repertoire of the host and gill-associated thioautotrophic bacterial endosymbiont from P. pectinatus Gmelin 1791 (syn = Tellina pectinata Gmelin 1791, Lucina pectinata (Gmelin 1791), Anodontia pectinatus (Gmelin 1791), Lucina jamaicensis Lamarck 1801, Lucina funiculata Reeve 1850). Possibly the only extant species of its genus, P. pectinatus possesses morphological features distinct from other lucinid bivalves, such as high levels of three types of hemoglobin in gill pigment granules, sulfur bodies, and large lysosomes [19, 20]. Molecular phylogeny studies place P. pectinatus as a deeply branching genus within the Lucinidae [21] and the thioautotrophic endosymbiont distant from seagrass- or other mangrove-associated lucinid endosymbionts [18, 22, 23]. This lucinid inhabits organic-rich seagrass and mangrove sediments [24] and has a widespread tropical geographic distribution that ranges from the Caribbean Sea and Gulf of Mexico to the Atlantic Ocean seaboard of South America to Brazil [25]. The unusual morphological features, phylogeny, and habitat distribution of P. pectinatus and its distinct thioautotrophic endosymbiont belonging to clade C have led to the hypothesis that symbiont metabolic pathways in this species are different than in other lucinid endosymbionts [8]. To test this hypothesis, we assessed gill microbiome diversity within P. pectinatus using 16S rRNA gene sequencing, quantitative PCR (qPCR), metagenomic sequencing, and metatranscriptomic sequencing and compared the expression profiles from P. pectinatus and its gill microbiome species to previously sequenced seagrass-associated lucinid endosymbiont species from clade A, including Ca. Thiodiazotropha endoloripes within Loripes orbiculatus [14] and Ca. Thiodiazotropha endolucinida within Codakia orbicularis [13].
Materials and methods
Sample collection
Phacoides pectinatus populations at Wildcat Cove, St. Lucie County, FL, USA (Figure S1), as well as their ecology, sediment geochemistry, and microbiology, have previously been investigated [23, 26] and briefly described in SI. For this study, research excursions were completed in February 2011, June 2013, July 2014, and November 2017, and live specimens were sieved from sediments hand-dug to 30 cm depth, approximately 3 m from the shoreline of Rhizophora mangle (red mangrove). Specimens were temporarily stored in Whirl-Pak® Bags (Nasco, Fort Atkinson, WI, USA) filled with surface water from the habitat and maintained at ambient temperature before dissection. During dissection, gill and foot tissues were separated from other body tissues. Tissues used for 16S rRNA gene sequencing and metagenomics were dissected within the same day of collection and fixed in 100% molecular-grade ethanol. Tissues used for metatranscriptomics were dissected within 30 min of collection and fixed in RNAlater. Tissues used for microscopy were fixed in 2% paraformaldehyde (pH 7) made with artificial sea water prepared using Difco™ Marine Broth 2216 formula (Becton Dickinson and Company, Franklin Lakes, NJ, USA) for 3 h at 4 °C prior to washing, sucrose infiltration, storage, hematoxylin–eosin staining, and fluorescence in situ hybridization (FISH) procedures described in Table S1 and SI. Total nucleic acids were extracted from partial gill and foot tissues using the Qiagen’s (Valencia, CA, USA) DNeasy Blood and Tissue Kit (2011 and 2013 sample collection) or Allprep DNA/RNA Mini Kit (2014 samples) after mechanical homogenization of the tissues with a motorized pestle and mortar (Argos Technologies, Elgin, IL, USA) or tissue grinder (Wheaton, Millville, NJ, USA). For further lysis, the sample was passed through a 21-gauge (0.8 mm) needle attached to a 3 mL syringe (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) at least ten times and incubated at 60 °C for at least 10 min. Extracted nucleic acid concentrations were quantified fluorometrically with Qubit™ dsDNA HS and RNA assays (Life Technologies, Austin, TX, USA).
16S rRNA gene, metagenomic, and metatranscriptomic sequencing
From the 2014 collection, 16S rRNA gene libraries of DNA extracted from 25 P. pectinatus gills, cDNA from the gills of four of these individuals, and DNA from the feet of three individuals were sequenced. From the 2017 collection, libraries of DNA and cDNA extracted from three gill samples were sequenced. All libraries were sequenced on Illumina Inc’s (San Diego, CA, USA) MiSeq 2 × 250 bp platform. 16S rRNA gene library preparation procedures are described in SI. Thirteen Illumina-compatible gill metagenomic libraries and one foot metagenomic library were prepared using the Nextera DNA Sample Preparation Kit (Illumina Inc., San Diego, CA, USA) on 50 ng of DNA per sample (2011 and 2013 collection) and NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® on DNA fragmented with NEBNext® dsDNA Fragmentase (New England Biolabs, Ipswich, MA, USA; 2014 collection). These libraries were sequenced on Illumina’s MiSeq 2 × 150 bp (2011 collection), 2 × 250 bp platforms (2013 collection), and HiSeq 2500 2 × 100 bp (2014 specimen) platforms. To generate long reads, metagenomic libraries were prepared from another 2014 gill specimen and two 2017 gill specimens using the Nanopore’s Rapid Sequencing Kit (Oxford Nanopore Technologies, Kidlington, Oxfordshire, UK) and sequenced on a MinIon flowcell (R9.4 nanopores) with a MinIon Mk1B sequencer. Three gill samples collected in 2014 within a 1 m2 quadrat were used for metatranscriptomic sequencing on Illumina’s HiSeq 4000 2 × 150 bp platform. RNAs extracted from these samples were treated with the Ambion® Turbo DNA-free™ DNase Kit (Life Technologies). Successful DNase treatment was confirmed by PCR amplification of the V9 region of bacterial 16S rRNA genes and subsequently through read-mapping analysis of sequenced metatranscriptomic libraries (SI). DNA-free RNAs were purified with the RNeasy MinElute Cleanup Kit (Qiagen). Illumina-compatible cDNA libraries were made from purified RNAs using Epicentre’s (Madison, WI, USA) Ribo-Zero rRNA removal Kit (bacteria) and ScriptSeq™ v2 RNA-Seq Library Preparation Kit, following the manufacturer’s low input protocol. The final concentration of each library was quantified with the Qubit® dsDNA HS assay (Life Technologies) and the average library insert size was determined with the Experion Automated Electrophoresis Station (Bio-Rad Laboratories, Hercules, CA, USA; 2011 and 2013 collections) and the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA; 2014 and 2017 collections). Sequencing service providers are listed in SI.
Data analysis
Mothur v1.39.5 [27] was used for data processing for the 16S rRNA gene dataset. Operational Taxonomic Unit (OTU) clustering was performed at 99% sequence identity for higher species resolution [28] and taxonomic classification was performed against the Silva v132 database [29]. The final dataset was subsampled to the library with the smallest four-digit number size (n = 1269). The 16S rRNA gene analysis pipeline and qPCR procedures used to validate analysis results are documented in Table S2 and SI. Trimmed Illumina-sequenced metagenomic reads from each sequenced sample were individually assembled using IDBA-UD v1.0.9 [30]. Additionally, reads from the most complete gammaproteobacterial assembly were co-assembled with unprocessed Nanopore reads using the hybridSPAdes algorithm [31] of the SPAdes genome assembler (v3.11.1). For each assembly, contigs >1500 bp long were binned with MetaBat v0.32.4 using the ensemble binning approach [32], after read mapping with Bowtie2 v2.2.7 [33] (very sensitive local and dovetail mode) and SAMtools v0.1.19 [34]. All metagenome-assembled genomes (MAGs) were annotated with NCBI’s Prokaryotic Genome Annotation Pipeline [35]. MAGs with >90% completeness were also annotated with Rapid Annotation using Subsystem Technology (RAST) FIGfam release 70 [36]. Methods for the evaluation of sequence and MAG quality, read trimming, sequencing depth analyses, sequence comparisons with published reference genomes, and PCR validation of sequencing results are in Table S2 and SI.
Metatranscriptomic assembly and downstream analyses were performed with Trinity v2.5.1 [37]. Trimmed reads from all three metatranscriptomic libraries were co-assembled into one metatransciptome de novo with Trinity’s default parameters (k = 20). The co-assembly standardizes transcript IDs, lengths, and clusters across libraries for efficient downstream quantification and cross-sample comparisons [37]. Trinity’s Chrysalis module clusters transcripts with at least k−1 bases overlap and with sufficient reads spanning the join across both transcripts, and the Butterfly module refines the clustering and uses these transcript clusters as proxy for genes [37, 38]. Reads were mapped to the co-assembly using Bowtie2 v2.2.7’s [33] no-mixed, no-discordant, end-to-end options reporting up to 200 alignments per read (-k 200) and disallowing gaps within 1000 nucleotides of read extremes (-gbar 1000). Isoform and gene-level abundances were estimated by RNA-Seq Expectation-Maximization (RSEM) that maximizes the probability of observed variables, including read lengths, quality scores, and sequences based on RSEM’s directed graph statistical model [39]. The probability value for each isoform/gene was divided by the effective transcript/gene length, which is the average number of possible start positions of a transcript of a given length or the abundance-weighted average effective transcript lengths of a gene’s isoforms [39]. The resulting length-normalized value for each transcript/gene was divided by the sum of length-normalized values for all transcripts/genes in each sample to calculate the transcript fraction value, which was then multiplied by 106 to derive the transcript per million (TPM) measure [39]. For cross-sample comparisons, TPM values were further normalized with the trimmed means of M-values (TMM) factor that minimizes log-fold changes across samples [40] using the edgeR Bioconductor package [41].
All assembled host and bacterial transcripts, as well as unbinned contigs from metagenomic assemblies, were annotated with Trinotate v3.1.1 (https://trinotate.github.io/), which uses the manually curated but less representative Swissprot [42] database as reference. rRNA transcripts were predicted with SortMeRNA v2 [43] using SILVA’s v119 [29] collection of archaeal, bacterial, and eukaryotic 16S rRNA, 23 S rRNA, 18 S rRNA, and 28 S rRNA gene sequences as references. Host and bacterial genes of interest were analyzed at the level of transcript clusters loosely equivalent to genes. To map transcript clusters to symbiont genes, a pan-genome for the thioautotrophic endosymbiont from P. pectinatus, named Candidatus Sedimenticola endophacoides (explained in the Results section), was created by extracting and concatenating nucleotide sequences of RAST-annotated PEGs and RNAs from six >90% complete MAGs, followed by de-duplication with CD-HIT v4.6 [44] at a global sequence identity threshold of 100%. The de-duplicated dataset was searched against the Trinity assembly using NCBI’s Basic Local Alignment Search Tool (BLAST) v2.6.0+ local blastn package [35, 45] and only the top hit was reported (-max_target_seqs 1). Similar local blastn searches were performed on other MAGs of interest for transcript cluster to gene mapping. Functions of transcript clusters of interest were inferred by comparing Trinotate’s transcript annotations with web blastp, blastn, or blastx search results [45] against the more representative NCBI’s non-redundant (nr) protein sequence or nucleotide (nt) databases [35] using the same 10−3 e-value threshold as Trinotate. For each transcript within a transcript cluster, a blastp search was performed if a likely peptide sequence was predicted by Transdecoder v5.1.0 (http://transdecoder.github.io/) based on a minimum open reading frame (ORF) length and a log-likelihood score related to the reading frame where the ORF was located. If the blastp search returned negative results or if no likely peptide sequence was predicted for a transcript, then blastn and blastx searches were performed instead. Functions of transcript clusters mapping to more than one gene were assigned based on annotations of transcript(s) within the cluster with the highest TMM-normalized TPM value(s). A transcript cluster was considered multi-mapping if more than one transcript within the cluster shared high TMM-normalized TPM values but different predicted functions and their corresponding genes were not adjacent to each other in the reference MAG.
Results
Site characterization
Live P. pectinatus had clumped distributions at Wildcat Cove in all sample years, with the highest concentrations being near the mangrove-lined coast where total organic carbon content in the sediment was highest [26]. Overall, live abundances averaged over 40 individuals per square meter [26]. Porewater dissolved sulfide and oxygen concentrations were measured and reported by Doty [26] from low-flow fluid sampling of piezometers installed near to where specimens were recovered, according to previously described methods [46]. Dissolved sulfide concentrations at Wildcat Cove (18–56 μmol/L) were an order of magnitude higher than concentrations measured from intertidal zone porewater occupied by the lucinid Lucinoma borealis [47]. Dissolved oxygen concentrations ranged from 78 to 125 μmol/L at quadrats adjacent to where P. pectinatus were collected [26].
Gill microbiome diversity
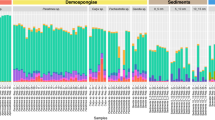
To examine P. pectinatus microbiome diversity, we sequenced 16S rRNA genes and used metagenomic and metatranscriptomic content from gill and foot samples collected in 2011, 2014, and 2017. All amplicon-sequenced DNA and cDNA samples were dominated by one gammaproteobacterial Sedimenticola-like species (OTU1), occurring at average 84 ± 11% relative abundance (Fig. 1a). MAGs of this species were binned from 14 separate assemblies and three co-assemblies (Table 1) and shared 100% sequence identity in the 16S rRNA gene V4 region with OTU1, as well as 99.8 ± 0.4% average nucleotide identity (ANI) and 99 ± 1% average amino acid identity (AAI; Figure S2a) with each other. These gammaproteobacterial MAGs were at least 20% smaller, and with at least 11% higher G+C content, than previously sequenced clade A thioautotrophic lucinid endosymbiont species Ca. Thiodiazotropha endoloripes [14] and Ca. Thiodiazotropha endolucinida [13] and Sedimenticola spp. [48, 49, 118] (Table 1). FISH using a newly designed SED642 probe targeting the 16S rRNA gene of this Sedimenticola-like species confirmed that the P. pectinatus gill bacteriocytes contained cells that matched the gammaproteobacterial MAGs (Figure S3). Results of phylogenetic analyses using 16S rRNA gene sequences (Figure S4) and ten single-copy marker genes (Figure S2b) corroborated previous reports on the distinct phylogenetic position of the thioautotrophic P. pectinatus endosymbiont in relation to other lucinid symbiont species [18, 22, 23]. The Sedimenticola-like MAGs shared 71 ± 4% ANI and 64 ± 1% AAI with sequenced clade A lucinid symbiont species, 76 ± 7% ANI and 59 ± 5% AAI with other marine thioautotrophic symbionts, and 76 ± 2% ANI and 69 ± 1% AAI to free-living Sedimenticola spp. [48, 49, 118] (Figure S2a). Based on the 93–95% ANI and 85–90% AAI boundaries proposed in Rodriguez-R and Konstantinidis [50], the Sedimenticola-like MAGs were likely a species separate from sequenced clade A lucinid symbionts, marine thioautotrophic symbionts, and Sedimenticola spp. Because the Sedimenticola-like MAGs shared the highest AAI with Sedimenticola spp. and the observed AAI values fall within the proposed genus boundary (55–60%) [50], we propose the name Candidatus Sedimenticola endophacoides for the P. pectinatus endosymbiont, where “endophacoides” refers the host association (“endo-” meaning “within”).

Relative abundances of a bacterial 16S rRNA gene OTUs and Good’s estimator of coverage [114], b copy numbers per ng of DNA or cDNA (%) of Sedimenticola-like OTU1, Kistimonas-like OTU2, and Spirochaeta-like OTU5 determined by qPCR, and c normalized average coverage depths with standard error bars mapped to Ca. Sedimenticola endophacoides, Kistimonas-like, and Spirochaeta-like MAGs in P. pectinatus foot and gill specimens/libraries. “R” denotes RNA-derived cDNA specimens in a, b and metatranscriptomic libraries in c. Foot-associated Christensenella-like OTU3 indicated with “#” in a was classified using 0% bootstrap confidence
Besides the thioautotrophic symbiont species, we also observed lower relative abundances of a gammaproteobacterial Kistimonas-like OTU (average 13 ± 12%; OTU2) belonging to the order Oceanospirillales in all amplicon-sequenced DNA and cDNA samples and a Spirochaeta-like OTU (average 0.2 ± 0.2%; OTU5) in 25 out of the 33 gill DNA and cDNA samples (Fig. 1a). The transcriptional activity of the Sedimenticola-like, Kistimonas-like, and Spirochaeta-like species was confirmed by absolute qPCR quantification, where copy numbers of the OTUs in matched DNA and cDNA samples were consistent with their OTU relative abundances (Fig. 1b). Deep metagenomic sequencing of one 2014 P. pectinatus gill sample also binned a Kistimonas-like MAG (3% of reads), a Spirochaeta-like MAG (0.4% of reads), a Ca. Sedimenticola endophacoides MAG (58% of reads; Table 1), and 12 other bins with 0% completeness and no taxonomic classification. These three MAGs contained 16S rRNA gene sequences with perfect matches to their corresponding OTU sequences. Unbinned contigs comprised 89% (527,385/591,741) of all assembled contigs from this sample, out of which only 11% (59,232/527,385) had predicted protein-coding regions (SI). Reads from all sequenced metagenomic and metatranscriptomic libraries could be mapped to MAGs of the Kistimonas-like (0.4 ± 0.4% of MiSeq metagenomic reads and 0.1 ± 0.04% of metatranscriptomic reads) and Spirochaeta-like species (1 ± 0.3% of MiSeq metagenomic reads and 0.008 ± 0.003% of metatranscriptomic reads) at lower sequencing depths compared to the Ca. Sedimenticola endophacoides MAG (8 ± 4% of MiSeq metagenomic reads and 1 ± 0.6% of metatranscriptomic reads; Fig. 1c).
MAGs of Ca. Sedimenticola endophacoides, the Kistimonas-like species, and the Spirochaeta-like species shared <70% ANI and <56% AAI with each other (Figure S2a). Phylogenetic analyses using 16S rRNA gene sequences clustered the Kistimonas-like OTU sequences with potentially pathogenic K. scapharcae from a dead ark clam Anadara broughtonii [51], skin-associated K. asteriae from the starfish Asterias amurensis [52], and gill-associated Oceanospirillales from the limid bivalve Acesta excavata [53] (Figure S4). The Spirochaeta-like OTU sequence was most closely related to spirochete endosymbionts in the gutless marine worm Olavius [54, 55] and loosely associated with spirochete sequences retrieved from a L. kazani-like lucinid [56] (Figure S4). Genomic sequences of these closest relatives of both species are not yet available in public databases. Foot microbiome diversity in P. pectinatus is described in SI.
Metagenomic and metatranscriptomic analyses
Sequenced gill cDNA libraries showed consistent read coverages of the co-assembled metatranscriptome and pairwise Pearson correlations of >0.8 across replicates (Figure S5). A total of 1,563,787 transcripts were assembled, out of which 85% (average length 364 ± 262 bp) were without protein-coding region and functional annotation (SI). In all, 11% of the total transcripts (average length 989 ± 1181 bp) could be mapped to gene/protein homologs. These were grouped into 91,465 transcript clusters (loosely equivalent to genes), from which a subset (3%) mapped to the bacterial MAGs of interest. As such, it should be noted that the quality of gene/transcript annotations is heavily dependent on the completeness of the MAGs and the reference databases used. Although we made every effort to search for absent genes and pathways in the unbinned gill metagenomes, incompletely binned MAGs used to make inferences may still contain missing genes and functions. The lack of host genomic data and the high abundances of unclassifiable sequences in the gill metagenomes and metatranscriptomes imply that functional analyses can be skewed toward annotated genes/transcripts that would overlook novel genes [57]. Also, gene/transcript annotations based on homology may not be accurate predictors of reaction mechanisms [57] and even function (in the case of novel paralogs). Transcript quantification can also be influenced by swift changes in mRNA expression occurring between sample collection and fixation, as well as mRNA turnover that causes rapidly degrading mRNAs to exhibit inaccurately low transcripts per million (TPM) values.
Host-related functions
Host-related rRNA gene transcript clusters made up two thirds of the 30 most abundantly expressed transcripts in the gill metatranscriptomes (Figure S6). Highly expressed eukaryotic and/or molluscan protein-coding genes included those encoding the respiratory cytochrome c oxidase subunits, hemoglobins 1 and 2, and actin (Fig. 2). A carbonic anhydrase transcript cluster related to the mangrove killifish (Kryptolebias marmoratus) was the 11th most abundantly expressed in the gill metatranscriptome (average 696 ± 260 TPM; Fig. 2a), while another molluscan transcript cluster encoding for a nacrein-like protein with putative carbonic anhydrase function [58] was expressed in only 1 out of the 3 specimens at 0.1 TPM. The top 30 most abundant molluscan transcript clusters also included transcripts encoding hemoglobin 3 (average 104 ± 25 TPM), ribosomal proteins (average 32 ± 18 TPM), other cytoskeletal proteins (tubulin and tropomyosin; average 33 ± 19 TPM), and lysozyme 3 (average 19 ± 20 TPM; Fig. 2b). Similarly, transcript clusters matching gene ontology [59] terms relevant to hemoglobin, cytoskeletal, and ribosomal functions were among the most abundant in the phylum Mollusca (Figure S7). Transcript clusters involved in the defense response to bacteria (GO:0042742; Figure S7) were potentially relevant to symbiosis. These included molluscan transcript clusters encoding lysozyme 1 (average 6 ± 4 TPM), lysozyme 3, an antibacterial glycoprotein aplysianin-A [60]/muscosal glycoprotein achacin [61] (average 7 ± 2 TPM), the H2O2-generating flavoenzyme L-amino oxidase [62] (average 7 ± 1 TPM), and nitric oxide synthase (average 0.6 ± 1 TPM).

Log2-transformed TMM-normalized TPM of gene products of the 30 most abundantly expressed protein-coding transcript clusters a mapped to any species and b mapped to the phylum Mollusca in sequenced P. pectinatus gill metatranscriptomes (specimens R1, R2, and R3). UDP uridine diphosphate, ORF open reading frame
Endosymbiont functions
Sixteen of the 30 most abundant bacteria-related transcript clusters could be mapped to Ca. Sedimenticola endophacoides, while 8 mapped to the species’ relatives (Fig. 3a). Many of these were housekeeping and stress response genes (Fig. 3 and SI). Candidatus Sedimenticola endophacoides expressed lithoautotrophic genes involved in sulfur oxidation, hydrogen oxidation, and carbon fixation (Figs. 3–5). Transcript clusters involved in thiotrophic sulfur oxidation (sox) and reverse dissimilatory sulfite reductase enzyme system-adenylylsulfate reductase-sulfate adenylyltransferase (dsr-apr-sat) pathways [63, 64] were detected in the transcriptome at TPM values between 0.07 (DsrK) and 55 (SoxZ; Figs. 3–4). Variants of sulfide:quinone oxidoreductase (Sqr), hydrogenases, and ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO) genes utilized by chemosynthetic marine symbionts differed across lineages (Table 2), and Ca. Sedimenticola endophacoides expressed a unique combination of type VI Sqr (average 0.09 ± 0.1 TPM), group 1 membrane-bound (average 0.2 ± 0.2 TPM) and group 2b soluble NAD-dependent (average 2 ± 2 TPM) Ni-Fe hydrogenases, and type II RuBisCO (average 0.08 ± 0.06 TPM) genes (Figs. 4 and 5). Expressed heterotrophy-related genes included those involved in dicarboxylate transport (average 0.2 ± 0.3 TPM) and a complete tricarboxylic acid (TCA) cycle (average 0.4 ± 0.8 TPM; Fig. 5a). Candidatus Sedimenticola endophacoides is capable of respiration on oxygen and nitrogenous compounds (average 0.2 ± 0.4 TPM; Fig. 3b). However, compared to other chemosynthetic marine symbionts that utilize a variety of terminal oxidases for aerobic respiration, we only detected genes and transcript clusters encoding subunits for the cbb3-type terminal oxidase (average 0.4 ± 0.5 TPM) in Ca. Sedimenticola endophacoides (Table 2). Candidatus Thiodiazotropha spp. are capable of nitrogen fixation and assimilatory nitrate and nitrite reduction [13, 14], and relevant transcripts mapped to Ca. Thiodiazotropha endoloripes, but not Ca. Sedimenticola endophacoides, were identified in the gill metatranscriptomes at average 0.3 ± 0.3 TPM (Table S4 and SI). Key genes in these pathways were, however, not detected in the sequenced P. pectinatus gill metagenomes (Tables S4–S5 and SI), suggesting that the transcripts were rare. MAGs of Ca. Sedimenticola endophacides encoded and expressed genes for urease and the urease accessory protein UreE (0.09 ± 0.1 TPM), urea ABC transporter (0.2 ± 0.2 TPM), and ammonium transporter (average 0.06 ± 0.1 TPM; Figure S8). Transcripts involved in type I, II, and possibly type III and VI, secretion systems were also observed in this species (Figures S9 and SI). Particularly, like Ca. Thiodiazotropha spp., Ca. Sedimenticola endophacoides may utilize the type I secretion system [65] potentially for the secretion of hemolysin A (average 1 ± 1 TPM), colicin V (average 0.4 ± 0.4 TPM), and repeats in toxin (average 0.2 ± 0.03 TPM; Figure S9). A transcript cluster encoding a hypothetical filamentous hemagglutinin N-terminal domain-containing iron-responsive protein (average 104 ± 80 TPM) secreted by the two-partner secretion system [66] was also the fifth most abundant in the bacterial metatranscriptomes (Fig. 3a). Genes to combat H2O2 stress, including those encoding the hydrogen peroxide-inducible genes activator (average 0.3 ± 0.3 TPM), superoxide dismutase (average 0.05 ± 0.06 TPM), and an alkyl hydroperoxide reductase subunit C-like protein (average 0.7 ± 0.8 TPM) were also expressed in Ca. Sedimenticola endophacoides. Other genetic functions in Ca. Sedimenticola endophacoides are presented in Table S6 and SI.

Log2-transformed TMM-normalized TPM of gene products of a the 30 most abundantly expressed protein-coding transcript clusters mapped to species (square brackets) from the domain Bacteria and b morphological features and major metabolic pathways predicted in Ca. Sedimenticola endophacoides. In b, the transcript cluster mapped to a non-thioautotrophic gammaproteobacterial species (Endozoicomonas numazuensis) is highlighted in green while transcript clusters mapped to non-gammaproteobacterial taxa are highlighted in pink. UbiE ubiquinone/menaquinone biosynthesis C-methyltransferase, FtsH ATP-dependent zinc metalloprotease, Hyb membrane bound [Ni-Fe] hydrogenase 2, Hup uptake hydrogenase, Hox soluble NAD-dependent hydrogenase, S0 elemental sulfur, Fcc flavocytochrome c-sulfide dehydrogenase, Sqr sulfide:quinone oxidoreductase, Sox sulfur oxidation enzyme complex, Dsr reverse dissimilatory sulfite reductase enzyme system, Apr adenylylsulfate reductase, APS adenosine-5’-phosphosulfate, Sat sulfate adenylyltransferase, ABC ATP-binding cassette transporters, GS glutamine synthetase, GOGAT glutamine oxoglutarate aminotransferase (glutamate synthase), Nap periplasmic dissimilatory nitrate reductase, Nir cytochrome nitrite reductase cd1, Nor nitric oxide reductase, Nos nitrous oxide reductase, TBDT TonB-dependent transporter, TonB TonB-ExbB-ExbD complex, FeoB ferrous iron transport protein, Pst phosphate specific transport, Pho phosphate regulon, PolyP polyphosphate granule, ActP acetate permease, TRAP tripartite ATP-independent periplasmic transport, RuBisCO ribulose-1,5-bisphosphate carboxylase/oxygenase, TCA cycle tricarboxylic acid cycle, IM inner membrane, OM outer membrane

Log2-transformed TMM-normalized TPM of gene products of lithotrophy-related transcript clusters mapped to Ca. Sedimenticola endophacoides. Transcript clusters with zero TPM values are represented as white cells. Sqr sulfide:quinone oxidoreductase, Fcc flavocytochrome c-sulfide, Sat sulfate adenylyltransferase, Apr adenylylsulfate reductase, Tus sulfur carrier proteins homologous to some Dsr proteins, Hyp hydrogenase pleiotropy operon involved in the biosynthesis and maturation of [Ni-Fe] hydrogenases, Hup regulatory uptake hydrogenase, Hox soluble NAD-dependent hydrogenase, Hyd periplasmic Ni-Fe hydrogenase, HdrA/MVH heterodisulfide reductase/methylviologen reducing hydrogenase, Hyb membrane-bound Ni-Fe hydrogenase 2, HyaC membrane-bound Ni-Fe-hydrogenase I cytochrome b subunit

Log2-transformed TMM-normalized TPM of transcript clusters encoding gene products involved in carbon metabolism mapped to a Ca. Sedimenticola endophacoides, b the Kistimonas-like species, and c the Spirochaeta-like species. Transcript clusters with zero TPM values are represented as white cells. TRAP tripartite ATP-independent periplasmic transport, TCA cycle tricarboxylic acid cycle, HMP hexose monophosphate shunt, Dct dicarboxylate transport proteins, Cbb proteins encoded by the Calvin–Bassham–Benson cycle operon, BCKDH complex branched-chain alpha-keto acid dehydrogenase complex, LacI lactose operon repressor
Other gill microbiome functions
Highly expressed protein-coding transcript clusters homologous to protein sequences from other non-thioautotrophic bacterial taxa, including Tepidimonas spp., Persicobacter sp., and Bacillus ginsengihumi, were also observed in the gill metatranscriptomes (Fig. 3a). A transcript cluster encoding a hypothetical DNA starvation/stationary phase protection protein from Endozoicomonas numazuensis, a relative of the Kistimonas-like species, was also identified (Fig. 3a). Seven of the 30 most abundant transcript clusters mapped to the Kistimonas-like species encoded transposases (average 3 ± 4 TPM; Fig. 6a). Two transcript clusters encoding poly(hydroxyalcanoate) granule-associated protein (phasin) involved in the fermentative synthesis of polyhydroxyalkanoate storage granules [67] were also highly expressed in the species (average 1 ± 1 TPM; Fig. 6a). Heterotrophy-related genes associated with other fermentation processes were expressed by the species at lower average TPM values of 0.09 ± 0.08, along with TCA cycle genes (average 0.2 ± 0.3 TPM; Fig. 5b and Fig. 6c). Transcript clusters linked to fatty acid catabolism and synthesis, including those involved in the glyoxylate cycle (average 0.4 ± 0.5 TPM) [68], methylcitrate cycle (average 0.08 ± 0.2 TPM) [69, 70], and the branched-chain alpha-keto acid dehydrogenase complex (BCKDH complex; average 0.1 ± 0.1 TPM) [71, 72], were also observed (Fig. 5b). A transcript cluster encoding a type VI secretion system-associated protein (average 0.5 ± 0.5 TPM) was among the most abundant in the species’ transcriptomes (Fig. 6a). The Kistimonas-like species likely respires aerobically with both cbb3-type cytochrome c oxidase (average 0.07 ± 0.07 TPM) and cytochrome bd ubiquinol oxidase (average 0.2 ± 0.3 TPM). For nitrogen assimilation (Fig. 6c), only two genes encoding NAD(P)H-dependent assimilatory nitrite reductase (average 0.02 ± 0.03 TPM; consistent with PCR results in SI) and type I glutamine synthetase (average 0.06 ± 0.1 TPM) were expressed in the species.

Log2-transformed TMM-normalized TPM of gene products of the 30 most abundantly expressed protein-coding transcript clusters mapped to a the Kistimonas-like species and b the Spirochaeta-like species and major metabolic pathways predicted in c the Kistimonas-like species and d the Spirochaeta-like species. Transcript clusters with zero TPM values in a, b are represented as white cells. MFS major facilitator superfamily transporter, Nas assimilatory nitrate reductase, Nit assimilatory nitrite reductase, GS glutamine synthetase, GOGAT glutamine oxoglutarate aminotransferase (glutamate synthase), Fd-GOGAT ferrodoxin-dependent glutamate synthase, Pst phosphate-specific transport, Pho phosphate regulon, PolyP polyphosphate granule, TBDT TonB-dependent transporter, ABC ATP-binding cassette transporters, DcuB C4-dicarboxylate uptake family transporter, SDH succinate dehydrogenase, FRD fumarate reductase, TCA cycle tricarboxylic acid cycle, TRAP tripartite ATP-independent periplasmic transport, ECF energy-coupling factor transporter
The most abundant transcript clusters mapped to the ~78% complete MAG of the lower abundance Spirochaeta-like species encoded transporters for ribose (average 0.2 ± 0.2 TPM) and oligopeptide (average 0.1 ± 0.2 TPM; Fig. 6b). Besides ribose, the species could potentially utilize other carbon sources through transcripts encoding sugar ABC transporter substrate-binding protein (average 0.09 ± 0.02 TPM), chitinase (average 0.06 ± 0.1 TPM), glycoside hydrolase (average 0.05 ± 0.08 TPM), and C4-dicarboxylate ABC transporter substrate-binding protein (average 0.05 ± 0.08 TPM; Fig. 5c). Other genetic functions in the Kistimonas-like species and Spirochaeta-like species are presented in Fig. 6, Tables S6 and SI.
Discussion
Systems-level approaches utilizing next-generation sequencing technologies successfully reveal host–microbe and microbe–microbe interactions in different invertebrate symbioses [73,74,75,76] but have not been widely applied to lucinid–bacteria chemosymbioses. Currently, the lack of genomic, transcriptomic, and proteomic data for lucinids hosting gammaproteobacterial clades B and C thioautotrophic endosymbionts results in a poor understanding of the metabolism, inter- and intra-species diversity, and molecular interactions between these partners that may impact their surrounding coastal habitat and other organisms in the environment. In this study, we focused on describing the gill microbiomes of the mangrove-dwelling P. pectinatus that hosts the poorly characterized clade C lucinid endosymbiont species. This is the first investigation to describe the functional repertoire of (1) a lucinid symbiont species belonging to clade C, (2) a lucinid clam, and (3) other bacterial species in a lucinid gill microbiome. Our comparative genomics analyses showed thioautotrophy, respiration, and nitrogen assimilation metabolic differences among the clade C P. pectinatus endosymbionts, clade A lucinid symbionts, and other thioautotrophic marine symbionts, while host transcriptomes revealed candidate genes putatively involved in symbiont/microbiome selection, regulation, and nutrient transfer. Metagenomic and metatranscriptomic analyses also uncovered consistency among members of the gill microbiome, including a Kistimonas-like species and a Spirochaeta-like species that have previously been associated with a variety of marine invertebrates but not yet been comprehensively studied in lucinid clams. Additional insights into the lucinid-bacteria chemosymbiosis is now possible, and these findings may help in species conservation, habitat management [77,78,79], and even in fisheries productivity [80], which are areas of ongoing research.
Compared to previously sequenced lucinid clade A endosymbiont species and other thioautotrophic symbionts, Ca. Sedimenticola endophacoides encoded a unique combination of low-affinity type VI Sqr that functions best at high sulfide concentrations [81, 82], form II RuBisCO that is less efficient at discriminating between oxygen and CO2 [83], and the high affinity cbb3-type terminal oxidase that performs best at low oxygen concentrations [84]. These genomic differences suggest that Ca. Sedimenticola endophacoides experiences a more oxygen-poor extracellular and/or intracellular environment compared to Ca. Thiodiazotropha spp. Although pore water sulfide concentrations at Wildcat Cove were higher than previous studies [47], pore water dissolved oxygen concentrations were similar to those from sub-tropical coastal mangroves [85] and seagrass rhizomes [86] that have the potential to harbor lucinids. Sulfide and oxygen levels in the clam gills are likely regulated through hemoglobins, which can be partially saturated with oxygen [87]. As such, sulfide-reactive hemoglobin 1, which has a higher oxygen dissociation rate than oxygen-reactive hemoglobins 2 and 3, may be confined to the symbiotic mollusc gills [88]. In support of previous literature, we observed high expression levels of host-related hemoglobin 1, 2, and 3 genes responsible for sulfide and oxygen transport [88,89,90]. Despite genomic evidence for the maintenance of low intracellular oxygen that would be conducive for nitrogen fixation, which can contribute to the lucinid’s diet and seagrass health [13, 14, 91], Ca. Sedimenticola endophacoides, unlike Ca. Thiodiazotropha spp., is likely incapable of diazotrophy. In lieu of nitrogen fixation, we speculate that Ca. Sedimenticola endophacoides may utilize urea and ammonium as its nitrogen source because these transcripts were detected.
Expression levels of autotrophy-related transcripts encoding RuBisCO and Calvin cycle enzymes in relation to other transcripts were much lower for Ca. Sedimenticola endophacoides than previously reported in Ca. Thiodiazotropha endoloripes [14] and other symbiotic bivalve species that expressed RuBisCO form Iaq [75, 92], where these transcripts were among the most abundant in the transcriptomes. Low RuBisCO protein levels (~1%) were similarly observed in the tubeworm Riftia pachyptila thioautotrophic symbiont, which was discovered to produce proteins involved in an additional oxygen-sensitive reductive TCA cycle [93,94,95]. Although Ca. Sedimenticola endophacoides expressed genes encoding 2-oxoglutarate oxidoreductase that may reverse the 2-oxoglutarate to succinyl-CoA step in the TCA cycle, we did not identify any gene for citrate lyase or citryl–coenzyme A synthetase subunit that potentially converts citrate to oxaloacetate or acetate [93, 95]. Mixotrophy has previously been inferred in Ca. Thiodiazotropha endoloripes [14], as well as thioautotrophic symbionts in a variety of other marine organisms [96,97,98], and is a likely possibility for Ca. Sedimenticola endophacoides because of encoded and expressed genes associated with the dicarboxylate transport and TCA cycle, as well as the correlation of P. pectinatus live abundances to sediment organic carbon content [26]. However, gene expression and geochemical data are insufficient support for proven mixotrophy, and more carbon assimilation experiments will be needed to determine such mechanisms in Ca. Sedimenticola endophacoides.
Besides Ca. Sedimenticola endophacoides, we also identified genes and transcripts belonging to other bacterial taxa in the P. pectinatus gill metagenomes and metatranscriptomes. Transcripts mapped to Ca. Thiodiazotropha endoloripes were noted in the gill metatransciptomes and could originate from unbinned contigs in the gill metagenomes or closely related species co-occurring in the gill microbial population. In all sequenced gill samples, we observed the consistent presence of a Kistimonas-like species related to the metabolically versatile Oceanospirillales species that can be symbiotic [99,100,101,102], parasitic [103], or pathogenic [51, 104]. In bivalves, parasitic Oceanospirillales have been identified from nuclei in the vent mussel Bathymodiolus spp. [103]. Another Oceanospirillales species with unknown functions was also reported in gills from A. excavata [53]. Consistent with previous genomic reports on Oceanospirillales species, we observed high expression of various families of transposases in the Kistimonas-like species, which may facilitate rapid adaptation to new hosts or environments [105,106,107]. We also identified lower relative abundances of a Spirochaeta-like species in most gill samples, as well as transcriptional evidence of their activity. Spirochete species have been associated with a L. kazani-like lucinid [56], the symbiotic gutless oligochete worm Olavius [55, 108], and episymbionts of the hydrothermal vent worm Alvinella pompejana [109].
Metatranscriptomic analyses showed that these three bacterial species may utilize distinct carbon sources. Specifically, Ca. Sedimenticola endophacoides may participate in mixotrophy in addition to thioautotrophy, whereas the Kistimonas-like species performs fermentation and fatty acid catabolism, and the Spirochaeta-like species breaks down chitin, sugars, and dicarboxylate compounds. To identify cellular locations of the Kistimonas-like and the Spirochaeta-like species within the host gill tissue, we designed multiple FISH probes targeting various 16S rRNA gene regions of the Kistimonas-like and Spirochaeta-like species, as these species showed positive DNA and cDNA amplification from gill specimens. However, in contrast to positive FISH signals for Ca. Sedimenticola endophacoides, we repeatedly failed to get unambiguous true positive signals for the Kistimonas-like and Spirochaeta-like species. This could be because of the low abundances of these species within the tissue samples, the hybridization efficiency of the designed probes, the resolution of the confocal microscopy, and/or other technical issues. Without microscopic data, we are unable to determine the location of these species and entirely rule out that they could be environmental contaminants, transient gill-filtered bacteria, pathogens, or parasites. More sensitive techniques, such as catalyzed reporter deposition–FISH [110] and hybridization chain reaction [111], should be performed to validate the presence of these bacteria species in the gills of P. pectinatus.
Our gill metatranscriptomic analyses also revealed potential host–microbiota interactions involved in the establishment and maintenance of lucinid–bacteria relationships. In P. pectinatus, transfer of nutrients, including carbon and possibly B vitamins and cofactors, from symbiont to host may predominantly occur via host lysosomal digestion. The high abundances of host-associated lysozyme-encoding transcripts observed in this study may indicate the presence of active lysosomes, supporting previous reports of lysosomes in the host gills [19] and in the vent mussel Bathymodiolus azoricus [75]. We speculate that host selection may include the secretion of bactericidal lysozyme and other compounds (SI), which can be countered by gill microbiome species. Presumably to decrease competition from closely related species/strains, as speculated in the Eupyrmna–Vibrio symbiosis [112], Ca. Sedimenticola endophacoides encoded and expressed genes for the production and secretion of bactericidal colicin [113], which were also annotated in the Kistimonas-like species MAG. A strongly expressed transcript cluster encoding a hypothetical filamentous hemagglutinin N-terminal domain-containing iron-responsive protein responsible for adhesion to host tissues [66] was also observed in Ca. Sedimenticola endophacoides, while fatty acid synthesis and catabolism-related genes encoding isocitrate lyase, BCKDH, and proteins within the methylcitrate cycle in Kistimonas-like species have been attributed to growth and virulence in other bacterial taxa [68,69,70,71,72]. Other genes associated with virulence and bacterial secretion systems were also detected in the genomes and transcriptomes of Ca. Sedimenticola endophacoides. However, their significance in the lucinid–bacteria chemosymbiosis is unclear. Nevertheless, the speculated roles of bactericidal, adhesion, and virulence compounds would have to be tested using experimental approaches to better understand host selection and microbiome persistence.
Overall, this study provides insight into the metabolic functions and interactions of P. pectinatus, its thioautotrophic symbiont, and other gill microbiome species. Our discovery of distinct metabolic differences between the clade C endosymbiont, clade A lucinid symbionts, and other marine thioautotrophic symbionts, as well as the consistent presence and activity of other bacterial taxa in the gills, suggests that lucinid gill microbiome diversity is currently underrepresented in the literature and should warrant more investigative efforts, including additional host–microbiome meta-omics, imaging, and experimental studies. It is well established that the lucinid gill microbiome and their interactions with the host and/or the environment contribute to nutrient cycles in coastal marine sediments; however, many details have been lacking. Our metagenomic and metatranscriptomic analyses of mangrove-associated lucinid host and gill microbiome functions provide a systems biology perspective of host and microbiome physiology that is relevant to host–microbe and microbe–microbe interactions.
Change history
15 January 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Taylor JD, Glover EA. Chemosymbiotic bivalves. In: Kiel S, editor. The vent and seep biota: aspects from microbes to ecosystems. Netherlands: Springer; 2010. p. 107–35.
Dubilier N, Bergin C, Lott C. Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat Rev Microbiol. 2008;6:725–40. https://doi.org/10.1038/nrmicro1992
Taylor JD, Glover EA. Functional anatomy, chemosymbiosis and evolution of the Lucinidae. In: Harper EM, Taylor JD, Crame JA. The evolutionary biology of the bivalvia. London: Geological Society of London; 2000. p. 207–27.
Liljedahl L. The Silurian Ilionia prisca, oldest known deep-burrowing suspension feeding bivalve. J Paleontol. 1992;66:206–10.
Gros O, Darrasse A, Durand P, Frenkiel L, Mouëza M. Environmental transmission of a sulfur-oxidizing bacterial gill endosymbiont in the tropical lucinid bivalve Codakia orbicularis. Appl Environ Microbiol. 1996;62:2324–30.
Gros O, Liberge M, Heddi A, Khatchadourian C, Felbeck H. Detection of the free-living forms of sulfide-oxidizing gill endosymbionts in the lucinid habitat (Thalassia testudinum environment). Appl Environ Microbiol. 2003;69:6264–7. https://doi.org/10.1128/AEM.69.10.6264-6267.2003
Gros O, Duplessis MR, Felbeck H. Embryonic development and endosymbiont transmission mode in the symbiotic clam Lucinoma aequizonata (Bivalvia: Lucinidae). Invertebr Reprod Dev. 1999;36:93–103.
Gros O, De Wulf-Durand P, Frenkiel L, Moueza M. Putative environmental transmission of sulfur-oxidizing bacterial symbionts in tropical lucinid bivalves inhabiting various environments. FEMS Microbiol Lett. 1998;160:257–62. https://doi.org/10.1016/S0378-1097(98)00041-X
Brissac T, Gros O, Mercot H. Lack of endosymbiont release by two Lucinidae (Bivalvia) of the genus Codakia: consequences for symbiotic relationships. FEMS Microbiol Ecol. 2009;67:261–7. https://doi.org/10.1111/j.1574-6941.2008.00626.x
Cavanaugh CM, McKiness ZP, Newton ILG, Stewart FJ. Marine chemosynthetic symbioses. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, editors. Prokaryotes. New York, NY: Springer; 2006. p. 475–507.
Fisher MR, Hand SC. Chemoautotrophic symbionts in the bivalve Lucina floridana from seagrass beds. Biol Bull. 1984;167:445–59. https://doi.org/10.2307/1541289
Hentschel U, Felbeck H. Nitrate respiration in chemoautotrophic symbionts of the bivalve Lucinoma aequizonata is not regulated by oxygen. Appl Environ Microbiol. 1995;61:1630–3.
König S, Gros O, Heiden SE, Hinzke T, Thurmer A, Poehlein A, et al. Nitrogen fixation in a chemoautotrophic lucinid symbiosis. Nat Microbiol. 2016;2:16193 https://doi.org/10.1038/nmicrobiol.2016.193
Petersen JM, Kemper A, Gruber-Vodicka H, Cardini U, van der Geest M, Kleiner M, et al. Chemosynthetic symbionts of marine invertebrate animals are capable of nitrogen fixation. Nat Microbiol. 2016;2:16195 https://doi.org/10.1038/nmicrobiol.2016.195.
Duperron S, Rodrigues CF, Leger N, Szafranski K, Decker C, Olu K, et al. Diversity of symbioses between chemosynthetic bacteria and metazoans at the Guiness cold seep site (Gulf of Guinea, West Africa). Microbiologyopen. 2012;1:467–80.
Ball AD, Purdy KJ, Glover EA, Taylor JD. Ctenidial structure and three bacterial symbiont morphotypes in Anodontia (Euanodontia) ovum (Reeve, 1850) from the Great Barrier Reef, Australia (Bivalvia: Lucinidae). J Mollusca Stud. 2009;75:175–85. https://doi.org/10.1093/mollus/eyp009
Pales-Espinosa E, Tanguy A, Le Panse S, Lallier F, Allam B, Boutet I. Endosymbiotic bacteria in the bivalve Loripes lacteus: localization, characterization and aspects of symbiont regulation. J Exp Mar Biol Ecol. 2013;448:327–36. https://doi.org/10.1016/j.jembe.2013.07.015
Brissac T, Mercot H, Gros O. Lucinidae/sulfur-oxidizing bacteria: ancestral heritage or opportunistic association? Further insights from the Bohol Sea (the Philippines). FEMS Microbiol Ecol. 2011;75:63–76. https://doi.org/10.1111/j.1574-6941.2010.00989.x
Liberge M, Gros O, Frenkiel L. Lysosomes and sulfide-oxidizing bodies in the bacteriocytes of Lucina pectinata, a cytochemical and microanalysis approach. Mar Biol. 2001;139:401–9. https://doi.org/10.1007/s002270000526
Read KRH. The characterization of the hemoglobins of the bivalve mollusc Phacoides pectinatus (Gmelin). Comp Biochem Physiol. 1965;15:137–57.
Williams ST, Taylor JD, Glover EA. Molecular phylogeny of the Lucinoidea (Bivalvia): non-monophyly and separate acquisition of bacterial chemosymbiosis. J Mollusca Stud. 2004;70:187–202. https://doi.org/10.1093/mollus/70.2.187
Durand P, Gros O, Frenkiel L, Prieur D. Phylogenetic characterization of sulfur-oxidizing bacterial endosymbionts in three tropical Lucinidae by 16S rDNA sequence analysis. Mol Mar Biol Biotech. 1996;5:37–42.
Green-García AM. Characterization of the lucinid bivalve-bacteria symbiotic system: the significance of the geochemical habitat on bacterial symbiont diversity and phylogeny. Master’s thesis (Louisiana State University, Baton Rouge, Louisiana, USA). 2008. https://digitalcommons.lsu.edu/gradschool_theses/1970/.
Frenkiel L, Gros O, Mouëza M. Storage tissue and reproductive strategy in Lucina pectinata (Gmelin), a tropical lucinid bivalve adapted to a reducing sulfur-rich, mangrove environment. Invertebr Reprod Dev. 1997;31:199–210. https://doi.org/10.1080/07924259.1997.9672577
Christo SW, Ivachuk CS, Ferreira-Junior AL, Absher TM. Reproductive periods of Lucina pectinata (Bivalve; Lucinidae) in the Paranagua Estuarine Complex, Parana - Brazil. Braz J Biol. 2016;76:300–6. https://doi.org/10.1590/1519-6984.12514
Doty TW. Environmental controls on the diversity and distribution of endosymbionts associated with Phacoides pectinatus (Bivalvia: Lucinidae) from shallow mangrove and seagrass sediments, St. Lucie County, Florida. MSc thesis (University of Tennessee, Knoxville, Tennessee, USA). 2015. http://trace.tennessee.edu/utk_gradthes/3548/.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41. https://doi.org/10.1128/AEM.01541-09
Nguyen N, Warnow T, Pop M, White B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Micro. 2016;2:16004.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6. https://doi.org/10.1093/nar/gks1219
Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–8. https://doi.org/10.1093/bioinformatics/bts174
Antipov D, Korobeynikov A, McLean JS, Pevzner PA. hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics. 2016;32:1009–15. https://doi.org/10.1093/bioinformatics/btv688
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165 https://doi.org/10.7717/peerj.1165
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. https://doi.org/10.1038/nmeth.1923
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. https://doi.org/10.1093/bioinformatics/btp352
NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016;44:D7–19. https://doi.org/10.1093/nar/gkv1290
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. https://doi.org/10.1186/1471-2164-9-75.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-Seq: Reference generation and analysis with Trinity. Nat Protoc. 2013;8:1494–512. https://doi.org/10.1038/nprot.2013.084
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 2011;29:644–52. https://doi.org/10.1038/nbt.1883
Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500. https://doi.org/10.1093/bioinformatics/btp692
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25 https://doi.org/10.1186/gb-2010-11-3-r25
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. https://doi.org/10.1093/bioinformatics/btp616
The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–12. https://doi.org/10.1093/nar/gku989
Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–7. https://doi.org/10.1093/bioinformatics/bts611
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2. https://doi.org/10.1093/bioinformatics/bts565
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. https://doi.org/10.1016/S0022-2836(05)80360-2
Green-García AM, Engel AS. Bacterial diversity of siliciclastic sediments in a Thalassia testudinum meadow and the implications for Lucinisca nassula chemosymbiosis. Estuar Coast Shelf Sci. 2012;112:153–61. https://doi.org/10.1016/j.ecss.2012.07.010
Dando PR, Southward AJ, Southward EC. Chemoautotrophic symbionts in the gills of the bivalve mollusc Lucinoma borealis and the sediment chemistry of its habitat. Proc R Soc Lond [Biol]. 1986;227:227–47.
Flood BE, Jones DS, Bailey JV. Sedimenticola thiotaurini sp. nov., a sulfur-oxidizing bacterium isolated from salt marsh sediments, and emended descriptions of the genus Sedimenticola and Sedimenticola selenatireducens. Int J Syst Evol Microbiol. 2015;65:2522–30. https://doi.org/10.1099/ijs.0.000295
Narasingarao P, Haggblom MM. Sedimenticola selenatireducens, gen. nov., sp. nov., an anaerobic selenate-respiring bacterium isolated from estuarine sediment. Syst Appl Microbiol. 2006;29:382–8. https://doi.org/10.1016/j.syapm.2005.12.011
Rodriguez-R LM, Konstantinidis KT. Bypassing cultivation to identify bacterial species. Microbe. 2014;9:111–8.
Lee J, Shin NR, Lee HW, Roh SW, Kim MS, Kim YO, et al. Kistimonas scapharcae sp. nov., isolated from a dead ark clam (Scapharca broughtonii), and emended description of the genus Kistimonas. Int J Syst Evol Microbiol. 2012;62:2865–9. https://doi.org/10.1099/ijs.0.038422-0
Choi EJ, Kwon HC, Sohn YC, Yang HO. Kistimonas asteriae gen. nov., sp. nov., a gammaproteobacterium isolated from Asterias amurensis. Int J Syst Evol Microbiol. 2010;60:938–43. https://doi.org/10.1099/ijs.0.014282-0
Jensen S, Duperron S, Birkeland NK, Hovland M. Intracellular Oceanospirillales bacteria inhabit gills of Acesta bivalves. FEMS Microbiol Ecol. 2010;74:523–33. https://doi.org/10.1111/j.1574-6941.2010.00981.x
Dubilier N, Amann R, Erseus C, Muyzer G, Park S, Giere O, et al. Phylogenetic diversity of bacterial endosymbionts in the gutless marine oligochete Olavius loisae (Annelida). Mar Ecol Prog Ser. 1999;178:271–80.
Blazejak A, Erseus C, Amann R, Dubilier N. Coexistence of bacterial sulfide oxidizers, sulfate reducers, and spirochetes in a gutless worm (Oligochaeta) from the Peru margin. Appl Environ Microbiol. 2005;71:1553–61. https://doi.org/10.1128/AEM.71.3.1553-1561.2005
Duperron S, Fiala-Medioni A, Caprais J, Olu K, Sibuet M. Evidence for chemoautotrophic symbiosis in a Mediterranean cold seep clam (Bivalvia: Lucinidae): comparative sequence analysis of bacterial 16S rRNA, APS reductase and RuBisCO genes. FEMS Microbiol Ecol. 2007;59:64–70. https://doi.org/10.1111/j.1574-6941.2006.00194.x
Raina J, Eme L, Pollock FJ, Spang A, Archibald JM, Williams TA. Symbiosis in the microbial world: from ecology to genome evolution. Biol Open. 2018;7:bio032524 https://doi.org/10.1242/bio.032524
Marie B, Le Roy N, Zanella-Cleon I, Becchi M, Marin F. Molecular evolution of mollusc shell proteins: insights from proteomic analysis of the edible mussel Mytilus. J Mol Evol. 2011;72:531–46. https://doi.org/10.1007/s00239-011-9451-6
Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–61. https://doi.org/10.1093/nar/gkh036
Takamatsu N, Shiba T, Muramoto K, Kamiya H. Molecular cloning of the defense factor in the albumen gland of the sea hare Aplysia kurodai. FEBS Lett. 1995;377:373–6. https://doi.org/10.1016/0014-5793(95)01375-X
Ehara T, Kitajima S, Kanzawa N, Tamiya T, Tsuchiya T. Antimicrobial action of achacin is mediated by L-amino acid oxidase activity. FEBS Lett. 2002;531:509–12. https://doi.org/10.1016/S0014-5793(02)03608-6
Guo C, Liu S, Yao Y, Zhang Q, Sun MZ. Past decade study of snake venom L-amino acid oxidase. Toxicon. 2012;60:302–11. https://doi.org/10.1016/j.toxicon.2012.05.001
Friedrich CG, Rother D, Bardischewsky F, Quentmeier A, Fischer J. Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl Environ Microbiol. 2001;67:2873–82. https://doi.org/10.1128/AEM.67.7.2873-2882.2001
Ghosh W, Dam B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol Rev. 2009;33:999–1043. https://doi.org/10.1111/j.1574-6976.2009.00187.x
Delepelaire P. Type I secretion in gram-negative bacteria. Biochim Biophys Acta. 2004;1694:149–61. https://doi.org/10.1016/j.bbamcr.2004.05.001
Sun YY, Chi H, Sun L. Pseudomonas fluorescens filamentous hemagglutinin, an iron-regulated protein, is an important virulence factor that modulates bacterial pathogenicity. Front Microbiol. 2016;7:1320 https://doi.org/10.3389/fmicb.2016.01320
de Almeida A, Nikel PI, Giordano AM, Pettinari MJ. Effects of granule-associated protein PhaP on glycerol-dependent growth and polymer production in poly(3-hydroxybutyrate)-producing Escherichia coli. Appl Environ Microbiol. 2007;73:7912–6. https://doi.org/10.1128/AEM.01900-07
Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–44. https://doi.org/10.1038/nm1252
Dolan SK, Wijaya A, Geddis SM, Spring DR, Silva-Rocha R, Welch M. Loving the poison: the methylcitrate cycle and bacterial pathogenesis. Microbiology. 2018;164:251–9. https://doi.org/10.1099/mic.0.000604
Munoz-Elias EJ, Upton AM, Cherian J, McKinney JD. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. 2006;60:1109–22. https://doi.org/10.1111/j.1365-2958.2006.05155.x
Sun Y, O’Riordan MX. Branched-chain fatty acids promote Listeria monocytogenes intracellular infection and virulence. Infect Immun. 2010;78:4667–73. https://doi.org/10.1128/IAI.00546-10
Surger MJ, Angelov A, Stier P, Ubelacker M, Liebl W. Impact of branched-chain amino acid catabolism on fatty acid and alkene biosynthesis in Micrococcus luteus. Front Microbiol. 2018;9:374 https://doi.org/10.3389/fmicb.2018.00374
Ankrah NY, Luan J, Douglas AE. Cooperative metabolism in a three-partner insect-bacterial symbiosis revealed by metabolic modeling. J Bacteriol. 2017. https://doi.org/10.1128/JB.00872-16.
Hansen AK, Moran NA. Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc Natl Acad Sci USA. 2011;108:2849–54. https://doi.org/10.1073/pnas.1013465108
Ponnudurai R, Kleiner M, Sayavedra L, Petersen JM, Moche M, Otto A, et al. Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis. ISME J. 2017;11:463–77. https://doi.org/10.1038/ismej.2016.124
Rader BA, Nyholm SV. Host/microbe interactions revealed through “omics” in the symbiosis between the Hawaiian bobtail squid Euprymna scolopes and the bioluminescent bacterium Vibrio fischeri. Biol Bull. 2012;223:103–11. https://doi.org/10.1086/BBLv223n1p103
Johnson MA, Fernandez C, Pergent G. The ecological importance of an invertebrate chemoautotrophic symbiosis to phanerogam seagrass beds. Bull Mar Sci. 2002;71:1343–51.
Meyer EL, Nilkerd B, Glover EA, Taylor JD. Ecological importance of chemoautotrophic lucinid bivalves in a peri–mangrove community in Eastern Thailand. RBZ. 2008;18:41–55.
Reynolds LK, Berg P, Zieman JC. Lucinid clam influence on the biogeochemistry of the seagrass Thalassia testudinum sediments. Estuar Coast. 2014;30:482–90.
Higgs ND, Newton J, Attrill MJ. Caribbean spiny lobster fishery is underpinned by trophic subsidies from chemosynthetic primary production. Curr Biol. 2016;26:3393–8. https://doi.org/10.1016/j.cub.2016.10.034
Shuman KE, Hanson TE. A sulfide:quinone oxidoreductase from Chlorobaculum tepidum displays unusual kinetic properties. FEMS Microbiol Lett. 2016;363:fnw100 https://doi.org/10.1093/femsle/fnw100
Eddie BJ, Hanson TE. Chlorobaculum tepidum TLS displays a complex transcriptional response to sulfide addition. J Bacteriol. 2013;195:399–408. https://doi.org/10.1128/JB.01342-12
Tabita FR, Satagopan S, Hanson TE, Kreel NE, Scott SS. Distinct form I, II, III, and IV RuBisCO proteins from the three kingdoms of life provide clues about RuBisCO evolution and structure/function relationships. J Exp Bot. 2008;59:1515–24. https://doi.org/10.1093/jxb/erm361
Pitcher RS, Watmough NJ. The bacterial cytochrome cbb3 oxidases. Biochim Biophys Acta. 2004;1655:388–99. https://doi.org/10.1016/j.bbabio.2003.09.017
Knight JM, Griffin L, Dale PER, Sheaves M. Short-term dissolved oxygen patterns in sub-tropical mangroves. Estuar Coast Shelf Sci. 2013;131:290–6. https://doi.org/10.1016/j.ecss.2013.06.024
Jensen SI, Kuhl M, Glud RN, Jorgensen LB, Prieme A. Oxic microzones and radial oxygen loss from roots of Zostera marina. Mar Ecol Prog Ser. 2005;293:49–58. https://doi.org/10.3354/meps293049
Wittenberg JB, Wittenberg BA. Mechanisms of cytoplasmic hemoglobin and myoglobin function. Annu Rev Biophys Biophys Chem. 1990;19:217–41. https://doi.org/10.1146/annurev.bb.19.060190.001245
Kraus DW, Wittenberg JB. Hemoglobins of the Lucina pectinata/bacteria symbiosis. I. Molecular properties, kinetics and equilibria of reactions with ligands. J Biol Chem. 1990;27:16043–53.
Frenkiel L, Gros O, Mouëza M. Gill structure in Lucina pectinata (Bivalvia: Lucinidae) with reference to hemoglobin in bivalves with symbiotic sulphur-oxidizing bacteria. Mar Biol. 1996;125:511–24. https://doi.org/10.1007/BF00353264
Rizzi M, Wittenberg JB, Coda A, Ascenzi P, Bolognesi M. Structural bases for sulfide recognition in Lucina pectinata hemoglobin I. J Mol Biol. 1996;258:1–5. https://doi.org/10.1006/jmbi.1996.0228
van der Heide T, Govers LL, de Fouw J, Olff H, van der Geest M, van Katwijk MM, et al. A three-stage symbiosis forms the foundation of seagrass ecosystems. Science. 2012;336:1432–4. https://doi.org/10.1126/science.1219973
Stewart FJ, Dmytrenko O, Delong EF, Cavanaugh CM. Metatranscriptomic analysis of sulfur oxidation genes in the endosymbiont of Solemya Velum. Front Microbiol. 2011;2:134 https://doi.org/10.3389/fmicb.2011.00134
Gardebrecht A, Markert S, Sievert SM, Felbeck H, Thurmer A, Albrecht D, et al. Physiological homogeneity among the endosymbionts of Riftia pachyptila and Tevnia jerichonana revealed by proteogenomics. ISME J. 2012;6:766–76. https://doi.org/10.1038/ismej.2011.137
Markert S, Gardebrecht A, Felbeck H, Sievert SM, Klose J, Becher D, et al. Status quo in physiological proteomics of the uncultured Riftia pachyptila endosymbiont. Proteomics. 2011;11:3106–17. https://doi.org/10.1002/pmic.201100059
Markert S, Arndt C, Felbeck H, Becher D, Sievert SM, Hugler M, et al. Physiological proteomics of the uncultured endosymbiont of Riftia pachyptila. Science. 2007;315:247–50. https://doi.org/10.1126/science.1132913
Woyke T, Teeling H, Ivanova NN, Huntemann M, Richter M, Gloeckner FO, et al. Symbiosis insights through metagenomic analysis of a microbial consortium. Nature. 2006;443:950–5. https://doi.org/10.1038/nature05192
Dmytrenko O, Russell SL, Loo WT, Fontanez KM, Liao L, Roeselers G, et al. The genome of the intracellular bacterium of the coastal bivalve, Solemya velum: a blueprint for thriving in and out of symbiosis. BMC Genomics. 2014;15:924 https://doi.org/10.1186/1471-2164-15-924
Nakagawa S, Shimamura S, Takaki Y, Suzuki Y, Murakami S, Watanabe T, et al. Allying with armored snails: the complete genome of gammaproteobacterial endosymbiont. ISME J. 2014;8:40–51. https://doi.org/10.1038/ismej.2013.131
Schreiber L, Kjeldsen KU, Funch P, Jensen J, Obst M, Lopez-Legentil S, et al. Endozoicomonas are specific, facultative symbionts of sea squirts. Front Microbiol. 2016;7:1042 https://doi.org/10.3389/fmicb.2016.01042
Ding JY, Shiu JH, Chen WM, Chiang YR, Tang SL. Genomic insight into the host-endosymbiont relationship of Endozoicomonas montiporae CL-33(T) with its coral host. Front Microbiol. 2016;7:251 https://doi.org/10.3389/fmicb.2016.00251
Neave MJ, Apprill A, Ferrier-Pages C, Voolstra CR. Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl Microbiol Biotechnol. 2016a;100:8315–24. https://doi.org/10.1007/s00253-016-7777-0
Neave MJ, Rachmawati R, Xun L, Michell CT, Bourne DG, Apprill A, et al. Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 2016b. https://doi.org/10.1038/ismej.2016.95
Zielinski FU, Pernthaler A, Duperron S, Raggi L, Giere O, Borowski C, et al. Widespread occurrence of an intranuclear bacterial parasite in vent and seep Bathymodiolin mussels. Environ Microbiol. 2009;11:1150–67. https://doi.org/10.1111/j.1462-2920.2008.01847.x
Mendoza M, Guiza L, Martinez X, Caraballo X, Rojas J, Aranguren LF, et al. A novel agent (Endozoicomonas elysicola) responsible for epitheliocystis in cobia Rachycentrum canadum larvae. Dis Aquat Organ. 2013;106:31–7. https://doi.org/10.3354/dao02636
Toshchakov SV, Korzhenkov AA, Chernikova TN, Ferrer M, Golyshina OV, Yakimov MM, et al. The genome analysis of Oleiphilus messinensis ME102 (DSM 13489T) reveals backgrounds of its obligate alkane-devouring marine lifestyle. Mar Genomics. 2017;36:41–7. https://doi.org/10.1016/j.margen.2017.07.005
Katharios P, Seth-Smith HM, Fehr A, Mateos JM, Qi W, Richter D, et al. Environmental marine pathogen isolation using mesocosm culture of sharpsnout seabream: striking genomic and morphological features of novel Endozoicomonas sp. Sci Rep. 2015;5:17609 https://doi.org/10.1038/srep17609
Neave MJ, Michell CT, Apprill A, Voolstra CR. Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci Rep. 2017;7:40579 https://doi.org/10.1038/srep40579
Ruehland C, Blazejak A, Lott C, Loy A, Erseus C, Dubilier N. Multiple bacterial symbionts in two species of co-occurring gutless oligochaete worms from Mediterranean sea grass sediments. Environ Microbiol. 2008;10:3404–16. https://doi.org/10.1111/j.1462-2920.2008.01728.x
Campbell BJ, Cary SC. Characterization of a novel spirochete associated with the hydrothermal vent polychaete annelid, Alvinella pompejana. Appl Environ Microbiol. 2001;67:110–7. https://doi.org/10.1128/AEM.67.1.110-117.2001
DeLong EF, Wickham GS, Pace NR. Phylogenetic stains: ribosomal RNA-based probes for the identification of single cells. Science. 1989;243:1360–3. https://doi.org/10.1126/science.2466341
Dirks RM, Pierce NA. Triggered amplification by hybridization chain reaction. Proc Natl Acad Sci USA. 2004;101:15275–8. https://doi.org/10.1073/pnas.0407024101
Soto W, Nishiguchi MK. Microbial experimental evolution as a novel research approach in the Vibrionaceae and squid-Vibrio symbiosis. Front Microbiol. 2014;5:593 https://doi.org/10.3389/fmicb.2014.00593
Cascales E, Buchanan SK, Duche D, Kleanthous C, Lloubes R, Postle K, et al. Colicin biology. Microbiol Mol Biol Rev. 2007;71:158–229. https://doi.org/10.1128/MMBR.00036-06
Good IJ. The population frequencies of species and the estimation of population parameters. Biometrika. 1953;40:237–64.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. https://doi.org/10.1101/gr.186072.114
Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2. https://doi.org/10.1093/bioinformatics/btv351
Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–31. https://doi.org/10.1038/nbt.3893
Louie TS, Giovannelli D, Yee N, Narasingarao P, Starovoytov V, Goker M, et al. High-quality draft genome sequence of Sedimenticola selenatireducens strain AK4OH1(T), a gammaproteobacterium isolated from estuarine sediment. Stand Genomic Sci. 2016;11:66 https://doi.org/10.1186/s40793-016-0191-5
Roeselers G, Newton IL, Woyke T, Auchtung TA, Dilly GF, Dutton RJ, et al. Complete genome sequence of Candidatus Ruthia magnifica. Stand Genomic Sci. 2010;3:163–73. https://doi.org/10.4056/sigs.1103048
Ikuta T, Takaki Y, Nagai Y, Shimamura S, Tsuda M, Kawagucci S, et al. Heterogeneous composition of key metabolic gene clusters in a vent mussel symbiont population. ISME J. 2016;10:990–1001. https://doi.org/10.1038/ismej.2015.176
Perez M, Juniper SK. Insights into symbiont population structure among three vestimentiferan tubeworm host species at eastern Pacific spreading centers. Appl Environ Microbiol. 2016;82:5197–205. https://doi.org/10.1128/AEM.00953-16
Acknowledgements
The research was permitted by the Florida Fish and Wildlife Conservation Commission through Special Activity Licenses to ASE. This work is supported through the Dimensions of Biodiversity Program of the National Science Foundation (DEB-1342785 to ASE, DEB-1242721 to LCA, and DEB-1342763 to BJC). We thank the following: Audrey Paterson, Brooke Long, Aaron Goemann, and Thomas Walters Doty for their contributions in field work and data collection; Chad L. McMahan for tissue sectioning and histology staining; Dr. Terri Bruce and Rhonda Reigers Powell at the Clemson Light Imaging Facility for microscopy advice; Louie Alexander for assistance with FISH; Dr. Vincent Richards for MiSeq sequencing resources and advice on RNA-seq; and Clemson University’s Palmetto cluster for computing resources.
Author contributions
ASE, BJC, and LCA secured the funding for this study, supervised sample collection, and put in research efforts; SJL, BJC, ASE, and LCA collected the samples used in the study; SJL and BJC conceived the experiments; SJL performed most of the experiments, software implementation, data analyses, and wrote the manuscript. BGD performed qPCR analyses on the thioautotrophic symbiont and Kistimonas-like species; DEG and JW performed qPCR and PCR analyses on the Spirochaeta-like species; JW performed transcriptomic analyses on the Spirochaeta-like species; EN performed PCR analyses on the Kistimonas-like species; SJL maintains the NCBI sequence data and LCA curates the metadata and maintains specimens of dissected tissues and valves. BJC, ASE, and LCA reviewed and edited the manuscript; and all authors approved the final manuscript.
Data and specimen availability
Sequence data were deposited in the National Center for Biotechnology Information [35] under the BioProject ID PRJNA368737. Accession numbers are listed in Table S3. Dissected specimen tissues and valves are cataloged at the South Dakota School of Mines and Technology, Museum of Geology, with details provided through the iDigBio portal (https://www.idigbio.org/portal/recordsets/db3181c9-48dd-489f-96ab-a5888f5a938c).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lim, S.J., Davis, B.G., Gill, D.E. et al. Taxonomic and functional heterogeneity of the gill microbiome in a symbiotic coastal mangrove lucinid species. ISME J 13, 902–920 (2019). https://doi.org/10.1038/s41396-018-0318-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0318-3
This article is cited by
-
Sulfur oxidation and reduction are coupled to nitrogen fixation in the roots of the salt marsh foundation plant Spartina alterniflora
Nature Communications (2024)
-
Gill-associated bacteria are homogeneously selected in amphibious mangrove crabs to sustain host intertidal adaptation
Microbiome (2023)
-
Gene loss and symbiont switching during adaptation to the deep sea in a globally distributed symbiosis
The ISME Journal (2023)
-
A Study of Sponge Symbionts from Different Light Habitats
Microbial Ecology (2023)
-
Microbiome differences between wild and aquarium whitespotted eagle rays (Aetobatus narinari)
Animal Microbiome (2022)
