
Abstract
An inter-kingdom beach warfare between a Streptomyces sp. and Aspergillus sp. co-isolated from shallow water beach sand, collected off Heron Island, Queensland, Australia, saw the bacteriostatic Aspergillus metabolite cyclo-(l-Phe-trans-4-hydroxy-l-Pro) (3) stimulate the Streptomyces to produce nitric oxide (NO), which in turn mediated transcriptional activation of a silent biosynthetic gene cluster (BGC) for fungistatic heronapyrrole B (1). Structure activity relationship studies, coupled with the use of NO synthase inhibitors, donors and scavangers, and both genomic and transcriptomic analyses, confirmed the extraordinary chemical cue specificity of 3, and its NO-mediated mechanism of transcriptional action. Our findings reveal the importance of inter-kingdom (fungal-bacterial) chemical communication in the regulation of silent BGCs coding for chemical defenses. We propose that the detection and characterisation of NO mediated transcriptional activation (NOMETA) of silent chemical defences in the environment, may inspire broader application in the field of microbial biodiscovery.
Similar content being viewed by others
Introduction
Natural selection relentlessly drives adaptations in microbial secondary metabolism, with successive generations of bacteria and fungi acquiring ever more diverse defensive chemicals, enhancing survival in complex and highly competitive ecosystems. As a result, the global microbiome has evolved to encompass a vast arsenal of ecologically and biologically potent natural products, encoded within a myriad of biosynthetic gene clusters (BGCs). For over 70 years readily accessible microbial natural products fuelled a revolution in science, inspiring new pharmaceuticals and agrochemicals, improving the quality of life for millions, and driving global commerce. This success is perhaps most clearly exemplified in modern antibiotics, which are overwhelmingly inspired by microbial natural products.
Notwithstanding past success, and confronted by a near exhausted accessible microbial resource and ever lower returns on investment, late last century the pharmaceutical industry turned elsewhere for inspiration [1]. Nearly two decades on, and faced by the challenge of drug resistance, and community demand for better, safer, cheaper drugs, the need for inspiration remains as urgent as ever. Reinvigorated by scientific and technical advances, there is a strong case that microbial biodiscovery can once again assume a prominent role. Such optimism arises on multiple fronts, including genomic discoveries documenting a near limitless reservoir of transcriptionally inactive (i.e., cryptic or silent) BGCs, dominant in virtually all microbial genomes [2]. Previously overlooked by both science and industry, this hidden resource has the potential to fuel a microbe-inspired renaissance in drug discovery. To deliver this bonanza requires robust, cost-effective methods for routine transcriptional activation of silent BGCs. While a range of approaches have proved promising [2,3,4,5,6,7] there is a compelling case that greater knowledge of microbial chemical ecology could inform new (and improved) methods for activation of silent BGCs in the native producing strain. Such knowledge could, for example, render to practice the strategy of microbial co-cultivation, in which natural microbial chemical cues prompt the on-demand activation of silent BGCs encoding for defensive secondary metabolites [8, 9]. One particularly intriguing implementation of this approach is that of inter-Kingdom co-cultivation between bacteria and fungi. For example, co-cultivation of Streptomyces rapamycinicus with Aspergillus nidulans led to transcriptional activation of silent BGCs and the production of several known fungal metabolites [10], while co-cultivation of S. rapamycinicus with A. fumigatus produced the two new fungal metabolites fumicyclines A-B [11]. In both cases, activation required intimate cell-cell contact, and was mediated by inhibition of the acetyltransferase (HAT) complex Saga/Ada. In another study, co-cultivation of ×58 soil-dwelling actinomycetes with A. nidulans revealed the activation of several known fungal metabolites [12], as did co-cultivation of S. bullii and A. fumigatus MBC-F1-10, co-isolated from soil samples from the northern Atacama Desert, Chile [13]. Likewise, co-cultivation of type strains of either Bacillus subtilis or S. lividans with A. austroafricanus, a fungal endophyte isolated from Egyptian water hyacinth, resulted in production of the new fungal metabolite austramide [14], while co-cultivation of mine drainage-derived isolates of the bacteria Sphingomonas sp. KMK-001 and fungus A. fumigatus KMC-901, resulted in production of two new nitrated diketopiperazines [15, 16].Co-cultivation of the two model microbes S. coelicolor A3(2) M145 and A. niger N402 produced known fungal metabolites [17], as did co-cultivation of a terrestrial soil derived S. fradiae 007, with a marine-derived Penicillium sp. WC-29-5, isolated from Shandong and Hainan Provinces, China, respectively [3]. Notwithstanding achievements to date, the current literature on bacteria-fungi co-cultivation microbial biodiscovery is dominated by the isolation and structure elucidation of the activated metabolites (known and new), rather than the chemical cues responsible for transcriptional activation, and by instances where bacteria stimulate fungal secondary metabolism, rather than visa versa. This leaves considerable scope to improve our understanding of the chemical ecology that underpins inter-Kingdom microbial co-cultivation, including identifying natural chemical cues that could be used as media additives to improve the productivity of modern microbial biodiscovery.
Alert to this challenge, we turned to a rare class of farnesylated 2-nitropyrroles, exemplified by heronapyrrole B (1) (Fig. 1, Supplementary Information Section 3), originally isolated from an Australian shallow water beach sand-derived Streptomyces sp. CMB-M0423 [18]. In this report we reveal, for the first time, that a quiescent fungus hidden within the original CMB-M0423 isolate produced a chemical cue that was a highly selective transcriptional activator of the silent BGC encoding for 1 and other biosynthetically related meroterpenes, such as debromomarinone (2) (Fig. 1, Supplementary Information Section 3). This unexpected and covert example of inter-kingdom chemical communication was seen as an excellent model to explore bacteria-fungi co-cultivation mediated transcriptional activation of silent BGCs, and in particular the structure and mechanism of action of the chemical cue(s) that drives this process.

Streptomyces sp. CMB-StM0423 and Aspergillus sp. CMB-StM0423 natural products 1-2 and 3, respectively
Materials and methods
Chemical characterisation
See Supplementary Information Section 1.
Microbioreactor microbial cultivations
Following a published protocol [19], analytical scale microbial cultivations were performed using a 24-well plate microbioreactor system employing a sandwich cover consisting of a stainless steel lid, microfiber layer, extruded Teflon with 0.2 µm pores and soft silicone layer (Supplementary Information Section 2). Microbial optical density measurements were acquired on a POLARstar Omega plate reader at an excitation wavelength of 490 nm and emission of 510 nm. Data were analysed by Prism 5.
Streptomyces sp. CMB-M0423 cultivation in the presence of aminoguanidine (AG)
Six Erlenmeyer flasks (2 L) containing M1 marine broth (500 mL; 1% starch, 0.4% yeast extract and 0.2% peptone) were inoculated with a starter culture (5 mL) of Streptomyces sp. CMB-M0423, and incubated at 27 °C on a rotary shaker at 190 rpm for 7 days. The flasks were then extracted with EtOAc (2 × 250 mL per flask) and the combined organic phase concentrated in vacuo to yield a crude extract (95.6 mg). The crude extract was subjected to sequential 25 mL solvent triturations to afford, after in vacuo concentration, hexane (5.3 mg) and CH2Cl2 (60 mg) soluble fractions. The CH2Cl2 solubles were further fractionated by HPLC (Zorbax C8 column, 250 × 9.4 mm, 5 μm, 3 mL/min, gradient elution from 90% H2O/MeOH to 100% MeOH over 30 min) to afford pseurotin A (tR = 21.3 min; 5.1 mg, 7.3%), gliotoxin (tR = 22.0 min; 4.5 mg, 6.4%), bisdethiobisgliotoxin (tR = 22.7 min; 3 mg, 4.3%), fumitremorgin C (tR = 25 min; 2.4 mg, 3.4%), cyclo-(L-Phe-trans-4-hydroxy-L-Pro) (tR = 15.7 min; 3 mg, 3.6%) (3) and cyclo-(L-Phe-L-Pro) (tR = 17.2 min; 1.5 mg, 2.3%). [Note - % yields are determined on a mass-to-mass basis against the EtOAc crude extract] (Supplementary Sections 5–6).
Growth curves for Aspergillus sp. CMB-AsM0423 in the presence of heronapyrrole B (1)
CMB-AsM0423 was streaked onto an M1 3.3% artificial sea salt agar plate and was incubated at 26.5 °C for 10 days. The spores from one colony were transferred to M1 3.3% artificial sea salt agar and the colony forming unit (CFU) count adjusted to ~1 × 104 spores/mL using a hemacytometer cell counter. Heronapyrrole B (1) was dissolved in DMSO and diluted with H2O to give a 1200 μM stock solution (20% DMSO) and the stock solution was then serially diluted with 20% DMSO to give concentrations from 1200 to 6 μM in 20% DMSO. An aliquot (10 μL) of each dilution was transferred to 96-well microtiter plate wells inoculated with CMB-AsM0423 broth (190 μL) to give final concentrations of 60 μM to 0.3 µM in 1% DMSO. The plates were incubated at 26.5 °C for 7 days and the optical density of each well was measured spectrophotometrically at 600 nm using POLARstar Omega plate (BMG LABTECH, Offenburg, Germany). Amphotericin B was used as a positive control to give final concentrations of 60 μM to 0.3 µM in 1% DMSO. All optical density measurements were obtained from triplicates from two independent experiments.
Growth curves for Streptomyces sp. CMB-StM0423 in the presence of cyclo-(L-Phe-trans-4-hydroxy-L-Pro) (3)
CMB-StM0423 was streaked onto an M1 3.3% artificial sea salt agar plate and was incubated at 27 °C for two weeks. The spores from one colony were transferred to M1 3.3% artificial sea salt broth and the CFU count adjusted to ~104 to 105 spores/mL using a hemacytometer cell counter. Cyclo-(L-Phe-trans-4-hydroxy-L-Pro) (3) was dissolved in DMSO and diluted with H2O to give a stock concentration of 200 µM in 20% DMSO. An aliquot (100 µL) was transferred to 24-well microtiter plate wells inoculated with CMB-StM0423 broth (1.9 mL) to give a final concentration of 10 µM in 1% DMSO. The plate was incubated at 27 °C for 7–10 days. Serial dilutions were plated on M1 3.3% artificial sea salt agar and colonies enumerated after 2 weeks of incubation at 27 °C to calculate CFU counts. Rifampicin (10 µM) was used as a positive control. CFU counts were determined from triplicates from two independent experiments.
Synthesis of diketopiperazines
Combinations of amino acid methyl esters in H2O in the presence of triethylamine (2.5 eq.) were subjected to microwave irradiation (140 °C, 300 WATT, 3 min) to afford mixtures of diketopiperazines, which were purified by HPLC (Supplementary Section 11).
Electron Microscopy
Coverslips coated with poly-l-lysine (1 mg/mL) were inverted onto plates, and left for 5 mins for samples to adhere. Coverslips were then immersed in 3% glutaraldehyde in 0.1 M sodium cacodylate buffer for 1 h before being washed twice in the same buffer for 5 min. Samples were then dehydrated in EtOH before being dried in a critical point dryer (Autosamdri-815, Tousimis) according to manufacturer’s instructions. Coverslips were attached to stubs with double-sided carbon tabs and coated with gold using an SPI-Module sputter coater (SPI) following manufacturer’s instructions. Samples were imaged in a Jeol Neoscope JCM 5000 at an accelerating voltage of 10 kV.
Genome sequencing and mining
Genomic DNA was extracted using the UltraClean microbial DNA isolation kit and sequenced using Single Molecule Real Time sequencing technology. A single contig was obtained using the Hierarchical Genome Assembly Process pipeline [20]. General genome annotation was performed using RAST [21]. ABYS and our in house pipeline for annotation. Annotation of biosynthetic gene clusters (BGCs) for natural products was performed using AntiSMASH [22] and PRISM [23], with BGC annotation manually curated using available literature and MIBiG [24]. For mining of specific enzymes we used a database of actinobacterial genomes available at GenBank including MAR4 Streptomyces genomes. Homologs of RNA polymerase subunit beta (RPOB), nitric oxide synthase (NOS) and xanthine oxidoreductase (XOR) were retrieved from the database using blastP with e-value cut-off of 1E9 and bit score cut-off of 200. Retrieved sequences were aligned with Muscle [25] manually trimmed and used for Bayesian phylogenetic reconstruction with MrBayes [26].
Transcriptomic analyses
For transcriptomic analysis, Streptomyces sp. CMB-StM0243 was cultivated in M1 media, with and without cyclo-(L-Phe-trans-4-hydroxy-L-Pro) (3) (10 µM) in two biological replicates. RNA extraction was performed as described [27], and ribosomal RNA depletion performed. The extracted RNA was sequenced using RNAseq sequencing (Illumina) in the 2 × 125 base pairs format. The quality of the reads was assessed using FastQC [28]. Trimming and filtering of the reads was performed using Trimmomatic [29]. Finally, differential expression of the transcriptomes was performed using the limma R/Bioconductor-package [30], and expressed as the logarithm base 2 of fold-changes in expression. The expression of BGCs and selected genes was also determined as the average of the sum of the Reads Per Kilobases of transcript per Million mapped reads (RPKM) of the genes annotated in each BGC (Supplementary Information Section 18).
Nitric oxide mediated transcriptional activation by sodium nitroprusside (SNP)
Microbioreactor shaken broth cultures of target isolates in preferred media (i.e., YES or M1 in 3.3% artificial sea salt) were treated with SNP (15 μL, 2 μM in water) on days 4 and 7. Negative controls comprised culture broths not treated with SNP, or treated with spent SNP (generated by heating SNP at 300 °C for 24 h to quench its capacity to release NO). All micro-bioreactor plates were incubated at 27 °C for 10 days at 190 rpm (Supplementary Information Section 19).
Nitric oxide mediated transcriptional activation by cyclo-l-Phe-trans-4-hydroxy-l-Pro (3)
Microbioreactor shaken broth cultures of target isolates in preferred media were treated with 3 (15 μL, 10 μM in 1% DMSO) on day 0. Negative controls comprised culture broths treated with a sham of 1% DMSO/H2O. All micro-bioreactor plates were incubated at 27 °C for 10 days at 190 rpm (Supplementary Information Section 19).
Nitric oxide mediated transcriptional activation by NO
Microbioreactor shaken broth cultures of target isolates in preferred media were treated with 1% NO in N2 (15 s pulse at a flow rate of 0.2 L/min) on days 4 and 7. Negative controls comprised culture broths either untreated or treated with 15 s pulses of either N2 or air. All micro-bioreactor plates were incubated at 27 °C for 10 days at 190 rpm (Supplementary Information Section 19).
Nitric oxide detected by fluorescent microscopy
Inocula were prepared by growing target isolates in the preferred shaken broth media in microbioreactor wells for 5 days at 190 rpm, 27 °C. Cells were recovered with phosphate buffered saline (pH 7.3, 14 mL), after which they were centrifuged at 14,000 rpm for 1 min. An aliquot (100 μL) of the NO detection reagent (2.5 μL in 1 mL) was added and cells were transferred to coverglass (12 mm) bottom plates. After being incubated for 2 h at 27 °C plates were treated with 3 (10 μM) and incubated for a further 30 min. Surplus NO detection solution was gently removed by absorbing with paper tissues, and the recovered cells washed with phosphate buffered saline, and observed under a fluorescent microscope. Negative controls comprised cells not treated with 3, as well as cells co-treated with 3 and the NO scavenger c-PTIO (40 μM). Streptomyces sp. CMB-St0423 cell images were acquired on a Zeiss 880 Inverted LSM confocal microscope with a 63 × 1.4 NA C-Apochromat water immersion objective. Laser brightfield images were acquired with a transmitted light photon multiple tube (T-PMT) at 561 nm wavelength through a LWD 0.55 NA condenser.
Nitric oxide detected by Griess reagent
Aliquots (200 μL) of a 5 d, 27 °C microbioreactor shaken broth culture in preferred media (i.e., YES or M1 in 3.3% artificial sea salt) were washed with sterile water (2 × 200 μL), after which the cells were centrifuged, resuspended in media (200 μL), treated with NOMETA stimuli and incubated for 3 h. Aliquots (50 μL) were sampled from individual wells at set timepoints (0, 15, 30, 60, 120, and 360 min), and the supernatant from centrigued cells transferred to wells in a clear bottom 96 well plate. Griess reagent (50 μL) (sulphanilamide and N-1-naphylethylenediamine dihydrochloride) was added to all wells, incubated in the dark for 5-10 min at room temperature, and its 520 – 550 nm absorbance measured within 30 min on an POLARstar Omega plate reader.
Nitric oxide mediated transcriptional activation detected by UPLC-DAD
Microbioreactor shaken broth cultures were extracted in situ with EtOAc (2 mL), and the decanted organic layer dried under N2 to yield individual crude extracts which were resuspended in MeOH (100 μL). Aliquots (1 μL) were analyzed by UPLC-DAD (Zorbax C8 RRHD 1.8 μm (2.1 × 50 mm) column, with a 2.5 min gradient elution at 0.417 mL/min from 90% H2O/MeCN to 100% MeCN, and inclusive of an isocratic 0.01% trifluoroacetic acid (TFA) modifier, with online detection at 210, 254 and 360 nm).
Results and discussion
An inter-kingdom conflict fuelled by a secret (NO) code
The presence of an aromatic nitro moiety in the heronapyrroles led us to hypothesise that cultivation in the presence of a nitric oxide synthase (NOS) inhibitor would suppress nitric oxide (NO) production and generate de-nitro analogues. We reasoned that de-nitro analogues would be excellent models to explore the antibacterial structure activity relationship of the heronapyrroles. In pursuit of this goal we exposed Streptomyces sp. CMB-M0423 cultures to amino guanidine (AG), a known NOS inhibitor. Rather than yielding de-nitro analogues this treatment unexpectedly suppressed heronapyrrole biosynthesis and appeared to stimulate the production of six fungal metabolites, namely pseurotin A, gliotoxin, bisdethiobis(methylthio)gliotoxin, fumitremorgin C, cyclo-(L-Phe-L-Pro) and cyclo-(L-Phe-trans-4-hydroxy-L-Pro) (3) (Fig. 1, and Supplementary Information Section 4). This effect, which could be replicated by treatment with the alternative NOS inhibitors N-nitro-L-arginine methyl ester (L-NAME) and N-methyl-L-arginine (L-NMMA), suggested to us the presence of a quiescent fungus. It was reasoned (and subsequently confirmed) that heronapyrroles exerted a fungistatic effect (Fig. 2B) that kept the fungal strain in a quiescent state, with AG suppression of heronapyrrole biosynthesis allowing the fungus (and its metabolites) to dominate co-cultivations. Intrigued by the prospect of a natural bacterial-fungal inter-kingdom chemical warfare possibly mediated by NO, we elected to investigate this phenomenum.

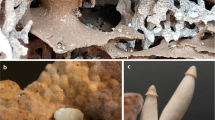
A. Growth inhibition of Streptomyces sp. CMB-StM0423 broth cultures exposed to (i) medium and (ii) 1% DMSO (-ve controls), (iii) 3 (10 μM), and (iv) rifampicin (10 μM, +ve control). Data are colony forming units (CFU ± SD) recovered from duplicates from two independent experiments. B. Growth inhibition of Aspergillus sp. CMB-AsM0423 broth cultures exposed to (i) 1% DMSO (−ve control), (ii) 1 and (iii) amphotericin B (+ve control), at the indicated concentrations. Data are optical density (ODS ± SD) from triplicates from two independent experiments. Note - all cultivations A and B were carried out in M1 3.3% seawater media. Electron microscope images of cells of C. Aspergillus sp. CMB-AsM0423 and D. Streptomyces sp. CMB-StM0423, decanted from M1 3.3% seawater media cultivations, respectively
Combatants exposed: Streptomyces vs Aspergillus
Serial cultivations of Streptomyces sp. CMB-M0423 in the presence of antifungal cyclohexamide and antibacterial rifampicin enabled recovery of pure isolates of constituent microbes—identified as Streptomyces sp. CMB-StM0423 and Aspergillus sp. CMB-AsM0423 (Fig. 2C, D, Supplementary Information Section 7). As predicted, Aspergillus sp. CMB-AsM0423 cultivations produced the six fungal metabolites detected following AG treatment (Supporting Information Sections 5–6), however, and quite unexpectedly, Streptomyces sp. CMB-StM0423 cultivations did not produce heronapyrroles (Supplementary Information Section 10, Figure S23). Notwithstanding this suprising development, the fungal metabolite 3 exhibited bacteriostatic activity against Streptomyces sp. CMB-StM0423 (Fig. 2A), and the bacterial metabolite 1 exhibited fungistatic activity against Aspergillus sp. CMB-AsM0423 (Fig. 2B), consistent with the no-growth conflict zone observed on agar plate co-cultivations (Fig. 3A).

A. Co-cultivation agar plate of Streptomyces sp. CMB-StM0423 (inner) and Aspergillus sp. CMB-AsM0423 (outer). B. HPLC-DAD (360 nm) chromatogram of Streptomyces sp. CMB-StM0423 broth cultures (i) with and (ii) without addition of 3 (10 μM), and (iii) medium only (−ve control), with 1 highlighted in blue. C. HPLC-DAD (210 nm) chromatogram of Aspergillus sp. CMB-AsM0423 broth cultures (i) with and (ii) without addition of 1 (10 μM), with 3 highlighted in red, with a ×10 expanded inset
Covert operations: Cyclo-(L-Phe-trans-5-hydroxy-L-Pro) (3) as a double agent
As Streptomyces sp. CMB-StM0423 cultivations failed to produce 1 we speculated that one (or more) of the metabolites produced by Aspergillus sp. CMB-AsM0423 was acting as a natural chemical cue, activating transcription of an otherwise silent heronapyrrole BGC. To test this hypothesis, we treated Streptomyces sp. cultivations with each of the six Aspergillus sp. metabolites in turn, and established that 3 alone stimulated the biosynthesis of 1 (Fig. 3B) (Supplementary Information Section 10-11). This effect was optimal at 10 μM, but dropped away below 0.5 μM and above 580 μM. Consistent with this observation, quantitative analysis of M1 3.3% seawater cultivations confirmed that ~10% of 3 was secreted into the broth, with the remaining 90% retained within the fungal mycelia (Supplementary Information Section 8). While a remarkable example of diketopiperazine mediated transcriptional activation of a silent BGC, we were alert to the fact that diketopiperazines are common bacterial and fungal natural products that are known to be upregulated during Aspergillus-Streptomyces co-cultivations [17, 31], which raised the question of the specificity of this activation response. To address this issue we synthesized a library of diketopiperazines (Fig. 4), including 3 and its stereoisomers 4–6, hydroxylated isomers 7–8, and structure isomers 9–10, as well the analogues 11–13, and 14 (the latter as a mixture of stereoisomers). All of 3–14 were evaluated for their ability to activate the biosynthesis of 1 (Supplementary Information Sections 14-15), and remarkably, only 3 exhibited activation (Fig. 3B). This level of specificity prompted an investigation into the mechanism of action.

Synthetic diketopiperazines 4–14
Informant in the ranks: NO signals a counter attack
As our initial study demonstrated heronapyrrole biosynthesis was quenched by the NOS inhibitor AG, it was hypothesized that transcriptional activation by 3 was mediated by NO. To test this hypothesis, a fluorescent microscopy method for NO detection in bacterial/fungal cells was implemented, and used together with high performance liquid chromatography coupled to diode array and mass spectrometric detection (HPLC-DAD-MS) to carry out a series of experiments (Supplementary Information Section 13, 16). For example, we demonstrated that Streptomyces sp. CMB-StM0423 cultured for 10 days in the presence of 3 (10 μM) produced NO (Fig. 5 A1-A4) and 1 (Supplementary Information Sections 13 and 16), whereas cultivations in the absence of 3 failed to produce either NO (Fig. 5 C1-AC4) or 1 (Supplementary Information Section 16). Likewise, Streptomyces sp. CMB-StM0423 cultured in the presence of 3 (10 μM) and AG (7.5 μM), failed to produce either NO or 1 (Supplementary Information Section 16). By contrast, co-cultures of Streptomyces sp. CMB-StM0423 and Aspergillus sp. CMB-AsM0423 led to the production of NO (Fig. 5 B1-B4) and 1 (Supplementary Information Section 9–10).

1. 3D fluorescent, 2. fluorescent, 3. brightfield and 4. combined microscopy images of M1 3.3% seawater media cultivations, dispensed onto slides and treated with an NO (fluorescent) detection reagent. A1-A4. Cultivation of Streptomyces sp. CMB-StM0423 with addition of 3 (10 μM), B1-B4. Co-cultivation of Streptomyces sp. CMB-StM0423 and Aspergillus sp. CMB-AsM0423, C1-C4. Cultivation of Streptomyces sp. CMB-StM0423 without addition of 3, and D1-D4. Cultivation of Aspergillus sp. CMB-AsM0423 without addition of 1. A4-D4 scale bar (10 μm) applies equally to A2-D2 and A3-D3, which are ×2-5-fold enlargements of A1-D1. Links to video images of A1-D1 are provided in Supplementary Information
While these observations confirm that 3 activates the production of both NO and 1, they do not determine whether 3 acts with NO in a mutually dependent manner to produce 1, or whether 3 or NO are independently capable of stimulating the production of 1. To differentiate between these two options, we carried out a series of additional experiments (Supplementary Information Section 17). In the first experiment, Streptomyces sp. CMB-StM0423 cultures treated with 3 (10 μM) and the NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (CPTIO) (1 mM), with sampling at days 1, 5, 6, and 10, failed to produce 1—confirming that 3 without NO was insufficient to activate the production of 1. In a second experiment, Streptomyces sp. CMB-StM0423 cultures treated with the NO donor dimethylamine nitric oxide (DEANO) (1 mM) on day 5, with sampling on day 10, produced 1—confirming that NO without 3 was sufficient to produce 1. In third and fourth experiments, we confirmed that DEANO (1 mM) could reactivate the biosynthesis of 1 in (i) Streptomyces sp. CMB-StM0423 cultures stimulated by 3 and, (ii) co-cultures of Streptomyces sp. CMB-StM0423 and Aspergillus sp. CMB-AsM0423, where endogenous NO was quenched by AG—confirming that exogenous NO alone could activate the production of 1.
Taken together these experiments confirm that 3 induces the production of NO, and that NO is essential for activating the production of 1, but that exogenous NO can bypass the role played by 3. To better understand the biosynthetic relationship between 3, NO and 1 we turned to genome mining and transcriptomics.
NO Command and Control
Using single molecule real time (SMRT) sequencing (Supplementary Information Section 18) we obtained a single contig, high quality, linear 8 Mb genome for Streptomyces sp. CMB-StM0423 (Fig. 6A, Supplementary Information Section 18), with strong homology to the marine-derived Streptomyces sp. CNQ 509 known to produce farnesylated-2-nitropyrroles closely related to 1 [32]. Further genomic analysis confirmed that CMB-StM0423 belonged to the MAR4 family of streptomycetes (Supplementary Information Section 18), a group that is remarkably proficient in producing isoprenoids [33]. The Streptomyces sp. CMB-StM0423 genome encodes for 27 BGCs (Fig. 6A, Supplementary Information Section 15) of which 9 feature isoprenoid characteristics. While inhouse annotation protocols, together with phylogenetic analysis of prenyl transferases, comparative genomics, and available literature [32, 34, 35], permitted prediction for the natural product classes encoded in some of the 27 putative BGCs (Supplementary Information Section 18), the heronapyrrole BGC proved elusive. Likewise, we were surprised when homology searches failed to detect a bona fide nitric oxide synthase (NOS). Instead, rapid annotation using subsystems technology (RAST) [21] revealed a protein annotated as flavodoxin/nitric oxide synthase (PEG 657). The gene was not located within any BGC, and therefore the protein annotation did not assist in identifying the heronapyrrole BGC. Further protein domain analysis revealed that the protein lacked the oxidation domain, suggesting that the enzyme was likely not responsible for the NO production. Transcriptional comparison showed however that the enzyme was down regulation in response to 3 (Supplementary Information Section 18).

A. Circular representation of the linear genome map for Streptomyces CMB-StM0423 including Protein Encoding Genes (PEGs) PEGs in the 5–3′ (light blue ring) and 3–5′ (purple ring) strands, with localization of predicted BGCs (black boxes and numbers). Inner-circles show coverage of RNA sequencing reads across the chromosome without (blue inner ring) and with (red inner ring) exposure to 3 (10 μM). B. Gene organization and transcriptional profile (RNA sequencing reads) aligning to BGC11. BGC11 features enzymes involved the biosynthesis of meroterpeoids and shows a clear upregulation in the presence of DKP (see Figure S52). The transcriptional response to treatment with 3 is shown in red and without treatment is shown in blue. Light blue genes correspond to genes in the isoprenoid biosynthetic pathway. Regulation genes are shown in yellow. Black genes are involved in resistance and green genes are involved in other pathways. C. Bar Chart of the top five transcribed BGCs in the Streptomyces CMB-StM0423 genome. Out of the 27 BGCs, BGCs 3, 6, 11, 14, and 27 display high level of transcription (above 1000 RPKM = Reads Per Kilobase of transcript per Million mapped reads). Blue bars show RPKM for cell without treatment and red bars show RPKM when cells grew in the presence of 3
Others have suggested that a xanthine oxidoreductase (XOR) (PEG 6836) [36] (Fig. 7D) with nitrate/nitrite reductases (NaR and NiR) [37] could result in NO production and that aminoguandine (AG) inhibits XOR in vitro at concentrations above 5 mM [38]. In fact, NaR and NiR have been shown to be involved in NO production in Streptomyces coelicolor, a model organism lacking a NOS [39]. Further mining of the CMB-StM0423 genome identified XOR, NiR/NaR homologs (Supplementary Information Section 18, PEGS 6836 and 5129-5130/5125, respectively). Although analysis of the three enzymes confirmed that XOR, NiR, and NaR are conserved among MAR4 streptomycetes, based on genomic context we could not establish a direct link between these enzymes and the biosynthesis of 1. To gain insights into the link between NO metabolic enzymes and 1 we analysed the transcriptional response to 3 at the genome scale level using RNA sequencing.

A. Proposed biosynthetic pathway for the NO cycle in CMB-StM0423. Arrows correspond to upregulated (green), and downregulated (red) genes on exposure to 3 (10 μM) and numbers indicate PEGs. B. Gene organization and transcriptional profile aligning to PEG 6836 (black) annotated as a Xanthine oxidoreductase (XOR), PEG 5125 annotated as a Nitrate reductase (NaR) and PEG 5129-5130 annotated as Nitrite reductase (NiR). Aligning RNA seq reads as a transcriptional response to treatment with 3 and represented in red and blue. Red colours shows reads in response to 3 and blue in response to without 3. PEG 6827 is a transcriptional regulator. C. Bar charts showing RPKM for relevant NO cycle genes. RPKM bars for cell without (blue) and with (red) 3. PEG 1176 annotated as nitric dioxygenase which is transcriptionally suppressed in the presence of 3 to accumulate NO
RNA samples from Streptomyces sp. CMB-StM0423 cultured without and with exposure to 3 (10 μM) (Supplementary Information Section 18) were sent for sequencing. RNA sequencing established that 3 activates transcription of NiR, NaR and XOR (Fig. 7A–D) [36]. The RNA sequencing analysis also showed that treatment with 3 changed the transcriptional status of ×10 of the ×27 BGCs (shown with red numbers in Fig. 6A). The data also showed that BGC 11, belonging to the top five transcribed BGCs (Fig. 6C), was significantly up-regulated (Fig. 6B). The transcriptional up-regulation was further confirmed by RT-PCR (Supplementary Information Section 18, Figure S57). Of note, BGC 11 contains various genes involved in the biosynthesis of isoprenoids (shown in light blue in Fig. 6B) and two prenyltransferases, one previously linked to debromomarinone biosynthesis [32]. Based on these observations BGC 11 is a promising candidate for the biosynthesis of 1 (Fig. 6B, C). Importantly, these genomic and transcriptomic studies validate the role of 3 in transcriptional activation of NO, and an array of silent BGCs.
NO MEdiated Transcriptional Activation (NOMETA)
Even though NO diffuses rapidly across biological membranes and has been postulated as a possible endogenous and transmissible chemical cue in Streptomyces [40], in the absence of a link to silent BGCs it has yet to find application as a tool in microbial biodiscovery. Our discovery that NO can act as a transcriptional regulator of silent BGCs in Streptomyces sp. CMB-StM0423 led us to speculate that NO MEdiated Transcriptional Activation (NOMETA) may have application in the activation of silent BGCs in other microbes.
To test this hypothesis, we challenged a panel of broth cultures prepared from ×10 Streptomyces spp. reference strains, to ×3 different modes of NO exposure (Supplementary Information Section 19–20). First we used the chemical cue 3, speculating that Streptomyces other than CMB-StM0423 may incorporate the requisite response elements. This approach successfully detected NOMETA in 1/10 strains tested, namely Streptomyces noursei. Of note, S. noursei is the source of the commercial antifungal agent nystatin [41], which is on the World Health Organisation List of Essential Medicines. We next used sodium nitroprusside (SNP), speculating that addition of this relatively cheap, vintage blood pressure medication to culture broths would lead to a sustained pulse of exogenous NO, bypassing the need for a chemical cue such as 3. This approach successfully detected NOMETA in 3/10 strains tested, including Streptomyces roseosporus (Figure S59). Of note, S. roseosporus is the source of the commercial antibiotic daptomycin [42], and its silent BGCs has been the subject of recent efforts at genome mining [43] and genetic manipulation [44]. Finally, we administered 1% NO in N2 directly into microbioreactor cultivations, reasoning that short, repeated exogenous NO pulses would deliver outcomes similar to SNP. This approach resulted in NOMETA in 1/10 strains tested, namely Streptomyces griseochromogenes. Of note, S. griseochromogenes is the source of the commercial antibiotic blasticidin S, coincidentally used in genetic engineering to assist in the selection of transformed cells. In all these instances, the NOMETA responsive chemistry detected in 5/10 Streptomyces spp. tested, was not detected under multiple (×33) culture conditions in the absence of NO. While still preliminary, our success with NOMETA is encouraging, and reinforces the view that knowledge of chemical communication between microbes has the capacity to inform and advance future practices in microbial biodiscovery.
Our observations on the inter-Kingdom beach warfare and chemical ecology between Aspergillus sp. CMB-AsM0423 and Streptomyces sp. CMB-M0423 revealed an unexpected NO mediated transcriptional activation of silent chemical defences. We determined that the rare fungistatic farnesylated 2-nitropyrrole heronapyrrole B (1), initially described from a beach sand-derived Streptomyces sp., is in fact the product of a silent BGC under the tight regulatory control of the bacteriostatic chemical cue cyclo-(l-Phe-trans-4-hydroxy-l-Pro) (3) produced by a co-isolated fungus. We applied a range of complementary chemistry, biochemistry, spectroscopic, and microscopy technologies to demonstrate the extraordinary specificity of 3 and its mechanism of action as an activator of NO biosynthesis. Genomic and transcriptomic approaches further validate the relationship between 3, NO and 1. We applied NO synthase inhibitors (AG, L-NAME or L-NMMA), NO donors (DEANO, SNP, NO gas), and an NO scavenger (CPITO), to confirm that transcriptional activation of the silent BGC for 1 can be achieved by either endogenous NO (i.e., that stimulated by the presence of 3), or exogenous NO (i.e., NO delivered directly from external sources) (Fig. 8). Finally, building on discoveries in a specific case study, we speculate and provide evidence that nitric oxide mediated transcription activation (NOMETA) may have broader significance in the control of chemical defences, with potential as an exciting new microbial biodiscovery tool for in situ activation of silent BGCs.

Aspergillus sp. CMB-AsM0423 constitutively produces a selection of natural products which are retained within the mycelia, with a. ~10% of the bacteriostatic diketopiperazine 3 being secreted into the culture broth. In co-culture, b. secreted 3 activates an XOR, NaR, NiR cycle in Streptomyces sp. CMB-StM0423 leading to c. the production of NO, which in turn d. activates transcription of the silent BGC encoding for heronapyrroles (i.e. 1). Heronapyrroles are then e. secreted into the broth where they stimulate the fungus to f. upregulate production of 3, and g. suppress the biosynthesis of all other co-metabolites. This a–g cycle sets up a confrontation between bacteriostatic 3 and fungistatic 1, ultimately driving the fungus into a quiescent state. This cycle can be disrupted by h. addition of an NO inhibitor (i.e., aminoguanidine), which shuts down the production of NO and heronapyrroles. This disruption can be overturned by i. addition of an NO donor, or can be reinstated by j. addition of an NO scavenger
References
Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14:111–29.
Rutledge PJ, Challis GL. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Microbiol. 2015;13:509–23.
Abdelmohsen UR, Grkovic T, Balasubramanian S, Kamel MS, Quinn RJ, Hentschel U. Elicitation of secondary metabolism in actinomycetes. Biotechnol Adv. 2015;33:798–811.
Luo Y, Huang H, Liang J, Wang M, Lu L, Shao Z, et al. Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat Commun. 2013;4:3804/1–3894/8.
Netzker T, Fischer J, Weber J, Mattern Derek J, Konig Claudia C, Brakhage Axel A, et al. Microbial communication leading to the activation of silent fungal secondary metabolite gene clusters. Front Microbiol. 2015;6:299.
Onaka H. Novel antibiotic screening methods to awaken silent or cryptic secondary metabolic pathways in actinomycetes. J Antibiot. 2017;70:865–70.
Zarins-Tutt JS, Barberi TT, Gao H, Mearns-Spragg A, Zhang L, Newman DJ, et al. Prospecting for new bacterial metabolites: a glossary of approaches for inducing, activating and upregulating the biosynthesis of bacterial cryptic or silent natural products. Nat Prod Rep. 2016;33:54–72.
Marmann A, Aly AH, Lin W, Wang B, Proksch P. Co-cultivation-a powerful emerging tool for enhancing the chemical diversity of microorganisms. Mar Drugs. 2014;12:1043–65.
Van der MA, Van WP, Worsley SF, Hutchings MI, Van WP. Chemical ecology of antibiotic production by actinomycetes. FEMS Microbiol Rev. 2017;41:392–416.
Nutzmann H-W, Reyes-Dominguez Y, Scherlach K, Schroeckh V, Horn F, Gacek A, et al. Bacteria-induced natural product formation in the fungus Aspergillus nidulans requires Saga/Ada-mediated histone acetylation. Proc Natl Acad Sci USA. 2011;108:14282–7. S14282/1–12.
Koenig CC, Scherlach K, Schroeckh V, Horn F, Nietzsche S, Brakhage AA, et al. Bacterium induces cryptic meroterpenoid pathway in the pathogenic fungus Aspergillus fumigatus. Chembiochem. 2013;14:938–42.
Schroeckh V, Scherlach K, Nutzmann H-W, Shelest E, Schmidt-Heck W, Schuemann J, et al. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Natl Acad Sci USA. 2009;106:14558–63.
Rateb ME, Hallyburton I, Houssen WE, Bull AT, Goodfellow M, Santhanam R, et al. Induction of diverse secondary metabolites in Aspergillus fumigatus by microbial co-culture. RSC Adv. 2013;3:14444–50.
Ebrahim W, El-Neketi M, Lewald L-I, Orfali RS, Lin W, Rehberg N, et al. Metabolites from the Fungal Endophyte Aspergillus austroafricanus in axenic culture and in fungal-bacterial mixed cultures. J Nat Prod. 2016;79:914–22.
Park HB, Kim Y-J, Park J-S, Yang HO, Lee KR, Kwon HC. Glionitrin B, a cancer invasion inhibitory diketopiperazine produced by microbial coculture. J Nat Prod. 2011;74:2309–12.
Park HB, Kwon HC, Lee C-H, Yang HO. Glionitrin A, an antibiotic-antitumor metabolite derived from competitive interaction between abandoned mine microbes. J Nat Prod. 2009;72:248–52.
Wu C, Zacchetti B, Ram AFJ, van Wezel GP, Claessen D, Hae CY. Expanding the chemical space for natural products by Aspergillus-Streptomyces co-cultivation and biotransformation. Sci Rep. 2015;5:10868.
Raju R, Piggott AM, Barrientos DLX, Khalil Z, Capon RJ. Heronapyrroles A-C: Farnesylated 2-Nitropyrroles from an Australian Marine-Derived Streptomyces sp. Org Lett. 2010;12:5158–61.
Khalil ZG, Kalansuriya P, Capon RJ. Lipopolysaccharide (LPS) stimulation of fungal secondary metabolism. Mycology. 2014;5:168–78.
Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–9.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genom. 2008;9:75.
Blin K, Lee SY, Weber T, Medema MH, Kottmann R, Lee SY. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017;45:D555–9.
Skinnider MA, Dejong CA, Rees PN, Johnston CW, Li H, Webster ALH, et al. Genomes to natural products prediction informatics for secondary metabolomes (PRISM). Nucleic Acids Res. 2015;43:9645–62.
Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, et al. Minimum information about a biosynthetic gene cluster. Nat Chem Biol. 2015;11:625–31.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Ronquist F, Teslenko M, van der Mark P, Ayres Daniel L, Darling A, Hohna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Marcellin E, Mercer TR, Licona-Cassani C, Palfreyman RW, Dinger ME, Steen JA, et al. Saccharopolyspora erythraea’s genome is organized in high-order transcriptional regions mediated by targeted degradation at the metabolic switch. BMC Genom. 2013;14:15.
Brown J, Pirrung M, McCue LA. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics. 2017;33:3137–9.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47/1–e47/13.
Wakefield J, Jaspars M, Ebel R, Hassan HM, Rateb ME. Dual induction of new microbial secondary metabolites by fungal bacterial co-cultivation. Front Microbiol. 2017;8:1284.
Leipoldt F, Zeyhle P, Kulik A, Kalinowski J, Heide L, Kaysser L. Diversity of ABBA prenyltransferases in marine Streptomyces sp. CNQ-509: promiscuous enzymes for the biosynthesis of mixed terpenoid compounds. PLoS ONE. 2015;10:e0143237/1–15.
Gallagher KA, Jensen PR. Genomic insights into the evolution of hybrid isoprenoid biosynthetic gene clusters in the MAR4 marine streptomycete clade. BMC Genom. 2015;16:960/1–13.
Kuzuyama T, Noel JP, Richard SB. Structural basis for the promiscuous biosynthetic prenylation of aromatic natural products. Nature. 2005;435:983–7.
Demydchuk Y, Sun Y, Hong H, Staunton J, Spencer JB, Leadlay PF. Analysis of the tetronomycin gene cluster: insights into the biosynthesis of a polyether tetronate antibiotic. Chembiochem. 2008;9:1136–45.
Godber BLJ, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, et al. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J Biol Chem. 2000;275:7757–63.
Maia LB, Moura JJG. Nitrite reduction by molybdoenzymes: a new class of nitric oxide-forming nitrite reductases. J Biol Inorg Chem. 2015;20:403–33.
Courderot-Masuyer C, Dalloz F, Maupoil V, Rochette L. Antioxidant properties of aminoguanidine. Fundam Clin Pharmacol. 1999;13:535–40.
Yukioka Y, Tanahashi T, Shida K, Oguchi H, Ogawa S, Saito C, et al. A role of nitrite reductase (NirBD) for NO homeostatic regulation in Streptomyces coelicolor A3(2). FEMS Microbiol Lett. 2017;364:fnw241.
Sasaki Y, Oguchi H, Kobayashi T, Kusama S, Sugiura R, Moriya K, et al. Nitrogen oxide cycle regulates nitric oxide levels and bacterial cell signaling. Sci Rep. 2016;6:22038.
Fjaervik E, Zotchev SB. Biosynthesis of the polyene macrolide antibiotic nystatin in Streptomyces noursei. Appl Microbiol Biotechnol. 2005;67:436–43.
Debono M, Abbott BJ, Molloy RM, Fukuda DS, Hunt AH, Daupert VM, et al. Enzymic and chemical modifications of lipopeptide antibiotic A21978C: the synthesis and evaluation of daptomycin (LY146032). J Antibiot. 1988;41:1093–105.
Liu W-T, Peng Y, Lamsa A, Pogliano K, Wong Weng R, Linington Roger G, et al. MS/MS-based networking and peptidogenomics guided genome mining revealed the stenothricin gene cluster in Streptomyces roseosporus. J Antibiot. 2014;67:99–104.
Jiang L, Wang L, Zhang J, Liu H, Hong B, Tan H, et al. Identification of novel mureidomycin analogs via rational activation of a cryptic gene cluster in Streptomyces roseosporus NRRL 15998. Sci Rep. 2015;5:14111.
Acknowledgements
ZK acknowledges the University of Queensland for a postgraduate scholarship, and PCM the Australian Department of Education and Training for an Endeavour Fellowship. We thank R Ritesh for original isolation of Streptomyces sp CMB-M0423, R Palfreyman for bioinformatic support, and the Queensland (UQ) node of Proteomics and Metabolomics Australia, a NCRIS initiative under Bioplatforms Australia Pty Ltd. Microscopy was performed at the Australian Cancer Research Foundation (ACRF) Institute for Molecular Bioscience, Cancer Biology Imaging Facility, with support from N Condon, J Springfield, M. Scott, J Griffin and K Green. This research was funded in part by the University of Queensland, Institute for Molecular Bioscience and Australian Institute for Bioengineering and Nanotechnology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Khalil, Z.G., Cruz-Morales, P., Licona-Cassani, C. et al. Inter-Kingdom beach warfare: Microbial chemical communication activates natural chemical defences. ISME J 13, 147–158 (2019). https://doi.org/10.1038/s41396-018-0265-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0265-z
This article is cited by
-
Coculturing of Mucor plumbeus and Bacillus subtilis bacterium as an efficient fermentation strategy to enhance fungal lipid and gamma-linolenic acid (GLA) production
Scientific Reports (2022)
-
A bioprocess perspective on the production of secondary metabolites by Streptomyces in submerged co-cultures
World Journal of Microbiology and Biotechnology (2021)
-
Microbe-driven chemical ecology: past, present and future
The ISME Journal (2019)
