
Abstract
Over the past decade several studies have reported that the gut microbiomes of mammals with similar dietary niches exhibit similar compositional and functional traits. However, these studies rely heavily on samples from captive individuals and often confound host phylogeny, gut morphology, and diet. To more explicitly test the influence of host dietary niche on the mammalian gut microbiome we use 16S rRNA gene amplicon sequencing and shotgun metagenomics to compare the gut microbiota of 18 species of wild non-human primates classified as either folivores or closely related non-folivores, evenly distributed throughout the primate order and representing a range of gut morphological specializations. While folivory results in some convergent microbial traits, collectively we show that the influence of host phylogeny on both gut microbial composition and function is much stronger than that of host dietary niche. This pattern does not result from differences in host geographic location or actual dietary intake at the time of sampling, but instead appears to result from differences in host physiology. These findings indicate that mammalian gut microbiome plasticity in response to dietary shifts over both the lifespan of an individual host and the evolutionary history of a given host species is constrained by host physiological evolution. Therefore, the gut microbiome cannot be considered separately from host physiology when describing host nutritional strategies and the emergence of host dietary niches.
Similar content being viewed by others


Introduction
The mammalian gut microbiota has a wide range of effects on host physiology and behavior. Therefore, a key focus of gut microbiota research over the past decade has been determining what factors shape its composition and function. Several comparative papers report that host dietary niches play a major role in determining the gut microbiota of a given host species, with diet specializations, such as carnivory, herbivory, and ant-eating resulting in similar gut microbial traits across diverse host species [1,2,3]. These findings coincide with studies of individual host species that demonstrate the strong impact of diet on the gut microbiota across days, months, and years, [4,5,6] and they support the hypothesis that gut microbes contribute to host dietary plasticity by providing specific metabolic services tailored for the breakdown of certain foods [7, 8]. However, several factors such as the inclusion of data from both wild and captive animals, as well as confounds between host phylogeny, anatomical specializations, and dietary niche complicate these comparative studies. Captivity has a strong effect on the gut microbiota [9,10,11], making it unlikely that all data from captive individuals are representative of the true evolutionary relationship between host and microbe. Additionally, gut morphology impacts the gut microbiota [12], and the gut microbiota can co-diversify with hosts, creating strong associations between host physiology, host phylogenetic similarity, and gut microbial similarity [13,14,15]. Published data describing the microbial signals of herbivory and carnivory rely heavily (albeit not exclusively) on closely related mammal species to represent these diets [2, 3], and a description of the impact of ant-eating on the gut microbiota encompasses both dietary and physiological factors since highly divergent myrmecophagus hosts share convergent physiological adaptations, such as muscular stomachs and short small intestines [1]. Therefore, the associations between the gut microbiota and host dietary niche reported by these studies may not be attributable only to host dietary niche, and thus may be over-interpreted. In fact, a recent study suggests that host phylogeny and physiology impact microbiome divergence rates in mammals more strongly than host diet [16]. Nevertheless, this study relies on existing data that incorporate many of the same biases described above. More robust tests of whether host dietary niche shape the mammalian gut microbiota independently of other host factors should focus exclusively on wild hosts while controlling for both host phylogeny and gut morphology.
Here, we capitalize on the remarkable dietary and anatomical diversity of non-human primates (primates hereon) to understand the effect of host dietary niche on the composition and function of the colonic gut microbiota. We use 16S rRNA gene amplicon and shotgun metagenomic sequencing to analyze the colonic gut microbiota of nine folivorous, wild primate species and nine closely related, non-folivorous, wild primate species representing the four major clades of the primate phylogeny, many of which overlap in geographic range (Fig. 1).

Host dietary niche has a weak effect on primate gut microbiota composition. Principal coordinates analysis (PCoA; a unweighted and b weighted UniFrac distances) of 16S rRNA gene amplicon data illustrates stronger clustering of non-human primate fecal samples by host phylogenetic clade (unweighted UniFrac: PERMANOVA F3,153 = 26.4, r2 = 0.29, p < 0.01; weighted UniFrac: PERMANOVA F3,153 = 21.7, r2 = 0.27, p < 0.01) than diet (unweighted UniFrac: PERMANOVA F1,153 = 13.1, r2 = 0.05, p < 0.01; weighted UniFrac: PERMANOVA F1,153 = 9.2, r2 = 0.04, p < 0.01). Large spheres represent folivorous primates and small spheres represent non-folivorous primates. Folivores are shaded in the phylogenetic tree. (Note that T. gelada consumes grass, which shares many nutritional challenges with leaves.)
Folivory—the ability to consume large amounts of leaves either seasonally or year-round—has evolved independently multiple times throughout the primate Order (e.g., in Malagasy strepsirrhines: sifaka, indriids; in platyrrhines, or New World monkeys: howler monkeys; in catarrhines, or Old World monkeys: colobines; and in hominoids, or apes: gorillas). Compared to other food resources such as fruit and insects, leaves generally have high amounts of structural carbohydrates and secondary metabolites, which make them difficult to digest [17]. In addition to food selectivity, folivorous primates are believed to rely heavily on the gut microbiota to utilize this challenging diet [5, 18]. Additionally, in each primate clade, unique anatomical specializations evolved in parallel to folivory. Gorillas have a large body size that maximizes gut volume and retention time, and colobines have a sacculated foregut. Howler monkeys have a slightly enlarged colon, and sifaka have an enlarged caecum. Therefore, it is possible to directly test whether all folivorous primates share gut microbial taxonomic and functional characteristics independently of host phylogenetic and morphological confounds. We hypothesized that despite an effect of host phylogeny, gut microbiota composition and function would be shaped by host dietary niche. In particular, we predicted that a subset of gut microbial taxa and functions related to cellulose and secondary metabolite degradation (e.g., tannins, phenols, etc.) would be enriched among all folivorous primates since some quantity of these compounds is likely to reach the colon, regardless of host physiology, and gut morphology.
Materials and methods
Sample collection
All samples were collected from wild non-human primates by collaborators at field sites around the world (Table S1). In every case, bulk fecal samples were collected immediately after defecation with a sterile utensil (e.g., plastic spoon) and stored in a collection tube with either 95% ethanol or RNALater. Samples were stored and transported to the United States by collaborators. Table S1 lists the responsible collaborator, sampling site, sample size, and preservation method for each non-human primate species. Appropriate government permits and IACUC protocols were obtained by each collaborator independently.
Sample processing for 16S rRNA gene amplicon sequencing
We began our analyses by describing the microbial taxonomic composition of all samples. To do this, we followed the Earth Microbiome Project protocol [19]. We extracted microbial DNA from all samples using the MO BIO PowerSoil DNA extraction kit. PCR targeting the V4 region of the 16S rRNA bacterial was performed with the 515F/806R primers, utilizing the protocol described in Caporaso et al. [20]. We barcoded and pooled amplicons in equal concentrations for sequencing. We then purified the amplicon pool with the MO BIO UltraClean PCR Clean-up kit and sequenced on the Illumina MiSeq sequencing platform (MiSeq Control Software 2.0.5 and Real-Time Analysis software 1.16.18) at the BioFrontiers Institute Next-Generation Genomics Facility at the University of Colorado, Boulder, USA. Samples were pseudo-randomly assigned to three different MiSeq runs as they were accumulated so that samples representing a given host clade or diet type were never all sequenced on the same run. In several cases, samples from the same host species were assigned to different runs.
Quality filtering and OTU-picking
The single-end sequencing reads from the 515f primer were quality-checked using the default settings for the split_libraries_fastq.py function in QIIME v1.9.0 [21]. After quality filtering we obtained 12,178,012 reads associated with these samples with an average of 23,152 reads/sample (range: 0–80,714 reads/sample).
Following common practice in microbiome research, sequences were initially clustered into representative bacterial operational taxonomic units (OTUs) using the sortmerna/sumaclust implementation of open-reference OTU-picking at 97% sequence similarity [22]. Sequences were aligned [23], and taxonomy was assigned using UCLUST [24] and the Green Genes 13_8 database [25, 26]. Sequences representing chloroplasts and mitochondria were filtered out, and any OTUs representing <0.00005% of the total dataset were filtered out as recommended for Illumina-generated sequencing data [27]. A subset of samples were randomly selected for analysis for each host species (Table S1). The data for these samples were rarefied to 15,012 reads/sample (single_rarefaction.py).
To increase our ability to describe patterns of microbial community structure at finer taxonomic resolution, we also processed sequences using Deblur [28], which bypasses the OTU clustering algorithm described above. Briefly, this algorithm uses Illumina error profiles to obtain putative error-free sequences that describe microbial community composition at the sub-OTU (sOTU) level. To place deblurred sequences into a phylogenetic context, we used SEPP [29] to insert unique deblurred V4 fragments into the most recent available Greengenes phylogeny of representative 97% clustered full-length 16S sequences (Greengenes v. 13_8). SEPP was run with an alignment subset of 100 and placement subset set of 500. Reference sequences were then trimmed from the tree, leaving the subsequent phylogeny for downstream applications, including alpha- and beta-diversity calculations and balance tree analysis. Taxonomy was also inferred from the SEPP insertions. For each deblurred sequence, SEPP returns a set of (at most) seven highest-likelihood candidate placements in the reference phylogeny, each of which includes an attaching branch from the reference tree along with a probability. For each branch in the reference tree, a taxonomic label was assigned at each rank if and only if at least 95% of the leaves below that branch share the same label. The root of the tree was located on a branch that splits the kingdoms Bacteria and Archaea perfectly, so every rank of the taxonomy is well contained on one side of the root or the other. For a given deblurred sequence at a given taxonomic rank, each candidate placement inherits the label of its attaching branch and a label is assigned to the sequence if candidate placements representing at least 80% cumulative probability share that label. Effectively, labels were assigned to internal branches of the Greengenes tree by a de-facto voting of child leaves with a quorum of 95%, and query sequences were labeled by SEPP if it assigned at least 80% probability to branches with a common label.
All subsequent statistical analyses were performed on both the OTU-clustered dataset and the deblurred dataset, and results were consistent across methods. The analyses of microbial community taxonomic composition presented in the main text utilize the deblurred data unless otherwise noted.
Analysis of sample taxonomic composition
Once processed, we used sequence data to compare richness, diversity, and microbial community composition among samples. We generated beta diversity distance matrices using QIIME (beta_diversity_through_plots.py), and we visualized clustering patterns among samples using principal coordinates analysis (PCoA, Emperor v 0.9.51 [30] and non-metric multi-dimensional scaling (vegan package, R software, version 3.0.2)). We calculated pairwise distances between samples using unweighted UniFrac and weighted UniFrac similarity indices [31]. We tested for significant differences in sample clustering patterns and microbial community composition across host clades (Old World monkey, New World monkey, ape, lemur) and diet type (folivore, non-folivore, controlling for host clade) for each species using permutational analysis of variance (PERMANOVA, adonis function in the vegan package, R software, version 3.0.2). Because PERMANOVA is sensitive to differences in dispersion between groups, we also tested for these differences. Host phylogenetic groups exhibited significantly different dispersion (F3,150 = 13.4, p < 0.01) while host diet groups did not (F3,150 = 3.5, p = 0.06). However, visual inspection of clustering plots suggests that differing dispersion is not driving patterns of significance across host phylogenetic groups. We calculated the number of observed sOTUs and the Faith’s phylogenetic diversity [32] to describe the alpha diversity in each sample using QIIME (alpha_rarefaction.py). We then identified core sOTUs shared by 90% of the samples for each host clade and in 80% of the samples for each host diet type (compute_core.py).
To detect sOTUs that were significantly different in relative abundance among the four clades of primates we utilized a linear discriminant analysis of effect size (LEfSe; [33]). We assigned primate families as the class vector and kept features with a logarithmic LDA score of >3 using default parameters. We reran this analysis using diet as the class vector and primate family as the subclass vector to detect sOTUs different in abundance between folivores non-folivores; however, no features were detected even with a low LDA score cutoff of 0.8.
Because, we observed high levels of variation in the distribution of bacterial taxa among primate clades, we also used a more sensitive method for detecting differences in microbial community composition using a concept known as balance trees [34]. These balances are the log-ratios of phylogenetic clades and analyzing these balances alleviates the common problems associated with compositionality in microbial sequence data [35]. The specific methodology used for constructing and analyzing can be found at https://github.com/biocore/gneiss. Briefly, a pseudocount of 1 was added to all of the values in the deblurred sequence table to account for zeroes and then transformed using the isometric log-ratio calculation [36]. Using the microbial phylogenetic tree built during processing, balances were calculated by computing the log-ratio of proportions between adjacent phylogenetic clades at each internal node of the tree. A linear mixed effects model was then run on each balance, to test for significant differences in the ratios of bacterial taxa among folivorous and non-folivorous lineages while accounting for phylogeny and variability among individuals as random effects.
Testing the effect of host geography
Because host phylogeny, geographic location, and local diet are often confounded, we wanted to more closely explore the potential influence of host geography and local diet on our dataset. Therefore, we created two additional sets of PCoA plots (based on 97% OTUs) with new samples included. First, we examined only New World monkeys but utilized additional howler monkey samples collected from different sites with different forest types [37]. Specifically, we included Alouatta pigra samples from a semi-deciduous forest (El Tormento, Mexico) and Alouatta palliata samples from an evergreen rainforest (La Suerte, Costa Rica), which represent markedly distinct environments and diets. Additionally, because the Alouatta diet varies in leaf intake seasonally, we also included samples collected in the same forest during both periods of high fruit intake and high leaf intake when possible.
We also examined the effect of host geography on a larger scale by comparing the gut microbiota of African and Asian colobines. While all of these colobines have similar gut morphology and dietary niches, they inhabit distinct continents with different environments and local diets. To perform this comparison, we integrated published data from twelve wild Asian colobines (red-shanked doucs, Pygathrix nemaeus) with our original data [38]. In both cases, open-reference OTUs were re-picked for the entire dataset using the same methods described above. The resulting data were filtered, rarefied, and analyzed the same way as well.
Cophylogenetic analyses
Given that codiversification of hosts and gut microbes has been emphasized as an important process contributing to the composition of the primate gut microbiome [13, 14], we wanted to explicitly explore the relationship between the host phylogeny and diversity of microbial 16S sequences in this dataset. Therefore, we performed two analyses: one, a reimplementation of the beta-diversity clustering sensitivity analysis in Sanders et al. [39] to assess whether patterns of microbial community similarity that are correlated with host phylogeny are likely to indicate codiversification; and two, an application of the permutation test of cophylogeny from Hommola et al. [40] to sequence diversity within OTUs to test for codiversification in individual bacterial lineages. Both analyses were implemented in a Snakemake [41] workflow available at https://github.com/tanaes/snakemake_codiversification. Briefly, deblurred 16S sequences were clustered using the USEARCH [24] pipeline in QIIME 1.8.1 at similarity thresholds of 85, 88, 91, 94, 97, and 99% identity. For beta-diversity clustering sensitivity analysis, beta-diversity distances among four randomly selected samples per species were calculated from 100 OTU tables jackknifed to 7200 sequences. Each jackknifed distance matrix was UPGMA-clustered, and the resulting similarity dendrogram compared against the actual host phylogeny. A summary figure was then created to illustrate the number of times each actual host clade was recovered from each parameter combination.
For per-lineage codiversification analysis, the deblurred 16S rRNA sequences composing each 97% OTU were realigned using MUSCLE, a phylogeny estimated with FastTree [42], and the pairwise distances among unique bacterial sequences compared to the pairwise patristic distances of their host taxa using an adaptation of the Hommola et al. [40] permutation test of cospeciation, with 10,000 permutations. This test is an extension of the Mantel test of distance matrix correlation, modified to allow multiple symbionts per host (and vice versa). p-values were corrected using the Benjamini–Hochberg False Discovery Rate, and OTUs estimated to be significantly codiversifying with their hosts illustrated by mapping host information onto the intra-OTU phylogeny.
Sample processing for shotgun metagenomic sequencing
In addition to describing the taxonomic composition of the sampled primate gut microbiomes, we also wanted to assess the functional capacity of these microbiomes. To do this, a subset of 95 samples was randomly selected for shotgun metagenomic sequencing (Table S1). Sequencing libraries were robotically prepared with the Kapa Hyper Library Preparation kit (Kapa Biosystems) at the Roy J. Carver Biotechnology Center at the University of Illinois at Champaign-Urbana. Library insert sizes ranged from 80 to 700 bp. Libraries were combined into four pools, each of which was sequenced on one lane of the Illumina HiSeq2500 using TruSeq SBS sequencing chemistry version 4. A total of 160-nt paired-end reads were generated using 161 cycles for each end of the fragment. Fastq files were generated and demultiplexed using the bcl2fastq v1.8.4 Conversion Software (Illumina). The run produced a total of 1,472,869,654 reads (average: 7,671,196 ± 2,966,770 reads/sample) with average quality scores of 32 and above.
Gene ortholog group and pathway relative abundance
Shotgun metagenomic data were quality filtered with Trimmomatic v.0.32 with parameters ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36. This yielded 94 metagenomes with sufficient sequencing depth for quantitative analysis, which were subsampled to the size of smallest one (3,589,870 reads) using seqtk v.1.0. Forward and reverse reads were concatenated (seqtk was run with the same seed for both to maintain read pairs). The 94 metagenomes were analyzed for the relative abundance of gene ortholog groups and biochemical pathways using HUMAnN2 v.0.2.2 with the following workflow to ensure comparable results across samples: MetaPhlAn2 was run on each metagenome using MetaPhlAn2 database v.20. Lists of matched strains in 94 results were merged to a single ‘bugs list’. A single HUMAnN2 run was done using this bugs list with the sole purpose of generating a custom Bowtie2 database from the subset of the ChocoPhlAn v.0.1.1 centroid genomes corresponding to the bugs list. HUMAnN2 (http://huttenhower.sph.harvard.edu/humann2) was then run on all 94 samples using this pre-compiled database (first step: Bowtie 2 on all reads) and then with the default UniRef50 database (second step: DIAMOND translated search on leftover reads) with options--bypass-prescreen--bypass-nucleotide-index. Each of the three types of HUMAnN2 output tables (genefamilies, pathabundance, pathcoverage) were then merged across the 94 samples (humann2_join_tables), then normalized to counts per million (cpm) and relative abundance (relab) (humann2_renorm_table). The genefamilies tables were regrouped (humann2_regroup_table) from UniRef50 families to KEGG Orthology (KO), Gene Ontology (GO), MetaCyc reaction (rxn), and Enzyme Classification number (EC). Finally, tables were split into two versions: stratified by taxonomy, and unstratified (sum of all strains).
Analysis of sample functional composition
Once sequence data were processed, we used them to compare richness, diversity, and gene composition among samples. Beta diversity distance matrices were generated using QIIME (beta_diversity_through_plots.py), and clustering patterns among samples were visualized using principal coordinates analysis (PCoA, Emperor v 0.9.51 [30]) and non-metric multi-dimensional scaling (vegan package, R software, version 3.0.2). Pairwise distances between samples were calculated using Bray-Curtis similarity indices. We tested for significant differences in sample clustering patterns and microbial community composition across host clades (Old World monkey, New World monkey, ape, lemur) and diet type (folivore, non-folivore, controlling for host clade) for each species using permutational analysis of variance (PERMANOVA, adonis function in the vegan package, R software, version 3.0.2).
To investigate whether taxonomic and gene abundance patterns were similar in our samples, we compared beta-diversity patterns between 16S rRNA (unweighted UniFrac) and shotgun metagenomics Metacyc reaction pathway (Bray-Curtis) datasets. We used a Procrustes analysis (least-squares orthogonal mapping) to transform the first three principal coordinates for each dataset (QIIME, transform_coordinate_matrices.py) and estimate m2 value (sum of the squared deviations). We then shuffled sample identifiers and recalculated m2 999 times and reported the p-value as the proportion of m2 values lower than the actual m2 value. We also directly compared the distance matrices using a mantel test with 999 permutations.
In addition to assessing overall functional capacity, we also wanted to examine differences in the relative abundances of specific enzymes associated with functions such as cellulose degradation and plant secondary metabolite degradation. To do this, we extracted information about CAZyme relative abundances from the metagenomic dataset. The HUMAnN2 analyses described above were reimplemented, but using the dbCAN [43] version of the CAZyDB [44] as a custom translated alignment database, skipping the Bowtie2 nucleotide alignment step. Substrate specificities of particular CAZyme families used to produce Figure S8 were derived from Table S1 of [45] (Cantarel et al. 2012). Summed counts for CAZymes in each of these categories were compared using a 2-way ANOVA in R, with diet category and host phylogenetic group modeled as additive effects. p-values for an effect of diet were corrected for multiple hypothesis tests using the Bonferroni method.
We also examined whether other microbial metabolic pathways differed in relative abundance among samples. We used a linear mixed effects model to assess the abundance of specific MetaCyc pathways in folivorous versus non-folivorous lineages. Model comparisons were performed between one that simply accounted for the random effects of each species nested within the four phylogenetic groups and a second which incorporated diet as an additional fixed effect. Pathways were identified as associated with diet category when inclusion significantly improved the fit of the second model over the first with a p-value of <0.01.
For similar reasons, we also utilized a linear discriminant analysis of effect size (LEfSe; [33]) to detect pathways that were significantly different in relative abundance among clades of primates. We assigned primate phylogenetic group as the class vector and diet as the subclass vector. Features with a logarithmic LDA score of >3.0 using default parameters were kept.
Results
Gut microbial composition
Using deblurred 16S rRNA amplicon data [28] and controlling for host phylogeny by comparing folivorous and non-folivorous primates across the entire primate order, we found that folivory had a small but significant effect on gut microbiota composition at the sub-OTU level (sOTU; Fig. 1, S1; unweighted UniFrac: PERMANOVA F1,153 = 13.1, r2 = 0.05, p < 0.01; weighted UniFrac: PERMANOVA F1,153 = 9.2, r2 = 0.04, p < 0.01). However, it was difficult to clearly define a characteristic ‘folivorous primate gut microbiota.’ There were no consistent differences in gut microbial richness or diversity between diet types at the sOTU level (Fig. S2). Additionally, neither diet type was associated with a core gut microbiota, and LefSe analysis did not indicate strong differences in the relative abundances of sOTUs between diet types across the primate phylogeny. Given the possibility that existing taxonomic labels do not correspond to the specific bacterial clades most associated with folivory, we also performed a balance tree analysis to find nodes of the bacterial phylogeny for which daughter lineages were present in different ratios in folivorous and non-folivorous primates after controlling for host phylogenetic group [34]. This analysis revealed several such bacterial groups within the Clostridia, for which a significant effect of folivory could be observed in the aggregate; although, to some extent, these patterns were still specific to a subset of primate clades (Fig. 2).

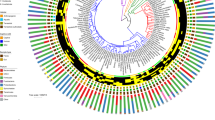
Folivorous primates share few gut microbiota traits at the taxonomic level. A phylogenetic tree summarizes the results of the linear mixed effects analysis applied to balances. The circular heatmaps surrounding the tree plot the proportions of microbes across all of the samples, with the outmost ring containing samples from folivorous species and the inner ring containing samples from non-folivorous species. Three significant balances (p-value <0.01) differentiate the gut microbiota of folivorous primates from non-folivorous primates. Darker shades represent enrichment of that particular microbial clade
Given that we detected a weak effect of host dietary niche on the composition and function of the primate gut microbiota, we set out to determine whether other host traits are more important for shaping the primate gut microbiota. Our analysis indicated that host phylogenetic relationships were the strongest determinants of primate gut microbiota composition at the sOTU level (Fig. 1, S1; unweighted UniFrac: PERMANOVA- F3,153 = 26.4, r2 = 0.29, p < 0.01, weighted UniFrac: PERMANOVA F3,153 = 21.7, r2 = 0.27, p < 0.01). Microbial community richness and diversity differed significantly across the four primate clades, with lemurs exhibiting significantly lower sOTU richness and diversity than all other primates (Fig. S2). Each primate clade exhibited a distinct core gut microbiota (Table S2), and LDA Effect Size analysis (LEfSe) [33] indicated that several sOTUs, particularly in the bacterial class Clostridia, significantly differed in relative abundance across the primate clades (Table S3). Additionally, 56% of the sequence reads generated did not match the GreenGenes database at 97%, and an average of 15% of the reads in each sample could not be classified past the kingdom level (range 2–44%; Table S4). While there was a trend for more unclassified reads associated with folivorous primates (Table S5), stronger patterns were observed in relation to host clades. The majority of unclassified reads were detected in lemurs, both in terms of total proportion of reads and fraction of observed sOTUs (Figs. S3, S4).
Notably, the observed effect of host phylogeny on gut microbiota composition did not appear to reflect codiversification of host and gut microbiome (a pattern of concordant phylogenetic histories, potentially resulting from cospeciation over time [46]), as is sometimes assumed. Patterns of phylosymbiosis, or congruence between host phylogeny and whole gut microbial community similarity patterns, were maintained regardless of OTU clustering widths (Fig. S5). This observation suggests that these patterns did not arise from recent microbial evolutionary processes, as would have been the case if the patterns arose via microbial lineage splitting concurrent with host lineage splitting [39]. Furthermore, within individual OTUs picked at 97% similarity, 16S rRNA gene sequence variation was not associated with hosts in a way that indicated strong codiversification; the strongest, most consistent pattern was a division between Old World monkeys and New World monkeys (Fig. S6). Patterns of codiversification may indeed be present in these communities, but if so the majority of the signal is likely found at a finer resolution than can be resolved using the short portion of 16S rRNA gene sequenced here [13].
After controlling for host phylogeny, host geography explained a substantial proportion of variation in gut microbiota composition, especially at narrow clustering widths (Fig. S7). Because host geography is often confounded with host phylogeny, we closely examined the New World monkeys at the OTU level using additional samples (Materials and methods). Our findings revealed that neither host geographic location nor the proportion of fruits and leaves in the diet at the time of sampling drove the observed patterns (Fig. S8). Samples clustered by host genus and species independently of both intra-specific differences in sampling location or percent folivory of the diet (Fig. S8). Integrating previously published Asian colobine data (Pygathrix nemaeus) [38] with our larger dataset also indicated stronger clustering of African colobines with Asian colobines compared to other African primates (Fig. S9), suggesting an important role for host physiology in shaping the gut microbiota given the specialized gut morphology of colobines.
Gut microbial function
Compared to gut microbial taxonomic composition, we detected a slightly more robust signal of gut microbial functional similarity (measured using shotgun metagenomics) among folivorous primates (Fig. 3a, MetaCyc reactions, Bray-Curtis: PERMANOVA F1,93 = 8.7, r2 = 0.07, p < 0.01). Nevertheless, Procrustes analysis demonstrated strong concordance between patterns in gut microbiota composition and function (Fig. 3b, S10), as previously demonstrated for other mammals on a much broader phylogenetic scale [3], and again, there were few specific microbial characteristics driving overall patterns. Controlling for host phylogeny, folivorous primates were enriched for microbial biosynthesis of arginine and chorismate (a precursor to tryptophan), as well as aminoimidazole ribonucleotide biosynthesis (precursor to adenine; Fig. 4). Non-folivorous primates were enriched for purine degradation and for multiple pathways involved in aerobic energy production and sugar degradation (Fig. 4). There was no difference in pathways for cellulose degradation or plant secondary metabolite degradation between the two diet groups. However, non-folivorous primates were enriched for CAZymes involved in starch and sucrose degradation (Fig. S11).

Host dietary niche has a weak effect on primate gut microbiota functional potential. a Principal coordinates analysis (PCoA; Bray-Curtis dissimilarity) of MetaCyc reaction pathway data illustrates weak clustering of non-human primate fecal samples by diet (PERMANOVA F1,93 = 8.7, r2 = 0.07, p < 0.01). b PCoA illustrating Procrustes analysis of 16S rrNA gene amplicon data (unweighted UniFrac distance) and Metacyc reaction pathway data (Bray-Curtis dissimilarity). For both datasets, host phylogenetic clade is the strongest driver of sample clustering patterns. Sample sizes indicated in parentheses

Folivorous primates share few gut microbiota traits at the functional level. MetaCyc reaction pathways with differential relative abundances between folivorous and non-folivorous primates according to linear mixed effects models show few patterns. Positive values illustrate enrichment in folivorous primates while negative values illustrate enrichment in non-folivorous primates
Similar to taxonomic composition, the functional profile of the primate gut microbiota was most strongly influenced by host phylogeny and physiology (Fig. 3b, S5; Bray-Curtis: PERMANOVA F3,93 = 11.5, r2 = 0.28, p < 0.01). The richness of MetaCyc reaction pathways associated with each primate clade was similar, but LefSe analysis revealed differences in the relative abundance of several pathways across clades (Table S6). Furthermore, CAZyme analysis highlighted an increased relative abundance of enzymes for degrading peptidoglycans and plant cell walls in New World monkeys (Fig. S11). These findings suggest that physiological similarities between closely related primate species result in requirements for similar microbial services regardless of recent divergence in host dietary niches.
Discussion
Collectively, our results demonstrate that the influence of host phylogeny and physiology on the primate gut microbiota is substantially greater than that of host dietary niche. While some shared traits in microbial taxonomy and function are apparent among folivorous primates, the evolution of folivory in each primate clade seems to have been more strongly characterized by unique changes in the distal gut microbiome. For example, at the taxonomic level, even when gut microbial changes in response to folivory appear to involve the same clades of bacteria, different members of those clades fill what we predict are similar niches in different host lineages. These results are consistent with the observation that very ancient splits in bacterial evolution are associated with microbial signatures observed in much more recently evolved mammalian dietary specializations [47].
Whether host gut morphology, immune system function, or other physiological factors are most important in shaping the gut microbiota in the context of host phylogeny remains to be seen, but we speculate that a combination of these physiological factors interact to determine the gut microbiota. For example, in addition to influencing the volume and surface area of different parts of the gut occupied by microbes, anatomical specializations such as a sacculated foregut or an enlarged caecum may alter signaling molecules such as toll-like receptors that act as selective filters on gut microbiota composition. These changes could encourage the acquisition or evolution of new microbial taxa that contribute important metabolic functions to hosts. Shifts in dietary substrates provided to gut microbes as a result of host diet changes could result in the same processes, explaining the dual impact of host phylogeny and dietary niche that we observed. Our discovery of novel microbial taxa in each primate clade, many of which were associated with folivory, provides evidence for this process, as well as expanding our understanding of the gut microbial diversity contained within the primate order [14].
These evolutionary and ecological processes also likely feed back positively among diet, the gut microbiota, and host physiology, intensifying the microbial signal of diet over time. In fact, our data clearly demonstrate a stronger signal of folivory in the gut microbiota of those primate clades in which folivory has been established for longer (e.g. lemurs: ~40 mya; compared to Old World monkeys: ~20 mya; New World monkeys: ~17 mya; apes: ~10 mya) [48]) (Fig. 4). This pattern parallels the pattern reported in a study of 24 animal species in which the effect of host phylogeny on the gut microbiota increased in accordance with the time since host species divergence [15]. It may also explain why diet-associated signals of microbial convergence have been more difficult to detect in host clades with more recent evolutionary diet shifts (e.g., bears, ~5 mya; [2]).
Finally, although, we report a weak influence of host diet on the gut microbiota, given the range of habitats, behaviors, and physiological adaptations represented by these primate species, and the fact that primates have been diverging for ~65 million years (with folivory emerging at different points during this period) [48], our ability to detect any similarities in the gut microbiota of folivorous primates is striking. Future studies targeting the microbial taxa and functions associated with folivory in primates are therefore warranted. A folivorous primate microbiome enriched in pathways for the production of essential or conditionally essential amino acids, such as tryptophan and arginine [49], and a non-folivorous primate microbiome enriched in pathways for starch and sucrose degradation, suggest that microbial services such as vitamin and nutrient biosynthesis and energy metabolism may be especially important to understanding adaptations to folivory.
It is also important to note that this study relied solely on fecal samples despite a range of gut morphological specializations being represented by the sampled primates. A recent study of sloths indicates that microbial signals of diet between species are greater in the foregut compared to the hindgut [50]. Therefore, a comparison of the microbiota of gut chambers where most leaf degradation occurs in each primate clade could reveal more marked patterns than those reported here. However, these data require invasive sampling and are vastly more difficult to obtain from wild individuals.
This analysis provides important insight into the processes behind the evolution of both hosts and their gut microbes. While the flexibility of the mammalian gut microbiota in response to host diet has been a dominant theme in the field, by utilizing independent contrasts of dietary niche across multiple primate clades with distinct gut morphologies, we demonstrate clear limits to the ability of the mammalian gut microbiota to shift in response to changes in host diet. While differences in diet across space and time have a strong effect on the gut microbiome of any given host species when considered in isolation [4, 5, 51,52,53,54,55,56], their effect is much smaller than that of host phylogeny and physiology and is difficult to detect in the context of cross-host species comparisons. In this sense, the importance of diet in shaping the gut microbiome is influenced by study design and scale. Although, gut microbes likely play a critical role in supporting host dietary specializations and facilitating individual host dietary plasticity, our data indicate that the bidirectional interactions of host physiology and gut microbiota over evolutionary time ultimately dictate the host nutritional outcomes resulting from a given dietary strategy.
References
Delsuc F, Metcalf JL, Parfrey LW, Song SJ, Gonzalez A, Knight R. Convergence of gut microbiomes in myrmecophagous mammals. Mol Ecol. 2014;23:1301–17.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647–51.
Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–4.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–66.
Amato KR, Leigh SR, Kent A, Mackie RI, Yeoman CJ, Stumpf RM, et al. The gut microbiota appears to compensate for seasonal diet variation in the wild black howler monkey (Alouatta pigra). Microb Ecol. 2015;69:434–43.
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–8.
Kohl KD, Weiss RB, Cox J, Dale C, Dearing MD. Gut microbes of mammalian herbivores facilitate intake of plant toxins. Ecol Lett. 2014b;17:1238–46.
Scharf ME, Karl ZJ, Sethi A, Boucias DG. Multiple levels of synergistic collaboration in termite lignocellulose digestion. PLoS ONE. 2011;6:e21709.
Amato KR, Yeoman CJ, Kent A, Carbonero F, Righini N, Estrada AE, et al. Habitat degradation impacts primate gastrointestinal microbiomes. ISME J. 2013;7:1344–53.
Clayton JB, Vangay P, Huan H, Ward T, Hillmann BM, Al-Ghalith GA, et al. Captivity humanizes the primate microbiome. Proc Natl Acad Sci USA. 2016;113:10376–81.
Kohl KD, Skopec MM, Dearing MD. Captivity results in disparate loss of gut microbial diversity in closely related hosts. Conserv Physiol. 2014a;2:cou009.
Sanders JG, Beichman AC, Roman J, Scott JJ, Emerson D, McCarthy JJ, et al. Baleen whales host a unique gut microbiome with similarities to both carnivores and herbivores. Nat Commun. 2015;6:8285.
Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, et al. Cospeciation of gut microbiota with hominids. Science. 2016;353:380–2.
Ochman H, Worobey M, Kuo CH, Ndjango JBN, Peeters M, Hahn BH, et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 2010;8:e1000546.
Brooks AW, Kohl KD, Brucker RM, van Opstal EJ, Bordenstein SR. Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 2016;14:e2000225.
Nishida A, Ochman H. Rates of gut microbiome divergence in mammals. Mol Ecol. 2018;27:1884–97.
Lambert JE. Primate nutritional ecology: feeding biology and diet at ecological and evolutionary scales. In: Campbell C, Fuentes A, MacKinnon KC, Panger M, Bearder SK, (eds). Primates in Perspective. Second edition. New York: Oxford University Press; 2011. p. 512–22. edn
Gomez A, Rothman JM, Petrzelkova KJ, Yeoman CJ, Vlckova K, Umana JD, et al. Temporal variation selects for diet-microbe co-metabolic traits in the gut of Gorilla spp. ISME J. 2016;10:514–26.
Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nat Biotech. 2017;551:457–63.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010b;7:335–6.
Kopylova E, Noe L, Touzet H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–7.
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010a;26:266–7.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72.
Price MN, Dehal PS, Arkin AP. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490.
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10:57–59.
Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Xu ZZ, et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems. 2017;2:e00191–00116.
Mirarab S, Nguyen N, Warnow T. SEPP: SAT-enabled phylogenetic placement. Pac Symp Biocomput. 2012;247–58.
Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. Emperor: a tool for visualizing high-throughput microbial community data. Gigascience. 2013;2:16.
Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–35.
Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61:1–10.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Morton JT, Sanders J, Quinn RA, McDonald D, Gonzalez A, Vazquez-Baeza Y, et al. Balance trees reveal microbial niche differentiation. mSystems. 2017;2:e00162–00116.
Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8:e1002687.
Egozcue JJ, Pawlowsky-Glahn V. Groups of parts and their balances in compositional data analysis. Math Geol. 2005;37:795.
Amato KR, Martinez-Mota R, Righini N, Raguet-Schofield ML, Corcione FP, Marini E, et al. Phylogenetic and ecological factors impact the gut microbiota of Neotropical primate species. Oecologia. 2016a;180:717–33.
Amato KR, Metcalf JL, Song SJ, Hale VL, Clayton JB, Ackermann G, et al. Using the gut microbiota as a novel tool for examining colobine primate GI health. Glob Ecol Conserv. 2016b;7:225–37.
Sanders JG, Powell S, Kronauer DJC, Vasconcelos HL, Fredrickson ME, Pierce NE. Stability and phylgenetic correlation in gut microbiota: lessons from ants and apes. Mol Ecol. 2014;23:1268–83.
Hommola K, Smith JE, Qiu Y, Gilks WR. A permutation test of host-parasite co-speciation. Mol Biol Evol. 2009;26:1457–68.
Koster J, Rahmann S. Snakemake- a scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Yin Y, Mao X, Yang JC, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–451.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495.
Cantarel BL, Lombard V, Henrissat B. Complex carbohydrate utilization by the healthy human microbiome. PLoS ONE. 2012;7:e28742.
Davenport ER, Sanders JG, Song SJ, Amato KR, Clark AG, Knight R. The human microbiome in evolution. BMC Biol. 2017;15:127.
Groussin M, Mazel F, Sanders JG, Smillie CS, Lavergne S, Thuiller W, et al. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat Commun. 2017;8:14319.
Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MAM, et al. A molecular phylogeny of living primates. PLoS Genet. 2011;7:e1001342.
National Research Council. Nutrient Requirements of Nonhuman Primates: Second Revised Edition, Second edn. Washington, D.C: The National Academies Press; 2003.
Dill-McFarland KA, Weimer PJ, Pauli JN, Peery MZ, Suen G. Diet specialization selects for an unusual and simplified gut microbiota in two- and three-toed sloths. Environ Microbiol. 2016;18:1391–402.
Nelson TM, Rogers TL, Carlini AR, Brown MV. Diet and phylogeny shape the gut microbiota of Antarctic seals: a comparison of wild and captive animals. Environ Microbiol. 2012;15:1132–45.
Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science. 2017;357:802–6.
Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529:212–5.
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon HA. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14.
Williams CL, Willard S, Kouba A, Sparks D, Holmes W, Falcone J, et al. Dietary shifts affect the gastrointestinal microflora of the giant panda (Ailuropoda melanoleuca). J Anim Physiol Anim Nutr. 2012;97:577–85.
Zhang J, Guo Z, Lim AAQ, Zheng Y, Koh EY, Ho D, et al. Mongolians core gut microbiota and its correlation with seasonal dietary changes. Sci Rep. 2014;4:5001.
Acknowledgements
This project was funded by the NSF (HOMINID, grant #0935347), the Earth Microbiome Project (W.M. Keck Foundation DT061413), and the John Templeton Foundation (Grant ID 44000, Convergent Evolution of the Vertebrate Microbiome). Field collection of samples was funded through a variety of individual grants awarded to the co-authors and collaborators listed in Table S1. The authors are grateful for the support with logistics and permits provided by the US government, as well as the governments and local agencies associated with each nation listed in Table S1, as well as countless field assistants at these locations. Raw sequence data generated by 16S rRNA amplicon sequencing and shotgun metagenomics are located in EBI under accession ERP104379.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Amato, K.R., G. Sanders, J., Song, S.J. et al. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J 13, 576–587 (2019). https://doi.org/10.1038/s41396-018-0175-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0175-0
This article is cited by
-
Comparing the gut microbiota of Sichuan golden monkeys across multiple captive and wild settings: roles of anthropogenic activities and host factors
BMC Genomics (2024)
-
Eco-evolutionary dynamics of gut phageome in wild gibbons (Hoolock tianxing) with seasonal diet variations
Nature Communications (2024)
-
Effects of Antimicrobial Peptides on Antioxidant Properties, Non-specific Immune Response and Gut Microbes of Tsinling Lenok Trout (Brachymystax lenok tsinlingensis)
Biochemical Genetics (2024)
-
Wildlife gut microbiomes of sympatric generalist species respond differently to anthropogenic landscape disturbances
Animal Microbiome (2023)
-
Gut microbiome variations in Rhinopithecus roxellanae caused by changes in the environment
BMC Genomics (2023)
