
Abstract
Disease suppressive soils typically develop after a disease outbreak due to the subsequent assembly of protective microbiota in the rhizosphere. The role of the plant immune system in the assemblage of a protective rhizosphere microbiome is largely unknown. In this study, we demonstrate that Arabidopsis thaliana specifically promotes three bacterial species in the rhizosphere upon foliar defense activation by the downy mildew pathogen Hyaloperonospora arabidopsidis. The promoted bacteria were isolated and found to interact synergistically in biofilm formation in vitro. Although separately these bacteria did not affect the plant significantly, together they induced systemic resistance against downy mildew and promoted growth of the plant. Moreover, we show that the soil-mediated legacy of a primary population of downy mildew infected plants confers enhanced protection against this pathogen in a second population of plants growing in the same soil. Together our results indicate that plants can adjust their root microbiome upon pathogen infection and specifically recruit a group of disease resistance-inducing and growth-promoting beneficial microbes, therewith potentially maximizing the chance of survival of their offspring that will grow in the same soil.
Similar content being viewed by others


Introduction
In nature, plants accumulate pathogens in their surrounding soil, which ultimately negatively influences their performance, a phenomenon called negative soil-feedback [1, 2]. Negative soil-feedback can promote plant biodiversity by tempering the success of dominant plant species [3]. Such buildup of soil-borne pathogens can be devastating in agricultural monocultures, but is alleviated through crop rotation. Interestingly, continuous cultivation of important crops like wheat and sugar beet can induce soil suppressiveness to disease [4,5,6]. In such disease-suppressive soils, plants remain healthy despite the presence of a virulent pathogen. Disease suppressiveness is caused by specific microbes or microbial consortia that inhibit growth and activity of soil-borne pathogens [4, 6]. This phenomenon is often attributed to the production of antimicrobial compounds that selectively inhibit pathogen growth [4, 7]. However, stimulation of the host’s immune system by protective rhizosphere microbes, a phenomenon called induced systemic resistance (ISR), may also contribute to disease suppressiveness [8].
Buildup of disease suppressiveness in soils typically follows after a disease outbreak [6, 9], suggesting that, upon pathogen attack, plants recruit a community of protective microbiota. The capacity of plants to exploit protective benefits from their root microbiome is plant genotype dependent [10,11,12,13], implying that plants can manipulate protective rhizosphere processes to their advantage. The defense-related phytohormones salicylic acid (SA) and jasmonic acid (JA) are important modulators of the rhizosphere microbiome assembly of Arabidopsis thaliana (hereafter called Arabidopsis) [14, 15]. SA and JA are major hormonal regulators of the plant immune signaling network in which SA is typically effective against infection by biotrophic pathogens, whereas JA is typically effective against attack by necrotrophic pathogens [16]. Because SA or JA systemically accumulates in response to infection by biotrophs or necrotrophs, respectively, and can affect rhizosphere microbiome assembly, we hypothesized that foliar infection by pathogens with these contrasting lifestyles would result in differential stimulation of specific microbiota in the rhizosphere.
To test this hypothesis, Arabidopsis accession Col-0 plants were grown in soil collected from a natural field site at Reijerscamp (the Netherlands) that supports an abundant endemic Arabidopsis population. Five-week-old Arabidopsis plants were leaf inoculated with the biotrophic pathogen Hyaloperonospora arabidopsidis [17], or the necrotrophic pathogen Botrytis cinerea [18], or treated repeatedly with 1 mM SA (mimicking biotroph-triggered immunity), or 100 µM methyl JA (MeJA, mimicking necrotroph-triggered immunity), or not treated (control). To identify key bacterial and archaeal rhizosphere community members upon activation of the different foliar defense responses, DNA was isolated from rhizosphere and bulk soil at one and 2 weeks after the start of the treatments, and analyzed using a high-density 16S ribosomal DNA (rDNA) oligonucleotide microarray, referred to as the PhyloChip [4, 19, 20]. We subsequently characterized the bacteria that were promoted in the rhizosphere upon foliar defense activation and investigated the biological relevance of their promotion.
Materials and methods
Soil and soil preparation
The soil used in this study was taken from a field in the Reijerscamp nature reserve, the Netherlands (52°01′02.55″, 5°77′99.83″) in April 2012. An abundant endemic Arabidopsis population was found at the site. The field had been used for crop production for more than six decades, before it was given back to nature in 2000. Since then, the site was grazed by free-living cattle and deer and tree seedlings were periodically removed. The soil, a gleyic placic podzol, consisted of coarse sand and gravel covered by a 30–50 cm top layer. The top layer consisted of 88% sand, 8% silt, 2% clay and 2.3% organic matter and had a C/N ratio of 24 at pH 5.4 (soil chemical analysis performed by Eurofins Agro, Wageningen, the Netherlands). The top 20 cm of soil was collected, air dried and sieved (3-mm sieve) to remove plant debris and rocks and subsequently stored at room temperature. To revive the microbial community prior to the experiment, a 5-cm thick layer of soil was placed on a bench in a greenhouse, watered to saturation and sown with Arabidopsis accession Col-0. Seedlings were allowed to grow for 3 weeks on the soil after which the soil was sieved again to remove plants. The soil was stored at 4 °C and was then used within 2 weeks for the experiment.
Plant growth conditions
Arabidopsis thaliana accession Col-0 was sown on sterilized river sand (Hubun Inc., the Netherlands) saturated with modified ½ strength Hoagland solution [21]. After 2 days of stratification in the dark at 4 °C, the seeds were allowed to germinate in a growth chamber (21 °C, 70% relative humidity, 10 h light/14 h dark, light intensity 100 μmol m−2 s−1). After 2 weeks, single seedlings were transferred to 60-mL pots with approximately 110 g of Reijerscamp soil (described above). Bulk soil pots were left unplanted. Spontaneously developing seedlings from plant seeds that came with the soil were removed with tweezers upon detection. Pots were watered with ½ strength Hoagland every two weeks and watered when required.
Experimental treatments
Five-week-old plants were inoculated with Hyaloperonospora arabidopsidis (Hpa) strain Noco2 by spraying a spore suspension (104 sporangia/mL water) onto all the leaves [22] or with Botrytis cinerea (Bc) strain B0510 by applying a 5-μL drop of half-strength potato dextrose broth containing 5 × 105 spores mL−1 to a true leaf of the rosette [18]. For hormonal treatments, plants were dipped in a 0.015% (v/v) Silwet L-77 solution with either 1 mM SA or 100 µM MeJA. These concentrations are typically used to mimic activation of the respective hormonal signaling pathways [23]. The hormone treatments were repeated every 4th day until the end of the experiment. Pathogens were inoculated twice with an 8-day interval to ensure continued pathogen pressure for the duration of the experiment. Pots were randomly placed in a climate chamber (21 °C, 70% relative humidity, 10 h light/14 h dark, light intensity 100 μmol m−2 s−1), but were covered with transparent lids to increase humidity for a 72-h period following inoculation of the pathogens. Hpa started to sporulate on Hpa-inoculated plants 8 days after inoculation. Downy mildew disease symptoms progressed to heavy yellowing of the leaves at day 15. Bc-inoculated plants developed lesions on the inoculated leaves, which on primary-inoculated leaves had spread to the leaf edges by day 15.
Plant RNA extraction and qRT-PCR analysis
For RNA extraction, 2 leaves were harvested from each of 4 plants of every treatment at 1, 3, 5, 7, 9, 11, 13, and 15 days after the start of the treatment and snap frozen in liquid nitrogen. Total RNA was extracted from plant leaves and treated with Ambion DNase I (ThermoFischer Scientific). RevertAid H Minus Reverse Transcriptase (Thermo Scientific) was used to convert DNA-free total RNA into cDNA using oligo (dT) primers. Two-step qRT–PCR reactions were performed in optical 96-well plates with a ViiA 7 real time PCR system (Applied Biosystems), using Power SYBR® Green PCR Master Mix (Applied Biosystems) with 10 pmol µL−1 primers (Table S1). A standard thermal profile was used: 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Amplicon dissociation curves were recorded after cycle 40 by heating from 60 to 95 °C with a ramp speed of 1.0 °C min−1. Expression levels were normalized to the reference gene At1g13320, which encodes PROTEIN PHOSPHATASE 2A SUBUNIT A3 (PP2A-A3) [24] using the 2−ΔΔCT method described previously [25, 26].
Microbiome analysis
To analyze the microbiome composition, samples from unplanted soil and plant root systems with adhering soil (50–250 mg) from 16 pots per treatment were harvested at day 8 and at day 15 after the start of the treatment. Pots containing soil and plants were carefully turned over and most soil was gently removed from the roots, keeping roots intact as much as possible. Root systems were then lifted by the shoot and slightly shaken to remove loosely adhering soil. Shoots were subsequently removed with a razor blade and total microbial genomic DNA was isolated from 160 root systems with adhering soil and 32 bulk soil samples using the PowerSoil® DNA Isolation Kit (Mobio). For each microbiome determination, DNA of four samples was pooled to form one replicate. For each treatment and time point, four replicates (with DNA from 4 soil/rhizosphere samples each) were subjected to PhyloChip analysis by Second Genome Inc. (San Francisco, USA) as described previously [19, 20, 27, 28]. Briefly, PhyloChip analysis is a microarray-based approach that can quantify the relative abundance of over 60,000 microbial taxa. Bacterial and archaeal 16S rRNA gene amplicons were hybridized to the 1,016,064 probes of the PhyloChip G3 spiked with a determined amount of non-16S rRNA genes. Fluorescence intensity (FI) was measured to quantify the hybridization of the amplicons to the probes and scaled to the spiked-in quantitative standards. Fluorescence intensity observed from perfectly matching probes (PM) were compared to mismatching probes (MM) and were considered positive if PM/MM > 1.5 and PM-MM > 50*N and r > 0.95 where N indicates the array specific noise [27] and r represents the response score [19]. Probes were clustered into probe-sets based on both correlations in FI across all biological samples and taxonomic relatedness [20]. The empirical OTU (eOTU) tracked by a probe set was taxonomically annotated from the combination of the 9-mers contained in all probes of the set. The mean log2 FI for each eOTU and each sample was calculated and then are referred to as the hybridization score (HybScore) used in abundance-based analysis. eOTUs were considered present if >80% of their probes were positive. Of 379 eOTUs, 341 achieved this threshold in at least one sample. Principal component analyses were applied to the HybScores of the 75% most abundant eOTUs based on highest HybScore of all biological samples, using XLSTAT Version 2015.6.01.25740 (Addinsoft) add-in for Excel (Microsoft). False discovery rates were determined using the Phyloprofiler tool [28].
Selection of bacterial isolates
Rhizospheres of Hpa-infected plants were stored in 1 mL of 5 mM MgSO4 with 25% glycerol (v/v) at −80 °C at the end of the experiment described above. Rhizospheres were thawed at room temperature and suspended by vortexing for 120 s. In order to maximize the isolation of culturable bacterial species, dilution series were plated on the following media: (1) 1/10 strength tryptic soy agar (TSA; Difco); (2) TSA amended with 10 mg L−1 colistin; (3) TSA amended with 5 mg L−1 colistin and 10 mg L−1 naladixic acid; (4) TSA amended with 5 g L−1 NaCl and 64 mg L−1 polymyxin B; (5) TSA amended with 5 g L−1 NaCl, 64 mg L−1 polymyxin B and 10 mg L−1 gentamycin; (6) Nutrient Agar (Merck) amended with water yeast agar (per L: 5 g NaCl, 1 g KH2PO4, 0.1 g yeast extract (Difco), 15 g agar (Difco)); (7) peptone yeast agar (per L: 10 g peptone (Difco), 2 g yeast extract (Difco), 2 g NaCl, 2 g MgSO4·7H2O and 15 g agar (Difco)); (8) R2A agar (Difco); (9) Xanthomonas selective medium [29]; (10) Stenotrophomonas selective medium [30]; and (11) King’s medium B agar [31] amended with 13 mg L−1 chloramphenicol and 40 mg L−1 ampicillin. All media were amended with 200 mg L−1 Delvocid (DSM; active compound: natamycin) to prevent fungal growth. The plates were incubated for 3–5 days at 20 °C. A total of 288 bacterial colonies with unique morphologies were selected and streaked on TSA. Single colonies from pure cultures were inoculated in 1/10 strength tryptic soy broth (TSB; Difco), incubated overnight at 20 °C at 180 rpm, and stored at −80 °C in 25% (v/v) glycerol. Pure cultures were labeled WCS2014 for “Willie Commelin Scholten” and the year of isolation and numbered.
Identification and characterization of bacterial isolates
A loop of bacterial cells was added to 20 µL of water, incubated for 15 min at 95 °C and immediately cooled on ice. This bacterial lysate was diluted ten times with water and cell debris was removed by centrifugation (1 min, 10,000×g). The 16S rRNA gene was amplified with primers F27 and R1492 [32] (Table S1). Two microliter of the colony lysate was added to a total volume of 50 µL PCR reaction mixture (5 µL 10× Dreamtaq buffer (Thermo Scientific), 1 µL 10 mM dNTP’s, 2.5 µL 10 µM forward primer F27, 2.5 µL 10 µM reverse primer R1492, 1 µL Dreamtaq polymerase (Thermo Scientific), 36 µL H2O). PCR conditions used in the thermocycler (Hybaid, Ashford, UK) were 5 min at 94 °C, followed by 30 cycles of 1 min at 94 °C, 1 min at 55 °C and 1 min at 72 °C, and a final extension of 10 min at 72 °C. The PCR products were checked by electrophoresis on 1.5% agarose gels in 1x TAE buffer (40 mM Tris–acetate/1 mM EDTA, pH 8). PCR products were sequenced by Macrogen (Seoul, Korea). Taxonomy of isolates was determined through the Sequence Match function of the ribosomal database project [33].
BOX-PCR [34] was performed with primer BOXA1R to determine if the isolated strains were identical on strain level and most likely isogenic (Table S1). For BOX-PCR, bacterial genomic DNA was isolated with the GenElute™ Bacterial Genomic DNA Kit (Sigma) according to the manufacturer’s instructions. A volume of 1 µL of the genomic DNA was added to a BOX-PCR-reaction mix (2.5 µL 10× Dreamtaq buffer (Thermo Scientific), 0.5 µL 10 mM dNTP’s, 0.25 µL 10 µM primer BOXA1R, 0.125 µL Dreamtaq polymerase (Thermo Scientific), 20.625 µL H2O) to a total volume of 25 µL. PCR conditions used in the thermocycler (Hybaid, Ashford, UK) were 7 min at 95 °C, followed by 30 cycles of 30 s at 90 °C, 1 min at 95 °C, 1 min at 52 °C and 8 min at 65 °C, and a final extension of 16 min at 65 °C. Amplified PCR fragments were analyzed by electrophoresis on 1.5% agarose gels in 1x TAE buffer.
Genome sequencing and genetic comparison
Whole-genome sequencing libraries for Xanthomonas sp. WCS2014-23, Stenotrophomonas sp. WCS2014-113 and Microbacterium sp. WCS2014-259 were prepared using the Nextera XT kit (Illumina, USA) according to the manufacturer’s recommendations and then sequenced using the Illumina MiSeq (Illumina, USA) technology. The 2250 bp paired-end reads were cleaned, trimmed and assembled using the A5-MiSeq pipeline [35] and the assembled draft genomes were uploaded to Integrated Microbial Genomes Expert Review system for gene calling and annotation [36]. The genomes are available through the Joint Genome Institute genome portal (https://genome.jgi.doe.gov/; IMG Genome IDs 2747842429, 2747842501 and 2747842428). The genomic distance of the sequenced isolates to sequences of the Refseq database (https://www.ncbi.nlm.nih.gov/refseq/) were estimated using MASH [37].
Quantification of biofilm formation
Biofilm formation by the selected bacterial isolates was quantified as described by Ren and coworkers [48]. Briefly, Xanthomonas sp. WCS2014-23, Stenotrophomonas sp. WCS2014-113 and Microbacterium sp. WCS2014-259 were inoculated in 5 mL TSB and incubated for 24 h at 20°C at 180 rpm. Dilution series of the stationary phase bacterial cultures were subsequently transferred to 5 mL TSB and again incubated overnight at 20 °C at 180 rpm. The exponentially growing dilution with an optical density at 590 nm (OD590) of ca. 0.5 was then selected and diluted to 0.15 in TSB. The bacterial cultures were then added separately or mixed with the other cultures in equal quantities to the wells of Nunc-TSP 96-wells plates (ThermoFisher Scientific, Waltham, USA) and to a total volume of 160 µL. Final bacterial densities in the separate and mixed suspensions were equal. The plates were sealed with Parafilm and incubated for 48 h at 20 °C without shaking. Planktonic cells were then washed off the peg lids of the Nunc-TSP plates by transferring the lids successively to microtiter plates containing 200 µL phosphate-buffered saline (PBS). Biofilms formed on the peg lids were then stained for 20 min in 180 µL of aqueous 1% (W/V) crystal violet. The pegs were again rinsed three times in PBS and then incubated in 200 µL 96% ethanol to release the crystal violet absorbed by the biofilm. Biofilm thickness was then quantified by measuring the OD590 of the crystal violet-ethanol solution with a plate spectrophotometer (BioTek Synergy HT).
Attraction between colonies
Xanthomonas sp. WCS2014-23, Stenotrophomonas sp. WCS2014-113, and Microbacterium sp. WCS2014-259 were inoculated in 5 mL King’s medium B and incubated overnight at 20 ℃ at 180 rpm. The optical density of the bacterial cultures was adjusted to 0.1 at 600 nm. Seven times 1 µL of these dilutions were inoculated in a diagonal row on both sides of a square petri-dish with King’s medium B agar with a multichannel pipet, creating a V-shape of increasingly closer inoculation sites. The plates were sealed with Parafilm and incubated for 15 days at 20 ℃. Colony diameters were measured on a line orthogonal to the line dividing the V-shape.

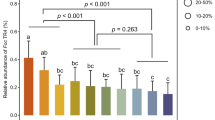
Downy mildew infection promotes growth of specific microbiota in the rhizosphere of Arabidopsis. a Principal component (PC) analysis of microbial communities in unplanted soil and the rhizospheres of pathogen-infected or defense hormone-treated Arabidopsis plants. Microbial communities were isolated from unplanted soil (brown symbols), rhizospheres of untreated control plants (green), rhizospheres of plants of which the leaves were inoculated with the biotroph Hpa (red), or the necrotroph Bc (blue), or rhizospheres of plants of which the leaves were repeatedly treated with SA (yellow), or MeJA (purple). Microbial communities were analyzed 1 week (squares) and 2 weeks (triangles) after the start of the foliar treatments. Eigenvalues of PC1 and PC2 are expressed on the X- and Y-axis, respectively. b Biplot of eOTU correlations (PC scores) to the same PC1 and PC2. Each red dot represents an eOTU. PC1 separates bulk soil eOTUs (left) from rhizosphere eOTUs (right). Three eOTUs that strongly correspond to PC2 are designated with a number. c–e Quantification of eOTU abundance in the different treatments. Boxplots of PhyloChip HybScore per treatment and time point are shown for c Stenotrophomonas sp. eOTU 97, d Xanthomonas sp. eOTU 106 and e Microbacterium sp. eOTU 107. Black dots represent the values of the 4 replicates per treatment. Red plus signs signify the averages. Red asterisks denotes significant differences from control rhizospheres in the same time point (false discovery rate <0.05). U, unplanted soil; C, rhizosphere of control-treated plants; B, rhizosphere of Bc-inoculated plants; H, rhizosphere of Hpa-inoculated plants; S, rhizosphere of SA-treated plants; J, rhizosphere of MeJA-treated plants
Selection of rifampicin-resistant mutants
In order to quantify bacterial numbers in soil during plant growth promotion and ISR assays, spontaneous rifampicin-resistant mutants of Xanthomonas sp. WCS2014-23, Stenotrophomonas sp. WCS2014-113 and Microbacterium sp. WCS2014-259 were obtained as described by Glandorf et al. [38]. Briefly, colonies of these strains were transferred to TSA agar plates containing increasing concentrations (50, 100, 150, 200, and 250 µg mL−1) of rifampicin. The stability of the rifampicin resistance in the mutants was confirmed and growth rate of the rifampicin-resistant mutants was found to be similar as their respective wild types.
Growth promotion assay
Arabidopsis accession Col-0 was sown on sand and cultivated as describe above. After two weeks, seedlings were transferred to 60-mL pots filled with a potting soil-sand mixture pre-inoculated with Pseudomonas simiae WCS417r ([39]; formerly known as Pseudomonas fluorescens WCS417r [40], rifampicin-resistant mutants of Xanthomonas sp. WCS2014-23, Stenotrophomonas sp. WCS2014-113 or Microbacterium sp. WCS2014-259, or a combination of the three strains, in all cases to a final density of 1 × 108 cfu/g of soil. Soils were prepared as described by [21]. The strains were thoroughly washed before being suspended and added to the soil. Initial microbial densities in the soil were determined by suspending approximately 0.1 g of soil of five replicate pots per treatment and plating a dilution series on King’s medium B amended with 150 µg mL−1 rifampicin. After plants had grown for 4 weeks in microbe-amended soil, the then 6-week-old plants were harvested and shoot fresh weight was determined.
ISR assay
In the ISR assay, plants were grown similarly and on similarly prepared soil as for the growth promotion assay. Assessment of downy mildew disease resistance was performed essentially as described [22]. In brief, 5-week-old plants were inoculated with Hpa Noco2 as described above. Plants were grown in pots placed in trays and covered with transparent lids to increase relative humidity. Six days after Hpa inoculation, the number of Hpa spores were determined. To this end, shoots of infected plants were harvested, collected in 15-mL tubes with 3 mL H2O and vortexed for 30 s. Subsequently, spores were counted using a phase-contrast microscope (Axioskope, Zeiss, Jena, Germany) after which the number of Hpa spores produced per plant was calculated. These experiments were repeated with similar results. Analysis of variance was performed with XLSTAT Version 2015.6.01.25740 (Addinsoft) add-in for Excel (Microsoft).
Soil-mediated legacy of downy mildew-infected plants
In June 2016, soil was collected from the same site in the Reijerscamp nature reserve after which it was sieved, dried, and stored as described above. Eighty 60-mL pots were filled with approximately 110 g of soil watered to field capacity. To prevent growth of moss and algae, the soil surface was covered by a circular plastic cut-out of micro pipette tip holder (Greiner Bio-One, 0.5–10 µL, Item No.: 771280). Arabidopsis Col-0 seeds were suspended in a 0.2% (w/v) water agar solution and imbibed in the dark at 4 ˚C for 2 days. Approximately 40 seeds were sown on each of 40 pots by pipetting 1-2 seeds in each hole of the cover, the other 40 pots were left unplanted. The pots were placed on small saucers, watered with modified ½ strength Hoagland solution, randomly placed in trays, covered by transparent lids and transferred to a growth chamber (21 °C, 70% relative humidity, 10 h light/14 h dark, light intensity 100 μmol m−2 s−1). After a week, the lids were replaced by lids with a mesh to reduce the humidity in the trays. After two weeks, the pots were sprayed with either a Hpa spore suspension (50 spores µL−1 as described above) or mock-treated by spraying with water. Trays were again covered with transparent lids to increase humidity after which downy mildew infections were allowed to develop for a week. Subsequently, all above-ground parts of the first population of plants were cut off and removed and new seeds were sown on all pots as described above. After 2 weeks of growth, this second population of plants was either inoculated with Hpa or mock treated with water after which downy mildew infections were allowed to develop for 1 week. Downy mildew disease severity was then quantified by determining the number of Hpa spores produced per pot on above ground plant tissue as described above.
Results
Root microbiome changes on immune-stimulated plants
In order to investigate the effect of foliar pathogen infection or treatment of the leaves with SA or MeJA on root microbiome assemblage, we monitored the changes in the microbial communities over a period of 2 weeks after defense activation. PhyloChip analysis of the microbial communities revealed a total of 341 eOTUs, the majority belonging to the phyla Proteobacteria (43%), Firmicutes (20%), and Bacteroidetes (20%) (Fig. S1). To identify key drivers of variability in abundance of the eOTUs among samples, we used principal component analyses (Fig. 1a). The first principal component (PC1) separates the unplanted soil samples from the rhizosphere samples, confirming the rhizosphere effect for Arabidopsis [41, 42] in which specific microbiota are promoted in the rhizosphere compared to bulk soil. The second principal component (PC2) visually distinguishes the rhizospheres of plants infected by Hpa from the other rhizospheres. Most eOTUs correlate with PC1 and are either promoted or suppressed by the plant rhizosphere (Fig. 1b; Fig. S2). Only three eOTUs strongly correlate with PC2 (Fig. 1b). These eOTUs, designated eOTU 97, 106 and 107, were significantly more abundant in rhizospheres of Hpa-infected plants compared to that of untreated plants at both one and 2 weeks after inoculation (Fig. 1c–e; Fig. S3). These eOTUs were recognized as Xanthomonas, Microbacterium, and Stenotrophomonas sp., respectively. Moreover, at one of the two timepoints eOTU97, 106 and 107 were also significantly more abundant in the rhizosphere of SA-treated plants, albeit at a lower abundance than in Hpa-inoculated plants (Fig. 1c–e; Fig. S3). In the rhizospheres of Bc-inoculated and MeJA-treated plants, eOTU107 and/or eOTU97 also showed a small but significant increase in abundance at one of the timepoints tested, suggesting that the enhanced abundance of these eOTUs in the rhizosphere is related to the activation of plant defense responses, in particular those induced by Hpa infection.

Synergistic interactions between recruited Xanthomonas, Stenotrophomonas, and Microbacterium spp. strains. a Boxplot of biofilm formation by single Xanthomonas sp. WCS2014-23 (X; eOTU 106), Stenotrophomonas sp. WCS2014-113 (S; eOTU 97) and Microbacterium sp. WCS2014-259 (M; eOTU 107) or the double (XS, XM, and SM) and triple combinations (XSM) thereof in Nunc-TSP lid plates. After 24 h of incubation, the biofilm formation was quantified by staining with crystal violet. b Attraction between colonies of X, S, and M grown at increasing proximity on King’s medium B agar
Hpa infection progressed to heavy sporulation in 7 days after inoculation, and triggered SA-dependent defense responses in the leaves, as exemplified by the strongly induced expression of the SA-responsive marker gene PATHOGENESIS RELATED-1 (PR-1) [16] (Fig. S4A). The SA treatment also induced PR-1 (831 fold-induction cumulative over time), but to a lesser extent than Hpa (4369 fold-induction cumulative over time). Bc infection and MeJA treatment lead to a relatively low, but detectable induction of PR-1. These results show that the observed increased abundance of eOTUs 97, 106, and 107 in the root microbiome correlated with the activation of the SA response. Although Bc infections and repeated MeJA treatment of Arabidopsis leaves resulted in strong upregulation of the JA-responsive marker genes PLANT DEFENSIN1.2 (PDF1.2) and VEGETATIVE STORAGE PROTEIN2 (VSP2), respectively ([16]; Fig. S4B-C), significant microbiome changes mediated by these JA-related treatments were not observed.
Characterization of recruited rhizosphere microbes of Hpa-infected plants
To further characterize eOTUs 97, 106, and 107, we isolated candidate microbes from the rhizosphere of the Hpa-infected plants. Using both broad-spectrum media and media selective for Xanthomonas, Stenotrophomonas, or Microbacterium spp., 279 rhizobacterial strains were isolated and identified based on 16S rRNA gene sequences. The 279 isolates represented 35 genera among which were three isolates belonging to the genus Xanthomonas, three to the genus Microbacterium and one to the genus Stenotrophomonas (Table S2). BOX PCR-generated DNA fingerprints indicated that each of the three representatives of Xanthomonas and of Microbacterium were identical (Fig. S5). The probe sets that defined eOTUs 97, 106, and 107 perfectly matched the 16S rRNA gene sequence of the respective Xanthomonas, Stenotrophomonas and Microbacterium spp. isolates (Fig. S6), indicating that we likely isolated the actual eOTUs that were promoted in the rhizosphere by Hpa infection.
The 16S rRNA gene sequences of the isolated Xanthomonas, Stenotrophomonas, and Microbacterium spp. did not conclusively distinguish the species level taxonomy of these strains. To resolve species level identification we proceeded to sequence the genomes of each isolate by means of Illumina sequencing. The genomic distances of Xanthomonas sp. WCS2014-23, Stenotrophomonas sp. WCS2014-113 and Microbacterium sp. WCS2014-259 were estimated pairwise to all genomes in the Refseq database belonging to respectively the Xanthomonas, Stenotrophomonas or Microbacterium genus (Additional data S3). Average nucleotide identity (ANI) values between 94 and 96% have been proposed for defining the boundaries between prokaryotic species [43, 44]. Based on the ANI, the closest relative to Xanthomonas sp. WCS2014-23 in the Refseq database is Xanthomonas sp. Leaf148 (97.5% ANI), which has been isolated from A. thaliana leaves [45]. All of the genomes in the Refseq database belonging to Xanthomonas type strains share 91.5% or less of their ANI, indicating that WCS2014-23 belongs to an undescribed species or a species for which the type strain has not been completely sequenced. The closest relative to Stenotrophomonas sp. WCS2014-113 is Stenotrophomonas maltophilia strain AA1 (94.5% ANI), a strain isolated from maize roots [46]. However, the ANI shared with the Stenotrophomonas maltophilia type strain NBRC 14161 of 90.5% indicates that strain WCS2014-23 is not Stenotrophomonas maltophilia, but a new species or a species of which the type strain has not been completely sequenced. The closest neighbor to Microbacterium sp. WCS2014-259 was Microbacterium foliorum strain 122, an endophyte of Dactylis glomerata. The ANI of 88.8% shared with this strain indicates that none of the Microbacterium genomes in the Refseq database derive from the same Microbacterium species as WCS2014-259. In conclusion, Xanthomonas sp. WCS2014-23, Stenotrophomonas sp. WCS2014-113 and Microbacterium sp. WCS2014-259 cannot be taxonomically classified on the species level based on their full genome sequence, but are closely related to other plant-associated strains within their respective genera.
Plant-beneficial effects of recruited rhizosphere microbes
The recruitment of the three eOTUs as a consortium suggests that they exist in close association in the rhizosphere. Rhizosphere-colonizing bacteria form biofilms on the root surface in which diverse microbial processes, such as the production of plant growth regulators, defense elicitors, and antibiotics are triggered and may affect both the host and microbes in their surrounding [47]. Bacterial interspecific cooperation has been suggested in the synergistic biofilm formation by different bacterial strains isolated from agricultural soil [48]. Whether the three identified eOTUs act synergistically in biofilm formation was investigated in an in vitro biofilm formation assay. Indeed, the three bacterial rhizosphere isolates showed synergy in biofilm formation, as the combination of three consistently formed more biofilm than the separate strains (Fig. 2a). This suggests that the three rhizobacterial species are synergistically assembled as a consortium in the rhizosphere of Arabidopsis upon Hpa infection of the foliar tissue. In agreement with this, the three strains were found to attract each other on plate (Fig. 2b; Fig. S7). Colony growth of the Stenotrophomonas isolate was stimulated towards both the Xanthomonas and the Microbacterium isolate. Similarly, growth of the Microbacterium colonies were induced in the vicinity of both the Stenotrophomonas and the Xanthomonas isolate.
To investigate the biological relevance of the promotion of this bacterial consortium, we grew Arabidopsis plants in soils pre-inoculated with the single strains or mixture of the three to test for their ability to induce ISR against foliar infection with Hpa. Pseudomonas simiae WCS417r (hereafter called WCS417) is a well-studied plant-beneficial rhizobacterium that induces systemic resistance and promotes growth upon colonization of the roots [39, 49]. Colonization of the rhizosphere of Arabidopsis plants by WCS417 indeed induced ISR as reflected by the reduced production of Hpa spores on downy mildew infected leaves (Fig. 3a). None of the single strains reduced the number of Hpa spores produced on infected plants significantly. However, the consortium of three strains had a more robust effect than the single strains, as together they significantly reduced the number of Hpa spores on infected leaves in comparison to non-colonized control plants, while the single strains did not (Fig. 3a). Collectively, these results show that the Xanthomonas, Stenotrophomonas, and Microbacterium isolates that are promoted in the rhizosphere of Arabidopsis upon foliar infection with Hpa, can systemically enhance the level of protection against Hpa. These results support the notion that the rhizobacteria act as a consortium, resulting in the promotion of plant health.

Synergistic effects of recruited rhizobacteria on systemic immunity against Hpa and plant growth promotion. a Spore production by Hpa on Arabidopsis plants growing on soil pre-inoculated with Pseudomonas simiae WCS417, Xanthomonas sp. WCS2014-23 (X; eOTU 106), Stenotrophomonas sp. WCS2014-113 (S; eOTU 97) and Microbacterium sp. WCS2014-259 (M; eOTU 107or a mixture of X, S and M (XSM). b Boxplot showing shoot fresh weight of 6-week-old Arabidopsis plants grown in soil pre-inoculated with WCS417, X, S, M, or a mixture of X, S, and M. Italic letters depict statistically significant (P < 0.05) differences according to analysis of variance with Tukey’s posthoc test. Black dots represent the respective replicate values. All experiments were repeated at least three times with similar results. c Picture showing 6-week-old Arabidopsis plants grown in control soil or soil pre-inoculated with a mixture of X, S, and M
Direct activation of defenses by pathogens will generally result in an increased level of disease resistance, but comes at a fitness cost for the plant as evidenced by reduced plant growth and seed production [23, 50]. ISR, however, as induced by the model strain WCS417, has limited fitness cost that are mostly outweighed by the plant growth promoting effects of this bacterium [8, 49]. We therefore also inoculated soils with the three strains to test their effects on plant growth. None of the bacterial strains, including WCS417, negatively affected shoot fresh weight (Fig. 3b). Moreover, inoculation of the soil with the mixture of the three isolates even increased shoot fresh weight significantly (Fig. 3b, c).
Soil-mediated legacy of Hpa-infected plants protects a successive plant population
ISR benefits plants as it primes the plant for enhanced defense against future pathogen or insect attack [8]. Nonetheless, it is not likely to rescue an already infected plant. We therefore hypothesized that recruitment of beneficial microbes to the root upon pathogen attack benefits a following population of plants, such as the plant’s offspring, growing in the same soil. To test this hypothesis, we pre-conditioned the Reijerscamp soil by growing Hpa-infected or mock-treated Arabidopsis plants. After removal of this first population of plants, we sowed a second population of Arabidopsis plants on the differently pre-conditioned soils and challenged them with Hpa (Fig. 4). We included control soils that were left unplanted in the conditioning phase but were otherwise treated the same, including spray treatment with water or Hpa spores. In line with our hypothesis, the second population of Arabidopsis seedlings growing in soil pre-conditioned by downy mildew-infected plants were more resistant to foliar downy mildew infection than plants growing in soil pre-conditioned by control plants (Fig. 4b). Pre-conditioning of soil by control plants or spraying unplanted soil with Hpa spores did not affect resistance of a subsequent population of plants. Together these results suggest that foliar downy mildew infections lead to a soil-mediated legacy that renders a subsequent population of plants growing in the same soil more resistant against downy mildew infection.

Soil-mediated effects of Hpa-infected plants on disease resistance in a subsequent population of plants. a Schematic representation of the experiment. A first population (1°) of Arabidopsis Col-0 seedlings or unplanted soil (U) was inoculated with a Hpa (H) spore suspension or mock treated (M). Seven days after inoculation, aboveground plant parts were removed, after which a second population (2°) of Arabidopsis plants was sown and grown on the remaining soils. After 2 weeks of growth in the pre-conditioned or unconditioned soils, Arabidopsis plants were inoculated or not with Hpa. Disease severity was quantified by counting the number of Hpa spores that were produced at 7 days after inoculation. b Boxplot showing the number of Hpa spores produced on plants growing on the indicated pre-conditioned soils. Different letters indicate statistically significant (P < 0.05) differences according to analysis of variance with Tukey’s posthoc test. Red plus signs signify the averages of the 10 replicates per treatment. Black dots represent the respective replicate values. The experiment was repeated 4 times with similar results [57].
Discussion
This study shows that downy mildew infection in Arabidopsis leaves leads to promotion of a bacterial consortium consisting of a Microbacterium, a Stenotrophomonas, and a Xanthomonas sp. in the rhizosphere. Our results suggest that foliar infection with a biotrophic pathogen systemically signals to the roots to promote growth of specific microbial species in the rhizosphere. The promoted microbial species interact synergistically in biofilm formation, suggesting that they are also synergistically assembled at the root-microbiome interface. As a consortium, the increased microbes are beneficial to the plant as together they induce resistance against Hpa but also promote plant growth. Together these findings show that Arabidopsis plants infected with a biotrophic pathogen can promote specific members of their microbiome to aid in their defense. Recent studies with wheat and pepper plants support this finding as pathogen or insect attack resulted in plant-mediated changes in rhizosphere microbial communities [51, 52]. Interestingly Stenotrophomonas is also one of the few genera that increased in abundance on pepper plant roots upon infestation of foliar tissue by white fly [52]. This shows that this genus can be promoted upon defense activation in various plant species. In contrast to Hpa, infection by the necrotrophic fungus Botrytis cinerea strongly induced the JA-dependent marker gene PDF1.2 in above ground plant tissue, but this did not lead to clear promotion of microbes in the rhizosphere. This suggests that different foliar pathogens differently affect the rhizosphere microbiome. A better understanding of the plant genetic basis of disease-induced recruitment of beneficial root-associated microbes could unlock new possibilities for breeding of crop plants that are better able to employ their microbiomes in their defense and have enhanced capacities for controlling disease.
In our study, we used PhyloChip for the analysis of root microbial communities. PhyloChip can more sensitively detect differences in taxon abundance [53] and PhyloChip analyses have more reproducible output than sequencing-based methods ([19, 54, 55]). However, PhyloChip cannot provide accurate estimates of the relative amounts of a taxon as a proportion of the total community. It is therefore difficult to assess the actual populations of the recruited Microbacterium, Stenotrophomonas, and Xanthomonas spp. in our experiment. For Pseudomonas spp., a threshold population level of 105 CFU/g root is required for the onset of ISR [56]. Regardless, it is unlikely that recruitment of ISR-inducing bacteria can arrest or cure an already strongly developed infection. Thus, we tested the hypothesis that recruitment of root-associated protective microbes upon foliar infection by a pathogen benefits the plant’s offspring that subsequently germinates in the same soil. We demonstrated that downy mildew infections in a first population of plants indeed confer a soil-mediated legacy that provides increased resistance against this pathogen in a subsequent population of plants that grow in the same soil. This legacy effect is likely of microbial origin, although we cannot exclude a direct effect of plant-produced chemicals. Together, our results suggest that infection of aboveground plant tissue results in the recruitment of beneficial root-associated microbes that have the potential to protect a subsequent population of plants growing in the same soil against the pathogen that initiated the recruitment.
References
Van der Putten WH, Van Dijk C, Peters BAM. Plant-specific soil-borne diseases contribute to succession in foredune vegetation. Nature. 1993;362:53–56.
Bever JD, Platt TG, Morton ER. Microbial population and community dynamics on plant roots and their feedbacks on plant communities. Annu Rev Microbiol. 2012;66:265–83.
Klironomos JN. Feedback with soil biota contributes to plant rarity and invasiveness in communities. Nature. 2002;417:67–70.
Mendes R, Kruijt M, De Bruijn I, Dekkers E, Van der Voort M, Schneider JH, et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science. 2011;332:1097–1100.
Raaijmakers JM, Mazzola M. Soil immune responses. Science. 2016;352:1392–3.
Weller DM, Raaijmakers JM, McSpadden Gardener BB, Thomashow LS. Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu Rev Phytopathol. 2002;40:309–48.
Raaijmakers JM, Weller DM. Natural plant protection by 2,4-diacetylphloroglucinol-producing Pseudomonas spp. in take-all decline soils. Mol Plant Microbe Interact. 1998;11:144–52.
Pieterse CMJ, Zamioudis C, Berendsen RL, Weller DM, Van Wees SCM, Bakker PAHM. Induced systemic resistance by beneficial microbes. Annu Rev Phytopathol. 2014;52:347–75.
Berendsen RL, Pieterse CMJ, Bakker PAHM. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012;17:478–86.
Haney CH, Samuel BS, Bush J, Ausubel FM. Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nat Plants. 2015;1:1–9.
Perez-Jaramillo JE, Carrion VJ, Bosse M, Ferrao LFV, De Hollander M, Garcia AAF, et al. Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 2017;11:2244–57.
Pieterse CMJ, De Jonge R, Berendsen RL. The soil-borne supremacy. Trends Plant Sci. 2016;21:171–3.
Wintermans PCA, Bakker PAHM, Pieterse CMJ. Natural genetic variation in Arabidopsis for responsiveness to plant growth-promoting rhizobacteria. Plant Mol Biol. 2016;60:623–34.
Lebeis SL, Paredes SH, Lundberg DS, Breakfield N, Gehring J, McDonald M, et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science. 2015;349:860–4.
Carvalhais LC, Dennis PG, Badri DV, Kidd BN, Vivanco JM, Schenk PM. Linking jasmonic acid signaling, root exudates, and rhizosphere microbiomes. Mol Plant Microbe Interact. 2015;28:1049–58.
Pieterse CMJ, Van der Does D, Zamioudis C, Leon-Reyes A, Van Wees SCM. Hormonal modulation of plant immunity. Annu Rev Cell Dev Biol. 2012;28:489–521.
Lapin D, Meyer RC, Takahashi H, Bechtold U, Van den Ackerveken G. Broad-spectrum resistance of Arabidopsis C24 to downy mildew is mediated by different combinations of isolate-specific loci. New Phytol. 2012;196:1171–81.
Coolen S, Proietti S, Hickman R, Davila Olivas NH, Huang P-P, Van Verk MC, et al. Transcriptome dynamics of Arabidopsis during sequential biotic and abiotic stresses. Plant J. 2016;86:249–67.
Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, Singh N, et al. Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science. 2010;330:204–8.
Probst AJ, Birarda G, Holman H-YN, DeSantis TZ, Wanner G, Andersen GL, et al. Coupling genetic and chemical microbiome profiling reveals heterogeneity of archaeome and bacteriome in subsurface biofilms that are dominated by the same archaeal species. PLoS One. 2014;9:e99801.
Pieterse CMJ, Van Wees SCM, Hoffland E, Van Pelt JA, Van Loon LC. Systemic resistance in Arabidopsis induced by biocontrol bacteria is independent of salicylic acid accumulation and pathogenesis-related gene expression. Plant Cell. 1996;8:1225–37.
Van Damme M, Andel A, Huibers RP, Panstruga R, Weisbeek PJ, Van den Ackerveken G. Identification of Arabidopsis loci required for susceptibility to the downy mildew pathogen Hyaloperonospora parasitica. Mol Plant Microbe Interact. 2005;19:583–92.
Vos IA, Moritz L, Pieterse CMJ, Van Wees SCM. Impact of hormonal crosstalk on plant resistance and fitness under multi-attacker conditions. Front Plant Sci. 2015;6:639.
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible W-R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005;139:5–17.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–8.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C-T method. Nat Protoc. 2008;3:1101–8.
DeSantis TZ, Stone CE, Murray SR, Moberg JP, Andersen GL. Rapid quantification and taxonomic classification of environmental DNA from both prokaryotic and eukaryotic origins using a microarray. FEMS Microbiol Lett. 2005;245:271–8.
Van der Voort M, Kempenaar M, Van Driel M, Raaijmakers JM, Mendes R. Impact of soil heat on reassembly of bacterial communities in the rhizosphere microbiome and plant disease suppression. Ecol Lett. 2016;19:375–82.
Kado CI. Selective media for isolation of Agrobacterium, Corynebacterium, Erwinia, Pseudomonas, and Xanthomonas. Phytopathology. 1970;60:969.
Juhnke ME, Des Jardin E. Selective medium for isolation of Xanthomonas maltophilia from soil and rhizosphere environments. Appl Environ Microbiol. 1989;55:747–50.
King EO, Ward MK, Raney DE. Two simple media for the demonstration of pyocyanin and fluorescin. J Lab Clin Med. 1954;44:301–7.
Thomas MC, Thomas DK, Kalmokoff ML, Brooks SPJ, Selinger LB, Inglis, et al. Molecular methods to measure intestinal bacteria: a review. J AOAC Int. 2012;95:5–23.
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:633–42.
Versalovic J, Schneider M, De Bruijn FJ, Lupski JR. Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Method Mol Cell Biol. 1994;5:25–40.
Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinform Oxf Engl. 2015;31:587–9.
Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40:D115–D122.
Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17:132.
Glandorf DCM, Brand I, Bakker PAHM, Schippers B. Stability of rifampicin resistance as a marker for root colonization studies of Pseudomonas putida in the field. Plant Soil. 1992;147:135–42.
Berendsen RL, Van Verk MC, Stringlis IA, Zamioudis C, Tommassen J, Pieterse CMJ, et al. Unearthing the genomes of plant-beneficial Pseudomonas model strains WCS358, WCS374 and WCS417. BMC Genom. 2015;16:539.
Lamers JG, Schippers B, Geels FP. Soil-borne diseases of wheat in the Netherlands and results of seed bacterization with Pseudomonas against Gaeumannomyces graminis var. tritici. In: Jorna ML, Slootmaker LAJ, editors. Cereal breeding related to integrated cereal production. Wageningen: PUDOC; 1988. p. 134–139.
Bakker PAHM, Berendsen RL, Doornbos RF, Wintermans PCA, Pieterse CMJ. The rhizospere revisited: root microbiomics. Front Plant Sci. 2013;4:165.
Reinhold-Hurek B, Bünger W, Burbano CS, Sabale M, Hurek T. Roots shaping their microbiome: global hotspots for microbial activity. Annu Rev Phytopathol. 2015;53:403–24.
Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA. 2005;102:2567–72.
Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 2009;106:19126–31.
Bai Y, Müller DB, Srinivas G, Garrido-Oter R, Potthoff E, Rott M, et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature. 2015;528:364–9.
Niu B, Paulson JN, Zheng X, Kolter R. Simplified and representative bacterial community of maize roots. Proc Natl Acad Sci. 2017;114:E2450–9.
Beauregard PB, Chai YR, Vlamakis H, Losick R, Kolter R. Bacillus subtilis biofilm induction by plant polysaccharides. Proc Natl Acad Sci USA. 2013;110:E1621–30.
Ren D, Madsen JS, Sørensen SJ, Burmølle M. High prevalence of biofilm synergy among bacterial soil isolates in cocultures indicates bacterial interspecific cooperation. ISME J. 2015;9:81–89.
Zamioudis C, Mastranesti P, Dhonukshe P, Blilou I, Pieterse CMJ. Unraveling root developmental programs initiated by beneficial Pseudomonas spp. bacteria. Plant Physiol. 2013;162:304–18.
Van Hulten M, Pelser M, Van Loon LC, Pieterse CMJ, Ton J. Costs and benefits of priming for defense in Arabidopsis. Proc Natl Acad Sci USA. 2006;103:5602–7.
Dudenhöffer J-H, Scheu S, Jousset A. Systemic enrichment of antifungal traits in the rhizosphere microbiome after pathogen attack. J Ecol. 2016;104:1566–75.
Kong HG, Kim BK, Song GC, Lee S, Ryu C-M. Aboveground whitefly infestation-mediated reshaping of the root microbiota. Front Microbiol. 2016;7:1314.
Miezeiewski M, Schnaufer T, Muravsky M, Wang S, Caro-Aguilar I, Secore S, et al. An in vitro culture model to study the dynamics of colonic microbiota in Syrian golden hamsters and their susceptibility to infection with Clostridium difficile. ISME J. 2015;9:321–32.
Wen C, Wu L, Qin Y, Nostrand JDV, Ning D, Sun B, et al. Evaluation of the reproducibility of amplicon sequencing with Illumina MiSeq platform. PLoS One. 2017;12:e0176716.
Zhou J, Wu L, Deng Y, Zhi X, Jiang Y-H, Tu Q, et al. Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 2011;5:1303–13.
Raaijmakers JM, Leeman M, Van Oorschot MMP, Van der Sluis I, Schippers B, Bakker PAHM. Dose-response relationships in biological control of fusarium wilt of radish by Pseudomonas spp. Phytopathology. 1995;85:1075–81.
Zamioudis C, Korteland J, Van Pelt JA, Van Hamersveld M, Dombrowski N, Bai Y, et al. Rhizobacterial volatiles and photosynthesis-related signals coordinate MYB72 in Arabidopsis roots during onset of induced systemic resistance and iron deficiency responses. Plant J. 2015;84:308–22.
Acknowledgements
This work was financially supported by ERC Advanced Grant no 269072 of the European Research Council and by the Dutch Technology Foundation STW, which is part of the Netherlands Organization of Scientific Research (NWO) and partly funded by the Ministry of Economic Affairs (Back2Roots Grant 14219) and by a postdoctoral fellowship of the Research Foundation Flanders to R.d.J. (FWO 12B8116N). We would like to thank Ioannis Stringlis, Marrieke Bos, Iliass Bouallal and Diana van Houten for experimental assistance, Annemiek van Andel for propagation of Hpa in planta, Hans van Pelt for taking photographs, Menno van der Voort for help with the Phyloprofiler, Stephen Barnett for advice on the isolation of Microbacterium spp. and Alexandre Jousset for his valuable comments on the manuscript. Furthermore, we would like to thank Machiel Bosch and Natuurmonumenten for permitting the sampling of soil in the Reijerscamp reserve.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Berendsen, R.L., Vismans, G., Yu, K. et al. Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J 12, 1496–1507 (2018). https://doi.org/10.1038/s41396-018-0093-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0093-1
This article is cited by
-
Synthetic community derived from grafted watermelon rhizosphere provides protection for ungrafted watermelon against Fusarium oxysporum via microbial synergistic effects
Microbiome (2024)
-
Redundancy in microbiota-mediated suppression of the soybean cyst nematode
Microbiome (2024)
-
Bacterial wilt affects the structure and assembly of microbial communities along the soil-root continuum
Environmental Microbiome (2024)
-
The impacts of ecological disturbances on the diversity of biosynthetic gene clusters in kauri (Agathis australis) soil
Environmental Microbiome (2024)
-
Bacillus velezensis YC89-mediated recruitment of rhizosphere bacteria improves resistance against sugarcane red rot
Chemical and Biological Technologies in Agriculture (2024)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}