
Abstract
The dark ocean microbiota represents the unknown majority in the global ocean waters. The SAR202 cluster belonging to the phylum Chloroflexi was the first microbial lineage discovered to specifically inhabit the aphotic realm, where they are abundant and globally distributed. The absence of SAR202 cultured representatives is a significant bottleneck towards understanding their metabolic capacities and role in the marine environment. In this work, we use a combination of metagenome-assembled genomes from deep-sea datasets and publicly available single-cell genomes to construct a genomic perspective of SAR202 phylogeny, metabolism and biogeography. Our results suggest that SAR202 cluster members are medium sized, free-living cells with a heterotrophic lifestyle, broadly divided into two distinct clades. We present the first evidence of vertical stratification of these microbes along the meso- and bathypelagic ocean layers. Remarkably, two distinct species of SAR202 cluster are highly abundant in nearly all deep bathypelagic metagenomic datasets available so far. SAR202 members metabolize multiple organosulfur compounds, many appear to be sulfite-oxidizers and are predicted to play a major role in sulfur turnover in the dark water column. This concomitantly suggests an unsuspected availability of these nutrient sources to allow for the high abundance of these microbes in the deep sea.
Similar content being viewed by others


Introduction
The marine habitat is the largest on Earth and marine microbes play fundamental roles in global biogeochemical nutrient cycling [1]. Planktonic communities are permanently stratified by light into two different zones with remarkably different microbial compositions. The photic zone occupies the uppermost ca. 200 m and is characterized by intense photosynthetic activity amidst steep gradients of light intensity, nutrients and temperature. The far larger aphotic zone extends all the way down to the sea floor and is permanently dark, cold, oligotrophic and heavily dependent upon organic input from the more productive layers above. Microbial communities are further stratified within these two zones. For instance, a clear stratification is observed among several planktonic cyanobacterial genera, Prochlorococcus being located below the Synechococcus maxima in the photic zone [2, 3]. When spatio-temporal distributions of microbes in the water column of the marine habitat began to be explored by 16S rRNA sequences, novel and widespread microbial groups were discovered. e.g., SAR11 [4] and mesophilic group I (now Thaumarchaeota) and II archaea [5, 6]. Among the early discoveries was a group of 16S rRNA sequences named SAR202 cluster that affiliated to the phylum Chloroflexi and had no previously described representatives in the marine habitat [7]. These sequences were found preferentially at the lowermost bounds of the photic zone (depth 250 m) in the North Atlantic and Pacific oceans. Additional SAR202 sequences were retrieved in 16S rRNA clone surveys in deep waters of the Pacific Ocean (3000 m), extending the presence of this group to the deep- sea [8]. Similar sequences were also retrieved from the deep, cold hypolimnion (500 m) of an oligotrophic lake (Crater Lake) [9]. Unequivocal evidence for the widespread prevalence of the SAR202 throughout the mesopelagic and bathypelagic realms was provided by fluorescence in-situ hybridization (FISH), revealing high numbers of SAR202 cells throughout the aphotic water column ( > 10% of total prokaryotic community), down to nearly 3500–4000 m in the Atlantic and Pacific oceans [10, 11].
Despite the SAR202 group being among the first widespread microbial lineages discovered in the aphotic zones [7] we still lack cultured representatives for this group and their physiological characteristics remain completely unknown. In-situ substrate uptake experiments suggested that a large fraction of SAR202 cells utilize L-aspartic acid at all depths in favor of the more refractory D-asp. This is in contrast to the global bacterial and archaeal community that shows decreased uptake rates for L-Asp with increasing depth, suggesting adaptability of SAR202 to available dissolved organic matter in the deep sea [12]. A metagenomic analysis of the deep Mediterranean Sea water column also reported a high abundance of Chloroflexi in the deep sea and suggested the potential for carbon-monoxide oxidation in these microbes [13, 14]. FISH analyzes showed that SAR202 cells have a coccoid morphology with a diameter of > 1 µm [10] that does not vary with depth. This is unlike SAR11, whose cell-size generally increases with depth [11], something that correlates with a slight increase in genome size for deep SAR11 ecotypes [15]. SAR202 does not show strong correlation to environmental parameters (except perhaps to lower temperatures and increasing depth). Reports are sometimes conflicting in this respect, e.g., correlation to high oxygen concentrations found by Giovannoni et al. [7] and lack thereof by Schattenhofer et al. [11].
This in itself is quite remarkable for a group that at some sites may comprise > 40% of the total bacterial community in the deep oceans [12]. Phylogenetic analysis of 16S rRNA genes in general provides little information regarding metabolic traits. This is particularly true for Chloroflexi, which display a broad spectrum of lifestyle characteristics, e.g., anoxygenic photosynthesizers [16, 17], obligate aerobic/anaerobic heterotrophs [18,19,20,21], thermophiles [18, 20, 22], halophiles [23], nitrite oxidizers [24, 25], predators with gliding motility [26], some with capacity for reductive dehalogenation [27, 28] and even endospore-forming Gram-positive bacteria in a largely Gram-negative phylum [29, 30]. The only genomic information from the SAR202 group at present is from an incomplete single cell amplified genome (SAGs), Pac-SCGC-AAA240-N13, obtained from a depth of 770 m from the ALOHA station in the Pacific Ocean north of Hawaii [31] but no genomic analyzes have been reported.
In this work, we reconstructed several SAR202 genomes from deep marine and brackish metagenomics datasets from the Ionian, Aegean and the Caspian Sea as well as Pacific, Atlantic and Indian oceans. By screening publicly available unclassified Chloroflexi genomes we also identified another SAG, Atl-SCGC-AB-629-P13, (obtained from a depth of 553 m in the mid-Atlantic) as belonging to the SAR202 lineage. These metagenome-assembled SAR202 genomes (MAGs), together with the two SAGs provide a first insight into the metabolic capabilities of this group and suggest a significant role in the deep-ocean sulfur cycle. Moreover, metagenomic fragment recruitment analyzes reveal that highly related SAR202 genomes are prevalent at geographically distant locations (from the Mediterranean to the Pacific) implying their global distribution as a deep stratified cluster with a sizeable contribution to global ocean biogeochemical cycles, particularly the sulfur cycle.
Material and methods
Metagenomic datasets used in this study
The metagenomic samples from the Caspian Sea were taken at the peak of stratification in October 2013 at three depths (15 m, 40 m and 150 m, bottom depth 230 m). Sample collection, DNA extraction, sequencing and assembly of these datasets are described in more detail in Mehrshad et al. Mehrshad et al. [32] and the sequence data are available from NCBI SRA (Bioproject PRJNA279271). The two deep sea samples from the Mediterranean were collected in the Aegean Sea (600 m, bottom depth 699 m) and Ionian Sea (3500 m, bottom depth 3633 m) in October 2010. These datasets and additional deep chlorophyll maximum samples used in this study are available in NCBI SRA (Bioprojects PRJNA305355 and PRJNA257723). Three representative deep, marine metagenomes from the MALASPINA expedition were also used for assembly (SRR3965592, SRR3963457, and SRR3961935). The Caspian and the Mediterranean metagenomes from all samples analyzed in this study are from the 0.22–5.0 µm fraction and were sequenced by HiSeq2000 (paired end reads of length 100 bp). The MALASPINA datasets are from the 0.2 to 0.8 µm fraction and were sequenced using HiSeq2000 (paired end reads of length 150 bp). Basic metadata (sampling date, latitude, longitude, depth, bioproject identifiers, SRA accessions), sequence statistics (number of reads, read length, dataset size) and % of Chloroflexi assigned 16S rRNA reads identified in all metagenomes used for assembly are provided in Supplementary Table S1.
Unassembled 16S rRNA read classification
A non-redundant version of the RDP database [33] was created by clustering its ca. 2.3 million 16S rRNA gene sequences into approximately 800,000 sequences at 90% nucleotide identity level using UCLUST [34]. A 20 million subset of the reads from the Illumina datasets was compared to this reduced set and an e-value cutoff of 1e-5 was used to identify candidate 16S rRNA gene sequences. The candidate sequences were further examined using ssu-align, to separate them into archaeal, bacterial, and eukaryotic 16S/18S rRNA or non-16S rRNA gene sequences [35]. Only bona fide 16S rRNA sequences were finally compared to the complete RDP database and classified into a high level taxon if the sequence identity was ≥ 80% and the alignment length was ≥ 90 bp. Sequences failing these thresholds were discarded. To get an overview of the distribution of 16S rRNA reads assigned to Chloroflexi across the global ocean, in addition to the Mediterranean (DCM and Deep), Caspian [32] and MALASPINA [36] datasets, we also used surface, DCM and mesopelagic datasets generated by the TARA expedition [37].
Metagenome assembly and annotation
All three datasets retrieved from depth profile of the Caspian Sea (15 m, 40 m and 150 m) were assembled together (using IDBA_UD) [38] as described previously [32]. The Mediterranean DCM datasets, the Deep Mediterranean and the MALASPINA datasets were quality trimmed using sickle (https://github.com/najoshi/sickle, default parameters) and assembled using the megahit assembler (--k-min 39 --k-max 99 --k-step 10 --min-count 2) [39]. The MALASPINA datasets were chosen because they had a high percentage of Chloroflexi 16S rRNA reads (9–11%) (Supplementary Table S1), and also because they represented geographically distant locations (South Atlantic, Indian Ocean and North Pacific).
Prodigal (in metagenomic mode) was used for predicting protein coding genes in the assembled contigs [40]. tRNA prediction was performed using tRNAscan-SE [41] and ribosomal rRNA genes were identified with meta_rna [42]. Functional gene annotation was performed by comparisons against COG hmms [43] using and e-value cutoff of 1e-5, and the TIGRfams models [44] (using trusted score cutoffs --cut_tc) using the hmmer package [45]. The assembled genomes were also annotated using the RAST server [46] and BlastKOALA [47]. Additionally, all predicted proteins were also compared to the NCBI-NR database using BLASTP at e-value 1e-5.
Genome reconstruction
Only contigs longer than 10 kb were used in the genome reconstructions. A contig was considered to belong to the phylum Chloroflexi if a majority of its genes gave best BLAST hits to this phylum. Contigs within each dataset were grouped using taxonomic affiliation, principal component analysis of tetranucleotide frequencies, GC% and coverage in different metagenomes as described previously [48,49,50]. Tetranucleotide frequencies were computed using the compseq program (-word 4 -reverse) in the EMBOSS package (Rice et al. 2000). Principal components analysis was performed using the FactoMineR package (function res.pca()) in R [104]. Enzyme EC numbers were predicted using PRIAM [51] and metabolic pathway reconstruction was performed using Pathway Tools [52].
Metagenomic fragment recruitment
To avoid bias in recruitment results owing to the presence of highly related rRNA sequences, rRNA sequences in genomes were masked. After masking, recruitments were performed using BLASTN [53], and a hit was considered only when it was at least 50 nucleotides long, had a nucleotide identity of > 95% and an e-value of ≤ 1e-5. These cutoffs approximate species-level divergence [54]. These hits were used to compute the RPKG (reads recruited per kilobase of genome per gigabase of metagenome) values that reflect abundance values and are normalized and comparable across different genomes and metagenomes (RPKG = Number of reads matching a contig or genome per Kb/Total number of bases in the metagenome expressed in Gb). In order to confirm that the reads assigned to SAR202 genomes using these cutoffs did not contribute to abundance of other microbes, we also compared the SAR202 assigned reads from three bathypelagic MALASPINA datasets (10 million reads each from SRR3961935, SRR3963457, SRR3965592) to the complete RefSeq collection (ca. 5000 microbial genomes). None of the SAR202 assigned reads matched any genome in RefSeq confirming that these cutoffs are specific for detecting only reads originating from genomes of the SAR202 cluster.
Screening for SAR202 cluster genomes. All publicly available Chloroflexi genomes from NCBI and JGI were downloaded. Phylogenetic analyzes of 16S rRNA sequence and conserved concatenated proteins suggested that two single cell amplified genomes SCGC-AB-629-P13 (Mid-Altlantic Ocean, 553 m depth) and SCGC-AAA240-N13 (Pacific Ocean, ALOHA station, Hawaii, 770 m depth) also belonged to the SAR202 group and were included in all analyzes (Supplementary Figs. 3 and 5).
Phylogenomics and genome size estimation
A reference phylogenomic tree was made using PhyloPhlAN [55], the complete genomes of representatives from all known Chloroflexi classes and reconstructed MAGs of this study were added to the built-in tree of life in PhyloPhlAN (Supplementary Fig. S5). The two single cell amplified genomes SCGC-AB-629-P13 (Mid-Altlantic Ocean, 553 m depth) and SCGC-AAA240-N13 (Pacific Ocean, ALOHA station, Hawaii, 770 m depth) were also added as confirmed SAR202 representatives verified by 16S rRNA analysis (Supplementary Fig. S3). PhyloPhlAN uses USEARCH [34] to identify the conserved proteins and subsequent alignments against the built-in database are performed using MUSCLE [56]. Finally, an approximate maximum-likelihood tree is generated using FastTree [57] with local support values using Shimodaira–Hasegawa test [58]. This analysis confirmed that all MAGs belong to the phylum Chloroflexi and are phylogenetically most related to the SAR202 cluster branching together with the two confirmed SAR202 SAGs. For creating the sub-tree (Fig. 1) of MAGs with higher than 50% completeness, only Dehalococcoidia genomes were used as outgroups (as they are the closest neighbors of SAR202 (Supplementary Fig. S5) to reveal better the internal phylogenetic relationships within the incomplete genomes by maximizing the number of common proteins. For creating the sub-tree phylogeny shown in Fig. 1, 50 conserved proteins in the reconstructed genomes and the reference genomes were identified using the COG database [43]. These proteins were concatenated and aligned using Kalign (default parameters) [59] and the alignment was trimmed using trimAL (--gappyout) [60]. A maximum-likelihood tree was constructed with FastTree2 [57], using a JTT + CAT model, a gamma approximation, and 100 bootstrap replicates.

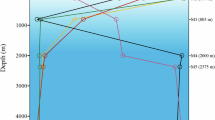
a Maximum likelihood phylogeny of the 12 almost complete reconstructed genomes from Caspian, Aegean, Ionian Seas and MALASPINA datasets in the SAR202 cluster together with the single cell amplified genomes of the SAR202 cluster. The tree was made using a concatenate of 50 conserved proteins. Genomes of the class Dehalococcoidia were used to root the tree. The reconstructed genomes from this study are highlighted in red. The subcluster designation for genomes containing 16S rRNA is shown in blue (SC III and SC V). Bootstrap values (%) are indicated at the base of each node. Legends for lifestyle hints are on top left. b Metagenomic recruitment of almost complete reconstructed genomes together with available single cell genomes of SAR202 cluster in different deep datasets from brackish and marine environments. Brackish datasets include two aphotic depths of the Caspian Sea. Marine datasets include Aegean, Ionian, and Marmara Sea deep datasets, and Puerto Rico Trench deep dataset, together with three of the deep MALASPINA datasets also used for assembly. The depth of samples for each dataset is mentioned inside parenthesis
Two sets of previously described genes, one with 35 single copy orthologous genes [61], and another with 112 essential genes [48] found in bacteria and also a set of 140 common genes (identified using TIGRFams [62] between 41 complete Chloroflexi genomes) were used to estimate genome completeness (Supplementary Table S2). Additionally, CheckM [63] was also used to estimate genome completeness and provided very similar results to the genome size estimation performed using 140 common Chloroflexi genes (Supplementary Table S3).
Single gene phylogeny and ANI
16S rRNA sequences from Chloroflexi genomes, clone libraries, SAGs and MAGs were aligned using MUSCLE [56], and a maximum-likelihood tree was constructed with FastTree2 [57], using a GTR model, a gamma approximation, and 100 bootstrap replicates. The apr protein sequence alignments were also performed using MUSCLE [56], and FastTree2 [57], was used for creating the maximum-likelihood tree (JTT + CAT model, gamma approximation, 100 bootstrap replicates). Average Nucleotide Identity (ANI) was calculated as defined in Ref. [54].
Accession numbers
The assembled genomic bins have been deposited to DDBJ/EMBL/GenBank and can be accessed under the Bioproject PRJNA356693 and the accession numbers MUCH00000000, MUCI00000000, MUCJ00000000, MUCK00000000 (for Casp-Chloro-G1-4), MUCL00000000, MUCM00000000, MUCN00000000 (for Ae2-Chloro-G1-3), MUCO00000000, MUCP00000000, MUCQ00000000, MUCR00000000, MUCS00000000, MUCT00000000, MUCU00000000, MUCV00000000, MUCW00000000 (for Io17-Chloro-G1-9), MUCX00000000, MUCY00000000 (for MP-SAtl-SRR3965592-G1 and –G2), MUCZ00000000, MUDA00000000 (for MP-SInd-SRR3963457-G1 and –G2), MUDB00000000 (for MP-NPac-SRR3961935-G1).
Results and discussion
SAR202 abundance in marine and brackish environments
A broad overview of presence of phylum Chloroflexi representatives in the water column of marine and brackish environments using 16S rRNA metagenomic reads is shown in Supplementary Fig. S1A (See also Supplementary Table S4). While SAR202 were barely detectable in the photic zone and deep chlorophyll maximum (DCM) datasets (Western and Eastern Mediterranean, Caspian and Red Sea, HOTs and BATs), they comprised a substantial fraction of the microbial community in the datasets from the deep aphotic zones of eastern Mediterranean (Aegean ca. 5% and Ionian ca. 10%). The Caspian datasets also showed a progressive increase in the percentage of reads assigned to Chloroflexi with increasing depth, reaching a maximum of ca. 4% at 150 m, suggesting the existence of novel brackish SAR202 representatives. In comparison, the Red Sea depth profile did not show any significant amounts of Chloroflexi members (maximum sample depth 500 m), suggesting that SAR202 does not show an ecological preference for the particular physico-chemical conditions characterizing the dark Red Sea [64, 65]. An integrated view of the distribution of 16S rRNA reads assigned to Chloroflexi in metagenomic datasets from the surface, DCM, mesopelagic and bathypelagic ocean layers shows clearly an increasing abundance with depth (Supplementary Fig. S1B). There are statistically significant differences between the surface-mesopelagic, surface-bathypelagic datasets, DCM- mesopelagic and DCM-bathypelagic datasets (Kruskal–Wallis test, P = <0.001). Apart from the deep Mediterranean, the high abundance of Chloroflexi across different geographical locations in the global ocean and depths in the Pacific, Atlantic and Indian Oceans reaffirms the deep dark waters of global oceans as their preferred niche (Supplementary Fig. S1 and Supplementary Table S3). Chloroflexi even comprised up to 14% of microbial community in some deep ocean datasets of the MALASPINA expedition. They appear to be present in variable proportions at all deep aphotic zones for which data are available (largely −40 N to 44 S) without obvious preferences. Only one deep ocean dataset is available from the Antarctic Province that is outside these latitudinal bounds (TARA expedition, 790 m depth, −61.98 S, −49.36 W) and it also contained 3.35 percent of 16S rRNA reads of Chloroflexi (Supplementary Fig. S2). Overall, these analyzes recapitulate previous findings indicating that the SAR202 abundance increases with depth [7, 10,11,12, 14] and has a global distribution in the marine water column.
Genome reconstruction and phylogenomics
In order to obtain a first insight into the genomic repertoire and diversity of the SAR202 group, we chose a number of marine and brackish metagenomic datasets for assembly, annotation and taxonomic binning (Supplementary Table S1, see also Methods). These include two deep datasets from the Eastern Mediterranean (Aegean and Ionian Sea) and three representative deep datasets from the MALASPINA expedition (one each from South Atlantic Ocean, Indian Ocean and North Pacific Ocean). To identify brackish SAR202 representatives, existing assemblies from the Caspian Sea were used [32]. We also included for assembly five deep chlorophyll maximum datasets from the Mediterranean. After the assembly, we selected long contigs ( > 10 Kb) in which the majority of predicted proteins gave best hits to Chloroflexi genomes. As expected from the 16S rRNA results, very few Chloroflexi contigs could be identified in the DCM datasets as Chloroflexi are only minor constituents of the photic zone communities (Supplementary Fig. S1 and Supplementary Table S5) and they were not considered further. The Ionian Sea (Io17) datasets from 3500 m depth had the highest number of assembled Chloroflexi affiliated genomic fragments (709 contigs) with total size of 17.8 Mb (Supplementary Table S5). Using differences in GC content, coverage in different datasets, and principal components analysis of tetranucleotide frequencies, (see Methods) these contigs were further segregated to 21 genomic bins (nine from the Ionian, three from Aegean, four from the Caspian, two each from South Atlantic and South Indian Ocean and one from the North Pacific Ocean, Supplementary Table S5). Although, like all other metagenomic assemblies, these bins are not “clonal”, we will refer to them as “genomes” or MAGs (metagenome-assembled genomes). We also identified two single-cell amplified genomes (SAGs) that also belong to the SAR202 cluster in a comparison with these metagenome-assembled genomes (Atl-SCGC-AB-629-P13, Mid-Atlantic Ocean, 553 m depth and Pac-SCGC-AAA240-N13, Pacific Ocean, ALOHA Station, Hawaii, 770 m depth). Detailed statistics of these reconstructed Chloroflexi genomic bins and the two SAGs are shown in Table 1. Three genomic bins contained contigs harboring partial 16S rRNA genes (Io17-Chloro-G6 – 333 bp, MP-SInd-SRR3963457-G1 – 947 bp, and MP-SAtl-SRR3965592-G2 – 926 bp) and all could be unequivocally affiliated to the SAR202 cluster (subcluster III) within the phylum Chloroflexi (Supplementary Fig. S3) to which the original SAR202 clones also belong [10]. A nearly complete 16S rRNA gene was also found in the SAG Pac-SCGC-AAA240-N13 (1508 bp), which also affiliated to subcluster III. Only a single genome (SAG Atl-SCGC-AB-629-P13, 16S rRNA 1202 bp) was found to belong to subcluster V. The average nucleotide identity (ANI) comparison of all the MAGs and SAGs of SAR202 cluster shows that the five MAGs from MALASPINA datasets belong to two different species (Supplementary Fig. S4).
The MAGs, SAGs and the previously available Chloroflexi genomes were inserted into a reference tree of life (3174 microbial genomes) using PhyloPhlAn [55]. All MAGs and SAGs formed a large “Class” level group with two distinct sub-clusters within the phylum Chloroflexi (Supplementary Fig. S5 and Fig. 1). This was possible for all genomes except Caspian-Chloro-G1 because of the small size (only 0.35 Mb) and lack of common conserved proteins although the predicted proteins in this MAG gave best blast hits to SAR202 members. The closest neighbors of the SAR202 group appear to be the class Dehalococcoidia, whose cultivated representatives are mostly sediment-dwelling anaerobes (freshwater or marine) [28, 66, 67]. This relatedness has been seen in the 16S rRNA phylogeny as well (Supplementary Fig. S3) [10]. Of the few sequenced genomes that are available from the genus Dehalococcoidea, it appears they have streamlined, small genomes with size in the range 1.5–1.8 Mb (genomic GC 45-65%). Several studies showed that they are capable of organohalide respiration (particularly reductive dechlorination) [28, 68,69,70]. However, recently this ability has been also expanded to some members of the class Anaerolineae of the phylum Chloroflexi [71]. In what follows, we focused our analyzes largely on the twelve most complete MAGs (seven genomes from deep Ionian Sea, two genomes from the Caspian Sea, one genome each from assembled deep MALASPINA datasets, MP-NPac-SRR3961935, MP-SInd-SRR3963457, and MP-SAtl-SRR3965592) and the two SAR202 cluster SAGs.
SAR202 global distribution and vertical stratification
All MAGs reconstructed in this study showed highest recruitment values in their environment of origin along with higher abundances in the deeper strata of the water column (Fig. 1 and Supplementary Fig. S6). Similar trends were observed in the global ocean, where the surface and deep chlorophyll maximum datasets were largely devoid of SAR202 representatives, but the deep aphotic datasets showed a far higher abundance for reconstructed MAGs and SAGs of the SAR202 cluster (Supplementary Fig. S7A).
Nine SAR202 genomes, including three from the Ionian (Io17-G1, Io17-G5, Io17-G8, 3500 m), two each from the South Atlantic (MP-SAtl-SRR3965592-G1 and –G2, 3199 m) and Indian Ocean (MP-SInd-SRR3963457-G1 and –G2, 4000 m), one from North Pacific (MP-NPac-SRR3961935-G14004 m depth) and Pac-SCGC-AAA240-N13 (770 m depth, Pacific Ocean) appear to be widely distributed in several deep aphotic datasets (Supplementary Figs. S6 and S7B).
However, on segregating datasets into mesopelagic (up to 1000 m deep, 19 datasets) and bathypelagic (1500–6000 m, 31 datasets) a different recruitment pattern is evident. While Ae2-Chloro-G1, Ae2-Chloro-G3, Io17-Chloro-G5 recruited more reads in the mesopelagic datasets, Io17-Chloro-G1, Io17-Chloro-G8, all MALASPINA genomes and Pac-SCGC-AAA240-N13 recruited even more from the bathypelagic datasets (Fig. 2). This differential recruitment in meso- and bathypelagic layers of the ocean suggests a vertical stratification of SAR202 in the aphotic zone. It has been suggested that the mesopelagic layer is more influenced by the upper layers and the bathypelagic realm has a comparatively more stable status regarding different physicochemical features [72,73,74,75]. Despite the distant origin of the genomes from the MALASPINA datasets, they show high recruitment values in almost all available deep bathypelagic datasets suggesting the presence of the same species in widely different geographic areas (Southern Indian Ocean, South Atlantic, North Pacific Ocean). Remarkably, a differential vertical stratification was also seen for currently available deep specific SAR11 SAGs, (isolated from the mesopelagic zone, 770 m depth), that are more abundant in the mesopelagic than in bathypelagic datasets (Fig. 2). These deep specific SAR11 SAGs appear also to be much more abundant in the mesopelagic samples than the cultured isolates in the photic zone (Supplementary Fig. S8).

Overview of the recruitment (RPKG) distribution of all genomes in SAR202 cluster together with deep specific single cell amplified genomes of the SAR11 clade. a Recruitment RPKG distribution in marine mesopelagic datasets together with the deep Caspian dataset from 150 m depth. Datasets cover the depths from 150 m to 1000 m (n = 19) in the mesopelagic layer and they include Caspian Sea (150 m), TARA mesopelagic (550–1000 m), Aegean Sea (600 m) and Marmara Sea (1000 m). b Recruitment (RPKG) distribution in marine deep datasets in the range of 1500–6000 m (n = 31) including deep MALASPINA datasets together with the deep sea datasets of the Ionian Sea (3500 m) and Puerto Rico deep trench (6000 m). The black dots in the box plots are outlier values and the line inside the boxes indicates the median value
However, another pattern is also evident for some SAR202 genomes, with high abundances at the place where the original metagenome was obtained from, and lower at others. These observations likely reflect biogeographic patterns resulting from local environmental selection. Nevertheless, these results suggest that at least some SAR202 genomes recovered here are primarily bathypelagic, extremely abundant, much more than the currently available SAR11 representative genomes are (either from deep or surface), being among the most abundant microbial genomes retrieved from the aphotic zone at large. The brackish SAR202 MAGs reconstructed from metagenomes of the Caspian Sea also display a clear vertical stratification similar to the marine habitat along the Caspian depth profile datasets (Supplementary fig. S6) although at shallower depths than their marine bathypelagic counterparts.
Metabolism
Genome annotation of SAR202 members suggests they are capable of organo and lithotrophic metabolisms. Representative genes for central carbohydrate metabolism like tricarboxylic acid cycle, pentose phosphate, glycolysis and gluconeogenesis pathways were present (see Fig. 3 and Fig. 4). However, both aerobic and anaerobic routes for pyruvate oxidation to acetyl CoA, via pyruvate dehydrogenase (PDH; aerobic) and pyruvate ferredoxin oxidoreductase (PFOR; anaerobic) were found, similar to other Chloroflexi genomes [76] [102]. No evidence for autotrophic carbon fixation was found in any of these genomes.

Mosaic schematic view of the metabolic traits in all available genomes from SAR202 cluster. The parts affiliated with sulfur compound metabolism are highlighted in gray

Overview of the metabolic highlights of the MAGs and SAGs of the SAR202 cluster. Filled squares indicate the presence of the pathway/gene
Almost all MAGs and SAGs of the SAR202 cluster encode genes responsible for ammonia assimilation as a part of their nitrogen metabolism (e.g., ammonium transporter, glutamate synthase, glutamine synthase, all key genes for ammonia assimilation) (See Figs. 3 and 4). In addition, SAR202 seem to utilize other potential nitrogen sources in the deep sea such as hypotaurine and taurine, that function as high concentration osmolytes in some certain deep-sea invertebrates and can also be used as carbon, nitrogen, or energy sources by bacteria [77, 78]. Taurine/hypotaurine transporter genes and degradation pathways for hypotaurine and taurine were found that release L-alanine (which may be converted to pyruvate or degraded to yield ammonium) and pyruvate that can be fed directly into central carbon metabolism. The sulfoacetaldehyde and sulfinoacetaldehyde produced as end products of taurine/hypotaurine degradation may also be further metabolized to acetyl-CoA and acetate with the concomitant production of sulfite. It has been shown that SAR11 can grow on taurine alone [79], and taurine has been suggested as a major source of C, N and S for SAR11 in the marine habitat [80]. The presence of these pathways suggests that at least some SAR202 members are also capable of utilizing taurine and hypotaurine as carbon sources.
One of the most readily available form of reduced sulfur in the euphotic zone of the open ocean is dimethylsulfoniopropionate (DMSP), an organosulfur compound produced by marine algae as compatible solute [81]. the main source of DMSP in the deeper parts of the water column is sedimentation of particulate DMSP and DMSP containing cells [81, 82]. While most DMSP is demethylated and utilized for amino acid biosynthesis (e.g., SAR11 and Roseobacter clade) [83], an alternative degradation pathway allows cleavage to 3-hydroxypropionate and dimethylsulfide (DMS) by the action of DMSP lyase (dddD) (e.g., SAR11) [84]. Several bacterial genera, e.g., Rhodococcus, Acinetobacter, Pseudomonas etc. are able to use DMS as sole sulfur source. As only one SAG encoded a DMSP lyase (Alt-SCGC-AB629-P13), and coupled with the uncertain knowledge on availability of DMSP in deeper waters, this process is unlikely to be widespread in the deep strata. However, several key downstream genes for oxidation of DMS itself, leading to the production of methanesulfonates, sulfite and formaldehyde were found (See Figs. 3 and 4). Apart from the biological production of methanesulfonates, chemical oxidation of atmospheric DMS can also lead to methanesulfonate production (several megatons), which is ultimately deposited in the oceans. Methanesulfonate transporters, methanesulfonate monoxygenases and formate dehydrogenases, which would allow the use of methanesulfonates as an energy source, were found (Figs. 3 and 4). However genes for formaldehyde assimilation (found in methylotrophs) were absent in SAR202 genomes.
While the deep marine habitat is considered to be largely oxic, local oxygen-deprived conditions favoring anaerobic respiration can exist in, e.g., particulate matter, oxygen minimum zones, and deep-sea vents and cold seeps characterized by the emission of reduced fluids. While most components of the aerobic respiratory chain were detected in SAR202 genomes (see Fig. 3), the widespread presence of tetrathionate reductases (at least 15 MAGs and 1 SAG) that can reduce tetrathionate to thiosulfate under anaerobic conditions, suggests the possibility for facultative anaerobic respiration [85]. Furthermore, thiosulfate/sulfate transporters were found, that may help import thiosulfate, opening a possibility for the enzyme rhodanese to metabolize thiosulfate [86, 87]. The enzyme rhodanese is widely distributed in prokaryotes [88] but its actual role is still debated, and suggestions span from involvement in cyanide detoxification to sulfur metabolism. However, its activity also results in the production of sulfite [86].
The degradation of taurine, hypotaurine, methanosulfonate, DMS and rhodanese activity all would release sulfite, which is highly reactive. Sulfite may be oxidized by two distinct dissimilatory oxidation pathways, either via the SOX system [89] or by apr (adenylylsulfate reductase) [78, 90]. No genes for the SOX system were found, but several genomes encoded the apr gene (Figs. 3 and 4) that converts sulfite to adenosine 5’-phosphosulfate (APS). It has been suggested that the phylogeny of the aprA gene can discriminate sulfur-oxidizing (SOB) and sulfate-reducing (SRB) bacteria [91, 92]. However, all SAR202 aprA genes form a separate clade, which also includes an aprA gene found in a euryarchaeal fosmid (KM3-67-G08), originating from metagenomic fosmid libraries of the Ionian Sea at 3000 m depth [93] (Supplementary Fig. S9). The closest relatives of this clade all originate from MAGs of sediment microorganisms (Chloroflexi bacterium CSP1-4 [94] and the Candidatus Rokubacteria, representative of a new phylum [94, 95]. In our case, given that the deep ocean is largely oxic, and that we found no evidence of dissimilatory sulfite reductases (dsr) genes in these genomes, the aprA genes from SAR202 likely represent a new group of sulfur-oxidizing aprA genes. While aprA genes have also been reported in deep specific SAR11 SAGs [15], they cluster together with the aprA genes of surface SAR11 genomes in the sulfur-oxidizing proteobacteria lineage I of aprA phylogeny (Supplementary Fig. S9). These genes were proposed to participate in taurine metabolism to detoxify the highly reactive sulfite (producing APS) and play a key role in the sulfur metabolism in deep specific SAR11 bacteria [15].
However, to produce ATP from sulfite via the activity of aprBA genes, a second step involving conversion of APS to sulfate is required. The gene encoding the corresponding enzyme (sat, sulfate adenylyltransferase) was also identified in SAR202 genomes. This would allow SAR202 to utilize the excess sulfite produced as a by-product of organosulfur compound metabolism as an energy source, producing sulfate as a final product. It is unclear if the complete sulfate assimilation pathways are present in SAR202, as only a single phosphoadenosine phosphosulfate (PAPS) reductase was found in one MAG (Casp-Chloro-G4) and no sulfite reductases were found. It may be that SAR202, similar to SAR11, rely on availability of reduced sulfur compounds [15, 96,97,98].
Conclusions
The dark ocean is several times larger than the photic zone in sheer size and the majority of its microbial inhabitants are unknown. In this work, we have reconstructed genomes of the uncultivated SAR202 cluster (phylum Chloroflexi), which appears particularly abundant in the aphotic zone of global oceans. Moreover, the distribution of SAR202 sequences in the currently available deep metagenomic datasets (n = 50) suggests that SAR202 populations are vertically stratified, with distinct mesopelagic and bathypelagic groups. In particular, at least five genomes (belonging to only two distinct species) appear to be widely distributed in geographically distant regions in the bathypelagic zone of the world’s oceans.
Based on the higher abundance of the SAR202 cluster in the metagenomes of 0.–0.8 µm size fraction in the MALASPINA datasets, it is expected that these are free living microbes. Genome size estimations suggested sizes in the range of 2.5–3 Mb, that is in line with the large cell sizes observed by FISH [10]. Their larger cell-sizes coupled with higher abundances in the deep ocean suggest they alone make up for a significant fraction of the total biomass in this oligotrophic habitat. It must be considered however that total cell numbers are an order of magnitude lower in the deeper oceans than at the surface [99]. The genomes of the SAR202 group described here contain several pathways for metabolizing multiple organosulfur compounds as carbon, nitrogen and sulfur sources, which in turn implies that at least some of these compounds (e.g., DMS, DMSP, taurine, hypotaurine) may be more widely available in the deep ocean than suspected before [100, 101]. There are multiple sources of sulfur containing compounds, e.g., DMSP in sinking particulate cells (algal or microbial) from the upper layers, release of osmolytes from deep marine invertebrates (e.g., taurine, hypotaurine), production by heterotrophic microbes [103], hydrogen sulfide emissions from deep-sea vents, cold seeps and ocean sediments, especially at continental margins, or others that are as yet unknown. Similar disparities also exist in the measurements of total carbon input into this realm that do not match the higher estimated respiration rates [99], illustrating our as yet incomplete understanding of the remineralization processes and the participating entities at work in the dark ocean. In this work, we present initial evidence that hints at the presence of multiple, broadly available sulfur compounds in the deep sea that may be used by the abundant SAR202 group both as carbon and energy sources. Moreover, multiple degradation pathways found in these genomes appear to converge upon the production of sulfite, that may be oxidized to sulfate by adenylylsulfate reductase, suggesting that SAR202 are sulfite-oxidizers, making them key players in the sulfur cycle at the deep marine environment.
References
Azam F, Fenchel T, Field JG, Gray JC, Meyer-Reil LA, Thingstad F. The ecological role of water-column microbes in the sea. Mar Ecol Prog Ser 1983;10:257–64.
Biller SJ, Berube PM, Lindell D, Chisholm SW. Prochlorococcus: the structure and function of collective diversity. Nat Rev Micro 2015;13:13–27.
Chisholm SW, Olson RJ, Zettler ER, Goericke R, Waterbury JB, Welschmeyer NA. A novel free-living prochlorophyte abundant in the oceanic euphotic zone. Nature 1988;334:340–3.
Britschgi TB, Giovannoni SJ. Phylogenetic analysis of a natural marine bacterioplankton population by rRNA gene cloning and sequencing. Appl Environ Microbiol 1991;57:1707–13.
Delong EF. Archaea in coastal marine environments. Proc Natl Acad Sci USA 1992;89:5685–9.
Fuhrman JA, Mccallum K, Davis AA. Phylogenetic diversity of subsurface marine microbial communities from the Atlantic and Pacific Oceans. Appl Environ Microbiol 1993;59:1294–302.
Giovannoni SJ, Rappé MS, Vergin KL, Adair NL. 16S rRNA genes reveal stratified open ocean bacterioplankton populations related to the green non-sulfur bacteria. Proc Natl Acad Sci USA 1996;93:7979–84.
Fuhrman Ja, Davis Aa. Widespread archaea and novel bacteria from the deep sea as shown by 16S rRNA gene sequences. Mar Ecol Prog Ser 1997;150:275–85.
Urbach E, Vergin KL, Young L, Morse A, Larson GL, Giovannoni SJ. Unusual bacterioplankton community structure in ultra-oligotrophic Crater Lake. Limnol Oceanogr 2001;46:557–72.
Morris RM, Rappé MS, Urbach E, Connon SA, Rappe MS, Giovannoni SJ. Prevalence of the Chloroflexi-related SAR202 bacterioplankton cluster throughout the mesopelagic zone and deep ocean. Appl Environ Microbiol 2004;70:2836–42.
Schattenhofer M, Fuchs BM, Amann R, Zubkov MV, Tarran GA, Pernthaler J. Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic Ocean. Environ Microbiol 2009;11:2078–93.
Varela MM, Van Aken HM, Herndl GJ. Abundance and activity of chloroflexi-type SAR202 bacterioplankton in the meso- and bathypelagic waters of the (sub)tropical Atlantic. Environ Microbiol 2008;10:1903–11.
Martín-Cuadrado A-B, López-García P, Alba J-C, Moreira D, Monticelli L, Strittmatter A, et al. Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PLoS ONE 2007;2:e914
Martin-Cuadrado AB, Ghai R, Gonzaga A, Rodriguez-Valera F. CO dehydrogenase genes found in metagenomic fosmid clones from the deep Mediterranean Sea. Appl Environ Microbiol 2009;75:7436–44.
Thrash CJ, Temperton B, Swan BK, Landry ZC, Woyke T, DeLong EF, et al. Single-cell enabled comparative genomics of a deep ocean SAR11 bathytype. ISME J 2014;8:1440–51.
Klatt CG, Bryant DA, Ward DM. Comparative genomics provides evidence for the 3-hydroxypropionate autotrophic pathway in filamentous anoxygenic phototrophic bacteria and in hot spring microbial mats. Environ Microbiol 2007;9:2067–78.
Thiel V, Hamilton TL, Tomsho LP, Burhans R, Gay SE, Schuster SC, et al. Draft genome sequence of a sulfide-oxidizing, autotrophic filamentous anoxygenic phototrophic bacterium, Chloroflexus sp. strain MS-G (Chloroflexi). Genome Announc 2014;2:9–10.
Wu D, Raymond J, Wu M, Chatterji S, Ren Q, Graham JE, et al. Complete genome sequence of the aerobic CO-oxidizing thermophile Thermomicrobium roseum. PLoS ONE 2009;4:e4207.
Hemp J, Ward LM, Pace LA, Fischer W. Draft genome sequence of Levilinea saccharolytica KIBI-1, a member of the chloroflexi class Anaerolineae. Genome Announc 2015;3:2618.
Hugenholtz P, Stackebrandt E. Reclassification of Sphaerobacter thermophilus from the subclass Sphaerobacteridae in the phylum Actinobacteria to the class Thermomicrobia (emended description) in the phylum Chloroflexi (emended description). Int J Syst Evol Microbiol 2004;54:2049–51.
Sekiguchi Y, Yamada T, Hanada S, Ohashi A, Harada H, Kamagata Y. Anaerolinea thermophila gen. nov., sp. nov. and Caldilinea aerophila gen. nov., sp. nov., novel filamentous thermophiles that represent a previously uncultured lineage of the domain bacteria at the subphylum level. Int J Syst Evol Microbiol 2003;53:1843–51.
Jackson TJ, Ramaley RF, Meinschein WG. Thermomicrobium, a new genus of extremely thermophilic bacteria. Int J Syst Bacteriol 1973;23:28–36.
Van De Meene AML, Olson TL, Collins AM, Blankenship RE. Initial characterization of the photosynthetic apparatus of ‘Candidatus Chlorothrix halophila,’ a filamentous, anoxygenic photoautotroph. J Bacteriol 2007;189:4196–203.
Sorokin DY, Lücker S, Vejmelkova D, Kostrikina Na, Kleerebezem R, Rijpstra WIC, et al. Nitrification expanded: discovery, physiology and genomics of a nitrite-oxidizing bacterium from the phylum Chloroflexi. ISME J 2012;6:2245–56.
Sorokin DY, Vejmelkova D, Lücker S, Streshinskaya GM, Rijpstra WIC, Sinninghe Damsté JS, et al. Nitrolancea hollandica gen. nov., sp. nov., a chemolithoautotrophic nitrite-oxidizing bacterium isolated from a bioreactor belonging to the phylum Chloroflexi. Int J Syst Evol Microbiol 2014;64:1859–65.
Kiss H, Nett M, Domin N, Martin K, Maresca JA, Copeland A, et al. Complete genome sequence of the filamentous gliding predatory bacterium Herpetosiphon aurantiacus type strain (114-95T). Stand Genom Sci 2011;5:356–70.
Yan J, Rash BA, Rainey FA, Moe WM. Isolation of novel bacteria within the Chloroflexi capable of reductive dechlorination of 1,2,3-trichloropropane. Environ Microbiol 2009;11:833–43.
Löffler FE, Yan J, Ritalahti KM, Adrian L, Edwards EA, Konstantinidis KT, et al. Dehalococcoides mccartyi gen. nov., sp. nov., obligately organohalide-respiring anaerobic bacteria relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidia classis nov., order Dehalococcoidales ord. nov. and famil. Int J Syst Evol Microbiol 2013;63:625–35.
Cavaletti L, Monciardini P, Bamonte R, Schumann P, Ronde M, Sosio M, et al. New lineage of filamentous, spore-forming, gram-positive bacteria from soil. Appl Environ Microbiol 2006;72:4360–9.
Chang Y-J, Land M, Hauser L, Chertkov O, Del Rio TG, Nolan M, et al. Non-contiguous finished genome sequence and contextual data of the filamentous soil bacterium Ktedonobacter racemifer type strain (SOSP1-21). Stand Genom Sci 2011;5:97–11.
Swan BK, Martinez-Garcia M, Preston CM, Sczyrba A, Woyke T, Lamy D, et al. Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science 2011;333:1296–9.
Mehrshad M, Amoozegar MA, Ghai R, Shahzadeh Fazeli SA, Rodriguez-Valera F. Genome reconstruction from metagenomic datasets reveals novel microbes in the brackish waters of the Caspian Sea. Appl Environ Microbiol 2016;82:AEM.03381–15.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucl Acid Res 2009;37:D141–5.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010;26:2460–1.
Nawrocki E. Structural RNA homology search and alignment using covariance models. Washington University in ST. Louis 2009.
Salazar G, Cornejo-Castillo FM, Benítez-Barrios V, Fraile-Nuez E, Álvarez-Salgado XA, Duarte CM et al. Global diversity and biogeography of deep-sea pelagic prokaryotes. ISME J 2015;10:596–608.
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Ocean plankton. Structure and function of the global ocean microbiome. Science 2015;348:1261359.
Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012;28:1420–8.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2014;31:1674–6.
Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma 2010;11:119.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acid Res 1997;25:0955–64.
Huang Y, Gilna P, Li W. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 2009;25:1338–40.
Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 2001;29:22–8.
Haft DH, Loftus BJ, Richardson DL, Yang F, Eisen JA, Paulsen IT et al. TIGRFAMs: a protein family resource for the functional identification of proteins. Nucleic Acids Res 2001;29:41–43.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol 2011;7. e1002195, doi: https://doi.org/10.1371/journal.pcbi.1002195.
Aziz RK, Bartels D, Best Aa, DeJongh M, Disz T, Edwards Ra, et al. The RAST server: rapid annotations using subsystems technology. BMC Genom 2008;9:75.
Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol 2016;428:726–31.
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotech 2013;31:533–38.
Ghai R, Pašić L, Fernández AB, Martin-Cuadrado A-B, Mizuno CM, McMahon KD, et al. New abundant microbial groups in aquatic hypersaline environments. Sci Rep 2011;1:135.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J-F, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013;499:431–7.
Claudel-Renard C, Chevalet C, Faraut T, Kahn D. Enzyme-specific profiles for genome annotation: PRIAM. Nucl Acid Res 2003;31:6633–9.
Karp PD, Paley S, Romero P. The Pathway Tools software. Bioinformatics 2002;18:S225–32.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–402.
Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA 2005;102:2567–72.
Segata N, Börnigen D, Morgan XC, Huttenhower C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun 2013;4:2304.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acid Res 2004;32:1792–7.
Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 2010;5:e9490.
Shimodaira H, Hasegawa M, Multiple comparisons of log-likelihoods with applications to phylogenetic inference.Mol Biol Evol. 1999;16:1114–16
Lassmann T, Sonnhammer ELL. Kalign—an accurate and fast multiple sequence alignment algorithm. BMC Bioinforma 2005;6:298.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009;25:1972–3.
Raes J, Korbel JO, Lercher MJ, von Mering C, Bork P. Prediction of effective genome size in metagenomic samples. Genome Biol 2007;8:R10.
Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucl Acid Res 2003;31:371–73.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 2015;25:1043–55.
Haroon MF, Thompson LR, Parks DH, Hugenholtz P, Stingl U. Data descriptor: a catalogue of 136 microbial draft genomes from Red Sea metagenomes. Sci Data 2016;1–6.
Wang Y, Yang JK, Lee OO, Li TG, Al-Suwailem A, Danchin A, et al. Bacterial niche-specific genome expansion is coupled with highly frequent gene disruptions in deep-sea sediments. PLoS ONE 2011;6:e29149.
Biderre-petit C, Dugat-bony E, Mege M, Parisot N. Distribution of Dehalococcoidia in the anaerobic deep water of a remote meromictic crater lake and detection of dehalococcoidia -derived reductive dehalogenase homologous genes. PLoS One 2016;11:e0145558.
Wasmund K, Schreiber L, Lloyd KG, Petersen DG, Schramm A, Stepanauskas R et al. Genome sequencing of a single cell of the widely distributed marine subsurface Dehalococcoidia, phylum Chloroflexi. ISME J 2014;8:383–97.
Lee PKH, Cheng D, West KA, Alvarez-Cohen L, He J. Isolation of two new Dehalococcoides mccartyi strains with dissimilar dechlorination functions and their characterization by comparative genomics via microarray analysis. Environ Microbiol 2013;15:2293–305.
Poritz M, Schiffmann CL, Hause G, Heinemann U, Seifert J, Jehmlich N, et al. Dehalococcoides mccartyi strain DCMB5 respires a broad spectrum of chlorinated aromatic compounds. Appl Environ Microbiol 2015;81:587–96.
Schneidewind U, Haest PJ, Atashgahi S, Maphosa F, Hamonts K, Maesen M, et al. Kinetics of dechlorination by Dehalococcoides mccartyi using different carbon sources. J Contam Hydrol 2014;157:25–36.
Fullerton H, Moyer CL. Comparative single-cell genomics of Chloroflexi from the Okinawa Trough deep-subsurface biosphere. Appl Environ Microbiol 2016;82:3000–08.
Buesseler KO, Lamborg CH, Boyd PW, Lam PJ, Trull TW, Bidigare RR, et al. Revisiting Carbon Flux Through the Ocean’ s Twilight Zone. Science (80-) 2007;316:567–70.
Hioki N, Kuma K, Morita Y, Sasayama R, Ooki A, Kondo Y, et al. Laterally spreading iron, humic-like dissolved organic matter and nutrients in cold, dense subsurface water of the Arctic Ocean. Sci Rep 2014;4:6775.
Knauer GA, Martin JH, Bruland KW. Fluxes of particulate carbon, nitrogen, and phosphorus in the upper water column of the northeast Pacific. Deep Sea Res Part A Oceanogr Res Pap 1979;26:97–108.
Martin JH, Knauer GA, Karl DM, Broenkow WW. VERTEX: carbon cycling in the northeast Pacific. Deep Sea Res Part A Oceanogr Res Pap 1987;34:267–85.
Hug La, Castelle CJ, Wrighton KC, Thomas BC, Sharon I, Frischkorn KR, et al. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 2013;1:22.
Huxtable RJ. Actions of Taurine. Physiol Rev 1992;72:101–63.
Thus N. Metabolism of Nitrogen and Sulfur. Struct Funct Relationships Prokaryotes 1951;680–20.
Schwalbach MS, Tripp HJ, Steindler L, Smith DP, Giovannoni SJ. The presence of the glycolysis operon in SAR11 genomes is positively correlated with ocean productivity. Environ Microbiol 2010;12:490–500.
Williams TJ, Long E, Evans F, Demaere MZ, Lauro FM, Raftery MJ, et al. A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. ISME J 2012;6:1883–900.
Yoch DC, Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide.Appl Environ Microbiol. 2002;68:5804–15
Rellinger AN, Kiene RP, del Valle DA, Kieber DJ, Slezak D, Harada H, et al. Occurrence and turnover of DMSP and DMS in deep waters of the Ross Sea, Antarctica. Deep Res Part I Oceanogr Res Pap 2009;56:686–702.
Howard EC, Henriksen JR, Buchan A, Reisch CR, Bürgmann H, Welsh R, et al. Flux from the Ocean. Science 2006;649:649–52.
Reisch CR, Moran MA, Whitman WB. Dimethylsulfoniopropionate-dependent demethylase (DmdA) from Pelagibacter ubique and Silicibacter pomeroyi. J Bacteriol 2008;190:8018–24.
Friedrich CG 1998. Physiology and genetics of sulfur-oxidizing bacteria. In: Vol. 39. Advances in microbial physiology. Edited by R. K. Poole. San Diego, CA: Academic Press. 1998. p. 235–89.
Cipollone R, Ascenzi P, Visca P. Common themes and variations in the rhodanese superfamily. IUBMB Life 2007;59:51–59.
Cypionka H, Smock A, Bottcher M. A combined pathway of sulfur compound disproportionation in Desulfovibrio desulfuricans. FEMS Microbiology Letters 1998;166:181–6.
Aussignargues C, Giuliani MC, Infossi P, Lojou E, Guiral M, Giudici-Orticoni MT, et al. Rhodanese functions as sulfur supplier for key enzymes in sulfur energy metabolism. J Biol Chem 2012;287:19936–48.
Ghosh W, Dam B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol Rev 2009;33:999–43.
Meyer B, Kuever J. Homology modeling of dissimilatory APS reductases (AprBA) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE 2008;3. e1514, doi: https://doi.org/10.1371/journal.pone.0001514.
Meyer B, Kuevert J. Phylogeny of the alpha and beta subunits of the dissimilatory adenosine-5??? -phosphosulfate (APS) reductase from sulfate-reducing prokaryotes—origin and evolution of the dissimilatory sulfate-reduction pathway. Microbiology 2007;153:2026–44.
Watanabe T, Kojima H, Fukui M. Identity of major sulfur-cycle prokaryotes in freshwater lake ecosystems revealed by a comprehensive phylogenetic study of the dissimilatory adenylylsulfate reductase. Sci Rep 2016;6:36262.
Deschamps P, Zivanovic Y, Moreira D, Rodriguez-Valera F, López-García P. Pangenome evidence for extensive interdomain horizontal transfer affecting lineage core and shell genes in uncultured planktonic thaumarchaeota and euryarchaeota. Genome Biol Evol 2014;6:1549–63.
Hug LA, Thomas BC, Sharon I, Brown CT, Sharma R, Hettich RL, et al. Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ Microbiol 2016;18:159–73.
Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun 2016;7:1–11.
Giovannoni SJ. SAR11 bacteria: the most abundant plankton in the oceans. Ann Rev Mar Sci. 2017;9.
Smith DP, Nicora CD, Carini P, Lipton MS, Norbeck AD, Smith RD, et al. Proteome remodeling in response to sulfur limitation in ‘Candidatus Pelagibacter ubique’. mSystems 2016;1:e00068–16.
Tripp HJ, Kitner JB, Schwalbach MS, Dacey JWH, Wilhelm LJ, Giovannoni SJ. SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature 2008;452:741–4.
Arístegui J, Gasol JM, Duarte CM, Herndl GJ. Microbial oceanography of the dark ocean’s pelagic realm. Limnol Oceanogr 2009;54:1501–29.
Asher EC, Dacey JWH, Mills MM, Arrigo KR, Tortell PD. High concentrations and turnover rates of DMS, DMSP and DMSO in Antarctic sea ice. Geophys Res Lett 2011;38:1–5.
Schmidt S, Reigstad M, Belviso S, Wassmann P, Arashkevich E, Stefels J et al. Significance of vertical flux as a sink for surface water DMSP and as a source for the sediment surface in coastal zones of northern Europe. Estuar Coast Shelf Sci 2006;68: 473–88.
Tang K. Functional genomics of anoxygenic green bacteria Chloroflexi Species and evolution of photosynthesis. Biochemistry, Prof. Deniz Ekinci (Ed.), InTech, DOI: 10.5772/32496. Available from: https://www.intechopen.com/books/biochemistry/functional-genomics-of-anoxygenic-green-bacteria-chloroflexi-species-and-evolution-of-photosynthesis 2012.
Andrew R. J. Curson, Ji Liu, Ana Bermejo Martínez, Robert T. Green, Yohan Chan, Ornella Carrión, Beth T. Williams, Sheng-Hui Zhang, Gui-Peng Yang, Philip C. Bulman Page, Xiao-Hua Zhang, Jonathan D. Todd, (2017) Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nature Microbiology 2:17009
Lê S, Josse J, Husson F. FactoMineR : An R Package for Multivariate Analysis. J Stat Softw 2008;25:1–18.
Acknowledgements
MM was supported by the Czech Academy of Sciences (Postdoc program PPPLZ application number L200961651). RG was supported by the research grants 13-00243 S and 17-04828 S from the Grant Agency of the Czech Republic. MAA was supported by the grant MI-1391-20 from the Iranian Biological Resource Centre (IBRC). FRV was supported by projects “VIREVO” CGL2016-76273-P [AEI/FEDER, UE], from Agencia Estatal de Investigación (AEI), “REDES DE EXCELENCIA” CONSOLIDER- CGL2015-71523-REDC from the Spanish Ministerio de Economía, Industria y Competitividad and PROMETEO II/2014/012 “AQUAMET” from Conselleria d’Educació, Cultura i Esport, Generalitat Valenciana. Work in PLG’s lab was funded by the French National Agency for Research (ANR-08-GENM-024-002) and the European Research Council (ERC) under the European Commission 7th Framework Program (ERC Grant Agreement 322669). We are thankful to L Gasperini and G Bortoluzzi of the Istituto di Geologia Marina (ISMAR), CNR, Bologna (Italy) for allowing PLG to participate in the Marmara2010 R/V Urania cruise during which part of the samples analyzed in this study were collected. The authors would like to thank Gerard Muyzer for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mehrshad, M., Rodriguez-Valera, F., Amoozegar, M. et al. The enigmatic SAR202 cluster up close: shedding light on a globally distributed dark ocean lineage involved in sulfur cycling. ISME J 12, 655–668 (2018). https://doi.org/10.1038/s41396-017-0009-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-017-0009-5
