
Abstract
Cancer, a complex and multifactorial disease, presents a significant challenge to global health. Despite significant advances in surgical, radiotherapeutic and immunological approaches, which have improved cancer treatment outcomes, drug therapy continues to serve as a key therapeutic strategy. However, the clinical efficacy of drug therapy is often constrained by drug resistance and severe toxic side effects, and thus there remains a critical need to develop novel cancer therapeutics. One promising strategy that has received widespread attention in recent years is drug repurposing: the identification of new applications for existing, clinically approved drugs. Drug repurposing possesses several inherent advantages in the context of cancer treatment since repurposed drugs are typically cost-effective, proven to be safe, and can significantly expedite the drug development process due to their already established safety profiles. In light of this, the present review offers a comprehensive overview of the various methods employed in drug repurposing, specifically focusing on the repurposing of drugs to treat cancer. We describe the antitumor properties of candidate drugs, and discuss in detail how they target both the hallmarks of cancer in tumor cells and the surrounding tumor microenvironment. In addition, we examine the innovative strategy of integrating drug repurposing with nanotechnology to enhance topical drug delivery. We also emphasize the critical role that repurposed drugs can play when used as part of a combination therapy regimen. To conclude, we outline the challenges associated with repurposing drugs and consider the future prospects of these repurposed drugs transitioning into clinical application.
Similar content being viewed by others

Introduction
Cancer remains a leading cause of death worldwide, posing a significant burden on global health.1,2 The high incidence of cancer may be caused by several factors, such as genetic mutations, environmental factors, insufficient physical activity, diverse lifestyles, unstable behaviors related to diet, smoking, and alcohol consumption.3,4,5,6,7 The current treatment methods for different stages of various cancers include chemotherapy, radiation therapy, and surgical procedures for solid tumors, or a combination of the above.8 Although these different treatment modalities can effectively reduce cancer, patients may also experience side effects. Radiation therapy runs the risk of causing DNA damage in surrounding healthy cells, which could potentially lead to new incidences of cancer.9 Similarly, although surgical intervention—the primary treatment for solid tumors—significantly improves patient survival, its success rate depends on the expertise of the surgeon and the availability of screening methods, including hospital imaging equipment.10 The introduction of chemotherapy was a milestone in cancer treatment. However, prolonged use of chemotherapy drugs, especially those affecting tumor cell metabolic pathways and signal transduction, can influence tumor occurrence, metastasis, drug response, recurrence, drug resistance, and cancer stem cells (CSCs).11,12 Therefore, there remains an urgent need to develop novel treatment strategies with high anti-tumor efficacy and minimal side effects.
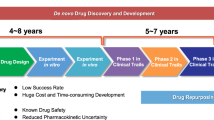
Traditionally, drug development involves preclinical research and clinical trials. Preclinical studies involve testing the efficacy, toxicity, pharmacokinetics, and pharmacodynamics of drugs in human tumor cells and animal models. Once the therapeutic efficacy of a drug has been determined, the drug moves into the clinical trial phase, which includes Phase I, II, and III human clinical trials, to determine the safety and effectiveness of the drug. As such, it takes 10–15 years and costs $1–2 billion to produce a new drug approved for clinical use. Despite these investments, less than 1% of compounds are expected to enter clinical trials, let alone reach the market.13,14,15 The strategies of drug repurposing involve exploring new therapeutic applications for drugs that have already been approved. Drugs that were originally approved for one indication and have since been studied and used to treat different medical conditions are gaining prominence. This approach is exemplified in the comprehensive review by Kirtonia et al., which underscores the innovative methodologies and potential transformative impact of drug repurposing specifically in the field of oncology.16 Drug repurposing has several inherent advantages including a faster and more cost-efficient drug development time due to prior knowledge about the safety, dosage, and toxicity profiles of existing medications. In recent years, the interest in drug repurposing has risen. Successful candidates including chlorambucil and bufulfone were originally developed as alkylating agents based on the toxic chemical warfare agent mustard gas but were later found to be effective for treating leukemias.17 Similarly, thalidomide, despite its infamous history of causing severe birth defects, has been repurposed to treat conditions such as leprosy and multiple myeloma.18 In addition, arsenic trioxide (a poison) and all-trans retinoic acid (a metabolite of vitamin A) are examples of other chemical compounds19 that were approved by the FDA in 2000 for the treatment of acute promyelocytic leukemia. Thus, drug repurposing may be a compelling and viable strategy for enhancing cancer treatment options.20 In this review, we embark on a comprehensive examination of drug repurposing as a potential strategy for the treatment of cancer. We begin with an introduction to the definition and background of drug repurposing, setting the stage for its potential use in cancer treatment. To provide a context for the application of repurposed drugs, we offer an in-depth discussion of the epidemiology and current treatment landscape of cancer, including a detailed overview of the 14 updated hallmarks of cancer, which serve as critical targets for novel therapeutic strategies. We then discuss the molecular mechanisms through which repurposed drugs exert their antitumor effects, focusing on their role in regulating different aspects of the tumor microenvironment (TME). Next, we summarize the innovative application of nanomaterials to enhance the delivery of repurposed drugs, shedding light on this expanding area of research. We also identify and discuss the many obstacles and challenges faced during the bench-to-bedside translation of repurposed drugs. Finally, we emphasize the transformative potential of drug repurposing in advancing the field of tumor treatment and highlight its ability to introduce more effective and less toxic therapeutic options for cancer patients.
Drug repurposing strategies
In summary, drug repurposing can be divided into three stages: identifying the core targets of the disease (hypothesis generation), determining the efficacy of the drug through in vitro and in vivo models, and proceeding to phase II clinical trials in cases where phase I trials have yielded adequate data.21,22,23 The inception stage is critical since hypothesis generation is the key to any drug repurposing endeavor.24 Historically, drug repurposing in oncology has largely been driven by either an understanding of the disease pathways or through serendipitous findings. Thus, designing innovative strategies to match existing drugs with newfound applications could increase the success of drug repurposing. Identification of a potential repurposed drug can be made using computational and experimental methods. The experimental approach considers tools such as induced pluripotent stem cell models and function-first phenotypic screenings (or reverse chemical biology),25,26 while computational methods use target-centric, knowledge-driven, signature-aligned, pathway-focused, and mechanism-specific strategies.27,28 More often, these techniques are synergistically utilized. Notably, high-throughput screening using sophisticated models can identify compounds that mitigate disease symptoms without necessitating pre-existing knowledge about the drug-target interactions.29,30 Current computational methodologies, such as merging drug effects with clinical disease signatures and model systems that predict disease-modifying effects, are available for the selection of drug candidates suitable for drug repurposing in cancer. These tools can identify ligands, decode drug ingredient binding schemas, and highlight promising candidates from an expansive list of potential compounds.27,31,32 In summary, although the idea of drug repurposing is long-established, it is only recently that technological advances, such as the ones outlined in this article, have led to the development of cutting-edge strategies that can be consciously paired with novel indications.
Experimental approaches
Organoid models of cancer
Organoids are classified as “stem cell-containing self-organizing structures”, while tumoroids are a special type of cancer organoid.33 Organoids are in vitro tissues that originate from human stem cells, organ-specific progenitor cells, or even disassociated tumor tissues and are cultured in specialized ECM-based medium with relatively high success rates. Tumoroids mimic the primary tissue in both architecture and function and retain the histopathological features, genetic profile, mutational landscape, and even responses to therapy.34 The use of tumoroids is expanding, and their utility for basic research and early steps of drug development has been recognized.35 Cisplatin, for example, has been found to be less effective in patient-derived organoids (PDOs) generated from non-small cell lung cancer (NSCLC) tissues than from cell lines, highlighting the ability of patient-derived material to provide important information on potential resistance mechanisms.36 With respect to gastrointestinal cancers, several investigations have harnessed PDOs as tools to evaluate drugs and identify potential therapeutic routes.37,38 Such models have adeptly mirrored the feasibility of tumoroids in the accurate recapitulation of KRAS-mutant metastatic rectal cancer with microsatellite stability after hepatic resection and treatment with neoadjuvant combination chemotherapies in colorectal cancer (CRC),39 as well as gauged drug reactions in hepatocellular carcinoma (HCC)40,41 and mimicked treatment resistance patterns observed in esophageal squamous cell carcinoma.42
In addition, tumoroid models present a distinct advantage in cancer drug screening due to their ability to emulate the structure, gene expression patterns, and essential characteristics and functionalities of their originating organs (Fig. 1). For example, SMAC mimetics such as LCL161 have been studied in hepatic metastatic rectal cancer,39 while novel CDK7 inhibitors like YPN-005 have been analyzed in SCLC tumoroid systems.43 A high-throughput screening, based on the interaction between patient-derived breast cancer organoids and tumor-specific cytotoxic T cells, identified three epigenetic inhibitors - BML-210, GSK-LSD1, and CUDC-101—that displayed significant antitumor effects.44 In addition, the drug atorvastatin was found to inhibit angiogenesis in a dose-dependent manner through the downregulation of vascular endothelial growth factor (VEGF), CD31, and Bcl-2 in a co-culture of glioblastoma organoids and human umbilical vein endothelial cells in fibrin gels, indicating that atorvastatin may be a promising drug for the treatment of glioblastoma.45 Finally, a 2019 research study demonstrated the predictive potential of PDOs for personalized medicine, using a biobank of PDOs sourced from cancer patients participating in phase I/II clinical trials.46

Tumoroids model in drug repurposing. a Schematic showing the generation of patient-derived organoids (PDOs) from a cancer biopsy: enzymatic digestion, embedding in extracellular matrix, addition of growth medium and cancer tumoroids enrichment by media compound withdrawal and/or addition of mutation related inhibitors. b, c The tumoroid model is used to screen drug repurposing candidates, resulting in the identification of drugs for preclinical and clinical testing. This figure was created with Biorender.com
Tumoroids accurately model human primary tumors, positioning them as an invaluable platform for both foundational research and translational medicine. This includes their use in cancer models to study tumorigenesis and cancer progression, as well as in the prediction of drug responses, treatment optimization, and the discovery of novel anticancer therapeutics. Despite these advantages, current tumoroid systems are not without limitations. A primary concern is that the tissue samples used to create organoids represent only a fraction of the entire tumor. Given the substantial heterogeneity inherent in tumors, the reliability of using small tissue samples to effectively represent the entire tumor mass is questionable. Thus, caution is required when extrapolating tumoroid data to the whole tumor. In addition, tumoroid models often lack key non-tumorous cellular components, such as mesenchymal tissues, neural cells, and immune cells. The absence of these cell types in tumoroids limits their ability to fully mimic the complex structure and functionality of their corresponding organs. In particular, the imprecise modeling of the tumor immune environment significantly impedes the utility of tumoroids in both translational and precision medicine applications. Another major challenge is the vascularization of tumoroids.47 Effective vascularization is crucial for accurately replicating tumor biology, yet it remains an unresolved issue in the development of tumoroids. Furthermore, standardization of specific tumoroid culture conditions is essential for enhancing reproducibility on a large scale and facilitating the application of tumoroid technology in high-throughput drug screening. The specific culture conditions required for cancer organoids, if not meticulously managed, can lead to a reduction in the intrinsic diversity within tumors over prolonged cultivation periods. In summary, while tumoroid models offer significant insights and advances in cancer research and treatment development, addressing these challenges is crucial for maximizing their potential and applicability in advanced cancer research and personalized medicine.48,49
Phenotypic analysis
Several drugs that exhibit potent off-target effects (side effects) in cancer are also worth exploring. These off-target effects can be viewed as new indications of the drug for other diseases (phenotypic analysis).25 Phenotypic screening is a strategy that analyzes biology-associated (phenotypic) effects in given models such as animals, cells, or organisms to help identify the targets of candidate drugs.25,50 A 96-well or 384-well format is typically utilized for in vitro phenotypic screening.51 Previous studies have identified repurposed drugs by conducting high-throughput cell-based screening, using some ‘classical’ hallmarks of in vitro phenotypes including sustained proliferation, increased angiogenesis, and resistance to cell death.25 For example, Jacquemet et al. utilized a phenotypic screen to identify FDA-approved calcium channel blockers as potent inhibitors of filopodia formation in cancer cells. Cancer cells expressing MYO10-GEP were treated with different drugs from the compound library. From this screen, L-type calcium channel blockers, such as amlodipine besylate, felodipine, diclomanidipine, and cilidipine, were found to inhibit filopodia formation and prevent cancer cell invasion, thereby highlighting the importance of L-type calcium channels in regulating calcium entry and filamentous pore stabilization.52 Thus, phenotypic screening and identifying drug candidates with yet-to-be-identified targets can economize both time and resources in the drug discovery process, as well as minimize premature clinical trial setbacks.
Computational approaches
Computational methodology has emerged as a powerful tool in the field of drug repurposing.53,54 Our understanding of the mechanisms and modes of action within oncology has deepened substantially with the increase in omics technologies coupled with breakthroughs in big data analytics, machine learning, and computational algorithms. These computational techniques grant expansive access to both disease-centric and drug-centric data.55,56 Several computer-assisted drug repurposing strategies such as molecular docking, network analysis, data mining, similarity analysis, machine learning, and transcriptional signature techniques, are readily available to researchers.57,58 Through these computational approaches, we can delve further into the anticancer prospects of drug repurposing and provide disease-related data for the repurposing of drugs.59,60 The identification of oncogenic pathway inhibitor activity via computer-aided drug repurposing approaches also represents a robust method.61,62 Researchers can exploit multiple databases for extensive analysis of drug bioinformatics (Table 1). And repurposed drugs identified by network-centric systems biological approaches are shown in Table 2. These repositories not only amplify the therapeutic potential of repurposed drugs across various diseases,63,64 but also strengthen chemotherapeutic strategies, providing novel strategies to reduce the development of resistance and tailor treatments to maximize patient-specific outcomes.65,66
Drug repurposing: candidates for the therapeutic targeting of hallmarks of cancer
The hallmarks of cancer are fundamental characteristics that drive the development and progression of cancer. Initially proposed by Hanahan and Weinberg in 2000, this concept has since been expanded to encompass 14 distinct hallmarks of cancer.67,68 Understanding these hallmarks is pivotal for developing effective strategies for cancer prevention, diagnosis, and treatment. Indeed, targeting the hallmarks of cancer has emerged as a promising approach towards the development of novel therapies that strike at the root causes of the disease, offering the potential for durable and transformative patient benefits.69 This approach uses the known safety and pharmacologic profiles of existing drugs to potentially accelerate the development of effective and affordable cancer therapeutics. Here, we systematically review the current progress in this area of research and provide representative examples for each of the hallmarks of cancer (Fig. 2), specifically describing how drug repurposing could be used to target these hallmarks. This provides a comprehensive background for further investigations into the potential of drug repurposing in cancer treatment, and a strong theoretical foundation that could guide the identification of promising new drug candidates.

Diverse cancer hallmarks targeted by repurposed non-oncology drugs. Repurposed non-oncology candidates have shown great promise against cancer by targeting different hallmarks of cancer including sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, genome instability and mutation, tumor-promoting inflammation, reprogramming energy metabolism, evading immune destruction, unlocking phenotypic plasticity, non-mutational epigenetic regulation, polymorphic microbiomes, and senescent cells. This figure was created with Biorender.com
Inhibiting proliferative signaling
Cancer cells are characterized by their inherent ability to sustain chronic proliferation, which effectively enables them to become self-sufficient in growth signaling and to control their own fate. Uncontrolled proliferation is primarily facilitated by deregulation of the production and release of growth-promoting signals.69 Growth-promoting signals are predominantly transmitted via growth factors that bind to cell-surface receptors, which typically contain intracellular tyrosine kinase domains.70 The phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), mammalian target of rapamycin (mTOR),71 and mitogen-activated protein kinases/extracellular signal-regulated kinase (MAPK/ERK) pathways have all been implicated in sustaining proliferative signaling. However, while these pathways are significant, they represent only a subset of the pathways involved in cancer cell proliferation.72,73 In the current landscape of cancer treatment, an increasing number of drugs, originally developed for non-oncological conditions, are being repurposed to target these signaling pathways. Such approaches exemplify the creative and adaptive strategies being undertaken to combat the complexity and adaptability of cancer.
Salidroside
Salidroside, an active compound isolated from the dried roots, rhizomes, and entire plants of Rhodiola rosea, has attracted recent attention due to its wide-ranging pharmacological activities, including anti-hypoxic, anti-aging, immune-enhancing, and anti-fibrotic properties.74,75,76,77,78 Among these diverse effects, the ability of salidroside to act as an anticancer agent is of interest. For example, salidroside treatment has been shown to inhibit the proliferation of nasopharyngeal carcinoma (NPC) cells, including the CNE2 and HONE cell lines, through regulation of the miR-4262/GRP78 axis.79 Similarly, Liu et al. showed that salidroside suppressed proliferation, colony formation, and migration of the PC3 and DU145 prostate cancer cell lines in a dose-dependent manner by inhibiting the PI3K/AKT pathway.80 In addition, salidroside was found to impede cancer cell proliferation in HeLa cells and a subcutaneous HeLa-ADR-luc (doxorubicin-resistant derived HeLa-cell lines) cell xenograft mouse model through the activation of apoptosis and inhibition of the PI3K/ Akt/HIF-1α signaling pathway.81 These preclinical findings suggest that repurposing salidroside as a potential anticancer agent is worthwhile. However, due to the limited clinical research on salidroside, there remains a critical need for further clinical trials to validate these effects and facilitate the translation of salidroside into a viable treatment option in clinical practice.
Inducing cell death
Cell death is a critical process in biological systems that is not only essential for the maintenance of correct physiological development and tissue homeostasis, but also acts as a natural defense mechanism against tumor formation. Currently, cell death can be classified as accidental cell death (ACD) and regulated cell death (RCD). Unlike ACD, which is generally an uncontrolled process, RCD is more organized and involves genetically encoded molecular mechanisms that help to maintain a stable internal environment.82 RCD can be further subdivided into apoptotic and non-apoptotic subcategories.83 Since RCD has a fundamental role in cellular regulation and can act as a barrier to tumorigenesis, targeting RCD pathways through drug repurposing may be a promising strategy to impede the development and progression of tumors (Fig. 3). Accumulating evidence has highlighted the potential of using repurposed drugs to restrict tumor growth. These drugs have been shown to activate various RCD pathways, including apoptosis, necroptosis, pyroptosis, autophagy, ferroptosis, and cuproptosis. It is important to note, however, that since cuproptosis is a newer area of study, no associated preclinical trials are currently underway.

Inducing cell death in cancers by repurposed non-oncology drugs. Regulated cell death (RCD) is a critical and active process that is controlled by specific signal transduction pathways and can be regulated by drug interventions. Repurposed non-oncology candidates can exert anticancer effects by inducing classical apoptosis and other RCD processes, such as ferroptosis, autophagy, necroptosis and pyroptosis. This figure was created with Biorender.com
Triptolide
Triptolide (TPL) was first isolated in 1972 from a perennial vine-like herb called Thunder God Vine.84 Since then, its mechanism of action and pharmacological properties have been extensively researched to reveal prominent anti-rheumatic, anti-bacterial, anti-inflammatory, and immunomodulatory activities. More recent studies have also demonstrated that TPL possesses anti-tumor properties.85,86,87,88 Specifically, cells exposed to TPL were found to undergo non-apoptotic cell death. Treatment with TPL resulted in morphological alterations characterized by cytoplasmic swelling and membrane disruption, as well as a marked elevation in the mRNA and protein expression of gasdermin E (GSDME) and GSDMB. Selective inhibition of GSDME was found to counteract TPL-induced cell death and mitigated cytoplasmic swelling and membrane disruption.89 In addition to pyroptosis, Wang and colleagues also found that the use of TPL-loaded polyethylene glycol (PEG) nanocarriers induced necrosis and enhanced the sensitivity of MIA PaCa-2 (pancreatic ductal adenocarcinoma) tumors to gemcitabine.90 Furthermore, TPL has been shown to induce autophagy-mediated caspase-independent cell death in tumor cells. One study showed that in prostate cancer cells, TPL-based drugs stimulated the release of free calcium, promoted endoplasmic reticulum stress and induced activation of the CAMKKb-AMPK signaling pathway, leading to inhibition of mTOR and activation of beclin-1 and Unc-51-like kinase 1 (ULK1), thereby promoting autophagy.91 Currently, a clinical trial has investigated the safety and anticancer efficacy of the TPL derivative F60008 in patients with advanced solid tumors. However, due to significant variability among patients and high toxicity in some, F60008 cannot be considered as an appropriate derivative of TPL for cancer patients.92 Minnelide, another derivative of TPL, is presently under investigation for the treatment of refractory advanced pancreatic adenocarcinoma, either alone or in combination with paclitaxel (PAX) (NCT04896073 and NCT03117920).
Tanshinone IIA
Tanshinone IIA, an active compound extracted from Salvia Miltiorrhiza (Danshen), is traditionally used as a low-cost and safe treatment for various ailments, including cardiovascular and cerebrovascular diseases.93 However, more recently, Tanshinone IIA has been shown to induce various types of cell death in cancer cells and may therefore be a potential anti-tumor agent. For example, Tanshinone IIA has been shown to exert anti-neoplastic effects in renal cell carcinoma (RCC) cells through the down-regulation of β-catenin, which results in the formation of autophagic vacuoles such as autophagosomes and autolysosomes, increased apoptosis, and the induction of autophagic cell death.94 In addition, Tanshinone IIA has been shown to induce ferroptosis in gastric cancer (GC) cells through p53-mediated down-regulation of solute carrier family 7 member 11 (SLC7A11), leading to elevated levels of reactive oxygen species (ROS), lipid peroxidation, and iron accumulation, and effectively inhibiting tumor cell growth both in vitro and in vivo.95 Interestingly, Lin et al. demonstrated that Tanshinone IIA might also induce necroptosis in human HCC by promoting the formation of a necrosomal complex composed of receptor-interacting protein 1 (RIP1)/RIP3.96 Due to its multifaceted effects on cell death pathways, minimal resistance to its targets, and traditional use as a safe compound, Tanshinone IIA is emerging as a compelling candidate for anti-tumor therapies. Thus, a deeper understanding of its mechanisms of action in the context of cancer treatment is critical.
Regulation of cellular metabolism
The rapid proliferation of cancer cells is sustained through corresponding adaptations in tumor metabolism, which involve the activation or modification of metabolic pathways to harness more energy.97 Deregulating cellular energetics has recently been added as a new hallmark of cancer.98 Current research on metabolic reprogramming has primarily focused on the aberrant activation of the PI3K/AKT/mTOR pathway, as well as activation of oncoproteins such as MYC, RAS, pyruvate kinase M2 (PKM2), and hypoxia-inducible factor 1 (HIF-1). In addition, the role of mutations or deactivation of tumor suppressor genes, including P53 and phosphatase and tensin homolog (PTEN) are being explored.97,99,100,101 In the past decade, only a few metabolism-based cancer drugs have been successfully developed, some of which are in or nearing clinical trials. Meanwhile, repurposed drugs have been extensively examined in preclinical studies as a potential means of targeting key pathways in malignant metabolism. Thus, using repurposed drugs to target essential pathways in tumor metabolism presents a promising therapeutic strategy.
Leflunomide
Leflunomide, an immunomodulatory drug primarily prescribed for rheumatoid arthritis and psoriatic arthritis, has seen a resurgence in research interest due to its potential anticancer properties.102 Mechanistically, the metabolic effects of leflunomide stem from its active metabolite, A77 1726, which inhibits the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH). In the 1990s, leflunomide was evaluated as an epidermal growth factor receptor (EGFR) inhibitor with potential anticancer applications.103,104 Recent studies have highlighted its potential in treating breast and prostate cancer.105,106 In addition, Yamaguchi and colleagues proposed that leflunomide-mediated pyrimidine synthesis could be a therapeutic target for mitigating the metastatic progression of CRC. Further studies have suggested that the action of leflunomide on DHODH, combined with its disruption of de novo pyrimidine biosynthesis, can induce apoptosis in CRC cells that express transcriptionally active P53. These effects appear to be linked to inhibition of the electron transport chain complex III.107 Leflunomide has also been shown to suppress melanoma growth by impeding the effective transcription elongation of requisite genes. In human A375 melanoma cells, for example, nucleotide depletion by leflunomide reduced the chromatin occupancy of the RNA helicase protein DDX21. Combination therapy using leflunomide with checkpoint kinase 1 (CHK1) inhibitors has shown enhanced efficacy in reducing the growth of P53-deficient breast tumors and inducing cell apoptosis compared to treatment with leflunomide alone.108 Furthermore, in a phase I clinical trial aimed at refractory multiple myeloma, leflunomide displayed manageable side effects, with disease stabilization occurring in 9 of the 11 patients (NCT: NCT02509052). Collectively, these findings indicate that leflunomide is a promising candidate for broader cancer therapy applications.
Disulfiram
Disulfiram (DSF), also known by its trade name Antabuse, was originally approved by the FDA in 1951 as a treatment for alcoholism.109 The anticancer properties of DSF were serendipitously discovered in 1977 when Lewison reported the drug’s potential to inhibit bone metastasis in breast cancer patients.110 This initial observation has since been substantiated through extensive research, including analyses of Danish demographic and health registries that observed lower mortality rates for colon, prostate, and breast cancers among ongoing DSF users compared to former users.111 Notably, DSF has recently attracted attention for its potent anticancer effects and its capacity to modulate cellular energy metabolism. For example, Du et al. found that DSF inhibited the glycolysis of cancer cells in a copper-dependent manner. Furthermore, a combination of DSF and copper was shown to significantly reduce the expression levels of key molecules, including S6K1, MYC, and their downstream targets, glucose transporter 1 (GLUT1), PKM2, and lactate dehydrogenase A (LDHA), which are integral to the regulation of critical cellular processes, such as apoptosis, cell differentiation, and metabolism.112 DSF has also been reported to augment oxidative metabolism within thyroid cancer cells, primarily by increasing ROS production, which, in turn, triggers apoptosis in an ROS-dependent manner.113 Given its anti-tumor properties and metabolic modulating potential, DSF is a promising drug to include in combination therapy strategies using repurposed drugs.
Activating antitumor immunity
In the early stages of tumorigenesis, the body’s lymphocytes, including cytotoxic T cells and natural killer (NK) cells, actively target and aim to eliminate emerging cancer cells through the secretion of perforin and granzyme to induce apoptosis or through activation of the death ligand/death receptor pathway. Perforin creates pores in the membranes of the target cells, allowing granzymes to enter and subsequently initiate apoptosis.114 However, during tumor progression, the immunosuppressive mechanisms within the TME become more pronounced. For example, tumor cells may begin to express programmed death ligand 1 (PD-L1), a ligand that binds to the PD-1 protein on NK cells and T cells, thereby inhibiting their activity. In addition, the emergence of suppressive immune cellular populations further limits the effectiveness of the body’s natural antitumor immunity.115,116,117 In light of these challenges, an increasing number of immunotherapies have been developed and used to treat tumors. Notable among these are adoptive cell transfer (ACT) and immune checkpoint inhibitors (ICIs), both of which aim to strengthen the body’s immune response to cancer. Despite their potential, these strategies are not universally effective across all patient populations. Thus, there is growing interest in identifying repurposed drugs that have the potential to activate antitumor immunity, thereby providing additional strategies to enhance cancer treatment outcomes.
Oleanolic acid
Oleanolic acid (OA, 3β-hydroxyolean-12-en-28-oic acid) is a pentacyclic triterpenoid derived from the Oleaceae family that is prevalent in many dietary and medicinal plants and possesses anti-diabetic,118 anti-bacterial,119 anti-parasitic,120 and anticancer properties.121 The recent interest in OA is largely due to its effects on antitumor immunity, which position it as a potential candidate for cancer treatment through drug repurposing. For example, OA was found to promote the balance of regulatory T cells (Tregs)/Th17 cells in GC by targeting interleukin-6 (IL-6) via the miR-98-5p pathway.122 OA has also emerged as an epigenetic modulator in immunotherapy for cancer. OA was shown to inhibit the IL-1/NF-κB/TET3 axis in cancer cells, resulting in DNA hypomethylation and the suppression of PD-L1, thereby strengthening the robust T-cell defense mechanism.123 In addition, synthetic derivatives of OA, such as CDDO-Im, have been shown to block the EGFR/signal transducer and activator of transcription 3 (STAT3)/Sox-2 signaling pathway in tumor-associated macrophages (TAMs), which have been implicated in promoting breast cancer proliferation and metastasis.124 Thus, OA may be a potential therapeutic agent for the treatment of cancer. In conclusion, although clinical studies examining the role of OA in cancer are limited, existing preclinical evidence indicates that OA may be a suitable drug candidate to treat cancer, and may have the potential to increase the therapeutic outcomes of present-day immunotherapies.
Reactivating growth suppressors
Tumor suppressor genes, such as P53 and retinoblastoma protein (RB), are crucial regulators in cancer progression.125,126,127 Specifically, P53 acts as a sentinel, responding to intracellular disturbances like metabolic and oxidative stress, and inducing cell cycle arrest until the TME returns to a balanced state.128,129 However, some tumors are able to bypass these suppressor genes or deactivate pivotal tumor suppressors. In these cases, repurposed drugs may be able to target cancer cells that have avoided suppressor gene regulation, providing potential therapeutic benefits.
Statins
Statins lower circulating blood lipids including low-density lipoprotein (LDL) cholesterol through the competitive inhibition of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), an enzyme that facilitates the conversion of HMG-CoA to mevalonic acid, which is a crucial step in cholesterol biosynthesis. By inhibiting this process, statins not only reduce cholesterol production but also impact by-products essential for cancer cell growth, thereby demonstrating their potential as anticancer agents.130,131 Of the statins, simvastatin is particularly interesting due to its anticancer applications, which have been observed in various cancer types and are largely mediated through activation of mutant P53. Specifically, simvastatin was found to reduce the migratory and invasive abilities of human epithelial MDA-MB-231 breast cancer cells in vitro by increasing mutant P53 expression and repressing expression of the stem cell marker CD44, which is essential for cell migration. Similarly, in MDA-MB231 mouse xenograft models, simvastatin treatment led to elevated P53 and reduced CD44 expression levels.132 In parallel studies, Miyajima and colleagues found that simvastatin and fluvastatin activated the transcriptional function of P53 by suppressing TAZ protein expression. When used in combination with nutlin-3, simvastatin was found to reduce cell viability through the suppression of MDM2 and activation of wild-type P53.133 Moreover, certain statins, notably simvastatin and atorvastatin, have been shown to downregulate MCM7 and RB expression. These statins have been linked to an increase in chromosomal abnormalities in RB-deficient tumor cells, hinting at their potential application for the treatment of RB-deficient tumors.134 Ongoing clinical trials are currently examining the efficacy of simvastatin against various cancers, including breast (NCT00807950, NCT05550415), gastric (NCT01099085, NCT03086291), colorectal (NCT01238094), and bladder (NCT02360618) cancer. Although the specifics of its mechanism, optimal dosage, and compatibility with other anticancer drugs remain unclear, we anticipate the emergence of novel strategies employing statins in cancer treatment in the future.
Interfering with replication
Unlike normal cells, cancer cells display elevated levels of the specialized DNA polymerase, telomerase, which provides tumor cells with the unique ability to evade the usual cellular growth and division constraints dictated by senescence and crisis/apoptosis mechanisms.135,136 In addition, human telomerase reverse transcriptase (hTERT) plays an integral role in cancer signaling pathways as a transcriptional regulator, orchestrating the activation of crucial genes that are vital for tumor proliferation and survival. The noncanonical roles of hTERT in cancer progression involve the WNT/β‐catenin137,138 and nuclear factor‐κB (NF-κB) signaling pathways.139,140 hTERT-dependent transcription mediated through these pathways allows tumor cells to have unlimited replication capabilities. Thus, telomerase has emerged as a compelling target for repurposed drugs, potentially bolstering the efficacy of cancer therapies.
Epigallocatechin 3-gallate
Epigallocatechin 3-gallate (EGCG), a compound synthesized from epicatechin and gallic acid, has garnered considerable attention in the scientific community due to its multifaceted biological and pharmacological properties. These include its anti-oxidant, anti-inflammatory, anti-angiogenic, anti-proliferative, pro-apoptotic, and anti-metastatic functions. Numerous studies, both in vitro using various cancer cell lines and in vivo in animal models, have consistently demonstrated the ability of EGCG to inhibit the initiation, promotion, and progression of different types of tumors.141,142,143,144,145 The ability of EGCG to act as a potent inhibitor of telomerase, an enzyme associated with cellular aging and cancer, is of particular interest. For example, EGCG was shown to induce apoptosis in Hep-2 cells in a dose-dependent manner through the inhibition of telomerase. Similarly, EGCG was found to induce apoptosis in T47D breast cancer cells via inhibition of the telomerase and PI3K/AKT pathways and simultaneous upregulation of the P53 and Bax/Bcl-2 pathways. Impressively, EGCG was able to induce apoptosis in cancer cells without manifesting notable toxicity to healthy cells.146 Recently, Dong and colleagues reported the nuanced relationship between EGCG and the WNT/β-catenin signaling pathway by demonstrating that overexpression of β-catenin could either augment or reduce the anticancer effects of EGCG.147 In addition, another study found that EGCG was involved in reducing the mRNA expression and transcriptional activity of β-catenin in wild-type P53-expressing KB cells. When used in combination with gemcitabine, EGCG was found to exert stronger inhibitory effects on β-catenin and N-cadherin in pancreatic cancer cells.145 Given these promising insights, a phase I clinical trial (NCT00516243) has been initiated that targets women with hormone receptor-negative stages I-III breast cancer and aims to explore the safety and effectiveness of EGCG. Concurrently, several clinical trials for CRC (NCT02321969 and NCT01360320) are also in progress. However, while these studies are promising, the potential therapeutic application of EGCG in cancer treatment is still restricted by its limited bioavailability.
Decreasing angiogenesis
An “angiogenic switch” is activated temporarily in normal tissue during physiological processes such as wound healing to promote angiogenesis, which accelerates tissue repair. In contrast, during tumor progression, the “angiogenic switch” is activated and remains switched on. Persistent activation promotes the formation of new blood vessels, which not only sustain the growth of neoplastic tissues but also paves the way for tumor invasion and metastasis.148,149 The induction of chronic angiogenesis is now acknowledged as a hallmark of cancer. Many of the antiangiogenic agents currently in use, such as bevacizumab, target VEGF or are tyrosine kinase inhibitors that target VEGF receptors (VEGFRs). However, these agents have limitations. For example, sometimes these drugs induce states of stress resistance, which reduce their efficacy. Overall, the strategy of inhibiting angiogenesis via drug repurposing has recently become an area of interest and is worthwhile exploring as a potential target.
Artemisinin
Artemisinin, derived from the plant Artemisia annua, is a sesquiterpene lactone with a distinct peroxide linkage that has been used as an anti-malarial agent.150 Dihydroartemisinin (DHA), a reduced lactol semi-synthetic derivative of artemisinin, boasts an impressive safety record. Interestingly, preclinical and clinical studies have shown that both artemisinin and DHA possess promising anticancer properties in various therapeutic strategies. The endoperoxide bridge, the active moiety of artemisinin derivatives, is cleaved in DHA in the presence of iron ions, leading to the release of cytotoxic ROS, which is thought to be the pivotal mechanism behind the anti-malarial and antitumor properties of DHA.151 In addition, neither artemisinin nor DHA have shown substantial toxicity towards normal cells, highlighting their potential to act as suitable anticancer candidates. Recently, an increasing number of studies have demonstrated that DHA also exhibits multi-target anti-angiogenic effects by modulating various angiogenesis signaling pathways. The ability of DHA to markedly inhibit proliferation, migration, and tube formation in human umbilical vein endothelial cells (HUVECs) has been recently reported, while Wang and colleagues found that DHA not only reduced angiogenesis in pancreatic cancer cells but also suppressed NF-κB-DNA binding activity, subsequently downregulating pro-angiogenic genes.152 In addition, DHA has been shown to trigger autophagy in HUVECs through suppression of the AKT/mTOR signaling pathway. Interestingly, DHA was also found to enhance VEGFR1 expression through upregulation of ETS-1 transcription factor. With such promising preliminary findings, a Phase II clinical trial (NCT03402464) is currently underway to examine the combined efficacy of DHA with a standard chemotherapy agent, erlotinib, specifically for patients diagnosed with EGFR-mutated lung adenocarcinoma. While these studies have shed new light on the potential of DHA to act as an anticancer agent, more comprehensive clinical trials are necessary in the future to solidify its place in cancer therapy.
Suppressing invasion and metastasis
Tumor invasion and metastasis contribute to increased tumor-related mortality. Central to these processes is the aberrant activation of the epithelial-mesenchymal transition (EMT) in cancer cells. EMT is regulated by a complex interplay of signaling pathways, including but not limited to the transforming growth factor beta (TGF-β), WNT, NOTCH, and PI3K-AKT pathways.153,154 Accumulating evidence has indicated that repurposed drugs may have a significant role in targeting these pivotal signaling pathways, and may, therefore, exhibit potential antitumor effects across diverse metastatic cancer models.
Mebendazole
Mebendazole (MBZ; 5-benzoyl-1H-benzimidazol-2-ylcarbamate) first described in 1968, was initially recognized as a broad-spectrum anthelmintic agent and was applied to humans in 1971.155 Fast forward two decades, and the focus on anthelmintics shifted towards their potential anticancer properties, primarily due to their interactions with microtubules.156,157,158,159 MBZ has been shown to potentially suppress tumor growth in various cancer cell lines and animal models through the inhibition of microtubule polymerization, a process that, when interrupted, can lead to the death of rapidly dividing cells. Significantly, the anticancer effects of MBZ extend to inhibiting the invasion and metastasis of malignant tumors. One study, in particular, highlighted the ability of MBZ to decrease integrin β4 expression and reduce CSC-like properties, which led to the shrinkage of primary tumors and a reduced risk of metastases, notably to the lungs and liver.160 MBZ has also been shown to inhibit tumor growth via the JAK2/STAT3/Bcl-2 signaling pathway, while other studies have indicated that MBZ might modulate cancer cell migration through the S1P/FAK/vimentin pathway.161 MBZ has also been found to restrict the migratory and invasive tendencies of glioblastoma cells, and concurrently modulate pivotal markers in the EMT, suggesting a potential role for MBZ in mitigating glioblastoma metastasis. In oral squamous cell carcinoma, MBZ was found to downregulate specific proteins and enzymes, including FAK, Rho-A, and Rac1 GTPase. Moreover, in the TGF-β-induced dysplastic oral keratinocyte (DOK) cell line, which models EMT, MBZ was shown to disrupt the cadherin equilibrium, further accentuating its potential as an anticancer agent.162,163 Recent anecdotal evidence from two case reports has further supported the possibility of MBZ being repurposed as an anticancer drug by documenting its success in managing metastatic patients.164,165 Together, these findings demonstrate the critical need to learn more about the therapeutic profile of MBZ to ensure its safety in oncological applications and determine its efficacy as a groundbreaking anticancer treatment.
DNA damage response
The DNA damage response (DDR), which is activated in response to numerous DNA damage events including germline or somatic defects in DNA repair,166 oncogene-induced replication stress,69 flawed mitotic chromosome segregation,167 clashes between replication and transcription machinery168 or even as a consequence of genotoxic anticancer treatments,169 is a crucial hallmark of cancer. The DDR is instrumental not just during the onset and development of cancer, but also in its treatment. Although the majority of studies on DDR have focused on the role of poly (ADP-ribose) polymerase (PARP), the development of resistance to PARP inhibitors is becoming increasingly problematic in clinical settings since resistance exacerbates disease recurrence and worsens patient prognosis. Based on these findings, studies have shifted focus to other DDR targets, including ataxia-telangiectasia mutated kinase, ataxia telangiectasia and Rad3-related kinase, CHK1, and protein kinase, membrane-associated tyrosine/threonine 1. The use of repurposed drugs to target these entities presents a promising alternative strategy to combat genomic instability.
Genistein
Genistein (4’,5,7-trihydroxyisoflavone) is a naturally occurring isoflavone that is found in a vast range of foods.170 Notably, the median daily intake of isoflavones for adults in Japan and China is estimated to be between 25 and 50 mg, which is significantly higher than the intake levels of Western females. Interestingly, several epidemiological studies have reported that Asian countries exhibit significantly lower incidence rates of certain types of cancer, such as breast and prostate cancer, than Western countries.171,172 These discrepancies have fueled a surge of interest within the scientific community, which has prompted rigorous studies into the potential role of genistein in cancer prevention and suppression of tumor growth. The anticancer effects of genistein are thought to be intricately associated with its ability to modulate the mechanisms of DDR proteins, and thus position genistein as a central player in cancer research and prevention. Genistein has been identified as a potent inhibitor of DNA topoisomerase II, and is known for its ability to induce double-strand breaks in DNA by inhibiting the activity of this critical enzyme. One pivotal study demonstrated that genistein induced DNA damage in human lymphoblastoid TK6 cells. Furthermore, cells that lacked Ligase4 (a critical enzyme in the non-homologous end joining (NHEJ) pathway) displayed increased sensitivity to genistein. This heightened susceptibility was manifested through increased accumulation of γ‐H2AX foci and an increased number of chromosomal aberrations. These findings not only highlighted the collaborative roles of NHEJ and homologous recombination (HR) in the repair of genistein-induced DNA damage but also suggested that genistein has the potential to amplify the activity of drugs targeting DNA damage including inhibitors of the NHEJ and HR pathways. Thus, genistein may be a potential adjuvant in therapeutic strategies that exploit the DDR for cancer treatment.173 Similarly, Liu et al. found that genistein inhibited the phosphorylation of DNA-PKcs, subsequently suppressing the NHEJ repair pathway and delaying the HR repair process. In addition, they demonstrated that genistein sensitized DNA-PKcs-proficient glioblastoma cells to carbon ion radiotherapy. Genistein has also been shown to activate key proteins involved in the DDR, such as JNK and Ask1, which play crucial roles in various cellular processes, including DNA damage, caspase-3 activation, DNA fragmentation, and the downregulation of thioredoxin-1. Together, these studies highlight the multifaceted impact of genistein on cellular responses to DNA damage, as well as its potential to enhance the effectiveness of radiotherapy in specific cancer contexts. In addition, these findings provide valuable insights into the mechanisms underlying the anticancer properties of genistein and solidify its standing as a promising therapeutic agent.174 Finally, genistein has also been shown to reduce NNKAc-induced ROS and DNA damage through the activation of Nrf2, demonstrating its protective role against oxidative stress and DNA damage.175 To date, numerous clinical trials have been initiated to determine the therapeutic potential of genistein against various types of cancer, including NSCLC (NCT01628471), pancreatic cancer (NCT00376948), and breast cancer (NCT00769990). Predominantly, these studies have aimed to investigate the synergistic effects of combining genistein with radiotherapy and chemotherapy, thereby elucidating its potential as a chemopreventive agent, particularly when used in conjunction with other treatment modalities. Although initial findings have been promising, it is imperative to acknowledge that translating the therapeutic benefits of genistein from preclinical models to clinical application necessitates exhaustive and meticulous research to validate its efficacy and safety in human subjects.
Targeting tumor-promoting inflammation
Inflammation significantly contributes to the progression of cancer through the generation of angiogenic factors and metastasis-associated proteins that can intensify or promote tumor invasion, migration, and malignancy via interactions with the TME.176,177 Consequently, using repurposed drugs to modulate inflammation has emerged as a potent anticancer strategy.
Celecoxib
Celecoxib, a selective cyclooxygenase-2 (COX-2) inhibitor, is conventionally prescribed for adult arthritis.178 However, recent studies have described the anticancer properties of celecoxib, which are mediated through the suppression of COX-2, a factor that is closely associated with cancer-related inflammation by promoting the synthesis of various prostaglandins, such as prostaglandin E2 (PGE2). Celecoxib has been shown to enhance the chemosensitivity of platinum-treated GC cells through the inhibition of prostaglandin-endoperoxide synthase 2 (PTGS2) and Bcl2 expression via the ERK1/2 and P38 signaling axis. PTGS2 has also been shown to reduce the cytotoxic effects of cisplatin (DDP) on GC cells through the PGE2/EP4/MAPKs (ERK1/2 and P38) axis.179 The significant role of inflammation in the malignant evolution of multiple cancers, mediated largely through the NF-κB signaling pathway, has been consistently emphasized in multiple studies.180,181,182 Celecoxib has been shown to enhance the efficacy of BRAF/MEKi treatments in melanoma through suppression of the NF-κB pathway. Since COX-2 promotes the resistance of melanoma cells to kinase inhibitors through the regulation of NF-κB -mediated inflammatory mediators, celecoxib can effectively counter the tumor-promoting actions of COX-2 by inhibiting its expression.183 Interestingly, Zhang et al. reported that celecoxib increased PTEN protein expression while inhibiting NF-κB and phosphatase of regenerating liver-3 expression levels in HCC-afflicted mouse livers.184 A compelling study by Guo et al. demonstrated that administration of celecoxib post-diagnosis led to better overall survival rates in cancer patients, particularly those exhibiting positive PTGS2 expression combined with a phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) mutation.185 Overall, the momentum of clinical trials investigating the role of celecoxib in cancer therapy is intensifying. In particular, the potential synergistic combination of celecoxib with chemotherapy or immunotherapy could improve cancer treatment outcomes, and confirm the importance of modulating inflammation as a potential therapeutic strategy against cancer.
Locking phenotypic plasticity
During organismal development, cells often undergo terminal differentiation as they are organized into specific tissues. Cellular differentiation typically leads to anti-proliferative outcomes, which form a vital component of the body’s defense against tumor formation. However, when the usually restricted capabilities for phenotypic plasticity become unrestrained, tumor cells might bypass terminal differentiation.186 There are three manifestations of phenotypic plasticity: dedifferentiation, blocked differentiation, and transdifferentiation, each representing a distinct disruption to cellular differentiation.67 From a conceptual standpoint, a tumor’s resistance to differentiation can be counteracted by increasing expression of developmental transcription factors like mothers against decapentaplegic homolog 4 (SMAD4) and homeobox A5.187,188 Specifically, SMAD4 can promote differentiation, thereby suppressing WNT-induced proliferation.189 Exploiting these mechanisms through drug repurposing might offer novel groundbreaking therapeutic approaches.
Metformin
Metformin, a well-established oral hypoglycemic agent from the biguanide class, has gained prominence over the years due to its cost-effectiveness and favorable safety profile.190,191 Interestingly, long-term use of metformin in type 2 diabetes patients has been linked to a decline in tumor incidence and a reduced rate of cancer-related deaths.192,193,194,195,196 Recent studies have indicated that metformin might exert direct anticancer effects on a range of tumor cells, including the elusive CSCs,197,198 thereby highlighting the multifaceted impact of metformin on cancer cells, as well as its potential to reduce phenotypic plasticity. Chang et al., for example, demonstrated that metformin reduced hepatocyte nuclear factor 4α (HNF4α) levels via activation of AMPKα, which subsequently modulated the WNT signaling pathway.199 A different study reported that metformin inhibited HNF4G activity via AMPK-driven phosphorylation and ubiquitin-mediated degradation, which suppressed the invasive abilities and metastasis of SMAD4-deficient pancreatic ductal adenocarcinoma cells.200 Intriguingly, metformin also appears to suppress the self-renewal capacities of CSCs, as evidenced by diminished expression of CSC markers (such as CD44 and CD133) and the growth of tumor spheroids through activation of AMP-activated protein kinase (AMPK) and inhibition of protein prenylation in the mevalonate pathway.201 Consistent with these findings, a meta-analysis revealed a 45% risk reduction in thyroid cancer among metformin users in Eastern countries, a phenomenon which was more evident in Asian populations than their Western counterparts.202 Another extensive meta-analysis ascertained a notably lower gynecological cancer occurrence in those undergoing metformin treatment compared to alternative therapies (gynecological cancer: HR = 0.60, 95% CI: 0.49–0.74; endometrial cancer: HR = 0.65, 95% CI: 0.50–0.85; ovarian cancer: HR = 0.47, 95% CI: 0.27–0.82).203 Thus, metformin has emerged as a paradigm of successful drug repurposing for oncological applications. Preclinical, epidemiological, and clinical insights have confirmed that metformin may act as a metabolic modulator by targeting various molecular pathways. Thus, metformin is a promising potent adjunct in anticancer regimens that could potentially synergize with chemotherapy, targeted agents, and immunomodulators.
Suppressing nonmutational epigenetic regulation
Recent studies confirming the significance of non-mutational epigenetic regulation in oncology have introduced an intriguing perspective on genome reprogramming that appears to operate independent of mutations, thus emphasizing a role for mutation-free pathways in cancer evolution. Many of these mechanisms are deeply interconnected with microenvironmental cues that govern epigenetic reprogramming. A prime example is hypoxia, a common trait within tumors, which profoundly modifies the TME. A direct consequence of hypoxia is the diminished activity of ten eleven translocation (TET) demethylases, which leads to marked changes in the methylome, especially elevated methylation levels.204 In addition, metabolic shifts that occur within the TME also play pivotal roles. For example, acetyl-CoA, derived from butyrate, drives histone acetylation, which subsequently modulates gene expression by activating histone acetyltransferases and suppressing histone deacetylases (HDACs) in both acyl-dependent and -independent manners.205 Furthermore, mounting evidence has suggested that repurposed drugs that target the TME can produce potent antitumor effects. Thus, drug repurposing aimed at the TME is a promising approach for counteracting non-mutational epigenetic shifts.
Baicalein
The roots, seeds and bark of Oroxylum indicum (L.), a traditional herbal medicine in China, India and other countries, has been used to treat a wide range of ailments including dysentery, rheumatic discomfort, diarrhea, pharyngitis, and persistent coughs, as well as more severe respiratory conditions like bronchitis.206 Baicalein (BE), a prominent flavonoid derived from the roots of O. indicum, possesses anti-oxidant, anti-inflammatory, anti-hepatotoxic, anti-viral, and antitumor properties.207,208 More recently, a role for BE in non-mutational epigenetic regulation has been described. Specifically, BE has been shown to reverse hypoxia-induced resistance to tamoxifen (TAM) through the downregulation of HIF-1α levels in breast cancer cells.209 From a molecular perspective, the 6-phosphogluconate dehydrogenase-driven oxidative pentose phosphate pathway is thought to facilitate the reshaping of histone H3K9 and DNA methylation patterns during tumor progression. Such changes lead to upregulation of N-cadherin transcription, a hallmark of EMT, and subsequent promotion of N-cadherin-induced distant metastasis.210 Interestingly, the intriguing interactions between BE and HIF-1α, the glycolytic regulator hexokinase I, and other glycolysis-associated genes suggest a role for BE in controlling the glycolytic pathway and subsequently regulating the energy dynamics of gastrointestinal cancer cells.211 BE is a major component of PC-SPES, a herbal concoction enriched with Scutellaria baicalensis, which was meticulously formulated under strict quality controls, and investigated in a phase 1 clinical trial in 2008. The trial was aimed at hormone-refractory prostate cancer patients and revealed encouraging outcomes both in terms of therapeutic efficacy and safety.212 Despite these promising results, further studies are required to elucidate the intricate mechanisms and better understand the broader implications of using BE as a potential therapeutic agent for the treatment of cancer.
Decreasing polymorphic microbiomes
The human microbiota predominantly colonizes epithelial surfaces, with the most significant concentration found within the gastrointestinal tract.213,214 Gut microbiota orchestrate a range of vital physiological functions, from nutrient and drug metabolism and vitamin synthesis to immune regulation and preservation of gastrointestinal structure.215,216 Intriguingly, although many of these microorganisms play benign or even beneficial roles, some can also contribute to disease progression, including cancers. An imbalance in microbial ecology, characterized by a shift in microbiota composition and disrupted homeostasis, is often observed during tumor development.217,218 For example, the Fusobacterium adhesin A antigen found in Fusobacterium nucleatum (F. nucleatum) has been associated with the promotion of CRC progression via the E-cadherin/WNT-β-catenin signaling pathway.219 While some microorganisms affect tumorigenesis directly, others enhance antitumor immune responses, effectively serving as immune adjuvants. This interaction between microbes and immunity in the context of cancer has been coined the ‘immune-oncology-microbiome axis’.214 Emerging evidence has highlighted the intricate relationship between the gut microbiota and conventional anticancer treatments, such as chemotherapy, radiotherapy, targeted therapy, and immunotherapy. Thus, a thorough understanding of the multifaceted biological roles of the gut microbiome, along with the associated molecular pathways, is imperative. Such insights would allow for the discovery of new targets for cancer interventions and subsequent clinical evaluations.220
Inulin
Inulin (C17H19NO3), a quintessential soluble dietary fiber predominantly sourced from plants such as chicory, ginger, garlic, onion, and asparagus,221 has emerged as a multifaceted ingredient in the culinary and pharmaceutical sectors. Known for its versatility, inulin functions as a prebiotic, a salubrious substitute for fats and sugars, a texture enhancer, and a cornerstone in the formulation of functional foods.222 Contemporary research has highlighted the potential efficacy of inulin in oncological interventions, particularly due to its capacity to modulate the polymorphic microbiome of the gut. An exemplary application has been observed in the amalgamation of inulin, cellulose, and their derivatives in preventing liver metastasis associated with CRC, primarily through the modulation of gut microbiota. Further scrutiny has revealed that inulin’s putative anticancer properties may emanate from its influential role in reshaping the composition of the intestinal microbiota. This involves not only the synthesis of short-chain fatty acids but also the nuanced regulation of the gut microbiota’s dynamics and their metabolic offshoots, thereby offering a holistic strategy for cancer prophylaxis.223 Intriguingly, experimental studies on animal models have revealed that oral administration of inulin gel significantly amplifies the efficacy of immune checkpoint therapy while maintaining an admirable safety profile. This gel has the propensity to reformulate the composition of the gut microbiota and its metabolic outputs, consequently invigorating the immune system and potentiating an antitumor-immune response.224 Moreover, inulin has shown a pronounced ability to inhibit tumor proliferation and extend the latency phase of oncogenesis. In a landmark study by Wu et al., rodents fed an inulin-enriched diet exhibited a markedly diverse and robust gut microbiome compared to their counterparts. The inclusion of inulin in the diet resulted in a significant increase in plasma propionate levels and a concomitant decline in the expression of pivotal epigenetic regulatory proteins such as HDAC2, HDAC8, and DNA methyltransferase 3b. Concurrently, there was a discernible decrease in the expression of proteins pivotal to tumor cell proliferation and survival, such as Akt, phospho-PI3K, and NF-kB. This study highlights the potential of dietary inulin as an avant-garde tactic in the preventative arsenal against breast cancer, potentially leveraging epigenetic mechanisms to manifest its prophylactic effects.225 Furthermore, inulin has been credited with fostering the proliferation of beneficial gut flora while impeding the growth of deleterious bacteria in an animal model harboring the murine pks + E. coli strain NC101. This prebiotic agent not only recalibrates the equilibrium of the gut microbiota but also augments the functionality of intestinal immune cells, thereby magnifying the efficacy of immune responses. Inulin has also been shown to modulate the expression of genes associated with colon cancer, effectively curbing tumor growth and metastasis.226 In summary, inulin possesses a range of physiological benefits, with a particularly pronounced impact on cancer therapeutics. Nevertheless, the integration of inulin into clinical practice necessitates further human trials to affirm its safety and to elucidate the nature of its decomposition products within the human body.
Targeting senescent cells
The four hallmarks of cellular senescence include: (i) a consistent, often irrevocable, cell-cycle arrest; (ii) the emergence of a senescence-associated secretory phenotype (SASP); (iii) macromolecular damage; and (iv) metabolic shifts.227 Serving as a countermeasure to programmed cell death, the primary function of cellular senescence is to remove damaged cells, including those predisposed to malignant transformation, thereby providing a safeguard against cancer.228 As such, the initiation of cellular senescence can pose a barrier to tumor development, presenting itself as a potentially favorable outcome for anticancer therapies. However, paradoxically, over the past decade, senescent cells have been shown to promote tumor growth and malignancy through a variety of mechanisms under certain circumstances.229,230,231 For example, DOX-induced systemic senescence was found to promote metastasis in an orthotopic mouse model of breast cancer. However, these harmful effects were mitigated either through genetic manipulation or pharmacological removal of senescent cells.232 Pioneering research has shown that senescent cells possess the capability to initiate malignancy in benign cells both in culture and in animal models. Moreover, in immunocompromised mice, senescent cells were shown to promote the growth of fully malignant breast cancer cells.233,234 Given these insights, the ability of repurposed drugs to act as both inducers of senescence and therapeutic agents targeting senescence has been acknowledged. Thus, the development of senescence-based therapy using repurposed drugs is an innovative therapeutic approach.
Quercetin
Quercetin, a potent flavonoid, is abundant in various plants, fruits, and vegetables, predominantly in glycoside forms found in onions, apples, blueberries, and broccoli.235 Its anti-inflammatory and antioxidant properties, as well as its ability to modulate the TME, have led to its inclusion in functional foods as a commercial dietary supplement.235 Recent studies have delved deep into its myriad of biological functions, particularly highlighting its anti-inflammatory, antioxidant, and anticancer properties.236 One of the most promising avenues for quercetin lies in its potential as a senotherapeutic agent. For example, in T24 bladder cancer cells, nuclear morphology analysis (NMA) revealed that quercetin treatment led to a marked increase in the percentage of cell nuclei during cellular senescence.237 Similarly, in Colo-320 and Colo-741 cells, quercetin treatment led to increased expression of several senescence markers including lamin B1, p16, and cyclin B1.238 Furthermore, quercetin was found to promote senescence in glioma cells by inhibiting the activity of HDACs. Moreover, in HepG2 liver cancer cells, quercetin was shown to reactivate P53, thereby inhibiting RNA degradation and protein ubiquitination, leading to the upregulation of P21 expression and concurrent downregulation of cyclin D1, a crucial player in cell cycle arrest.239 Thus, quercetin stands out as a potent tool to induce senescence in cancer cells. In the burgeoning field of senolytic treatments, combining quercetin with other agents has also shown promise in efficiently targeting senescent cells. Notably, a study by Zhu et al. revealed that quercetin (10 μM) could induce cell death in mouse bone marrow-derived senescent MSCs and radiation-induced senescent endothelial cells. In addition, pre-treatment with quercetin protected against DOX-induced normal cell senescence by reducing the number of senescent cells and suppressing the release of SASP factors.240 The ability of cancer cells to exploit senescence as a defense mechanism against therapies necessitates innovative approaches. Repurposing quercetin as an oncological drug has emerged as a strategic solution, as evidenced by a Phase II clinical trial that combined dasatinib and quercetin for the treatment of head and neck squamous cell carcinoma (NCT05724329). However, the broader application of quercetin in oncology has several challenges, and issues such as its limited bioavailability, instability, and lack of precise tumor targeting need to be addressed. Thus, novel strategies that augment the bioavailability of quercetin using lipid nanoparticles and chitosan nanoparticles have been developed. For example, a recent Phase II clinical trial assessed the therapeutic efficacy of both quercetin and its nanoparticle variant against oral squamous cell carcinoma cell lines (NCT05456022). In summary, the potential repurposing of quercetin is full of promise, highlighting the need for continued exploration and clinical validation.
Summary
During the early stages of cancer management, various chemotherapy agents and targeted therapies often yield promising results. However, as treatment progresses, tumors display remarkable resilience, developing adaptive resistance through mutations in treatment targets or by activating alternative signaling pathways. This adaptability often undermines or even neutralizes the effectiveness of therapeutic interventions. Furthermore, activation of survival pathways or suppression of death signals further promotes resistance. Current research has indicated that repurposed drugs, which target a range of malignant features, might enhance the potency of existing anticancer agents. In this section, we described repurposed drugs that target multiple malignancies in cancer, and revealed their potential for combination therapies, including immunotherapy, chemotherapy, and targeted treatments. While only a few repurposed drugs have been recognized for their direct anticancer effects, their multifaceted therapeutic targets are noteworthy. Looking ahead, integrating these drugs either as supportive agents in cancer care or in tandem with established anticancer agents may set the stage for more sustainable and effective cancer treatments. Such an approach has the potential to increase the impact of current cancer therapeutics.
Drug repurposing: candidates for TME-targeting therapy
The TME contains various immune cells, such as helper T (Th) cells,241 Tregs,242 dendritic cells,243 TAMs,244 and mesenchymal stem cells (MSCs),245 as well as fibroblastic stromal cells, including cancer-associated fibroblasts (CAFs), which surround tumor cells and are sustained by adjacent blood vessels. By secreting a range of molecules, these cells can either directly stimulate cancer cell proliferation or modify the molecules within their surroundings, which promotes tumor growth.246,247 The TME supports the survival and migration of cancer cells throughout the organism in response to internal or external stimuli, including treatments.248 Thus, an accurate and detailed understanding of the TME and other specialized TMEs will be beneficial in the development of potential cancer therapies.249 With respect to the different hallmarks of the TME, in the current section we will elaborate on the tumor immune microenvironment (TIME), metabolism microenvironment, hypoxic microenvironment, acidic niche, mechanical microenvironment, and innerved niche (Fig. 4), since they represent the critical aspects of the TME. Gut microbiota and their metabolites also play an important role in tumorigenesis. Various metabolic by-products of bacteria such as lactic acid,250 adenosine, nitric oxide (NO),251 potassium ions (K+),252 and ROS253 accumulate in the microenvironment, resulting in abnormal pH and oxygen levels, which promote cancer growth.246 To date, multiple studies have demonstrated that non-oncological repurposed drugs can produce unique antitumor effects by targeting one or more of the specialized microenvironments outlined above.

Classification of the tumor microenvironment. Underlying mechanisms of repurposed drugs targeting the specialized tumor microenvironments (TMEs). The TME can be divided into seven specialized microenvironments: hypoxic niche, immune microenvironment, metabolic microenvironment, acidic niche, innervated niche, mechanical microenvironment, and microbial microenvironment. Repurposed drugs with multi-targeted effects may reverse the effects of tumor-promoting microenvironments. TAM tumor-associated macrophage, CAF cancer-associated fibroblast, MDSC myeloid-derived suppressor cell, PNI perineural invasion, NK natural killer. This figure was created with Biorender.com
Immune microenvironment
Targeting and reversing the immunosuppressive attributes of the TME is critical for exploiting the TIME for therapeutic gain. Numerous studies have reported that immunosuppressive cells, ranging from Tregs and B cells, are recruited during tumor progression.254,255 These cells have been found to suppress the immune system by inhibiting both the trafficking and functionality of T cells through direct and indirect mechanisms.256 Moreover, other components of the TIME, such as TAMs, monocytes, and granulocytes, have also been reported to inhibit the antitumor activity of T cells and NK cells via various mechanisms, thereby leading to resistance to immunotherapy.244,257 Given this highly intricate milieu, a synergistic approach integrating ICIs with therapies that specifically target the TIME such as cytokine therapy, oncolytic viruses, and anti-angiogenic treatments could potentially increase antitumor immune responses. Concurrently, emerging research has explored the repurposing of traditional drugs to target pivotal facets of the TIME, such as CAFs and the extracellular matrix (ECM), offering innovative avenues to strengthen the potency of immunotherapeutic interventions.
Apigenin
Apigenin is a flavonoid ubiquitously found in numerous fruits and vegetables. Due to its anti-inflammatory, antioxidant, antitumor, anti-microbial, anti-viral, and cardiovascular protective properties, interest in apigenin has recently increased in the field of immune microenvironment research.258 Recent studies have revealed the potential of apigenin to act as an immunomodulator, positioning it as a promising candidate for cancer therapy repurposing. For example, in specific breast cancer (MDA‐MB‐468, SK‐BR‐3, and 4T1) and melanoma (A375, A2058, and RPMI‐7951) cell lines, apigenin has shown efficacy in negating the upregulation of PD‐L1, an effect induced by IFN‐γ. This capacity to modulate PD‐L1 has been further confirmed in co-culture settings with Jurkat T cells, where apigenin was shown to not only increase T lymphocyte proliferation, but also promote their apoptosis in breast cancer (MDA‐MB‐468) and melanoma (A375) cells.259,260 These findings highlight the intertwined relationship between T cell activation, apigenin-dependent PD-L1 regulation, and subsequent cancer cell demise. Apigenin has also been shown to modulate the transcription of SHIP-155 through the suppression of miRNA-1, demonstrating its pivotal role in orchestrating the antitumor immune dynamics within both the bone marrow and the TME of mice with pancreatic cancer.261 To date, only one clinical trial (NCT00609310) has examined the clinical application of apigenin in CRC. The lack of clinical trials may be due to its limited bioavailability and stability. Nonetheless, the development of advanced delivery mechanisms designed to overcome these obstacles may broaden the clinical application of apigenin in the future.
Infectious disease vaccines
Many non-oncological drugs and vaccines have emerged as potential candidates for repurposing in cancer treatment, with some already incorporated into clinical practice. The FDA-approved anticancer infectious disease vaccines currently in use include Bacillus Calmette-Guerin (BCG; bacterial-based), Talimogene laherparepvec (TVEC; an oncolytic virus), and Provenge® (Sipuleucel T; a dendritic cell-based vaccine).262 Thus, vaccines pose a promising approach to target the TIME in cancer treatment. Yao et al. reported that BCG heightened the cytotoxicity of GC cells by inducing lymphocytes to secrete IFN-g. In addition, BCG was found to augment the phagocytic capacity of human THP-1 monocytes/macrophages against H. pylori, by increasing the expression of the surface integrins CD11b, CD11d and CD18, as well as the membrane and soluble lipopolysaccharide receptors CD14 and sCD14, ultimately reducing the incidence of GC.263 Another study reported that intratumoral delivery of the prophylactic yellow fever vaccine (live 17D) into mice stimulated immune-mediated antitumor effects including the orchestration of cytotoxic T lymphocytes and suppression of Tregs.264 Intriguingly, Tai et al. found that administering a single dose of the influenza vaccine intramuscularly a day before surgery optimally activated NK cells in specific murine models.265 Based on these findings, a phase 1 clinical trial (NCT02998736) was initiated to evaluate the safety and NK cell killing activity of perioperative administration of the intramuscular influenza vaccine. The trial was carried out in conjunction with the phosphodiesterase-5 inhibitor, tadalafil, to counteract myeloid-derived suppressor cell (MDSC) inhibition of NK cells in patients undergoing abdominal cancer surgery.266 Currently, multiple infectious disease vaccines are the focus of oncology-based preclinical and clinical trials. Repurposing such vaccines to target the TIME could be a cost-efficient approach to expand the therapeutic alternatives available to patients.
Metabolism microenvironment
As previously discussed, the reprogramming of metabolism is regarded as one of the hallmarks of cancer. The reconfiguration of catabolic and anabolic processes in cancer cells contributes significantly to tumorigenesis and disease progression.99 In cancer cells, tailored modification of metabolic flux across diverse pathways caters to the heightened energy and synthesis demands intrinsic to tumor proliferation.267 More recently, it has become apparent that not only the metabolism of cancer cells but also that of stromal and immune cells within the TME plays a pivotal role in tumor maintenance. Cells that support tumor maintenance include endothelial cells (both vascular and lymphatic) and their affiliated pericytes, CAFs, and a range of immune cells including tumor-infiltrating lymphocytes (TILs) such as T cells, B cells, NK cells, TAMs, and mast cells.268,269 Endothelial cells are among the most studied stromal cells within the TME. Lactate uptake by endothelial cells has been shown to induce angiogenesis by increasing the expression of IL-8, VEGF, VEGFR2 and basic fibroblast growth factor (bFGF), as well as the phosphorylation of AKT, thereby promoting tumor growth. Moreover, CAFs reportedly secrete lactate, which tumor cells can use as a metabolic fuel by incorporating it into oxidative phosphorylation (OXPHOS) in the mitochondria, thereby supporting their high proliferation rates. This phenomenon is known as the “reverse Warburg effect”.270 Tumor metabolic reprogramming also plays a pivotal role in shaping the immune response, specifically antitumor immunity. For example, high expression of hexokinase 2 (HK2) in tumor cells can suppress transcription of the gene encoding IFN-γ, thereby contributing to immune response evasion.271 This concept of “oncometabolites” has ushered in a new era for cancer treatment. In terms of potential therapies, targeting the metabolic characteristics of different cells within the TME will enable the development of novel drug-repurposing strategies to combat cancer, which can be used in combination with traditional metabolic approaches.
Emodin
Emodin, a quintessential anthraquinone derivative from traditional Chinese herbs, has been increasingly recognized for its diverse biological activities, which include anticancer, antibacterial, liver-protective, and anti-inflammatory properties.272,273 More recently, the role of emodin in metabolic reprogramming has become an area of interest, and its role as a promising metabolic inhibitor has expanded its potential applications for cancer treatment. Emodin has been shown to suppress cholesterol biosynthesis in human HCC cells by attenuating the transcriptional activity of SREBP2 and inhibiting AKT signaling.274 Similarly, other studies have demonstrated that emodin regulates SREBP1 leading to a reduction in triglyceride levels and fatty acid desaturation, and ultimately inducing apoptosis in liver cancer cells.275 Furthermore, Xing et al. demonstrated that the cytotoxic effects of emodin on HepG2 cells were associated with disruptions in metabolic homeostasis including oxidative stress and imbalances in amino acids.276 Moreover, in addition to its lipid metabolism modulatory functions, emodin has also been shown to induce apoptosis in cervical cancer cells through the suppression of HPV E6/E7 expression and modulation of glucose metabolism.277 Despite these encouraging results, few clinical trials have focused on emodin due to its limited bioavailability. In the future, greater emphasis needs to be placed on exploring the clinical implications of emodin, as well as its therapeutic applications.
Mechanical microenvironment
During tumor development and progression, dynamic and heterogeneous interactions between tumor cells and the surrounding microenvironment occur, such that cells within the TME are subjected to both external and internal forces. In response to these mechanical pressures, including solid stress, shear stress, increased matrix stiffness, and topological changes, cells experience mechanosensing and mechanotransduction. These processes promote tumor progression by influencing phenomena such as the EMT and enhancing cell survival through autophagy.278,279 The formation of a mechanical microenvironment primarily depends on collagen and fibrin, integrins and fibroblasts. These components play important roles in regulating cell adhesion,280 morphology,281 motility, proliferation, differentiation, and migration.282 CAFs reportedly secrete matrix metalloproteinases (MMPs), including MMP-2, MMP-3, and MMP-9, or activate Yes-associated protein to promote ECM degradation and remodeling, EMT, and CSC stemness.283,284,285,286,287 Many studies have focused on the cellular proteins involved in mechanosensing, such as integrins and focal adhesion proteins, and their associated molecular mechanisms including cytoskeleton remodeling, integrin signaling, Rho signaling, and Hippo signaling.281 Consequently, treatments targeting the mechanical TME represent an emerging strategy in targeted therapy.
Curcumin
Curcumin, a vibrant yellow polyphenol derived from the turmeric plant (Curcuma longa), boasts an impressive range of bioactive properties and has been used to treat dermatological conditions.288,289 Interestingly, curcumin has displayed antitumor properties both in vitro and in vivo, and has been shown to act through multiple cellular pathways. It is one of the few compounds that has progressed to clinical trials.290 Of note, curcumin has recently been found to exert antitumor effects by targeting the mechanical microenvironment. Jalilian et al. showed that curcumin attenuated expression of the genes α-SMA and COX-2 and the production of PGE2 in CAFs. This study further demonstrated that curcumin effectively altered the pro-tumor characteristics of CAFs by suppressing PGE2 gene expression, leading to upregulation of the T-bet gene and increased production of interferon-gamma, which collectively contribute to a marked decrease in inflammation within the TME. As a result, these changes significantly enhance the antitumor capabilities of immune cells.291 The potential of curcumin to reduce metastasis of breast cancer is also evident in its inhibition of CXCL12/CXCR4 axis-mediated activation of the differentiation of adipose-derived MSCs (ADMSCs) into CAFs.292 In addition, Mao et al. demonstrated that curcumin suppressed proliferation of LGR5(+) colorectal CSCs by not only promoting autophagy but also through transcriptional repression of the oncogenic transcription factor activating enhancer-binding protein 2A (TFAP2A)-regulated ECM pathway.293 Although the antitumor properties of curcumin have been confirmed by multiple pre-clinical and clinical studies, its transition into mainstream cancer treatment has been limited by its subpar bioavailability. Furthermore, clinical investigations on curcumin have often involved small cohorts, which can lead to clinical variability. As a result, more expansive and well-structured clinical trials are required in the future to confirm that curcumin is a potential therapeutic agent for the treatment of cancer.
Hypoxic microenvironment
Hypoxia, a deficiency in the amount of oxygen reaching the tissues, is a defining feature of the TME and acts as an important negative prognostic indicator across numerous solid tumors.294 Hypoxia occurs when there is an imbalance between the oxygen supply and oxygen requirements of cancerous and stromal cells, and often involves a malfunctioning microvascular system. This oxygen-deprived environment plays a pivotal role in shaping the biological characteristics and aggressive nature of cancer cells.295 HIF-1α serves as a primary marker of the hypoxic environment. Under low-oxygen conditions, elevated HIF-1α activity leads to the upregulation of Snail and Twist, transcriptional regulators that suppress E-cadherin expression resulting in EMT. The adverse effect of hypoxia is evident in multiple types of cancer, while its intricate role in modulating the efficacy of chemotherapy, radiotherapy, and immunotherapy cannot be overstated. Notably, Wenger et al. examined the impact of hypoxia on mouse embryonic fibroblast proliferation, and found that the suppressive effects of agents like carboplatin and etoposide on cell growth were significantly increased when HIF-1α was deactivated.296 Given these insights, targeting the hypoxic TME via drug repurposing may be beneficial in the development of oncological agents.
Ascorbic acid
Ascorbic acid, commonly known as vitamin C, is an essential water-soluble vitamin that humans are unable to synthesize endogenously, and must therefore obtain from dietary sources.297,298 As an antioxidant, ascorbic acid protects against free radicals, and is involved in several vital physiological functions including collagen formation, wound healing, tissue repair, and maintaining the health of bones, cartilage, and teeth.299 More recently, groundbreaking preclinical studies have shed light on the novel roles of vitamin C as a supplementary agent in a range of innovative cancer treatment approaches that involve epigenetics, immunomodulation, and selective cytotoxicity. Recent studies have established a compelling link between the anticancer properties of ascorbic acid and the hypoxic microenvironment within tumors, which has placed significant emphasis on the role of HIF-1α. For example, in human endometrial tumors, elevated levels of ascorbic acid within the tumor have been shown to correlate with decreased expression of HIF-1α, VEGF, and GLUT1 proteins, leading to a marked reduction in malignancy.300 Furthermore, ascorbic acid has been shown to induce a dose-dependent decrease in the expression of HIF-1α and GLUT1, both identified as downstream targets of HIF-1, in thyroid cancer cells in vitro. Similarly, low concentrations of ascorbic acid (25 mM) have been shown to suppress HIF-1α expression in hypoxic conditions, subsequently impeding tumor growth.301 Moreover, in an in vivo subcutaneous lung tumor transplantation model in rats, intraperitoneal injections of ascorbic acid (1 g/kg) were found to reduce HIF-1α expression within the tumor, while simultaneously suppressing tumor growth and reducing vascular density.302 Furthermore, a retrospective analysis involving human patient tumor samples paired with controls for endometrial cancer, RCC, and CRC revealed that tumors exhibiting the highest HIF-1 activity were generally associated with patients who had a notable deficiency in ascorbic acid.303 Thus, the vast amount of preclinical and clinical data currently available indicates that ascorbic acid may be a promising agent for therapeutic repurposing, particularly with respect to its applications in targeted therapy and dietary treatment.
Acidic niche
The typical pH of the interstitial space in solid tumors ranges from 6.4 to 7, which is more acidic than that of normal tissues, which maintain a pH between 7.3 and 7.4.304 During acidic selection, cancer cells acquire enhanced acid extrusion capacity and other properties that confer a high degree of fitness in evolutionary terms, leading to an increased propensity for proliferation, survival, and invasiveness.305 For example, exposure of melanoma cells to extracellular acidosis for 24 hours led to increased expression of EMT markers and enhanced in vitro invasiveness.306 In addition, the functionality of antitumor agents, such as T cells and NK cells, is often reduced under acidic conditions. In contrast, the actions of tumor-promoting cells such as MDSCs and Tregs are often increased under acidic conditions, leading to enhanced tumor growth and suppression of antitumor immune responses. Notably, in one study, a pH of 6.5 was found to reversibly impair T cell function, characterized by a reduced expression of T cell receptor components in melanoma patients.307 Thus, drug repurposing aimed at neutralizing acidic niches might offer a viable approach to stabilize pH levels within the TME.
Pump proton inhibitors
Proton pump inhibitors (PPIs) are the preferred drugs for inhibiting gastric acid secretion. They are commonly prescribed to treat conditions like peptic ulcers, gastroesophageal reflux disease, Zollinger-Ellison syndrome, and upper gastrointestinal bleeding.308,309 Over the years, PPIs have been administered to billions of people worldwide and have shown a remarkable safety profile even at high dosages. The ability of PPIs to counteract tumor acidity and reduce acid-induced chemoresistance has been the focus of recent studies. Multiple reports have demonstrated that PPIs modulate tumor acidification and renew chemosensitivity in resistant cancer cells both in vitro and in vivo.310 PPIs have been shown to reduce V-ATPase activity in the acidic niche of cancer cells, thereby disrupting proton transport, and subsequently shifting the pH balance within these cells resulting in increased intracellular concentrations of cytotoxic drugs.311 Similar findings have been reported in other in vitro studies, indicating that potential downstream effectors, such as the dephosphorylation of low-density lipoprotein receptor-related protein 6 and the inhibition of WNT/β-catenin312 or PI3K/AKT/mTOR/HIF-1α signaling pathways,313 follow the inhibition of V-type H+ ATPase by PPIs in GC cells. PPIs have also been found to counteract the effects of an acidic microenvironment on the ability of tumor cells to evade immune surveillance. For example, pantoprazole reportedly increased the number of TAMs in the TME, as well as enhanced CD11c expression and phagocytosis, and influenced macrophage shape.314 Interestingly, a recent meta-analysis suggested a heightened risk of cancer in PPI users versus non-users.315 This might be due to the inhibition of H+/K+ ATPase by PPIs in parietal cells, which leads to increased release of gastrin from G-cells.316 Gastrin has long been suspected to be a potential risk factor for GC. However, this analysis did not stratify the risk of GC by dose due to a lack of data. Given the historical association between gastrin and potential risks of GC, this correlation warrants closer examination, especially with respect to dosage. Moreover, a study of 6754 breast cancer patients revealed that the use of PPIs not only markedly improved their overall survival rates but also reduced the risk of disease recurrence.317 Notably, these PPIs also mitigated resistance commonly seen with conventional chemotherapy drugs and radiation treatments. Given these findings, PPIs have emerged as a promising adjunctive treatment strategy, especially when combined with other therapeutic drugs. Various PPIs are currently being utilized in the treatment of different cancers, including liver and breast cancer. As we look to the future, there is a compelling case for more clinical trials to accelerate the wider adoption of PPIs in cancer therapy.
Innervated niche
The role of the nervous system in the development of solid tumors has often been underestimated. Although the potential significance of the peripheral nervous system in oncogenesis was suggested by pioneers in the field last century, the functional contribution of nerves to cancer development and modulation of the TME have remained under-explored for a long time.318 More recently, groundbreaking research has revealed that tumors can actively recruit nerves into the TME, which can then directly promote tumor growth—a phenomenon known as “tumor innervation”.319,320 These findings have highlighted novel perspectives on tumor–nerve interactions. Emerging evidence has suggested that certain neurochemicals, such as dopamine, catecholamines, and acetylcholine, have direct roles in tumor initiation and progression. For example, β-adrenergic signaling has been implicated in promoting EMT and increasing the metastatic potential in cancer cells via the VEGF/MMP and STAT3/ERK/MAPK pathways.321 Furthermore, Sloan et al. found that both stress-induced and pharmacological β-adrenergic stimulation promoted macrophage migration into tumor tissue in a mouse model of breast cancer. These macrophages then adopted an immunosuppressive M2 phenotype, leading to increased production of TGF-β, VEGF, and MMP-9, which in turn promoted angiogenesis and metastasis.322 Thus, a deeper understanding of the nerve-rich regions within tumors could be beneficial for the development of innovative cancer therapeutic strategies.
β-adrenergic antagonists
β-adrenergic antagonists, commonly known as β-blockers, are primarily used to manage cardiovascular conditions such as hypertension and coronary artery disease, and act by inhibiting β-adrenergic receptors (β-ARs) within the adrenergic system.323,324,325 Recent studies have suggested that the antitumor effects of β-blockers might be due to disruption of the innervated niche. Catecholamines, for example, have been shown to activate macrophages via adrenergic signaling, leading to a shift towards the M2-polarized phenotype and increased VEGF production, which ultimately promotes tumor angiogenesis.326,327 This catecholamine-driven effect was found to be mitigated by the β-AR antagonist, propranolol. Furthermore, elevated catecholamine levels, often present during depressive episodes, have been shown to interact with β2-ARs on GC cells, thus promoting expression of metastasis-associated in colon cancer 1 (MACC1). MACC1 then binds to synaptophysin to preserve the neuroendocrine phenotype, which is associated with increased invasiveness and the metastatic potential of GC. This malignant transformation induced by catecholamines was effectively blocked by the β2-AR antagonist, ICI-118,551. In addition, Shi et al. demonstrated that ICI-118,551 could reverse catecholamine-induced resistance to trastuzumab in cancer cells, thereby improving the efficacy of targeted cancer treatments.328 In conclusion, β-adrenergic antagonists, repurposed as anticancer drugs, might be suitable candidates for combination therapies targeting disparate neoplastic processes. To date, most of the clinical trials on β-adrenergic antagonists have a relatively small sample size, or are still underway, with results yet to be published. Thus, there remains a critical need for extensive randomized controlled clinical trials with a specific focus on β-adrenergic antagonists.
Microbial microenvironment
The microbial microenvironment is primarily comprised of intestinal and intra-tumoral microorganisms and their metabolites, which possess the ability to either facilitate or inhibit tumor progression and impact the effectiveness and toxicity of cancer treatments.329,330,331,332 For example, beneficial gut microbes have been shown to improve the antitumor effects of certain therapies such as ICIs by modulating the immune response.333,334,335 In contrast, other microbes can promote tumor initiation and progression directly by generating toxic or tumorigenic products or indirectly by shaping a pro- or anti-inflammatory microenvironment. Parhi et al. found that F. nucleatum accumulated specifically within tumors and reduced the number of tumor-infiltrating CD4+ and CD8+ T cells, indicating that F. nucleatum-mediated changes in the TIME contributed significantly to tumor growth.336 In addition, the F. nucleatum DNA load in MSI-high CRC tumors was found to be inversely correlated with the density of tumor-infiltrating FOXP3+ T cells, and positively correlated with the ratio of M2-like TAMs to total TAMs, suggesting that F. nucleatum primarily promotes tumor progression through the proliferation of suppressive immune cells.337 Therefore, studies into the causal relationship between the tumor microbial microenvironment and the overall TME may provide novel insights that contribute to the development of new drug research within the era of precision medicine.
Evodiamine
Evodiamine (EVO) is a natural alkaloid compound derived from the fruit of Evodiae fructus, a member of the Rutaceae family, that is primarily used for the treatment of various ailments such as vomiting, diarrhea, headaches, and challenging menstrual cycles.338 Beyond its traditional medicinal applications, EVO is also a popular dietary supplement.339,340 Recent studies have revealed a role for EVO in targeting the microbial microenvironment, which may provide potential anticancer benefits in a spectrum of malignancies. For example, Yang et al. found that EVO impeded the growth of H. pylori reference strains and clinical isolates, as well as reduced the translocation of CagA and VacA proteins into AGS GC cells.341 EVO appears to exert its protective effects against CRC through modulation of the gut microbiota and reduction of intestinal inflammation, potentially through inhibition of the IL6/STAT3/P65 signaling pathway.342 In addition to its direct anticancer effects, EVO has also been shown to temper inflammatory reactions and tumor-associated immune responses. Further analysis has revealed that EVO augments the presence of bacteria that produce short-chain fatty acids while reducing the number of pro-inflammatory bacteria.343 Such modulations affect microbiota metabolism through regulation of the tryptophan pathways. However, few clinical studies have focused on EVO, despite these promising preclinical insights and the substantial evidence outlining the safe dosages and pharmacological implications for tumor management. Thus, it is likely that EVO-infused medications may become integral in preventive and therapeutic healthcare strategies in the future.
Summary
The TME is a dynamic and multifaceted milieu that consists of multiple cellular entities and molecular constituents that often dictate the behavior of nearby cancer cells. Given its intricate nature and profound influence on cancer progression, the TME has attracted interest recently as a potential therapeutic target.344 In this section, we evaluated the anticancer activity of repurposed drugs from multiple perspectives including the TIME, metabolism microenvironment, mechanical microenvironment, hypoxic microenvironment, acidic niche, innervated niche, and microbial microenvironment. Presently, there is a lack of clinical trial evidence describing the efficacies of these repurposed agents in modulating the TME, which probably reflects our nascent understanding of the unique anticancer mechanisms each drug embodies. Thus, it is crucial to understand that cancer is not a monolithic entity, but a continuum, evolving through multiple stages. Accordingly, repurposed drugs might manifest distinct effects contingent on the phase of the disease. In the future, it is necessary to conduct more nuanced preclinical studies and tailored clinical trials that identify the most efficacious therapeutic strategies. The re-emergence of repurposed drugs for oncological purposes signals a renaissance in their applicability, and paves the way for future studies aiming to harness their full potential against both the TME and the malignancies they harbor.
Drug repurposing: candidates for nanomaterial-based therapy
The efficacy of repurposed therapeutic agents is chiefly determined by their side effects and resistance profiles.345 While the ultimate goal is to pinpoint and precisely target cancer cells, prevailing systemic drug administration techniques struggle with challenges such as uneven distribution and suboptimal pharmacokinetics, which can translate into unwanted side effects. Moreover, the non-specific nature of drug delivery might result in a disproportionate accumulation of repurposed agents in organs such as the kidneys, liver, and spleen. Such uneven distribution often culminates in inadequate drug concentrations at the tumor site, thereby reducing the drug’s potential to effectively inhibit tumor growth.346 Thus, it is crucial to devise the right therapeutic strategies to enhance their impact on tumor growth, while minimizing side effects and drug resistance. Here, our emphasis lies primarily on nano-delivery systems for topical drug delivery, such as polylactic-co-glycolic acid (PLGA) nanoparticles, mesoporous silica nanoparticles, polymeric micelles (PMs), liposomes, and metallic nanoparticles (Fig. 5). These nanoparticles have demonstrated their potential for drug delivery and imaging in biomedicine.347 Nanoparticles can minimize the systemic toxicity of traditional therapies, thereby significantly enhancing the ability of repurposed drugs to eliminate tumors without harming normal cells.348 In this section, we catalog the various nano-carriers tailored for the targeted delivery of repurposed drugs against tumors, alongside ongoing clinical trials that are using nanomaterials to treat cancer.

Nanocarriers in repurposed drug delivery in cancer. Multiple nanocarriers including polymeric nanocarriers, mesoporous silica nanoparticles, polymeric micelles, liposomes, and metallic nanoparticles have shown promising effects in the delivery of repurposed therapeutics in cancer. Targeted cancer NP drug delivery systems: a Diversity of NPs in thedrug delivery platform. b Active-targeting ligand-decorated repurposed drug-loaded NPs for cancers. c Passive tumor targeting via the enhanced permeability and retention (EPR) effect. d Internalization of active-targeting NPs driven by receptor activation. This figure was created with Biorender.com
Polymeric nanoparticles
PLGA polymers have emerged as a focal point in drug delivery research due to their349 outstanding biodegradability and biocompatibility, as well as their easy modifiability for precise drug control and release.350,351 This adaptability is particularly impactful when encapsulating drugs and elevating their targeted delivery, especially in oncology. For example, Güor e et al. found a significant reduction in cellular proliferation in A-549 cells treated with ibuprofen-loaded unique EudragitVR RS 100 and/or octadecylamine modified PLGA nanoparticles compared to cells treated with the equivalent dosage of unencapsulated drug. Such outcomes accentuate the transformative potential of PLGA nanoparticles in augmenting cancer therapeutics.352
Silk fibroin nanoparticles
Silk fibroin (SF), derived from silkworms, is a biocompatible and biodegradable protein, making it an ideal candidate for various biomedical applications. Its unique scaffolding and matrix properties have been shown to exhibit exceptional cell compatibility both in vivo and in vitro.353,354 Given these attributes, SF is gaining traction as a potential material for drug delivery systems, and could therefore play a transformative role in treating a variety of diseases. One example of the potential application of SF involves celastrol, a bioactive compound extracted from the traditional Chinese herb, clover. Celastrol possesses a distinct chemical structure and a range of bioactivities including its anticancer properties. However, its direct clinical use has been limited by its poor water solubility and marked toxicity.355,356 To circumvent these challenges, Ding et al. innovatively encapsulated celastrol into SF nanoparticles (CL-SFNPs) using a modified desolvation method and found that CL-SFNPs outperformed free-form celastrol in terms of antitumor efficacy, inhibition of colony formation, and induction of cancer cell apoptosis.354 Thus, these findings highlight the potential of SFNPs to act as a delivery platform for celastrol to advance cancer therapies.
Polymeric micelles
PMs were one of the first polymeric self-assemblies and possess many advantages over other nano-assemblies357 including ease of processing, a streamlined architecture, and superior drug solubilization. In addition, PMs exhibit enhanced biocompatibility, advanced pharmacokinetics, biodistribution properties, and increased adaptability for engineering customizations.358,359 As a result, PMs are excellent vehicles for drug delivery, displaying better absorption and reduced side effects through varied administration methods, which range from parenteral to oral, nasal, and ocular routes.360,361,362,363 Thus, PMs could act as potential carriers for repurposed drugs. A case in point is rapamycin (RAPA), a naturally-derived macrolide drug lauded for its anti-fungal, anti-angiogenesis, and immunosuppressive traits. In order to exploit the full potential of RAPA, Shin et al. pioneered the development of fenbendazole/rapamycin-loaded mPEG-b-PCL micelles (M-FR) via freeze-drying. Notably, the M-FR system was found to increase the cytotoxicity and reduce the proliferation abilities of ovarian cancer cells compared to treatment with the unconjugated fenbendazole/rapamycin combination. Moreover, in vivo pharmacokinetic studies revealed that M-FR offered improved bioavailability and significant antitumor effects.352 This collaborative application of a nanocarrier system, therefore, has the potential to re-engineer RAPA, initially developed as a non-tumoricidal drug, into a formidable tumoricidal agent, thus making it a potent strategy in the chemoprevention of cancer.
Liposomes
Liposomes are lipid bilayer nanovesicles designed for the transport of diverse molecules such as DNA, proteins, antigens, and drugs.364 Their inherent ability to transport both hydrophobic and hydrophilic agents,347 combined with their ease of synthesis, extensive surface area, and adaptability for targeted modifications, positions them as a pivotal vehicle for drug delivery.365 Niclosamide (NIC), recognized as an anthelmintic drug for over 50 years, also serves as an effective inhibitor of STAT3. NIC has been shown to induce apoptosis in breast, lung, and colon cancer.366,367 However, the clinical application of NIC has been constrained due to its poor water solubility and bioavailability. Shah and colleagues addressed this issue using a design-of-experiments approach to develop NIC-loaded liposomes, which were found to enhance antitumor efficacy against melanoma cells in vitro.368 Similarly, Hatamipour et al. found that NIC-loaded nanoliposomes markedly inhibited the growth of CT26 colon cancer cells, leading to a pronounced delay in tumor progression and increased survival.369 These promising outcomes highlight the potential of using a reinvigorated strategy such as NIC-loaded liposomes in cancer therapeutics.
Metallic nanoparticles
Metallic nanoparticles, notably gold and silver, possess unique properties, such as their small size and large specific surface area, which enhance their surface affinity towards specific antibodies and ligands, and increase their efficiency in targeted drug delivery for various ailments.370,371 Their prominence as anticancer therapeutic agents arises from their unparalleled stability, substantial drug-loading capacities, potent targeting capabilities, and precise drug-release mechanisms.372 Among repurposed phytochemicals, ginsenoside is of interest to oncologists due to its potential tumor-suppressing properties. However, its therapeutic potential may be limited by the hydrophobic nature of its aglycone backbone, suboptimal bioavailability, and cytotoxicity to benign cells. In an attempt to address these issues, Kim et al pioneered the synthesis of gold nanoparticles carried by Lactobacillus kimchicus DCY51T as a potential nano-pharmaceutical against solid tumors. This novel nano-delivery system not only displayed increased cytotoxicity but also increased apoptotic rates within tumors compared to application of ginsenoside alone,373 suggesting that metal nanoparticle-imbued repurposed drugs may have the ability to inhibit tumor proliferation and potentially induce regression of established tumors.
Challenges of drug repurposing
Although systematic drug repurposing has provided new opportunities, to date, few repurposed drugs in cancer or even oncology have successfully transitioned into clinical application. Even though the drug repurposing process is perceived to be significantly quicker and less expensive than traditional drug discovery, rushing into clinical trials could potentially impede the search for more precise treatments. Furthermore, like all drug development, there still exists a risk of failure in the later stages of clinical trials. Other hurdles include legal and regulatory issues, as well as challenges related to pharmacology and dosing.374,375 It is our hope that such barriers can be overcome to fully realize the potential of drug repurposing.
Pharmacological challenges and high effective concentrations may not be clinically achievable
Drug repurposing, while promising, presents several pharmacological challenges. Drugs initially tailored for specific receptors, cells, or organs might not have the same efficacy when rerouted for different therapeutic purposes. Consequently, higher doses or augmented drug interactions might be necessary to achieve therapeutic levels, which could, in turn, introduce novel mechanisms of action, distinct from their intended use.376 This shift could lead to unexpected binding to off-target molecules, and the introduction of unforeseen side effects when the drug is administered to humans. Unfortunately, the nuances of dosage and achievable blood serum concentrations are often overlooked, rendering the clinical translation of such drugs problematic. Alterations in dosage can escalate the risk of adverse events. For example, repurposing simvastatin and metformin for cancer could raise questions about their potential to cause hypolipidemia and hypoglycemia, respectively. Likewise, could aspirin repurposed in this manner increase the risk of gastrointestinal bleeding? The feasibility of these repurposed treatments in a clinical setting will depend on the tolerability of side effects during their administration period.
Patent considerations and regulatory considerations
Intellectual property (IP) rights present another significant issue that needs to be addressed. It is possible to secure IP protection for a newly repurposed medical use of a known drug molecule in most of the major pharmaceutical markets, provided that the new medical use is novel and inventive (i.e., non-obvious). However, many potential repurposing uses are already known in the scientific literature, which limits the potential for securing patent protection for the repurposed context.58 In certain cases, even when a drug has demonstrated promising results, its market entry can be hindered due to conflicts associated with IP rights. Furthermore, in cases of repurposing an off-patent drug, the prospect for a return on investment is limited, which makes industries less inclined to fund a trial. In addition to patent issues, there are numerous legal and regulatory barriers to drug repurposing. Regulatory incentives or formal guidance encouraging companies to invest in research and development for further uses of existing drugs are often lacking, or if present, are inadequate.375
Conclusion and future perspectives
Theoretically, repurposed drugs can partially alleviate the shortage of new drugs and resistance to existing chemotherapeutic drugs.377 In this review, we have drawn on an expansive exploration of drug repurposing research over the years to collate and appraise potential drug candidates for cancer therapy, the relevant clinical studies of which are presented in Table 3. We have explored the mechanisms by which non-oncological drugs target the hallmarks of cancer and the specialized TME. However, it remains unclear whether these drugs will translate into clinical medications. For patients with advanced disease or chemotherapy resistance who lack alternative treatment options, combination therapy is a promising and valuable treatment option. Combining repurposed therapeutic drugs with approved anticancer drugs can achieve synergy and improve therapeutic effectiveness and safety. In addition, as with other drug strategies for cancer treatment, new drug delivery technologies are necessary for treating cancer cells. In this regard, nanotechnology represents a potential strategy to improve the current prognosis and treatment of tumors.378 Currently, large clinical studies are examining the use of established safety nanomedicines such as nab-paclitaxel (Abraxane), liposomal DOX, liposomal verteporfin (Visudine), and gadolinium nanoparticles (AGuIX)379 in cancers such as pancreatic cancer,380 advanced squamous NSCLC,381 breast cancer,382 platinum-refractory metastatic urothelial cancer,383 and gastrointestinal cancer.384 However, many components (materials) of the nanocarriers that have exhibited excellent tumor targeting and therapeutic properties in a xenograft tumor model have not been tested for safety and their long-term toxicity is not known. This has resulted in few clinical trials of nanoparticles loaded with repurposed drugs for cancer treatment. Although these target-based nano-formulation therapies have seen minimal translation from pre-clinical research to clinical usage, the increasing number of studies on the synergistic combination of drugs targeting new mechanisms with traditional therapeutics to eliminate cancer survival pathways suggests that the emerging field of combining repurposed drugs loaded with nanomaterials with first-line anticancer drugs for cancer treatment will attract more attention from researchers in the future.
Drug repurposing has, to date, attracted considerable attention from researchers and pharmaceutical industries worldwide.385 Several promising strategies based on clinical symptoms, genome and transcriptome data, as well as various databases, have been devised to advance the development of repurposed drugs for tumor treatment.386 This article provides a summary of the most frequently employed approaches to drug repurposing, including both experimental and computational methodologies. The emergence of cutting-edge technologies has resulted in the production of a significant amount of data that includes genomics, proteomics, drug-disease associations, drug chemical structure profiles, and phenotypes. These substantial data resources are pivotal in comprehending the intricacies of cancer mechanisms and enabling large-scale screening of repurposed drugs. Widely used data resources have been compiled and are presented in Table 3 for further reference.
However, approved drugs are encumbered by legal and safety liabilities, IP rights issues, patent and regulatory barriers, and the relative lack of funding for clinical trials by pharmaceutical companies due to expected low returns on investment. Moreover, phase II and phase III clinical trials of repurposed drugs also require substantial resources in terms of money and time.387 Clinical trials are essential for addressing questions about whether different pharmacodynamic and pharmacokinetic properties are required for their activity in specific settings also remain.388
In addition, several key points warrant particular attention. Due to the multiple mechanisms of resistance and complex oncogenic signaling pathways of cancers, monotherapy may be relatively ineffective for cancer patients. This may explain why few repurposed drugs can be used in cancer treatment, and why, in the era of precision medicine, drug combination therapies are a more promising strategy. Drug combination therapies typically target multiple mechanisms, including downstream off-target, parallel pathways, or compensatory signaling. They can also provide alternative strategies for apoptosis-resistant cancers by targeting other modes of cell death such as ferroptosis and pyroptosis. Concurrently, with the rapid development of molecular profiling, the use of non-oncological drugs that have the potential to target multiple hallmarks of cancer and specialized TME could be a vital complement to personalized/precision treatment in the near future. Furthermore, combining repurposed drugs with first-line anticancer drugs will offer cancer patients new treatment opportunities.
Furthermore, taking into account the commonalities and differences between various cancers can also enhance research in drug repurposing. The commonality among different cancers lies in their potential sharing of key molecular and cellular mechanisms. For instance, many types of cancers display dysregulation in cell metabolism,389,390 and impaired apoptotic pathways.391 In terms of drug repurposing, this implies that certain drugs might show efficacy across multiple cancer types, especially those targeting these common mechanisms. For example, simvastatin, a statin class drug for lowering blood lipids, works by competitively inhibiting the enzyme HMG-CoA reductase, and blocking the synthesis of cholesterol, an end product of mevalonate metabolism.392 Cancers such as gastric cancer, pancreatic cancer, and colon cancer, which exhibit dysregulated lipid metabolism and rely on the mevalonate pathway, highlight the potential of repurposing statin drugs for treating various cancers.393,394,395 However, the diversity of cancers is equally important. Different cancer types have distinct genetic backgrounds, biomarkers, and microenvironment interactions, leading to varying responses to the same drug. For example, EGFR inhibitors show significant efficacy in NSCLC with specific EGFR mutations but are less effective in cancers without these mutations.396 Therefore, when considering drug repurposing, the specific characteristics and needs of each cancer type must be taken into account.
Moreover, in the process of drug repurposing, it is necessary to conduct in-depth studies of the pharmacodynamics and pharmacokinetics of drugs to determine optimal dosages, administration frequencies, and potential drug interactions. For instance, drugs originally used for cardiovascular diseases might require dosage adjustments when repurposed for cancer treatment to maximize their antitumor effects while minimizing toxic side effects.13,397 In addition, the design and execution of clinical trials are crucial for drug repurposing strategies. These include not only traditional assessments of drug safety and efficacy but also a deep understanding of the mechanisms of drugs in specific cancer types. By designing clinical trials that include various cancer types and patient groups, we can more comprehensively evaluate the potential of repurposed drugs.
In summary, drug repurposing offers significant opportunities in the field of cancer treatment, and despite the many challenges, these efforts are undoubtedly worthwhile. Drug repurposing provides a window of opportunity for drug discovery and represents a developing trend in cancer treatment. Despite the numerous challenges, these efforts are undoubtedly worthwhile.398,399
References
Renzi, C., Odelli, S., Morani, F., Benitez Majano, S. & Signorelli, C. Delays in cancer diagnosis: challenges and opportunities in Europe. Acta Biomed. 94, e2023161 (2023).
Mima, K. et al. The microbiome and rise of early-onset cancers: knowledge gaps and research opportunities. Gut Microbes 15, 2269623 (2023).
Ajmeera, D. & Ajumeera, R. Drug repurposing: a novel strategy to target cancer stem cells and therapeutic resistance. Genes Dis. 11, 148–175 (2024).
Wang, Z., McLoone, P. & Morrison, D. S. Diet, exercise, obesity, smoking and alcohol consumption in cancer survivors and the general population: a comparative study of 16,282 individuals. Br. J. Cancer 112, 572–575 (2015).
Donaldson, M. S. Nutrition and cancer: a review of the evidence for an anti-cancer diet. Nutr. J. 3, 19 (2004).
Ruiz-Núñez, B., Pruimboom, L., Dijck-Brouwer, D. A. J. & Muskiet, F. A. J. Lifestyle and nutritional imbalances associated with Western diseases: causes and consequences of chronic systemic low-grade inflammation in an evolutionary context. J. Nutr. Biochem. 24, 1183–1201 (2013).
Espina, C. et al. European code against cancer 4th edition: environment, occupation and cancer. Cancer Epidemiol. 39, S84–S92 (2015).
Debela, D. T. et al. New approaches and procedures for cancer treatment: current perspectives. SAGE Open Med. 9, 20503121211034366 (2021).
Barker, H. E., Paget, J. T. E., Khan, A. A. & Harrington, K. J. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425 (2015).
Arruebo, M. et al. Assessment of the evolution of cancer treatment therapies. Cancers 3, 3279–3330 (2011).
Poonpanichakul, T. et al. Capturing tumour heterogeneity in pre- and post-chemotherapy colorectal cancer ascites-derived cells using single-cell RNA-sequencing. Biosci. Rep. 41, BSR20212093 (2021).
Holohan, C., Van Schaeybroeck, S., Longley, D. B. & Johnston, P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726 (2013).
Fu, L. et al. Repurposing non-oncology small-molecule drugs to improve cancer therapy: current situation and future directions. Acta Pharm. Sin. B 12, 532–557 (2022).
Pillai U, J., Ray, A., Maan, M. & Dutta, M. Repurposing drugs targeting metabolic diseases for cancer therapeutics. Drug Discov. Today 28, 103684 (2023).
Sun, D., Gao, W., Hu, H. & Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 12, 3049–3062 (2022).
Kirtonia, A. et al. Repurposing of drugs: an attractive pharmacological strategy for cancer therapeutics. Semin. Cancer Biol. 68, 258–278 (2021).
Schein, C. H. Repurposing approved drugs for cancer therapy. Br. Med. Bull. 137, 13–27 (2021).
Gao, S., Wang, S., Fan, R. & Hu, J. Recent advances in the molecular mechanism of thalidomide teratogenicity. Biomed. Pharmacother. 127, 110114 (2020).
Rao, Y., Li, R. & Zhang, D. A drug from poison: how the therapeutic effect of arsenic trioxide on acute promyelocytic leukemia was discovered. Sci. China Life Sci. 56, 495–502 (2013).
Sanz, M. A. et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood 133, 1630–1643 (2019).
Pantziarka, P., Verbaanderd, C., Huys, I., Bouche, G. & Meheus, L. Repurposing drugs in oncology: from candidate selection to clinical adoption. Semin. Cancer Biol. 68, 186–191 (2021).
Moffat, J. G., Vincent, F., Lee, J. A., Eder, J. & Prunotto, M. Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat. Rev. Drug Discov. 16, 531–543 (2017).
Shim, J. S. & Liu, J. O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 10, 654–663 (2014).
Pillaiyar, T., Meenakshisundaram, S., Manickam, M. & Sankaranarayanan, M. A medicinal chemistry perspective of drug repositioning: recent advances and challenges in drug discovery. Eur. J. Med. Chem. 195, 112275 (2020).
Moffat, J. G., Rudolph, J. & Bailey, D. Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discov. 13, 588–602 (2014).
Rabben, H. L. et al. Computational drug repositioning and experimental validation of ivermectin in treatment of gastric cancer. Front. Pharmacol. 12, 625991 (2021).
Parvathaneni, V., Kulkarni, N. S., Muth, A. & Gupta, V. Drug repurposing: a promising tool to accelerate the drug discovery process. Drug Discov. Today 24, 2076–2085 (2019).
Tanoli, Z. et al. Exploration of databases and methods supporting drug repurposing: a comprehensive survey. Brief. Bioinform. 22, 1656–1678 (2021).
Huang, H., Zhang, P., Qu, X. A., Sanseau, P. & Yang, L. Systematic prediction of drug combinations based on clinical side-effects. Sci. Rep. 4, 7160 (2014).
Kuhn, M., Campillos, M., Letunic, I., Jensen, L. J. & Bork, P. A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol. 6, 343 (2010).
Celebi, R., Bear Don’t Walk, O. 4th, Movva, R., Alpsoy, S. & Dumontier, M. In-silico prediction of synergistic anti-cancer drug combinations using Multi-omics Data. Sci. Rep. 9, 8949 (2019).
Waldron, D. Cancer genomics: a multi-layer omics approach to cancer. Nat. Rev. Genet. 17, 436–437 (2016).
Meijer, R. P. Urothelial cancer organoids: a tool for bladder cancer research. Pathologe 42, 165–169 (2021).
Xu, H., Jiao, D., Liu, A. & Wu, K. Tumor organoids: applications in cancer modeling and potentials in precision medicine. J. Hematol. Oncol. 15, 58 (2022).
Kim, M. et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 10, 3991 (2019).
Zhang, Z. et al. Establishment of patient-derived tumor spheroids for non-small cell lung cancer. PLoS One 13, e0194016 (2018).
Kasagi, Y. et al. The esophageal organoid system reveals functional interplay between notch and cytokines in reactive epithelial changes. Cell Mol. Gastroenterol. Hepatol. 5, 333–352 (2018).
Nanki, K. et al. Divergent routes toward Wnt and R-spondin niche independency during human gastric carcinogenesis. Cell 174, 856–869.e17 (2018).
Kryeziu, K. et al. Increased sensitivity to SMAC mimetic LCL161 identified by longitudinal ex vivo pharmacogenomics of recurrent, KRAS mutated rectal cancer liver metastases. J. Transl. Med. 19, 384 (2021).
Cao, W. et al. Modeling liver cancer and therapy responsiveness using organoids derived from primary mouse liver tumors. Carcinogenesis 40, 145–154 (2019).
Nuciforo, S. et al. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep. 24, 1363–1376 (2018).
Uemura, N. et al. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 345, 784–789 (2001).
Choi, Y. J. et al. Discovery of a novel CDK7 inhibitor YPN-005 in small cell lung cancer. Eur. J. Pharmacol. 907, 174298 (2021).
Zhou, Z. et al. An organoid-based screen for epigenetic inhibitors that stimulate antigen presentation and potentiate T-cell-mediated cytotoxicity. Nat. Biomed. Eng. 5, 1320–1335 (2021).
Bayat, N. et al. The anti-angiogenic effect of atorvastatin in glioblastoma spheroids tumor cultured in fibrin gel: in 3D in vitro model. Asian Pac. J. Cancer Prev. 19, 2553–2560 (2018).
Vlachogiannis, G. et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 (2018).
Huang, Y. et al. Research progress, challenges, and breakthroughs of organoids as disease models. Front. Cell. Dev. Biol. 9, 740574 (2021).
Lau, H. C. H., Kranenburg, O., Xiao, H. & Yu, J. Organoid models of gastrointestinal cancers in basic and translational research. Nat. Rev. Gastroenterol. Hepatol. 17, 203–222 (2020).
Aberle, M. R. et al. Patient-derived organoid models help define personalized management of gastrointestinal cancer. Br. J. Surg. 105, e48–e60 (2018).
Yoshida, G. J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 13, 4 (2020).
Xue, H., Li, J., Xie, H. & Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 14, 1232–1244 (2018).
Jacquemet, G. et al. L-type calcium channels regulate filopodia stability and cancer cell invasion downstream of integrin signalling. Nat. Commun. 7, 13297 (2016).
Vanhaelen, Q. et al. Design of efficient computational workflows for in silico drug repurposing. Drug Discov. Today 22, 210–222 (2017).
Masoudi-Sobhanzadeh, Y., Omidi, Y., Amanlou, M. & Masoudi-Nejad, A. Drug databases and their contributions to drug repurposing. Genomics 112, 1087–1095 (2020).
Mottini, C., Napolitano, F., Li, Z., Gao, X. & Cardone, L. Computer-aided drug repurposing for cancer therapy: approaches and opportunities to challenge anticancer targets. Semin. Cancer Biol. 68, 59–74 (2021).
Issa, N. T., Stathias, V., Schürer, S. & Dakshanamurthy, S. Machine and deep learning approaches for cancer drug repurposing. Semin. Cancer Biol. 68, 132–142 (2021).
Lotfi Shahreza, M., Ghadiri, N., Mousavi, S. R., Varshosaz, J. & Green, J. R. A review of network-based approaches to drug repositioning. Brief. Bioinform. 19, 878–892 (2018).
Jarada, T. N., Rokne, J. G. & Alhajj, R. A review of computational drug repositioning: strategies, approaches, opportunities, challenges, and directions. J. Cheminform. 12, 46 (2020).
Montalvo-Casimiro, M. et al. Epidrug repurposing: discovering new faces of old acquaintances in cancer therapy. Front. Oncol. 10, 605386 (2020).
Serafin, M. B. et al. Drug repositioning in oncology. Am. J. Ther. 28, e111–e117 (2021).
Hurle, M. R. et al. Computational drug repositioning: from data to therapeutics. Clin. Pharmacol. Ther. 93, 335–341 (2013).
Khaladkar, M. et al. Uncovering novel repositioning opportunities using the Open Targets platform. Drug Discov. Today 22, 1800–1807 (2017).
Pushpakom, S. et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58 (2019).
Adasme, M. F., Parisi, D., Sveshnikova, A. & Schroeder, M. Structure-based drug repositioning: potential and limits. Semin. Cancer Biol. 68, 192–198 (2021).
Würth, R. et al. Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds. Drug Discov. Today 21, 190–199 (2016).
Jin, G. & Wong, S. T. C. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 19, 637–644 (2014).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Du, Z. & Lovly, C. M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 17, 58 (2018).
Panwar, V. et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target Ther. 8, 375 (2023).
Rodrik-Outmezguine, V. S. et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 1, 248–259 (2011).
Ahronian, L. G. et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 5, 358–367 (2015).
Chen, X., Liu, J., Gu, X. & Ding, F. Salidroside attenuates glutamate-induced apoptotic cell death in primary cultured hippocampal neurons of rats. Brain Res. 1238, 189–198 (2008).
Chen, T. et al. Suppressing receptor-interacting protein 140: a new sight for salidroside to treat cerebral ischemia. Mol. Neurobiol. 53, 6240–6250 (2016).
Wang, S. et al. Protective effects of salidroside in the MPTP/MPP(+)-induced model of Parkinson’s disease through ROS-NO-related mitochondrion pathway. Mol. Neurobiol. 51, 718–728 (2015).
Zheng, T. et al. Salidroside ameliorates insulin resistance through activation of a mitochondria-associated AMPK/PI3K/Akt/GSK3β pathway. Br. J. Pharmacol. 172, 3284–3301 (2015).
Tang, Y. et al. Salidroside exerts angiogenic and cytoprotective effects on human bone marrow-derived endothelial progenitor cells via Akt/mTOR/p70S6K and MAPK signalling pathways. Br. J. Pharmacol. 171, 2440–2456 (2014).
Liu, S., Li, Y. & Li, Z. Salidroside suppresses the activation of nasopharyngeal carcinoma cells via targeting miR-4262/GRP78 axis. Cell Cycle 21, 720–729 (2022).
Liu, R.-H. et al. Salidroside suppresses proliferation and migration in prostate cancer via the PI3K/AKT pathway. Cancer Biomark. 38, 321–332 (2023).
Zeng, Q. et al. Salidroside promotes sensitization to doxorubicin in human cancer cells by affecting the PI3K/Akt/HIF signal pathway and inhibiting the expression of tumor-resistance-related proteins. J. Nat. Prod. 85, 196–204 (2022).
Zeng, Q. et al. Targeting regulated cell death in tumor nanomedicines. Theranostics 12, 817–841 (2022).
Peng, F. et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct. Target. Ther. 7, 286 (2022).
Kupchan, S. M., Court, W. A., Dailey, R. G., Gilmore, C. J. & Bryan, R. F. Triptolide and tripdiolide, novel antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J. Am. Chem. Soc. 94, 7194–7195 (1972).
Qiu, D. & Kao, P. N. Immunosuppressive and anti-inflammatory mechanisms of triptolide, the principal active diterpenoid from the Chinese medicinal herb Tripterygium wilfordii Hook. f. Drugs R. D. 4, 1–18 (2003).
Li, X.-J., Jiang, Z.-Z. & Zhang, L.-Y. Triptolide: progress on research in pharmacodynamics and toxicology. J. Ethnopharmacol. 155, 67–79 (2014).
Ziaei, S. & Halaby, R. Immunosuppressive, anti-inflammatory and anti-cancer properties of triptolide: a mini review. Avicenna J. Phytomed. 6, 149–164 (2016).
Zhang, L. & Lu, S. Overview of medicinally important diterpenoids derived from plastids. Mini. Rev. Med. Chem. 17, 988–1001 (2017).
Cai, J. et al. Natural product triptolide induces GSDME-mediated pyroptosis in head and neck cancer through suppressing mitochondrial hexokinase-ΙΙ. J. Exp. Clin. Cancer Res. 40, 190 (2021).
Wang, C. et al. Toward targeted therapy in chemotherapy-resistant pancreatic cancer with a smart triptolide nanomedicine. Oncotarget 7, 8360–8372 (2016).
Zhao, F. et al. Triptolide induces protective autophagy through activation of the CaMKKβ-AMPK signaling pathway in prostate cancer cells. Oncotarget 7, 5366–5382 (2016).
Kitzen, J. J. E. M. et al. Phase I dose-escalation study of F60008, a novel apoptosis inducer, in patients with advanced solid tumours. Eur. J. Cancer 45, 1764–1772 (2009).
Gao, S. et al. Cardiovascular actions and therapeutic potential of tanshinone IIA. Atherosclerosis 220, 3–10 (2012).
Kim, N. Y. et al. Tanshinone IIA exerts autophagic cell death through down-regulation of β-catenin in renal cell carcinoma cells. Biochimie 200, 119–130 (2022).
Ni, H. et al. Tanshinone IIA inhibits gastric cancer cell stemness through inducing ferroptosis. Environ. Toxicol. 37, 192–200 (2022).
Lin, C. Y. et al. Simultaneous induction of apoptosis and necroptosis by Tanshinone IIA in human hepatocellular carcinoma HepG2 cells. Cell Death Discov. 2, 16065 (2016).
Sainero-Alcolado, L., Liaño-Pons, J., Ruiz-Pérez, M. V. & Arsenian-Henriksson, M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. 29, 1304–1317 (2022).
Kansara, S. et al. The emerging regulatory roles of non-coding RNAs associated with glucose metabolism in breast cancer. Semin. Cancer Biol. 95, https://doi.org/10.1016/j.semcancer.2023.06.007 (2023).
Stine, Z. E., Schug, Z. T., Salvino, J. M. & Dang, C. V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 21, 141–162 (2022).
Sun, L., Zhang, H. & Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 13, 877–919 (2022).
Pavlova, N. N., Zhu, J. & Thompson, C. B. The hallmarks of cancer metabolism: still emerging. Cell Metab. 34, 355–377 (2022).
Prakash, A. & Jarvis, B. Leflunomide: a review of its use in active rheumatoid arthritis. Drugs 58, 1137–1164 (1999).
Mattar, T., Kochhar, K., Bartlett, R., Bremer, E. G. & Finnegan, A. Inhibition of the epidermal growth factor receptor tyrosine kinase activity by leflunomide. FEBS Lett. 334, 161–164 (1993).
Ghosh, S. et al. Structure-based design of potent inhibitors of EGF-receptor tyrosine kinase as anti-cancer agents. Anticancer Drug Des. 14, 403–410 (1999).
Brown, K. K., Spinelli, J. B., Asara, J. M. & Toker, A. Adaptive reprogramming of de novo pyrimidine synthesis is a metabolic vulnerability in triple-negative breast cancer. Cancer Discov. 7, 391–399 (2017).
Mathur, D. et al. PTEN regulates glutamine flux to pyrimidine synthesis and sensitivity to dihydroorotate dehydrogenase inhibition. Cancer Discov. 7, 380–390 (2017).
Khutornenko, A. A., Dalina, A. A., Chernyak, B. V., Chumakov, P. M. & Evstafieva, A. G. The role of dihydroorotate dehydrogenase in apoptosis induction in response to inhibition of the mitochondrial respiratory chain complex III. Acta Nat. 6, 69–75 (2014).
Hubackova, S. et al. Replication and ribosomal stress induced by targeting pyrimidine synthesis and cellular checkpoints suppress p53-deficient tumors. Cell Death Dis. 11, 110 (2020).
Li, H. et al. The combination of disulfiram and copper for cancer treatment. Drug Discov. Today 25, 1099–1108 (2020).
Lewison, E. F. Spontaneous regression of breast cancer. Prog. Clin. Biol. Res. 12, 47–53 (1977).
Skrott, Z. et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 552, 194–199 (2017).
Du, C. et al. Disulfiram/copper induces antitumor activity against gastric cancer cells in vitro and in vivo by inhibiting S6K1 and c-Myc. Cancer Chemother. Pharmacol. 89, 451–458 (2022).
Xie, J. et al. Disulfiram/Cu kills and sensitizes BRAF-mutant thyroid cancer cells to BRAF kinase inhibitor by ROS-dependently relieving feedback activation of MAPK/ERK and PI3K/AKT pathways. Int. J. Mol. Sci. 24, https://doi.org/10.3390/ijms24043418 (2023).
Martínez-Lostao, L., Anel, A. & Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 21, 5047–5056 (2015).
Garner, H. & de Visser, K. E. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat. Rev. Immunol. 20, 483–497 (2020).
Vinay, D. S. et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 35, S185–S198 (2015).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011).
Liu, Y. et al. Renoprotective effects of oleanolic acid and its possible mechanisms in rats with diabetic kidney disease. Biochem. Biophys. Res. Commun. 636, 1–9 (2022).
Kim, S., Lee, H., Lee, S., Yoon, Y. & Choi, K.-H. Antimicrobial action of oleanolic acid on Listeria monocytogenes, Enterococcus faecium, and Enterococcus faecalis. PloS One 10, e0118800 (2015).
Pertino, M. W. et al. Antiprotozoal activity of triazole derivatives of dehydroabietic acid and oleanolic acid. Molecules 22, 369 (2017).
Baer-Dubowska, W., Narożna, M. & Krajka-Kuźniak, V. Anti-cancer potential of synthetic oleanolic acid derivatives and their conjugates with NSAIDs. Molecules 26, 4957 (2021).
Xu, Q.-F. et al. Oleanolic acid regulates the Treg/Th17 imbalance in gastric cancer by targeting IL-6 with miR-98-5p. Cytokine 148, 155656 (2021).
Lu, X. et al. Inhibition of NF-κB is required for oleanolic acid to downregulate PD-L1 by promoting DNA demethylation in gastric cancer cells. J. Biochem. Mol. Toxicol. 35, e22621 (2021).
Yang, J. et al. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 31, 248–258 (2013).
Sherr, C. J. & McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2, 103–112 (2002).
Sherr, C. J. Cancer cell cycles. Science 274, 1672–1677 (1996).
Williams, B. O. et al. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat. Genet. 7, 480–484 (1994).
Manning, A. L. & Dyson, N. J. RB: mitotic implications of a tumour suppressor. Nat. Rev. Cancer 12, 220–226 (2012).
Bykov, V. J. N., Eriksson, S. E., Bianchi, J. & Wiman, K. G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 18, 89–102 (2018).
Jiang, W., Hu, J. W., He, X. R., Jin, W. L. & He, X. Y. Statins: a repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 40, 241 (2021).
Di Bello, E., Zwergel, C., Mai, A. & Valente, S. The innovative potential of statins in cancer: new targets for new therapies. Front. Chem. 8, 516 (2020).
Mandal, C. C., Ghosh-Choudhury, N., Yoneda, T., Choudhury, G. G. & Ghosh-Choudhury, N. Simvastatin prevents skeletal metastasis of breast cancer by an antagonistic interplay between p53 and CD44. J. Biol. Chem. 286, 11314–11327 (2011).
Miyajima, C., Hayakawa, Y., Inoue, Y., Nagasaka, M. & Hayashi, H. HMG-CoA reductase inhibitor statins activate the transcriptional activity of p53 by regulating the expression of TAZ. Pharmacology 15, 1015 (2022).
Li, J. et al. Simvastatin and Atorvastatin inhibit DNA replication licensing factor MCM7 and effectively suppress RB-deficient tumors growth. Cell Death Dis. 8, e2673 (2017).
Gao, J. & Pickett, H. A. Targeting telomeres: advances in telomere maintenance mechanism-specific cancer therapies. Nat. Rev. Cancer 22, 515–532 (2022).
Vishwakarma, K., Dey, R. & Bhatt, H. Telomerase: a prominent oncological target for development of chemotherapeutic agents. Eur. J. Med. Chem. 249, 115121 (2023).
Hoffmeyer, K. et al. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science 336, 1549–1554 (2012).
Park, J.-I. et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460, 66–72 (2009).
Ghosh, A. et al. Telomerase directly regulates NF-κB-dependent transcription. Nat. Cell. Biol. 14, 1270–1281 (2012).
Perkins, N. D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 12, 121–132 (2012).
Yu, C. et al. Metformin sensitizes non-small cell lung cancer cells to an epigallocatechin-3-gallate (EGCG) treatment by suppressing the Nrf2/HO-1 signaling pathway. Int. J. Biol. Sci. 13, 1560–1569 (2017).
Fujiki, H., Sueoka, E., Rawangkan, A. & Suganuma, M. Human cancer stem cells are a target for cancer prevention using (-)-epigallocatechin gallate. J. Cancer Res. Clin. Oncol. 143, 2401–2412 (2017).
Luo, K.-W. et al. EGCG inhibited bladder cancer SW780 cell proliferation and migration both in vitro and in vivo via down-regulation of NF-κB and MMP-9. J. Nutr. Biochem. 41, 56–64 (2017).
Wang, J., Man, G. C. W., Chan, T. H., Kwong, J. & Wang, C. C. A prodrug of green tea polyphenol (-)-epigallocatechin-3-gallate (Pro-EGCG) serves as a novel angiogenesis inhibitor in endometrial cancer. Cancer Lett. 412, 10–20 (2018).
Wei, R., Penso, N. E. C., Hackman, R. M., Wang, Y. & Mackenzie, G. G. Epigallocatechin-3-Gallate (EGCG) suppresses pancreatic cancer cell growth, invasion, and migration partly through the inhibition of Akt pathway and epithelial-mesenchymal transition: enhanced efficacy when combined with gemcitabine. Nutrients 11, 1856 (2019).
Moradzadeh, M., Hosseini, A., Erfanian, S. & Rezaei, H. Epigallocatechin-3-gallate promotes apoptosis in human breast cancer T47D cells through down-regulation of PI3K/AKT and Telomerase. Pharmacol. Rep. 69, 924–928 (2017).
Dong, C. et al. Epigallocatechin-3-gallate suppresses the growth of human osteosarcoma by inhibiting the Wnt/β-catenin signalling pathway. Bioengineered 13, 8490–8502 (2022).
Hanahan, D. & Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364 (1996).
Bergers, G. & Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603 (2008).
Zeng, Z.-W., Chen, D., Chen, L., He, B. & Li, Y. A comprehensive overview of Artemisinin and its derivatives as anticancer agents. Eur. J. Med. Chem. 247, 115000 (2023).
Xu, C.-C. et al. Synthesis and in vitro antitumor evaluation of dihydroartemisinin-cinnamic acid ester derivatives. Eur. J. Med. Chem. 107, 192–203 (2016).
Wang, S.-J. et al. Dihydroartemisinin inhibits angiogenesis in pancreatic cancer by targeting the NF-κB pathway. Cancer Chemother. Pharmacol. 68, 1421–1430 (2011).
Huang, Y., Hong, W. & Wei, X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 15, 129 (2022).
Stuelten, C. H., Parent, C. A. & Montell, D. J. Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat. Rev. Cancer 18, 296–312 (2018).
Brugmans, J. P. et al. Mebendazole in enterobiasis. Radiochemical and pilot clinical study in 1278 subjects. JAMA 217, 313–316 (1971).
Guerini, A. E. et al. Mebendazole as a candidate for drug repurposing in oncology: an extensive review of current literature. Cancers 11, 1284 (2019).
Son, D.-S., Lee, E.-S. & Adunyah, S. E. The antitumor potentials of benzimidazole anthelmintics as repurposing drugs. Immune Netw. 20, e29 (2020).
Armando, R. G., Mengual Gómez, D. L. & Gomez, D. E. New drugs are not enough‑drug repositioning in oncology: an update. Int. J. Oncol. 56, 651–684 (2020).
Nath, J. et al. Drug repurposing and relabeling for cancer therapy: emerging benzimidazole antihelminthics with potent anticancer effects. Life Sci. 258, 118189 (2020).
Joe, N. S. et al. Mebendazole prevents distant organ metastases in part by decreasing ITGβ4 expression and cancer stemness. Breast Cancer Res. 24, 98 (2022).
Limbu, K. R., Chhetri, R. B., Oh, Y. S., Baek, D. J. & Park, E.-Y. Mebendazole impedes the proliferation and migration of pancreatic cancer cells through SK1 inhibition dependent pathway. Molecules 27, 8127 (2022).
Zhang, J. et al. Budding uninhibited by benzimidazoles 1 promotes cell proliferation, invasion, and epithelial-mesenchymal transition via the Wnt/β-catenin signaling in glioblastoma. Heliyon 9, e16996 (2023).
Kralova, V. et al. Flubendazole and mebendazole impair migration and epithelial to mesenchymal transition in oral cell lines. Chem. Biol. Interact. 293, 124–132 (2018).
Dobrosotskaya, I. Y., Hammer, G. D., Schteingart, D. E., Maturen, K. E. & Worden, F. P. Mebendazole monotherapy and long-term disease control in metastatic adrenocortical carcinoma. Endocr. Pract. 17, e59–e62 (2011).
Nygren, P. & Larsson, R. Drug repositioning from bench to bedside: tumour remission by the antihelmintic drug mebendazole in refractory metastatic colon cancer. Acta Oncol. 53, 427–428 (2014).
Aguilera, A. & García-Muse, T. Causes of genome instability. Annu. Rev. Genet. 47, 1–32 (2013).
Lawrence, K. S., Chau, T. & Engebrecht, J. DNA damage response and spindle assembly checkpoint function throughout the cell cycle to ensure genomic integrity. PLoS Genet. 11, e1005150 (2015).
Brambati, A., Colosio, A., Zardoni, L., Galanti, L. & Liberi, G. Replication and transcription on a collision course: eukaryotic regulation mechanisms and implications for DNA stability. Front. Genet. 6, 166 (2015).
Christensen, S. et al. 5-Fluorouracil treatment induces characteristic T>G mutations in human cancer. Nat. Commun. 10, 4571 (2019).
Ronis, M. J. J. Effects of soy containing diet and isoflavones on cytochrome P450 enzyme expression and activity. Drug Metab. Rev. 48, 331–341 (2016).
Sak, K. Current epidemiological knowledge about the role of flavonoids in prostate carcinogenesis. Exp. Oncol. 39, 98–105 (2017).
Sak, K. Epidemiological evidences on dietary flavonoids and breast cancer risk: a narrative review. Asian Pac. J. Cancer Prev. 18, 2309–2328 (2017).
Hu, X. et al. Genistein-induced DNA damage is repaired by nonhomologous end joining and homologous recombination in TK6 cells. J. Cell. Physiol. 234, 2683–2692 (2019).
Liu, X. et al. Genistein sensitizes glioblastoma cells to carbon ions via inhibiting DNA-PKcs phosphorylation and subsequently repressing NHEJ and delaying HR repair pathways. Radiother. Oncol. 129, 84–94 (2018).
Suraweera, T. L., Merlin, J. P. J., Dellaire, G., Xu, Z. & Rupasinghe, H. P. V. Genistein and procyanidin B2 reduce carcinogen-induced reactive oxygen species and DNA damage through the activation of Nrf2/ARE cell signaling in Bronchial Epithelial cells in vitro. Int. J. Mol. Sci. 24, 3676 (2023).
Diakos, C. I., Charles, K. A., McMillan, D. C. & Clarke, S. J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 15, e493–e503 (2014).
Greten, F. R. & Grivennikov, S. I. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 51, 27–41 (2019).
Wang, J. et al. Celecoxib could reverse the hypoxia-induced angiopoietin-2 upregulation in gastric cancer. Cancer Lett. 242, 20–27 (2006).
Lin, X.-M. et al. Cisplatin induces chemoresistance through the PTGS2-mediated anti-apoptosis in gastric cancer. Int. J. Biochem. Cell Biol. 116, 105610 (2019).
Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 1, a000141 (2009).
Capece, D. et al. NF-κB: blending metabolism, immunity, and inflammation. Trends Immunol. 43, 757–775 (2022).
Chauhan, A., Islam, A. U., Prakash, H. & Singh, S. Phytochemicals targeting NF-κB signaling: potential anti-cancer interventions. J. Pharm. Anal. 12, 394–405 (2022).
Vergani, E. et al. miR-146a-5p impairs melanoma resistance to kinase inhibitors by targeting COX2 and regulating NFkB-mediated inflammatory mediators. Cell Commun. Signal 18, 156 (2020).
Zhang, C. et al. Celecoxib attenuates hepatocellular proliferative capacity during hepatocarcinogenesis by modulating a PTEN/NF-κB/PRL-3 pathway. RSC Adv. 9, 20624–20632 (2019).
Guo, Q. et al. A comprehensive evaluation of clinical efficacy and safety of celecoxib in combination with chemotherapy in metastatic or postoperative recurrent gastric cancer patients: a preliminary, three-center, clinical trial study. Medicines 98, e16234 (2019).
Yuan, S., Norgard, R. J. & Stanger, B. Z. Cellular plasticity in cancer. Cancer Discov. 9, 837–851 (2019).
Tan, S. H. & Barker, N. Stemming colorectal cancer growth and metastasis: HOXA5 forces cancer stem cells to differentiate. Cancer Cell 28, 683–685 (2015).
Perekatt, A. O. et al. SMAD4 Suppresses WNT-driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res. 78, 4878–4890 (2018).
Ordóñez-Morán, P., Dafflon, C., Imajo, M., Nishida, E. & Huelsken, J. HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 28, 815–829 (2015).
Giovannucci, E. et al. Diabetes and cancer: a consensus report. CA Cancer J. Clin. 60, 207–221 (2010).
Lv, Z. & Guo, Y. Metformin and its benefits for various diseases. Front. Endocrinol. 11, 191 (2020).
Gandini, S. et al. Metformin and cancer risk and mortality: a systematic review and meta-analysis taking into account biases and confounders. Cancer Prev. Res. 7, 867–885 (2014).
Franciosi, M. et al. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One 8, e71583 (2013).
Zhang, K., Bai, P., Dai, H. & Deng, Z. Metformin and risk of cancer among patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Prim. Care Diabetes 15, 52–58 (2021).
Decensi, A. et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev. Res. 3, 1451–1461 (2010).
Evans, J. M. M., Donnelly, L. A., Emslie-Smith, A. M., Alessi, D. R. & Morris, A. D. Metformin and reduced risk of cancer in diabetic patients. Br. Med. J. 330, 1304–1305 (2005).
Bednar, F. & Simeone, D. M. Metformin and cancer stem cells: old drug, new targets. Cancer Prev. Res. 5, 351–354 (2012).
Chen, G., Xu, S., Renko, K. & Derwahl, M. Metformin inhibits growth of thyroid carcinoma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the effect of chemotherapeutic agents. J. Clin. Endocrinol. Metab. 97, E510–E520 (2012).
Chang, H. R. et al. HNF4α is a therapeutic target that links AMPK to WNT signalling in early-stage gastric cancer. Gut 65, 19–32 (2016).
Wang, C. et al. Metformin inhibits pancreatic cancer metastasis caused by SMAD4 deficiency and consequent HNF4G upregulation. Protein Cell 12, 128–144 (2021).
Seo, Y. et al. Metformin suppresses cancer stem cells through AMPK activation and inhibition of protein prenylation of the mevalonate pathway in colorectal cancer. Cancers 12, 2554 (2020).
Li, H. et al. Will metformin use lead to a decreased risk of thyroid cancer? A systematic review and meta-analyses. Eur. J. Med. Res. 28, 392 (2023).
Yao, K., Zheng, H. & Li, T. Association between metformin use and the risk, prognosis of gynecologic cancer. Front. Oncol. 12, 942380 (2022).
Thienpont, B., Van Dyck, L. & Lambrechts, D. Tumors smother their epigenome. Mol. Cell Oncol. 3, e1240549 (2016).
Donohoe, D. R. et al. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 48, 612–626 (2012).
Dinda, B., SilSarma, I., Dinda, M. & Rudrapaul, P. Oroxylum indicum (L.) Kurz, an important Asian traditional medicine: from traditional uses to scientific data for its commercial exploitation. J. Ethnopharmacol. 161, 255–278 (2015).
Gasiorowski, K. et al. Flavones from root of Scutellaria baicalensis Georgi: drugs of the future in neurodegeneration? CNS Neurol. Disord. Drug Targ. 10, 184–191 (2011).
Dinda, B. et al. Therapeutic potentials of baicalin and its aglycone, baicalein against inflammatory disorders. Eur. J. Med. Chem. 131, 68–80 (2017).
Chen, Y. et al. Baicalein resensitizes tamoxifen-resistant breast cancer cells by reducing aerobic glycolysis and reversing mitochondrial dysfunction via inhibition of hypoxia-inducible factor-1α. Clin. Transl. Med. 11, e577 (2021).
McDonald, O. G. et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 49, 367–376 (2017).
Guo, D. et al. Baicalein inhibits the progression and promotes radiosensitivity of esophageal squamous cell carcinoma by targeting HIF-1A. Drug Des. Dev. Ther. 16, 2423–2436 (2022).
Shabbir, M., Love, J. & Montgomery, B. Phase I trial of PC-Spes2 in advanced hormone refractory prostate cancer. Oncol. Rep. 19, 831–835 (2008).
Sender, R., Fuchs, S. & Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14, e1002533 (2016).
Sepich-Poore, G. D. et al. The microbiome and human cancer. Science 371, eabc4552 (2021).
Jandhyala, S. M. et al. Role of the normal gut microbiota. World J. Gastroenterol. 21, 8787–8803 (2015).
Akshintala, V. S., Talukdar, R., Singh, V. K. & Goggins, M. The gut microbiome in pancreatic disease. Clin. Gastroenterol. Hepatol. 17, 290–295 (2019).
Zhuang, L. et al. Intestinal microbiota in early life and its implications on childhood health. Genom. Proteom. Bioinform. 17, 13–25 (2019).
Fan, Y. & Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 19, 55–71 (2021).
Tilg, H., Adolph, T. E., Gerner, R. R. & Moschen, A. R. The intestinal microbiota in colorectal cancer. Cancer Cell 33, 954–964 (2018).
Sholl, J., Sepich-Poore, G. D., Knight, R. & Pradeu, T. Redrawing therapeutic boundaries: microbiota and cancer. Trends Cancer 8, 87–97 (2022).
Sheng, W., Ji, G. & Zhang, L. Immunomodulatory effects of inulin and its intestinal metabolites. Front. Immunol. 14, 1224092 (2023).
Shoaib, M. et al. Inulin: properties, health benefits and food applications. Carbohydr. Polym. 147, 444–454 (2016).
Wang, C. et al. The preventive effects of inulin, cellulose, and their mixture on colorectal cancer liver metastasis in mice by regulating gut microbiota. J. Food Sci. 88, 4705–4717 (2023).
Han, K. et al. Generation of systemic antitumour immunity via the in situ modulation of the gut microbiome by an orally administered inulin gel. Nat. Biomed. Eng. 5, 1377–1388 (2021).
Wu, H., Van Der Pol, W. J., Dubois, L. G., Morrow, C. D. & Tollefsbol, T. O. Dietary supplementation of inulin contributes to the prevention of estrogen receptor-negative mammary cancer by alteration of gut microbial communities and epigenetic regulations. Int. J. Mol. Sci. 24, 9015 (2023).
Oliero, M. et al. Inulin impacts tumorigenesis promotion by colibactin-producing Escherichia coli in ApcMin/+ mice. Front. Microbiol. 14, 1067505 (2023).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Basu, A. The interplay between apoptosis and cellular senescence: Bcl-2 family proteins as targets for cancer therapy. Pharmacol. Ther. 230, 107943 (2022).
Moiseeva, O., Guillon, J. & Ferbeyre, G. Senescence: a program in the road to cell elimination and cancer. Semin. Cancer Biol. 81, 48–53 (2022).
Domen, A. et al. Cellular senescence in cancer: clinical detection and prognostic implications. J. Exp. Clin. Cancer Res. 41, 360 (2022).
Pérez, R. F., Tejedor, J. R., Fernández, A. F. & Fraga, M. F. Aging and cancer epigenetics: where do the paths fork? Aging Cell 21, e13709 (2022).
Demaria, M. et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 (2017).
Liu, D. & Hornsby, P. J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67, 3117–3126 (2007).
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P. Y. & Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl Acad. Sci. Usa. 98, 12072–12077 (2001).
Batiha, G. E.-S. et al. The pharmacological activity, biochemical properties, and pharmacokinetics of the major natural polyphenolic flavonoid: quercetin. Foods 9, https://doi.org/10.3390/foods9030374 (2020).
Li, Y. et al. Quercetin, inflammation and immunity. Nutrients 8, 167 (2016).
Adami, B. S. et al. Morphological and mechanical changes induced by quercetin in human T24 bladder cancer cells. Micron 151, 103152 (2021).
Özsoy, S., Becer, E., Kabadayı, H., Vatansever, H. S. & Yücecan, S. Quercetin-mediated apoptosis and cellular senescence in human colon cancer. Anticancer Agents Med. Chem. 20, 1387–1396 (2020).
Tanigawa, S., Fujii, M. & Hou, D.-X. Stabilization of p53 is involved in quercetin-induced cell cycle arrest and apoptosis in HepG2 cells. Biosci. Biotechnol. Biochem. 72, 797–804 (2008).
Bientinesi, E. et al. Doxorubicin-induced senescence in normal fibroblasts promotes in vitro tumour cell growth and invasiveness: the role of Quercetin in modulating these processes. Mech. Ageing Dev. 206, 111689 (2022).
Saravia, J., Chapman, N. M. & Chi, H. Helper T cell differentiation. Cell. Mol. Immunol. 16, 634–643 (2019).
Ohue, Y. & Nishikawa, H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 110, 2080–2089 (2019).
Marciscano, A. E. & Anandasabapathy, N. The role of dendritic cells in cancer and anti-tumor immunity. Semin. Immunol. 52, 101481 (2021).
Vitale, I., Manic, G., Coussens, L. M., Kroemer, G. & Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 30, 36–50 (2019).
Papait, A. et al. The multifaceted roles of MSCs in the tumor microenvironment: interactions with immune cells and exploitation for therapy. Front. Cell Dev. Biol. 8, 447 (2020).
Zhao, L. et al. Impacts and mechanisms of metabolic reprogramming of tumor microenvironment for immunotherapy in gastric cancer. Cell Death Dis. 13, 378 (2022).
Oya, Y., Hayakawa, Y. & Koike, K. Tumor microenvironment in gastric cancers. Cancer Sci. 111, 2696–2707 (2020).
Jin, M.-Z. & Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 5, 166 (2020).
Hinshaw, D. C. & Shevde, L. A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 79, 4557–4566 (2019).
Wang, J. X. et al. Lactic acid and an acidic tumor microenvironment suppress anticancer immunity. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21218363 (2020).
Kamm, A. et al. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 93, 102–114 (2019).
Vaupel, P. & Multhoff, G. Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front. Immunol. 8, 1887 (2017).
Moldogazieva, N. T., Lutsenko, S. V. & Terentiev, A. A. Reactive oxygen and nitrogen species-induced protein modifications: implication in carcinogenesis and anticancer therapy. Cancer Res. 78, 6040–6047 (2018).
Gajewski, T. F., Schreiber, H. & Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
Fu, T. et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J. Hematol. Oncol. 14, 98 (2021).
Haddad, R. & Saldanha-Araujo, F. Mechanisms of T-cell immunosuppression by mesenchymal stromal cells: what do we know so far? Biomed. Res. Int. 2014, 216806 (2014).
Chen, Y. et al. Tumor-associated macrophages: an accomplice in solid tumor progression. J. Biomed. Sci. 26, 78 (2019).
Singh, D., Gupta, M., Sarwat, M. & Siddique, H. R. Apigenin in cancer prevention and therapy: a systematic review and meta-analysis of animal models. Crit. Rev. Oncol. Hematol. 176, 103751 (2022).
Coombs, M. R. P., Harrison, M. E. & Hoskin, D. W. Apigenin inhibits the inducible expression of programmed death ligand 1 by human and mouse mammary carcinoma cells. Cancer Lett. 380, 424–433 (2016).
Xu, L. et al. Apigenin suppresses PD-L1 expression in melanoma and host dendritic cells to elicit synergistic therapeutic effects. J. Exp. Clin. Cancer Res. 37, 261 (2018).
Husain, K. et al. Apigenin targets MicroRNA-155, enhances SHIP-1 expression, and augments anti-tumor responses in pancreatic cancer. Cancers 14, https://doi.org/10.3390/cancers14153613 (2022).
DeMaria, P. J. & Bilusic, M. Cancer vaccines. Hematol. Oncol. Clin. North Am. 33, 199–214 (2019).
Gonciarz, W., Chyb, M. & Chmiela, M. Mycobacterium bovis BCG increase the selected determinants of monocyte/macrophage activity, which were diminished in response to gastric pathogen Helicobacter pylori. Sci. Rep. 13, 3107 (2023).
Aznar, M. A. et al. Repurposing the yellow fever vaccine for intratumoral immunotherapy. EMBO Mol. Med. 12, e10375 (2020).
Tai, L.-H. et al. Perioperative influenza vaccination reduces postoperative metastatic disease by reversing surgery-induced dysfunction in natural killer cells. Clin. Cancer Res. 19, 5104–5115 (2013).
Tai, L.-H. et al. Phosphodiesterase-5 inhibition reduces postoperative metastatic disease by targeting surgery-induced myeloid derived suppressor cell-dependent inhibition of Natural Killer cell cytotoxicity. Oncoimmunology 7, e1431082 (2018).
Martínez-Reyes, I. & Chandel, N. S. Cancer metabolism: looking forward. Nat. Rev. Cancer 21, 669–680 (2021).
Nyberg, P., Salo, T. & Kalluri, R. Tumor microenvironment and angiogenesis. Front. Biosci. 13, 6537–6553 (2008).
Quesada, A. R., Medina, M. A. & Alba, E. Playing only one instrument may be not enough: limitations and future of the antiangiogenic treatment of cancer. Bioessays 29, 1159–1168 (2007).
Pavlides, S. et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 8, 3984–4001 (2009).
Ho, P.-C. et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228 (2015).
Dong, X. et al. Emodin: a review of its pharmacology, toxicity and pharmacokinetics. Phytother. Res. 30, 1207–1218 (2016).
Peng, W., Qin, R., Li, X. & Zhou, H. Botany, phytochemistry, pharmacology, and potential application of Polygonum cuspidatum Sieb.et Zucc.: a review. J. Ethnopharmacol. 148, 729–745 (2013).
Kim, Y.-S. et al. Emodin sensitizes hepatocellular carcinoma cells to the anti-cancer effect of sorafenib through suppression of cholesterol metabolism. Int. J. Mol. Sci. 19, https://doi.org/10.3390/ijms19103127 (2018).
Yang, N. et al. Emodin induced SREBP1-dependent and SREBP1-independent apoptosis in hepatocellular carcinoma cells. Front. Pharmacol. 10, 709 (2019).
Xing, Y.-X. et al. Anti-cancer effects of emodin on HepG2 cells as revealed by 1H NMR based metabolic profiling. J. Proteome Res. 17, 1943–1952 (2018).
Gao, R. et al. Anti-tumor effect of aloe-emodin on cervical cancer cells was associated with human papillomavirus E6/E7 and glucose metabolism. Onco-Targets Ther. 12, 3713–3721 (2019).
Nagelkerke, A., Bussink, J., Rowan, A. E. & Span, P. N. The mechanical microenvironment in cancer: how physics affects tumours. Semin. Cancer Biol. 35, 62–70 (2015).
Taber, L. A. & Humphrey, J. D. Stress-modulated growth, residual stress, and vascular heterogeneity. J. Biomech. Eng. 123, 528–535 (2001).
Yeung, T. et al. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskeleton 60, 24–34 (2005).
Iskratsch, T., Wolfenson, H. & Sheetz, M. P. Appreciating force and shape—the rise of mechanotransduction in cell biology. Nat. Rev. Mol. Cell Biol. 15, 825–833 (2014).
Qin, X. et al. The tumor biochemical and biophysical microenvironments synergistically contribute to cancer cell malignancy. Cell Mol. Immunol. 17, 1186–1187 (2020).
Junttila, M. R. & de Sauvage, F. J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354 (2013).
Calvo, F. et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 15, 637–646 (2013).
Vermeulen, L. et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 12, 468–476 (2010).
Plaks, V., Kong, N. & Werb, Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16, 225–238 (2015).
Panciera, T., Azzolin, L., Cordenonsi, M. & Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 18, 758–770 (2017).
Fowler, J. F., Woolery-Lloyd, H., Waldorf, H. & Saini, R. Innovations in natural ingredients and their use in skin care. J. Drugs Dermatol. 9, S72–S83 (2010).
Fu, Y.-S. et al. Pharmacological properties and underlying mechanisms of curcumin and prospects in medicinal potential. Biomed. Pharmacother. 141, https://doi.org/10.1016/j.biopha.2021.111888 (2021).
Wang, M. et al. Potential mechanisms of action of curcumin for cancer prevention: focus on cellular signaling pathways and miRNAs. Int. J. Biol. Sci. 15, 1200–1214 (2019).
Jalilian, E. et al. Neutralizing tumor-related inflammation and reprogramming of cancer-associated fibroblasts by Curcumin in breast cancer therapy. Sci. Rep. 13, https://doi.org/10.1038/s41598-023-48073-w (2023).
Jang, B.-Y., Shin, M. K., Han, D.-H. & Sung, J.-S. Curcumin disrupts a positive feedback loop between ADMSCs and cancer cells in the breast tumor microenvironment via the CXCL12/CXCR4 Axis. Pharmaceutics 15, https://doi.org/10.3390/pharmaceutics15112627 (2023).
Mao, X. et al. Curcumin suppresses LGR5(+) colorectal cancer stem cells by inducing autophagy and via repressing TFAP2A-mediated ECM pathway. J. Nat. Med. 75, 590–601 (2021).
Jing, X. et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 18, https://doi.org/10.1186/s12943-019-1089-9 (2019).
Vaupel, P., Höckel, M. & Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal 9, 1221–1235 (2007).
Daponte, A. et al. Prognostic significance of Hypoxia-Inducible Factor 1 alpha(HIF-1 alpha) expression in serous ovarian cancer: an immunohistochemical study. BMC Cancer 8, https://doi.org/10.1186/1471-2407-8-335 (2008).
Nishikimi, M., Koshizaka, T., Ozawa, T. & Yagi, K. Occurrence in humans and guinea pigs of the gene related to their missing enzyme L-gulono-gamma-lactone oxidase. Arch. Biochem. Biophys. 267, 842–846 (1988).
Padayatty, S. J. et al. Vitamin C as an antioxidant: evaluation of its role in disease prevention. J. Am. Coll. Nutr. 22, 18–35 (2003).
Gęgotek, A. & Skrzydlewska, E. Ascorbic acid as antioxidant. Vitam. Horm. 121, 247–270 (2023).
Ozbudak, I. H., Karaveli, S., Simsek, T., Erdogan, G. & Pestereli, E. Neoangiogenesis and expression of hypoxia-inducible factor 1alpha, vascular endothelial growth factor, and glucose transporter-1 in endometrioid type endometrium adenocarcinomas. Gynecol. Oncol. 108, 603–608 (2008).
Jóźwiak, P. et al. Expression of hypoxia inducible factor 1α and 2α and its association with vitamin C level in thyroid lesions. J. Biomed. Sci. 24, https://doi.org/10.1186/s12929-017-0388-y (2017).
Nakanishi, K., Hiramoto, K. & Ooi, K. High-dose vitamin C exerts its anti-cancer effects in a xenograft model of colon cancer by suppressing angiogenesis. Biol. Pharm. Bull. 44, 884–887 (2021).
Campbell, E. J. et al. Activation of the hypoxia pathway in breast cancer tissue and patient survival are inversely associated with tumor ascorbate levels. BMC Cancer 19, https://doi.org/10.1186/s12885-019-5503-x (2019).
Vaupel, P., Kallinowski, F. & Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 49, 6449–6465 (1989).
Huber, V. et al. Cancer acidity: an ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 43, 74–89 (2017).
Peppicelli, S., Bianchini, F., Torre, E. & Calorini, L. Contribution of acidic melanoma cells undergoing epithelial-to-mesenchymal transition to aggressiveness of non-acidic melanoma cells. Clin. Exp. Metastasis 31, 423–433 (2014).
Calcinotto, A. et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 72, 2746–2756 (2012).
Clarke, K. et al. Indications for the use of proton pump inhibitors for stress ulcer prophylaxis and peptic ulcer bleeding in hospitalized patients. Am. J. Med. 135, 313–317 (2022).
Tyron, J. M., Eliasen, A., Dalhoff, K. & Nørgaard, M. M. The efficacy and safety of proton pump inhibitors in infants with reflux. Ugeskr Laeger 184 (2022).
Luciani, F. et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J. Natl Cancer Inst. 96, 1702–1713 (2004).
Yeo, M. et al. Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin. Cancer Res. 10, 8687–8696 (2004).
Shen, W. et al. Effect of pantoprazole on human gastric adenocarcinoma SGC7901 cells through regulation of phospho‑LRP6 expression in Wnt/β-catenin signaling. Oncol. Rep. 30, 851–855 (2013).
Chen, M. et al. Effects of proton pump inhibitors on reversing multidrug resistance via downregulating V-ATPases/PI3K/Akt/mTOR/HIF-1α signaling pathway through TSC1/2 complex and Rheb in human gastric adenocarcinoma cells in vitro and in vivo. Onco Targets Ther. 11, 6705–6722 (2018).
Katara, G. K. et al. Inhibition of vacuolar ATPase subunit in tumor cells delays tumor growth by decreasing the essential macrophage population in the tumor microenvironment. Oncogene 35, 1058–1065 (2016).
Segna, D., Brusselaers, N., Glaus, D., Krupka, N. & Misselwitz, B. Association between proton-pump inhibitors and the risk of gastric cancer: a systematic review with meta-analysis. Ther. Adv. Gastroenterol. 14, 17562848211051463 (2021).
Dacha, S., Razvi, M., Massaad, J., Cai, Q. & Wehbi, M. Hypergastrinemia. Gastroenterol. Rep. 3, 201–208 (2015).
Wang, C. J. et al. Proton pump inhibitors suppress DNA damage repair and sensitize treatment resistance in breast cancer by targeting fatty acid synthase. Cancer Lett. 509, https://doi.org/10.1016/j.canlet.2021.03.026 (2021).
De Sousa Pereira, A. A basis for sympathectomy for cancer of the cervix uteri. Arch. Surg. 52, 260–285 (1946).
Zahalka, A. H. & Frenette, P. S. Nerves in cancer. Nat. Rev. Cancer 20, 143–157 (2020).
Jobling, P. et al. Nerve-cancer cell cross-talk: a novel promoter of tumor progression. Cancer Res. 75, 1777–1781 (2015).
Zhang, X. et al. Chronic stress promotes gastric cancer progression and metastasis: an essential role for ADRB2. Cell Death Dis. 10, 788 (2019).
Sloan, E. K. et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 70, 7042–7052 (2010).
Del Pinto, R., Ferri, C. & Parati, G. Reduction of blood pressure variability: an additional protective cardiovascular effect of vasodilating beta-blockers? J. Hypertens. 38, 405–407 (2020).
Prins, K. W., Neill, J. M., Tyler, J. O., Eckman, P. M. & Duval, S. Effects of beta-blocker withdrawal in acute decompensated heart failure: a systematic review and meta-analysis. JACC Heart Fail. 3, 647–653 (2015).
Ong, H. T. Beta blockers in hypertension and cardiovascular disease. BMJ 334, 946–949 (2007).
Fumagalli, C., Maurizi, N., Marchionni, N. & Fornasari, D. β-blockers: Their new life from hypertension to cancer and migraine. Pharm. Res 151, 104587 (2020).
Xia, Y. et al. Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages. Brain Behav. Immun. 81, 111–121 (2019).
Shi, M. et al. Catecholamine-Induced β2-adrenergic receptor activation mediates desensitization of gastric cancer cells to trastuzumab by upregulating MUC4 expression. J. Immunol. 190, 5600–5608 (2013).
Zmora, N., Soffer, E. & Elinav, E. Transforming medicine with the microbiome. Sci. Transl. Med. 11, https://doi.org/10.1126/scitranslmed.aaw1815 (2019).
Elinav, E., Garrett, W. S., Trinchieri, G. & Wargo, J. The cancer microbiome. Nat. Rev. Cancer 19, 371–376 (2019).
Roy, S. & Trinchieri, G. Microbiota: a key orchestrator of cancer therapy. Nat. Rev. Cancer 17, 271–285 (2017).
Schwabe, R. F. & Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 13, 800–812 (2013).
Gopalakrishnan, V., Helmink, B. A., Spencer, C. N., Reuben, A. & Wargo, J. A. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 33, 570–580 (2018).
Zitvogel, L., Ma, Y., Raoult, D., Kroemer, G. & Gajewski, T. F. The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 359, 1366–1370 (2018).
Helmink, B. A., Khan, M. A. W., Hermann, A., Gopalakrishnan, V. & Wargo, J. A. The microbiome, cancer, and cancer therapy. Nat. Med 25, 377–388 (2019).
Parhi, L. et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat. Commun. 11, 3259 (2020).
Lee, J. A. et al. Differential immune microenvironmental features of microsatellite-unstable colorectal cancers according to Fusobacterium nucleatum status. Cancer Immunol. Immunother. 70, 47–59 (2021).
Liao, J.-F., Chiou, W.-F., Shen, Y.-C., Wang, G.-J. & Chen, C.-F. Anti-inflammatory and anti-infectious effects of Evodia rutaecarpa (Wuzhuyu) and its major bioactive components. Chin. Med. 6, 6 (2011).
Kim, H.-J., Park, J.-M., Kim, J.-A. & Ko, B.-P. Effect of herbal Ephedra sinica and Evodia rutaecarpa on body composition and resting metabolic rate: a randomized, double-blind clinical trial in Korean premenopausal women. J. Acupunct. Meridian Stud. 1, 128–138 (2008).
Bak, E. J. et al. Inhibitory effect of evodiamine alone and in combination with rosiglitazone on in vitro adipocyte differentiation and in vivo obesity related to diabetes. Int J. Obes. (Lond.) 34, 250–260 (2010).
Yang, J. Y., Kim, J.-B., Lee, P. & Kim, S.-H. Evodiamine inhibits Helicobacter pylori growth and helicobacter pylori-induced inflammation. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22073385 (2021).
Zhu, L.-Q. et al. Evodiamine inhibits high-fat diet-induced colitis-associated cancer in mice through regulating the gut microbiota. J. Integr. Med. 19, 56–65 (2021).
Wang, M. et al. Amelioration of AOM/DSS-induced murine colitis-associated cancer by evodiamine intervention is primarily associated with gut microbiota-metabolism-inflammatory signaling axis. Front. Pharmacol. 12, 797605 (2021).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550 (2018).
Nagaraju, G. P. et al. Nanoparticles guided drug delivery and imaging in gastric cancer. Semin. Cancer Biol. 69, 69–76 (2021).
Khodaei, T., Inamdar, S., Suresh, A. P. & Acharya, A. P. Drug delivery for metabolism targeted cancer immunotherapy. Adv. Drug Deliv. Rev. 184, 114242 (2022).
Jain, P., Kathuria, H. & Momin, M. Clinical therapies and nano drug delivery systems for urinary bladder cancer. Pharmacol. Ther. 226, 107871 (2021).
Kumbhar, P. et al. Drug repurposing: an emerging strategy in alleviating skin cancer. Eur. J. Pharm. 926, 175031 (2022).
Danhier, F. et al. PLGA-based nanoparticles: an overview of biomedical applications. J. Control. Release 161, 505–522 (2012).
Hua, X. et al. Externally controlled triggered-release of drug from PLGA micro and nanoparticles. PloS One 9, e114271 (2014).
Shen, X. et al. PLGA-based drug delivery systems for remotely triggered cancer therapeutic and diagnostic applications. Front Bioeng. Biotechnol. 8, 381 (2020).
Shin, Y. B., Choi, J.-Y., Shin, D. H. & Lee, J.-W. Anticancer evaluation of methoxy poly(ethylene glycol)-b-poly(caprolactone) polymeric micelles encapsulating fenbendazole and rapamycin in ovarian cancer. Int J. Nanomed. 18, 2209–2223 (2023).
Ma, Y., Canup, B. S. B., Tong, X., Dai, F. & Xiao, B. Multi-responsive silk fibroin-based nanoparticles for drug delivery. Front Chem. 8, 585077 (2020).
Florczak, A. et al. Silk particles as carriers of therapeutic molecules for cancer treatment. Materials 13, https://doi.org/10.3390/ma13214946 (2020).
Wang, C. et al. Celastrol as an emerging anticancer agent: current status, challenges and therapeutic strategies. Biomed. Pharmacother. 163, 114882 (2023).
Gu, J. et al. Celastrol functions as an emerging manager of lipid metabolism: mechanism and therapeutic potential. Biomed. Pharmacother. 164, 114981 (2023).
Zhou, Q., Zhang, L., Yang, T. & Wu, H. Stimuli-responsive polymeric micelles for drug delivery and cancer therapy. Int. J. Nanomed. 13, 2921–2942 (2018).
Cho, H., Lai, T. C., Tomoda, K. & Kwon, G. S. Polymeric micelles for multi-drug delivery in cancer. AAPS PharmSciTech 16, 10–20 (2015).
Yousefpour Marzbali, M. & Yari Khosroushahi, A. Polymeric micelles as mighty nanocarriers for cancer gene therapy: a review. Cancer Chemother. Pharm. 79, 637–649 (2017).
Richter, A., Olbrich, C., Krause, M., Hoffmann, J. & Kissel, T. Polymeric micelles for parenteral delivery of sagopilone: physicochemical characterization, novel formulation approaches and their toxicity assessment in vitro as well as in vivo. Eur. J. Pharm. Biopharm. 75, 80–89 (2010).
Kanazawa, T., Morisaki, K., Suzuki, S. & Takashima, Y. Prolongation of life in rats with malignant glioma by intranasal siRNA/drug codelivery to the brain with cell-penetrating peptide-modified micelles. Mol. Pharm. 11, 1471–1478 (2014).
Luschmann, C. et al. Self-assembling colloidal system for the ocular administration of cyclosporine A. Cornea 33, 77–81 (2014).
Gaucher, G., Satturwar, P., Jones, M.-C., Furtos, A. & Leroux, J.-C. Polymeric micelles for oral drug delivery. Eur. J. Pharm. Biopharm. 76, 147–158 (2010).
Zahednezhad, F., Saadat, M., Valizadeh, H., Zakeri-Milani, P. & Baradaran, B. Liposome and immune system interplay: challenges and potentials. J. Control. Release 305, 194–209 (2019).
Taléns-Visconti, R., Díez-Sales, O., de Julián-Ortiz, J. V. & Nácher, A. Nanoliposomes in cancer therapy: marketed products and current clinical trials. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms23084249 (2022).
Dinić, J. et al. Repurposing old drugs to fight multidrug resistant cancers. Drug Resist. Update 52, 100713 (2020).
Barbosa, E. J., Löbenberg, R., de Araujo, G. L. B. & Bou-Chacra, N. A. Niclosamide repositioning for treating cancer: challenges and nano-based drug delivery opportunities. Eur. J. Pharm. Biopharm. 141, 58–69 (2019).
Shah, S. et al. Quality by design steered development of Niclosamide loaded liposomal thermogel for Melanoma: in vitro and ex vivo evaluation. Eur. J. Pharm. Biopharm. 180, 119–136 (2022).
Hatamipour, M., Jaafari, M. R., Momtazi-Borojeni, A. A., Ramezani, M. & Sahebkar, A. Nanoliposomal encapsulation enhances in vivo anti-tumor activity of niclosamide against melanoma. Anticancer Agents Med. Chem. 19, 1618–1626 (2019).
Singh, P. et al. Gold nanoparticles in diagnostics and therapeutics for human cancer. Int. J. Mol. Sci. 19, https://doi.org/10.3390/ijms19071979 (2018).
Conde, J., Doria, G. & Baptista, P. Noble metal nanoparticles applications in cancer. J. Drug Deliv. 2012, 751075 (2012).
Zheng, X., Wu, Y., Zuo, H., Chen, W. & Wang, K. Metal nanoparticles as novel agents for lung cancer diagnosis and therapy. Small 19, e2206624 (2023).
Kim, Y.-J. et al. Development of lactobacillus kimchicus DCY51T-mediated gold nanoparticles for delivery of ginsenoside compound K: in vitro photothermal effects and apoptosis detection in cancer cells. Artif. Cells Nanomed. Biotechnol. 47, 30–44 (2019).
Gonzalez-Fierro, A. & Dueñas-González, A. Drug repurposing for cancer therapy, easier said than done. Semin. Cancer Biol. 68, 123–131 (2021).
Breckenridge, A. & Jacob, R. Overcoming the legal and regulatory barriers to drug repurposing. Nat. Rev. Drug Discov. 18, 1–2 (2019).
Schein, C. H. Repurposing approved drugs on the pathway to novel therapies. Med. Res. Rev. 40, 586–605 (2020).
Zhang, Z., Ji, J. & Liu, H. Drug repurposing in oncology: current evidence and future direction. Curr. Med. Chem. 28, 2175–2194 (2021).
Bhatia, S. N., Chen, X., Dobrovolskaia, M. A. & Lammers, T. Cancer nanomedicine. Nat. Rev. Cancer 22, 550–556 (2022).
Papaioannou, L. & Avgoustakis, K. Responsive nanomedicines enhanced by or enhancing physical modalities to treat solid cancer tumors: preclinical and clinical evidence of safety and efficacy. Adv. Drug Deliv. Rev. 181, 114075 (2022).
Kunzmann, V. et al. Nab-paclitaxel plus gemcitabine versus nab-paclitaxel plus gemcitabine followed by FOLFIRINOX induction chemotherapy in locally advanced pancreatic cancer (NEOLAP-AIO-PAK-0113): a multicentre, randomised, phase 2 trial. Lancet Gastroenterol. Hepatol. 6, 128–138 (2021).
Jotte, R. et al. Atezolizumab in combination with carboplatin and nab-paclitaxel in advanced squamous NSCLC (IMpower131): results from a randomized phase III trial. J. Thorac. Oncol. 15, 1351–1360 (2020).
Untch, M. et al. Nab-paclitaxel versus solvent-based paclitaxel in neoadjuvant chemotherapy for early breast cancer (GeparSepto-GBG 69): a randomised, phase 3 trial. Lancet Oncol. 17, 345–356 (2016).
Sridhar, S. S., Ding, K. & Parulekar, W. R. Evaluation of efficacy and safety of nab-paclitaxel vs paclitaxel in platinum-refractory metastatic urothelial cancer-reply. JAMA Oncol. 7, 634–635 (2021).
Joshi, S. S. et al. Clinical assessment of 5-fluorouracil/leucovorin, nab-aclitaxel, and irinotecan (FOLFIRABRAX) in untreated patients with gastrointestinal cancer using genotype-guided dosing. Clin. Cancer Res. 26, 18–24 (2020).
Sonaye, H. V., Sheikh, R. Y. & Doifode, C. A. Drug repurposing: iron in the fire for older drugs. Biomed. Pharmacother. 141, 111638 (2021).
Wojcicki, A. V. et al. Repurposing drugs for acute myeloid leukemia: a worthy cause or a futile pursuit? Cancers 12, https://doi.org/10.3390/cancers12020441 (2020).
De Lellis, L. et al. Drug repurposing, an attractive strategy in pancreatic cancer treatment: preclinical and clinical updates. Cancers 13, https://doi.org/10.3390/cancers13163946 (2021).
El Zarif, T. et al. Overcoming therapy resistance in colon cancer by drug repurposing. Cancers 14, https://doi.org/10.3390/cancers14092105 (2022).
Ruiz-Iglesias, A. & Mañes, S. The importance of mitochondrial pyruvate carrier in cancer cell metabolism and tumorigenesis. Cancers 13, https://doi.org/10.3390/cancers13071488 (2021).
Qiao, X. et al. Lipid metabolism reprogramming in tumor-associated macrophages and implications for therapy. Lipids Health Dis. 22, 45 (2023).
Li, W. & Xu, X. Advances in mitophagy and mitochondrial apoptosis pathway-related drugs in glioblastoma treatment. Front. Pharmacol. 14, 1211719 (2023).
Soma, M. R., Corsini, A. & Paoletti, R. Cholesterol and mevalonic acid modulation in cell metabolism and multiplication. Toxicol. Lett. 64-65, 1–15 (1992).
Corcos, L. & Le Jossic-Corcos, C. Statins: perspectives in cancer therapeutics. Dig. Liver Dis. 45, 795–802 (2013).
Lu, J. et al. Targeting cholesterol metabolism in cancer: from molecular mechanisms to therapeutic implications. Biochem. Pharmacol. 218, 115907 (2023).
Alizadehasl, A. et al. Lipid-lowering drugs and cancer: an updated perspective. Pharmacol. Rep. 76, 1–24 (2023).
Hsu, R. & Benjamin, D. J. A narrative review of antibody-drug conjugates in EGFR-mutated non-small cell lung cancer. Front. Oncol. 13, 1252652 (2023).
Akbulut, M. & Urun, Y. Onco-cardiology: drug-drug interactions of antineoplastic and cardiovascular drugs. Crit. Rev. Oncol. Hematol. 145, 102822 (2020).
Zhang, Z. et al. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 5, 113 (2020).
Jourdan, J.-P., Bureau, R., Rochais, C. & Dallemagne, P. Drug repositioning: a brief overview. J. Pharm. Pharmacol. 72, 1145–1151 (2020).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Benson, D. A. et al. GenBank. Nucleic Acids Res. 46, D41–D47 (2018).
Lamb, J. et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006).
Flicek, P. et al. Ensembl 2011. Nucleic Acids Res. 39, D800–D806 (2011).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212 (2015).
Zhuo, D. et al. Assembly, annotation, and integration of UNIGENE clusters into the human genome draft. Genome Res. 11, 904–918 (2001).
Suzek, B. E., Huang, H., McGarvey, P., Mazumder, R. & Wu, C. H. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–1288 (2007).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Szklarczyk, D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
Chatr-Aryamontri, A. et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 41, D816–D823 (2013).
Chen, J. Y., Pandey, R. & Nguyen, T. M. HAPPI-2: a comprehensive and high-quality map of human annotated and predicted protein interactions. BMC Genom. 18, 182 (2017).
Croft, D. et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 39, D691–D697 (2011).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Wang, Z., Jensen, M. A. & Zenklusen, J. C. A practical guide to The Cancer Genome Atlas (TCGA). Methods Mol. Biol. 1418, 111–141 (2016).
Barretina, J. et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Baxevanis, A. D. Searching online mendelian inheritance in man (OMIM) for information on genetic loci involved in human disease. Curr. Protoc. Hum. Genet. 73, 9.13.1–9.13.10 https://doi.org/10.1002/0471142905.hg0913s73 (2012).
Goh, K.-I. et al. The human disease network. Proc. Natl Acad. Sci. USA 104, 8685–8690 (2007).
Iorio, F., Tagliaferri, R. & di Bernardo, D. Identifying network of drug mode of action by gene expression profiling. J. Comput. Biol. 16, 241–251 (2009).
Brown, A. S. & Patel, C. J. A standard database for drug repositioning. Sci. Data 4, 170029 (2017).
Gillen, J. E., Tse, T., Ide, N. C. & McCray, A. T. Design, implementation and management of a web-based data entry system for ClinicalTrials.gov. Stud. Health Technol. Inform. 107, 1466–1470 (2004).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018).
Szklarczyk, D. et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 44, D380–D384 (2016).
Andersson, M. L. et al. Evaluation of usage patterns and user perception of the drug-drug interaction database SFINX. Int. J. Med. Inform. 84, 327–333 (2015).
Wang, Y. et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 48, D1031–D1041 (2020).
Kuhn, M., Letunic, I., Jensen, L. J. & Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 44, D1075–D1079 (2016).
Pacini, C. et al. DvD: an R/Cytoscape pipeline for drug repurposing using public repositories of gene expression data. Bioinformatics 29, 132–134 (2013).
Sun, J. et al. Deciphering signaling pathway networks to understand the molecular mechanisms of metformin action. PLoS Comput. Biol. 11, e1004202 (2015).
Sacco, F. et al. Deep proteomics of breast cancer cells reveals that metformin rewires signaling networks away from a Pro-growth state. Cell Syst. 2, 159–171 (2016).
Hart, T. et al. Toward repurposing metformin as a precision anti-cancer therapy using structural systems pharmacology. Sci. Rep. 6, 20441 (2016).
Li, J. et al. Network-based identification of microRNAs as potential pharmacogenomic biomarkers for anticancer drugs. Oncotarget 7, 45584–45596 (2016).
Wu, Z., Li, W., Liu, G. & Tang, Y. Network-based methods for prediction of drug-target interactions. Front. Pharmacol. 9, 1134 (2018).
Zhang, L. et al. Weighted gene co-expression network analysis and connectivity map identifies lovastatin as a treatment option of gastric cancer by inhibiting HDAC2. Gene 681, 15–25 (2019).
Lo, Y.-C. et al. Computational cell cycle profiling of cancer cells for prioritizing FDA-approved drugs with repurposing potential. Sci. Rep. 7, 11261 (2017).
Garmhausen, M. et al. Virtual pathway explorer (viPEr) and pathway enrichment analysis tool (PEANuT): creating and analyzing focus networks to identify cross-talk between molecules and pathways. BMC Genom. 16, 790 (2015).
Taccioli, C. et al. MDP, a database linking drug response data to genomic information, identifies dasatinib and statins as a combinatorial strategy to inhibit YAP/TAZ in cancer cells. Oncotarget 6, 38854–38865 (2015).
Zeng, X. et al. Pantoprazole, an FDA-approved proton-pump inhibitor, suppresses colorectal cancer growth by targeting T-cell-originated protein kinase. Oncotarget 7, 22460–22473 (2016).
Fako, V. E., Wu, X., Pflug, B., Liu, J.-Y. & Zhang, J.-T. Repositioning proton pump inhibitors as anticancer drugs by targeting the thioesterase domain of human fatty acid synthase. J. Med. Chem. 58, 778–784 (2015).
Fang, J. et al. Quantitative and systems pharmacology. 1. In silico prediction of drug-target interactions of natural products enables new targeted cancer therapy. J. Chem. Inf. Model 57, 2657–2671 (2017).
Huang, J. et al. A multicenter phase II study of temozolomide plus disulfiram and copper for recurrent temozolomide-resistant glioblastoma. J. Neurooncol. 142, 537–544 (2019).
Hundley, W. G. et al. Statins and left ventricular ejection fraction following doxorubicin treatment. NEJM Evid. 1, https://doi.org/10.1056/evidoa2200097 (2022).
Asari, K. et al. Simvastatin enhances the efficacy of nilotinib in chronic myeloid leukaemia by post-translational modification and drug transporter modulation. Anticancer Drugs 32, 526–536 (2021).
Marlatt, K. L. et al. The effect of atorvastatin on vascular function and structure in young adult survivors of childhood cancer: a randomized, placebo-controlled pilot clinical trial. J. Adolesc. Young-. Adult Oncol. 8, 442–450 (2019).
Cooper, J. P. et al. Allogeneic hematopoietic cell transplantation with non-myeloablative conditioning for patients with hematologic malignancies: improved outcomes over two decades. Haematologica 106, 1599–1607 (2021).
Manni, A. et al. Stearoyl-CoA desaturase-1, a novel target of omega-3 fatty acids for reducing breast cancer risk in obese postmenopausal women. Eur. J. Clin. Nutr. 71, 762–765 (2017).
Harvey, R. D. et al. Effect of multiple-dose osimertinib on the pharmacokinetics of simvastatin and rosuvastatin. Br. J. Clin. Pharmacol. 84, 2877–2888 (2018).
Chataway, J. et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet 383, 2213–2221 (2014).
Payne, J. M. et al. Randomized placebo-controlled study of lovastatin in children with neurofibromatosis type 1. Neurology 87, 2575–2584 (2016).
Statler, A. et al. Comparable outcomes of patients eligible vs ineligible for SWOG leukemia studies. Blood 131, 2782–2788 (2018).
Raja-Khan, N. et al. Effects of atorvastatin on vascular function, inflammation, and androgens in women with polycystic ovary syndrome: a double-blind, randomized, placebo-controlled trial. Fertil. Steril. 95, 1849–1852 (2011).
Siblini, H. et al. Assessing the hepatic safety of epigallocatechin gallate (EGCG) in reproductive-aged women. Nutrients 15, https://doi.org/10.3390/nu15020320 (2023).
Kumar, N. B. et al. Randomized, placebo-controlled trial of green tea catechins for prostate cancer prevention. Cancer Prev. Res. 8, 879–887 (2015).
Bathgate, J. R., Radler, D. R., Kurzer, M. & Samavat, H. Green tea extract supplementation does not modify plasma concentration of F2-isoprostanes in women who are postmenopause: findings from a randomized controlled trial. Nutr. Res. 113, 29–38 (2023).
Zhang, Z. et al. Effects of ω-3 fatty acids and catechins on fatty acid synthase in the prostate: a randomized controlled trial. Nutr. Cancer 68, 1309–1319 (2016).
Jo, U. et al. Novel and highly potent ATR inhibitor M4344 kills cancer cells with replication stress, and enhances the chemotherapeutic activity of widely used DNA damaging agents. Mol. Cancer Ther. 20, 1431–1441 (2021).
D’Amico, A. V. et al. Radiation and androgen deprivation therapy with or without docetaxel in the management of nonmetastatic unfavorable-risk prostate cancer: a prospective randomized trial. J. Clin. Oncol. 39, 2938–2947 (2021).
Coombes, R. C. et al. Effect of celecoxib vs placebo as adjuvant therapy on disease-free survival among patients with breast cancer: the REACT randomized clinical trial. JAMA Oncol. 7, 1291–1301 (2021).
Rader, J. S. et al. A stratified randomized double-blind phase II trial of celecoxib for treating patients with cervical intraepithelial neoplasia: the potential predictive value of VEGF serum levels: an NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 145, 291–297 (2017).
Gaitskell, K. et al. Angiogenesis inhibitors for the treatment of epithelial ovarian cancer. Cochrane Database Syst. Rev. 4, CD007930 (2023).
Penas-Prado, M. et al. Randomized phase II adjuvant factorial study of dose-dense temozolomide alone and in combination with isotretinoin, celecoxib, and/or thalidomide for glioblastoma. Neuro. Oncol. 17, 266–273 (2015).
Gatti-Mays, M. E. et al. Exemestane use in postmenopausal women at high risk for invasive breast cancer: evaluating biomarkers of efficacy and safety. Cancer Prev. Res. 9, 225–233 (2016).
El-Chemaly, S. et al. Celecoxib in lymphangioleiomyomatosis: results of a phase I clinical trial. Eur. Respir. J. 55, https://doi.org/10.1183/13993003.02370-2019 (2020).
Lynch, P. M. et al. An international randomised trial of celecoxib versus celecoxib plus difluoromethylornithine in patients with familial adenomatous polyposis. Gut 65, 286–295 (2016).
Felgenhauer, J. L. et al. A pilot study of low-dose anti-angiogenic chemotherapy in combination with standard multiagent chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma family of tumors: a Children’s Oncology Group (COG) Phase II study NCT00061893. Pediatr. Blood Cancer 60, 409–414 (2013).
Lalla, R. V. et al. Randomized double-blind placebo-controlled trial of celecoxib for oral mucositis in patients receiving radiation therapy for head and neck cancer. Oral. Oncol. 50, 1098–1103 (2014).
Tong, C. C. L. et al. Phase II trial of concurrent sunitinib and image-guided radiotherapy for oligometastases. PloS One 7, e36979 (2012).
Roy, S., Malone, S., Grimes, S. & Morgan, S. C. Impact of concomitant medications on biochemical outcome in localised prostate cancer treated with radiotherapy and androgen deprivation therapy. Clin. Oncol. (R. Coll. Radiol.) 33, 181–190 (2021).
Kalinsky, K. et al. Presurgical trial of metformin in overweight and obese patients with newly diagnosed breast cancer. Cancer Invest. 32, 150–157 (2014).
Chak, A. et al. Metformin does not reduce markers of cell proliferation in esophageal tissues of patients with Barrett’s esophagus. Clin. Gastroenterol. Hepatol. 13, 665–672.e1-4 (2015).
Martinez, J. A. et al. Phase II study of metformin for reduction of obesity-associated breast cancer risk: a randomized controlled trial protocol. BMC Cancer 16, 500 (2016).
Brown, J. R. et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight. 5, https://doi.org/10.1172/jci.insight.133247 (2020).
Crist, M. et al. Metformin increases natural killer cell functions in head and neck squamous cell carcinoma through CXCL1 inhibition. J. Immunother. Cancer 10, https://doi.org/10.1136/jitc-2022-005632 (2022).
Pimentel, I. et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 48, 17–23 (2019).
Nwanaji-Enwerem, J. C. et al. An epigenetic aging analysis of randomized metformin and weight loss interventions in overweight postmenopausal breast cancer survivors. Clin. Epigenet. 13, 224 (2021).
Goodwin, P. J. et al. Effect of metformin vs placebo on invasive disease-free survival in patients with breast cancer: the MA.32 randomized clinical trial. JAMA 327, 1963–1973 (2022).
Mueller, N. T. et al. Metformin affects gut microbiome composition and function and circulating short-chain fatty acids: a randomized trial. Diabetes Care 44, 1462–1471 (2021).
Kemnade, J. O. et al. Phase I/II trial of metformin as a chemo-radiosensitizer in a head and neck cancer patient population. Oral. Oncol. 145, 106536 (2023).
Elkind-Hirsch, K. E., Paterson, M. S., Seidemann, E. L. & Gutowski, H. C. Short-term therapy with combination dipeptidyl peptidase-4 inhibitor saxagliptin/metformin extended release (XR) is superior to saxagliptin or metformin XR monotherapy in prediabetic women with polycystic ovary syndrome: a single-blind, randomized, pilot study. Fertil. Steril. 107, 253–260.e1 (2017).
Evia-Viscarra, M. L. et al. The effects of metformin on inflammatory mediators in obese adolescents with insulin resistance: controlled randomized clinical trial. J. Pediatr. Endocrinol. Metab. 25, 41–49 (2012).
Marsh, C. A. et al. Functional neuroimaging of emotional processing in women with polycystic ovary syndrome: a case-control pilot study. Fertil. Steril. 100, 200–207.e1 (2013).
Pernicova, I. et al. Metformin to reduce metabolic complications and inflammation in patients on systemic glucocorticoid therapy: a randomised, double-blind, placebo-controlled, proof-of-concept, phase 2 trial. Lancet Diabetes Endocrinol. 8, 278–291 (2020).
Lundgren, J. A., Kim, S. H., Burt Solorzano, C. M., McCartney, C. R. & Marshall, J. C. Progesterone suppression of luteinizing hormone pulse frequency in adolescent girls with hyperandrogenism: effects of metformin. J. Clin. Endocrinol. Metab. 103, 263–270 (2018).
Ladson, G. et al. Effects of metformin in adolescents with polycystic ovary syndrome undertaking lifestyle therapy: a pilot randomized double-blind study. Fertil. Steril. 95, 2595–2598.e2591 (2011).
Ladson, G. et al. The effects of metformin with lifestyle therapy in polycystic ovary syndrome: a randomized double-blind study. Fertil. Steril. 95, 1059–1066.e1-7 (2011).
Elkind-Hirsch, K. E., Chappell, N., Seidemann, E., Storment, J. & Bellanger, D. Exenatide, dapagliflozin, or phentermine/topiramate differentially affect metabolic profiles in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 106, 3019–3033 (2021).
Samson, S. L. et al. Managing pasireotide-associated hyperglycemia: a randomized, open-label, Phase IV study. Pituitary 24, 887–903 (2021).
Cruz-Correa, M. et al. Efficacy and safety of curcumin in treatment of intestinal adenomas in patients with familial adenomatous polyposis. Gastroenterology 155, 668–673 (2018).
Vishwanathan, K. et al. The effect of food or omeprazole on the pharmacokinetics of osimertinib in patients with non-small-cell lung cancer and in healthy volunteers. J. Clin. Pharmacol. 58, 474–484 (2018).
Wilcox, C. M., Seay, T., Arcury, J. & Hirschowitz, B. I. Presentation, response to lansoprazole therapy, and outcome of Zollinger-Ellison syndrome-like gastric acid hypersecretors. Scand. J. Gastroenterol. 46, 277–280 (2011).
Steeghs, N. et al. A phase 1 open-label study to assess the relative bioavailability of TAK-931 tablets in reference to powder-in-capsule in patients with advanced solid tumors. Invest. N. Drugs 41, 53–59 (2023).
Falk, G. W. et al. A combination of esomeprazole and aspirin reduces tissue concentrations of prostaglandin E(2) in patients with Barrett’s esophagus. Gastroenterology 143, 917–926.e1 (2012).
Fox, E. et al. A pharmacologically-based approach to high dose methotrexate administration to investigate nephrotoxicity and acute kidney injury biomarkers in children and adolescents with newly diagnosed osteosarcoma. Cancer Chemother. Pharmacol. 87, 807–815 (2021).
Knight, J. M. et al. Repurposing existing medications as cancer therapy: design and feasibility of a randomized pilot investigating propranolol administration in patients receiving hematopoietic cell transplantation. BMC Cancer 18, 593 (2018).
Yu, A. F. et al. Association of circulating cardiomyocyte cell-free DNA with cancer therapy-related cardiac dysfunction in patients undergoing treatment for ERBB2-positive breast cancer. JAMA Cardiol. 8, 697–702 (2023).
Guglin, M. et al. Randomized trial of lisinopril versus carvedilol to prevent trastuzumab cardiotoxicity in patients with breast cancer. J. Am. Coll. Cardiol. 73, 2859–2868 (2019).
Ji, Y. et al. Efficacy and safety of propranolol vs atenolol in infants with problematic infantile hemangiomas: a randomized clinical trial. JAMA Otolaryngol. Head. Neck Surg. 147, 599–607 (2021).
Davies, O. M. T. et al. Cutaneous vascular anomalies associated with a mosaic variant of AKT3: genetic analysis continues to refine the diagnosis, nomenclature, and classification of vascular anomalies. J. Am. Acad. Dermatol. 87, 162–164 (2022).
Léauté-Labrèze, C. et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N. Engl. J. Med. 372, 735–746 (2015).
Kim, K. H. et al. Comparison of efficacy and safety between propranolol and steroid for infantile hemangioma: a randomized clinical trial. JAMA Dermatol. 153, 529–536 (2017).
Shepherd, D. M., Jahnke, H., White, W. L. & Little, A. S. Randomized, double-blinded, placebo-controlled trial comparing two multimodal opioid-minimizing pain management regimens following transsphenoidal surgery. J. Neurosurg. 128, 444–451 (2018).
Schott, A. F. et al. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-negative metastatic breast cancer. Clin. Cancer Res. 23, 5358–5365 (2017).
Blumenstein, B. et al. Reduction in serum clusterin is a potential therapeutic biomarker in patients with castration-resistant prostate cancer treated with custirsen. Cancer Med. 2, 468–477 (2013).
Pintova, S. et al. Genistein combined with FOLFOX or FOLFOX-Bevacizumab for the treatment of metastatic colorectal cancer: phase I/II pilot study. Cancer Chemother. Pharmacol. 84, 591–598 (2019).
Khan, S. A. et al. Soy isoflavone supplementation for breast cancer risk reduction: a randomized phase II trial. Cancer Prev. Res. 5, 309–319 (2012).
Messing, E. et al. A phase 2 cancer chemoprevention biomarker trial of isoflavone G-2535 (genistein) in presurgical bladder cancer patients. Cancer Prev. Res. 5, 621–630 (2012).
Acknowledgements
This study was supported by research grants from the National Natural Science Foundation of China to H.H. (82060442), the Foundation of the Education Department of Guizhou Province (no. [2022]214) and the Undergraduate Teaching Engineering Construction Project of Guizhou College of Traditional Chinese Medicine to Y.X. (no. GZY-JG (2018)6). This work was funded in part by the China Scholarship Council (No. 202008520053 to Y.X.).
Author information
Authors and Affiliations
Contributions
W.L.J., Y.X., and M.S. were involved in the conception and design of the review. Y.X. and M.S. wrote the paper and prepared the figures and tables. W.L.J. and H.H. critically reviewed and edited the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xia, Y., Sun, M., Huang, H. et al. Drug repurposing for cancer therapy. Sig Transduct Target Ther 9, 92 (2024). https://doi.org/10.1038/s41392-024-01808-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01808-1
