
Abstract
G protein-coupled receptors (GPCRs), the largest family of human membrane proteins and an important class of drug targets, play a role in maintaining numerous physiological processes. Agonist or antagonist, orthosteric effects or allosteric effects, and biased signaling or balanced signaling, characterize the complexity of GPCR dynamic features. In this study, we first review the structural advancements, activation mechanisms, and functional diversity of GPCRs. We then focus on GPCR drug discovery by revealing the detailed drug-target interactions and the underlying mechanisms of orthosteric drugs approved by the US Food and Drug Administration in the past five years. Particularly, an up-to-date analysis is performed on available GPCR structures complexed with synthetic small-molecule allosteric modulators to elucidate key receptor-ligand interactions and allosteric mechanisms. Finally, we highlight how the widespread GPCR-druggable allosteric sites can guide structure- or mechanism-based drug design and propose prospects of designing bitopic ligands for the future therapeutic potential of targeting this receptor family.
Similar content being viewed by others
Introduction
G protein-coupled receptors (GPCRs) are the largest superfamily of cell surface membrane receptors and are encoded by approximately 1000 genes, sharing conserved seven-transmembrane (7TM) helices connected by three intra- and three extra-cellular loops.1,2,3 GPCRs are conformationally dynamic proteins that mediate vital biological functions of signal transduction triggered by various extracellular signals such as photons, ions, lipids, neurotransmitters, hormones, peptides, and odorants.4,5,6,7,8 Due to the distinct topography between the binding sites of extracellular stimuli and the subsequent signaling events at the intracellular site (approximately 40 Å), GPCR signal transduction is allosteric.9,10,11,12,13 Advances in protein engineering, X-ray crystallography, and cryo-electron microscopy (cryo-EM), coupled with innovative technologies such as X-ray free electron lasers (XFELs) and nuclear magnetic resonance (NMR) spectroscopy, have revolutionized our understanding of GPCR structures and dynamics. These studies provide insights into ligand-receptor interactions, conformational changes, and signaling complexes, offering unprecedented opportunities for in-depth investigations into receptor activation, orthosteric/allosteric modulation, biased signaling, and dimerization.
Once activated by exogenous stimuli, GPCRs primarily employ heterotrimeric G-proteins and arrestins as transducers to produce second messengers and further initiate the downstream signaling, resulting in promiscuous signaling profiles within cells.11 Such spectrum of signaling is the prerequisite for function diversity of GPCRs and is fundamental in regulating physiological processes, including sensory perception, neurotransmission, and endocrine processes.14,15 The mutations and truncation of GPCRs; however, can dysregulate GPCR functionality by altering constitutive activity, influencing membrane expression and affecting post-translational behaviors.16 Unraveling the mechanisms of stimuli-GPCR-effector coupling, as well as the concise regulation of GPCR dysfunction will bring about valuable therapeutic potentials and inspire the design of modulators with high potency, selectivity, or biased signaling.
Till date, approximately 34% of the US Food and Drug Administration (FDA)-approved drugs are targeted to GPCRs, with modulators in clinical trials or preclinical stages experiencing exponential growth.17,18 Among them, orthosteric ligands impose an effective alteration on GPCR activity and signaling process by competitively preventing the binding of endogenous ligands.19 However, due to the sequence conservation of orthosteric sites, in most cases, subtype selectivity remains an intractable issue, which implies the inevitable side effects of orthosteric drugs.20 As an alternative or complementary option, targeting allosteric sites alone or targeting both orthosteric and allosteric sites can overcome these major hurdles.21,22,23,24,25 Allosteric modulators are highlighted for their high subtype selectivity and low side effects. A progressive structural understanding of the detailed receptor-ligand interactions is paving the way for fragment-to-lead optimization in structure-based drug design (SBDD) (Fig. 1). Moreover, the knowledge of allosteric sites is useful for the design of bitopic ligands by creating a molecule attached to both an allosteric and orthosteric site. Bitopic ligands have several advantages of improved affinity and enhanced selectivity over a single allosteric or orthosteric ligand. In addition, elucidating allosteric mechanisms of GPCRs provides a viable strategy to develop biased ligands such as G protein- or β-arrestin-based allosteric modulators.26 Bitopic modulators have higher selectivity to reduce side effects since they exert pathway-specific effects on GPCR signaling.26,27

Phylogenetic tree of GPCRs indicating GPCR structures that have been solved in complex with modulators. Nodes represent GPCRs named according to their UniProt gene name and are organized according to the GPCR database. GPCR structures bound to modulators are highlighted by color
In this review, we first summarize the structural progression, activation mechanisms, and functional diversity of GPCRs. To delve into the advancement of GPCR drug discovery, we investigate the detailed drug-target interactions at the orthosteric sites, focusing on GPCR structures in complex with recent FDA-approved orthosteric drugs. Subsequently, allosteric modulators are extensively discussed, with a focus on recent breakthroughs in GPCR structures that bind to synthetic small molecules. Notably, peptides and antibodies are excluded from our analysis. Such investigation systematically clusters the location of allosteric sites in the extracellular vestibule, transmembrane domain, and intracellular surface, highlighting the key binding modes with their target receptors and allosteric mechanisms. This review aims to provide a deeper understanding of GPCR structures, mechanisms, and drug discovery, which has important implications for structure- or mechanism-based drug design and the design of bitopic ligands for the future therapeutic potential of targeting this receptor family.
Structure advances in GPCRS
The low expression of membrane protein GPCRs, combined with their conformational flexibility, initially posed great challenges for high-resolution diffraction.28 The initial crystal structures of rhodopsin and the ligand-activated β2 adrenergic receptor (β2AR) were resolved in 2000 and 2007, respectively.29,30 Over the past two decades, considerable progress has been made in the engineering of proteins and the technique of X-ray crystallography.31 Notably, the use of GPCR engineering with fusion proteins,32,33 antibody fragment crystallization34,35, and thermostabilizing mutations36, has produced numerous antagonist- or agonist-bound GPCR structures. However, only agonist-bound GPCRs frequently exist in an intermediate conformation because the fully active conformation requires stabilizing chaperones, including G proteins, G protein mimetics, conformationally specific nanobodies, and mini-G proteins.37
The first GPCR-G protein complex was determined in 2011 using X-ray diffraction;38 however, the demanding nature of X-ray crystallography has rendered GPCR-G protein complex crystallization a difficult undertaking. Cryo-EM has developed to be an alternative technique, driving a novel trend in GPCR structural biology. Unlike X-ray crystallography, cryo-EM does not rely on crystals and has considerably superior potential to directly visualize detergent- or nanodisc-solubilized GPCRs. This capability enables the determination of previously intractable fully active states and larger protein complexes, including GPCR-G protein complexes.39 Since then, the number of cryo-EM structures depicting GPCRs in complex with intracellular partners has experienced exponential growth (Fig. 2). As of November 2023, the Protein Data Bank has accumulated 554 complex structures, of which 523 are resolved using cryo-EM.40 However, both crystallography and cryo-EM are limited to capturing the most stable and lowest energy conformations under crystallization conditions.4 Moreover, the comprehensive characterization of intermediate states and transition kinetics remains elusive. Crystallographic, spectroscopic, and simulation techniques have offered complementary information on the conformational dynamics of GPCRs.

Timeline of major advancements in GPCR structure study using X-ray crystallography and cryo-EM
The advanced XFELs possess the potential to solve the missing information. The exceptional properties of XFELs, characterized by extreme brilliance and femtosecond short pulses, allow them to overcome radiation damage, facilitating the determination of GPCR structures with atomic-level information at femtosecond timescales.41 NMR spectroscopy offers a valuable technique to detect dynamic features of GPCRs in liquid environments.42,43 The number, position, and shape of signals in the NMR spectra are sensitive to changes in the micro-environment of stable-isotope “probes” incorporated into receptors. Double electron-electron resonance (DEER) spectroscopy enables the assessment of a distance distribution between two different probes. Fluorescence resonance energy transfer (FRET), a technique based on fluorescence, functions as an “atomic ruler” to detect the proximity between two labels, providing valuable data about the number of states and their relative populations.44,45 Among these, DEER and FRET provide only localized details regarding the chemical probes that have been inserted. In addition, molecular dynamics (MD) simulations offer a comprehensive, time-resolved view of complete protein structures, capturing intermediate states along the transition pathway.46,47,48 Advances in the structural biology of GPCRs have revealed key information on ligand-receptor interactions, conformational changes, and signaling complexes, opening the opportunity for exploration of receptor activation, orthosteric/allosteric modulation, biased signaling, and dimerization.
Mechanism of GPCR activation and signaling
Although the nature of GPCRs and activating stimuli may vary significantly, GPCRs primarily coordinate distinct downstream signaling responses through two types of transducers: heterotrimeric G proteins and arrestins. Human G proteins comprise four major families (Gs, Gi/o, Gq/11, and G12/13) and more than half of GPCRs activate two or more G proteins, each of which exhibits distinct efficacies and kinetics.49,50 The promiscuous coupling leads to fingerprint-like signaling profiles inside the cell, which contributes to the complexity of GPCR signaling.
When bound to GDP, the Gαβγ heterotrimer is inactive. Agonist binding leads to the formation of an active conformation of GPCRs, which initiates signaling cascades involving the recruitment and activation of G-proteins. The activated GPCR catalyzes the GDP/GTP exchange on the Gα subunit, causing the dissociation of Gα from the Gβγ dimer. Due to high cellular concentrations of GTP, Gα rapidly binds a molecule of GTP at the nucleotide-binding site. Both Gα-GTP and Gβγ can modulate subsequent effector proteins. Gα-GTP can activate or inhibit enzymes such as adenylyl cyclase (AC), phospholipase C (PLC), or ion channels, depending on the specific G protein type. Gβγ can also modulate various signaling pathways and interact with target proteins. Activation of effector proteins by Gα-GTP or Gβγ generates second messengers, such as cyclic AMP (cAMP). The cellular response concludes with the Gα subunit hydrolyzing GTP to GDP, leading to its reassociation with Gβγ and G protein inactivation. Subsequently, the Gα subunit completes the G-protein activation cycle by reassociating with Gβγ.
To prevent sustained signaling, activated GPCRs may also undergo C-terminal phosphorylation facilitated by G-protein-coupled receptor kinases (GRKs). This multi-site GPCR phosphorylation determines β-arrestin binding affinity and induces receptor desensitization via steric hindrance, followed by clathrin-mediated endocytosis and ubiquitination of the receptor (Fig. 3a).11,51,52 The receptor-arrestin complex also serves as a scaffold for over 20 different kinases, including mitogen-activated protein (MAP) kinases, ERK1/2, p38 kinases, and c-Jun N-terminal kinases, activating G-protein independent signaling pathway. Four isoforms of arrestin (arrestins 1-4) and multiple GRK isoforms were discovered, with arrestins 1 and 4 being only found in the visual system. β-arrestins 1 and 2, also referred to as arrestins 2 and 3, interact with and regulate numerous non-visual GPCRs.34

a Schematic representation of GPCR activation process. Upon agonist (red circle) binding, the receptor proceeds into a pre-activation state coupling with the G protein heterotrimer, where the exchange of GDP and GTP in G protein α subunit leads to G protein dissociation and mediate G protein signaling pathway. The phosphorylation of the receptor C-terminal tail by GRK binding promotes arrestin recruitment and signaling. When the antagonists (blue circle) bind, the receptor stabilizes in an inactive state. b Crosstalk of downstream pathway of Gs, Gq, Gi and arrestin
Originally classified as monomers, GPCRs were subsequently recognized to engage in homo- or hetero-dimerization, displaying distinct properties in receptor activation, pharmacological cascades, and biological functions.53,54 Recent research indicates that GPCRs can bind to various single transmembrane accessory proteins to regulate their biological functions such as ligand binding, transducer coupling, and intracellular signaling.55,56 Prominent examples include the family of receptor activity-modifying proteins (RAMPs) that majorly regulate the glucagon receptor (GCGR) and the melanocortin receptor accessory proteins (MRAPs) that regulate the melanocortin receptors (MC1R-MC5R).57,58 Currently, the interactions between the negative allosteric modulator RAMP2 and GCGR as well as the positive allosteric modulator MRAP1 and MC2R have been elucidated by cryo-EM.59,60
Structural changes within GPCRs facilitate their function as molecular conduits that transmit extracellular signals across membranes to elicit cellular responses. A distinctive feature of GPCR activation involves notable outward movement of the cytoplasmic end of TM6, creating an intracellular pocket to accommodate the downstream transducers. GPCRs contain several conserved structural motifs relevant to their activation, including the CWxP motif of TM6, the NPxxY motif of TM7, and the ionic lock that involves TM3-TM6, as well as TM3-TM7.61,62,63 Additionally, Na+ acts as an endogenous negative allosteric modulator (NAM) of class A GPCR activation, stabilizing the inactive state through direct interactions.64,65 High resolution structures reveal that Na+ interacts mainly with residues from TM1, TM2, TM3, and TM7 and these interactions vary across GPCRs.66,67
Ligands can regulate receptor activity by stabilizing distinct conformations. Since the diverse signaling pathways elicit distinct physiological effects, ligands that selectively induce beneficial pathways hold promising therapeutic value. These drugs are commonly referred to as “biased ligands.” For instance, G protein-biased μ-opioid receptor (μOR) agonists are of remarkable clinical relevance as they enhance analgesia effects and reduce adverse reactions associated with the activation of β-arrestin pathways, in contrast to morphine. Several novel biased ligands are currently in clinical use or under investigation, such as TRV130, PZM21, and SR-17018.68,69,70 Hence, unraveling the coupling mechanisms governing G proteins, GRKs, and arrestins will establish a robust foundation for designing biased ligands tailored to selectively activate or inhibit specific pathways.
In the absence of agonists, GPCRs may display different levels of constitutive activities. The efficacy of diverse ligands acting on a single GPCR in terms of activation or inactivation also varies widely. Considering both receptor constitutive activity and drug efficacy, GPCR ligands are categorized as (full) agonists, partial agonists, antagonists, and inverse agonists. These variations in efficacy significantly influence their therapeutic properties.
Functional diversity of GPCRS
Overview of GPCR subfamilies and their physiological functions
GPCRs can be categorized into class A, class B, class C, class F, and class T according to their structural and functional characteristics. Class A GPCRs, namely the rhodopsin-like family, is the superfamily with the largest proportion and the most extensive research.71 Class A GPCRs can further be divided by function into aminergic, peptide, protein, lipid, melatonin, nucleotide, steroid, dicarboxylic acid, sensory, and orphan subgroups,72 with their corresponding indications ranging from hypertension, cardiovascular diseases, and pulmonary diseases, to depression and psychiatric disorders.17 Class B GPCRs are divided into secretin (B1) and adhesion (B2) subfamilies, with the former characteristic of large extracellular domains (ECD) and the latter possessing a unique long N-terminal motif and autoproteolysis-inducing domain.73 While glucagon-like peptide-1 receptor (GLP-1R) and glucagon receptor (GCGR) are emerging as the famous B1 GPCR targets in regulating blood glucose homeostasis and lipid metabolism;74,75 the B2 subfamily is critical in modulating sensory, endocrine, and gastrointestinal systems.76 Class C GPCRs, the glutamate receptors, are unique in their large ECDs, conserved venus fly traps (VFTs), cysteine-rich domains (CRDs) on the ligand binding sites, and constitutive dimers for receptor activation.77 With mGluRs (metabotropic glutamate receptors) taking the lead in clinical transformation, the physiological functions of class C GPCRs are implicated in cancer, migraine, schizophrenia, and movement disorders.77 Class F GPCRs, comprising 10 frizzled receptors (FZDs) and one smoothened receptor (SMO), are distinctive in their conserved CRD regions and involvement in Hedgehog and Wnt signaling pathways. Therefore, they are mainly associated with cancer, fibrosis, and embryonic development.78 The current drug discovery is only focused on SMO,79 leaving broad exploration space for the therapeutic potential of FZDs. Particularly, although taste 2 receptors (TAS2Rs), the receptors modulating taste perception of humans, show structure similarity with class A GPCRs, their low sequence homology (<20%) with the existing types of GPCRs isolates them to a novel category of class T GPCRs,76 deepening our understanding of the entire GPCR family.
Involvement of GPCRs in sensory perception, neurotransmission, and endocrine regulation
Rhodopsin, TAARs, and TASRs in sensory perception
One of the most significant physiological functions GPCRs exercise is mediating sensory information such as light perception, taste, olfaction, and pheromone sensation. Rhodopsin, which contributes to the first stage of visual activation in vertebrates, exhibits the typical and representative features of class A GPCRs. Upon absorbing photons, the orthosteric ligand of rhodopsin, retinal, experiences conformational flipping within picoseconds, thus rapidly triggering signal propagation from the receptor to G proteins, cGMP phosphodiesterase, or cGMP-gated ion channel.80 The covalent linkage of retinal with the receptor, and the instantaneous overturning and signaling serve as a paradigm for elucidating the efficiency of GPCRs in sensory perception.
Olfactory sensory receptors, which can be categorized into odorant receptors (ORs) and trace amine-associated receptors (TAARs), are a valuable medium for researchers to understand olfactory information encoding. Guo et al.81 has recently revealed the universal mechanism of TAARs recognition of amine odor molecules and the structural basis of “combinatorial coding” of the olfactory receptor in ligand recognition. Notably, the selective coupling of mTAAR9 with Gs and Golf is also delineated, which serves as a pioneer in the field of mammalian olfactory recognition. Apart from selective G-proteins, the downstream transduction mechanism of olfactory receptors is also associated with adenylyl cyclase and cAMP-gated ion channel,82 leaving favorable exploration opportunities.
To regulate the sensory function of taste, which is one of the most important sensations in human life, taste receptors (TASRs) are extensively studied from physiological and pharmacological perspectives. Among them, type I taste GPCRs function by forming heterodimeric complexes to stimulate sweet (TAS1R2/TAS1R3) and umami (TAS1R1/TAS1R3) sensation, whereas Type II are monomeric TAS2Rs that regulate bitter flavor.83 Tastant binding to the receptor activates downstream secondary messengers, resulting in depolarization and sensitizing the transient receptor potential (TRP) channel, which in turn innervates the gustatory cortex in the brain.84 Given the inapplicability of the previous GPCR expression techniques in TAS2Rs,85 overcoming difficulties in the structural determination of taste receptors will further facilitate their physiological research.
μOR and CBR in neurotransmission
Currently, neurological therapeutic demands mainly revolve around neuropathic pain alleviation, treatment of depression, psychiatric disorders, and Parkinson’s diseases. μ-Opioid receptors (μORs), possessing a research history of over 50 years, have been extensively researched about their mechanism of analgesic action in the peripheral nervous system (PNS) and the central nervous system (CNS). For instance, μORs reduce the release of nociceptive substances and decrease Ca2+ production following nerve injury by interacting with TRPV1, H1R, and NK1R in nociceptive receptors,86 whereas in spinal dorsal horn neurons, μORs modulate 5-HT receptors, glycine receptors, and norepinephrine receptors to activate pain inhibitory pathway.87 Orthosteric biased modulators, allosteric modulators, and bitopic modulators have been successively developed to exert analgesic effects while alleviating side effects like respiratory depression and addiction.88 Cannabinoid receptors (CBRs) are also representative targets involved in neurotransmission and neuropathic pain pathophysiology. The subtype CB1R is primarily found in presynaptic terminals of neurons in CNS, the activation of which inhibits neurotransmitter release and algesthesia transmission,89 while CB2R is highly expressed in immune cells, the activation of which can inhibit inflammatory factors that promote pain sensitization.90 No-selective orthosteric CB1R and CB2R activators can produce an antinociceptive effect and improve sleep in several animal models, while selective positive allosteric modulators (PAMs) like ZCZ011 (40) are rising as more promising ligands without inducing cannabis-like side effects.91
GLP-1R and GPR120 in endocrine regulation
Endocrine syndrome has been rising as one of the most critical health issues in the 21st century. Numerous metabolism-related GPCRs, which are usually activated by energy metabolites or substrates, are pivotal sensors of endocrine dysregulation. GLP-1R and GPR120 (also known as free fatty acid receptor 4), for example, are both promising therapeutic targets for the treatment of type 2 diabetes and obesity.74,92 Mechanistically, the endogenous ligand of GLP-1R, GLP-1, can reduce the secretion of glucagon in pancreatic α cells and promote insulin secretion in pancreatic β cells. For GPR120, however, the binding of omega-3 polyunsaturated fatty acids (ω3-FAs) and receptor activation can reduce inflammation of adipose tissue and protect against insulin resistance.93 The receptor’s coupling with Gq/11 subsequently stimulates the PI3K/Akt pathway, resulting in the uptake of glucose in adipocytes.94 As GLP-1R agonist liraglutide takes the lead in FDA-approved drugs treating type 2 diabetes and obesity,95 drug development of more endocrine-related targets such as GPR35, GPR40, GPR41, GPR43, GPR81, and GPR119 are supposed to come into our view.
Receptor promiscuity and cross-talk between different signaling pathways
GPCR receptors convert the extracellular stimuli to intracellular signals to control cellular function and phenotype. GPCR receptors convert the extracellular stimuli to intracellular signals to control cellular phenotype and function. These intracellular signaling pathways intersect with each other to enhance or downgrade relevant responses in a phenomenon known as “cross-talk.” The promiscuity of the GPCR signaling network is consequently outlined, resulting in more extensive regulation, low selectivity, and possible adverse effects.
Promiscuity and cross-talk can occur at three levels, including the GPCR receptors, G-proteins/β-arrestins, and the downstream effectors. The receptor promiscuity lies in the formation of heterodimers, which can either be constituted of subtypes of the same receptor family or those of different families. A compelling case is the heterodimerization of GABAb(1) and GABAb(2) which leads to the functionality of modulating GIRK (G-protein gate inward rectifying channel) potassium channels, whilst neither of them is functional when expressed as a monomer.96 Another well-established example is the plentiful interrelationship of adenosine receptors and dopamine receptors, where the activation of A1A and A2A adenosine receptors decreases dopamine binding to D1 and D2 dopamine receptors.97 The bivalent ligands that bind adenosine receptors and dopamine receptors at each end further demonstrate the occurrence and functionality of heterodimerization.98 The participants of heterodimerization are assumed to share a common G-protein pool, thus contributing to the redistribution of their interaction of G-proteins and reshaping the signaling landscape.99 Given this, by direct cross-talk between two GPCR receptors, ligands can be designed towards one receptor to modulate the affinity and efficacy of the other target, although certain pharmacological profiles remains unclear.
At the second stage of the hierarchical signaling of GPCRs, namely the recruitment of Gs, Gi, Gq, G12, β-arrestin 1, and β-arrestin 2, a spectrum of coupling strengths ranging from highly selective coupling to promiscuous coupling is exhibited. MD simulations performed by Sandhu et al. revealed that engineered mutant GPCRs can alter the coupling of non-cognate G-proteins by reshaping the intracellular interface,99 demonstrating that “dynamic structural plasticity” of the GPCR cytosolic pockets is the foundation of G-protein promiscuity. Mutants, orthosteric and allosteric modulators that exert long-range and delicate effects towards the cytosolic binding interface are therefore principal strategies to achieve selectivity of G-protein signaling.
The promiscuity of distinct downstream effectors, known as the third stage of signaling, is highly correlated with the cross-talk of G-proteins. Normally, stimulation of Gs, Gi, and Gq results in the activation of AC, the inhibition of AC, and the stimulation of PLC, respectively.100 However, once distinct G-proteins are recruited near the membrane at a similar time, βγ subunits released from respective G-protein activation are “exchangeable” between diverse signaling pathways and can potentiate responses mediated by other G-proteins.101 The second messengers then phosphorylate, activate, or deactivate each other to construct a fine-tuning network (Fig. 3b). Albeit conducting a great deal of research, the precise control of GPCR promiscuity remains obscure.
Impact of GPCR mutations on human diseases and therapeutic implications
Besides being involved in numerous physiological processes, mutations in GPCRs can be linked to manifold human diseases, underlying the necessity of GPCR genomics, and imposing therapeutic implications. Till date, over 2350 mutations in GPCR genes have been identified as the major causes of more than 60 inherited monogenic diseases in humans (Fig. 4a), with missense mutations harboring the maximum proportion (>60%) and small inserts/deletions ranking the second (>15%).16

a Categories of Representative human diseases caused by GPCR dysfunctions. b Classification of the effects of mutations on GPCR dysfunctions
Classification of the effects of mutations on GPCR dysfunctions
The effects of mutations in GPCRs can be categorized into gain-of-function (GoF) and loss-of-function (LoF), corresponding to physiological hyperfunction and hypofunction, respectively. Recent studies have provided a more detailed explanation of the diverse underlying mechanisms of GoF and LoF mutations. Compared with the wild-type (WT) GPCR activation, the common pharmacological mechanisms of activating and inactivating mutations lie in three aspects: (1) Mutations transform micro-switch cascades within the receptors and induce active/inactive conformations, thus altering the constitutive activity of GPCRs and affecting the recruitment of downstream effectors. (2) Mutations influence receptor expression directly or indirectly increasing/decreasing receptors’ intracellular transport, degradation, and recycling. (3) Some mutations affect ligand potency, specificity, or promiscuous recognition, thereby exerting regulatory functions by shifting the conformational population, redistributing the downstream couplings, or altering receptor dimerization. Furthermore, all variants are not pathogenic. This provides robust evidence for another classification of “driver” and “passenger” mutations (Fig. 4b).102 Computational approaches are recently emerging to predict the driver ability of mutations in GPCR-related diseases, based on abundant clinical data of mutations and relevant GPCR dysfunctions.
Correlation of GPCR mutations and human diseases
The genomic alterations induced by GPCR mutations serve as the major driver of various monogenic diseases. Some well-established examples include missense mutations in SMO receptor causing basal cell carcinoma,103 missense and nonsense mutations in MC4R causing obesity104, and missense mutations in FSHR inducing ovarian hyperstimulation syndrome. The majority of mutations are highly conserved and thus in an advantageous position during evolution.105 Therefore, the pathological relevance between GPCR mutations and human diseases may be more effectively predicted taking the evolutionary conservation of a certain residue into consideration.
Therapeutic implications and approaches of GPCR pathologies
Terapeutic approaches of GPCR pathologies mainly include symptomatic and etiological treatment. As many GPCR dysfunctions ultimately result in endocrine diseases with end-organ resistance or cancer, the administration of hormones or chemotherapeutics may be considered to reduce pathologic phenotypes.106,107 More state-of-the-art therapeutic implications, however, are oriented towards etiological treatment. Missense mutations in GPCRs can mislead protein folding and post-translational modifications to cause trafficking alterations, in which pharmacological chaperones are applicative therapeutic regimens.108 For receptor truncation resulting from nonsense mutations or frame-shifting mutations, RNA interference, gene replacement approaches, and the genome editing approach CRISPR/Cas9 may rescue the receptor integrality, provided that at least the first three transmembrane helices remain in the mutant receptor.109 Designing peptides or small molecule modulators is the most straightforward means for the restoration of receptor pharmacology, though high expenditure in multiple mutations remain an intractable issue.
Advances in GPCR drug discovery
Overview of traditional and emerging approaches for GPCR drug discovery
Since enkephalin was first recognized as the endogenous ligand of opioid receptors,110 the discovery of modulators with diverse regulatory effects is constantly endowing meaning in the research of GPCRs. Several decades have witnessed the transformation from serendipity to rational design in the field of GPCR drug discovery, and the ligands have been expanded from natural products to synthesized compounds and engineered antibodies. Currently, apart from the traditional molecular docking and SBDD, more screening methodologies of wet experiments have been established to facilitate the selection of high-quality hits, including FRET/ BRET (Bioluminescence Resonance Energy Transfer) assay, NanoBiT (NanoLuc Binary Interaction Technology) assay, Tango assay, and 19F NMR.111 Once the hits were obtained, structure-activity relationship (SAR) optimization in synergistic application of computational methodologies such as fragment-growing, property prediction, and MD simulations, was conducted to initiate the hit-to-lead and lead-to-drug campaign.99
Herein, we specially emphasize on the interaction and signaling mechanism of synthetic small-molecule modulators bound to GPCRs, with the aim of enlightening the discovery of more ingenious molecules with high potency, selectivity, and potential biased effects.
Structure-based drug design targeting the orthosteric sites of GPCRs
Orthosteric small molecule modulators are the most universal non-peptide regulators of GPCRs. By competing with endogenous ligands, they interact with the orthosteric binding pocket (OBP) and exert a full agonistic112,113/partial agonistic114/antagonistic function115,116 by triggering the conformational displacement of GPCR internal structures.117,118 Despite their relatively mature development, side effects derived from low subtype selectivity and promiscuous signaling remain the major hurdle.49,119
Over the past 30 years, the widespread use of X-ray and Cryo-EM has facilitated the characterization of GPCR-orthosteric ligand complexes, with 657 class A, 16 class B1, 6 class B2, 19 class C, 18 class F, and 1 class T structures solved (supplementary Table 1–5).120 Here, we meticulously selected five representative complexes in which ligands have been launched recently to elucidate the mechanisms of ligand recognition, specificity, and elaborate signaling transduction. Furthermore, we exemplified two cases to demonstrate the beneficial engagement of structural information in exploiting not only SAR but also the structure-functional selectivity relationship (SFSR). Considering these seven cases as a paradigm, we aimed to condense valuable hints based on a detailed analysis of approved drugs or selective compounds and provide a constructive outlook for the high-quality discovery of GPCR orthosteric modulators that may overcome the current dilemma.
μOR in complex with oliceridine
With morphine and fentanyl (1) the most effective drugs treating acute or chronic pain,121,122 their common receptor μOR was revealed to be responsible for both analgesic and adverse effects.123,124,125 To attenuate side effects and broaden the therapeutic window, modulators that can abolish β-arrestin activity while maintaining relatively intact G-protein signaling are of intense pharmaceutical interest.126,127,128 Oliceridine (2), a partial agonist binding at the orthosteric site of μOR, was approved by the FDA in 2020 for its ability to biased signaling via the G protein pathway and thus alleviating side effects (Fig. 5a, b).129 Therefore, casting light on the oliceridine-μOR complex structure and the underlying mechanism of biased signaling will provide insight in developing a novel generation of analgesic drugs.

a A bridged general view of fentanyl and oliceridine inducing distinct pharmacological profiles. b 2D structure of fentanyl and oliceridine shown for clarity. c Superimposed views of μOR–fentanyl (gray cartoon, gray sticks; PDB: 8EF5) and μOR–oliceridine (light green cartoon, light green sticks; PDB: 8EFB) complex structure, together with the comparison of ligand binding modes and arrestin coupling interfaces, are presented. 2D structures of two designed biased modulators are also presented
By aligning the complex structures of μOR–oliceridine and μOR–fentanyl, a well superimposed binding mode in OBP above Trp2956.48 was found. The only exception was that the pyridine ring of oliceridine tilts 35° toward TM2 relative to the n-aniline group of fentanyl, resulting in weaker hydrophobic interactions with TM6/7 than that with fentanyl. Based on the results of MD performed by Zhang et al.,130 extended interactions with TM6/7 can be inferred to have elicited inward movement of TM6 and TM7-H8 toward the TM core, shaping adaptive intracellular pocket conformation for both G-protein and β-arrestin coupling and thus leading to neutral signaling, whereas reduced interactions may have kept the intracellular end of TM6/7 relatively away from the TM core and therefore stabilize an intracellular pocket preferential for G protein binding and signaling. Two fentanyl-derived μOR agonists (3-4), which substituted the aniline group on fentanyl with n-propyl or isopropyl to reduce hydrophobicity with TM6/7, were thereupon designed as “proof-of-concept” to successfully achieve biased signaling via the G protein pathway (Fig. 5c). Different from the “trial-and-error” mode when developing biased ligand oliceridine,131 the comprehensive study by Zhang et al. serves as a paradigm for dissecting co-crystallized complexes to understand the molecular basis of preferential signaling mechanisms initiated from the orthosteric pocket and broadens the avenue for designing biased modulators of ORs through SBDD strategies.
S1PR in complex with siponimod
Sphingosine-1-phosphate receptor (S1PR), a family of class A GPCR consisting of five subtypes, S1PR1-S1PR5, modulates diverse physiological functions, including lymphocyte trafficking, vascular development, endothelial integrity, and heart rate.132,133,134,135,136 Although Fingolimod received regulatory approval from the FDA in 2010 as a first-in-class S1PR agonist,137 its low subtype selectivity has led to several “off-target” effects, including bradycardia and atrioventricular blockade.138 Therefore, a second-generation, highly subtype-selective S1PR modulator is crucially needed. Siponimod (5) was globally approved in 2019 for the treatment of adults with relapsing MS by selectively targeting S1PR1 and S1PR5.139 Insights into the mechanisms of drug recognition and receptor activation will provide a framework for understanding ligand selectivity and signal transduction in GPCRs.140,141
Yuan et al. presented the cryo-EM structures of siponimod–S1PR1–Gi and siponimod-S1PR5 complexes, in which the ligands exhibited an identical linear conformation across a polar module and the deep hydrophobic cavity of the orthosteric pocket (Fig. 6a).142 Given that members of the S1PR family display different extracellular vestibules, distinct extracellular leaflets have been reported to have contributed to diverse access channels for ligand entry and thus relate to specificity among subtypes (Fig. 6b).143 Moreover, further careful comparison of the siponimod-S1PR1-Gi complex with antagonist ML056-bound S1PR1 structure underlines the “twin toggle mechanism” during receptor activation.144 Upon ligand binding, Leu1283.36 rotates 130° away from TM5 to form a direct interaction with the hydrophobic portion of siponimod, disrupting its previous interaction with Trp2696.48 and triggering a synergistic downward movement of Trp2696.48. The dramatic displacement of the two residues can therefore loosen the interaction between TM3 and TM6, inducing a consequent outward movement of TM6 that can accommodate G protein binding (Fig. 6c, d). Similar activation mechanism involving corresponding mechanical switches can also be found in CB1 and MC4R,145,146 which provides valuable hints that designing ligands forming elaborate hydrophobic interaction with residue 3.36 or directly inducing reconfiguration of 3.36-6.48 may contribute to enhanced activation efficacy.

a Detailed binding modes of S1PR5 in complex with siponimod. Labels of the residues engaged in polar contacts with siponimod are colored in blue, with hydrogen bonds presented by orange dashes. The residues of the hydrophobic pocket that stabilizes ligand binding are marked with green labels, while residues that are critical for signal transduction are labeled in red. b Superimposed views of S1PR1 (orange cartoon, PDB: 7T6B), S1PR2 (light green cartoon, PDB: 7C4S), S1PR3 (light purple cartoon, PDB: 7YXA), and S1PR5 (yellow cartoon, PDB: 7TD4) GPCR structures, where TM1 and TM7 of S1PR5 are highlighted for clarity. c Superimposed views of active S1PR1-siponimod complex (cyan cartoon, cyan stick, PDB: 7TD4) and inactive S1PR1 structure (gray cartoon, gray stick, PDB: 3V2Y) to illustrate the “toggle switch” activation mechanism. d 2D structure of siponimod is shown for clarity
OX2R in complex with lemborexant
Orexin receptors are expressed throughout the central nervous system and demonstrate therapeutic potential for insomnia by regulating the sleep-wake cycle.147,148,149 The two subtypes, OX1R and OX2R, dominate the respective regulatory behaviors, with OX1R involved in gating rapid eye movement (REM) sleep and OX2R involved in gating non-REM and REM sleep.150 Lemborexant (6), an orthosteric competitive antagonist approved by the FDA in 2019, exhibits outstanding inhibitory activity against OXRs.151,152 However, the most important features of lemborexants lie in two aspects: (1) Why lemborexants show moderate selectivity toward OX2R over OX1R,152 which will facilitate the design of OX1R/OX2R-selective modulators that can be applied to REM and non-REM functionality studies? 2) What is the basis of the dynamic parameters of lemborexant that may explain the relationship between drug-induced improvement of sleep onset and a decrease in wake time after sleep?
To elucidate the mechanism of lemborexant subtype selectivity and provide guidance for anti-insomnia drug development, Asada et al. presented the crystal structure of the OX2R–lemborexant complex and compared its ligand-binding mode with that of the previously solved OX1R–lemborexant complex structure.153 Despite the ligand’s shared hydrogen bonds with Gln1263.32 of OX1R and Gln1343.32 of OX2R, lemborexant binds OX1R as a mixture of two orientations owing to the small side chain of Ala1273.33, whereas lemborexant binds OX2R in only one configuration because of the steric hindrance of Thr1353.33, which is inferred to be the primary cause of the difference in its affinity for OX1R and OX2R (Fig. 7a, b). In contrast, by simulating lemborexant in solution, the intramolecular stacking of two aromatic rings was observed to play a vital role in shaping the conformation of lemborexant close to the bound state before receptor binding, which explains the high kon value of the ligand. In addition, the higher binding free energy of lemborexant compared to other OXR modulators may contribute to a higher koff value. Collectively, these observations highlight the possibility of obtaining a high kon by optimizing the conformation of free molecules via intramolecular interactions (Fig. 7c, d). By extension, separately modulating the enthalpy of molecular binding to the receptor and entropy derived from the intramolecular structure may be important strategies for designing drugs with enhanced kinetics and dynamics.

a Detailed binding mode of lemborexant in complex with OX2R (receptor: light orange, ligand: cyan, PDB: 7XRR), where steric hindrance of T1353.33 only allows one orientation of the ligand. b Detailed binding mode of lemborexant in complex with OX1R (receptor: light pink, ligand: yellow, PDB: 6TOT), where small side chain of A1273.33 accounts for two orientations of the ligand. c Abridged general view of employing MD simulation to predict the conformation of the ligand before receptor binding, to improve Kon values. d 2D structure of lemborexant is shown for clarity
5-HT1F in complex with lasmiditan
The 5-HT1 receptor subtypes, including 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, and 5-HT1F, are well-known class A GPCRs that respond to the endogenous neurotransmitter serotonin and have been proven to be promising targets for the treatment of migraine, depression, and schizophrenia.154,155,156 Although traditional targeted agonists have been clinically used as anti-migraine drugs for decades, side effects such as therapeutic vasoconstrictive actions owing to the non-selective activation of 5-HT1B and 5-HT1D remain a major hindrance.157 Lasmiditan (7), a potent and highly selective drug toward 5-HT1F was approved by the FDA in 2019 because of its vasoconstrictive side effects and high-penetration properties.158 Elucidation of the scaffold features of lasmiditan and the mechanism of 5-HT1F-selective activation will provide a template for the rational design of safer anti-migraine drugs.
Through the 5-HT1F-lasmiditan-Gi1 complex solved by Huang et al., an overview of the lasmiditan-binding mode was presented.159 In the orthosteric binding pocket, the primary amine on the methylpiperidine group largely contributes to the stability of lasmiditan by forming a canonical charge interaction with Asp1033.32 of the receptor while simultaneously forming a hydrogen bond with Tyr3377.42. Notably, in the extended binding pocket (EBP), the trifluorobenzene group of lasmiditan forms additional hydrophobic interactions with Ile174ECL2 and Pro1584.60 and forms hydrogen bonds with residue Glu3136.55, Asn3176.59, Thr1825.40, and His176ECL2. Structural alignment of 5-HT1F with other 5-HT1 receptor subtypes revealed that the TM4-TM5-ECL2 region, which is highly conserved in the other four subtypes, underwent a notable conformational change, thereby disrupting the interaction between lasmiditan and 5-HT1A, 5-HT1B, 5-HT1D, and 5-HT1E. Thus, designing ligands that accommodate EBP and form specific interactions with the TM4-TM5-ECL2 region may enable high 5-HT1F selectivity (Fig. 8a, b). Activation mechanical analysis by Huang et al. revealed that lasmiditan triggers the downward movement of the toggle switch residue Trp6.48 and then induces conformational rearrangement of the PIF, DRY, and NPxxY motifs. Particularly, structural comparison of 5-HT1F-Gi complex and other 5-HT1-Gi/o showed that the αN of 5-HT1F-bound Gi shifts away from other 5-HT1 receptor-bound Gi/o, suggesting unique Gi coupling and corresponding specific downstream effects (Fig. 8c). Therefore, designing modulators that interact with the toggle switch residue and optimize their blood-brain-barrier (BBB) penetration properties may yield effective and safer 5-HT1F agonists.

a Detailed binding mode of 5-HT1F in complex with lasmiditan. Hydrogen bonds are presented by orange dashes, while halogen bonds are presented by green dashes. b Superimposed views of 5-HT1A (light green cartoon, PDB: 7E2X), 5-HT1B (light orange cartoon, PDB: 5V54), 5-HT1D (light gray cartoon, PDB: 7E32), 5-HT1E (light pink cartoon, PDB: 7E33), and 5-HT1F (light purple cartoon, PDB: 7EXD). The TM4-ECL2-TM5 region of the 5-HT1F receptor is highlighted for clarity. c The structure alignment comparison of αN helices of G protein coupling with their corresponding 5-HT receptors. αN helix of Gi protein coupled with 5-HT1F is highlighted for clarity. d 2D structure of lasmiditan
GnRH1 in complex with elagolix
The representative class A GPCR, gonadotropin-releasing hormone 1 receptor (GnRH1R), once activated by its endogenous peptide activator, gonadotropin-releasing hormone (GnRH), can initiate the reproductive hormone cascade and release gonadotropins through the activation of the Gq protein pathway.160,161,162 With the first availability of GnRH1R non-peptidic antagonist elagolix (8) on the market in 2018,163 structural insights into the GnRH1R-elagolix complex have gained pharmaceutical interest.164 Additionally, unlike other class A GPCRs, GnRH1R lacks a C-terminal helix (helix 8) in the cytoplasmic region and harbors Asn2.50 instead of the highly conserved Asp2.50 present in other receptors,165 leaving a wide space for different microswitches along the signaling cascade within 7TMD.
The crystal structure of the GnRH1R-elagolix complex studied by Yan et al. revealed that polar network residues composed of Lys1213.32 and Asp982.61 play critical roles in forming polar interactions with the ligand, whereas Tyr2836.51 and Tyr2906.58 are engaged in ligand recognition by contributing to hydrophobic interactions (Fig. 9a). Notably, Elagolix is located closer to TM7, resulting in an enlarged orthosteric pocket that allows N-terminal entry and co-occupation of the site. Structural alignment and IP accumulation assays showed that, unlike some GPCRs in which ligands can contact residue Trp6.48 directly and trigger the toggle switch, the special motif Tyr2836.51-Tyr2846.52-Trp2806.48-Phe2766.44 in TM6 was suggested to be a critical structural motif involved in mediating the propagation of signal transmission (Fig. 9b). Moreover, only 4% of class A GPCRs, including GnRH1R, have asparagine at the 5.58 position, which is implicated in a polar interaction with Ser1363.47 GnRH1R, thus leading to TM6 packing tightly with TM3 and TM5 in GnRH1R and exercising an antagonistic function. Collectively, these analyses highlight the distinctive features of GnRH1R in the binding of a representative antagonist and provide insights for structural biologists.

a Detailed binding mode of GnRH1 in complex with elagolix (receptor: light gray, ligand: light pink, PDB: 7BR3), where N-termini of GnRH1R is highlighted in a light purple to present its co-occupation with elagolix in the orthosteric pocket. b Overview of the special signal transduction mechanism in GnRH1R. c 2D structure of elagolix for clarity
SFSR study utilizing structural information
While the development of crystallography over the last decade has revealed an attractive possibility of SBDD, the mainstream strategy of GPCR drug discovery remains extensive SAR study and fragment-based drug design (FBDD).166,167,168 This is partially due to the “activity-cliff” phenomenon which to some extent, undermines the profits from structural information. Nevertheless, the promising prospect still deserves expecting. Two examples are analyzed here to arouse future interest in SFSR studies utilizing structural information.
The first example is the efficient discovery and optimization of A2AR selective antagonist 1,2,4-triazine derivative 4d (9) via SBDD strategy. With Biophysical Mapping (BPM) approach and crystal structure analysis, compound 4d was revealed to be primarily stabilized by two hydrogen bonds between the triazine core and N2536.55, with ring A oriented towards TM2 and TM7 (Fig. 10a). Hence, the presence of a hydrogen bond acceptor at the para position of ring A to interact with His2787.43, as well as the introduction of one or more flanking lipophilic substituents on the same ring to interact with Ile662.64 was suggested as the focus of the SAR program. Introducing either a phenolic hydroxyl or 4-pyridyl nitrogen at the para position of ring A, and fine-tuning affinity by various combinations of small lipophilic substituents efficiently yielded compound 4k, which proves the best balance of potency and efficacy (Fig. 10b).169 Further research compared the binding pockets of A2AR in complex with adenosine (agonist), ZM241385 (antagonist), and compound 4e (antagonist). The hydrophobic sub-pocket in the lower chamber was observed to be occupied by the ribose ring system of adenosine analogs in agonist complexes, though was typically unoccupied when antagonists bound. The same region also allowed optimization of selectivity for A2AR over A1AR (Fig. 10c). Therefore, expanding chemotypes into this region may harvest a more efficient chemical series when designing selective and diverse functional modulators.170

a Detailed binding mode of A2AR in complex with compound 4d (receptor: light gray, ligand: orange, PDB: 3UZA). b SAR study of A2AR antagonist. c Comparison of the orthosteric binding site of A2AR–Adenosine complex (light blue, PDB: 2YDO), A2AR–ZM241385 complex (light yellow, PDB: 4EIY), A2AR–Compound 4e complex (light green, PDB: 3UZC), the difference in cavity occupation is highlighted by red circles and arrows
The second paradigm entails the structure-based drug design of novel β-arrestin-biased D2R agonists commencing with aripiprazole, so as to alleviate the movement disorders associated with the adverse effects of antipsychotics.171 The dichlorophenylpiperazine portion of aripiprazole was first replaced with an indolepiperazine, leading to 12 that displayed comparable activity in both Gi/o-mediated cAMP inhibition and β-arrestin2 recruitment assays. Molecular docking with a D2R homology model revealed that the indole NH of 12 formed a hydrogen bond with Ser5.42, which has been shown to mediate G-protein-dependent signaling in highly homologous β2 adrenergic receptors. A methyl group was thus attached to the NH of indole to fine-tune the binding conformation of 12 and thereby preclude TM5 engagement (Fig. 11a). Inspired by structural information from homologous 5-HT2B receptor, where ligand interactions with hydrophobic residues on ECL2 appear to promote β-arrestin recruitment (Fig. 11a), a second methyl was introduced to position 2 of the indole ring, yielding 13 with a β-arrestin bias factor of 20 and potentially reduced side effects (Fig. 11b).172 To our knowledge, this is the first successful attempt at using structural information for the rational design of GPCR-biased ligands, underlining the necessity of interactive structural comparison in SFSR study.

a Detailed binding mode of D2R in complex with compound 1 (12) (receptor: light gray, ligand: salmon, the receptor is modeled from PDB: 3PBL). TM5 of the receptor is colored in pink and ECL2 is colored in blue for clarity. b SAR study of β-arrestin biased agonists of D2R
Delineation of GPCR structures complexed with small-molecule allosteric modulators and allosteric signaling
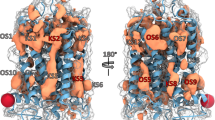
Over the past 10 years, allosteric drug discovery targeting GPCRs has witnessed significant progress in structural understanding, with the advance in knowledge of GPCR allostery.173,174 Till February 2024, the crystal structures of 59 allosteric small-molecule modulators bound to GPCRs have been solved, including 33 class A, 7 class B, 18 class C, and 1 class F modulators. These structures reveal that despite the intrinsic dynamic nature of GPCRs and the structural diversity among different GPCRs, only limited locations function as allosteric pockets, and the same pockets are present in GPCRs with different homologies.175 Even within a single receptor, more than one allosteric site has been identified. In addition, druggable allosteric hotspots spread throughout the receptor and can be divided into the following sections: extracellular vestibule, transmembrane domain, intracellular surface, outside 7TMD, and inside 7TMD domains.174,175 As will be discussed, allosteric binding sites in all GPCRs are currently known to be located at 11 distinct locations with some consensus,176 depicted in Fig. 12. In this figure, the locations of all pockets identified in different GPCRs are mapped onto the structure of an example GPCR to facilitate the comparison of these sites.

11 allosteric binding sites reported across GPCRs mapped onto representative class A GPCR CB1R. Gray pockets represent binding pockets within 7TMD, and white pockets represent binding pockets outside 7TMD. For each pocket, the number of unique ligands is indicated using boldface type, and the number of GPCRs containing the pocket is provided in parentheses. The boundary of the lipid bilayer is indicated by gray dashes
Based on the compounds’ ability to affect the stimulatory activity of orthosteric ligands, allosteric ligands can be classified into several categories, including positive allosteric modulators (PAMs), negative allosteric modulators (NAMs), allosteric modulators, and allosteric inverse agonists.22,177 A PAM, such as cinacalcet, targets the calcium-sensing receptor (CaSR) and potentiates the response of the receptor to its orthosteric agonist. Conversely, NAM attenuates the response of the receptor to its orthosteric agonist, mavoglurant, which targets the metabotropic glutamate receptor 5 (mGluR5).178 Ago allosteric modulators can activate or inhibit a receptor without an orthosteric agonist such as compound 2, which targets the glucagon-like peptide-1 receptor (GLP-1R).
Targeting of GPCR extracellular vestibule (outside and inside 7TMD)
After the first FDA approval of cinacalcet (a PAM of CaSR) in 2004 as a treatment for hyperparathyroidism,179 small-molecule allosteric modulators bound to the extracellular vestibule have developed rapidly. Till date, four of these modulators have been approved by the FDA, and one has entered clinical trials, as summarized in Table 1. Resolved crystal structures have revealed three extracellular binding sites: the pocket outside helices I and II, the pocket outside helices II and III, and the pocket inside 7TMD (Fig. 13).180,181 Due to their proximity to the traditional active sites of class A and B GPCRs, such allosteric modulators may exert their effects by directly altering the binding of orthosteric ligands to the receptor. As GPCRs evolved from a common ancestor, this allosteric site, found on receptors, may represent the ancestral orthosteric site.176,182

Three extracellular allosteric binding sites in GPCRs and the corresponding small-molecule allosteric modulators. Stick models of small-molecule ligands are mapped to representative members of outside 7TMD (I and II) (GLP-1R, PDB: 6VCB), outside 7TMD (II and III) (GPR101, PDB: 8W8S), and within 7TMD (M4R, PDB: 7V68) GPCRs. The position of an orthosteric ligand of M4R (shown in gray and sphere-and-stick representation) is mapped onto the overview of allosteric modulators for comparison. For each pocket, the number of unique modulators is indicated in boldface type, and the number of GPCRs containing the pocket is indicated in parentheses
1) Outside 7TMD (TM I-II):
GLP-1R–LSN3160440 structure
The glucagon-like peptide-1 receptor (GLP-1R) is a peptide hormone class B GPCR whose activation stimulates the glucose-dependent stimulation of insulin and decreases glucagon secretion.183,184,185 For such peptide receptors, allosteric pockets on GPCRs may be easier to target for small-molecule drugs than orthosteric drugs.186 Therefore, highly potent agonists and PAMs of GLP-1R must be developed to treat type 2 diabetes.187,188,189,190,191
LSN3160440 (14) (Fig. 14) is a small-molecule PAM targeted GLP-1R with an EC50 of 1 μM to enhance the potency and efficacy of GLP-1(9-36) becoming a full agonist.180 The cryo-EM structure of GLP-1R in complex with LSN3160440, the orthosteric ligand GLP-1, and the Gs protein revealed a clear depiction of the U-shaped binding mode of LSN3160440. The allosteric site is formed by residues on helices I and II in the extracellular vestibule (Fig. 15a).180 Within the binding pocket, the benzimidazole moiety of LSN3160440 (Fig. 15c) formed hydrophobic contacts with Leu1421.37 (the superscript represents the generic residue numbers of GPCRs) and engaged in aromatic interactions with Tyr1451.40 (Fig. 15b). Mutation and molecular dynamics (MD) simulation results also suggest that water-mediated hydrogen bonds may form between N3 of benzimidazole and Lys2022.72.192,193 Notably, LSN3160440 interacts with GLP-1 and acts as a molecular glue.194 The 2,6-dichloro-3-methoxyl phenyl moiety of LSN3160440 forms van der Waals interactions with Phe12GLP-1, Val16GLP-1 and Leu20GLP-1 simultaneously.

Two-dimensional (2D) chemical structures of synthetic allosteric ligands targeting the GPCR extracellular vestibule

a Schematic representation of PAM LSN3160440 and orthosteric GLP-1 bound to GLP-1R (PDB: 6VCB). GLP-1 is indicated in pink. b Detailed binding modes of GLP-1R bound to LSN3160440; π–π stacking is indicated in gray dashes. c 2D structure of small-molecule allosteric ligand LSN3160440 presented for clarity. d Superposition of orthosteric small-molecule agonists Boc5 (displayed with purple sticks), TT-OAD2 (displayed with salmon sticks), LY3502970 (displayed with yellow sticks), and CHU-128 (displayed with blue sticks) to LSN3160440–GLP-1–GLP-1R structure reveals a partial overlap in the TM1-TM2 cleft. The conserved residue Tyr1451.40 is highlighted
Several structures of orthosteric small-molecule agonists complexed with GLP-1R were resolved (Fig. 15d).195,196,197,198 Structural comparisons of these ligands with LSN3160440 revealed a shared region situated at the extracellular termini of the TM1-TM2 cleft, further suggesting that this is a promising area for lead optimization for both orthosteric and allosteric agonists. Within the binding site, the aromatic interactions with Tyr1451.40 are conserved.
2) Outside 7TMD (TM I-II)
GPR101–AA-14 structure
GPR101 is an orphan class A GPCR that is highly expressed in the nucleus accumbens and the hypothalamus and has constitutive Gs and Gq activity.199 GPR101 gene duplication or mutation modulates its constitutive activity, rendering GPR101 a promising target for metabolic diseases.200 Recent studies have identified AA-14 (15) (Fig. 14) as an allosteric agonist of GPR101, demonstrating robust Gs activation activity and high subtype selectivity.201 In vivo studies have shown that AA-14 exerts rejuvenating effects by activating GPR101 in the pituitary.
The cryo-EM structure of the AA-14–GPR101–Gs complex unveils two distinct binding sites for AA-14 (Fig. 16a): one located outside 7TMD, surrounded by helices I, VI, and VII, while the other is positioned outside TM2–TM3 and ECL1.201 Within the extracellular allosteric site, the 3-(trifluoromethyl) phenyl group (Fig. 16c) establishes polar interactions with Asn100ECL1 and hydrophobic interactions with Phe1033.24 and Trp872.60 (Fig. 16b). The 4-methyl-2-pyridinyl group packs against Phe96ECL1 and Leu99ECL1.

a Schematic representation of allosteric agonist AA-14 bound to GPR101 (PDB: 6VCB). b Detailed binding modes of GPR101 bound to AA-14. c 2D structure of small-molecule allosteric ligand AA-14 presented for clarity
3) Inside 7TMD:
GPR52–c17, MRGPRX1–ML382, PAR2–AZ8838, LHCGR–Org43553, M2R–LY2119620, M4R–LY2119620, M4R–compound-110, TSHR–ML109, CaSR–cinacalcet, CaSR–evocalcet, CaSR–NPS-2143, CaSR–R-568, mGluR1–FITM, SMO–vismodegib, mGluR2–JNJ-40411813, mGluR2–NAM563, mGluR2–NAM597, and CCR5–maraviroc structures
This allosteric site is located in an extracellular pocket surrounded by a 7TM helical bundle, directly above the traditional orthosteric site of family A and B GPCRs and the cholesterol-binding site of the SMO receptor (Fig. 13).202,203 Until now, this allosteric site has been the most frequently targeted binding site for drug-like allosteric modulators, mainly because allosteric modulators can enter from the extracellular region, allowing ligand binding without the need to penetrate the membrane.204 For these receptors, the pocket in the extracellular vestibule can be partitioned into two subpockets, namely the orthosteric and allosteric pockets. The N-terminal group and ECL2 regulate the sizes of the two sub-pockets by pushing the ligand to one side,205,206,207 thereby contributing to the creation of a new ligand pocket.
As prototypical class A GPCRs, muscarinic M1–M5 acetylcholine receptors (mAChRs) are responsible for the release of acetylcholine into the brain and play fundamental roles in the central and peripheral nervous system.208,209,210 Muscarinic receptors have garnered attention as potential drug targets to treat several pathophysiological disorders including Alzheimer’s disease, schizophrenia, and drug addiction.211,212,213,214 LY2119620 (20) acts as a PAM that has activity at both the M2 and M4 receptors (Fig. 14) but is inappropriate for treatment, probably because of cross-reactivity and cardiovascular liability.215
The structures of the M2 and M4 receptors bound to PAM LY2119620 have been solved.216,217,218 LY2119620 demonstrated a similar binding pattern to both the M2 and M4 receptors; nonetheless, subtle differences were noted (Fig. 17a). LY2119620 binds to a spacious extracellular vestibule just above the orthosteric pocket and is segregated from the orthosteric pocket via three tyrosine residues: Tyr3.33, Tyr6.51, and Tyr7.39. The thienopyridine ring of LY2119620 is sandwiched by π–π stacking between Tyr177ECL2 and Trp4227.35 in M2 receptor (Fig. 17b), Phe186ECL2 and Trp4357.35 in M4 receptor (Fig. 17c). Particularly, in the M2 receptor, the residues Tyr802.61, Asn4106.58, and Asn419ECL3 formed hydrogen bonds with the modulator, and Glu172ECL2 participated in ionic interactions with piperidine. Contrarily, in the M4 receptor, only Gln427ECL3 formed a hydrogen bond with the modulator.

a Superposition of PAM LY2119620 bound to M2 receptor (pink cartoon, pink sticks; PDB: 4MQT) and M4 receptor (yellow cartoon, yellow sticks; PDB: 7V68). b Detailed binding modes of M2 receptor bound to LY2119620. c Detailed binding modes of M4 receptor bound to LY2119620. Hydrogen bonds are presented as orange dashes and π–π stacking is presented as gray dashes. d Superimposed views of highlighted residue Trp4227.36 on M2 receptor–iperoxo–LY2119620 (pink cartoon, pink sticks; PDB: 6U1N), M2 receptor–LY2119620 (purple cartoon, purple sticks; PDB: 4MQT), and M2 receptor–iperoxo (blue cartoon, blue sticks; PDB: 4MQS) structures. The orthosteric agonist iperoxo is presented in orange
In addition, the structures of M2 receptor–iperoxo–LY2119620 (PDB: 4MQT) and M2 receptor–LY2119620 (PDB: 6U1N) are highly similar, with Trp4227.35 perpendicular to the horizontal plane and forming a π–π stacking with LY2119620 (Fig. 17d). In contrast, Trp4227.35 of the M2 receptor–iperoxo (PDB:4MQS), exhibits a parallel conformation, suggesting that the allosteric binding site is formed predominantly in the presence of an allosteric modulator. MD simulations have revealed that LY2119620 modulates the conformation of Trp4227.35, causing reorientation of Tyr4267.39 within the orthosteric site.219 This reorientation may explain the observed increase in affinity for iperoxo, thereby providing insight into the underlying allosteric mechanism.220
For GPCRs that use other sites to bind endogenous ligands, the traditional orthosteric pocket is potentially druggable for allosteric modulators.221 Calcium-sensing receptors (CaSR), members of the family C GPCR, are found primarily in the parathyroid glands and kidneys to ensure strict control of calcium homeostasis.222,223 Elevated Ca2+ levels trigger the activation of CaSR, leading to the inhibition of parathyroid hormone (PTH) secretion. Thus, CaSR has become a potential target for calcimimetic drugs to treat parathyroid disorders.224,225,226 Cinacalcet (27) (Fig. 14), an orally active allosteric agonist of CaSR, has been used for the treatment of secondary hyperparathyroidism227,228 whereas calcilytic NPS-2143 (29) (Fig. 14) is a potent NAM-targeting CaSR that exhibits favorable in vitro and in vivo activity.229
When bound to CaSR, PAM cinacalcet adopted extended and bent poses between CaSR homodimers (Fig. 18a). The naphthylethylamine moiety was bound to highly similar poses in both the extended and bent conformations (Fig. 18b, c). The naphthyl group engaged in hydrophobic interactions with Ile7775.44 on one side and formed edge-to-face π–π interactions with Phe6843.36 and Trp8186.50 on the other, thereby effectively securing the side chain of Trp8186.50 inside 7TM helical bundle. The NH group formed a hydrogen bond with Gln6813.33. In the extended conformation (Fig. 18b), the linker and phenyl group were parallel to TM VI, extending upward and driving Tyr8256.57 to orient downward. In the bent conformation (Fig. 18c), the phenyl group folded between TM V and TM VI to form a parallel-displaced π–π stacking with the naphthyl group, whereas Tyr8256.57 assumed a conformation perpendicular to TM VI and stabilized the ligand through a σ–π interaction.

a Schematic representation of PAM cinacalcet bound to CaSR (PDB: 7M3F). b Detailed binding modes of CaSR bound to cinacalcet in extended conformations. c Detailed binding modes of CaSR bound to cinacalcet in bent conformations. Hydrogen bonds are presented as orange dashes and π–π stackings are presented as gray dashes
In the CaSR–NPS-2143 complex, NPS-2143 exhibited the same crescent conformation as the homodimers (Fig. 19a). The naphthyl group at one end of NPS-2143 (Fig. 19c) was lined by residues Phe6843.36, Leu7765.43, Ile7775.44, Trp8186.50, and Ile8417.37 in the interior of the pocket (Fig. 19b). Conversely, the 3-chloro-2-cyano-phenyl ring of NPS-2143 protrudes out toward the lateral opening and forms hydrophobic contacts with Leu7735.40 and π–π stacking interactions with Tyr8256.57. A single hydrogen bond was established between the hydroxyl group and Tyr8256.57. Moreover, the conformation of the NAM-bound CaSR agrees well under both active (in the presence of Ca2+ and L-Trp) and inactive (no Ca2+) conditions.

a Schematic representation of NAM NPS-2143 bound to CaSR (PDB: 7M3E). b Detailed binding modes of CaSR bound to NPS-2143. Hydrogen bond is presented as orange dashes and π–π stacking is presented as gray dashes. c 2D structure of small-molecule allosteric ligand NPS-2143 provided for clarity. d Superimposed views of highlighted residues on CaSR–Cinacalcet–Ca2+–Trp (blue cartoon, blue sticks; PDB: 7M3F), CaSR–Ca2+–Trp (pink cartoon, pink sticks; PDB: 7DD6), and CaSR–NPS-2143–Ca2+–Trp (purple cartoon, purple sticks; PDB: 7M3E) structures. e Workflow of discovery of novel CaSR PAMs utilizing structural information
Despite having highly similar binding sites, NPS-2143 and cinacalcet exhibit quite different pharmacological properties, which may be explained by the conformation of the receptor residues. The conformations of NPS-2143-bound and Ca2+-bound CaSR were similar.230 Nevertheless, cinacalcet binding induced significant conformational changes in Trp8186.50, Phe8216.53, and Tyr8256.57 within the allosteric pocket (Fig. 19d). Trp8186.50 rotates inwardly from a vertical to a horizontal conformation, forming extensive π–π interactions with cinacalcet. Phe8216.53 underwent an outward shift and was inserted into a crevice between TM6 and TM7 facing the dimer interfacial area. Simultaneously, Tyr8256.57 flips down, driven by structural conflicts in the extended conformation of cinacalcet. In summary, cinacalcet induces a bent conformation of TM6 and stabilizes the homodimer interface, thereby contributing to receptor activation.231 Contrarily, NPS-2143 decreased agonist efficacy by enhancing TM VI helicity, which spatially hindered receptor activation.
Based on the special binding conformations of cinacalcet, Liu et al. conducted a virtual screening of 1.2 billion compounds to discover novel PAMs with potentially novel pharmacology. To respectively mimic the “extended” and “bent” conformation, extensive orientations and conformations of library molecules were sampled, which gave 682 trillion configurations overall and finally achieved a 3.8% and 13.6% hit rate. The hits were then optimized to a pharmacologically potent lead (36) via synergistic application of structural information, fragment hybridization, and stereochemistry separation (Fig. 19e).232 Such practice serves as a paradigm for its elaborate utility of solved GPCR structures and conformation sampling strategy and is generalizable in the discovery of CaSR NAMs and other allosteric modulators.
Targeting of GPCR transmembrane domain (outside 7TMD)
As shown by their structures, GPCRs utilize the domain outside 7TMD at the lipid interface to bind allosteric modulators. Till date, five different binding sites outside 7TMD in the transmembrane domain have been defined by their crystal structures (Table 2): the pocket outside helices I–III, the pocket outside helices II–IV, the pocket outside helices III–V, the pocket outside helices V–VI, and the pocket outside helices I, VI, and VII (Fig. 20a, c). Allosteric modulator binding to these regions targets class A GPCRs. These sites are typically shallow and not as well surrounded by the 7TM helical bundle as the traditional orthosteric sites. Polar functional groups are commonly found in allosteric modulators at these sites where they anchor themselves to the pocket. Thus, hydrogen atom donor or acceptor groups exposed between the receptor and lipid bilayer are more likely to mediate the binding of such allosteric ligands. These modulators are also required to preserve their overall hydrophobic character to enter the transmembrane domain. Allosteric modulators bound to the transmembrane domain outside 7TMD appear to regulate receptor signal transduction from outside the 7TM helices in a manner that stabilizes inactive or active interaction networks or impedes or facilitates the interhelical motions required for receptor activation.233,234,235

a Five allosteric binding sites in the transmembrane domain outside 7TMD of GPCRs and the corresponding small-molecule allosteric modulators. Stick models of small-molecule ligands are mapped to representative members of outside 7TMD (I–III) (P2Y1, PDB: 4XNV), outside 7TMD (II–IV) (CB1R, PDB: 6KQI), outside 7TMD (III and V) (C5aR1, PDB: 6C1R), outside 7TMD (V and VI) (CXCR3, PDB: 8HNN), and outside 7TMD (I, VI, and VII) (A1R, PDB: 7LD3) GPCRs. For each pocket, the number of unique modulators is indicated in boldface type, and the number of GPCRs containing the pocket is provided in parentheses. b 2D chemical structures of synthetic small-molecule allosteric ligands targeting the transmembrane domain outside 7TMD of GPCRs. c Extracellular view of the five allosteric sites
1) TM I–III:
P2Y1–BPTU Structure
To the best of our knowledge, one small molecule allosterically targets this relatively shallow pocket. Because of the flat TM helical bundle surface and relatively narrow cavity of the binding pocket, rational drug design in this area may be challenging.
In this instance, the protein target was the P2Y1 purinergic receptor. Agonists induce the activation of the P2Y1 receptor, leading to the potentiation of platelet aggregation that triggers platelet secretion;236,237 thus, antagonists targeting the P2Y1 receptor offer a prospective approach to treat thrombosis.238,239 BPTU (37) (Fig. 20b), a P2Y1 antagonist, has been gaining attention as an antithrombotic treatment and is the first allosteric GPCR modulator located outside the helical bundle.240 BPTU blocks the P2Y1-induced platelet aggregation with nanomolar potency and presents good selectivity for P2Y1 receptor and highly homologous P2Y12 receptor (P2Y1Ki = 75 nM, P2Y12Ki > 70 μM).241
The binary complex structure of P2Y1 − BPTU was determined and showed that the BPTU binding pocket consists mainly of residues in helices I − III (Fig. 21a).242 Notably, two crucial hydrogen bonds were formed between the two NH moieties of the urea group of BPTU and the main-chain carboxyl group of Leu1022.55 (Fig. 21b). In terms of hydrophobic interactions, the BPTU pyridyl group makes contact with the residues Ala1062.59 and Phe119ECL1. Hydrophobic interactions of the tert-butyl phenyl group occur within a distinct subpocket shaped by helices II and III, including residues Leu1022.55, Thr1032.56, Met1233.24, Leu1263.27, and Gln1273.28. On the opposite side of the ligand, the trifluoromethoxyphenyl group participated in hydrophobic interactions with Phe621.43 and Phe661.47.

a Schematic representation of the allosteric antagonist BPTU bound to P2Y1 receptor (PDB code 4XNV). b Detailed binding modes of P2Y1 receptor bound to BPTU. Hydrogen bonds are presented as orange dashes. c Superimposed views of the highlighted residues on 2MeSADP − P2Y1R − G11 (pink cartoon, pink sticks; PDB: 7XXH) and P2Y1R − BPTU structures (blue cartoon, blue sticks; PDB: 4XNV)
This case suggests an effective shape-complementary mechanism for allosteric ligands: bulges on 7TM helices can be utilized as anchors for fixation. In this instance, Pro1052.58 serves as the corresponding anchor, which is conserved in 74% of non-olfactory class A GPCRs.243 A comparison between the P2Y1 receptor bound to the agonist 2MeSADP and the allosteric antagonist BPTU revealed that the BPTU induces a 1.4 Å shift in Tyr1002.53 toward TM3 (Fig. 21c), preventing the conformational change of Phe1313.32.244 Thus, Phe1313.32 interacts with Phe2766.51 and limits the TM6 transition, which is required for the activation of class A GPCRs.245 In addition, MD simulations have demonstrated that the binding of BPTU stabilizes the helical bundle, leading to an increase in lipid order.246 This, in turn, stabilizes the ionic lock formed between Lys461.46 and Arg195ECL2 in the inactive receptor.
2) TM II−IV:
CB1R−ORG27569, CB1R − ZCZ011, and PAR2 − AZ3451 Structures
Cannabinoid receptors, which consist of two subtypes, CB1R and CB2R, are activated by neurotransmitter endocannabinoids and play key modulatory roles in synaptic transmission.247,248 Among them is the most abundant GPCR in the human brain is CB1R.249 Because of its widespread distribution and regulatory roles in various physiological functions, CB1R is considered an important target for the treatment of various central nervous system (CNS) disorders.250,251,252,253
ORG27569 (39) (Fig. 20b) is the first and most comprehensively studied NAM of CB1R.254,255 However, when tested in vivo, Org27569 was not sufficiently effective in modulating the effects of orthosteric cannabinoids.256 The co-crystal structure of CB1R and NAM ORG27569, together with its orthosteric CP55940 agonist, has been solved.257 ORG27569 occupied an allosteric site outside helices II and IV in the inner lobe of the phospholipid bilayer (Fig. 22a).258 Specifically, the chloro-indole ring of ORG27569 establishes a key aromatic interaction with the indole group of Trp2414.50 and buries in a hydrophobic pocket surrounded by His1542.41 and Val1612.48. Cys2384.47, Trp2414.50, Thr2424.51, and Ile2454.54 supply hydrophobic interactions for the amide-linked piperidinylphenyl chain (Fig. 22b).

a Superposition of the cocrystal structures of NAM ORG27569 (yellow, PDB: 6KQI) and PAM ZCZ011 (pink, PDB: 7FEE) with orthosteric ligand-bound CB1R. b Detailed binding modes of CB1R binding to ORG27569. c Detailed binding modes of CB1R binding to ZCZ011. Hydrogen bond is presented as orange dashes, and π–π stacking is presented as gray dashes. d Superimposed views of highlighted residues on CB1R–CP55940–ORG27569 (pink cartoon, pink sticks; PDB: 6KQI) and CB1R–AMG315–Gi structures (blue cartoon, blue sticks; PDB: 8GHV). e Superimposed views of highlighted residues on CB1R–AM6538 (pink cartoon, pink sticks; PDB: 5TGZ) and CB1R-CP55940–ZCZ011 structures (blue cartoon, blue sticks; PDB: 7FEE)
The recently developed ZCZ011 (Fig. 20b) is also an indole derivative that exerts PAM and partial agonist effects in vivo assays.259,260 ZCZ011 showed good shape complementarity with the pocket outside 7TMD comprising helices II−IV (Fig. 22a). The indole group in ZCZ011 is anchored by Phe1913.27 and forms a hydrogen bond with the main chain of Phe1913.27 while forming π−π stacking interactions with the side chain. (Fig. 22c).261 In addition, the indole group interacted with Leu1652.52, Ile1692.56, Ile2454.54, and Val2494.58 from TM II and TM IV, respectively.
NAM ORG27569 and AZ3451 (38), as well as PAM ZCZ011, bind to the same TM II−IV surface, however, exert opposite allosteric effects.262 Unlike traditional NAMs, ORG27569 enhances the affinity of the agonist though reduces the activity of Gi turnover.263 Structurally, the inactivating efficacy of ORG27569 acts by stabilizing the “activation switch” formed by Phe1552.42 and Phe2374.46 in CB1R (Fig. 22d).264,265 Furthermore, a hydrogen bond was formed between ORG27569 and Trp2414.50, which, together with the hydrogen bond formed between Trp2414.50, Ser1582.45, and Ser2063.42 (Fig. 22b), created a polar network that contributed to maintaining the inactive conformation.266 However, the precise mechanism through which ORG27569 augments agonist affinity remains to be elucidated. Upon comparing the CB1 structures bound to the PAM ZCZ011 and the antagonist AM6538, a notable shift of Ile1692.56 in TM2 towards TM3 was observed. This shift results in the contraction of the receptor“s active site (Fig. 22e), and is believed to be associated with activation.267,268 Additionally, Ser1732.60 undergoes notable inward movement and forms a hydrogen bond with CP55940, thereby stabilizing the agonist binding.
3) TM III–V:
GPR40–compound 1, GPR40–AP8, β2AR–AS408, β2AR–Cmpd-6FA, C5aR1–NDT9513727, C5aR1–avacopan, and DRD1–LY3154207 structures
A deep pocket is present outside the transmembrane helices III–V among GPCRs, which allows for the presence of a population of allosteric regulators bound to this pocket. In this case, allosteric agonists and PAMs (i.e., compound 1 (41), AP8 (42), Cmpd-6FA (43), and LY3154207 (47)) localize to regions near ICL2 and stabilize the ICL2 α helix through direct interactions, facilitating the inward movement of ProICL2. This movement leads to a ~3° inward displacement of TM III, which in turn determines the outward shift of TM V together with TM VI, which is a hallmark of GPCR activation.269,270 Thus, the binding of allosteric modulators increases the proportion of receptors that adopt active conformations, thereby increasing their affinity for agonists. Contrarily, allosteric antagonists and NAMs (i.e., AS408 (44), NDT9513727 (45), and avacopan (46)) bind to regions far from ICL2. As these ligands are bound close to the proline kink of TM V, the receptor-ligand interactions collectively inhibit the interhelical movements and rotations within TM III, TM IV, and TM V, which are required for receptor activation.271
The dopamine D1 receptor is a prototypical example. Dopamine functions as an essential catecholamine neurotransmitter that signals via the dopamine D1 to D5 receptors.272,273 The dopamine D1 receptor (DRD1) regulates neuronal growth, memory, and learning in the central nervous system.274,275 LY3154207 (Fig. 20b) is a selective PAM of the dopamine D1 receptor that improved motor symptoms associated with Lewy body dementia in a 2022 phase 2 clinical trial.276 Two distinct binding modes of LY3154207 to DRD1 have been reported, and structural comparisons have demonstrated upright and boat conformations (Fig. 23a).277,278,279,280 In both binding modes, LY3154207 was localized at the receptor–lipid bilayer interface surrounded by TM III, TM IV, and ICL2.

a Superposition of the cocrystal structures of PAM LY3154207 bound to dopamine D1 receptor in upright (pink, PDB: 7CKZ) and boat (yellow, PDB: 7LJC and 7X2F) conformations. b Detailed binding modes of dopamine D1 receptor binding to LY3154207 in upright conformations. c Detailed binding modes of dopamine D1 receptor binding to LY3154207 in boat conformations. Hydrogen bonds are presented as orange dashes; π–π stacking and π–cation stacking interactions are presented as gray dashes
In the upright conformation, the tertiary alcohol group of LY3154207 extends toward TM III, establishing a single hydrogen bond with Cys1153.44 (Fig. 23b). The central tetrahydroisoquinoline ring of LY3154207 engages in extensive hydrophobic interactions with Val1193.48, Trp1233.52, and Leu1434.45. Additionally, the dichlorophenyl group forms a π-cation interaction with the side chain of Arg130ICL2. In the boat conformation, the dichlorophenyl group participates in π–cation interactions with the side chains of Arg130ICL2, and interacts sandwich-like π–π stacking with Trp1233.52 (Fig. 23c). The tetrahydroisoquinoline ring of LY3154207 established hydrophobic interactions with neighboring residues, including Met135ICL2, Ala1394.41, Ile1424.44, Leu1434.45, and the alkyl chain of Lys134ICL2. In addition, two hydrogen bonds were observed between LY3154207 and the polar residues Arg130ICL2 and Lys1384.40.
Superposition of the D1R–LY3154207–dopamine and D1R–dopamine structures showed near-identical binding positions for dopamine and the surrounding residues. Instead, LY3154207 interacted with Arg130ICL2 and Lys134ICL2 and stabilized ICL2, which interacts directly with G-proteins, thereby increasing the population of D1R adopting active conformations (Fig. 23b, c).279
Another example of an allosteric modulator that binds outside 7TMD formed by helices III–V is the allosteric antagonist avacopan of C5a receptor 1. Human C5a receptor 1 (C5aR1), which binds to the pro-inflammatory mediator C5a, is primarily expressed on the surfaces of various immune cells, such as neutrophils, eosinophils, and dendritic cells.281,282 Overactivation of the C5aR1-C5a axis will cause uncontrolled inflammation;283 thus, C5aR1 antagonists are ideal candidates for treating various inflammatory conditions, including sepsis COVID-19, etc.284,285,286
Avacopan (Fig. 20b) is an orally administered allosteric antagonist of C5aR1 approved by the FDA in 2021 to treat severe autoantibody (ANCA)-ANCA-associated vasculitis (granulomatosis with polyangiitis and microscopic polyangiitis).287,288 Pharmacological studies have indicated the ability of avacopan for biased inhibition of β-arrestin coupling.289,290,291 The co-crystal structure of C5aR1 was reported with avacopan, highlighting the binding site outside 7TMD between helices III and V (Fig. 24a). The cyclopentane group of avacopan extended into the crevice between helices III and IV and occupied the hydrophobic pocket consisting of Leu1253.41, Val1594.48, Leu1634.52, and Leu1674.56 (Fig. 24b). The o-methyltrifluoromethylbenzene group exhibited hydrophobic and aromatic interactions mediated by residues Ile1243.40, Leu1253.41, Leu2095.45, Trp2135.49, Pro2145.50, and Leu2185.45 in the binding cleft between helices III and V. The m-methylfluorobenzene group lies deeper and forms non-polar interactions with residues Phe1353.52, Ile2205.56, Cys2215.57, and Phe2245.60 in C5aR1. Only one hydrogen bond was observed between the carbonyl substituent of the amide bond of avacopan and Trp2135.49. Additionally, there is a water-mediated polar interaction between avacopan and Thr2175.53.

a Schematic representation of the allosteric antagonist avacopan bound to C5a receptor 1 (PDB: 6C1R). b Detailed binding modes of C5a receptor 1 bound to avacopan. Hydrogen bond is presented as orange dashes. c Superimposed views of highlighted residue Trp2135.49 on C5aR1–PMX53–avacopan (pink cartoon, pink sticks; PDB: 6C1R) and C5aR1–C5a–Go (blue cartoon, blue sticks; PDB: 8IA2) structures
In the inactive C5aR1 structure, Trp2135.49 undergoes a conformational transition to accommodate avacopan binding, in contrast to the active C5aR1 structure (Fig. 24c). Avacopan may stabilize the conformation of the residues Ile1243.40, Pro2145.50, and Phe2516.44 in their inactive states through direct hydrophobic interactions (Fig. 24b). This stabilization, in turn, hinders conformational changes in transmembrane helices TM5 and TM6, which are necessary for receptor activation.292,293
4) TM V–VI:
CXCR3–SCH546738 structure
C-X-C chemokine receptor type 3 (CXCR3), a class A GPCR, is highly expressed on effector T cells and is activated by chemokines CXCL9, CXCL10 and CXCL11.294 Due to the critical role of CXCR3 in type 1 immunity, agonists and antagonists targeting CXCR3 have been synthesized to treat infection, autoimmune diseases, allograft rejection and cancers.295,296,297 Among these, SCH546738 (48) (Fig. 25c) has shown remarkable efficacy in several preclinical trials by effectively inhibiting the activation of T cell chemotaxis with an affinity of 0.4 nM.298

a Schematic representation of the allosteric antagonist SCH546738 bound to CXCR3 (PDB code 8HNN). b Detailed binding modes of CXCR3 binding to SCH546738. c 2D structure of small-molecule allosteric ligand SCH546738 provided for clarity
In the CXCR3–SCH546738 structure, SCH546738 is trapped in a narrow hydrophobic pocket surrounded by TM3, TM5 and TM6 (Fig. 25a).299 The head of SCH546738 is surrounded by a hydrophobic pocket formed by residues Phe1353.36, Ala1393.40, Phe2245.47, Leu2285.51, Met2315.54, Ile2616.41, Ala2736.53 (Fig. 25b). The tail of SCH546738 stretches out to the lipid bilayer and interacts with Tyr2355.58, Leu2395.62 and Leu2586.38. Given the unique allosteric site of SCH546738, the interposition of SCH546738 may weaken the repacking between TM5-TM6, maintaining the receptor in an inactive state.
5) TM I, VI, VII:
A1R–MIPS521, GPR101–AA-14, mGlu4–VU0364770, and mGlu4–ADX88178 Structures
The adenosine A1 receptor (A1R), a subtype of the adenosine receptor,300 has been a highly pursued non-opioid analgesic target for the treat chronic pain.301,302,303,304 Nonetheless, no selective clinically approved A1R agonists or antagonists are currently available.
MIPS521 (49) (Fig. 20b), a PAM of A1R, suppressed spinal nociceptive signaling and displayed an analgesic effect in a rat model with a pEC50 of 6.9 ± 0.4.305 Structural analysis revealed that MIPS521 binds outside 7TMD surrounded by helices I, VI, and VII (Fig. 26a). Residues Leu181.41, Ile191.42, Val221.45, Leu2426.43, Leu2456.46, Ser2466.47, Phe2757.40, Leu2767.41, and Met2837.48 formed shallow hydrophobic pockets at the allosteric site (Fig. 26b). The amino group of MIPS521 (Fig. 26c) was hydrogen-bonded to the main-chain carbonyl groups in Ser2466.47 and Leu2767.41. Comparing the ADO–A1R–Gi2 and MIPS521–ADO–A1R–Gi2 structures showed that MIPS521 has minimal impact on the binding position of ADO and receptor conformations. Mechanistically, the binding of MIPS521 may stabilize the active conformation by interacting with the allosteric site, which, in turn, promotes the collapse of the Na+ pocket (a nearby conserved class A activation motif).245,306

a Schematic representation of PAM MIPS521 bound to A1R (PDB code 7LD3). b Detailed binding modes of A1R binding to MIPS521. Hydrogen bonds are presented as orange dashes. c 2D structure of small-molecule allosteric ligand MIPS521 provided for clarity
Targeting of GPCR transmembrane domain (inside 7TMD)
An empty pocket is present in the middle of the GPCR transmembrane domain that serves as an allosteric site inside 7TMD, as shown in Fig. 27. Allosteric modulators bound to this pocket primarily interact with helices other than TM I and TM IV. To date, a PAM of FFAR3, an allosteric antagonist of CRF1R, an allosteric agonist of PTH1R, and five NAM and one PAM of mGluR5 have been reported to bind to this region (Table 3). In contrast, the binding of NAMs and an allosteric antagonist blocks the outward movement of TM VI, thereby acting as an antagonist. Notably, the binding of only the allosteric agonist stabilizes the G protein through direct interactions.

Allosteric binding sites in the transmembrane domain within 7TMD of GPCRs and corresponding small-molecule allosteric modulators. Stick models of small-molecule ligands are mapped to representative member CRF1R, PDB: 4K5Y. The number of unique modulators is indicated in boldface type, and the number of GPCRs containing the pocket is provided in parentheses
1) Inside 7TMD:
FFAR3–AR420626, CRF1R–CP-376395, mGluR5–mavoglurant, mGluR5–compound 14, mGluR5–HTL14242, mGluR5–Fenobam, mGluR5–M-MPEP, and PTH1R–PCO371 structures
The metabotropic glutamate (mGlu) receptor type 5 is among the eight most widely expressed mGlu receptors in the brain.307 Recently, mGluR5 has been shown to be involved in a growing number of cognitive and psychiatric disorders including schizophrenia.308,309,310 The mGlu receptor family is abundant in allosteric pharmacology among the class C GPCRs.311,312,313,314,315 In particular, allosteric regulation of mGlu5 receptors has received significant attention as a novel modality to treat diseases, including Parkinson’s disease and schizophrenia.316,317,318 NAMs of the mGlu5 receptors may serve to normalize excessive glutamate activity without blocking the physiological roles of the brain; thus, they are regarded as prospective agents for the treatment of multiple neurological disorders.319,320
The key binding determinants of these NAMs were identified in TM III, VI, and VII (Fig. 29a). Among these, HTL14242 (57) (Fig. 28) is an advanced and orally active NAM that progresses in early clinical testing for the potential treatment of amyotrophic lateral sclerosis (ALS) (Fig. 29). Crystal structures of mGluR5 with the lead compound of HTL14242, compound 14 (56), demonstrate the underlying basis for SAR optimization. With the pyrimidine linker traversing the narrow channel in the allosteric pocket formed by Tyr6593.44, Ser8097.39, Val8067.36, and Pro6553.40, the benzene ring and pyrazole ring of compound 56 respectively sit in two sub-pockets. The presence of the nitrile moiety on the benzene ring is crucial due to its delicate orientation that mediates the formation of hydrogen bonds with Val7405.40 via the bridging of a water molecule (Fig. 29b). Therefore, removal or replacement of the nitrile moiety caused a substantial drop in affinity, underlying the significance of maintaining this moiety during SAR studies (proved by compounds 57 and 58) (Fig. 29c). Notably, the conformation of the pyrazole end allows for slightly bulkier rings and small substitutes, as long as the hydrogen bond between the nitrogen atom and Ser8097.39, and the surrounding polar network is maintained (Fig. 29b). Such SBDD analysis thereupon yielded HTL14242, which harbors highly similar binding modes with 56 while simultaneously benefiting from its favorable physicochemical and pharmacokinetic properties (Fig. 29c).321

2D chemical structures of synthetic small-molecule allosteric ligands targeting the transmembrane domain within 7TMD of GPCRs

a Schematic representation of the NAM fenobam bound to mGlu5 receptor (PDB: 5CGC). b Detailed binding modes of mGlu5 receptor binding to compound 14; polar interactions are shown in orange dashes. c SAR optimization of mGlu5 receptor NAMs. d Superimposed views of the highlighted residues on mGlu5–mavoglurant (purple cartoon, purple sticks; PDB: 4OO9), mGlu5–HTL14242 (orange cartoon, orange sticks; PDB: 5CGD), mGlu5–fenobam (blue cartoon, blue sticks; PDB: 6FFH), and mGlu5–CDPPB (pink cartoon, pink sticks; PDB: 8TAO) structures
Furthermore, by aligning mGlu5-NAM structures, a common salt bridge interaction was found between the residue pair Lys6653.50 and Glu7706.35, whereas it was absent in the PAM-bound complex, suggesting that NAM hinders the outward motion of TM VI by enhancing the interactions between TM III and TM VI at the cytoplasmic end.322,323,324 Conformational transitions of the highly conserved Trp7856.50 occur in the allosteric modulator-bound mGluR5 structure to adjust the size of the allosteric pocket and accommodate binding (Fig. 29d).178,325 Trp7856.50 within the FxxCWxP6.50 motif serves as a “toggle switch” in class A receptors, undergoing a conformation change during activation.321,326 It is plausible that this residue may also play a role in class C GPCR activation, though further investigation is required to elucidate its involvement.178
Other allosteric inhibitors bound to the transmembrane domain include allosteric PTH1R agonists. The parathyroid hormone receptor (PTH1R), a class B1 GPCR,327 is activated by parathyroid hormone and parathyroid hormone-related peptides and plays a central role in maintaining mineral ion homeostasis and skeletal metabolism.328,329,330 Recently, a highly selective PTH1R agonist, the orally active non-peptidic small molecule PCO371 (54) (Fig. 28), was identified and is currently undergoing phase 1 clinical trials to treat hypoparathyroidism.331,332 PCO371 consists of four chemical modules, from left to right: trifluoromethoxyphenyl, spiro-imidazolone, dimethylphenyl, and dimethylhydantoin.333
The solved crystal structure complex of PTH1R–PCO371 reveals important information regarding the PCO371 binding cavity,334 which was observed to reside in a pocket surrounded by residues on TM II, TM III, TM VI, and TM VII, analogous to mGluR5 (Fig. 30a).335,336 In the PCO371 binding site, the NH moiety of the spiro-imidazolone in PCO371 is engaged in a hydrogen binding interacts with Tyr4597.57 (Fig. 30b). The carbonyl group of dimethylhydantoin in PCO371 forms a salt bridge interaction with the protonated nitrogen of Arg2192.46. In addition to polar contacts, the trifluoromethoxyphenyl group formed nonpolar interactions with Met4146.46, Leu4166.48, and Phe4547.52; the spiroimidazolone group formed hydrophobic interactions with Leu2262.53, Ile2993.47, Pro4156.47, and Phe4176.49; the dimethylphenyl group formed hydrophobic interactions with His2232.50, Leu3063.54, Val4126.44, Leu4136.45 and Tyr4597.57; and the dimethylhydantoin group formed hydrophobic interactions with Asn4638.47. Notably, Glu394G.H5.24 in the Gs protein was hydrogen-bonded to the carbonyl group of dimethylhydantoin, and Tyr393G.H5.23 formed a hydrophobic interaction with dimethylhydantoin, thus stabilizing the ternary PCO371–PTH1R–Gs complex.

a Schematic representation of the allosteric agonist PCO371 bound to PTH1R (PDB: 8GW8). b Detailed binding modes of PTH1R binding to PCO371. Hydrogen bonds are presented as orange dashes. c Superposition of the PTH1R bound to PCO371 (displayed in pink) and PTH (displayed in blue) reveals conformational changes upon PCO371 binding
As the binding of PCO371 precludes the endogenous ligand from occupying the core of the 7TM helical bundle, only structures in which PCO371 alone binds to PTH1R exist. The presence of PCO371 induced inward displacement of the extracellular and cytoplasmic termini of TM6 cells (Fig. 30c). Comparison of the structures of PCO371–PTH1R and CP-376395–CRF1R revealed that PCO371 has a deeper binding site and therefore does not impede the formation of the conserved Pro6.47-X-X-Gly6.50 kink, as CP-376395 does. In addition, the binding site of PCO371 is shallower than that of allosteric modulators that bind to the intracellular surface inside 7TMD and therefore does not impede G-protein or arrestin binding. Till date, PCO371 is probably the only receptor allosteric agonist reported to stabilize the active state through direct interactions with downstream transducers.
Targeting of GPCR Intracellular Surface (outside and inside 7TMD)
In these instances, allosteric modulators bind to interfaces located between the cytoplasmic terminus of the 7TM helical bundle and downstream transducers,337 including the outside and inside of 7TMD.46 Till date, only two different binding sites in the intracellular surface have been identified using crystal structures (Table 4), namely the pocket outside helices V − VII and the pocket inside 7TMD (Fig. 31).

Two intracellular allosteric binding sites in GPCRs and the corresponding small-molecule allosteric modulators. Stick models of small-molecule ligands are mapped to representative members of outside 7TMD (V−VII) (GCGR, PDB: 5EE7) and inside 7TMD (CCR2, PDB: 5T1A) GPCRs. For each pocket, the number of unique modulators is indicated in boldface type, and the number of GPCRs containing the pocket is provided in parentheses
1) Outside 7TMD (V−VII):
GPR88−2-PCCA, GCGR−MK-0893, GCGR−NNC0640, GLP-1R−PF-06372222, GLP-1R−compound 2, GABAB−GS39783, and GABAB−BHFF structures
The reported allosteric sites on the intracellular surface outside 7TMD are all attached to TM VI, located between helices V and VII, suggesting that TM VI plays a functional role in transmitting ligand-binding information to the orthosteric pocket.338 G-protein coupling requires outward motion of the intracellular end of TM VI, a key signature for activating GPCR,339 which inspired the design of small-molecule allosteric modulators that target this site.
The GABAB receptor, classified as a heterodimeric class C GPCR, comprises two distinct subunits, GB1 and GB2, which can be activated by γ-aminobutyric acid (GABA), a primary inhibitory neurotransmitter in the central nervous system.340,341 GABAB receptors are key therapeutic targets in the treatment of multiple neurological disorders, including depression, schizophrenia, and drug addiction.342,343,344 Dimerization of GABAB receptors results in the emergence of this new dimer-specific allosteric binding site (Fig. 33a),345,346,347,348,349 which prevents GS39783 (69) and BHFF (70) from acting on the monomer, but instead specifically controls GPCR dimer activity.
BHFF (Fig. 32), a potent PAM of the GABAB receptor that enhances receptor potency only when activated by orthosteric agonists, lacks efficacy in mouse experiments.350 Determination of the BHFF−GABAB receptor co-crystal structure clearly shows that BHFF is bound between TM V−TM VI of GB1 and TM VI of GB2 within the intracellular tips (Fig. 33a).351 The 3-hydroxy group and ketone of BHFF formed two hydrogen bonds with the Lys792ICL3 side chain of GB1 (Fig. 33b). For hydrophobic interactions, BHFF is buried in a cavity defined by the residues Ala7885.58, Tyr7895.59, Met8076.41, Tyr8106.44 of GB1, and Tyr6916.38 and Met6946.41 of GB2.

2D structures of synthetic allosteric ligands targeting the GPCR intracellular surface

a Schematic representation of PAM BHFF bound to GABAB receptor (PDB: 7C7Q). b Detailed binding modes of GABAB receptor binding to BHFF. Hydrogen bonds are presented by orange dashes. c Superimposed views of the N-terminal α-helix on compound 2–GLP-1R–Gs (blue cartoon, blue sticks; PDB: 7DUR) and GLP-1–GLP-1R–Gs (pink cartoon, pink sticks; PDB: 6×18) structures. d Schematic representation of ago-PAM compound 2 bound to GLP-1R (PDB: 7EVM). e Detailed binding modes of GLP-1R bound to compound 2. f Superimposed views of highlighted residues on compound 2–GLP-1R–Gs (blue cartoon, blue sticks; PDB: 7DUR), GLP-1–GLP-1R–Gs (pink cartoon, pink sticks; PDB: 6×18), and compound 2–GLP-1–GLP-1R–Gs (purple cartoon, purple sticks; PDB: 7DUQ) structures
Compared with GS39783, another PAM molecule for the GABAB receptor, both PAM molecules bind to highly similar locations within the dimer interface, and the overall complex structure is almost identical. In contrast to the binding of the orthosteric agonist baclofen alone, the PAM induces a straightening and inward shift of TM3 and TM5 in GB2 on the intracellular side and stabilizes the TM6-mediated dimerization. This alteration contributes to the stabilization of the receptor in its active state.352,353,354
Compound 2 (68) (Fig. 32) is a novel ago-PAM of GLP-1R that covalently binds to Cys3476.36 on helices VI and displays partial and almost full agonism.355,356 However, clinical trials of compound 2 failed because of its poor pharmacokinetic properties.357 In the co-crystal structure of GLP-1R complexed with compound 2, it was demonstrated that compound 2 was located on the TM VI membrane-facing surface and formed interactions only with residues on TM VI (Fig. 33d), among which a covalent disulfide bond was formed between the sulfonic group of compound 2 and Cys3476.36.358 In addition, the tert-butyl group of compound 2 extended toward TM VII and formed nonpolar interactions with Ala3506.39 and Lys3516.40 (Fig. 33e). The dichloroquinoxaline group was directed toward ICL3, forming van der Waals contacts with Lys3466.35 and Cys3476.36.
The binding of compound 2 alone, GLP-1 alone, and compound 2 − GLP-1 together all led to a similar extent of outward movement in the intracellular terminus of TM6 (Fig. 33c). Nonetheless, in the compound 2–GLP-1–GLP-1R–Gs structure, two additional salt bridge interactions have arisen (R1762.46 and E4088.49, E4238.64 and R46 in Gβ) (Fig. 33f), offering the potential to strengthen G-protein binding through long-range allosteric communications.358 Furthermore, the N-terminal α-helix of the GLP‐1 R extends downward into the orthosteric pocket upon binding to compound 2, which may serve to stabilize the active conformation (Fig. 33c).359
Covalent binding confers a higher potential energy barrier upon compound 2 dissociation, thus minimizing potential off-target effects.360,361,362 This suggests that the protruding free cysteine residues in the transmembrane helix provide an opportunity to develop irreversible GPCR allosteric modulators.
All allosteric antagonists and NAMs (MK-0893 (65), NNC0640 (66), and PF-06372222 (67)) bind to the other side of TM VI near TM VII and appear to be well conserved with respect to the allosteric site and binding modes.363,364,365 In this case, the protruding TM VI can serve as a base for the attachment of modulators in a clamp shape. The binding of such allosteric modulators requires certain conditions, such as a cleft between helices for the insertion of the pincers and adequate hydrophobic contacts with the surrounding residues for stable binding. The outward displacement within the TM VI required for transducer binding is restricted, preventing receptor activation.366
The glucagon receptor belongs to the class B GPCR and plays a critical role in the maintenance of glucose homeostasis.367,368 Given the key role of glucagon in elevating glycemia, small-molecule antagonists targeting GCGR are considered promising treatments for diabetes.369,370,371 In support of this concept, a novel allosteric antagonist targeting GCGR, MK-0893 (Fig. 32), has advanced into phase 2 clinical studies to treat type 2 diabetes mellitus;372 however, side effects, including increased LDL-C, have hindered its clinical use.373
The X-ray crystal structure of MK-0893 with GCGR was solved, revealing the atomic details of allosteric modulator binding.363 In this structure, MK-0893 is situated within an intracellular pocket outside helices V–VII (Fig. 34a). In terms of polar interactions, the terminal anionic carboxylic acid moiety of MK-0893 (Fig. 34c) established a salt-bridge interaction with Arg3466.37 (Fig. 34b).374 It contributes to the establishment of a hydrogen-bonding network involving the side chain of Asn4047.61 and the main-chain amine of Lys4057.62. It also forms a water-mediated hydrogen bond with the side chain of Ser3506.44. Additionally, the amide group of the ligand formed hydrogen bonds with the backbone carbonyl groups of Ser3506.41, Leu3997.56, and the side chain of Lys3496.40. Regarding hydrophobic interactions, the methoxynaphthalene moiety wedges into a cavity formed by helices V and VI and interacts with residues Leu3295.61, Phe3456.36, Leu3526.43, and Thr3536.44, and the alkyl chain of Lys3496.40. The phenylethylpyrazole core scaffold engages in hydrophobic interactions with residues Thr3536.44, and Leu3997.56, and forms a π−cation interaction with Lys3496.40. This structure closely resembles other inactive structures (PDB:5XEZ, 8JRV, and 8JRU)375 and does not exhibit specific conformations induced by MK-0893.

a Schematic representation of the allosteric antagonist MK-0893 bound to GCGR (PDB: 5EE7). b Detailed binding modes of GCGR binding to MK-0893. Hydrogen bonds are presented as orange dashes. c 2D structure of small-molecule allosteric ligand MK-0893 shown for clarity
2) Inside 7TMD:
β2AR–Cmp-15PA, CCR2–CCR2-RA-[R], CCR9–vercirnon, CCR7–Cmp2105, CXCR2–00767013, GPR61–Compound 1, and NTSR1–SBI-553 structures
In contrast to orthosteric ligands, GPCRs utilize their intracellular surface inside 7TMD to directly mediate downstream signaling via G proteins or arrestins. This site has also been found to act as a binding site for allosteric regulators and is quite similar in all solved complexes (a cavity composed of the cytoplasmic region of helices I, II, VI, and VII) (Fig. 31).24,376,377 Allosteric antagonists bound to this region inhibit GPCR-mediated signaling through a novel dual mechanism, which appears to be a powerful way to antagonize the receptor. These allosteric modulators not only compete with G-proteins or arrestins by occupying their binding sites but also display π-π interactions (parallel or T-shaped) with Tyr7.53 from the conserved NPxxY motif in TM VII, acting as a molecular glue to hold together the cytoplasmic ends of the helical bundle, further preventing conformational transitions associated with activation. Therefore, targeting this druggable allosteric pocket in receptors using small-molecule allosteric antagonists may provide new opportunities for the discovery of GPCR drugs.
Chemokine receptors and their ligands are involved in chemotactic trafficking during the inflammatory responses.378,379,380 CC chemokine receptor 9 (CCR9) activated by CCL25 has emerged as a potential therapeutic target for inflammatory bowel disease because it is essential for mediating leukocyte homing to the gut.381,382,383
Vercirnon (73) (Fig. 32), an allosteric antagonist of CCR9 used in Crohn’s treatment, has progressed to Phase 3 clinical studies. However, its efficacy is limited due to the requirement for significantly high doses to effectively block receptor activation.384 This uncertainty may stem from the fact that drug molecules might not necessarily bind to the intracellular G protein region upon entering the cytoplasm.271 This challenge could be common among this class of modulators, but only vercirnon has entered clinical trials at present.
The binding site of vercirnon has been shown via X-ray crystallography to be the center of the 7TM helical bundle on the intracellular side (Fig. 35a). The sulfone group (Fig. 35c) of vercirnon engages in a trivalent hydrogen bond with the main-chain amino groups of three neighboring residues: Glu3228.48, Arg3238.49, and Phe3248.50.385 Additionally, the pyridine-N-oxide group is hydrogen-bonded to the side chain of Thr81ICL1, and the ketone group forms a hydrogen bond with the side chain of Thr2566.37 (Fig. 35b). Regarding hydrophobic interactions, the tert-butylphenyl group is deeply buried in the hydrophobic pocket surrounded by Val691.53, Val721.56, Tyr731.57, Leu872.42, Tyr3177.53, and Phe3248.50. The chlorophenyl group was situated within a narrow crevice formed by the hydrophobic parts of residues Leu872.43, Ile1403.46, Val2596.40, and Tyr3177.53. Furthermore, the pyridine-N-oxide group is enclosed within a polar cavity formed by residues Thr81ICL1, Thr832.39, Asp842.40, Arg1443.50, and Arg3238.49.

a Schematic representation of the allosteric antagonist vercirnon bound to CCR9 (PDB: 5LWE). b Detailed binding modes of CCR9 binding to vercirnon. Hydrogen bonds are presented as orange dashes. c 2D structure of small-molecule allosteric ligand vercirnon shown for clarity
Conclusions and perspectives
Over the past few decades, crystallographic, biochemical, and computational studies have provided unprecedented atomic and structural insights into the comprehensive regulation of GPCR structures.386,387,388,389 Till date, 388 orthosteric modulators, as well as 717 complex structures of GPCRs bound to the orthosteric modulators, have been reported. In addition, 53 different allosteric small-molecule modulators bound to GPCR complex structures and nine different allosteric sites were also solved. In this study, we first review the structure advances, signaling mechanisms, and functional diversity of GPCRs to outline the current profile and up-to-date development in this field. Importantly, by means of the aforementioned crystallographic advancements, orthosteric and allosteric modulators are discussed separately in terms of their complex structures, signaling mechanisms, and consequential implications for drug discovery. Such in-depth investigation underlines the significance of understanding GPCR structures and mechanisms for developing effective therapeutics.
Because of the relatively large amount of orthosteric modulators and their shared functional mechanism of competing with the endogenous ligands, representative cases of orthosteic drugs launched within the past five years have been carefully selected and dissected, including μ-OR-oliceridine complex (stand-out for G-protein biased signaling), S1PR-siponimod complex (stand-out for subtype selectivity and “toggle switch” activation mechanism), OX2R-lemborexant complex (stand-out for kinetics and dynamics parameters), 5-HT1F-lasmiditan complex (distinctive for subtype selectivity), and GnRH1-elagolix complex (distinctive for signal transmission based on atypical receptor structure). Structural analysis revealed that modulators are stabilized within the orthosteric pockets by key polar interactions with residues on the TM bundles, in which residues that are unconserved among the family subtypes are often determinants of selectivity. Furthermore, the “toggle switch”, PIF, DRY, and NPxxY motifs are critical mechanical switches that transmit extracellular stimuli to the intracellular regions, and some specific polar interactions between TM3 and TM5/6 may also serve as a boost for the displacement of TM5/6, which is a hallmark of receptor activation/deactivation.
Since the binding patterns and action mechanisms of allosteric modulators tend to be unique, we placed special emphasis on the depiction of allosteric regulators.390 From the analysis of these structural complexes, the extracellular vestibule inside 7TMD was identified as the most prevalent binding site for allosteric modulators, and induced-fit shape matching and charge matching are the determinants of allosteric ligand accommodation. The binding of allosteric modulators alters the free-energy landscape and stabilizes the different dominant conformations of the receptor.391 Specifically, we established a novel classification of allosteric modulators into two categories. First, the allosteric effects of the modulators can be realized by directly modulating the binding of orthosteric ligands or intracellular transducer proteins to receptors. In this instance, allosteric modulators may occupy a pocket above the orthosteric binding pocket and interact directly with orthosteric ligands and/or surrounding residues to alter association or dissociation. Allosteric modulators may also occupy the binding pockets of G-proteins and arrestins to impede their binding or occupy the pockets above their binding sites to regulate binding. Second, allosteric modulators regulate the ability of receptor complexes to interact with intracellular transducer proteins by indirectly altering their activation pathways. In this instance, allosteric modulators appear to act as steric wedges, stabilizing or destabilizing certain interactions to restrict or facilitate conformational rearrangements of the receptor.
In addition to the commonalities summarized above, it is of broad pharmaceutical interest to understand how the drug recognition basis and delicate mechanistic regulation of orthosteric/allosteric modulators can expand upon drug design and why we need to pursue this line of research.117,392,393 From a structural and chemical biology perspective, understanding the mechanical switch cascade assists in acquiring insights into fine-tuning regulation and enables human control of different downstream functions of GPCRs.62 Therefore, GPCRs after mutagenesis may be employed as biosensors to initiate distinct intracellular signaling events.394
Medicinal chemists can be inspired by five perspectives based on the analysis presented in this review:
-
(1)
With orthosteric small-molecule modulators remaining the mainstream therapeutic agents for GPCRs, the rational design of orthosteric ligands with high affinity, efficacy, and selectivity has been a long-standing topic.395,396,397,398 Concerning with enhancement of affinity and efficacy, designing ligands that constitute “anchors” and “drivers” has been suggested.399,400 The “anchors” represent the primary parts of the ligands that contribute to affinity by binding with utmost residues and remain nearly unchanged during the transition between different states of the receptor. The “drivers” should be designed to form certain interactions with “toggle switches” and thus trigger activation/deactivation signaling. The “anchors” provide a foundation that allows the “drivers” to exert a “pull” and/or “push” action that shifts the receptor population, thereby enhancing efficacy. This “mechanism-based drug design” concept, expanding upon the traditional “structure-based drug design” strategy, may shed light on effective and efficient GPCR drug discovery.401,402 Regarding the improvement of selectivity, although the orthosteric sites are highly conserved in one family, capturing slight differences in loop/TM bundle/key residue conformation from other receptor subtypes and specifically interacting with these hotspots may result in a multifold increase in selectivity, thus alleviating side effects.
-
(2)
Uncovering the appropriate allosteric pockets and further conducting allosteric drug design are also durable and questionable research topics.403,404,405 With the development of crystallography offering credible initial structures, MD simulations, together with enhanced sampling methodologies, have enabled the characterization of various intermediate conformations where cryptic allosteric sites may emerge.406,407,408,409,410,411,412,413,414 Based on the identified allosteric sites, allosteric modulators can be designed using a three-dimensional (3D) molecular generation algorithm that uses the topological surface and geometric structure.415 Although the low conservation of allosteric sites may result in a lack of a general formula concerning the scaffold of allosteric modulators,416 it can be concluded that while the hydrophobic nature of allosteric ligands will contribute to receptor binding and regulation in most instances, polar groups can also interact, especially when the modulators bind outside 7TMD and require immobilization to the receptor. Moreover, an “allosteric-like” rule condensed by our group (molecular weight ≤600; 2≤ number of rotatable bonds ≤6; number of rings ≤5; number of rings in the largest ring system= 1 or 2, 3≤ SlogP ≤7) can serve as a filter criteria when designing molecules.417
-
(3)
With the GPCR-ligand complex structural information in hand, the topic of whether the kinetic and dynamic properties of ligands can also be modified by medicinal chemists is considered.177,418,419 Based on the desirable kon and koff values of lemborexant analyzed above, it is inferred that designing a ligand that can assemble to its receptor-bound state in advance may help achieve a high kon value, in which case a simple MD simulation of designed ligands in a solvent can acquire the preferential conformation of the ligand and guide our selection. In contrast, estimating the binding free energy of the designed ligands may help predict and rank their koff values, thus providing valuable guidance for ligand optimization from a kinetic perspective.420,421
-
(4)
For both orthosteric and allosteric modulators, achieving biased signaling is of scientific and clinical significance for any GPCR target.26,422,423,424,425,426,427,428 With the design of G-protein-biased modulators of μ-OR as a paradigm, it is suggested that comparison of the biased and unbiased ligands binding modes may help locate the key residues or regions that contribute to biased signaling. A “mechanism-based drug design” that designs ligand forming/avoiding interactions with hotspots may lead to the successful distinguishing of promiscuous signals.
-
(5)
In the regulation of PAM and NAM, orthosteric and allosteric sites are coupled through mutual signal perturbations within the protein.429,430,431 Thus, bitopic ligands, which connect the pharmacophores of both orthosteric and allosteric ligands through a linker, may facilitate a deeper investigation of site-site coupling within GPCRs and harvest ligands with higher selectivity and biased signaling abilities.432,433,434,435,436,437 With GPCR bitopic ligands occupying a relatively blank zone, linker optimization while immobilizing fragments at both ends, as well as fixing the orthosteric fragment while attaching a warhead to the allosteric fragment to anchor it in an allosteric pocket containing nucleophilic amino acids, may prove to be promising strategies for exploiting this field.
Despite significant technological advances and studies on GPCR structure and drug discovery,438,439,440,441,442 obstacles still exist. As GPCR crystallography is still a time-consuming and labor-intensive process,31 structure-dependent scenarios can somewhat hinder the study of GPCR mechanism and drug discovery, even with the use of AlphaFold and RoseTTAFold.443,444,445 For example, modeling from AlphaFold is not precise in residue orientation and can thus mislead the analysis of signaling mechanisms.446 Moreover, MD simulations do not guarantee the identification of novel cryptic allosteric sites in allosteric drug design.447 Therefore, other novel perspectives such as sequence, coarse-grained topology,448 and evolution are urgently required and may alleviate structural dependencies.449 One possibility is the use of available large data of sequences and end-to-end concepts in deep learning to develop “sequence-to-mechanism” or “sequence-to-drug discovery” methodologies,450 which can help avoid error accumulation from various models if the experimental strategies fail to provide the crystal structures.
In summary, we are at a stage of in-depth research on the orthosteric and allosteric modulation of GPCRs. Although our understanding of drug-target interactions, binding hotspots, and mechanisms for small-molecule modulators has become increasingly clear, some aspects still require further study. The present study contributes to a better understanding of the ligand recognition and regulatory mechanisms of GPCRs. Furthermore, by proposing a novel classification of the mechanism of allosteric modulators and an innovative concept of “mechanism-based drug discovery,” we aim to outline the latest landscape of GPCRs and interest researchers to facilitate this field. More effective, selective, and safer small-molecule therapeutics for GPCRs should be the focus of future studies in this field.
References
Eichel, K. & von Zastrow, M. Subcellular organization of GPCR signaling. Trends Pharmacol. Sci. 39, 200–208 (2018).
Rosenbaum, D. M., Rasmussen, S. G. F. & Kobilka, B. K. The structure and function of G-protein-coupled receptors. Nature 459, 356–363 (2009).
Katritch, V., Cherezov, V. & Stevens, R. C. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 33, 17–27 (2012).
Latorraca, N. R., Venkatakrishnan, A. J. & Dror, R. O. GPCR dynamics: structures in motion. Chem. Rev. 117, 139–155 (2017).
Mannes, M., Martin, C., Menet, C. & Ballet, S. Wandering beyond small molecules: peptides as allosteric protein modulators. Trends Pharmacol. Sci. 43, 406–423 (2022).
Nussinov, R. Introduction to protein ensembles and allostery. Chem. Rev. 116, 6263–6266 (2016).
Dunn, H. A., Orlandi, C. & Martemyanov, K. A. Beyond the ligand: extracellular and transcellular g protein-coupled receptor complexes in physiology and pharmacology. Pharmacol. Rev. 71, 503–519 (2019).
Zarzycka, B., Zaidi, S. A., Roth, B. L. & Katritch, V. Harnessing ion-binding sites for GPCR pharmacology. Pharmacol. Rev. 71, 571–595 (2019).
Flock, T. et al. Universal allosteric mechanism for Gα activation by GPCRs. Nature 524, 173–179 (2015).
Dawaliby, R. et al. Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat. Chem. Biol. 12, 35–39 (2016).
Hilger, D., Masureel, M. & Kobilka, B. K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12 (2018).
Dokholyan, N. V. Controlling allosteric networks in proteins. Chem. Rev. 116, 6463–6487 (2016).
Canals, M. et al. A Monod-Wyman-Changeux mechanism can explain G Protein-coupled Receptor (GPCR) allosteric modulation. J. Biol. Chem. 287, 650–659 (2012).
Hauser, A. S. et al. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017).
Sriram, K. & Insel, P. A. G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258 (2018).
Schöneberg, T. & Liebscher, I. Mutations in G protein-coupled receptors: mechanisms, pathophysiology and potential therapeutic approaches. Pharmacol. Rev. 73, 89–119 (2021).
Yang, D. H. et al. G protein-coupled receptors: structure- and function-based drug discovery. Signal Transduct. Target. Ther. 6, 7 (2021).
Casadó, V. & Casadó-Anguera, V. What are the current trends in G protein-coupled receptor targeted drug discovery? Expert Opin. Drug Discov. 18, 815–820 (2023).
Mohr, K. et al. Rational design of dualsteric GPCR ligands: quests and promise. Br. J. Pharmacol. 159, 997–1008 (2010).
Goupil, E., Laporte, S. A. & Hébert, T. E. Functional selectivity in GPCR signaling: understanding the full spectrum of receptor conformations. Mini Rev. Med. Chem. 12, 817–830 (2012).
Lu, S. Y., He, X. H., Ni, D. & Zhang, J. Allosteric modulator discovery: from serendipity to structure-based design. J. Med. Chem. 62, 6405–6421 (2019).
Wootten, D., Christopoulos, A. & Sexton, P. M. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discov. 12, 630–644 (2013).
Wang, Y. et al. Allosteric binding sites at the receptor-lipid bilayer interface: novel targets for GPCR drug discovery. Drug Discov. Today 26, 690–703 (2020).
Zhang, M., Lan, X., Li, X. & Lu, S. Pharmacologically targeting intracellular allosteric sites of GPCRs for drug discovery. Drug Discov. Today 28, 103803 (2023).
Smith, R. D., Lu, J. & Carlson, H. A. Are there physicochemical differences between allosteric and competitive ligands? Plos Comput. Biol. 13, e1005813 (2017).
Slosky, L. M., Caron, M. G. & Barak, L. S. Biased allosteric modulators: new frontiers in GPCR drug discovery. Trends Pharmacol. Sci. 42, 283–299 (2021).
Tan, L., Yan, W. Z., McCorvy, J. D. & Cheng, J. J. Biased ligands of G Protein-Coupled Receptors (GPCRs): Structure-Functional Selectivity Relationships (SFSRs) and therapeutic potential. J. Med. Chem. 61, 9841–9878 (2018).
Robertson, M. J., Meyerowitz, J. G. & Skiniotis, G. Drug discovery in the era of cryo-electron microscopy. Trends Biochem. Sci. 47, 124–135 (2022).
Palczewski, K. et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745 (2000).
Rasmussen, S. G. F. et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 (2007).
Ghosh, E., Kumari, P., Jaiman, D. & Shukla, A. K. Methodological advances: the unsung heroes of the GPCR structural revolution. Nat. Rev. Mol. Cell Biol. 16, 69–81 (2015).
Cherezov, V. et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 (2007).
Rosenbaum, D. M. et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 318, 1266–1273 (2007).
Chen, Q. Y. & Tesmer, J. J. G. G protein-coupled receptor interactions with arrestins and GPCR kinases: The unresolved issue of signal bias. J. Biol. Chem. 298, 102279 (2022).
Li, D. F. & Caffrey, M. Structure and functional characterization of membrane integral proteins in the lipid cubic phase. J. Mol. Biol. 432, 5104–5123 (2020).
Srivastava, A. et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 513, 124–127 (2014).
Manglik, A., Kobilka, B. K. & Steyaert, J. Nanobodies to study G protein-coupled receptor structure and function. Annu. Rev. Pharmacol. Toxicol. 57, 19–37 (2017).
Scheerer, P. et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502 (2008).
Shimada, I. et al. GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 18, 59–82 (2019).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res 28, 235–242 (2000).
Ishchenko, A., Gati, C. & Cherezov, V. Structural biology of G protein-coupled receptors: new opportunities from XFELs and cryoEM. Curr. Opin. Struct. Biol. 51, 44–52 (2018).
Bostock, M. J., Solt, A. S. & Nietlispach, D. The role of NMR spectroscopy in mapping the conformational landscape of GPCRs. Curr. Opin. Struct. Biol. 57, 145–156 (2019).
Park, S. H. & Lee, J. H. Dynamic G protein-coupled receptor signaling probed by solution NMR Spectroscopy. Biochemistry 59, 1065–1080 (2020).
Asher, W. B. et al. Single-molecule FRET imaging of GPCR dimers in living cells. Nat. Methods 18, 397–405 (2021).
Shi, P. et al. A genetically encoded small-size fluorescent pair reveals allosteric conformational changes of G proteins upon its interaction with GPCRs by fluorescence lifetime based FRET. Chem. Commun. 56, 6941–6944 (2020).
Sandhu, M. et al. Conformational plasticity of the intracellular cavity of GPCR-G-protein complexes leads to G-protein promiscuity and selectivity. Proc. Natl Acad. Sci. USA 116, 11956–11965 (2019).
Zhang, M. Y. et al. Decoding the conformational selective mechanism of FGFR Isoforms: A comparative molecular dynamics simulation. Molecules 28, 2709–2727 (2023).
Aydin, Y. et al. Structural details of a Class B GPCR-arrestin complex revealed by genetically encoded crosslinkers in living cells. Nat. Commun. 14, 1151 (2023).
Hauser, A. S. et al. Common coupling map advances GPCR-G protein selectivity. Elife 11, e74107 (2022).
Inoue, A. et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947 (2019).
Leftowitz, R. J. & Shenoy, S. K. Transduction of receptor signals by β-arrestins. Science 308, 512–517 (2005).
Ranjan, R. et al. Novel structural insights into GPCR-β-Arrestin interaction and signaling. Trends Cell Biol. 27, 861–872 (2017).
Milligan, G., Ward, R. J. & Marsango, S. GPCR homo-oligomerization. Curr. Opin. Cell Biol. 57, 40–47 (2019).
Bulenger, S., Marullo, S. & Bouvier, M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol. Sci. 26, 131–137 (2005).
Sato, M., Blumer, J. B., Simon, V. & Lanier, S. M. Accessory proteins for G proteins: Partners in signaling. Annu. Rev. Pharmacol. Toxicol. 46, 151–187 (2006).
Rouault, A. A. J. et al. The GPCR accessory protein MRAP2 regulates both biased signaling and constitutive activity of the ghrelin receptor GHSR1a. Sci. Signal. 13, eaax4569 (2020).
Serafin, D. S. et al. Dawn of a New RAMPage. Trends Pharmacol. Sci. 41, 249–265 (2020).
Wang, M., Lyu, J. J. & Zhang, C. Single transmembrane GPCR modulating proteins: neither single nor simple. Protein Cell pwad035 (2023).
Kumar, K. K. et al. Negative allosteric modulation of the glucagon receptor by RAMP2. Cell 186, 1465–1477 (2023).
Luo, P. et al. Structural basis of signaling regulation of the human melanocortin-2 receptor by MRAP1. Cell Res 33, 46–54 (2023).
Manglik, A. & Kruse, A. C. Structural basis for G protein-coupled receptor activation. Biochemistry 56, 5628–5634 (2017).
Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 55, 114–120 (2019).
Mafi, A., Kim, S.-K. & Goddard III, W. A. The mechanism for ligand activation of the GPCR–G protein complex. Proc. Natl Acad. Sci. USA 119, e2110085119 (2022).
Hori, T. et al. Na+-mimicking ligands stabilize the inactive state of leukotriene B4 receptor BLT1. Nat. Chem. Biol. 14, 262–269 (2018).
Selvam, B., Shamsi, Z. & Shukla, D. Universality of the sodium ion binding mechanism in Class A G-protein-coupled receptors. Angew. Chem. Int. Ed. 57, 3048–3053 (2018).
Liu, W. et al. Structural basis for allosteric regulation of GPCRs by Sodium ions. Science 337, 232–236 (2012).
Katritch, V. et al. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244 (2014).
DeWire, S. M. et al. AG protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 344, 708–717 (2013).
Manglik, A. et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190 (2016).
Schmid, C. L. et al. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 171, 1165–1175 (2017).
Hu, G. M., Mai, T. L. & Chen, C. M. Visualizing the GPCR network: classification and evolution. Sci. Rep. 7, 15495 (2017).
Foster, S. R. et al. Discovery of human signaling systems: pairing peptides to G protein-coupled receptors. Cell 179, 895–908 (2019).
Pal, K., Melcher, K. & Xu, H. E. Structure and mechanism for recognition of peptide hormones by Class B G-protein-coupled receptors. Acta Pharmacol. Sin. 33, 300–311 (2012).
Yu, M. Z. et al. Battle of GLP-1 delivery technologies. Adv. Drug Deliv. Rev. 130, 113–130 (2018).
Williams, D. M., Nawaz, A. & Evans, M. Drug therapy in obesity: a review of current and emerging treatments. Diabetes Ther. 11, 1199–1216 (2020).
Bondarev, A. D. et al. Opportunities and challenges for drug discovery in modulating Adhesion G protein-coupled receptor (GPCR) functions. Expert Opin. Drug Discov. 15, 1291–1307 (2020).
Pin, J. P. et al. Allosteric functioning of dimeric class C G-protein-coupled receptors. Febs J. 272, 2947–2955 (2005).
Schulte, G. & Wright, S. C. Frizzleds as GPCRs - More conventional than we thought! Trends Pharmacol. Sci. 39, 828–842 (2018).
Ruat, M., Hoch, L., Faure, H. & Rognan, D. Targeting of smoothened for therapeutic gain. Trends Pharmacol. Sci. 35, 237–246 (2014).
Gruhl, T. et al. Ultrafast structural changes direct the first molecular events of vision. Nature 615, 939–944 (2023).
Guo, L. L. et al. Structural basis of amine odorant perception by a mammal olfactory receptor. Nature 618, 193–200 (2023).
Dewan, A. Olfactory signaling via trace amine-associated receptors. Cell Tissue Res. 383, 395–407 (2021).
Ozeck, M., Brust, P., Xu, H. & Servant, G. Receptors for bitter, sweet and umami taste couple to inhibitory G protein signaling pathways. Eur. J. Pharm. 489, 139–149 (2004).
Ahmad, R. & Dalziel, J. E. G. Protein-coupled receptors in taste physiology and pharmacology. Front. Pharmacol. 11, 587664 (2020).
Xu, W. X. et al. Structural basis for strychnine activation of human bitter taste receptor TAS2R46. Science 377, 1298–1303 (2022).
Rubovitch, V., Gafni, M. & Sarne, Y. The mu opioid agonist DAMGO stimulates cAMP production in SK-N-SH cells through a PLC-PKC-Ca++ pathway. Mol. Brain Res. 110, 261–266 (2003).
Zhu, C. et al. Allosteric modulation of G protein-coupled receptors as a novel therapeutic strategy in neuropathic pain. Acta Pharm. Sin. B 14, 67–86 (2023).
Faouzi, A. et al. Structure-based design of bitopic ligands for the μ-opioid receptor. Nature 613, 767–774 (2023).
Katona, I. & Freund, T. F. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med. 14, 923–930 (2008).
Galve-Roperh, I. et al. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog. Lipid Res. 52, 633–650 (2013).
Ignatowska-Jankowska, B. M. et al. A Cannabinoid CB1 receptor-positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacol 40, 2948–2959 (2015).
Husted, A. S. et al. GPCR-mediated signaling of metabolites. Cell Metab. 25, 777–796 (2017).
Oh, D. Y. et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat. Med. 20, 942–947 (2014).
Oh, D. Y. et al. GPR120 Is an Omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698 (2010).
Divino, V. et al. Glucagon-like Peptide-1 receptor agonist treatment patterns among Type 2 diabetes patients in six European countries. Diabetes Ther. 5, 499–520 (2014).
Kaupmann, K. et al. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature 396, 683–687 (1998).
Ginés, S. et al. Dopamine D and adenosine A receptors form functionally interacting heteromeric complexes. Proc. Natl Acad. Sci. USA 97, 8606–8611 (2000).
Pulido, D. et al. Heterobivalent ligand for the Adenosine A2A-Dopamine D2 receptor heteromer. J. Med. Chem. 65, 616–632 (2022).
AbdAlla, S., Lother, H. & Quitterer, U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 407, 94–98 (2000).
Selbie, L. A. & Hill, S. J. G protein-coupled-receptor cross-talk: the fine-tuning of multiple receptor-signalling pathways. Trends Pharmacol. Sci. 19, 87–93 (1998).
Cordeaux, Y. & Hill, S. J. Mechanisms of cross-talk between G-protein-coupled receptors. Neurosignals 11, 45–57 (2002).
Maziarz, M. et al. Naturally occurring hotspot cancer mutations in Gα13 promote oncogenic signaling. J. Biol. Chem. 295, 16897–16904 (2020).
Khamaysi, Z. et al. Segmental basal cell naevus syndrome caused by an activating mutation in smoothened. Br. J. Dermatol. 175, 178–181 (2016).
Vaisse, C., Clement, K., Guy-Grand, B. & Froguel, P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet. 20, 113–114 (1998).
Schöneberg, T. et al. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol. Ther. 104, 173–206 (2004).
Bockenhauer, D. & Bichet, D. G. Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat. Rev. Nephrol. 11, 576–588 (2015).
Franco, R., Rivas-Santisteban, R., Navarro, G. & Reyes-Resina, I. Adenosine receptor antagonists to combat cancer and to boost anti-cancer chemotherapy and immunotherapy. Cells 10, 2831–2843 (2021).
Beerepoot, P., Nazari, R. & Salahpour, A. Pharmacological chaperone approaches for rescuing GPCR mutants: Current state, challenges, and screening strategies. Pharmacol. Res. 117, 242–251 (2017).
Schöneberg, T., Schultz, G. & Gudermann, T. Structural basis of G protein-coupled receptor function. Mol. Cell Endocrinol. 151, 181–193 (1999).
Sora, I., Funada, M. & Uhl, G. R. The mu-opioid receptor is necessary for [D-Pen(2),D-Pen(5)]enkephalin-induced analgesia. Eur. J. Pharm. 324, R1–R2 (1997).
Dogra, S., Sona, C., Kumar, A. & Yadav, P. N. Tango assay for ligand-induced GPCR-β-arrestin2 interaction: application in drug discovery. Method Cell Biol. 132, 233–254 (2016).
Di Fruscia, P. et al. The discovery of indole full agonists of the neurotensin receptor 1 (NTSR1). Bioorg. Med. Chem. Lett. 24, 3974–3978 (2014).
Hershberger, P. M. et al. Imidazole-derived agonists for the neurotensin 1 receptor. Bioorg. Med. Chem. Lett. 24, 262–267 (2014).
Fan, Y. et al. The identification of neurotensin NTS1 receptor partial agonists through a ligand-based virtual screening approach. Bioorg. Med. Chem. Lett. 18, 5789–5791 (2008).
Gully, D. et al. Biochemical and pharmacological profile of a potent and selective nonpeptide antagonist of the neurotensin receptor. Proc. Natl Acad. Sci. USA 90, 65–69 (1993).
Gully, D. et al. Biochemical and pharmacological activities of SR 142948A, a new potent neurotensin receptor antagonist. J. Pharmacol. Exp. Ther. 280, 802–812 (1997).
Wacker, D., Stevens, R. C. & Roth, B. L. How ligands illuminate GPCR molecular pharmacology. Cell 170, 414–427 (2017).
Lu, X. et al. Mechanistic elucidation of activation/deactivation signal transduction within neurotensin Receptor 1 triggered by ‘driver chemical groups’ of modulators: a comparative molecular dynamics simulation. Pharmaceutics 15, 2000–2015 (2023).
Avet, C. et al. Effector membrane translocation biosensors reveal G protein and βarrestin coupling profiles of 100 therapeutically relevant GPCRs. Elife 11, e74101 (2022).
Pándy-Szekeres, G. et al. GPCRdb in 2023: state-specific structure models using AlphaFold2 and new ligand resources. Nucleic Acids Res. 51, D395–D402 (2023).
Manglik, A. Molecular basis of opioid action: from structures to new leads. Biol. Psychiatry 87, 6–14 (2020).
Radoux-Mergault, A. et al. Subcellular location defines GPCR signal transduction. Sci. Adv. 9, eadf6059 (2023).
Matthes, H. W. D. et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383, 819–823 (1996).
Bartuzi, D., Kaczor, A. A. & Matosiuk, D. Activation and allosteric modulation of human μ opioid receptor in molecular dynamics. J. Chem. Inf. Model. 55, 2421–2434 (2015).
Raehal, K. M., Schmid, C. L., Groer, C. E. & Bohn, L. M. Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 63, 1001–1019 (2011).
Raehal, K. M., Walker, J. K. L. & Bohn, L. M. Morphine side effects in β-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 314, 1195–1201 (2005).
Bohn, L. M. et al. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286, 2495–2498 (1999).
Kaneko, S. et al. Activation mechanism of the μ-opioid receptor by an allosteric modulator. Proc. Natl Acad. Sci. USA 119, e2121918119 (2022).
Altarifi, A. A. et al. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol. 31, 730–739 (2017).
Zhuang, Y. W. et al. Molecular recognition of morphine and fentanyl by the human μ-opioid receptor. Cell 185, 4361–4375 (2022).
Chen, X. T. et al. Structure Activity Relationships and Discovery of a G Protein Biased μ Opioid Receptor Ligand, [(3-Methoxythiophen-2-yl)methyl]a2[(9R)-9-(pyridin-2-y1)-6-oxaspiro-[4.5]clecan-9-yl]ethylpamine (TRV130), for the Treatment of Acute Severe Pain. J. Med. Chem. 56, 8019–8031 (2013).
Proia, R. L. & Hla, T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J. Clin. Investig. 125, 1379–1387 (2015).
Rosen, H. et al. Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu. Rev. Biochem. 82, 637–662 (2013).
Wang, J. L., Gareri, C. & Rockman, H. A. G-protein-coupled receptors in heart disease. Circ. Res. 123, 716–735 (2018).
O’Sullivan, C. & Dev, K. K. The structure and function of the S1P1 receptor. Trends Pharmacol. Sci. 34, 401–412 (2013).
Liu, S. A. et al. Differential activation mechanisms of lipid GPCRs by lysophosphatidic acid and sphingosine 1-phosphate. Nat. Commun. 13, 731–741 (2022).
McGinley, M. P. & Cohen, J. A. Sphingosine 1-phosphate receptor modulators in multiple sclerosis and other conditions. Lancet 398, 1184–1194 (2021).
Strader, C. R., Pearce, C. J. & Oberlies, N. H. Fingolimod (FTY720): A recently approved multiple sclerosis drug based on a fungal secondary metabolite. J. Nat. Prod. 74, 900–907 (2011).
Al-Salama, Z. T. Siponimod: First global approval. Drugs 79, 1009–1015 (2019).
Yang, F. et al. Structural basis of GPBAR activation and bile acid recognition. Nature 587, 499–504 (2020).
Zhao, C. et al. Biased allosteric activation of ketone body receptor HCAR2 suppresses inflammation. Mol. Cell 83, 3171–3187 (2023).
Yuan, Y. et al. Structures of signaling complexes of lipid receptors S1PR1 and S1PR5 reveal mechanisms of activation and drug recognition. Cell Res 31, 1263–1274 (2021).
Xu, X. Y. et al. Binding pathway determines norepinephrine selectivity for the human β1AR over β2AR. Cell Res 31, 569–579 (2021).
Hanson, M. A. et al. Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855 (2012).
McAllister, S. D. et al. Structural mimicry in class A G protein-coupled receptor rotamer toggle switches -: The importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J. Biol. Chem. 279, 48024–48037 (2004).
Israeli, H. et al. Structure reveals the activation mechanism of the MC4 receptor to initiate satiation signaling. Science 372, 808–814 (2021).
Roecker, A. J., Cox, C. D. & Colemant, P. J. Orexin receptor antagonists: new therapeutic agents for the treatment of Insomnia. J. Med. Chem. 59, 504–530 (2016).
Scammell, T. E. & Winrow, C. J. Orexin receptors: pharmacology and therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 51, 243–266 (2011).
Tsuneki, H., Sasaoka, T. & Sakurai, T. Sleep control, GPCRs, and glucose metabolism. Trends Endocrinol. Metab. 27, 633–642 (2016).
Sakurai, T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat. Rev. Neurosci. 8, 171–181 (2007).
Landry, I. et al. Pharmacokinetics, pharmacodynamics, and safety of the dual orexin receptor antagonist lemborexant: findings from single-dose and multiple-ascending-dose Phase 1 studies in healthy adults. Clin. Pharmacol. Drug Dev. 10, 153–165 (2021).
Beuckmann, C. T. et al. In vitro and in silico characterization of Lemborexant (E2006), a novel dual orexin receptor antagonist. J. Pharmacol. Exp. Ther. 362, 287–295 (2017).
Asada, H. et al. Molecular basis for anti-insomnia drug design from structure of lemborexant-bound orexin 2 receptor. Structure 30, 1582–1589 (2022).
McCorvy, J. D. & Roth, B. L. Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther. 150, 129–142 (2015).
Pytliak, M., Vargová, V., Mechírová, V. & Felsöci, M. Serotonin receptors - from molecular biology to clinical applications. Physiol. Res. 60, 15–25 (2011).
Calderon, J. C. et al. General metadynamics protocol to simulate activation/deactivation of Class A GPCRs: Proof of principle for the Serotonin receptor. J. Chem. Inf. Model. 63, 3105–3117 (2023).
Negro, A., Koverech, A. & Martelletti, P. Serotonin receptor agonists in the acute treatment of migraine: a review on their therapeutic potential. J. Pain. Res. 11, 515–526 (2018).
Clemow, D. B. et al. Lasmiditan mechanism of action - review of a selective 5-HT1F agonist. J. Headache Pain. 21, 1–13 (2020).
Huang, S. J. et al. Structural basis for recognition of anti-migraine drug lasmiditan by the serotonin receptor 5-HT1F-G protein complex. Cell Res. 31, 1036–1038 (2021).
Wierman, M. E. et al. Molecular mechanisms of gonadotropin-releasing hormone neuronal migration. Trends Endocrinol. Metab. 15, 96–102 (2004).
Yang, X. et al. Discovery of small molecule agonist of Gonadotropin-Releasing Hormone Receptor (GnRH1R). J. Chem. Inf. Model. 62, 5009–5022 (2022).
Stojilkovic, S. S., Reinhart, J. & Catt, K. J. Gonadotropin-releasing-hormone receptors - structure and signal-transduction pathways. Endocr. Rev. 15, 462–499 (1994).
Lamb, Y. N. Elagolix: First global approval. Drugs 78, 1501–1508 (2018).
Zou, F. X. et al. Discovery of the thieno[2,3-d]pyrimidine-2,4-dione derivative 21a: A potent and orally bioavailable gonadotropin-releasing hormone receptor antagonist. Eur. J. Med. Chem. 242, 114679 (2022).
Yan, W. et al. Structure of the human gonadotropin-releasing hormone receptor GnRH1R reveals an unusual ligand binding mode. Nat. Commun. 11, 5287–5296 (2020).
Möller, D. et al. Discovery of G Protein-biased Dopaminergics with a Pyrazolo[1,5-]pyridine substructure. J. Med. Chem. 60, 2908–2929 (2017).
Szabo, M. et al. Structure-activity relationships of privileged structures lead to the discovery of novel biased ligands at the Dopamine D2 receptor. J. Med. Chem. 57, 4924–4939 (2014).
Zhang, G. P. et al. Discovery of -Substituted (2-Phenylcyclopropyl)methylamines as functionally selective Serotonin 2C receptor agonists for potential use as antipsychotic medications. J. Med. Chem. 60, 6273–6288 (2017).
Congreve, M. et al. Discovery of 1,2,4-Triazine derivatives as Adenosine A2A antagonists using structure based drug design. J. Med. Chem. 55, 1898–1903 (2012).
Congreve, M., de Graaf, C., Swain, N. A. & Tate, C. G. Impact of GPCR structures on drug discovery. Cell 181, 81–91 (2020).
Selfani, K., Soland, V. L., Chouinard, S. & Huot, P. Movement disorders induced by the “Atypical” antipsychotic Aripiprazole. Neurologist 22, 24–28 (2017).
McCorvy, J. D. et al. Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol. 14, 126 (2018).
Lu, S. Y. & Zhang, J. Small molecule allosteric modulators of G-protein-coupled receptors: drug-target interactions. J. Med. Chem. 62, 24–45 (2019).
Thal, D. M., Glukhova, A., Sexton, P. M. & Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53 (2018).
Wakefield, A. E. et al. Conservation of allosteric ligand binding sites in G-protein coupled receptors. J. Chem. Inf. Model. 62, 4937–4954 (2022).
Casadó-Anguera, V. & Casadó, V. Unmasking allosteric-binding sites: novel targets for GPCR drug discovery. Expert Opin. Drug Discov. 17, 897–923 (2022).
Lane, J. R. et al. A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol. 13, 929–937 (2017).
Dore, A. S. et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511, 557–562 (2014).
Barman Balfour, J. A. & Scott, L. J. Cinacalcet hydrochloride. Drugs 65, 271–281 (2005).
Bueno, A. B. et al. Structural insights into probe-dependent positive allosterism of the GLP-1 receptor. Nat. Chem. Biol. 16, 1105–1110 (2020).
Tan, Q. X. et al. Structure of the CCR5 Chemokine Receptor-HIV entry inhibitor Maraviroc Complex. Science 341, 1387–1390 (2013).
Congreve, M., Oswald, C. & Marshall, F. H. Applying structure-based drug design approaches to allosteric modulators of GPCRs. Trends Pharmacol. Sci. 38, 837–847 (2017).
de Graaf, C. et al. Glucagon-like Peptide-1 and its Class B G protein-coupled receptors: a long march to therapeutic successes. Pharmacol. Rev. 68, 954–1013 (2016).
de Graaf, C. et al. Extending the structural view of Class B GPCRs. Trends Biochem. Sci. 42, 946–960 (2017).
Baggio, L. L. & Drucker, D. J. Glucagon-like peptide-1 receptor co-agonists for treating metabolic disease. Mol. Metab. 46, 101090 (2020).
Muratspahic, E., Freissmuth, M. & Gruber, C. W. Nature-derived peptides: a growing niche for GPCR ligand discovery. Trends Pharmacol. Sci. 40, 309–326 (2019).
Frias, J. P. et al. Tirzepatide versus Semaglutide once weekly in patients with Type 2 Diabetes. N. Engl. J. Med. 385, 503–515 (2021).
Gerstein, H. C. et al. Cardiovascular and renal outcomes with Efpeglenatide in Type 2 Diabetes. N. Engl. J. Med. 385, 896–907 (2021).
Broichhagen, J. et al. Allosteric optical control of a Class B G-protein-coupled receptor. Angew. Chem. Int. Ed. 55, 5865–5868 (2016).
Willard, F. S. et al. Discovery of an orally efficacious positive allosteric modulator of the Glucagon-like Peptide-1 Receptor. J. Med. Chem. 64, 3439–3448 (2021).
Xin, Y. et al. Affinity selection of double-click triazole libraries for rapid discovery of allosteric modulators for GLP-1 receptor. Proc. Natl Acad. Sci. USA 120, e2220767120 (2023).
McRobb, F. M., Negri, A., Beuming, T. & Sherman, W. Molecular dynamics techniques for modeling G protein-coupled receptors. Curr. Opin. Pharmacol. 30, 69–75 (2016).
Ribeiro, J. M. L. & Filizola, M. Allostery in G protein-coupled receptors investigated by molecular dynamics simulations. Curr. Opin. Struct. Biol. 55, 121–128 (2019).
Wu, H. Y. et al. Molecular glues modulate protein functions by inducing protein aggregation: A promising therapeutic strategy of small molecules for disease treatment. Acta Pharm. Sin. B 12, 3548–3566 (2022).
Cong, Z. T. et al. Structural basis of peptidomimetic agonism revealed by small-molecule GLP-1R agonists Boc5 and WB4-24. Proc. Natl Acad. Sci. USA 119, e2200155119 (2022).
Zhao, P. S. et al. Activation of the GLP-1 receptor by a non-peptidic agonist. Nature 577, 432–436 (2020).
Kawai, T. et al. Structural basis for GLP-1 receptor activation by LY3502970, an orally active nonpeptide agonist. Proc. Natl Acad. Sci. USA 117, 29959–29967 (2020).
Zhang, X. et al. Differential GLP-1R binding and activation by peptide and non-peptide agonists. Mol. Cell 80, 485–500 (2020).
Abboud, D. et al. GPR101 drives growth hormone hypersecretion and gigantism in mice via constitutive activation of Gs and Gq/11. Nat. Commun. 11, 4752 (2020).
Garrett, L. et al. GPR101 loss promotes insulin resistance and diet-induced obesity risk. Neurosci. Appl. 2, 101126 (2023).
Yang, Z. et al. Structure of GPR101-Gs enables identification of ligands with rejuvenating potential. Nat. Chem. Biol. https://doi.org/10.1038/s41589-023-01456-6 (2023).
Xiao, X. et al. Cholesterol modification of smoothened is required for Hedgehog signaling. Mol. Cell 66, 154–162 (2017).
Huang, P. X. et al. Cellular cholesterol directly activates smoothened in Hedgehog signaling. Cell 166, 1176–1187 (2016).
Bokoch, M. P. et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463, 108–U121 (2010).
Peeters, M. C., van Westen, G. J. P., Li, Q. & IJzerman, A. P. Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol. Sci. 32, 35–42 (2011).
Xu, X. Y. et al. Constrained catecholamines gain β2AR selectivity through allosteric effects on pocket dynamics. Nat. Commun. 14, 2138–2150 (2023).
Nicoli, A. et al. Classification model for the second extracellular loop of Class A GPCRs. J. Chem. Inf. Model. 62, 511–522 (2022).
Wess, J., Eglen, R. M. & Gautam, D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat. Rev. Drug Discov. 6, 721–733 (2007).
Yohn, S. E., Weiden, P. J., Felder, C. C. & Stahl, S. M. Muscarinic acetylcholine receptors for psychotic disorders: bench-side to clinic. Trends Pharmacol. Sci. 43, 1098–1112 (2022).
Felder, C. C. Muscarinic Acetylcholine-receptors - signal-transduction through multiple effectors. FASEB J. 9, 619–625 (1995).
Melancon, B. J. et al. Allosteric modulation of the M-1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discov. Today 18, 1185–1199 (2013).
Moran, S. P., Maksymetz, J. & Conn, P. J. Targeting Muscarinic Acetylcholine receptors for the treatment of psychiatric and neurological disorders. Trends Pharm. Sci. 40, 1006–1020 (2019).
Miao, Y. L. et al. Accelerated structure-based design of chemically diverse allosteric modulators of a muscarinic G protein-coupled receptor. Proc. Natl Acad. Sci. USA 113, E5675–E5684 (2016).
Shivnaraine, R. V. et al. Allosteric modulation in monomers and oligomers of a G protein-coupled receptor. Elife 5, e11685 (2016).
Croy, C. H. et al. Characterization of the novel positive Allosteric Modulator, LY2119620, at the Muscarinic M-2 and M-4 receptors. Mol. Pharmacol. 86, 106–115 (2014).
Kruse, A. C. et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013).
Wang, J. J. et al. The unconventional activation of the muscarinic acetylcholine receptor M4R by diverse ligands. Nat. Commun. 13, 2855–2894 (2022).
Xu, J. et al. Structural and dynamic insights into supra-physiological activation and allosteric modulation of a muscarinic acetylcholine receptor. Nat. Commun. 14, 376–391 (2023).
Jiménez-Rosés, M., Matsoukas, M. T., Caltabiano, G. & Cordomí, A. Ligand-triggered structural changes in the M2 Muscarinic Acetylcholine receptor. J. Chem. Inf. Model. 58, 1074–1082 (2018).
Capelli, R. et al. Chasing the full free energy landscape of Neuroreceptor/Ligand unbinding by metadynamics simulations. J. Chem. Theory Comput. 15, 3354–3361 (2019).
Leach, K. & Gregory, K. J. Molecular insights into allosteric modulation of Class C G protein-coupled receptors. Pharmacol. Res. 116, 105–118 (2017).
Diaz-Soto, G. et al. The calcium-sensing receptor in health and disease. Int. Rev. Cell Mol. Biol. 327, 321–369 (2016).
Hannan, F. M. et al. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat. Rev. Endocrinol. 15, 33–51 (2018).
Conigrave, A. D. & Ward, D. T. Calcium-sensing receptor (CaSR): Pharmacological properties and signaling pathways. Best. Pract. Res. Clin. Endocrinol. Metab. 27, 315–331 (2013).
Leach, K., Conigrave, A. D., Sexton, P. M. & Christopoulos, A. Towards tissue-specific pharmacology: insights from the calcium-sensing receptor as a paradigm for GPCR (patho)physiological bias. Trends Pharmacol. Sci. 36, 215–225 (2015).
Leach, K. et al. Towards a structural understanding of allosteric drugs at the human calcium-sensing receptor. Cell Res 26, 574–592 (2016).
Block, G. A. et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N. Engl. J. Med. 350, 1516–1525 (2004).
Akizawa, T. et al. Evocalcet: A new oral calcimimetic for dialysis patients with secondary Hyperparathyroidism. Ther. Apher. Dial. 24, 248–257 (2020).
Hannan, F. M. et al. The calcilytic agent NPS 2143 rectifies hypocalcemia in a mouse model with an activating Calcium-Sensing Receptor (CaSR) mutation: relevance to Autosomal Dominant Hypocalcemia Type 1 (ADH1). Endocrinology 156, 3114–3121 (2015).
Gao, Y. et al. Asymmetric activation of the calcium-sensing receptor homodimer. Nature 595, 455–459 (2021).
Venkatakrishnan, A. J. et al. Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 (2013).
Liu, F. et al. Structure-based discovery of positive allosteric modulators for the calcium sensing receptor. bioRxiv 573448 (2023).
Wang, W. J., Qiao, Y. H. & Li, Z. J. New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci. 39, 367–386 (2018).
Weis, W. I. & Kobilka, B. K. The molecular basis of G protein-coupled receptor activation. Annu. Rev. Biochem. 87, 897–919 (2018).
Venkatakrishnan, A. J. et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 536, 484–487 (2016).
Cattaneo, M. Platelet P2 receptors: old and new targets for antithrombotic drugs. Expert Rev. Cardiovasc Ther. 5, 45–55 (2007).
Jacobson, K. A. et al. Update of P2Y receptor pharmacology: IUPHAR Review 27. Br. J. Pharmacol. 177, 2413–2433 (2020).
Jacobson, K. A. & Boeynaems, J. M. P2Y nucleotide receptors: promise of therapeutic applications. Drug Discov. Today 15, 570–578 (2010).
Cattaneo, M. New P2Y(12) Inhibitors. Circulation 121, 171–179 (2010).
Mane, N., Jimenez-Sabado, V. & Jimenez, M. BPTU, an allosteric antagonist of P2Y1 receptor, blocks nerve mediated inhibitory neuromuscular responses in the gastrointestinal tract of rodents. Neuropharmacology 110, 376–385 (2016).
Conroy, S., Kindon, N., Kellam, B. & Stocks, M. J. Drug-like antagonists of P2Y receptors-from lead identification to drug development. J. Med. Chem. 59, 9981–10005 (2016).
Zhang, D. D. et al. Two disparate ligand-binding sites in the human P2Y(1) receptor. Nature 520, 317–321 (2015).
Wu, Y. R. et al. GPCR allosteric modulator discovery. Protein Allostery Drug Discov. 1163, 225–251 (2019).
Li, B. B. et al. Structural insights into signal transduction of the purinergic receptors P2Y1R and P2Y12R. Protein Cell 14, 382–386 (2023).
Zhou, Q. T. et al. Common activation mechanism of class A GPCRs. Elife 8, e50279 (2019).
Yuan, S. G. et al. The molecular mechanism of P2Y1 receptor activation. Angew. Chem. Int. Ed. 55, 10331–10335 (2016).
Zou, S. L. & Kumar, U. Cannabinoid receptors and the endocannabinoid system: signaling and function in the central nervous system. Int. J. Mol. Sci. 19, 833–855 (2018).
Brown, A. J. Novel cannabinoid receptors. Br. J. Pharmacol. 152, 567–575 (2007).
Kofuji, P. & Araque, A. G-protein-coupled receptors in astrocyte-neuron communication. Neuroscience 456, 71–84 (2021).
Busquets-Garcia, A., Bains, J. & Marsicano, G. CB1 receptor signaling in the brain: extracting specificity from ubiquity. Neuropsychopharmacol 43, 4–20 (2018).
Nguyen, T. et al. Allosteric modulation: an alternate approach targeting the cannabinoid CB1 receptor. Med. Res. Rev. 37, 441–474 (2017).
Mackie, K. Cannabinoid receptors: Where they are and what they do. J. Neuroendocrinol. 20, 10–14 (2008).
Gado, F. et al. CB1 receptor binding sites for NAM and PAM: A first approach for studying, new n-butyl-diphenylcarboxamides as allosteric modulators. Eur. J. Pharm. Sci. 169, 106088 (2022).
Yuan, J. Y. et al. Targeting the endocannabinoid system: Structural determinants and molecular mechanism of allosteric modulation. Drug Discov. Today 28, 103615 (2023).
Janero, D. R. & Thakur, G. A. Leveraging allostery to improve G protein-coupled receptor (GPCR)-directed therapeutics: cannabinoid receptor 1 as discovery target. Expert Opin. Drug Discov. 11, 1223–1237 (2016).
Gamage, T. F. et al. In-vivo pharmacological evaluation of the CB1-receptor allosteric modulator Org-27569. Behav. Pharm. 25, 182–185 (2014).
Shao, Z. H. et al. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 15, 1199–1205 (2019).
Obi, P. & Natesan, S. Membrane lipids are an integral part of transmembrane allosteric sites in GPCRs: A case study of cannabinoid CB1 receptor bound to a negative allosteric modulator, ORG27569, and analogs. J. Med. Chem. 65, 12240–12255 (2022).
Green, H. M. et al. In vitro characterization of 6-Methyl-3-(2-nitro-1-(thiophen-2yl)ethyl)-2-phenyl-1H-indole (ZCZ011) at the Type 1 Cannabinoid receptor: allosteric agonist or allosteric modulator? ACS Pharm. 5, 1279–1291 (2022).
Saleh, N. et al. Multiple binding sites contribute to the mechanism of mixed agonistic and positive allosteric modulators of the Cannabinoid CB1 receptor. Angew. Chem. Int. Ed. 57, 2580–2585 (2018).
Yang, X. et al. Molecular mechanism of allosteric modulation for the cannabinoid receptor CB1. Nat. Chem. Biol. 18, 831–840 (2022).
Wold, E. A., Chen, J. P., Cunningham, K. A. & Zhou, J. Allosteric modulation of Class A GPCRs: Targets, agents, and emerging concepts. J. Med. Chem. 62, 88–127 (2019).
Ahn, K. H., Mahmoud, M. M. & Kendall, D. A. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and G protein-independent ERK1/2 Kinase activation. J. Biol. Chem. 287, 12070–12082 (2012).
Shao, Z. H. et al. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 540, 602–606 (2016).
Kumar, K. K. et al. Structural basis for activation of CB1 by an endocannabinoid analog. Nat. Commun. 14, 2672–2682 (2023).
Fay, J. F. & Farrens, D. L. Structural dynamics and energetics underlying allosteric inactivation of the cannabinoid receptor CB. Proc. Natl Acad. Sci. USA 112, 8469–8474 (2015).
Dalton, J. A. R., Lans, I. & Giraldo, J. Quantifying conformational changes in GPCRs: glimpse of a common functional mechanism. BMC Bioinforma. 16, 124–138 (2015).
Wheatley, M. et al. Lifting the lid on GPCRs: the role of extracellular loops. Br. J. Pharmacol. 165, 1688–1703 (2012).
Liu, X. Y. et al. Mechanism of beta(2)AR regulation by an intracellular positive allosteric modulator. Science 364, 1283–1287 (2019).
Hauser, A. S. et al. GPCR activation mechanisms across classes and macro/microscales. Nat. Struct. Mol. Biol. 28, 879–888 (2021).
Chan, H. C. S. et al. New BINDING SITES, NEW OPPORTUNities for GPCR drug discovery. Trends Biochem. Sci. 44, 312–330 (2019).
Beaulieu, J. M. & Gainetdinov, R. R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217 (2011).
Chen, K. Y. M., Keri, D. & Barth, P. Computational design of G Protein-Coupled Receptor allosteric signal transductions. Nat. Chem. Biol. 16, 77–86 (2020).
Missale, C. et al. Dopamine receptors: From structure to function. Physiol. Rev. 78, 189–225 (1998).
Kobayashi, K. et al. Noradrenaline activation of hippocampal dopamine D1 receptors promotes antidepressant effects. Proc. Natl Acad. Sci. USA 119, e2117903119 (2022).
Biglan, K. et al. Safety and efficacy of Mevidalen in Lewy Body Dementia: A Phase 2, randomized, placebo-controlled trial. Mov. Disord. 37, 513–524 (2022).
Xiao, P. et al. Ligand recognition and allosteric regulation of DRD1-Gs signaling complexes. Cell 184, 943–956 (2021).
Zhuang, Y. W. et al. Mechanism of dopamine binding and allosteric modulation of the human D1 dopamine receptor. Cell Res 31, 593–596 (2021).
Teng, X. et al. Ligand recognition and biased agonism of the D1 dopamine receptor. Nat. Commun. 13, 3186–3196 (2022).
Sibley, D. R., Luderman, K. D., Free, R. B. & Shi, L. Novel Cryo-EM structures of the D1 dopamine receptor unlock its therapeutic potential. Signal Transduct. Target. Ther. 6, 205 (2021).
Lee, H., Whitfeld, P. L. & Mackay, C. R. Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol. Cell Biol. 86, 153–160 (2008).
Guo, R. F. & Ward, P. A. Role of C5A in inflammatory responses. Annu. Rev. Immunol. 23, 821–852 (2005).
Sun, L. & Ye, R. D. Role of G protein-coupled receptors in inflammation. Acta Pharmacol. Sin. 33, 342–350 (2012).
Sadik, C. D., Miyabe, Y., Sezin, T. & Luster, A. D. The critical role of C5a as an initiator of neutrophil-mediated autoimmune inflammation of the joint and skin. Semin. Immunol. 37, 21–29 (2018).
Woodruff, T. M. & Shukla, A. K. The Complement C5a-C5aR1 GPCR Axis in COVID-19 Therapeutics. Trends Immunol. 41, 965–967 (2020).
Garred, P., Tenner, A. J. & Mollnes, T. E. Therapeutic targeting of the complement system: from rare diseases to pandemics. Pharmacol. Rev. 73, 792–827 (2021).
Lee, A. Avacopan: First approval. Drugs 82, 79–85 (2022).
Jayne, D. R. W. et al. Avacopan for the treatment of ANCA-associated Vasculitis. N. Engl. J. Med. 384, 599–609 (2021).
Feng, Y. Y. et al. Mechanism of activation and biased signaling in complement receptor C5aR1. Cell Res 33, 312–324 (2023).
Gurevich, V. V. & Gurevich, E. V. GPCR signaling regulation: the role of GRKs and Arrestins. Front. Pharmacol. 10, 125 (2019).
Maharana, J. et al. Structural snapshots uncover a key phosphorylation motif in GPCRs driving p-arrestin activation. Mol. Cell 83, 2091–2107 (2023).
Rasmussen, S. G. F. et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180 (2011).
Huang, W. J. et al. Structural insights into μ-opioid receptor activation. Nature 524, 315–321 (2015).
Groom, J. R. & Luster, A. D. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 89, 207–215 (2011).
Lacotte, S., Brun, S., Muller, S. & Dumortier, H. CXCR3, inflammation, and autoimmune diseases. Ann. Ny. Acad. Sci. 1173, 310–317 (2009).
Milanos, L. et al. Discovery and characterization of biased allosteric agonists of the chemokine Receptor CXCR3. J. Med. Chem. 59, 2222–2243 (2016).
Heise, C. E. et al. Pharmacological characterization of CXC chemokine receptor 3 ligands and a small molecule antagonist. J. Pharmacol. Exp. Ther. 313, 1263–1271 (2005).
Jenh, C. H. et al. A selective and potent CXCR3 antagonist SCH 546738 attenuates the development of autoimmune diseases and delays graft rejection. BMC Immunol. 13, 2 (2012).
Jiao, H. et al. Structural insights into the activation and inhibition of CXC chemokine receptor 3. Nat. Struct. Mol. Biol. 31, 1–11 (2024).
Rittiner, J. E. et al. AMP is an Adenosine A1 receptor agonist. J. Biol. Chem. 287, 5301–5309 (2012).
Zylka, M. J. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 17, 188–196 (2011).
Narlawar, R. et al. Hybrid ortho/allosteric ligands for the Adenosine A1 receptor. J. Med. Chem. 53, 3028–3037 (2010).
Lee, D. F., Geron, M. & Scherrer, G. A modulator-bound GPCR structure enables allosteric non-opioid analgesia. Nat. Struct. Mol. Biol. 28, 871–872 (2021).
Mattedi, G. et al. Understanding ligand binding selectivity in a prototypical GPCR family. J. Chem. Inf. Model. 59, 2830–2836 (2019).
Draper-Joyce, C. J. et al. Positive allosteric mechanisms of adenosine A(1) receptor-mediated analgesia. Nature 597, 571–576 (2021).
Li, Y., Sun, J. X., Li, D. M. & Lin, J. P. The full activation mechanism of the adenosine A1 receptor revealed by GaMD and Su-GaMD simulations. Proc. Natl Acad. Sci. USA 119, e2203702119 (2022).
Niswender, C. M. & Conn, P. J. Metabotropic Glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322 (2010).
Andre, M. A. E., Gunturkun, O. & Manahan-Vaughan, D. The metabotropic Glutamate receptor, mGlu5, is required for extinction learning that occurs in the absence of a context change. Hippocampus 25, 149–158 (2015).
Brambilla, C. R. et al. mGluR5 receptor availability is associated with lower levels of negative symptoms and better cognition in male patients with chronic schizophrenia. Hum. Brain Mapp. 41, 2762–2781 (2020).
Roth, B. L. Molecular pharmacology of metabotropic receptors targeted by neuropsychiatric drugs. Nat. Struct. Mol. Biol. 26, 535–544 (2019).
Lindsley, C. W. et al. Practical strategies and concepts in GPCR allosteric modulator discovery: recent advances with metabotropic glutamate receptors. Chem. Rev. 116, 6707–6741 (2016).
Liauw, B. W. H. et al. Conformational fingerprinting of allosteric modulators in metabotropic glutamate receptor 2. Elife 11, e78982 (2022).
Gutzeit, V. A. et al. Conformational dynamics between transmembrane domains and allosteric modulation of a metabotropic glutamate receptor. Elife 8, e45116 (2019).
Ellaithy, A., Younkin, J., González-Maeso, J. & Logothetis, D. E. Positive allosteric modulators of metabotropic glutamate 2 receptors in schizophrenia treatment. Trends Neurosci. 38, 506–516 (2015).
Vafabakhsh, R., Levitz, J. & Isacoff, E. Y. Conformational dynamics of a class C G-protein-coupled receptor. Nature 524, 497–501 (2015).
Gentry, P. R., Sexton, P. M. & Christopoulos, A. Novel allosteric modulators of G protein-coupled receptors. J. Biol. Chem. 290, 19478–19488 (2015).
Stansley, B. J. & Conn, P. J. Neuropharmacological insight from allosteric modulation of mGlu receptors. Trends Pharmacol. Sci. 40, 240–252 (2019).
Jones-Tabah, J. Targeting G protein-coupled receptors in the treatment of Parkinson’s disease. J. Mol. Biol. 435, 167927 (2023).
Luessen, D. J. & Conn, P. J. Allosteric modulators of metabotropic glutamate receptors as novel therapeutics for neuropsychiatric disease. Pharmacol. Rev. 74, 630–661 (2022).
Foster, D. J. & Conn, P. J. Allosteric modulation of GPCRs: New insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron 94, 431–446 (2017).
Christppher J. A. et al. Fragment and structure-based drug discovery for a Class C GPCR: Discovery of the mGlu5 negative allosteric modulator HTL14242 (3-Chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile). J. Med. Chem. 58, 6653–6664 (2015).
Cong, X. J. et al. Allosteric modulation mechanism of the mGIuR(5) Transmembrane domain. J. Chem. Inf. Model. 59, 2871–2878 (2019).
del Torrent, C. L. et al. Mechanisms underlying allosteric molecular switches of metabotropic glutamate Receptor 5. J. Chem. Inf. Model. 59, 2456–2466 (2019).
Grushevskyi, E. O. et al. Stepwise activation of a class C GPCR begins with millisecond dimer rearrangement. Proc. Natl Acad. Sci. USA 116, 10150–10155 (2019).
Kumar, K. K. et al. Step-wise activation of a Family C GPCR. Biorxiv. https://doi.org/10.1101/2023.08.29.555158 (2023).
Schwartz, T. W. et al. Molecular mechanism of 7TM receptor activation - A global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 46, 481–519 (2006).
Harmar, A. J. Family-B G-protein-coupled receptors. Genome Biol. 2, reviews3013.3011–3013.3010 (2001).
Vilardaga, J. P. et al. Molecular mechanisms of PTH/PTHrP Class B GPCR signaling and pharmacological implications. Endocr. Rev. 44, 474–491 (2023).
Wootten, D. et al. Allostery and biased agonism at Class B G protein-coupled receptors. Chem. Rev. 117, 111–138 (2017).
Sutkeviciute, I. et al. Precise druggability of the PTH type 1 receptor. Nat. Chem. Biol. 18, 272–280 (2022).
Tamura, T. et al. Identification of an orally active small-molecule PTHR1 agonist for the treatment of hypoparathyroidism. Nat. Commun. 7, 13384–13397 (2016).
Nishimura, Y. et al. Lead optimization and avoidance of reactive metabolite leading to PCO371, a potent, selective, and orally available human parathyroid hormone receptor 1 (hPTHR1) agonist. J. Med. Chem. 63, 5089–5099 (2020).
Hiesinger, K., Dar’in, D., Proschak, E. & Krasavin, M. Spirocyclic scaffolds in medicinal chemistry. J. Med. Chem. 64, 150–183 (2021).
Xiao, H., Sun, Q. & Sun, Q. A promising small molecule binding pocket in class B GPCRs: expanding potential for drug development. Signal Transduct. Target. Ther. 8, 313 (2023).
Kobayashi, K. et al. Class B1 GPCR activation by an intracellular agonist. Nature 618, 1085–1093 (2023).
Zhao, L. H. et al. Conserved class B GPCR activation by a biased intracellular agonist. Nature 621, 635–641 (2023).
Wootten, D. et al. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 19, 638–653 (2018).
Huang, S. J. et al. GPCRs steer Gi and Gs selectivity via TM5-TM6 switches as revealed by structures of serotonin receptors. Mol. Cell 82, 2681–2695 (2022).
Hilger, D. et al. Structural insights into differences in G protein activation by family A and family B GPCRs. Science 369, eaba3373 (2020).
Evenseth, L. S. M., Gabrielsen, M. & Sylte, I. The GABA(B)Receptor-structure, ligand binding and drug development. Molecules 25, 3093–3111 (2020).
Cryan, J. F. & Kaupmann, K. Don’t worry ‘B’ happy!:: a role for GABAB receptors in anxiety and depression. Trends Pharmacol. Sci. 26, 36–43 (2005).
Wieronska, J. M. & Pilc, A. Depression and schizophrenia viewed from the perspective of amino acidergic neurotransmission: Antipodes of psychiatric disorders. Pharmacol. Ther. 193, 75–82 (2019).
Tyacke, R. J., Lingford-Hughes, A., Reed, L. J. & Nutt, D. J. GABAB receptors in addiction and its treatment. Adv. Pharm. 58, 373–396 (2010).
Filip, M. et al. GABAB receptors as a therapeutic strategy in substance use disorders: Focus on positive allosteric modulators. Neuropharmacology 88, 36–47 (2015).
Xue, L. et al. Rearrangement of the transmembrane domain interfaces associated with the activation of a GPCR hetero-oligomer. Nat. Commun. 10, 2765–2776 (2019).
Farran, B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol. Res. 117, 303–327 (2017).
Liu, L. et al. Allosteric ligands control the activation of a class C GPCR heterodimer by acting at the transmembrane interface. Elife 10, e70188 (2021).
Galvez, T. et al. Allosteric interactions between GB1 and GB2 subunits are required for optimal GABAB receptor function. EMBO J. 20, 2152–2159 (2001).
Gusach, A., García-Nafría, J. & Tate, C. G. New insights into GPCR coupling and dimerisation from cryo-EM structures. Curr. Opin. Struct. Biol. 80, 102574 (2023).
Zemoura, K., Ralvenius, W. T., Malherbe, P. & Benke, D. The positive allosteric GABA(B) receptor modulator rac-BHFF enhances baclofen-mediated analgesia in neuropathic mice. Neuropharmacology 108, 172–178 (2016).
Mao, C. Y. et al. Cryo-EM structures of inactive and active GABA(B) receptor. Cell Res 30, 564–573 (2020).
Shaye, H. et al. Structural basis of the activation of a metabotropic GABA receptor. Nature 584, 298–303 (2020).
Kim, Y. et al. Structural basis for activation of the Heterodimeric GABA(B) receptor. J. Mol. Biol. 432, 5966–5984 (2020).
Monnier, C. et al. Trans-activation between 7TM domains: implication in heterodimeric GABAB receptor activation. EMBO J. 30, 32–42 (2011).
Knudsen, L. B. et al. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc. Natl Acad. Sci. USA 104, 937–942 (2007).
Wootten, D. et al. Differential activation and modulation of the Glucagon-Like Peptide-1 receptor by small molecule ligands. Mol. Pharmacol. 83, 822–834 (2013).
Willard, F. S., Ho, J. D. & Sloop, K. W. Discovery and pharmacology of the covalent GLP-1 receptor (GLP-1R) allosteric modulator BETP: A novel tool to probe GLP-1R pharmacology. Adv. Pharm. 88, 173–191 (2020).
Cong, Z. T. et al. Molecular insights into ago-allosteric modulation of the human glucagon-like peptide-1 receptor. Nat. Commun. 12, 3763 (2021).
Cong, Z. T. et al. Structural perspective of class B1 GPCR signaling. Trends Pharmacol. Sci. 43, 321–334 (2022).
Bian, Y. M., Jun, J. J., Cuyler, J. & Xie, X. Q. Covalent allosteric modulation: An emerging strategy for GPCRs drug discovery. Eur. J. Med. Chem. 206, 112690 (2020).
Lu, S. Y. & Zhang, J. Designed covalent allosteric modulators: an emerging paradigm in drug discovery. Drug Discov. Today 22, 447–453 (2017).
Guo, D., Hillger, J. M., IJzerman, A. P. & Heitman, L. H. Drug-target residence Time-A Case for G protein-coupled receptors. Med. Res. Rev. 34, 856–892 (2014).
Jazayeri, A. et al. Extra-helical binding site of a glucagon receptor antagonist. Nature 533, 274–277 (2016).
Zhang, H. N. et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 546, 259–264 (2017).
Song, G. J. et al. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 546, 312–315 (2017).
Hollenstein, K. et al. Insights into the structure of class B GPCRs. Trends Pharmacol. Sci. 35, 12–22 (2014).
Cho, Y. M., Merchant, C. E. & Kieffer, T. J. Targeting the glucagon receptor family for diabetes and obesity therapy. Pharmacol. Ther. 135, 247–278 (2012).
Kjolbye, L. R. et al. Lipid modulation of a Class B GPCR: Elucidating the modulatory role of PI(4,5)P2 lipids. J. Chem. Inf. Model. 62, 6788–6802 (2022).
Scheen, A. J., Paquot, N. & Lefebvre, P. J. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert Opin. Investig. Drugs 26, 1373–1389 (2017).
Pettus, J. et al. Glucagon receptor antagonist volagidemab in type 1 diabetes: a 12-week, randomized, double-blind, phase 2 trial. Nat. Med. 28, 2092–2099 (2022).
Oh, D. Y. & Olefsky, J. M. G protein-coupled receptors as targets for anti-diabetic therapeutics. Nat. Rev. Drug Discov. 15, 161–172 (2016).
Xiong, Y. S. et al. Discovery of a Novel Glucagon Receptor Antagonist N-[(4-{(1S)-1-[3-(3,5-Dichlorophenyl)-5-(6-methoxynaphthalen-2-yl)-1H-pyrazol-1-yl]ethyl}phenyl)carbonyl]-beta-alanine (MK-0893) for the Treatment of Type II Diabetes. J. Med. Chem. 55, 6137–6148 (2012).
Sammons, M. F. & Lee, E. C. Y. Recent progress in the development of small-molecule glucagon receptor antagonists. Bioorg. Med. Chem. Lett. 25, 4057–4064 (2015).
Ding, T. Y., Karlov, D. S., Pino-Angeles, A. & Tikhonova, I. G. Intermolecular interactions in G protein-coupled receptor allosteric sites at the membrane interface from molecular dynamics simulations and quantum chemical calculations. J. Chem. Inf. Model. 62, 4736–4747 (2022).
Chen, K. et al. Tail engagement of arrestin at the glucagon receptor. Nature 620, 904–910 (2023).
Zacarias, N. V. O. et al. Intracellular receptor modulation: novel approach to target GPCRs. Trends Pharmacol. Sci. 39, 547–559 (2018).
Krumm, B. E. et al. Neurotensin receptor allosterism revealed in complex with a biased allosteric modulator. Biochemistry 62, 1233–1248 (2023).
Vandercappellen, J., Van Damme, J. & Struyf, S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 267, 226–244 (2008).
Poeta, V. M., Massara, M., Capucetti, A. & Bonecchi, R. Chemokines and chemokine receptors: new targets for cancer immunotherapy. Front. Immunol. 10, 379 (2019).
Proudfoot, A. E. I. Chemokine receptors: Multifaceted therapeutic targets. Nat. Rev. Immunol. 2, 106–115 (2002).
Camba-Gomez, M., Arosa, L., Gualillo, O. & Conde-Aranda, J. Chemokines and chemokine receptors in inflammatory bowel disease: Recent findings and future perspectives. Drug Discov. Today 27, 1167–1175 (2022).
Wendland, M. et al. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc. Natl Acad. Sci. USA 104, 6347–6352 (2007).
Schall, T. J. & Proudfoot, A. E. I. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat. Rev. Immunol. 11, 355–363 (2011).
Feagan, B. G. et al. Randomised clinical trial: vercirnon, an oral CCR9 antagonist, vs. placebo as induction therapy in active Crohn’s disease. Aliment. Pharmacol. Ther. 42, 1170–1181 (2015).
Oswald, C. et al. Intracellular allosteric antagonism of the CCR9 receptor. Nature 540, 462–465 (2016).
Wagner, J. R. et al. Emerging computational methods for the rational discovery of allosteric drugs. Chem. Rev. 116, 6370–6390 (2016).
Erlandson, S. C., McMahon, C. & Kruse, A. C. Structural basis for G protein-coupled receptor signaling. Annu. Rev. Biophys. 47, 1–18 (2018).
Cooke, R. M., Brown, A. J. H., Marshall, F. H. & Mason, J. S. Structures of G protein-coupled receptors reveal new opportunities for drug discovery. Drug Discov. Today 20, 1355–1364 (2015).
Thal, D. M. et al. Recent advances in the determination of G protein-coupled receptor structures. Curr. Opin. Struct. Biol. 51, 28–34 (2018).
Dror, R. O. et al. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl Acad. Sci. USA 108, 13118–13123 (2011).
Changeux, J. P. & Christopoulos, A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Cell 166, 1084–1102 (2016).
Kenakin, T. Theoretical aspects of GPCR-ligand complex pharmacology. Chem. Rev. 117, 4–20 (2017).
Nussinov, R. & Tsai, C. J. The different ways through which specificity works in orthosteric and allosteric drugs. Curr. Pharm. Des. 18, 1311–1316 (2012).
Wright, S. C. & Bouvier, M. Illuminating the complexity of GPCR pathway selectivity - advances in biosensor development. Curr. Opin. Struct. Biol. 69, 142–149 (2021).
Cheng, L. et al. Orthosteric ligand selectivity and allosteric probe dependence at Hydroxycarboxylic acid receptor HCAR2. Signal Transduct. Target. Ther. 8, 364 (2023).
Powers, A. S. et al. Structural basis of efficacy-driven ligand selectivity at GPCRs (Feb, 10.1038/s41589-022-01247-5, 2023). Nat. Chem. Biol. 19, 529–529 (2023).
Liu, H. T. et al. Structure-guided development of selective M3 muscarinic acetylcholine receptor antagonists. Proc. Natl Acad. Sci. USA 115, 12046–12050 (2018).
Chatzigoulas, A. & Cournia, Z. Rational design of allosteric modulators: Challenges and successes. Wires Comput. Mol. Sci. 11, e1529 (2021).
Nussinov, R. & Tsai, C. J. Unraveling structural mechanisms of allosteric drug action. Trends Pharmacol. Sci. 35, 256–264 (2014).
Nussinov, R., Tsai, C. J. & Liu, J. Principles of allosteric interactions in cell signaling. J. Am. Chem. Soc. 136, 17692–17701 (2014).
Morimoto, J., Hayashi, Y. & Suga, H. Discovery of macrocyclic peptides armed with a mechanism-based warhead: Isoform-selective inhibition of human Deacetylase SIRT2. Angew. Chem. Int. Ed. 51, 3423–3427 (2012).
Orr, S. T. M. et al. Mechanism-Based Inactivation (MBI) of Cytochrome P450 Enzymes: Structure-activity relationships and discovery strategies to mitigate drug-drug interaction risks. J. Med. Chem. 55, 4896–4933 (2012).
Pan, Y. & Mader, M. M. Principles of Kinase allosteric inhibition and pocket validation. J. Med. Chem. 65, 5288–5299 (2022).
Ippolito, M. et al. Identification of a β-arrestin-biased negative allosteric modulator for the β2-adrenergic receptor. Proc. Natl Acad. Sci. USA 120, e2302668120 (2023).
Schuss, C. et al. Structure-activity relationship study of the high-affinity Neuropeptide Y-4 receptor positive allosteric modulator VU0506013. J. Med. Chem. 66, 8745–8766 (2023).
Wang, X. D. et al. Intermediate-state-trapped mutants pinpoint G protein-coupled receptor conformational allostery. Nat. Commun. 14, 1325–1334 (2023).
Lu, S. Y. et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat. Commun. 12, 4721–4735 (2021).
Ni, D. et al. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 12, 464–476 (2021).
Lu, X., Lan, X., Lu, S. & Zhang, J. Progressive computational approaches to facilitate decryption of allosteric mechanism and drug discovery. Curr. Opin. Struct. Biol. 83, 8 (2023).
Hollingsworth, S. A. et al. Cryptic pocket formation underlies allosteric modulator selectivity at muscarinic GPCRs. Nat. Commun. 10, 3289–3297 (2019).
Hertig, S., Latorraca, N. R. & Dror, R. O. Revealing atomic-level mechanisms of protein allostery with molecular dynamics simulations. Plos Comput. Biol. 12, e1004746 (2016).
Bartuzi, D., Kaczor, A. A. & Matosiuk, D. Interplay between two allosteric sites and their influence on agonist binding in human μ opioid receptor. J. Chem. Inf. Model. 56, 563–570 (2016).
Kumar, A. et al. Predicting allosteric pockets in protein biological assemblages. Bioinformatics 39, btad275 (2023).
Smith, R. D. & Carlson, H. A. Identification of cryptic binding sites using MixMD with standard and accelerated molecular dynamics. J. Chem. Inf. Model. 61, 1287–1299 (2021).
Zhang, O. D. et al. Learning on topological surface and geometric structure for 3D molecular generation. Nat. Comput. Sci. 3, 849–859 (2023).
Leander, M. et al. Functional plasticity and evolutionary adaptation of allosteric regulation. Proc. Natl Acad. Sci. USA 117, 25445–25454 (2020).
Wang, Q. et al. Toward understanding the molecular basis for chemical allosteric modulator design. J. Mol. Graph. Model. 38, 324–333 (2012).
Strasser, A., Wittmann, H. J. & Seifert, R. Binding kinetics and pathways of ligands to GPCRs. Trends Pharmacol. Sci. 38, 717–732 (2017).
Herenbrink, C. K. et al. The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun. 7, 10842–10855 (2016).
Diaz, O. et al. Allosteric binding cooperativity in a kinetic context. Drug Discov. Today 28, 103441 (2023).
Alhadeff, R., Vorobyov, I., Yoon, H. W. & Warshel, A. Exploring the free-energy landscape of GPCR activation. Proc. Natl Acad. Sci. USA 115, 10327–10332 (2018).
Shukla, A. K. Biasing GPCR Signaling from Inside. Sci. Signal. 7, pe3 (2014).
Gurevich, V. V. & Gurevich, E. V. Biased GPCR signaling: Possible mechanisms and inherent limitations. Pharmacol. Ther. 211, 107540 (2020).
Seyedabadi, M., Ghahremani, M. H. & Albert, P. R. Biased signaling of G protein coupled receptors (GPCRs): Molecular determinants of GPCR/transducer selectivity and therapeutic potential. Pharmacol. Therapeut. 200, 148–178 (2019).
Bermudez, M., Nguyen, T. N., Omieczynski, C. & Wolber, G. Strategies for the discovery of biased GPCR ligands. Drug Discov. Today 24, 1031–1037 (2019).
Seyedabad, M., Gharghabi, M., Gurevich, E. V. & Gurevich, V. V. Structural basis of GPCR coupling to distinct signal transducers: implications for biased signaling. Trends Biochem. Sci. 47, 570–581 (2022).
Wang, X. D. et al. The Potential of 19F NMR application in GPCR biased drug discovery. Trends Pharmacol. Sci. 42, 19–30 (2021).
Okashah, N. et al. Variable G protein determinants of GPCR coupling selectivity. Proc. Natl Acad. Sci. USA 116, 12054–12059 (2019).
Guarnera, E. & Berezovsky, I. N. On the perturbation nature of allostery: sites, mutations, and signal modulation. Curr. Opin. Struct. Biol. 56, 18–27 (2019).
Ni, D. et al. Along the allostery stream: Recent advances in computational methods for allosteric drug discovery. Wires Comput. Mol. Sci. 12, e1585 (2022).
Tee, W. V., Guarnera, E. & Berezovsky, I. N. Reversing allosteric communication: From detecting allosteric sites to inducing and tuning targeted allosteric response. Plos Comput. Biol. 14, e1006228 (2018).
Newman, A. H., Battiti, F. O. & Bonifazi, A. 2016 Philip S. Portoghese medicinal chemistry lectureship: designing bivalent or bitopic molecules for G-protein coupled receptors. the whole is greater than the sum of its parts. J. Med. Chem. 63, 1779–1797 (2020).
Aurelio, L. et al. A structure-activity relationship study of bitopic N6-substituted adenosine derivatives as biased Adenosine A1 receptor agonists. J. Med. Chem. 61, 2087–2103 (2018).
Feng, Z. W., Hu, G. X., Ma, S. F. & Xie, X. Q. Computational advances for the development of allosteric modulators and bitopic ligands in G protein-coupled receptors. AAPS J. 17, 1080–1095 (2015).
Ni, D. et al. Combining ALLOSTERIC AND ORTHOSTERIC DRUGS TO OVERCOME DRUG RESISTANce. Trends Pharmacol. Sci. 41, 336–348 (2020).
Huang, B. S., St Onge, C. M., Ma, H. G. & Zhang, Y. Design of bivalent ligands targeting putative GPCR dimers. Drug Discov. Today 26, 189–199 (2020).
Romantini, N. et al. Exploring the signaling space of a GPCR using bivalent ligands with a rigid oligoproline backbone. Proc. Natl Acad. Sci. USA 118, e2108776118 (2021).
Picard, L. P. & Prosser, R. S. Advances in the study of GPCRs by 19F NMR. Curr. Opin. Struct. Biol. 69, 169–176 (2021).
Calebiro, D. & Sungkaworn, T. Single-MOLECULE IMAGing of GPCR Interactions. Trends Pharmacol. Sci. 39, 109–122 (2018).
Kumari, P., Ghosh, E. & Shukla, A. K. Emerging Approaches to GPCR ligand screening for drug discovery. Trends Mol. Med. 21, 687–701 (2015).
Lee, H. M., Giguere, P. M. & Roth, B. L. DREADDs: novel tools for drug discovery and development. Drug Discov. Today 19, 469–473 (2014).
Jabeen, A. & Ranganathan, S. Applications of machine learning in GPCR bioactive ligand discovery. Curr. Opin. Struct. Biol. 55, 66–76 (2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Huang, X. P. et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 527, 477–483 (2015).
He, X. H. et al. AlphaFold2 versus experimental structures: evaluation on G protein-coupled receptors. Acta Pharmacol. Sin. 44, 1–7 (2023).
Kapla, J. et al. Can molecular dynamics simulations improve the structural accuracy and virtual screening performance of GPCR models? Plos Comput. Biol. 17, e1008936 (2021).
Xie, J., Pan, G. X., Li, Y. B. & Lai, L. H. How protein topology controls allosteric regulations. J. Chem. Phys. 158, 105102 (2023).
Xie, J. et al. Coevolution-based prediction of key allosteric residues for protein function regulation. Elife 12, e81850 (2023).
Chen, L. F. et al. Sequence-based drug design as a concept in computational drug design. Nat. Commun. 14, 4217–4237 (2023).
Cheng, R. K. Y. et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 545, 112–115 (2017).
Lin, X. et al. Structural basis of ligand recognition and self-activation of orphan GPR52. Nature 579, 152–157 (2020).
Duan, J. et al. Structures of full-length glycoprotein hormone receptor signalling complexes. Nature 598, 688–692 (2021).
Vuckovic, Z. et al. Pharmacological hallmarks of allostery at the M4 muscarinic receptor elucidated through structure and dynamics. Elife 12, e83477 (2023).
Duan, J. et al. Hormone- and antibody-mediated activation of the thyrotropin receptor. Nature 609, 854–859 (2022).
Liu, Y. F. et al. Ligand recognition and allosteric modulation of the human MRGPRX1 receptor. Nat. Chem. Biol. 19, 416–422 (2022).
Wu, H. X. et al. Structure of a Class C GPCR Metabotropic Glutamate Receptor 1 Bound to an Allosteric Modulator. Science 344, 58–64 (2014).
Wen, T. L. et al. Structural basis for activation and allosteric modulation of full-length calcium-sensing receptor. Sci. Adv. 7, eabg1483 (2021).
Park, J. et al. Symmetric activation and modulation of the human calcium-sensing receptor. Proc. Natl Acad. Sci. USA 118, e2115849118 (2021).
Lin, S. L. et al. Structures of G(i)-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 594, 583–588 (2021).
Du, J. et al. Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature 594, 589–593 (2021).
Byrne, E. F. X. et al. Structural basis of Smoothened regulation by its extracellular domains. Nature 535, 517–522 (2016).
Ho, J. D. et al. Structural basis for GPR40 allosteric agonism and incretin stimulation. Nat. Commun. 9, 1645–1655 (2018).
Lu, J. et al. Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nat. Struct. Mol. Biol. 24, 570–577 (2017).
Liu, X. Y. et al. An allosteric modulator binds to a conformational hub in the beta(2) adrenergic receptor. Nat. Chem. Biol. 16, 749–755 (2020).
Robertson, N. et al. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 553, 111–114 (2018).
Liu, H. et al. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 25, 472–481 (2018).
Wang, X. W. et al. Structural insights into dimerization and activation of the mGlu2-mGlu3 and mGlu2-mGlu4 heterodimers. Cell Res 33, 762–774 (2023).
Li, F. H. et al. Molecular recognition and activation mechanism of short-chain fatty acid receptors FFAR2/3. Cell Res. https://doi.org/10.1038/s41422-023-00914-z (2024).
Hollenstein, K. et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 (2013).
Christopher, J. A. et al. Structure-based optimization strategies for G Protein-Coupled Receptor (GPCR) allosteric modulators: a case study from analyses of new metabotropic Glutamate Receptor 5 (mGlu(5)) X-ray Structures. J. Med. Chem. 62, 207–222 (2019).
Chen, G. et al. Activation and allosteric regulation of the orphan GPR88-Gi1 signaling complex. Nat. Commun. 13, 2375–2386 (2022).
Liu, X. Y. et al. Mechanism of intracellular allosteric beta(2)AR antagonist revealed by X-ray crystal structure. Nature 548, 480–484 (2017).
Zheng, Y. et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540, 458–461 (2016).
Jaeger, K. et al. Structural basis for allosteric ligand recognition in the human CC chemokine receptor 7. Cell 178, 1222–1230 (2019).
Liu, K. W. et al. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 585, 135–140 (2020).
Lees, J. A. et al. An inverse agonist of orphan receptor GPR61 acts by a G protein-competitive allosteric mechanism. Nat. Commun. 14, 5938 (2023).
Duan, J. et al. GPCR activation and GRK2 assembly by a biased intracellular agonist. Nature 620, 676–681 (2023).
Acknowledgements
This study was supported by grants from the National Key R&D Program of the Ministry of Science and Technology (No. 2023YFC3404700), the National Natural Science Foundation of China (No. 22077082), the Shanghai Frontiers Science Center of Cellular Homeostasis and the Human Diseases, and the Innovative Research Team of High-Level Local Universities in Shanghai.
Author information
Authors and Affiliations
Contributions
S.L. and Z.C. conceived and designed the study. M.Z., T.C., X.L., and X.L. analyzed the results. M.Z. and X.L. made the figures and tables. M.Z., X.L., and T.C. wrote the original manuscript. S.L. revised the manuscript and supervised the project. All authors have discussed the results and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, M., Chen, T., Lu, X. et al. G protein-coupled receptors (GPCRs): advances in structures, mechanisms, and drug discovery. Sig Transduct Target Ther 9, 88 (2024). https://doi.org/10.1038/s41392-024-01803-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01803-6
