
Abstract
Emerging SARS-CoV-2 variants, particularly the Omicron variant and its sublineages, continually threaten the global public health. Small molecule antivirals are an effective treatment strategy to fight against the virus. However, the first-generation antivirals either show limited clinical efficacy and/or have some defects in pharmacokinetic (PK) properties. Moreover, with increased use of these drugs across the globe, they face great pressure of drug resistance. We herein present the discovery and characterization of a new generation antiviral drug candidate (SY110), which is a potent and selective inhibitor of SARS-CoV-2 main protease (Mpro). This compound displayed potent in vitro antiviral activity against not only the predominant SARS-CoV-2 Omicron sublineage BA.5, but also other highly pathogenic human coronaviruses including SARS-CoV-1 and MERS-CoV. In the Omicron-infected K18-hACE2 mouse model, oral treatment with SY110 significantly lowered the viral burdens in lung and alleviated the virus-induced pathology. Importantly, SY110 possesses favorable PK properties with high oral drug exposure and oral bioavailability, and also an outstanding safety profile. Furthermore, SY110 exhibited sensitivity to several drug-resistance Mpro mutations. Collectively, this investigation provides a promising new drug candidate against Omicron and other variants of SARS-CoV-2.
Similar content being viewed by others
Introduction
Since the outbreak of the Coronavirus Disease 2019 (COVID-19) pandemic due to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in late 2019, more than three years have passed.1,2 In the battle against this virus, vaccines have played a decisive role. However, continuous viral mutations, from Alpha, Beta, to the latest Omicron, bring challenges to the protection effect of existing vaccines.3,4,5,6,7 Especially, the Omicron variant and its sublineages have more than 30 mutations on the spike protein, which is the main target of vaccines, and have shown much higher transmissibility and escape of immune elicited by natural infection or vaccines than other SARS-CoV-2 variants.8,9,10,11 Furthermore, historical experiences tell us that the long-term existence of SARS-CoV-2 is perhaps inevitable.12 Therefore, besides vaccines, effective antivirals are also an important strategy to address the current epidemic and future threats.
SARS-CoV-2 is a positive-sense single stranded (+ss) RNA virus.2,13 It belongs to the genus β-coronavirus of the family Coronaviridae.2 The SARS-CoV-2 genome encodes 14 open reading frames (ORFs) with a length of around 30 kb. These ORFs encodes two long polyproteins, pp1a and pp1ab, four structural proteins and nine accessory proteins. The two polyproteins, pp1a and pp1ab, are self-catalyzed and cleaved into 16 non-structural proteins (NSPs) by two cysteine proteases, the main protease (Mpro, also referred to as 3CLpro) and the papain-like protease (PLpro).14,15 Mpro is a conserved gene in SARS-CoV-2 and its variants, as well as other highly pathogenic coronavirus, including SARS-CoV-1, MERS-CoV.15,16,17,18 This protease is responsible for the generation of 13 NSPs by cleaving pp1a and pp1ab, including RNA-dependent RNA polymerase (RdRp), helicase, exoribonucleases, 2′-O-methyltransferase and uridine-specific endoribonuclease.15,19 Of note is that Mpro recognizes and cleaves amino acid sequence of the substrate with specificity and its most cleavage sites at sequences Leu-Gln ↓ (Ser, Ala, or Gly) (↓ represents the cleavage site), while no known human proteases have the similar specificity.20 The critical role of Mpro in viral replication and its high conservation, combined with no known human proteases with similar cleavage specificity, render it an attractive target for the development of antivirals. The RdRp, a highly versatile enzyme that assists in RNA synthesis by catalyzing the RNA-template-dependent formation of phosphodiester bonds, is another important antiviral target, and has also been extensively studied.18,19
Currently, several small-molecule antivirals targeting Mpro or RdRp have been approved to use in some countries and regions worldwide. For example, three small-molecule antivirals, Remdesivir, Molnupiravir, and Paxlovid (a combination of Nirmatrelvir and Ritonavir), have been approved to use clinically by US Food and Drug Administration.21,22,23 Ensitrelvir has been approved by the Ministry of Health, Labour and Welfare of Japan (MHW).24 China National Medical Products Administration also approved three antivirals, Azvudine, Renmindevir, and Xiannuoxin (a combination of Simnotrelvir and Ritonavir).25,26 Of them, Remdesivir,27 Molnupiravir,28 Renmindevir,29 and Azvudine30 are inhibitors of SARS-CoV-2 RdRp, and Nirmatrelvir31 (the active ingredient of Paxlovid), Ensitrelvir,32 and Simnotrelvir26 (the active ingredient of Xiannuoxin) are inhibitors against SARS-CoV-2 Mpro. However, due to the need of emergency response to the pandemic, they have more or less defects, for example, suboptimal potency, toxicity, or imperfect pharmacokinetic (PK) properties, including low oral drug exposure, poor oral bioavailability, and moderate stability in human liver microsomes (HLM).26,27,28,29,31,33 Moreover, drug resistance variants against currently approved drugs have already emerged. For examples, variants bearing E166N/V, M165T, G143S, Q189E, A173V, H172F/Q/Y, or Q192S/T/V mutants in Mpro have been reported to be resistant to Nirmatrelvir treatment.34,35,36,37,38,39,40,41 Therefore, developing next-generation antivirals is urgent.
In this investigation, we report the discovery of a new potent and selective Mpro inhibitor (SY110). The X-ray crystal structure of the Mpro-SY110 complex indicates that SY110 occupies only the Mpro substrate binding sites S1′, S1, and S2 with an atypical binding mode. Meanwhile, SY110 showed potent antiviral activity against SARS-CoV-2 Omicron and its sublineages, as well as other highly pathogenic human coronaviruses including SARS-CoV-1 and MERS-CoV in vitro, and alleviated the virus-induced pathology in the Omicron-infected K18-hACE2 mouse model. Importantly, SY110 demonstrated favorable PK properties with high oral drug exposure and excellent oral bioavailability, as well as an outstanding safety profile, while can also partially overcome drug resistance mutations of Mpro. Taken together, our studies demonstrate that SY110 could be a therapeutic candidate for COVID-19.
Results
Retrieval of a hit compound and its binding mode with Mpro
To obtain a new starting active compound for the drug development targeting Mpro, we first screened an in-house chemical library containing about 30,000 compounds by fluorescence resonance energy transfer (FRET) assay. Four compounds were found to be able to inhibit the enzymatic activity of Mpro with 50% inhibition concentration (IC50) values less than 50 μM (Supplementary Fig. S1). Of them, the most potent compound, Hit-1, exhibited an IC50 value of 1.30 μM (Fig. 1a, b). The activity of Hit-1 was further validated by differential scanning fluorimetry (DSF) assay, which gave a thermal shift (ΔTm) value of 8.45 ± 0.35 °C (Fig. 1c), indicating a direct binding between Hit-1 and Mpro.

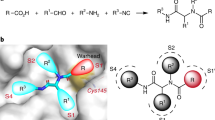
Discovery of a hit compound against SARS-CoV-2 Mpro. a The chemical structure of Hit-1. The P1′, P1, and P2 moieties of Hit-1 are labeled. The warhead carbon is marked with a black asterisk. b Dose-activity curve of Hit-1 against SARS-CoV-2 Mpro in the FRET assay. Data shown are the mean ± standard deviation (SD) from three independent experiments. c Differential scanning fluorimetry analysis of the effect of Hit-1 on SARS-CoV-2 Mpro stability. Exposure of hydrophobic residues monitored by an increase in relative fluorescence units (RFUs). Curves represent the average of three experiments. d Hit-1 (orange) is located at the substrate-binding pocket of Mpro (cyan). His41 of Mpro is in blue, Cys145 is yellow. Pockets (S1′, S1, S2, and S4) of Mpro and moieties (P1′, P1 and P2) of Hit-1 are both labeled. e Interactions between Hit-1 and Mpro. Fo – Fc density map is shown for Hit-1 (gray mesh, σ = 2.5) and Cys145. Covalent bond is shown by a black arrow and hydrogen bonds are displayed by red dashed lines. f Regions of Hit-1 for structural optimization. Images in d, e were processed by using PyMOL (https://pymol.org)
To facilitate the subsequent structural optimization, we solved the co-crystal structure of Mpro-Hit-1 complex (PDB ID: 8HHT, Supplementary Table S1). As shown in Fig. 1d, the S1′ and S1 pockets of Mpro were filled with benzyl (P1) and thiazole (P1′) moieties of Hit-1, respectively. Compared with other peptidomimetic Mpro inhibitors, the P1 and P1′ moieties present an inverted conformation, indicating an atypical binding mode to Mpro. The P2 group directly points to the solvent region, rather than occupying the expected S2 pocket. The warhead carbon of Hit-1 is covalently linked to the sulfur atom of the catalytic Cys145 (Fig. 1e). The hydroxyl (or oxyanion) moiety of the thiohemiketal group generates a 2.5 Å hydrogen bond with the side-chain of His41. The amide oxygen of Hit-1 points to the canonical “oxyanion hole” and generates two hydrogen bonds with the backbones of Gly143 (3.0 Å) and Cys145 (3.1 Å), respectively. In addition, the amide nitrogen of P1′ forms a hydrogen bond with the main-chain of His164 (2.7 Å). Whereas the thiazole nitrogen of P1′ forms the other 3.3 Å hydrogen bond with the side-chain of His163 (Fig. 1e).
Structural optimization of Hit-1
To improve the potency of Hit-1 (1a), we carried out a stepwise structural optimization, which focused on three regions: thiazole (P1′, R1), benzyl (P1, R2), and phenyl (P2, R3) (Fig. 1f). Firstly, we fixed R2 and R3 and optimized R1. A total of 9 new compounds (1b-j) with different R1 were synthesized. Unfortunately, the newly synthesized compounds did not exhibit improved activity against SARS-CoV-2 Mpro (Fig. 2). Secondly, we optimized R2 with R1 and R3 fixed as its original subgroups. Another 7 compounds (2a-g) with varied R2 were prepared and 1a was still the most potent compound (Fig. 2). We finally retained R1 and R2 and optimized R3. 25 new compounds (3a-y) were synthesized and 20 compounds showed elevated potency compared with 1a. Of special note are compounds 3q-3x, which showed IC50 < 30 nM (Fig. 2).

The stepwise structural optimization process of Hit-1. Bioactivities were measured by the FRET assay and are represented as the mean ± SD from three experiments. Molecules with IC50 < 30 nM are shown in red color
Selection of SY110 as the candidate compound for further studies
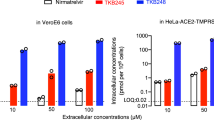
To select a better candidate compound for further studies, we investigated the metabolic stability, bioavailability and toxicity of the eight most potent compounds (3q-x, Fig. 2) from the enzymatic assay. Six compounds (3r, 3t-x) showed comparable or higher stability compared with Nirmatrelvir (Supplementary Table S2). We next measured the PK parameters of the six compounds in Sprague-Dawley (SD) rats. All six compounds exhibited good PK properties, and compound 3w (referred to as SY110 hereafter) displayed much higher oral area under the curve (AUC) (19018.08 h*ng*mL−1) and oral bioavailability (F: 82.75%) than others (Supplementary Table S3). Finally, we investigated the cytotoxicity of the six compounds in different cells by MTT assay, including BEAS-2B, VeroE6, and HUVEC cells. As shown in Supplementary Table S4, except compound 3v, which showed relatively weak cytotoxicity (CC50: 129.2 - 158.6 μM), all other compounds did not display cytotoxicity (CC50 [concentration cytotoxicity 50%] > 500 μM). We carried on to examine the antiviral activity of the six compounds using the authentic SARS-CoV-2 Omicron BA.1 (B.1.1.529.1). Among the compounds tested, SY110 showed the most potent antiviral activity in VeroE6-TMPRSS2 (Fig. 3a). Comparing with Nirmatrelvir, SY110 suppressed the viral genome copies to significantly lower level at 0.8, 4 and 20 µM concentration (viral genome RNA [Nirmatrelvir vs SY110]: 3- (p = 0.0001), 8- (p = 0.0356), 9-fold (p = 0.0101) higher) (Fig. 3a). We further determined the antiviral potency of SY110 with plaque reduction assay. Consistent with the in vitro screening results, the 50% effective concentration (EC50) of SY110 determined with plaque reduction assay was 4-fold lower (EC50 [Nirmatrelvir vs SY110]: 5.051 µM vs 1.264 µM) than Nirmatrelvir against Omicron BA.5 sublineage (Fig. 3b). Since Mpro is evolutionarily conserved, we further evaluated whether SY110 demonstrated pan-coronavirus antiviral efficacy. SY110 consistently showed improved antiviral potency compared with Nirmatrelvir (Fig. 3b) against not only SARS-CoV-2 variants of concern (VOCs) B.1.1.7, B.1.351, and BA.2 but also SARS-CoV-1 and MERS-CoV. Together, SY110 demonstrated highly potent pan-coronavirus antiviral efficacy in vitro with high metabolic stability and remarkable in vivo bioavailability. Therefore, further in-depth studies are carried out on SY110 in the follows.

In vitro cellular antiviral activity of SY110. a Cells were infected with Omicron BA.1 sublineage at 0.1 MOI and treated with 3r, 3 u, 3t, 3 v, 3w (SY110), 3x, Nirmatrelvir or DMSO at 1 hpi.. At 24 hpi., cell lysates were harvested. Viral genome copies were quantified by probe-based RT-qPCR (n = 4). b Cells were infected and treated with 3w (SY110) or Nirmatrelvir at 2 hpi. (n = 3). Cells were fixed at 72 hpi. for plaque formation units quantification by visualization with 0.5% crystal violet staining. Plaque formation units were normalized to that of the vehicle group. Statistical significance is evaluated by one-way ANOVA in a compared to vehicle group. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. ns, not statistically significant
Crystal structure of SARS-CoV-2 Omicron variant Mpro in complex with SY110
The crystal structure of Omicron variant Mpro-SY110 complex is in space group C2 with one protomer per asymmetric unit (PDB ID: 8HHU, Supplementary Table S1). The electron density map illustrates a unique interaction mode of SY110 with Mpro (Fig. 4a–c). The P1′ thiazole ring of SY110 inserts into the S1 pocket while the P1 benzyl group occupies the S1′ site. The P2 chiral ether chain points to the solvent region, and the P3 3,3-difluorocyclohexyl group is placed in the S2 pocket (Fig. 4a, b). In detail, the carbonyl carbon of the SY110 α-ketoamide motif generates a 1.8 Å reversible covalent bond with the Cys145 in the (R) configuration (Fig. 4c). The oxyanion of this thiohemiketal moiety forms a hydrogen bond with the side-chain of His41 (2.3 Å). Meanwhile, the amide oxygen of SY110 is located into the canonical “oxyanion hole” and generates two hydrogen bonds with the backbone amides of Cys145 (3.0 Å) and Gly143 (3.2 Å), respectively. The amide nitrogen of P1′ generates a 3.0 Å hydrogen bond with the main-chain oxygen of His164, and the thiazole nitrogen of P1′ adopts another hydrogen bond with the side-chain of His163 (3.0 Å). The P1 benzyl moiety displays the hydrophobic interactions with the Cα, Cβ, Cγ of Thr25 and the Cβ, Cδ of Leu27 (Fig. 4c), while the P2 chiral ether chain is near to the flexible loop (141Leu-Asn-Gly-Ser144). The P3 3,3-difluorocyclohexyl moiety generates the hydrophobic interactions with residues Met49, Met165 and Gln189 (Fig. 4c).

Crystal structure of SARS-CoV-2 Omicron variant Mpro in complex with SY110. a The chemical structure of SY110. The warhead carbon is marked with a black asterisk. b Close-up view of SY110 with the substrate-binding pocket of Mpro. SY110 is in orange, Mpro is aquamarine, His41 of Mpro blue, Cys145 yellow. Four sites S1′, S1, S2, and S4 of Mpro, and four moieties P1′, P1, P2, and P3 of SY110 are labeled, respectively. Fo – Fc density map of SY110 is shown (gray mesh, σ = 2.5). c Interactions between Mpro and SY110. The residues of Mpro involved in SY110 binding are displayed by sticks. The hydrogen bonds are displayed as red dashed lines. d Comparison of the binding modes of SY110 (orange, PDB ID: 8HHU), 11a (blue, PDB ID: 6LZE), MI-23 (wheat, PDB ID: 7D3I) and Nirmatrelvir (green, PDB ID: 7RFW). e Comparison of the binding modes between Mpro-SY110 and Mpro-calpain inhibitor XII (white, PDB: 6XFN). Images b–e were prepared using PyMOL (https://pymol.org)
The inhibitor SY110 P1′ and P1 moieties displays the inverted interaction mode with Mpro, which is dissimilar to the previously reported peptidomimetic inhibitors (Fig. 4d), such as 13b,20 11a,42 MI-23,43 and Nirmatrelvir.31 However, the similar inverted conformation of P1′ and P1 groups appeared in calpain inhibitor XII.44 We noticed that the Cα of P2 in the calpain inhibitor XII adopts the (S) configuration, which makes its P3 group point to the solvent region (Fig. 4e). In contrast, the Cα of P2 in SY110 adopts the (R) configuration, thus causing the SY110 P3 moiety to point into the S2 pocket and form the extensive hydrophobic interactions with Mpro (Fig. 4e).
Evaluations of the drug metabolism and pharmacokinetics (DMPK)
To evaluate the druggability of SY110, we examined the DMPK properties of SY110. Firstly, the inhibitory activity of SY110 against human cytochrome P450 enzymes was measured. In this assay, SY110 did not show obvious inhibitory activity against CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4 (IC50 values > 30 µM) (Supplementary Table S5), implying a low risk of drug-drug interaction (DDI). Then the human plasma protein binding of SY110 was examined under equilibrium dialysis conditions, which gave a plasma unbound fraction of 0.301, indicating moderate plasma protein binding (Supplementary Table S6). Next, we evaluated the pharmacokinetic properties of SY110 in beagle dogs, monkeys, and mice. SY110 displayed oral bioavailability of 131.99%, 24.36%, and 67.42%, respectively (Fig. 5a–c and Supplementary Table S7). Notably, SY110 demonstrated greater drug exposure, improved oral bioavailability, and a longer half-life in monkeys than Nirmatrelvir.31

In vivo pharmacokinetics and safety data of SY110.SY110 was administered by intravenous injection (i.v.) or oral gavage (p.o.) route, then the blood samples of the dogs (a), monkeys (b), and mice (c) were analyzed. The two dash lines in (c) indicate the EC50 and EC90 values for SY110 against BA.2 variant (Fig. 3b). d The weight change (percentage of initial weight) of mice treated with SY110 by oral administration for two weeks (n = 6)
Safety evaluation of SY110
We subsequently evaluated the safety of SY110. Before these evaluations, we measured the inhibitory activity of SY110 against human proteases. SY110 did not exhibit activity against the tested various human proteases (IC50 > 100 µM, Supplementary Table S8), suggesting lower off-target toxicity. For the safety evaluation, we firstly carried out an Ames mutagenicity test and a chromosomal aberrations assay, which showed negative results in both experiments. Then the hERG channel blockade assay was performed, which revealed that SY110 is unlikely to cause cardiotoxicity (IC50 > 30 µM). No mice died following treatment with 1000 mg/kg of SY110 by oral gavage (p.o.) (Supplementary Table S9) during the acute toxicity study. All mice tolerated oral administration of SY110 at 600 mg/kg twice daily for 14 days without death, significant body weight change, or pathological changes in the heart, liver, lung, kidney, or spleen, indicating low in vivo toxicity (Fig. 5d and Supplementary Table S9). We also evaluated the reproductive toxicity of SY110. Results of the embryo-fetal development studies in rats showed that there were no observed SY110-related maternal toxicity, reproductive toxicity, and effect on embryo-fetal viability (including fetal external, visceral or skeletal morphological development) at 1000 mg/kg/day (the highest dose tested), indicating lower teratogenic risk (Supplementary Table S10). Overall, SY110 showed outstanding preclinical safety.
In vivo antiviral activity of SY110 in the K18-hACE2 transgenic mouse model
To assess the in vivo antiviral efficacy of SY110 by oral delivery, we utilized the previously established K18-hACE2 transgenic mouse model infected with the Omicron BA.2 sublineage (Fig. 6a).45 Early therapeutic treatment with SY110 at 1 hpi. substantially lowered viral genomic RNA (vRNA) in the lung by 179 folds (p = 0.0009) at 4 dpi. (Fig. 6b). In parallel, production of the subgenomic mRNA of the envelope gene (sgE) was inhibited by 337 folds (p = 0.0009) under the same treatment condition (Fig. 6c). Even if SY110 treatment was postponed to 24 hpi., vRNA and sgRNA copies in the lung was reduced by 16- (p = 0.001) and 12- (p = 0.001) folds compared with mock-treated mice at 4 dpi., respectively (Fig. 6b, c). In comparison, vRNA and viral sgE copies of the Nirmatrelvir-treated mice was 3- (p = 0.9226) and 2-folds (p = 0.8916) higher that of the SY110-treated mice, respectively. Meanwhile, we measured the infectious viral burden in the lung with plaque assays. In accordance with viral gene copy quantification, production of the infectious virus particles in the lung was effectively suppressed by therapeutic treatment with SY110 (Fig. 6d). We and others previously reported co-administration with Ritonavir (RTV) delayed the clearance of antiviral compounds by liver microsomes, thus enhancing their pharmacokinetics and antiviral efficacy in vivo.31,46 Therefore, we also explored the synergistic therapeutic effect of co-administration with SY110 and RTV. Remarkably, therapeutic treatment initiated at 24 hpi. with SY110/RTV combined further alleviated viral burden in the lung by 15- (vRNA, p = 0.9997), 43- (sgRNA, p = 0.9983), and 6-folds (infectious viral titer, p = 0.0926) compared to single treatment, respectively. Importantly, infectious viral titers in the mice lung with SY110/RTV was significantly decreased by ~90% (p = 0.0127) compared to those co-treated with Nirmatrelvir/RTV (Fig. 6d). In addition, viral antigen expression in the lung tissue was visualized with immunohistochemistry staining against the SARS-CoV-2 nucleocapsid (N) protein. Consistent with the virological assessment findings, treatment with SY110 substantially reduced the viral N protein expression in the lung tissue compared with the mock-treated mice (Fig. 7a). Moreover, combination treatment of SY110/RTV further restricted the viral antigen expression to much lower level in the infected mouse lung tissue (Fig. 7a). Next, pathological lesions induced by SARS-CoV-2 infection in the lung tissue was examined with haematoxylin-eosin (H&E) staining (Fig. 7b). Despite robust virus replication in the airway, Omicron infection induced attenuated pathological lesions compared with other variants.47,48,49 The most prominent lung pathology of the vehicle control group was characterized by multi-focal inflammatory infiltrations in the alveolar septa, peribronchiolar regions and perivascular areas (Fig. 7b). In contrast, only scattered inflammatory cell infiltrates can be occasionally found in the alveolar interstitium of the mice therapeutically treated with SY110 (Fig. 7b). Evidently, SY110/RTV co-administration further improved the lung architecture compared with SY110 single treatment. Together, our in vivo results showed that therapeutic treatment with SY110, especially when co-administered with Ritonavir, potently suppressed Omicron infection in the airway and alleviated virus-induced lung pathology in the infected animals.

In vivo antiviral activity of SY110. a The schematic illustration of experiment design. b The viral RNA-dependent RNA polymerase (RdRp) gene copies were quantified by probe-based RT-qPCR (n = 5–7) or c subgenomic envelope (sgE) mRNA copies were quantified by probe-based RT-qPCR (n = 5–7) or d viral titer quantification with plaque assay (n = 5–7). All data were obtained from three times of independent experiments and shown as mean ± SD. Statistical differences were determined with one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test in b–d. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001

Immunohistochemistry and H&E analysis of the infected mice lung tissue. a Representative images of immunohistochemistry staining visualizing the SARS-CoV-2 nucleocapsid protein (brown, indicated by black arrows) in the lung of the infected mice at 4 dpi. (n = 5–7). Scale bar, 200 µm. b Representative images of H&E staining visualizing the virus-induced pathology in the lung of the infected mice at 4 dpi. (n = 5–7). Representative images of alveoli, bronchioles, and blood vessels of the lung indicated by yellow dashed circle were enlarged with numbers 1, 2, and 3, respectively. Scale bar, 500 µm
Inhibitory activities of SY110 against Nirmatrelvir-resistance Mpro mutants
Nirmatrelvir is the clinically-approved specific Mpro inhibitor for COVID-19 treatment currently. However, with increasing application of Nirmatrelvir, the emergence of drug-resistant mutant strains is posing an imminent challenge to the healthcare system. E166N/V M165T, G143S, Q189E, A173V, H172F/Q/Y or Q192S/T/V mutation in the Mpro was recently shown to be associated with Nirmatrelvir resistance.34,35,36,37,38,39,40,41 To evaluate the efficacy of SY110 against these mutated Mpro, we expressed a panel of 14 mutated recombinant Mpro and tested their enzymatic sensitivity to SY110 and Nirmatrelvir by FRET assay. As shown in Fig. 8, Nirmatrelvir displayed obviously reduced activity against all these Mpro mutants. Especially E166N and E166V are the most resistant mutants for Nirmatrelvir with IC50 values of 3.429 μM (336.3 folds) and 1.908 μM (187.3 folds), respectively. Although SY110 also displayed reduced activity against most of these mutants, it exhibited substantially elevated activity against the E166N and E166V mutants, indicating that SY110 can, at least partially, overcome these two kinds of Nirmatrelvir-resistances. A possible explanation for this might be as follows: E166 is a hotspot for Nirmatrelvir-resistant mutation,34,35,36,37,38,39,40,41 which locates at the S1 pocket. The side chain oxygen of E166 forms a 3.2 Å hydrogen bond with the lactam ring of Nirmatrelvir.31 The valine (E166V) or asparagine (E166N) substitution at this position directly impacted the ligand interactions of Nirmatrelvir in the S1 site, which results in the strong drug resistance observed. Unlike Nirmatrelvir, SY110 does not form any hydrogen bonding interaction with the E166 (Fig. 4c). Although E166 mutations also lead to a decrease in activity of SY110, the reduction is not too significant.

Effects of resistance mutations on the inhibition of SARS-CoV-2 Mpro by SY110 and Nirmatrelvir. a Inhibition of SY110 and Nirmatrelvir against SARS-CoV-2 Mpro polymorphisms. IC50 fold increase is relative to the WT. b IC50 determination of SY110 and Nirmatrelvir against Mpro mutants. Data are shown as mean ± SD based on three independent experiments
Discussion
Omicron and its sublineages were characterized as a variant of concern by WHO due to their higher transmissibility and altered virological features.50,51 Despite vaccination, mortality and morbidity were still observed due to the Omicron infection.52 Therefore, small-molecule antivirals targeting conserved viral genes are particularly important. Mpro is an appealing target for the development of antiviral compounds, due to its critical role in the viral life cycle and its high conservation among different coronaviruses and the continuously emerging mutants of SARS-CoV-2.15,16,17,18 Currently many Mpro inhibitors have been reported.20,31,32,42,43,53,54,55,56 Nevertheless, a majority of them have a low potency and/or a poor pharmacokinetic profile, which obstructs their further advance. The marketed Mpro inhibitor, Nirmatrelvir, showed high potency against Mpro, but still has some defects in PK properties.31 Moreover, Nirmatrelvir resistance has also been reported although it has been on the market for less than one year.34,35,36,37,38,39,40,41 Therefore, developing more Mpro inhibitors with new chemical structures and/or new action modes is necessary.
SY110 reported here is a new potent and selective Mpro inhibitor with IC50 of 14.4 nM against Mpro. This compound has good PK properties in mice, rats, dogs and cynomolgus monkeys, with oral bioavailability of 67.42%, 82.75%, 131.99%, and 24.36%, respectively. It displayed robust in vitro antiviral potency against SARS-CoV-2 Alpha (B.1.1.7), Beta (B.1.351), Omicron (B.1.1.529) BA.2 and BA.5 sublineages. As a potent pan-coronavirus antiviral inhibitor, SY110 also has notable efficacy on other coronaviruses like SARS-CoV-1 and MERS-CoV-1. In K18-hACE2 mice of Omicron (B.1.1.529) infection, oral treatment with SY110 significantly improved pathological damage in both turbinate and lung. Moreover, SY110 exhibited favorable in vitro safety profiles in the hERG test, the CYP assay, the Ames test, and chromosome aberration test. The maximal tolerated single doses of SY110 were at least 1.0 g/kg in rats and no observed adverse effect levels at 600 mg/kg in rats in the 14 days repeated dose toxicity studies. Collectively, the data presented here demonstrate that SY110 is an effective and safe antiviral drug candidate against SARS-CoV-2.
Antiviral drug resistance is an increasing concern, particularly in immunocompromised patient populations, which has been often observed in the therapy of influenza, hepatitis, and human immunodeficiency virus. Despite only very short time since the use of antivirals against SARS-CoV-2, drug resistance has also been found, mainly due to the mutations of viral genome. For example, SARS-CoV-2 viruses carrying Mpro mutants E166N/V, M165T, G143S, Q189E, A173V, H172F/Q/Y, or Q192S/T/V are found to be resistant to Paxlovid.34,35,36,37,38,39,40,41 To predict the ability of SY110 to overcome the drug-resistance, we in this investigation expressed a panel of 14 mutated recombinant Mpro; viruses carrying these Mpro mutants are reported to be resistant to Nirmatrelvir. Our data show that SY110 can, to some extent, overcome the Nirmatrelvir-resistances caused by mutations at E166N (IC50 folds: 54.5 vs 336.3) and E166V (IC50 folds: 17.8 vs 187.3). Pfizer reported in the Paxlovid label that in the EPIC-HR clinical trial, Mpro E166V was detected in more patients after Paxlovid treatment than those after placebo treatment.57 This means that SY110 can be used as an alternative drug of Paxlovid or in combination use with Paxlovid in future.
Compared with Nirmatrelvir and other reported peptidomimetic Mpro inhibitors, SY110 possesses the following unique features and advantages: 1) SY110 adopts a distinct binding mode with Mpro: the inverted conformation of P1′ and P1, the (R) configuration of Cα in P2 as well as the P3 pointing to the S2 pocket, present a unique binding to Mpro. Particularly, SY110 does not form a hydrogen bond with the side chain of E166, while this hydrogen bond is crucial for Nirmatrelvir to maintain its inhibition effect. With the E166V mutation in Mpro, the IC50 value of Nirmatrelvir reduces about 100 folds.39 Currently, SARS-CoV-2 variant with the E166V mutation has appeared (GenBank: OP814611.1). Therefore, SY110 could preserve the inhibition on such mutated variant because of its specific binding fashion. 2) SY110 possesses favorable PK properties, especially high oral drug exposure and oral bioavailability, and a long half-life period (T1/2). Of note is that the drug concentration in plasma after p.o. 150 mg/kg administration in mice could maintain EC90 for at least 6 h (Fig. 5c), indicating that a single use of SY110 by 3-4 times oral administration per day might achieve a better therapeutic effect. 3) It has excellent target selectivity and preclinical safety. Even so, the clinical therapeutic efficacy and safety need to be extensively evaluated in the subsequent clinical trial studies. Additionally, we have to mention that, despite possessing favorable PK properties, there is still a room to further improve the HLM stability for SY110. Overall, SY110 is a promising antiviral drug candidate against Omicron and other SARS-CoV-2 variants, deserves further clinical development.
Materials and methods
Ethics statements
All procedures related to animal in pharmacokinetic (PK) studies were performed according to the guidelines approved by the Institute Animal Care and Use Committee (IACUC) of Sichuan Greentech Biotechnology Co. Ltd, ZLA (Beijing) Pharmaceutical Technology Co. Ltd, and Shanghai Medicilon Inc. All procedures related to animal in toxicity study was approved by the IACUC (20211063A). All procedures related to animal in antiviral study was approved by the CULATR (5779-21).
Compounds, cell lines, and viruses
The compounds (1a-j, 2a-g, and 3a-y) shown in Fig. 2 were synthesized in our internal facilities. And the detailed synthesis methods and characterization data of these are listed in the supplementary materials. BEAS-2B cells (GNHu27), HUVEC (GDC0635), VeroE6 cells (CRL-1586) and VeroE6-TMPRSS2 (JCRB 1819) cells were cultured in DEME (Gibco) with fetal bovine serum (10%) and penicillin-streptomycin (1%). Cells were grown at 37 °C in a humidified atmosphere of 5% CO2.
SARS-CoV-2 B.1.1.7 (GISAID: EPI_ISL_1273444), B.1.351 (GISAID: EPI_ISL_2423556), B.1.1.529 BA.2 subvariant (GISAID: EPI_ISL_9845731) and B.1.1.529 BA.5 subvariant (GISAID: EPI_ISL_13777658) were derived from laboratory-confirmed COVID-19 patients in Hong Kong. R. Fouchier kindly provided the MERS-CoV strain (EMC/2012). SARS-CoV-1 GZ50 were archived clinical isolates at Department of Microbiology, HKU. Viruses were titrated by plaque assays in VeroE6-TMPRSS2 cells. All infectious studies followed the BSL-3 facility at the Department of Microbiology, HKU.
Production of authentic SARS-CoV-2 main protease and its mutants
Main protease (Mpro) of wide type SARS-CoV-2 (GenBank: MN908947.3) and Omicron variant (BA.5, GenBank: OP054053) was cloned into pET28b vector with E. coli codon optimization. Using the former plasmid as template, Mpro mutations were gained by site-directed mutagenesis PCR. And the corresponding primers are presented in Supplementary Table S11. After digested by DpnI (TakaRa), the PCR products were transformed into DH5α (Novagen) and the plasmids were extracted and verified.
The detailed Mpro and its mutant genes cloning, expression and purification used the same approach as we reported previously.43,46 Briefly, the cDNA sequence was designed as the Mpro cleavage-site at N-terminus and the PreScission cleavage-site (removing the hexa-histidine tag) at the C-terminus. The plasmid of WT (or each mutant) Mpro was transformed to E. coli BL21 cells and cultured with kanamycin (50 μg/mL, 37 °C). Until the E. coli BL21 cells were grown to an optical density at 600 nm of ~0.8, expression was induced by adding isopropyl-D-thiogalactoside [0.5 mM (final concentration)] to the cell culture at 18 °C, 16–18 h. The cell pellets were resuspended, lysed and centrifugation. Then, the supernatants were loaded onto HisTrap FF column (GE Healthcare) and the protein was eluted. PreScission protease cleaved the target protein to remove its hexa-histidine tag. The processed target protein dialyzed overnight. The flowthrough containing Mpro was dealt with GSTtrap FF (GE Healthcare) and HisTrap FF column, then purified through gel filtration (GE Healthcare).
Crystallization, data collection, phase determination, and refinement
Mpro (~5 mg/mL) incubated with Hit-1 or SY110 at a molar ratio of 1:10 for 12–14 h (4 °C). Crystallization was performed at 291 K employing the sitting-drop vapor-diffusion method, 1 μL + 1 μL drops. The best crystal of Mpro-Hit-1 was observed under condition No. 85 of IndexTM (Hampton research): 0.2 M Magnesium chloride hexahydrate, 25% w/v PEG3350, 0.1 M Tris pH 8.5. Mpro-SY110 was obtained under condition No. 42 of IndexTM: 25% w/v PEG3350, 0.2 M BIS-TRIS pH 5.5. Then, the fished crystals were flash-cooled in liquid nitrogen.
X-ray diffraction experiment for Mpro-Hit-1 was collected at 0.97852 Å at beamline BL18U1 (Shanghai Synchrotron Radiation Facility, Shanghai, China). Mpro-SY110 was carried out at 0.976246 Å at the beamline BioMax (MAX IV Laboratory, Sweden). After XDS58 processing, the datasets further scaled with Aimless59 in CCP4. And the structures of both SARS-CoV-2 Mpro-Hit-1 and SARS-CoV-2 Mpro-SY110 were determined by molecular replacement, using the Mpro (PDB ID: 7C7P) as a search model. Mpro-Hit-1 structure was refined with Phenix.refine,60 while Mpro-SY110 was refined with program BUSTER.61 The two models were inspected and rebuilt using Coot.62 In the final structures of Mpro-Hit-1 and Mpro-SY110, Rfactor/Rfree values are 0.20/0.25 and 0.19/0.23, respectively. 98.3% of Mpro-Hit-1 and 98.7% of Mpro-SY110 residues are in the the “Ramachandran plot” statistical regions. Supplementary Table S1 summarizes the diffraction dataset and statistics of final refinement.
FRET-based enzymatic assay
For in-house chemical library screen, 10 mM DMSO stock solutions of compounds were diluted in assay buffer with final concentration of 100 μM. The protease (final concentration: 100 nM) was incubated with diluted compounds at 37 °C for 10 min, followed by the addition of 25 μL of assay buffer containing substrate (MCA-AVLQSGFR-Lys (DNP) -Lys-NH2, final concentration: 20 μM). The fluorescence signal was read at 320 nm excitation and 405 nm emission on the microplate reader (BMG). By using the simple liner regression model in GraphPad Prism 8.0 software, the primal velocities were calculated. Inhibition percentage was calculated by normalizing to DMSO controls.
For IC50 measurement, recombinant protease with optimized concentrations was added with each compound for 10 min. Then, the process of adding fluorescent substrate to initiate the reaction, using the microplate reader (BMG) to read, and using a dose-response model to calculate the IC50 values.
Differential scanning fluorimetry (DSF) assay
Mpro was mixed with tested compounds and 5 × SYPRO Orange dye (Sigma) avoiding from light. The thermal denaturation spectra were run from 20 °C to 95 °C at a gradient of 1.5 °C/min (CFX96, BioRad). Data were analyzed by Boltzmann model to calculate the melt temperature (Tm). The equation ΔTm = Tm(compound) − Tm(DMSO) was used to calculate the thermal shift (ΔTm). The values are presented as mean ± SD (n = 3).
Mammalian proteases activity assay
The mammalian proteases activity assay was analyzed using the assay kit (Biovision, USA) in triplicate by following manufacturer’s manual.
Cytotoxicity assay
Cells were seeded and grown in 96 well plates overnight. Then various concentrations of compounds in fresh medium were added and incubated for 72 h. Using MTT (Sigma) assay to determine the cell viability.
Pharmacokinetic properties
The information of compounds, doses and administration scheme for PK studies are showed in Supplementary Table S3 and Supplementary Table S7. Briefly, male SD rats (n = 3), male ICR mice n = 3), male beagle dogs (n = 3), or male cynomolgus monkeys (n = 3) were treated by intravenous or gavage (p.o.). For male SD rats, the vehicle was consisted with 5% DMSO, 40% PEG400, 52% saline, and 3% Poloxamer-188. For the mice, dogs, and monkeys, the vehicle was consisted with 5% DMSO, 40% PEG400, 52% saline, and 3% Solutol HS-15.
After administration, the blood samples were collected at indicated time points and analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS).
Metabolic stability evaluation
Compounds were incubated with human liver microsomes (0.5 mg/mL) at room temperature. Samples were collected and analyzed by LC-MS/MS at specific time points. The equation: T1/2 = 0.693/K (K is the rate constant from a plot of ln [concentration] vs. incubation time) and CLint = (0.693/T1/2) × {1/[microsomal protein concentration (0.5 mg/mL)]} × Scaling Factors (1254.2) were used to calculate the substrate depletion half-life (T1/2) and intrinsic clearance (CLint).
Human plasma protein binding determination of SY110
The frozen plasma was thawed at 37 °C and centrifuged at 12,000 rpm for 5 min and collect the supernatant. Soak the dialysis membrane strips in distilled water and ethanol for 1 h and 20 min, respectively, and then rinse the membrane strips in distilled water three times to pretreatment the membrane strips. Spike 20 µL of test solutions containing the compound or the reference compound (final test concentration 1 µM) into the pre-loaded plasma in the 96-well plate. Apply aliquots of 100 µL of blank dialysis buffer to the receiver side of dialysis chambers. Then apply aliquots of 100 µL of the plasma spiked with test and reference compounds to the donor side of the dialysis chambers. Aliquot 25 µL of the plasma spiked with test and reference compounds into a 96-well sample preparation plate as T0 samples. The entire dialysis apparatus was placed in a shaker (600 rpm) at 37 °C for 5 h. Samples from the specified time point of 0–5 h were collected for LC/MS analysis.
Human CYP450 inhibition test of SY110
Preheat 0.1 M pH 7.4 K/Mg-buffer at 37 °C. The reference inhibitors for this assay are α-Naphthoflavone (1A2), Ticlopidine (2B6), Montelukast (2C8), Sulfaphenazole (2C9), Omeprazole (2C19), Quinidine (2D6), Ketoconazole (3A4). Test compound and reference inhibitors were dissolved in ACN. NADPH cofactor was dissolved in K/Mg-buffer. Each CYP450 isoform was prepared 2 mL substrate (1A2: Phenacetin, 2B6: Amfebutamone HCl, 2C8: Paclitaxel, 2C9: Diclofenac, 2C19: S-Mephenytoin, 2D6: Dextromethorphan, 3A4: Midazolam and Testosterone). Add 400 μL 0.2 mg/mL HLM and 2 μL of test compound set (serially diluted) to the assay wells. And then add 30 μL HLM-compound mixture and 15 μL substrate solution in the 96-well assay plate on ice. Then incubate the 96-well assay plate at 37 °C for 5 min. Add 15 µL of pre-warmed (37 °C) 8 mM NADPH solution to initiate the enzyme reaction. Incubate the assay plate at 37 °C for 5–45 min and quench the reaction. Then collect samples for LC/MS analysis.
In vivo toxicity study
Male and female ICR mice (6–8 weeks, 16–25 g) were used to assess the in vivo toxicity of SY110. SY110 were dissolved in 5:40:3:52 DMSO/PEG400/Solutol HS-15/saline. The specific information is showed in Supplementary Table S9. During the experiment, monitor the toxic signs (behaviors, food intake and weight) of the animals at least twice a day. At the last day, samples of visceral tissue were collected and analyzed by histological examine.
Embryo-fetal development study of SY110
SD rats (9–14 weeks and 230–320 g at study beginning) were randomly assigned to dose groups (at least seven successful mating animals in each group). Rats were administered SY110 (100, 300, or 1000 mg/kg, po) or vehicle from gestation day (GD) 6 through day 17. According to the ICH guidelines, the limit dose of SY110 is 1000 mg/kg/day. Observe the clinical signs, gestation body weights, weight changes and food consumption of animals during the study. On GD 20, the rats were weighed, euthanized by CO2 inhalation, exsanguinated, and necropsied.
In vitro antiviral screening
VeroE6-TMPRSS2 cells were infected with Omicron (B.1.1.529) BA.1 subvariant at 0.1 MOI and treated with 3r, 3u, 3t, 3v, 3w (SY110), 3× or Nirmatrelvir. Cell lysates were collected and extracted for RNA by RNeasy Mini kit (Qiagen) at 24 hpi. Quantify viral genome copies with one-step RT-qPCR kit (Qiagen) and Real-time PCR system (LightCycler480, Roche).63
Plaque reduction assay
Cells (VeroE6-TMPRSS2) were infected with MERS-CoV, SARS-CoV-1, SARS-CoV-2 Alpha (B.1.1.7), Beta (B.1.351), Omicron (B.1.1.529) BA.2 and BA.5 sublineages at 50-70 PFU. After 2 h inoculation, gently wash the cells with PBS 3–5 times. Then use 2 × DMEM/2% FBS-2% agarose/PBS mixture cover the cells followed by treatment with SY110 or Nirmatrelvir which were fivefold serially diluted from 20 µM to 0.0013 µM at 2 hpi.. At 48–72 hpi., cells were fixed and stained for 10 min by crystal violet.
In vivo SARS-CoV-2 infection
Female or male K18-hACE2 transgenic mice (6–8 weeks, The Jackson Laboratory) were used for the in vivo antiviral study. Mice were inoculated with Omicron BA.2 subvariant (2000 PFU) by intranasally. For early therapeutic treatment, mice were treated with SY110 (150 mg/kg, po, bid) from the day of infection until 4 dpi. For late therapeutic treatment, mice were administered with Nirmatrelvir (150 mg/kg, po, bid), SY110 (150 mg/kg, po, bid) with or without co-administration of Ritonavir (10 mg/kg, po, bid) until 4 dpi. Mice treated with vehicle (5% DMSO/3% Solutol HS-15/40% PEG400/normal saline) were included as controls. Mice were sacrificed at 4 dpi. and lung tissues were sampled for virological and histopathological analyses as previously described.64
RNA extraction and quantitative RT-PCR
Lung samples were homogenized using the TissueLyzer II (Qiagen). After lysed by RLT buffer (Qiagen), tissue lysates were extracted with the RNeasy Mini kit (Qiagen) following the manufacturer’s protocol. RNA was extracted, and RT-qPCR analysis was determined using the one-step RT-qPCR Kit (Qiagen) on the Real-Time PCR System (LightCycler 480, Roche).65
Plaque assays
Cells (VeroE6-TMPRSS2) were grown in 12-well plates overnight. Next day, cells were incubated with serially diluted samples for 1 h at room temperature. Then cells were washed 3–5 times by PBS and covered with 2 × DMEM/2% FBS, 2% agarose / PBS mixture at 1:1 ratio. The cells were incubated with the mixture at 37 °C for 72 h. After fixation, 10 min was stained with 25% (v/w) ethanol/ water by 0.5% crystal violet to show plaques.45
H&E staining and immunohistochemistry assays
The assays were conducted using previous protocols.43 Lung simples of the infected animals were fixed with 10% neutral-buffered paraformaldehyde solution and processed with semi-automatic tissue processor (Leica, Germany). Processed tissues are embedded with paraffin and cut at 5 μm thickness. Dewaxed and rehydrated tissues were treated for antigen exposure before being incubated with the rabbit anti-SARS-CoV-2-N immune serum (in-house) at 4 °C overnight. Next day, tissues were treated with the goat anti-rabbit IgG antibody (biotinylated, BA-1000-1.5, Vector aboratories) at room temperature for 1 h. Then DAB (3,3’-diaminobenzidine) substrate kit (Vector Laboratories) was used for visualization of signal. Tissue slides were counterstained with Gill’s Hematoxylin for visualization of the cell nuclei. For H&E staining, lung tissue slides were stained with Gill’s hematoxylin (Vector Laboratories) and eosin Y (Thermo Fisher Scientific). Images were obtained with the inverted light microscope (BX53, Olympus Life Science). Images shown represented unbiased description for the pathological damage in the lung which have been confirmed by an experienced pathologist.
Statistical analysis
All data were represented as means and standard deviations and analyzed with GraphPad Prism 8.0 software. Statistical comparison between different groups was performed by one-way ANOVA, and details are showed in the figure legends, respectively (Figs. 3, 6). Differences were considered statistically significant when p < 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Data availability
All data related to this research is listed in the main text or Supplementary Materials, and are available from the corresponding authors (S.Y., J.L., or H.C.) upon reasonable request. The coordinates and structure factors of SARS-CoV-2 Mpro in complex with Hit-1 and SY110 have been deposited into PDB with accession numbers 8HHT and 8HHU, respectively.
References
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020).
Planas, D. et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 596, 276–280 (2021).
Lopez Bernal, J. et al. Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 385, 585–594 (2021).
Tuekprakhon, A. et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 185, 2422–2433.e13 (2022).
Cao, Y. et al. BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 608, 593–602 (2022).
Andrews, N. et al. Covid-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 386, 1532–1546 (2022).
Garcia-Beltran, W. F. et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 185, 457–466.e4 (2022).
Mannar, D. et al. SARS-CoV-2 Omicron variant: antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 375, 760–764 (2022).
Liu, L. et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 602, 676–681 (2022).
Iketani, S. et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 604, 553–556 (2022).
Saad-Roy, C. M. et al. Immune life history, vaccination, and the dynamics of SARS-CoV-2 over the next 5 years. Science 370, 811–818 (2020).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Khailany, R. A., Safdar, M. & Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 19, 100682 (2020).
Hegyi, A. & Ziebuhr, J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 83, 595–599 (2002).
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300, 1763–1767 (2003).
Hilgenfeld, R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 281, 4085–4096 (2014).
Lu, L., Su, S., Yang, H. & Jiang, S. Antivirals with common targets against highly pathogenic viruses. Cell 184, 1604–1620 (2021).
Wu, C. R., Yin, W. C., Jiang, Y. & Xu, H. E. Structure genomics of SARS-CoV-2 and its Omicron variant: drug design templates for COVID-19. Acta Pharmacol. Sin. 43, 3021–3033 (2022).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368, 409–412 (2020).
US Food and Drug Administration (FDA), Coronavirus (COVID-19) Update: FDA Issues Emergency Use Authorization for Potential COVID-19 Treatment (2022); https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment.
U.S. Food and Drug Administration (FDA), Coronavirus (COVID-19) Update: FDA Authorizes Additional Oral Antiviral for Treatment of COVID-19 in Certain Adults (2022); https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-additional-oral-antiviral-treatment-covid-19-certain.
US Food and Drug Administration (FDA), Coronavirus (COVID-19) Update: FDA Authorizes First Oral Antiviral for Treatment of COVID-19 (2022); https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19.
Ministry of Health, Labour and Welfare of Japan (MHW): about urgent approval of new-style coronavirus medicine. (2022); https://www.mhlw.go.jp/stf/newpage_29320.html.
China National Medical Products Administration (NMPA): domestically developed drug joins virus battle (2022); http://english.nmpa.gov.cn/2022-08/15/c_797867.htm.
China National Medical Products Administration (NMPA): NMPA conditionally approved the marketing of the combined packaging of Simnotrelvir Tablets/Ritonavir Tablets and Renmindevir Hydrobromide Tablets for COVID-19 treatment (2023); https://www.nmpa.gov.cn/zhuanti/yqyjzxd/yqyjxd/20230129143814163.html.
Williamson, B. N. et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 585, 273–276 (2020).
Wahl, A. et al. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature 591, 451–457 (2021).
Xie, Y. et al. Design and development of an oral remdesivir derivative VV116 against SARS-CoV-2. Cell Res. 31, 1212–1214 (2021).
Zhang, J. L. et al. Azvudine is a thymus-homing anti-SARS-CoV-2 drug effective in treating COVID-19 patients. Signal Transduct. Target Ther. 6, 414 (2021).
Owen, D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Unoh, Y. et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 65, 6499–6512 (2022).
Yan, V. C. & Florian, L. M. Why remdesivir failed: preclinical assumptions overestimate the clinical efficacy of remdesivir for COVID-19 and Ebola. Antimicrob. Agents Chemother. 65, e0111721 (2021).
Heilmann, E. et al. SARS-CoV-2 3CLpro mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Sci. Transl. Med. 15, eabq7360 (2022).
Noske, G. D. et al. Structural basis of nirmatrelvir and ensitrelvir resistance profiles against SARS-CoV-2 Main Protease naturally occurring polymorphisms. J. Biol. Chem. 299, 103004 (2023).
Zhou, Y. et al. Nirmatrelvir resistant SARS-CoV-2 variants with high fitness in vitro. Sci Adv. 8, eadd7197 (2022).
Jochmans, D. et al. The substitutions L50F, E166A and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. mBio 14, e0281522 (2023).
Hu, Y. et al. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. bioRxiv 497978 (2022); https://doi.org/10.1101/2022.06.28.497978.
Iketani, S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564 (2023).
Moghadasi, S. A. et al. Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. bioRxiv 503099; https://doi.org/10.1101/2022.08.07.503099.
Sasi, V. M. et al. Predicting antiviral resistance mutations in SARS-CoV-2 main protease with computational and experimental screening. Biochemistry 61, 2495–2505 (2022).
Dai, W. et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331–1335 (2020).
Qiao, J. et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 371, 1374–1378 (2021).
Sacco, M. D. et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 6, eabe0751 (2020).
Chan, J. F. et al. Virological features and pathogenicity of SARS-CoV-2 Omicron BA.2. Cell Rep. Med. 3, 100743 (2022).
Quan, B. X. et al. An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbiol. 7, 716–725 (2022).
Shuai, H. et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 603, 693–699 (2022).
Halfmann, P. J. et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature 603, 687–692 (2022).
Suzuki, R. et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 603, 700–705 (2022).
World Health Organization (WHO), Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern (2021); https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern.
World Health Organization (WHO), Update 80: What we know about new COVID-19 Variants of Concern—the latest on the global situation & Omicron BA.4/5 (2021); https://www.who.int/publications/m/item/update-80-covid-19-omicron-ba-4-5-update.
Mohapatra, R. K. et al. Twin combination of Omicron and Delta variants triggering a tsunami wave of ever high surges in COVID-19 cases: a challenging global threat with a special focus on the Indian subcontinent. J. Med. Virol. 94, 1761–1765 (2022).
Jin, Z. et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 27, 529–532 (2020).
Fu, L. et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 11, 4417 (2020).
Rathnayake, A. D. et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl. Med. 12, eabc5332 (2020).
Han, S. H. et al. Structure-based optimization of ML300-derived, noncovalent inhibitors targeting the severe acute respiratory syndrome coronavirus 3CL protease (SARS-CoV-2 3CLpro). J. Med. Chem. 65, 2880–2904 (2022).
Pfizer Laboratories. 2022. Fact sheet for healthcare providers: emergency use authorization for Paxlovid. https://labeling.pfizer.com/ShowLabeling.aspx?id=16474.
Kabsch, W. XDS. Acta Crystallogr. D. Biol. Crystallogr 66, 125–132 (2010).
Evans, P. R. et al. How good are my data and what is the resolution? Acta Crystallogr. D. Biol. Crystallogr. 69, 1204–1214 (2013).
Afonine, P. V. et al. Towards automated crystallographic structure refnement with phenix.refne. Acta Crystallogr. D. Biol. Crystallogr. 68, 352–367 (2012).
Bricogne, G. et al. BUSTER version 2.10.3. Global Phasing Ltd (2017).
Emsley, P. et al. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Chu, H. et al. Host and viral determinants for efficient SARS-CoV-2 infection of the human lung. Nat. Commun. 12, 134 (2021).
Shuai, H. et al. Emerging SARS-CoV-2 variants expand species tropism to murines. EBioMedicine 73, 103643 (2021).
Hu, B. et al. Spike mutations contributing to the altered entry preference of SARS-CoV-2 Omicron BA.1 and BA.2. Emerg. Microbes Infect. 11, 2275–2287 (2022).
Acknowledgements
We thank Dr. Ana Gonzalez and Dr. Jie Nan at BioMAX beamline (Max IV Laboratory, Lund, Sweden), the staff of beamline BioMax (MAX IV Laboratory, Sweden) and Shanghai synchrotron radiation facility (SSRF) beamlines (Shanghai, China), and Qianlun Pu of the Core Facilities of West China Hospital (Sichuan, China) for great supports. We thank Prof. Minghai Tang for LC-MS analysis. This research was supported by National Natural Science Foundation of China [82130104, 81930125, T2221004 (S.Y.), and 22107081 (B.Q.)], National Key R&D Program of China [2022YFC2303701 and 2021YFF0702004 (J.L.)], 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University [ZYXY21001 (S.Y.) and ZYYC21008 (J.L.)], the fast-track grants of SARS-CoV-2 research, West China Hospital, Sichuan University [HX-2019-nCoV-053 (S.Y.)], National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University [Z2021JC008 (J.L.)], National Natural Science Foundation of China Excellent Young Scientists Fund (Hong Kong and Macau) [32122001 (H.C.)]; the Health and Medical Research Fund [CID-HKU1-5, COVID1903010-14, and 20190652 (H.C.)], the Food and Health Bureau, The Government of the Hong Kong Special Administrative Region; the General Research Fund [17118621, 17123920 and 17119122 (H.C.)] of Research Grants Council, the Government of the Hong Kong Special Administrative Region; the National Program on Key Research Project of China [2020YFA0707500 and 2020YFA0707504 (H.C.)].
Author information
Authors and Affiliations
Contributions
S.Y., H.C., and J.L. conceived the research; S.Y. and C.H. performed the drug design; C.H., A.X., and Z.F. with the assistance of Q.H., J.Y., B.Q., N.G., S.Z., R.M., J.Z., R.Y., S.Z., J.N., H.X., and F.W. performed the chemical synthesis; R.Z., L.X., J.Q., and X.Z. performed gene expression, protein purification and crystallization experiments; R.Z., L.X., and J.L. determined the crystal structures; J.Q. and Y.L. performed high-throughput screening, enzymatic activity and inhibition assays, IC50 measurements, DSF assays, cytotoxicity assays and in vivo toxicity assays; H.S., Y.H., C.Y., and B.H. performed in vitro and in vivo antiviral assays; S.Y., J.L., and H.C. with the assistance of C.H., H.S., J.Q., and R.Z. wrote and revised the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
All authors declared no competing interests. S.Y. is the member of editorial board, he has not been involved in the process of the manuscript handling.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, C., Shuai, H., Qiao, J. et al. A new generation Mpro inhibitor with potent activity against SARS-CoV-2 Omicron variants. Sig Transduct Target Ther 8, 128 (2023). https://doi.org/10.1038/s41392-023-01392-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01392-w
This article is cited by
-
Molecular mechanism of ensitrelvir inhibiting SARS-CoV-2 main protease and its variants
Communications Biology (2023)
-
Pathogen evolution, prevention/control strategy and clinical features of COVID-19: experiences from China
Frontiers of Medicine (2023)
