
Abstract
Despite the success of targeted therapies in cancer treatment, therapy-induced resistance remains a major obstacle to a complete cure. Tumor cells evade treatments and relapse via phenotypic switching driven by intrinsic or induced cell plasticity. Several reversible mechanisms have been proposed to circumvent tumor cell plasticity, including epigenetic modifications, regulation of transcription factors, activation or suppression of key signaling pathways, as well as modification of the tumor environment. Epithelial-to-mesenchymal transition, tumor cell and cancer stem cell formation also serve as roads towards tumor cell plasticity. Corresponding treatment strategies have recently been developed that either target plasticity-related mechanisms or employ combination treatments. In this review, we delineate the formation of tumor cell plasticity and its manipulation of tumor evasion from targeted therapy. We discuss the non-genetic mechanisms of targeted drug-induced tumor cell plasticity in various types of tumors and provide insights into the contribution of tumor cell plasticity to acquired drug resistance. New therapeutic strategies such as inhibition or reversal of tumor cell plasticity are also presented. We also discuss the multitude of clinical trials that are ongoing worldwide with the intention of improving clinical outcomes. These advances provide a direction for developing novel therapeutic strategies and combination therapy regimens that target tumor cell plasticity.
Similar content being viewed by others

Introduction to tumor cell plasticity (TCP)
Cellular plasticity is a fundamental feature of cells. During development, cells progressively differentiate into different hierarchies and obtain specific developmental commitments. Such processes are reversible, and the phenotypic changes can be diversely directed as needed. Genetic lineage tracing has demonstrated that functionally mature cells can undergo dedifferentiation (lineage reversion), transdifferentiation (cells converting into other distant lineages), or transdetermination (cells switching lineage commitment to a closely related cell type) upon injury or disease.1,2 As far back as 1956, Conrad Waddington stated that during development embryonic progenitor cells progressively differentiate into distinct cell types and are increasingly restricted within a specific lineage.3 The final specialized cells are then integrated into their respective tissues and conduct their specific invariable functions. However, later studies subverted this paradigm. Han Driesch isolated from a sea urchin embryo a single blastomere in an early multicellular state could develop into an entire larva.4 This finding suggested that partially developed cells are not fully determined but retain the potency to produce all types of cells found in an entire organism.
In fact, the states of cells are quite dynamic, especially when injury or disease occurs. An equilibrium between new cell generation and old cell loss is generally maintained in multicellular organisms. If perturbed by unexpected loss, stem cell replication and differentiation along with transformation of mature differentiated cells can restore equilibrium. For example, endothelial cells assume arterial, venous, or lymphatic identity in an early developmental stage. However, upon injury, endothelial cells can undergo lineage changes to become hematopoietic progenitor or cardiac valve cells.5,6,7 In liver homeostasis, the modest proliferation of hepatocytes in all zones contributes to the homeostatic hepatocyte pool, rather than a rare type of liver progenitor cell (LPC) subpopulation.8,9 In a genetic mouse model which mimics human Alagille syndrome and contributes to biliary epithelium regeneration, hepatocytes are able to transdifferentiate into biliary epithelial cells.10 Additionally, cellular plasticity can be “artificially” enhanced, as demonstrated by induced pluripotent stem cells (iPSCs), which were first generated in 2007,11 providing unprecedented opportunities for novel therapies.
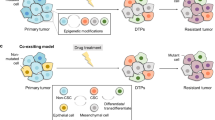
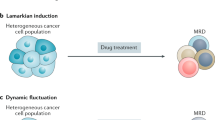
Cellular plasticity also contributes to pathological conditions. Similar to the cell plasticity observed during post-injury repair and regeneration, plasticity in tumor cells promotes survival against the immune attack and anti-tumor treatments. A major obstacle preventing complete remission, this evasion to therapy occurs as cancer cells play “hide and seek” by constant phenotypic switching. Some regard this phenotypic switching of cancer cells to result from passive Darwinian selection and enrichment upon drug exposure,12,13 while others argue it is an active adaptation to internal or external stimuli (Lamarckian induction).14 A coexisting model has also been formulated to illustrate that these two concepts complement and intertwine with each other (Fig. 1).14,15,16 The “hide and seek” behavior of cancer cells is known as TCP, which is a process that re-activates various developmental programs to achieve phenotypic switching. TCP enables transformation of tumor cell identity that allows the cells to adapt to different microenvironments and circumvent the drug-targeted pathway. This subpopulation of tumor cells is characterized by its dormant and stem-like trait and termed drug-tolerant persisters (DTPs).15 The “acquired inertia” of DTPs is regarded as a survival skill exposed by antitumor treatment that can fade away after a drug-free interval.17 Thus, with TCP, a reversible switching of phenotypes can be achieved between proliferative, metastatic tumor cells and dormant, drug-indifferent DTPs, thereby hindering the desired outcome of targeted therapies. By this universally plastic behavior, tumor cells transform in different but functionally overlapping processes, including epithelial-mesenchymal transition (EMT), transdifferentiation, and acquiring properties of cancer stem cells (CSCs).14,18,19

Changes in acquired therapy resistance induced by treatment initiation and discontinuation, with different scenarios employed to interpret the development of the therapy-resistant phenotype. According to the Darwinian selection model, drug-tolerant persistors (DTPs) that originated from the primary tumor tissue are selected and enriched by treatments. The Lamarckian induction model regards the acquired drug-indifferent phenotype as the result of tumor cell adaptation to treatments that leads to formation of induced DTPs. The coexisting model suggests that formation of both primary and induced DTPs occur and together contribute to the acquired therapy resistance
Epithelial–mesenchymal transition
EMT is a process by which tumor cells lose their epithelial features and acquire mesenchymal characteristics. EMT is characterized by loss of apical-basal polarity and disruption of intercellular interactions, accompanied by the acquisition of front-rear polarity and remodeling of the cytoskeleton.20,21 On the other hand, mesenchymal-epithelial transition (MET) is the inverse process of EMT. EMT and MET are thought to be regulated mainly at the transcriptional level by the activity of EMT transcription factors (EMT-TFs), including Snail, Slug, Zeb1/2, Twist, and microRNAs (miRs). Signaling pathways such as transforming growth factor (TGF-β), wingless/integrated (WNT), NOTCH, and HIPPO are also involved.15,22 Two double-negative feedback loops that include Snail/miR-34 and Zeb/miR-200 help to manipulate the initiation, transition, and maintenance of EMT program.23,24 Genetic mutations may also play a role in EMT regulation. As indicated by Martin et al.,25 a combined loss of the tumor suppressor gene P53 and PTEN in clonal prostate epithelial cells can lead to transformation of multipotent progenitors and ultimately EMT. Yamasaki et al.26 recently proposed that Kirsten rat sarcoma viral oncogene homolog (KRAS) mutation can also promote an EMT-like phenotypic change.
Both EMT and MET have been proposed to play key roles in cancer progression.21 Apart from a complete EMT/MET transition, tumor cells can exhibit a “partial EMT” state, a hybrid of epithelial and mesenchymal phenotypes.27 In fact, tumor cells can exhibit a spectrum of EMT states characterized by varying proportions of epithelial and mesenchymal traits, both in circulation and at a secondary site.28,29 Panchy et al.30 recently confirmed this epithelial-mesenchymal spectrum using transcriptomic analysis. This hybrid epithelial-mesenchymal state enables tumor cells to self-adapt and adjust quickly to changing environments, thus representing an ideal mechanism of TCP.28 Previous studies indicate that dynamic changes between epithelial and mesenchymal phenotype in tumor cells with partial EMT are critically important to the metastatic potential and therapy resistance of tumor cells.31
The contributions made by EMT programs to metastasis were first proposed by Yang et al.,32 who described that inhibition of Twist, a classic EMT-TF, can significantly restrain highly metastatic mammary carcinoma cells from metastasizing to the lung. The impact of other EMT-TFs on metastasis was then extensively investigated in different types of cancers.33 For example, depletion of Zeb1 was found to markedly inhibit pancreatic ductal adenocarcinoma progression towards metastatic type.34 Zeb1 was also found to function as a key master regulator of multiple EMT-TFs and metastasis in breast cancer.33 The MET program, in contrast, presumably functions during metastasis when the migratory CSCs settle down and adapt to a new microenvironment. In the process of colonization, the migratory CSCs regain self-renewal capacity and spawn fast-cycling epithelial progeny, thereby restoring epithelial characteristics.35 Such reversible EMT-MET programs seem to be a common feature shared by different cancer types during the seeding of a secondary tumor.35,36,37
Evidence also suggests that EMT can serve as a survival mechanism for cancer cells to escape immunosurveillance and resist therapy-induced death.38 Multiple studies have observed a strong correlation between the level of mesenchymal cell markers and therapeutic resistance.21,39,40,41,42 For example, Farmer et al.42 demonstrated that an upregulation of stromal gene expression predicted resistance to preoperative chemotherapy with 5-fluororacil, epirubicin, and cyclophosphamide in estrogen receptor (ER)-negative breast cancer. In ovarian cancer, inhibition of EMT can effectively eliminate cisplatin resistance.43 Byers et al.40 revealed a correlation between a 76-gene mesenchymal signature and non-small-cell lung cancer (NSCLC) resistance to epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) and phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) inhibitors. By proteomic screening, Paulitschke et al.41 also found that two EMT-related markers are effective in determining drug susceptibility in melanoma cells. Although the mechanisms underlying the therapeutic resistance conferred by EMT remain unclear, several hypotheses have been put forward, including reduced levels of proapoptotic proteins, increased drug efflux, and immunosuppressive tumor microenvironment (TME).44,45,46
Transdifferentiation
Transdifferentiation, also known as lineage plasticity or lineage switching, is a radical shift in cell identity that leads to cells acquiring new transcriptional or morphological characteristics.47,48 In the context of cancer, such lineage plasticity may serve as a promising therapeutic approach. For example, melanoma spheroid cells are able to transdifferentiate into benign cell lineages including melanocytes, adipocytes, osteocytes and chondrocytes.49 However, transdifferentiation in cancer cells can also be a double-edge sword. Transdifferentiation observed in basal cell carcinoma (BCC) contributes to BCC relapse following treatment discontinuation.50,51 When treated with vismodegib, some BCC cells transitioned from a phenotype featuring a transcriptional bulge-like signature to a mixed isthmus/interfollicular epidermis cell phenotype, thereby achieving drug resistance.52 A discontinuation of treatment led to tumor cell reactivation and resumption of proliferation.52,53 Another distinctive form of transdifferentiation is characterized by a drug-induced neuroendocrine transdifferentiation (NET). Observed in prostate cancer (PC) and lung adenocarcinoma (LUAD), NET induces drug resistance, thereby causing treatment failure and relapse.14,54,55 Here, we consider PC and LUAD as two typical examples of NET for further discussion.
Through NET, PC tumor cells gradually obtain the histomorphology of small cell prostate cancers (SCPC) that are “therapy indifferent” to androgen receptor signaling inhibitors (ARSi).54,56 The switch from PC to SCPC is characterized by the presence of neuroendocrine markers (chromogranin A, synaptophysin, and others), and the absence of androgen receptor (AR) and serum prostate-specific antigen.57 Functionally, SCPC cells have an elevated metastatic potential, enhanced stemness, heightened therapeutic resistance, and a worse prognosis.18,58 Inactivation of TP53, RB1, PTEN, and amplification of MYCN have been reported to be involved at the molecular level of the cell identity shift.18,59 Mutations in these candidate genes provide a specific genomic background that enables epigenetic regulation towards tumor plasticity.48,60
Moreover, epigenetic events are also indispensable. NET of PC cells towards SCPC is associated with the well-established undifferentiated cell marker SRY-Box Transcription Factor 2 (SOX2), epigenetic regulator histone methyltransferase enhancer of zeste homolog 2 (EZH2), the catalytic subunit of the polycomb repressive complex 2 (PRC2), and RE1-silencing transcription factor (REST).14,18,61,62,63,64 A recent study by Cyrta et al.65 suggested that the switch/sucrose non-fermentable (SWI/SNF) chromatin remodeling complexes may also be a potential regulator of SCPC lineage plasticity. Correspondingly, SCPC can be reverted to its original lineage by the re-establishment of TP53 and RB1 as well as re-exposure to androgens, or inhibition of SOX2, thus re-sensitized to ARSi treatment.61,62 This reversibility of lineage plasticity could be of critical importance as a future target in PC therapy.
Like SCPC, LUAD undergoes NET through LUAD – small-cell lung cancer (SCLC) transition, which enables the tumor to escape from EGFR-TKI treatment and gain a higher level of malignancy.48,54,66 Although NET is also identified in non-EGFR-mutant NSCLC, EGFR mutant LUADs are more likely to switch lineage.67 Oser et al.68 reported that 5–15% of EGFR mutant LUAD patients with TKI resistance showed small cell histology in relapsed tumors, and became sensitive to SCLC chemotherapy.48 The mechanism of LUAD NET at the molecular level is similar to that of PC NET. LUADs with loss of TP53 and RB1 expression are more likely to transdifferentiate into SCLC tumors, which suggests that these two genes may be critical in promoting transdifferentiation towards a neuroendocrine identity.60,68,69,70 The epigenetic regulator EZH2 is also regarded as a potential therapeutic target to reverse NET.71,72
PC and LUAD are currently the most extensively studied cancers exhibiting NET; however, transdifferentiation of epithelial cells is more common. According to a recent study, many epithelial cancers, including bladder, endometrial, and gastric cancer with neuroendocrine gene expression signatures tend to exhibit high-grade tumors and poor prognosis.66 This finding indicates that a latent neuroendocrine lineage plasticity may occur in multiple tissues.
Cancer stem cells
Accumulating evidence suggests that the existence of CSCs within tumor tissues is responsible for tumor initiation and intertumoral heterogeneity. CSCs have the potential to proliferate infinitely, self-renew, and differentiate.73,74 A recent study proposed that a CSC is a highly plastic state that results in fast adaptation of tumor cells to varying conditions in the TME.75 Previous studies revealed CSCs and adult stem cells share multiple signatures, including the surface markers CD133, CD44, CD24, CD26, CD166, and EPCAM, and intracellular proteins like ALDH1.76,77,78 Additionally, CSCs and stem cells also share similar developmental related signaling pathways, such as WNT, NOTCH, and Hedgehog (Hh).29,79
Like normal stem cells, CSCs propagate through asymmetric division, which produces one daughter cell that remains a CSC and one committed progenitor cell that differentiates into a non-CSC.80,81 Symmetric division can also occur simultaneously, with the production of either two CSCs (symmetric renewal) or two committed cells (symmetric commitment).80,82 The transition between CSC and non-CSC states seems irreversible and dynamically balanced. Under certain circumstances, however, the balance can shift in either direction. A shift towards renewal results in replenishment of the CSC pool and consequently produces a more aggressive, undifferentiated phenotype. A shift towards non-CSCs suggests tumor-formation.29 Equilibrium represents the plasticity of tumor cell fate and can be manipulated by various influencing factors. For example, a second mutation introduced in chronic-phase chronic myeloid leukemia (CML) has been found to trigger an imbalance that favors symmetric renewal and results in higher malignancy.83 EMT-TF Twist2 has been reported to induce stemness properties in lung CSCs by moving the balance towards symmetric division, while repressing asymmetric division.84 Other epigenetic regulations, including deoxyribonucleic acid (DNA) methylation, histone modification, and bivalent chromatin state, have been found to contribute to CSC plasticity.78,85 These studies indicate that manipulation of CSC division may be promising for cancer control.
CSCs and EMT share a high degree of consistency and may be a partially overlapping concept.86,87 As mentioned, the EMT program largely facilitates tumor cell dissemination. CSCs are also enriched in disseminated tumor cells and feature EMT expression signatures.88 Circulating tumor cells express a remarkably congruent transcriptomic profile with CSCs and have been found to overexpress both EMT and stem cell markers.89 CSCs led to therapeutic resistance through various mechanisms. For example, CSCs can specifically inherit effective drug-discharging ATP-binding cassette (ABC) transporters from asymmetrical division, which enable tumors to sustain long-term expansion.90,91 CSCs can also facilitate efficient DNA damage.92,93,94 Another mechanism of therapeutic resistance in CSCs is the formation of a protective niche, an immunosuppressive microenvironment characterized by low infiltration of immune cells and downregulated signaling by the programmed cell death ligand 1(PD-L1).79,95 Non-CSCs also help to maintain of a pool of CSCs by secreting supportive signals such as WNT and brain-derived neurotrophic factor (BDNF), as well as supportive factors such as interleukin (IL)-6 and IL-8.96,97,98
Mechanism of therapy-induced TCP
Various therapies have been developed to prolong survival and improve the quality of life of cancer patients, yet therapy-induced drug resistance remains a major challenge to achieving the desired clinical outcomes. Notably, up to 90% cancer-associated deaths are attributed to drug resistance.99 Acquired drug resistance can be induced by targeted treatments and is associated with TCP, the “hide-and-seek” behavior of cancer cells. The drug-resistant phenotype of cancer cells is not heritable but is a transient and reversible process.16 Therefore, a comprehensive understanding of the mechanism of such induced plasticity should greatly facilitate the development of novel therapies. Epigenetic alterations, transcription factors, key signaling pathways, and TME can each potentially contribute to the therapy-induced plasticity in multiple cancers.
Epigenetic mechanisms
Therapy-induced epigenetic plasticity largely contributes to the reprogramming of tumor cells and phenotypic switching towards therapy resistance. The epigenetic modifications can be either chemical (DNA methylation and histone modification) or structural (chromatin remodeling and inter/intrachromosomal interactions)100,101 (Figs. 2a and 3).

Therapeutic resistance involvement in epigenetic modifications and EMT/MET. a Schematic representation of therapy-induced drug resistance through chemical epigenetic modifications. Four core histone proteins (H2A, H2B, H3, and H4) can be diversely modified by multiple enzymes that result in methylation, acetylation, phosphorylation, or ubiquitylation. DNA can be methylated at the 5-carbon of the cytosine base to form 5-methylcytosine (5-mC), which can be further oxidized to form 5-hydroxymethyl cytosine (5-hmC). Enzymes involved in the epigenetic modifications of either histone or DNA can be utilized as potential future therapeutic targets against tumor cell plasticity. K, Lysine; S, Serine; R, Arginine; T, Threonine; me, methylation; ac, acetylation; ub, ubiquitination; P, phosphorylation; HAT, histone acetyltransferase; HMT, histone methyltransferase; HDAC, histone deacetylase; HDM, histone demethylase; TRIM, tripartite mortif; MSK1, mitogen- and stress-activated protein kinase 1; DNMT, DNA methyltransferase; TET, ten-eleven translocation enzymes. b Primary tumor cells undergo EMT and MET to achieve metastasis. Primary tumor cells undergo EMT by losing epithelial traits and acquiring mesenchymal characteristics, which enables tumor cells to migrate and invade into blood or lymphatic vessels. In circulation, tumor cells exhibit a “partial EMT” within the epithelial-mesenchymal spectrum. During colonization, migratory tumor cells with mesenchymal traits undergo MET to restore epithelial characteristics and proliferate to form a secondary tumor. Therapeutic resistance and invasiveness are higher in mesenchymal states, whereas a hybrid state of epithelial and mesenchymal indicates the highest level of stemness and capacity of self-adaptation. The process of EMT/MET is regulated by crosstalk among genomic factors, MET-TFs, and multiple signaling pathways. EM, epithelial-mesenchymal transition, MET mesenchymal-epithelial transition, TF transcription factor, miR microRNA

Mechanisms driving tumor cell plasticity and drug resistance. Both tumor microenvironment factors and EMT-related signaling pathways regulate the EMT process. The activation of EMT facilitates the dedifferentiation of non-cancer stem cells (CSCs) to CSC, which contributies to tumor heterogeneity
DNA methylation is widespread in the human genome and functions differently based on various methylated regions.99 The aberrant methylation status of gene promoters plays an instrumental role in acquired drug resistance in a variety of tumor types.102,103 The most common mechanisms of drug resistance include hypomethylation of gene promoters of drug efflux, hypermethylation of promoters of pro-apoptotic genes, and altered methylation levels of DNA-repair genes.99 Global loss of DNA methylation and histone acetylation has been observed in therapy-induced drug-resistant tumor cells under extended treatment with the neoadjuvant aromatase inhibitor (letrozole). Such epigenetic alterations result in tumor cells escaping from dormancy and acquiring resistance after targeted therapy in breast cancer patients.104
In contrast, indifference or resistance to endocrine therapy in ER-positive breast cancer is due to hypermethylation of estrogen-responsive enhancers.105 Moreover, effective therapies targeting the AR in PC inadvertently promote NET. The lineage switching into neuroendocrine PC was associated with increased DNA methylation, turning the original PC into a lethal, castration-resistant type. This process can be reversed by inhibition of DNA methyltransferase (DNMT).106 Histone modification, represented by euchromatin histone lysine methyltransferase 2 (EHMT2), has been implicated in acquired drug resistance following targeted therapy. EHMT2 overexpression was observed in erlotinib resistance in NSCLC, leading to transcriptional activation of PTEN and repressed Akt signaling, whereas inhibition of EHMT2 reverses the acquired indifference to therapy and re-sensitizes NSCLC cells to EGFR-TKI treatment.107 Histone modifications, either via EHMT2 or histone deacetylase 3 (HDAC3), can result in acquired resistance against mitogen-activated extracellular signal-regulated kinase inhibitors (MEKi) treatment in pancreatic ductal adenocarcinoma (PDAC).108
Structural epigenetic alterations, including changes in high-order structures of chromatin and interactions among distal regulatory elements, may also be involved in therapy-induced TCP. For example, targeted therapy using γ-secretase inhibitors in T cell acute lymphoblastic leukemias (T-ALL) can induce the expansion of a rare group of pre-existing persister cells. These persisters contain more compact chromatin and higher levels of repressive histone modifications compared to γ-secretase inhibitor-sensitive naïve cells.109 Similar to trametinib-resistant triple-negative breast cancer (TNBC), remodeling of enhancers and promoters was observed accompanied with adaptive transcriptomic responses.110
The role of transcription factors in controlling TCP
A network of transcription factors also plays an important role in TCP against targeted therapy. EMT-TFs like Twist and Snail actively respond to targeted therapy during the EMT process and result in enhanced plasticity and mesenchymal switching.111,112 In addition to multiple EMT-TFs, several other transcription factors function in controlling TCP.
AR is known as a ligand-activated transcription factor which regulates gene expression in the prostate epithelium.113 AR-targeted inhibitors, including bicalutamide and enzalutamide, have been developed for PC treatment.114,115 However, upon castration therapy, evading mechanisms are launched by AR amplification, point mutation, splicing variants, as well as substitution of AR functions by glucocorticoid receptors.116
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that has been studied extensively in multiple cancers. Previous studies have suggested that STAT3 activation is associated with trastuzumab as well as trastuzumab-emtansine resistance in human epidermal growth factor receptor 2 (HER2)-positive breast cancer.117,118 In triple-negative (ER-/PR-/HER2-) breast cancer (TNBC) patients, a strong transcriptomic response was observed after treatment with trametinib for seven continuous days, with up to 22% of the transcriptome either significantly up- or downregulated.110 The super transcription factor cellular MYC underwent rapid degradation during the treatment process and functioned as a controller of genomic reorganization and transcriptional complex recruitment in the phenotypic change of tumors.110,119
In melanoma, an assay for transposase-accessible chromatin using sequencing (ATAC-seq) revealed an initial loss of transcription factor binding sites, followed by an increase in accessible sites, during BRAF inhibition (BRAFi) treatment. These results indicated that targeted therapy altered transcription factors.120 According to a recent study by Yang et al.,121 activating transcription factor 4 (ATF4) stress signaling led to rapid tumor evasion within a few days after the initiation of mitogen-activated protein kinase (MAPK) pathway inhibition, and was accompanied by elevated levels of microphthalmia-associated transcription factor (MITF). This was followed by tumor cell dedifferentiation and a dormant phenotype marked with decrease in MITF expression. Additionally, the helix-loop-helix transcription factor inhibitor of DNA binding 3 (ID3) was upregulated after BRAFi therapy compared to that in the pretreatment state. ID3 acted as a transcriptional repressor of sex-determining region Y-box 10 (SOX10) and MITF, thereby promoting tumor cell transformation to a drug-resistant phenotype.122 Moreover, Sun et al.123 also observed an increase in the percentage of SOX10low EGFRhigh melanoma cells after BRAFi and MEKi treatment. This subset of tumor cells displayed a slow-cycling phenotype in response to these targeted drugs, highlighting the potential of transcription factors to mediate plasticity in melanoma.
The regulation patterns of the numerous known transcription factors are quite distinct in targeted therapy-induced tumor cell lines.120,124 This complexity indicates that tumors acquire resistance against targeted therapies in a patient- and drug-dependent manner. Hence, the transcription factors and their interaction modes remain to be explored.
Key signaling pathways in controlling TCP
Numerous signaling pathways are involved in embryonic development, and a re-activation of these signaling pathways is critical in tumor progression and therapy resistance. Interaction of these conserved signaling pathways (e.g., Hh, Wnt, and Notch pathways) and their crosstalk with other oncogenic pathways (e.g., Nuclear factor kappa B (NF-κB), PI3K/AKT/Mammalian target of rapamycin (mTOR), and MAPK pathways) contributes significantly to TCP by regulating the fluctuating expression of downstream effectors125,126,127,128 (Fig. 2b). Phenotypic switching driven by multiple signaling pathways is a huge challenge to successful tumor treatment.
Alterations of the Hh (Hedgehog) signaling pathway occur in one-third of cancers, and deregulation of the Hh signaling pathway plays an important role in therapeutic resistance.15,129,130,131,132 Aberrant activation of Hh signaling and subsequent upregulation of downstream effector glioma-associated transcription factor 2 (GLI2) was observed in bortezomib-resistant myeloma cells.133,134 Further studies demonstrated that Hh signaling is involved in CSC self-renewal in multiple myeloma, and blocking Hh signaling can lead to CSC differentiation.135 In TNBC, the Hh ligand produced by tumor cells can reprogram cancer-associated fibroblasts (CAFs) to promote a CSC-phenotype switching and the acquisition of chemo-resistant traits.136 Therefore, an effective Hh signaling pathway blockade is capable of lowering CSC proliferation, metastasis, and the EMT program, thereby controlling TCP.
Wnt signaling pathway is also highly associated with TCP. Wnt signaling is complex and can function through either an autocrine or a paracrine mode, by canonical (Wnt/β-catenin pathway) or non-canonical (the planar cell polarity pathway) pathways. Wnt signaling contributes to EMT regulation and TCP.137 In most cases, the Wnt/β-catenin signaling pathway is constitutively activated, which increases the expression of Snail, a regulator of EMT. Inhibition of the Wnt/β-catenin pathway inhibits EMT and tumor metastasis in a colorectal cancer model.138 Slug is another EMT modulator that is considered to be a downstream protein in Wnt/β-catenin-mediated tumor invasion. Moon et al. reported that concurrent expression of β-catenin and Slug predicts lymph node metastasis and survival rate in patients with head and neck squamous cell carcinoma.139 Overexpression of Slug can induce cancer cell stemness and enhance cell invasion and metastasis. Sun et al. reported that Slug overexpression is associated with the invasiveness of liver cancer both in vitro and in vivo.140 In contrast, overexpression of Snail did not have a similar effect in liver cancer cells. The Wnt pathway has been linked to increased expression of another EMT transcription factor, Twist, in mammary epithelial cells.141,142
Accumulating data show that TWIST not only induces EMT, but also promotes the formation of CSCs and enhances tumorigenesis.143 Therefore, inhibiting these EMT mediators may represent the strategy for inhibiting tumor metastasis and recurrence. Additionally, activation of the Wnt signaling pathway reportedly controls the stemness of CSCs in different types of cancers including hepatocellular carcinoma (HCC), and colon, lung, and other cancers.144,145,146,147,148 According to clinical practice, targeted treatment using MEKi (selumetinib, trametinib, and PD318088) inadvertently increased Wnt activity and led to enhanced stemness and tumor relapse in colorectal cancer.149 Similarly, valproic acid, which was originally regarded as a histone deacetylase (HDAC) inhibitor in breast cancer treatment, can activate Wnt signaling pathway and promote the pool of mammary CSCs, leading to tumor progression.150 Hence, attention should be paid to unexpected phenotypic switching regulated by Wnt signaling pathway in the processes of targeted therapy.
The Notch signaling pathway also enriches CSCs and regulates the EMT program in multiple cancers, including breast and lung cancer, pancreatic ductal adenocarcinoma, and malignant mesothelioma.151,152,153,154,155 A previous finding showed a change in EMT status in lung adenocarcinoma upon treatment with gefitinib.156 Subsequent studies further confirmed that the EMT process resulted from the activation of Notch-1 signaling pathway and led to gefitinib-acquired resistance.157 Notch signaling is one of the master regulators of neuroendocrine transdifferentiation in NSCLC, which enables NSCLC to transdifferentiate into SCLC and acquire resistance to EGFR-TKIs after initial targeted treatment.154
By extension, the Notch pathway has been discovered to play a crucial role in the EMT process in prostate cancer cells, promote a transformation towards basal stem-like properties, and lead to a castration-resistant phenotype.158,159 Components of the Notch signaling pathway are expressed at significant levels in tumor invasive regions, which suggests its crucial role in EMT regulation. A recent paper by Natsuizaka et al. reported that Notch1 signaling and EMT work closely to promote squamous cell carcinoma tumor initiation. Moreover, another important protein, TGF-β, guides Notch1 to drive EMT.160 A study by Zeng et al. showed that inhibition of Notch1 reverses the EMT process and overcomes cisplatin resistance in breast cancer cells.161 The data also suggested that high expression of Notch1 predicts a poor prognosis in TNBC patients. Saad et al. demonstrated that the Notch-mediated activation of EMT is associated with the regulation of EMT transcription factors.162 The two transcription factors, Snail and Slug, are involved in the inhibition of E-cadherin and β-catenin mediated by the Notch pathway. Inhibition of Notch activation limits the increase of MMP-2 and −9 expression, while it decreases Snail expression. The study provided direct evidence that Notch signaling induces EMT via the Snail pathway. Surprisingly, Notch signaling can synergistically work with other pathways to facilitate the EMT process. The crosstalk between these pathways is mediated by suppressor of mothers against decapentaplegic (SMADs) proteins that associate with other transcription factors for regulating the expression of genes required for the transition to mesenchymal characteristics. A single-cell sequencing study by Deshmukh et al. revealed both sequential and parallel activation of EMT signaling pathways. The Notch pathway also functions as a crucial regulator of TGF-β–induced EMT.163
The role of TME in controlling TCP
TME is a key player in TCP induced by targeted treatment. The characteristics of TME, including immune and stromal cells, cytokines, exosomes, hypoxia, pH change, nutritional deficiency, and angiogenesis, are key regulators of TCP.164,165,166,167
CAFs are recognized as major contributors in the crosstalk between tumor cells and adjacent stromal cells.168 By producing a variety of bioactive molecules such as growth factors and cytokines, CAFs exert a profound impact on TME and influence TCP,96,169,170 and have long been described as crucial players in the EMT program. CAFs also participate in CSC pool expansion.171,172,173 The effect of CAFs in manipulating therapy resistance was reported by Apicella et al.174 In this study, a prolonged treatment with TKIs generated resistance to erlotinib in vivo, but not in vitro. This intriguing phenomenon was explained by the existence of CAFs. Erlotinib treatment in vivo induces a metabolic rewiring towards aerobic glycolysis and increases lactate production; the secreted lactate triggers the overproduction of hepatocyte growth factor (HGF) in CAFs. Engulfed lactate and the subsequently increased HGF then activate the MET signaling pathway in cancer cells and cause resistance to erlotinib.175,176,177 Moreover, hypoxia-induced glycolysis represses natural killer (NK) cells via lactate accumulation and reduces cytotoxicity by decreased level of NKG2D, CD16, perforin, and granzyme B, thereby forming an immunosuppressive TME. Such hypoxia-related TME conditions were reported to play a role in resistance to therapeutic monoclonal antibodies.178,179
Cytokines secreted by immune and stromal cells also provide clues for acquired TCP induced by targeted therapy.180,181,182 According to Li et al.,183 IL-6 in EFGR-mutant NSCLC patients was significantly increased upon osimertinib therapy. This associated Upregulation of laminin α5, which is among the most widely distributed laminins and is a major component of extracellular matrix (ECM), also occurred. By upregulation of laminin α5, IL-6 remodeled ECM and activated a downstream target focal adhesion kinase (FAK), thus contributing to acquired resistance against Osimertinib.183,184,185 FAK activation was confirmed to be dispensable for IL-6 production in the maintenance of mesenchymal stem cells and can lead to acquired resistance of gefitinib as well.186,187
Contribution of TCP to targeted therapy resistance
Targeted therapy is one of the most successful treatments that strike at the heart of a wide range of cancers. Nonetheless, the emergence of drug resistance significantly impairs the efficacy of targeted drugs and contributes to cancer mortality. Drug target mutations and activation of bypass pathways are known to confer resistance in cancer cells to targeted inhibitors. With an increasing understanding of TCP, it is now recognized that TCP is an important factor in mediating drug resistance. In this section, we focus on the role of TCP in shaping tumor cell characteristics and mediating targeted therapy resistance (Fig. 4).

Therapeutic strategies targeting tumor cell plasticity. Three main strategies can be used in combination treatment against tumor cell plasticity: 1) prevent tumor cell plasticity; 2) reverse the phenotypic switching; and 3) direct therapy to target the induced therapy-resistant tumor cells
Epithelial–mesenchymal transition related factors in mediating drug resistance
As described above, EMT plays a critical role in mediating TCP and resistance to targeted therapy. The principal mechanisms of EMT-related drug resistance can be attributed to slow cell proliferation, increased drug efflux, and escape from apoptosis. Lung cancer is by far the most well-documented subtype of cancer that links EMT and targeted drug resistance. Namba et al. demonstrated that the upregulation of EMT-related gene AXL contributes to acquired resistance to osimertinib.188 Other EMT-related transcription factors, such as Snail, Slug, and ZEB1/2, are also reported to cause TKI resistance by regulation of EMT-related signaling pathways.189,190,191 Therefore, a comprehensive understanding of the EMT process is critical to combat targeted therapy resistance. In this subsection, we delineate the current research on signaling pathways and related factors that modulate EMT associated with drug resistance.
Signaling pathways including Notch, Wnt/β-catenin, and TGF-β play crucial roles in activating EMT-related transcription factors.192 Evidence supports that EMT transcription factors are involved in targeted therapy resistance, especially towards EGFR inhibitors. Consequently, signaling pathways that regulate the EMT process are important in mediating drug resistance. Chang et al. reported that Slug contributes to gefitinib resistance in NSCLC cells through downregulation of Bim and inhibition of caspase-9 activity.193 Among EMT inducers, TGF-β is significant because of its potency in inducing EMT and its roles in cancer-associated EMT.194 TGF-β-induced EMT is essential in cellular activities such as glycolysis and lipid/choline metabolism reprogramming.195 In response to TGF-β, cancer cells undergo complete or partial EMT, where epithelial and mesenchymal markers co-exist on cancer cells.37 The partial EMT status is characterized by an aggressive phenotype that renders cancer cells with high stemness and cell plasticity.196 Upon stimulation of TGF-β receptors, TGF-β signaling is divided into SMAD-dependent and -independent pathways. Both TGF-β/SMAD and non-SMAD pathways play a significant role in mediating EMT and promoting cell plasticity.197 Additionally, signaling pathways such as PI3K/AKT, mTOR, and MAPK are reported to play essential roles in TGF-β-induced EMT.198,199 Surprisingly, TGF-β-induced EMT can be reversed. A study conducted by Katsuno et al. found that upon TGF-β removal, mesenchymal cells revert to an epithelial cell phenotype.200 Moreover, prolonged TGF-β exposure promotes a stable EMT process in cancer cells, which is accompanied by increased tumor stemness and cancer drug resistance. The authors also identified that mTOR inhibitors can be a promising agent to target CSCs by antagonizing the TGF-β-induced EMT process.
MicroRNA (miRNAs) are small non-coding RNAs that play a pivotal role in the regulation of both EMT that promotes tumor metastasis and MET that facilitates metastatic colonization.201 EMT-related miRNAs are documented to impact cancer cell stemness and drug resistance. Recent evidence showed that miRNAs may affect the EMT process by targeting related transcription factors, including Snail, Slug, Twist, and ZEB1/2.202 The expression level of the Snail transcription factor may be controlled by multiple metastasis-related miRNAs, including miR-153, miR-203, miR-204, and miR-34c.203 The miR-30 family are well-documented mediators of EMT transcription factor Snail in different cancer subtypes, including lung and breast cancer.204 Liu et al. showed that miR-1 and miR-200 can inhibit EMT and mesenchymal differentiation via Slug-dependent mechanisms.205 Other newly identified EMT-related miRNAs include miR-27b-3p,206 which promotes the generation of circulating tumor cells, and miRNA-490-3p, which promotes the metastasis of invasive ductal carcinoma.207 Nonetheless, recent studies have reported that some miRNAs, such as miRNA-128 MiRNA-155-5p,208,209 are able to hinder the EMT process through inhibition of diverse EMT-related signaling pathways. With the increased understanding of miRNAs in EMT and TCP, different strategies focused on hindering miRNA function or miRNA delivery into tumor cells can be utilized to overcome cancer drug resistance.
Tumor heterogeneity
Tumor heterogeneity is defined as the existence of a subpopulation of cancer cells with different genotypes and phenotypes that exhibit various characteristics within a primary tumor. Heterogeneity may lead to divergent sensitivity to treatment and facilitate the development of drug resistance.210
The most recent tumor heterogeneity model is known as the CSC plasticity model.211 CSC is a subpopulation of cancer cells with self-renewal capacity to switch between stem and differentiated states. This dynamic transition can be attributed to gene mutations, epigenetic modification, as well as changes in TME. It should be noted that the EMT signaling pathway plays a central role in mediating CSC transition, which promotes cancer metastasis, recurrence, and drug resistance. Morel et al. reported that the induction of EMT can enhance human breast epithelial cell stemness and tumorigenic characteristics,212 which has also been confirmed in colorectal, pancreatic, and liver cancers.213,214,215 Quintana et al. demonstrated that most of the phenotypic heterogeneity in melanoma can be explained by the phenotypic plasticity model.216 Although they did not identify a marker that can significantly distinguish tumorigenic from non-tumorigenic cells, CD271 + cells were shown to have little tumorigenic activity. However, the results were inconsistent in different studies,217 suggesting that the role of cell plasticity in tumor heterogeneity may vary in cancer subtypes. Another study carried out by Charles et al. showed that nitric oxide can enhance the tumorigenic capacity of platelet-derived growth factor (PDGF)-driven glioma cells in vivo; therefore, it reversibly promotes their ability to form tumors.218 Nakano et al. reported the dynamic equilibrium between CSCs and non-CSCs in the development of colorectal cancer. Such equilibrium is mainly controlled by EMT-related TGF-β signaling pathway.219 One study demonstrated that, in colon cancer cells, the ablation of LGR5 + CSCs allowed the cancer cells to dedifferentiate into CSCs, which highlights the distinct CSC dependency for primary and metastatic tumor growth.220
Multiple studies showed that a high level of tumor heterogeneity is associated with poor response to targeted drugs, including osimertinib, rociletinib, and gefitinib.221,222,223 Chabon et al. demonstrated that multi-drug resistance was observed in 46% patients after targeted therapies, suggesting a frequent intra-tumor heterogeneity.222 TCP, i.e., phenotype switching between different signaling pathways, can potentially lead to heterogeneity under therapeutic selective pressure. As mentioned above, cell plasticity is essential in multiple crucial biological processes, including embryonic development, wound healing, and tissue regeneration. In tumors, the reactivation of these mechanisms allows tumor cells to switch between non-CSC and CSC-like phenotypes, leading to tumor progression, metastases, and drug resistance.224 Easwaran et al. performed a genome-wide analysis and found that silencing abnormal DNA methylation of promoters can decrease changes of plasticity of cell population and help in locking cell clones with abnormal retention of self-renewal capacity.225 Therefore, an effective approach to disrupt TCP may represent a promising strategy to tackle tumor heterogeneity and overcome drug resistance.
Interplay between TCP and TME
A tumor is not only a group of cancer cells, but also a complex interaction between cancer cells and their surrounding environment, known as the tumor microenvironment (TME), which has attracted substantial research and clinical interest in recent years.226 TME consists of tumor, immune (macrophages and lymphocytes), and stromal (stromal fibroblasts and endothelial cells) cells, blood vessels, and extracellular matrix (ECM).227 Accumulating data show that cancer cells are able to control the function of cellular and non-cellular components through complex signaling pathways.228 The cell-cell and cell-ECM interactions force non-malignant cells to acquire new phenotypes that facilitate the formation, progression, and metastasis of tumor cells. In addition, theinteractions of cancer cells with their TME are crucial to stimulate tumor heterogeneity, clonal evolution, and mediate drug resistance.229
TCP allows tumor cells to switch phenotypes and survive treatment through adaptive strategies, and the TME is involved in mediating TCP.165 For example, both cancer-associated fibroblasts (CAFs) and hypoxia contribute to oxidative stress, which is an important mediator of multiple adaptive strategies. Hypoxia is a hallmark of the TME and strongly linked to malignant progression, poor clinical response, and therapy resistance.230 Recent studies have shown that hypoxia may induce phenotypic switching, leading to drug resistance. Terry et al. reported that hypoxic stress induces phenotypic diversity of EMT in lung cancer, and leads to changes in EMT transcription factors such as SNAI1, SNAI2, and ZEB2, which ultimately result in the resistance to cell-mediated cytotoxicity. Paolicchi et al. suggested that tumor hypoxia is a major contributor to drug resistance and a predisposing factor for increased CSC formation and metastases.231 Therefore, one promising cancer treatment strategy is to block the proteins related to hypoxia-induced factor (HIF)-1α, EMT, and mitochondria functions, which in turn inhibits the response to hypoxia. In addition, cytokines such as interferon-gamma and tumor necrosis factor-alpha are believed to enhance stromal cell plasticity and contribute to TME.232
The interaction between TME and TCP can significantly contribute to the genetic heterogeneity.233 Reynolds et al. demonstrated that the number of genetic mutations arising in cells within tumors is much higher than that in the laboratory culture.234 Their results provide strong evidence that TME promotes genetic instability during tumor progression. On the other hand, tumor heterogeneity can lead to TME reprogramming and thus give rise to cancer drug resistance.235 Taken together, the interplay between TCP and TME serves an important role in mediating tumor progression, heterogeneity, and drug resistance.
New strategies for targeting TCP
Preventing initiation of TCP
Strategies that target TCP include intermittent treatments, combination therapies, and targeting the key nodes of phenotypic switching. A study showed that intermittent treatments and off dosing schedules may double the time of melanoma response to vemurafenib.74 However, intermittent treatment strategies are usually difficult to design as drug resistance may be complex and come from multiple mechanisms other than TCP.236,237 Combination therapies have also been applied to target parallel activation of key signaling pathways such as Hippo/yes-associated protein (YAP), PI3K, and EGFR,238,239 resulting in a significant decrease in the number of residual tumor cells. However, combination therapies usually require more optimized drug administration protocols to maximize responses and limit toxicity due to their complexity.
Other strategies to prevent TCP have focused on inhibiting the key nodes of phenotypic switching genes. Histone demethylases, KDM5A/B and KDM6A, were known to play significant roles in DTP survival.240,241,242 KDM inhibitors have been developed; however, they are still under assessment for their specificity and efficacy.243,244,245,246 DTPs undergo transcriptional adaptation through changing their global chromatin landscape. Another similar strategy that targets the bromodomain and extra terminal domain (BET) BRD4 gene overcame trametinib resistance in triple-negative breast cancer and was efficient both in vitro and in vivo.110 In basal cell carcinoma, the assessment of inhibition or activation of Wnt and Notch signaling pathways are underperforming in clinical trials to treat pathway-dependent malignancies.247 Notch-activated tumor cells tend to undergo apoptosis upon vismodegib treatment. Inhibitions of Retinoid X receptor (RXR) pathways using HX531 together with MAPK inhibitors can significantly delay development of resistance to MAPK inhibitors, compared to treatment with MAPK inhibitors alone.236 Discovery of more drug targets that prevent tumor plasticity will enable the design of treatment strategies.
Targeting the fate of new cells
In addition to preventing the formation of cell plasticity, targeting the emerging drug-resistant new cell identity holds great potential for overcoming tumor plasticity (Fig. 4). Transformation from the NSCLC to SCLC usually causes strong drug resistance due to cell plasticity.248 SCLC cells often lose EGFR expression; therefore, they are resistant to EGFR inhibitors, in contrast to the nontransformed NSCLC (specifically LUAD). Transformed SCLCs were more responsive to platinum-etoposide, similar to de novo SCLC.67 However, compared to de novo SCLC, transformed SCLC showed a higher response rate to taxanes yet were resistant to checkpoint inhibitor therapy, which was similar to EGFR-mutant LUAD.249 Recently, molecular studies showed that the growth of some mesenchymal cancer cells depends on the expression of phospholipid glutathione peroxidase 4 (GPX4), and inhibition of GPX4 can profoundly decrease DTP, thus preventing tumor relapse.250 However, current GPX4 inhibitors are chloroacetamide-containing compounds (such as RLS3), which have been identified to possess only poor pharmacokinetic properties, such as low stability, promiscuity, and bioavailability. The GPX-knockout mouse was lethal, indicating that choosing the therapeutic window that targets this protein is challenging.251,252,253 Thus, finding drugs that target the new cell identity is necessary for designing therapeutic strategies.
Reversing TCP
TCP can be driven by an inflamed and suppressive TME. Drugs and therapies have been developed against intracellular signaling kinases and extracellular signaling molecules in TME.254,255 Additionally, since epigenetic changes contribute to TCP, targeting those processes could potentially re-sensitize tumor cells to drug treatments.
IL-8
IL-8, a pro-inflammatory cytokine, is known to promote tumor cell remodeling and is involved in regulation of tumor cell stemness, EMT, and therapy resistance in multiple types of tumors.256,257 IL-8 production promotes tumor cells to transform to a mesenchymal phenotype and increase tumor cell migration, leading to metastases.258 C-X-C chemokine receptor 1 (CXCR1) antagonist therapies have been found to reverse the IL-8 related events.259 Resistance to PI3K/Akt/mTOR pathway inhibition in hepatocellular carcinoma has been associated with both liver cancer stem cells and a high level of IL-8 expression. Rapamycin, the mTOR inhibitor, was able to decrease the IL-8 expression, thus decrease the resistance to sorafenib.260 A neutralizing antibody, HuMAX-IL8, was used to deplete IL-8 secretion and treat human TNBC.261 Another study showed that neutralizing IL-8 can alleviate resistance to erlotinib in NSCLC, thereby enhancing sensitivity in chemotherapy-resistant cells.262 Inhibition of CXCR2 with SB225002 also alleviated the IL-8-related resistance in sorafenib-resistant ovarian tumor cells.263
TGF-β
Reversing EMT by blockage of TGF-β signaling is acknowledged as a promising way to reverse TCP, as TGF-β has been widely recognized as the major inducer of EMT.264 A recent single-cell transcriptome-based study showed that blocking TGF-β1 could restore stromal plasticity in tumors by affecting a subset of interferon-licensed fibroblasts.265 However, the pleiotropic functions of TGF-β1 in cancer render therapeutic inhibition of this pathway challenging.266 TGF-β1 inhibition in various combination therapies that target the drug-induced plasticity is under evaluation.267 Galunisertib (LY2157299 monohydrate), a small molecule inhibitor that inhibits TGFβRI ALK5 kinase, has been used in numerous preclinical trials.268 Forskolin and cholera toxin have been identified through a drug screen for E-cadherin promoter activation.269 JMF3086, a dual inhibitor that targets both HDAC and 3-hydroxy-3-methylglutaryl coenzyme A reductase, can restore the sensitivity of NSCLC to EGFR-TKIs (34). A preclinical study showed that galunisertib has the potential to regulate pancreatic cancer cell growth through prevention of TGF-β mediated E-cadherin downregulation.270 Galunisertib also demonstrated anti-tumor efficacy in patients with pancreatic cancer as well as in a subset of patients with advanced HCC.271 A recent study also demonstrated that inhibition of TGF-β and leukocyte-associated immunoglobin like receptor-1 (LAIR-1) can remodel TME, thereby enabling programmed-death ligand 1 (PD-L1)-mediated tumor eradication.272 Dual-blockade of CD73-TGFβ promotes a multifaceted inflammatory environment in TNBC, by diminishing myeloid-derived suppressor cells and M2-macrophages but increasing cytotoxic T cells and activated dendritic cells.273 These studies and novel drugs have provided strong evidence that blocking TGF-β can reverse TCP, thereby sensitizing tumor cells to targeted or immune-mediated cell death.
EZH2
EZH2, the catalytic subunit of polycomb repressive complex 2 (PRC2), is the central player in the process of epigenetic gene silencing. EZH2 promotes tumor cell survival, proliferation, EMT, and invasion, as well as drug resistance.274 Reversible transition between EMT and MET are key aspects of TCP. Interestingly, EZH2 was recently found to support TCP through MET facilitation and tumor colonization.275 Overexpression of EZH2 was associated with the conversion of prostate adenocarcinoma to neuroendocrine prostate cancer (NEPC), which is a more aggressive variant.61,276 Inhibition of EZH2 can reverse the cell lineage switch, leading to a decrease in neuroendocrine related key factors and restoring the sensitivity of NEPC cells to enzalutamide. Some EZH2 inhibitors (for example, CPI-1205 and Tazemetostat) have entered phase I and II clinical trials for lymphomas and solid tumors.277,278
Other targets and pathways
Other targets and pathways with potential ability to reverse TCP are emerging. Ferroptosis was identified as a new type of programmed cell death that can be harnessed to target drug resistance and EMT. Gagliardi et al. identified that EMT-associated gene expression reprogramming process that determines ferroptotic susceptibility cannot be applied to metastatic-derived cells, but SCL7A11 was a valuable marker to predict the susceptibility.279 Increased sensitivity to ferroptosis through crosstalk of BACH1 target genes can also impose programmed vulnerability upon cancer cells.280 Other than ferroptosis, HDAC inhibition was recently found to reverse EBV-induced dedifferentiation in nasopharyngeal carcinoma.281 The Yap-Sox9 axis was recently reported to determine hepatocyte plasticity in hepatocarcinogenesis and could be a promising target.282 Some other SOX factors have also been considered as promising candidates through proteolysis-targeting chimeric molecules or thalidomide analogues.283,284 An anti-CLDN6 antibody-drug conjugate has been reported to target tumor lineage plasticity.285 Another recent study showed that blocking the MNK1/2-eIF4E axis could inhibit melanoma phenotypic switching, thereby, sensitizing melanoma to anti–PD-1 immunotherapy.286 These studies suggest that reversing TCP through other targets could be a propitious future direction.
New therapies for overcoming TCP-induced drug resistance
TCP enables tumor cells to evade traditional anti-tumor therapies. Therefore, agents that block these escape routes can deprive the residual tumor cells of their hiding place and ensure positive treatment results (Fig. 5).

Combination treatments for overcoming the tumor cell plasticity induced-therapy resistance. Tumor cells evade targeted treatments by interacting with TME and via EMT programs. Inflammation, hypoxia, and immunosuppressive TME contribute to therapy evasion, and such induced-resistance could be inhibited by a combination treatment including anti-inflammation and anti-hypoxic drugs and immunosuppressive ICB regimens. EMT-modulating agents, on the other hand, could also deal with plasticity induced-therapy resistance as a combination treatment and lead to tumor cell death. TME tumor microenvironment, EMT epithelial-to-mesenchymal transition, ICB immune checkpoint blockade, Rg3 ginseng-extracted 20(R)-Ginsenoside
With immune checkpoint blockade (ICB)
An immunosuppressive TME is formed upon drug exposure as a consequence of the interaction between non-tumor and tumor cells. It is worth mentioning that the expression of immune checkpoint proteins, including programmed cell death 1 receptor (PD-1) and (PD-L1), cytotoxic T lymphocyte-associated protein 4 (CTLA-4), T cell immunoglobulin 3 (TIM-3), lymphocyte activation gene 3 (LAG-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT) contribute significantly to immunosuppression and serve as a bridge between EMT programs and immunosuppression.287,288,289,290,291 A positive correlation between EMT and immune checkpoint proteins was identified in multiple cancer types, including lung, breast, colorectum, oral, and esophageal cancers,289,292,293,294,295,296,297 and a higher expression of these checkpoint molecules indicates worse prognosis in a broad range of cancers.298,299,300 The functional relationship between immune checkpoint proteins and therapy evasion suggests that a combination treatment with immune checkpoint blockade (ICB) could be promising in developing a new approach against TCP.
Combining targeted therapy with ICB seems feasible according to current practice. Traditional EGFR-TKI was found to interfere with TME, thus laying a theoretical foundation for ICB application in EGFR-TKI-resistant NSCLC, especially in those with upregulated PD-L1 expression.301,302,303 Moreover, recent studies revealed that dynamic changes occur in TME in response to EGFR-TKIs and suggested that a combination treatment with immune-mediated anticancer approaches could be beneficial.302,304,305,306,307 EGFR-TKI-ICB combination treatment provided better clinical benefit for NSCLC patients with increased overall response rate (ORR) (41.7% vs 14.3%) and median progression free survival (PFS) (19.5 months vs 1.4 months) in comparison to monotherapy of either pembrolizumab or gefitinib alone.306 Patients with short EGFR-TKI-PFS may even benefit more from a combination therapy as a second-line treatment.308 A similar combination strategy was applied in multiple cancer types as a so-called basket trial design.309 A combination of ICB with antiangiogenic molecular targeted therapy is an attractive approach for treating HCC. According to the IMbrave150 trial, atezolizumab in combination with bevacizumab showed greater benefits compared with sorafenib in advanced HCC patients and suggested that this combination strategy could be used as the first-line standard of care.310,311,312,313 Additionally, a combination of ICB with other targeted therapeutic approaches, including ATR kinase inhibition and androgen receptor blockade resulted in synergistic therapeutic response in prostate cancer.314,315,316
Combining chemotherapy with ICB can be used to extend a synergistic therapeutic effect.317,318 A pooled analysis of three randomized clinical trials indicated that among patients with PD-L1-negative advanced / metastatic NSCLC, efficacy is better with pembrolizumab plus chemotherapy compared with chemotherapy alone.319 Similar results were obtained in PD-L1-positive NSCLC patients. As indicated by Zhang et al.,320 the combination regimen of toripalimab and pemetrexed/carboplatin was superior to chemotherapy alone. Moreover, an IMpower150 trial showed that a combination of atezolizumab, bevacizumab, carboplatin, and paclitaxel (ABCP) led to significant improvement in median overall survival (OS) in chemotherapy-naïve patients with NSCLC.321 Anti-CTLA-4 ipilimumab was also tested in the treatment for NSCLC in combination with nivolumab and platinum doublet chemotherapy and exhibited clinical value as a first-line treatment.322 Another phase III clinical trial compared the efficacy of chemotherapy with nivolumab and chemotherapy alone in treatment for advanced gastric, gastroesophageal junction (GEJ), and esophageal adenocarcinoma, and found that the combination strategy had a benefit of significantly superior OS and PFS.322 Janjigian et al.322 proposed that this combination treatment represented a new first-line therapy for patients with upper gastrointestinal tumors. A recent phase III study (KEYNOTE-811) also confirmed that adding pembrolizumab to standard first-line treatment (trastuzumab plus chemotherapy) in advanced gastric or GEJ adenocarcinoma could markedly improve prognosis.323
Though the basket trial design has greatly improved the clinical outcome of multiple cancer types, our understanding of the underlying mechanisms of the combination regimens remains limited. For example, nab-paclitaxel combined with atezolizumab are able to effectively prolong PFS in patients with metastatic TNBC as shown by the IMPASSION130 trial; however, the combination of paclitaxel with atezolizumab failed in the IMPASSION131 trial.324,325 Single-cell analyses revealed that baseline CXCL13+ T cell largely determined the clinical response in TNBC treatment.326 Further studies are warranted to explore the exact mechanisms of different combination strategies to be able to pin-point patients with higher therapeutic potential. Furthermore, novel combination methods are emerging. For example, a bioengineered platelet was designed that combines an inner-loaded doxorubicin and outer-anchored anti-PDL1-crosslinked nanogels. This composite design was reported to have the potential to reduce the recurrence and metastasis rate of postsurgical tumors.327
Despite some optimistic reports, the safety profile of combination therapies with ICB remains poorly understood and controversial. In NSCLC treatment, an increased incidence of interstitial lung disease-like events was observed in the combination treatment of osimertinib and durvalumab in the CAURAL (38%) and TATTON (22%) trial.328,329,330,331,332,333 Severe hepatotoxicity was observed in another clinical trial (CheckMate370) investigating the combined effect of nivolumab and crizotininb.306 Similar hepatic and immune-related adverse events were also observed in combined treatment with ICBs in melanoma.334,335,336
These described studies imply that a combination with ICB can improve therapeutic outcomes for therapy-resistant patients compared with either therapy alone. The clinical feasibility of the combined strategy warrants further exploration, and the safety profile should be carefully documented. Table 1 provides a list of some relevant clinical trials evaluating the combination therapy with ICB mentioned above.
With EMT-modulating agents
Because EMT plays an essential role in TCP and drug resistance, EMT-targeting compounds hold great potential as novel drugs in reversing drug resistance. However, investigation in this field is limited.
EMT-TFs play an essential role in TCP, and drugs directly targeting these transcription factors might have the potential to halt or reverse the plasticity and become a novel therapeutic approach to repress drug resistance. However, due to the redundancy of overlapping pathways and multitude of factors, it is hard to target and control TCP effectively. Direct targeting of these EMT-TFs is very challenging and there is not yet known EMT-TF-targeting molecule.19 Nevertheless, some related molecules involved in redundant EMT pathways have been investigated for clinical use. As a downstream target of YAP and EMT-TF Snail2, anexelekto (AXL) represents an ideal focus of EMT control.337,338 AXL-targeting inhibitors (SGI-7079 / Foretinib [XL-880] / Bemcentinib [BGB324] / 20G7-D9 / MYD1-72) have been reported to have an outstanding synergistic effect with erlotinib in NSCLC40,339,340 and advanced pancreatic and ovarian cancers,337 BRAF inhibitors in melanoma,341 and temozolomide with radiation in glioblastoma.342 Multitarget AXL inhibitors like SGI-7079 and sunitinib have also shown prominent clinical value in reversing therapeutic resistance.40,343,344 Moreover, secreted clusterin, an EMT-related protein which was shown to be significantly upregulated in EMT progression and HCC migration, is a candidate target for HCC therapy.345,346,347 An antibody against secreted clusterin, termed as AB-16B5, was assessed in a phase II clinical trial in combination with docetaxel for the treatment of metastatic NSCLC (Table 2).
Inhibition of EMT-related pathways is a theoretically feasible approach to control TCP. TGF-β, a widely accepted key promoter of EMT and sustainer of mesenchymal and CSC states, contributes to the induction of immunosuppressive TME via hijacking Tregs and NK cells.44,45,348 A small molecule inhibitor of TGF-β receptor 1 named galunisertib (LY2157299 monohydrate) is attracting attention. One preclinical evaluation of galunisertib showed an inhibition of TGF-β-mediated downregulation of E-cadherin in mouse pancreatic cancer cell line, indicating an anti-EMT effect.349 A recent study by Song et al.350 demonstrated a synergistic effect in castration resistant PC using enzalutamide combined with galunisertib. The combinatorial galunisertib treatment successfully sensitized PC cells to traditional androgen deprivation therapy (ADT) and provided a novel therapeutic strategy for control of lineage transdifferentiation. Other studies tested the efficacy of galunisertib in HCC and indicated a prolonged OS using a combination of galunisertib and sorafenib.351 Melisi et al.271 stated in a phase Ib/II clinical trial that a galunisertib-gemcitabine combination therapy could significantly improve OS in patients with unresectable pancreatic cancer compared with monotherapy with gemcitabine. The same result was found in myelodysplastic syndromes (MDS); galunsertib treatment was associated with hematologic improvement in patients with signs of blockage of early stem cell differentiation. However, some other clinical trials failed to prove the efficacy of galunisertib in HCC, metastatic pancreatic cancer, or glioma.352,353,354 Though such novel drugs can manipulate TCP and potentially re-sensitize tumor cells to immune-mediated cell death, further clinical trials are needed to validate their efficacy in different types of tumors. The ongoing clinical trials investigating the combination treatment with galunisertib are shown in Table 2.
Additionally, a combined immunotherapy of ICB and TGF-β blockade is also promising. In a murine model of metastatic intestinal tumor, the combination of anti-TGF-β and anti-PD-L1 showed an impressive antitumor response compared to either monotherapy.355 Currently, new approaches for simultaneous inhibition of TGF-β and PD-L1 are under evaluation. Bintrafusp alfa (also known as M7824) is a recently developed anti-PD-L1/ TGF-βRII fusion protein, which showed superior inhibition of tumor growth in multiple murine models.356,357 Other fusion drugs such as anti-CTLA4-TGF-βRII also showed promising efficacy in stimulating antitumor responses.358 Several early-phase trials showed encouraging clinical efficacy and manageable safety in different types of cancers.359,360,361 Several ongoing clinical trials are testing the efficacy and safety of bintrafusp alfa (Table 2).
Wnt inhibitors including ETC-1922159 and Ipafricept (also known as OMP-54F28) were also employed in clinical control of various types of solid tumors.362,363,364,365 Ipafricept combined with chemotherapy showed reasonable tolerance in early-phase clinical trials, and remains a therapeutic target of interest.363,364,366 ETC-1922159 is now under assessment in an ongoing clinical trial to test its efficacy as a pembrolizumab combination (Table 2).
With other therapy
The close interaction between inflammatory TME and EMT is well established and is regarded as a promising target for therapeutic use.15 Since TCP drives a highly inflamed TME, neutralizing monoclonal antibodies targeting IL-8 were developed to reverse TCP. HuMax-IL8 (also known as BMS-986253), a neutralizing anti-IL8 antibody that was designed to sequester IL-8 signaling, successfully inhibited the mesenchymal transition in human TNBC cell lines in vitro. Blockade of IL-8 enhanced the tumor cell-killing effect mediated by NK and T cells. Another in vivo study in NSCLC found that IL-8 neutralizing antibodies reversed the tumor plasticity and sensitized tumor cells to erlotinib. Currently, multiple ongoing clinical trials in phase I/II are examining the safety and efficacy of HuMax-IL8 as a combination therapy in various cancer types, including prostate and pancreatic cancer, HCC, NSCLC, and other solid tumors (Table 3). SX-682, another novel drug targeting the inflammatory EMT, was proposed to prevent polymorphonuclear (PMN)-MDSC migration and cooperate well with ICB treatment.367,368 Early-phase clinical trials of SX-682 in combination with ICBs (e.g., pembrolizumab and nivolumab) are currently recruiting participants (Table 3).
Hypoxia signaling contributes significantly to EMT and subsequent therapy resistance. Hypoxia-induced factor (HIF), a transcriptional regulator, was reported to be recruited to the hypoxia-responsive element of several EMT-related genes, including Twist, Snail, Vimentin, and TGF-β.369 HIF-1α promoted EMT via multiple signaling pathways, including PI3K/Akt, Notch, Wnt, and NF-κB.345,370 Treatments targeting hypoxia pathway are deemed as promising blockades against TCP. For example, by blocking the nuclear translocation of HIF-1α, 6-gingerol successfully inhibits the EMT pathway and suppresses proliferation and metastasis of lung cancer.369 Salidroside enhances antitumor efficacy of platinum drugs in HCC by promoting the degradation of HIF-1α, thereby inhibiting the EMT of HCC cells.371 Similarly in lung cancer, by inhibiting hypoxia-activated NF-κB signaling pathway and EMT, a ginseng-extracted 20(R)-Ginsenoside (Rg3) increases the susceptibility of cancer cells to cisplatin treatment.345
Conclusion and perspectives
Here, we present a comprehensive description of TCP development and cancer cell evasion from targeted therapy. We delineate the non-genetic mechanisms of targeted drug-induced TCP in various types of tumors and provide insights into TCP contribution to acquired drug resistance. Owing to the rapid development of experimental techniques, dynamic changes of tumor cells in response to treatments can be observed in high resolution images. The ceaseless change of tumor cells indicates that living things even as simple as single cells show amazing adaptability under ambient pressure. Continuous monitoring of phenotypic switching can be of great help in designing specific dynamic therapeutic regimens and developing such monitoring techniques is worth exploring. We have been able to look into some pathways, like EMT, CSC, and transdifferentiation, that facilitate transformation of tumor cells; however, we still cannot visualize all the mechanisms contributing to TCP.
Research and development for new treatment is never-ending. As an old saying goes, a coin has two sides. In spite of TCP-related therapy escape, TCP also allows therapeutic reprogramming and possibly reversal of tissue attrition. In this review, we summarize new therapeutic strategies that aim to inhibit or reverse TCP and discuss related combination treatment regimens. We note that regulation of plasticity-related TME with ICB or direct imposition of EMT-modulating agents will not bring revolutionary improvement in cancer treatment. However, each small step forward against cancer is worth trying, and the concept of TCP remains valuable for further development of novel therapeutic strategies. Inhibition of unexpected TCP is theoretically feasible by holding the tumor cell unchanged. On the other hand, effective manipulation of TCP also holds great potential, and converting diverse tumor cells into a unified and susceptible state could be an alternative. The basket trial design should be continued to probe new treatment opportunities for different types of cancers, and special attention should be given to searching for new targets to better manipulate TCP.
As for future research on TCP, modern technologies could be of great help. For example, omics technique and capacity of dealing with “big data” has largely broadened our knowledge of the molecular landscape of tumor tissues. Recently developed technologies could enable a simultaneous acquisition of multiple omics data at a single-cell level, and several analytic models have been established to interpret the high-dimensional multimodal data.372,373 Single-cell level multi-omics could precisely trace the therapy-induced responses of each cell in heterogeneous tumors from the aspects of transcriptomics, proteomics, epigenomics, even in combination with their spatial distribution. The innovative analytical modalities provide deep and comprehensive information at single-cell resolution and could help to explore yet unrecognized cellular activity both intracellularly and intercellularly. In addition, single-cell techniques could also help to identify biomarkers to determine TCP, tumor response to targeted therapy, as well as prognosis.374 Artificial intelligence could also be employed to capture the underlying regularities of such an unwieldly mass of data. It was expected that one day, with the help of big data and the corresponding interpreting capacity, complete cures could be achieved by personalized treatment strategies decided by molecules at single-cell level.
In the face of such capricious, unpredictable, and threatening disease, scientific research will be always on the way to distill the complexity of cancer into an increasingly logical science. The attempts to get a better understanding of TCP in targeted therapy hold great promise for future blockade against therapy evasion, which may promote realization of long-lasting curative effects and even complete cure in cancer patients. Ongoing clinical trials using conventional treatments combined with plasticity-regulating treatment also indicate new opportunities for achieving the goal of complete cancer cure.
References
Quintanal-Villalonga, A. et al. Comprehensive molecular characterization of lung tumors implicates AKT and MYC signaling in adenocarcinoma to squamous cell transdifferentiation. J. Hematol. Oncol. 14, 170 (2021).
Paksa, A. & Rajagopal, J. The epigenetic basis of cellular plasticity. Curr. Opin. Cell Biol. 49, 116–122 (2017).
Waddington, C. H. Embryology, epigenetics and biogenetics. Nature 177, 1241–1241 (1956).
Tata, P. R. & Rajagopal, J. Cellular plasticity: 1712 to the present day. Curr. Opin. Cell Biol. 43, 46–54 (2016).
Castranova, D. et al. Live Imaging of Intracranial Lymphatics in the Zebrafish. Circ. Res. 128, 42–58 (2021).
Bower, N. I. et al. Mural lymphatic endothelial cells regulate meningeal angiogenesis in the zebrafish. Nat. Neurosci. 20, 774–783 (2017).
Gunawan, F., Gentile, A., Gauvrit, S., Stainier, D. Y. R. & Bensimon-Brito, A. Nfatc1 Promotes Interstitial Cell Formation During Cardiac Valve Development in Zebrafish. Circ. Res. 126, 968–984 (2020).
Matsumoto, T., Wakefield, L., Tarlow, B. D. & Grompe, M. In Vivo Lineage Tracing of Polyploid Hepatocytes Reveals Extensive Proliferation during Liver Regeneration. Cell Stem Cell 26, 34–47 e33 (2020).
Chen, F. et al. Broad Distribution of Hepatocyte Proliferation in Liver Homeostasis and Regeneration. Cell Stem Cell 26, 27–33 e24 (2020).
Schaub, J. R. et al. De novo formation of the biliary system by TGFbeta-mediated hepatocyte transdifferentiation. Nature 557, 247–251 (2018).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Saha, S. et al. Aspirin Suppresses the Acquisition of Chemoresistance in Breast Cancer by Disrupting an NFkappaB-IL6 Signaling Axis Responsible for the Generation of Cancer Stem Cells. Cancer Res 76, 2000–2012 (2016).
Francescangeli, F. et al. A pre-existing population of ZEB2(+) quiescent cells with stemness and mesenchymal features dictate chemoresistance in colorectal cancer. J. Exp. Clin. Cancer Res. 39, 2 (2020).
Boumahdi, S. & de Sauvage, F. J. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 19, 39–56 (2020).
Qin, S. et al. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target Ther. 5, 228 (2020).
Shaffer, S. M. et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017).
Kurata, T. et al. Effect of re-treatment with gefitinib (‘Iressa’, ZD1839) after acquisition of resistance. Ann. Oncol. 15, 173–174 (2004).
Davies, A. H., Beltran, H. & Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 15, 271–286 (2018).
Horn, L. A., Fousek, K. & Palena, C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 6, 432–441 (2020).
Zhu, X., Chen, L., Liu, L. & Niu, X. EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front. Oncol. 9, 1044 (2019).
Shibue, T. & Weinberg, R. A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 14, 611–629 (2017).
Hass, R., von der Ohe, J. & Ungefroren, H. The Intimate Relationship Among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to Be Exploited for Therapeutic Purposes. Cancers (Basel) 12, 3674 (2020).
Gregory, P. A. et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 22, 1686–1698 (2011).
Siemens, H. et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 10, 4256–4271 (2011).
Martin, P. et al. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am. J. Pathol. 179, 422–435 (2011).
Yamasaki, J. et al. MEK inhibition suppresses metastatic progression of KRAS-mutated gastric cancer. Cancer Sci. 113, 916–925 (2022).
Luond, F. et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 56, 3203–3221 e3211 (2021).
Pastushenko, I. et al. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 (2018).
Lytle, N. K., Barber, A. G. & Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 18, 669–680 (2018).
Panchy, N., Azeredo-Tseng, C., Luo, M., Randall, N. & Hong, T. Integrative Transcriptomic Analysis Reveals a Multiphasic Epithelial-Mesenchymal Spectrum in Cancer and Non-tumorigenic Cells. Front. Oncol. 9, 1479 (2019).
Bakir, B., Chiarella, A. M., Pitarresi, J. R. & Rustgi, A. K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 30, 764–776 (2020).
Yang, J. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 (2004).
Addison, J. B. et al. Functional Hierarchy and Cooperation of EMT Master Transcription Factors in Breast Cancer Metastasis. Mol. Cancer Res 19, 784–798 (2021).
Krebs, A. M. et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 19, 518–529 (2017).
Lambert, A. W., Pattabiraman, D. R. & Weinberg, R. A. Emerging Biological Principles of Metastasis. Cell 168, 670–691 (2017).
Esposito, M. et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 21, 627–639 (2019).
Pastushenko, I. & Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 29, 212–226 (2019).
De Angelis, M. L., Francescangeli, F. & Zeuner, A. Breast Cancer Stem Cells as Drivers of Tumor Chemoresistance, Dormancy and Relapse: New Challenges and Therapeutic Opportunities. Cancers (Basel) 11, 1569 (2019).
Culig, Z. Epithelial mesenchymal transition and resistance in endocrine-related cancers. Biochim Biophys. Acta Mol. Cell Res 1866, 1368–1375 (2019).
Byers, L. A. et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res 19, 279–290 (2013).
Paulitschke, V. et al. Proteomic identification of a marker signature for MAPKi resistance in melanoma. EMBO J. 38, e95874 (2019).
Farmer, P. et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 15, 68–74 (2009).
Zhang, M. et al. Pyruvate dehydrogenase kinase 1 contributes to cisplatin resistance of ovarian cancer through EGFR activation. J. Cell. Physiol. 234, 6361–6370 (2019).
Liu, M., Li, S. & Li, M. O. TGF-beta Control of Adaptive Immune Tolerance: A Break From Treg Cells. Bioessays 40, e1800063 (2018).
Viel, S. et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal 9, ra19 (2016).
Saxena, M., Stephens, M. A., Pathak, H. & Rangarajan, A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2, e179 (2011).
Merrell, A. J. & Stanger, B. Z. Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 17, 413–425 (2016).
Rubin, M. A., Bristow, R. G., Thienger, P. D., Dive, C. & Imielinski, M. Impact of Lineage Plasticity to and from a Neuroendocrine Phenotype on Progression and Response in Prostate and Lung Cancers. Mol. Cell 80, 562–577 (2020).
Fang, D. et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res 65, 9328–9337 (2005).
Tang, J. Y. et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 17, 1720–1731 (2016).
Sofen, H. et al. A phase II, multicenter, open-label, 3-cohort trial evaluating the efficacy and safety of vismodegib in operable basal cell carcinoma. J. Am. Acad. Dermatol. 73, 99–105 e101 (2015).
Biehs, B. et al. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 562, 429–433 (2018).
Sanchez-Danes, A. et al. A slow-cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy. Nature 562, 434–438 (2018).
Konieczkowski, D. J., Johannessen, C. M. & Garraway, L. A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 33, 801–815 (2018).
Zou, M. et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Disco. 7, 736–749 (2017).
Conteduca, V. et al. Circulating tumor cell heterogeneity in neuroendocrine prostate cancer by single cell copy number analysis. NPJ Precis Oncol. 5, 76 (2021).
Epstein, J. I. et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 38, 756–767 (2014).
Conteduca, V. et al. Clinical features of neuroendocrine prostate cancer. Eur. J. Cancer 121, 7–18 (2019).
Wang, Y. et al. Molecular events in neuroendocrine prostate cancer development. Nat. Rev. Urol. 18, 581–596 (2021).
Quintanal-Villalonga, A. et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 17, 360–371 (2020).
Ku, S. Y. et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017).
Mu, P. et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88 (2017).
Svensson, C. et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res 42, 999–1015 (2014).
Merkens, L. et al. Aggressive variants of prostate cancer: underlying mechanisms of neuroendocrine transdifferentiation. J. Exp. Clin. Cancer Res. 41, 46 (2022).
Cyrta, J. et al. Role of specialized composition of SWI/SNF complexes in prostate cancer lineage plasticity. Nat. Commun. 11, 5549 (2020).
Balanis, N. G. et al. Pan-cancer Convergence to a Small-Cell Neuroendocrine Phenotype that Shares Susceptibilities with Hematological Malignancies. Cancer Cell 36, 17–34.e17 (2019).
Ferrer, L. et al. A Brief Report of Transformation From NSCLC to SCLC: Molecular and Therapeutic Characteristics. J. Thorac. Oncol. 14, 130–134 (2019).
Oser, M. G., Niederst, M. J., Sequist, L. V. & Engelman, J. A. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 16, e165–172 (2015).
Lee, J. K. et al. Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J. Clin. Oncol. 35, 3065–3074 (2017).
Offin, M. et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 14, 1784–1793 (2019).
Mahadevan, N. R. et al. Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Disco. 11, 1952–1969 (2021).
Shaurova, T., Zhang, L., Goodrich, D. W. & Hershberger, P. A. Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front Genet 11, 281 (2020).
Prasetyanti, P. R. & Medema, J. P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 16, 41 (2017).
Das, M. & Law, S. Role of tumor microenvironment in cancer stem cell chemoresistance and recurrence. Int. J. Biochem. Cell Biol. 103, 115–124 (2018).
Dirkse, A. et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 10, 1787 (2019).
Kusienicka, A. et al. Heme Oxygenase-1 Has a Greater Effect on Melanoma Stem Cell Properties Than the Expression of Melanoma-Initiating Cell Markers. Int. J. Mol. Sci. 23, 3596 (2022).
Lee, W. K. et al. Regulation of cancer stem cell activity by thyroid hormone receptor β. Oncogene 41, 2315–2325 (2022).
Thankamony, A. P., Saxena, K., Murali, R., Jolly, M. K. & Nair, R. Cancer Stem Cell Plasticity - A Deadly Deal. Front Mol. Biosci. 7, 79 (2020).
Correia, C. et al. Uncovering Pharmacological Opportunities for Cancer Stem Cells-A Systems Biology View. Front Cell Dev. Biol. 10, 752326 (2022).
Santoro, A., Vlachou, T., Carminati, M., Pelicci, P. G. & Mapelli, M. Molecular mechanisms of asymmetric divisions in mammary stem cells. EMBO Rep. 17, 1700–1720 (2016).
Peiris-Pagès, M., Martinez-Outschoorn, U. E., Pestell, R. G., Sotgia, F. & Lisanti, M. P. Cancer stem cell metabolism. Breast Cancer Res 18, 55 (2016).
Najafi, M., Mortezaee, K. & Ahadi, R. Cancer stem cell (a)symmetry & plasticity: Tumorigenesis and therapy relevance. Life Sci. 231, 116520 (2019).
Wu, M. et al. Imaging hematopoietic precursor division in real time. Cell Stem Cell 1, 541–554 (2007).
Liu, D. et al. Asymmetric Division Gene Neurl2 Mediates Twist2 Regulation of Self-Renewal of Mouse Lewis Lung Cancer Stem Cells. J. Cancer 10, 3381–3388 (2019).
Poli, V., Fagnocchi, L. & Zippo, A. Tumorigenic Cell Reprogramming and Cancer Plasticity: Interplay between Signaling, Microenvironment, and Epigenetics. Stem Cells Int 2018, 4598195 (2018).
Benboubker, V., Boivin, F., Dalle, S. & Caramel, J. Cancer Cell Phenotype Plasticity as a Driver of Immune Escape in Melanoma. Front. Immunol. 13, 873116 (2022).
Grillet, F. et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut 66, 1802–1810 (2017).
Lambert, A. W. & Weinberg, R. A. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer 21, 325–338 (2021).
Obermayr, E. et al. Cancer Stem Cell-Like Circulating Tumor Cells Are Prognostic in Non-Small Cell Lung Cancer. J. Pers. Med 11, 1225 (2021).
Jiang, W. et al. Combating multidrug resistance and metastasis of breast cancer by endoplasmic reticulum stress and cell-nucleus penetration enhanced immunochemotherapy. Theranostics 12, 2987–3006 (2022).
Hirschmann-Jax, C. et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl Acad. Sci. U. S. A. 101, 14228–14233 (2004).
Xu, J. et al. Disruption of DNA Repair and Survival Pathways through Heat Shock Protein Inhibition by Onalespib to Sensitize Malignant Gliomas to Chemoradiation Therapy. Clin. Cancer Res 28, 1979–1990 (2022).
Ouellette, M. M., Zhou, S. & Yan, Y. Cell Signaling Pathways That Promote Radioresistance of Cancer Cells. Diagnostics (Basel) 12, 656 (2022).
Wozny, A. S. et al. Involvement of HIF-1alpha in the Detection, Signaling, and Repair of DNA Double-Strand Breaks after Photon and Carbon-Ion Irradiation. Cancers (Basel) 13, 3833 (2021).
Zaretsky, J. M. et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 375, 819–829 (2016).
Su, S. et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 172, 841–856 e816 (2018).
Wang, X. et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 22, 514–528.e515 (2018).
Tammela, T. et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 545, 355–359 (2017).
Guo, L., Lee, Y. T., Zhou, Y. & Huang, Y. Targeting epigenetic regulatory machinery to overcome cancer therapy resistance. Semin. Cancer Biol. 83, 487–502 (2021).
Tan, T. et al. Epigenetic modification regulates tumor progression and metastasis through EMT (Review). Int. J. Oncol. 60, 70 (2022).
Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 (2016).
Yosifov, D. Y. et al. DNA methylation of chronic lymphocytic leukemia with differential response to chemotherapy. Sci. Data 7, 133 (2020).
Cardenas, H. et al. Methylomic Signatures of High Grade Serous Ovarian Cancer. Epigenetics 16, 1201–1216 (2021).
Selli, C. et al. Molecular changes during extended neoadjuvant letrozole treatment of breast cancer: distinguishing acquired resistance from dormant tumours. Breast Cancer Res 21, 2 (2019).
Stone, A. et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat. Commun. 6, 7758 (2015).
Reina-Campos, M. et al. Increased Serine and One-Carbon Pathway Metabolism by PKClambda/iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 35, 385–400 e389 (2019).
Nachiyappan, A., Gupta, N. & Taneja, R. EHMT1/EHMT2 in EMT, cancer stemness and drug resistance: emerging evidence and mechanisms. FEBS J. 289, 1329–1351 (2022).
Wang, Z. et al. SETD5-Coordinated Chromatin Reprogramming Regulates Adaptive Resistance to Targeted Pancreatic Cancer Therapy. Cancer Cell 37, 834–849 e813 (2020).
Knoechel, B. et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet. 46, 364–370 (2014).
Zawistowski, J. S. et al. Enhancer Remodeling during Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex. Cancer Disco. 7, 302–321 (2017).
Zheng, X. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015).
Yochum, Z. A. et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 38, 656–670 (2019).
Dai, C., Heemers, H. & Sharifi, N. Androgen Signaling in Prostate Cancer. Cold Spring Harb. Perspect. Med. 7, a030452 (2017).
Masiello, D., Cheng, S., Bubley, G. J., Lu, M. L. & Balk, S. P. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J. Biol. Chem. 277, 26321–26326 (2002).
Ito, Y. & Sadar, M. D. Enzalutamide and blocking androgen receptor in advanced prostate cancer: lessons learnt from the history of drug development of antiandrogens. Res Rep. Urol. 10, 23–32 (2018).
Fujita, K. & Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens. Health 37, 288–295 (2019).
Lee, H., Jeong, A. J. & Ye, S. K. Highlighted STAT3 as a potential drug target for cancer therapy. BMB Rep. 52, 415–423 (2019).
Wang, H. et al. MicroRNA-181d-5p-Containing Exosomes Derived from CAFs Promote EMT by Regulating CDX2/HOXA5 in Breast Cancer. Mol. Ther. Nucleic Acids 19, 654–667 (2020).
East, M. P. & Johnson, G. L. Adaptive chromatin remodeling and transcriptional changes of the functional kinome in tumor cells in response to targeted kinase inhibition. J. Biol. Chem. 298, 101525 (2022).
Wessely, A., Steeb, T., Berking, C. & Heppt, M. V. How Neural Crest Transcription Factors Contribute to Melanoma Heterogeneity, Cellular Plasticity, and Treatment Resistance. Int. J. Mol. Sci. 22, 5761 (2021).
Yang, C., Tian, C., Hoffman, T. E., Jacobsen, N. K. & Spencer, S. L. Melanoma subpopulations that rapidly escape MAPK pathway inhibition incur DNA damage and rely on stress signalling. Nat. Commun. 12, 1747 (2021).
Sachindra et al. New role of ID3 in melanoma adaptive drug-resistance. Oncotarget 8, 110166–110175 (2017).
Sun, C. et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122 (2014).
Hartman, M. L., Sztiller-Sikorska, M., Gajos-Michniewicz, A. & Czyz, M. Dissecting Mechanisms of Melanoma Resistance to BRAF and MEK Inhibitors Revealed Genetic and Non-Genetic Patient- and Drug-Specific Alterations and Remarkable Phenotypic Plasticity. Cells 9, 142 (2020).
Salaritabar, A. et al. Targeting Hedgehog signaling pathway: Paving the road for cancer therapy. Pharmacol. Res. 141, 466–480 (2019).
Choi, J. I. et al. Cancer-initiating cells in human pancreatic cancer organoids are maintained by interactions with endothelial cells. Cancer Lett. 498, 42–53 (2021).
Sacchetti, A. et al. Phenotypic plasticity underlies local invasion and distant metastasis in colon cancer. Elife 10, e61461 (2021).
Yang, X. et al. Wnt signaling in triple-negative breast cancers: Its roles in molecular subtyping and cancer cell stemness and its crosstalk with non-coding RNAs. Life Sci. 300, 120565 (2022).
Giroux-Leprieur, E., Costantini, A., Ding, V. W. & He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 19, 2835 (2018).
Bhateja, P., Cherian, M., Majumder, S. & Ramaswamy, B. The Hedgehog Signaling Pathway: A Viable Target in Breast Cancer? Cancers (Basel) 11, 1126 (2019).
Monkkonen, T. & Lewis, M. T. New paradigms for the Hedgehog signaling network in mammary gland development and breast Cancer. Biochim Biophys. Acta Rev. Cancer 1868, 315–332 (2017).
Song, J. et al. Noncoding RNAs related to the hedgehog pathway in cancer: clinical implications and future perspectives. Mol. Cancer 21, 115 (2022).
Cortes, J. E., Gutzmer, R., Kieran, M. W. & Solomon, J. A. Hedgehog signaling inhibitors in solid and hematological cancers. Cancer Treat. Rev. 76, 41–50 (2019).
Xie, Y. et al. Proteasome inhibitor induced SIRT1 deacetylates GLI2 to enhance hedgehog signaling activity and drug resistance in multiple myeloma. Oncogene 39, 922–934 (2020).
Merchant, A. A. & Matsui, W. Targeting Hedgehog-a cancer stem cell pathway. Clin. Cancer Res 16, 3130–3140 (2010).
Cazet, A. S. et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 9, 2897 (2018).
Barzegar Behrooz, A. et al. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 23, 1353 (2022).
Wang, J. et al. Cinobufacini Inhibits Colon Cancer Invasion and Metastasis via Suppressing Wnt/β-Catenin Signaling Pathway and EMT. Am. J. Chin. Med 48, 703–718 (2020).
Moon, J. H., Lee, S. H. & Lim, Y. C. Wnt/β-catenin/Slug pathway contributes to tumor invasion and lymph node metastasis in head and neck squamous cell carcinoma. Clin. Exp. Metastasis 38, 163–174 (2021).
Sun, Y., Song, G. D., Sun, N., Chen, J. Q. & Yang, S. S. Slug overexpression induces stemness and promotes hepatocellular carcinoma cell invasion and metastasis. Oncol. Lett. 7, 1936–1940 (2014).
Tian, S. et al. SERPINH1 regulates EMT and gastric cancer metastasis via the Wnt/β-catenin signaling pathway. Aging (Albany N. Y.) 12, 3574–3593 (2020).
Howe, L. R., Watanabe, O., Leonard, J. & Brown, A. M. Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res 63, 1906–1913 (2003).
Zhu, Q. Q., Ma, C., Wang, Q., Song, Y. & Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumour Biol. 37, 185–197 (2016).
Liu, X. et al. Cellular retinol binding protein-1 inhibits cancer stemness via upregulating WIF1 to suppress Wnt/beta-catenin pathway in hepatocellular carcinoma. BMC Cancer 21, 1224 (2021).
Suda, T. et al. Dickkopf-1 Promotes Angiogenesis and is a Biomarker for Hepatic Stem Cell-like Hepatocellular Carcinoma. Int. J. Mol. Sci. 23, 2801 (2022).
Rong, L., Xu, Y., Zhang, K., Jin, L. & Liu, X. HNRNPA2B1 inhibited SFRP2 and activated Wnt-beta/catenin via m6A-mediated miR-106b-5p processing to aggravate stemness in lung adenocarcinoma. Pathol. Res. Pract. 233, 153794 (2022).
Vermeulen, L. et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 12, 468–476 (2010).
Deldar Abad Paskeh, M., Mirzaei, S., Ashrafizadeh, M., Zarrabi, A. & Sethi, G. Wnt/β-Catenin Signaling as a Driver of Hepatocellular Carcinoma Progression: An Emphasis on Molecular Pathways. J. Hepatocell. Carcinoma 8, 1415–1444 (2021).
Zhan, T. et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 10, 2197 (2019).
Debeb, B. G. et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/β-catenin signaling. Stem Cells 30, 2366–2377 (2012).
De Santis, F. et al. BCL6 and the Notch pathway: a signaling axis leading to a novel druggable biotarget in triple negative breast cancer. Cell. Oncol. (Dordr.) 45, 257–274 (2022).
Buyuk, B., Jin, S. & Ye, K. Epithelial-to-Mesenchymal Transition Signaling Pathways Responsible for Breast Cancer Metastasis. Cell. Mol. Bioeng. 15, 1–13 (2022).
Wang, L. L., Wan, X. Y., Liu, C. Q. & Zheng, F. M. NDR1 increases NOTCH1 signaling activity by impairing Fbw7 mediated NICD degradation to enhance breast cancer stem cell properties. Mol. Med. 28, 49 (2022).
Koba, H. et al. NOTCH alteration in EGFR-mutated lung adenocarcinoma leads to histological small-cell carcinoma transformation under EGFR-TKI treatment. Transl. Lung Cancer Res 10, 4161–4173 (2021).
Anobile, D. P. et al. Splicing deregulation, microRNA and notch aberrations: fighting the three-headed dog to overcome drug resistance in malignant mesothelioma. Expert Rev. Clin. Pharmacol. 15, 305–322 (2022).
Uramoto, H., Shimokawa, H., Hanagiri, T., Kuwano, M. & Ono, M. Expression of selected gene for acquired drug resistance to EGFR-TKI in lung adenocarcinoma. Lung Cancer 73, 361–365 (2011).
Xie, M. et al. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 113, 1501–1513 (2012).
Shen, Y. et al. Estrogen receptor alpha-NOTCH1 axis enhances basal stem-like cells and epithelial-mesenchymal transition phenotypes in prostate cancer. Cell Commun. Signal 17, 50 (2019).
von Arx, C. et al. Updates on the Role of Molecular Alterations and NOTCH Signalling in the Development of Neuroendocrine Neoplasms. J. Clin. Med 8, 1277 (2019).
Natsuizaka, M. et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun. 8, 1758 (2017).
Zeng, D. et al. Inhibition of Notch1 reverses EMT and chemoresistance to cisplatin via direct downregulation of MCAM in triple-negative breast cancer cells. Int. J. Cancer 147, 490–504 (2020).
Saad, S., Stanners, S. R., Yong, R., Tang, O. & Pollock, C. A. Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the Snail transcription factor. Int. J. Biochem. Cell Biol. 42, 1115–1122 (2010).
Deshmukh, A. P. et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl Acad. Sci. U. S. A. 118, e2102050118 (2021).
Noman, M. Z. et al. Improving Cancer Immunotherapy by Targeting the Hypoxic Tumor Microenvironment: New Opportunities and Challenges. Cells 8, 1083 (2019).
Zheng, X., Yu, C. & Xu, M. Linking Tumor Microenvironment to Plasticity of Cancer Stem Cells: Mechanisms and Application in Cancer Therapy. Front. Oncol. 11, 678333 (2021).
Capparelli, C. et al. Targeting SOX10-deficient cells to reduce the dormant-invasive phenotype state in melanoma. Nat. Commun. 13, 1381 (2022).
Zhou, F. et al. Tumor Microenvironment-Activatable Prodrug Vesicles for Nanoenabled Cancer Chemoimmunotherapy Combining Immunogenic Cell Death Induction and CD47 Blockade. Adv. Mater. 31, e1805888 (2019).
Kalluri, R. & Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401 (2006).
Luque, M. et al. Targeted Therapy Modulates the Secretome of Cancer-Associated Fibroblasts to Induce Resistance in HER2-Positive Breast Cancer. Int. J. Mol. Sci. 22, 13297 (2021).
Huang, T. X., Guan, X. Y. & Fu, L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. Am. J. Cancer Res. 9, 1889–1904 (2019).
Doherty, M. R., Smigiel, J. M., Junk, D. J. & Jackson, M. W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers (Basel) 8, 8 (2016).
Yu, Y. et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 110, 724–732 (2014).
Luo, H., Tu, G., Liu, Z. & Liu, M. Cancer-associated fibroblasts: a multifaceted driver of breast cancer progression. Cancer Lett. 361, 155–163 (2015).
Apicella, M. et al. Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab. 28, 848–865 e846 (2018).
Gkountakos, A. et al. Identification of Targetable Liabilities in the Dynamic Metabolic Profile of EGFR-Mutant Lung Adenocarcinoma: Thinking beyond Genomics for Overcoming EGFR TKI Resistance. Biomedicines 10, 277 (2022).
Straussman, R. et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487, 500–504 (2012).
Wilson, T. R. et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487, 505–509 (2012).
Mahaweni, N. M., Bos, G. M. J., Mitsiades, C. S., Tilanus, M. G. J. & Wieten, L. Daratumumab augments alloreactive natural killer cell cytotoxicity towards CD38+ multiple myeloma cell lines in a biochemical context mimicking tumour microenvironment conditions. Cancer Immunol. Immunother. 67, 861–872 (2018).
Ikeda, S. & Tagawa, H. Impact of hypoxia on the pathogenesis and therapy resistance in multiple myeloma. Cancer Sci. 112, 3995–4004 (2021).
Barr, T. A. et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J. Exp. Med 209, 1001–1010 (2012).
Jung, B. G. et al. Early Secreted Antigenic Target of 6-kDa of Mycobacterium tuberculosis Stimulates IL-6 Production by Macrophages through Activation of STAT3. Sci. Rep. 7, 40984 (2017).
Shintani, Y. et al. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 11, 1482–1492 (2016).
Li, L. et al. Ibrutinib reverses IL-6-induced osimertinib resistance through inhibition of Laminin α5/FAK signaling. Commun. Biol. 5, 155 (2022).
Ju, X. et al. IL-6 regulates extracellular matrix remodeling associated with aortic dilation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J. Am. Heart Assoc. 3, e000476 (2014).
Zoeller, J. J., Bronson, R. T., Selfors, L. M., Mills, G. B. & Brugge, J. S. Niche-localized tumor cells are protected from HER2-targeted therapy via upregulation of an anti-apoptotic program in vivo. NPJ Breast Cancer 3, 18 (2017).
Fu, Y. et al. Abnormally activated OPN/integrin αVβ3/FAK signalling is responsible for EGFR-TKI resistance in EGFR mutant non-small-cell lung cancer. J. Hematol. Oncol. 13, 169 (2020).
Despeaux, M. et al. Critical features of FAK-expressing AML bone marrow microenvironment through leukemia stem cell hijacking of mesenchymal stromal cells. Leukemia 25, 1789–1793 (2011).
Namba, K. et al. Activation of AXL as a Preclinical Acquired Resistance Mechanism Against Osimertinib Treatment in EGFR-Mutant Non-Small Cell Lung Cancer Cells. Mol. Cancer Res 17, 499–507 (2019).
Lee, A.-F. et al. Reverse epithelial-mesenchymal transition contributes to the regain of drug sensitivity in tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. PLoS One 12, e0180383 (2017).
Wu, D. M. et al. The PAX6-ZEB2 axis promotes metastasis and cisplatin resistance in non-small cell lung cancer through PI3K/AKT signaling. Cell Death Dis. 10, 349 (2019).
Raoof, S. et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 38, 6399–6413 (2019).
Nieszporek, A., Skrzypek, K., Adamek, G. & Majka, M. Molecular mechanisms of epithelial to mesenchymal transition in tumor metastasis. Acta Biochim. Pol. 66, 509–520 (2019).
Chang, T. H. et al. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am. J. Respir. Crit. Care Med 183, 1071–1079 (2011).
Hao, Y., Baker, D. & Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 20, 2767 (2019).
Hua, W., Ten Dijke, P., Kostidis, S., Giera, M. & Hornsveld, M. TGFbeta-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol. Life Sci. 77, 2103–2123 (2020).
Santamaria, P. G., Moreno-Bueno, G. & Cano, A. Contribution of Epithelial Plasticity to Therapy Resistance. J. Clin. Med 8, 676 (2019).
Colak, S. & Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 3, 56–71 (2017).
Xu, W., Yang, Z. & Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. 9, 317–324 (2015).
Lee, M. K. et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957–3967 (2007).
Katsuno, Y. et al. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal 12, eaau8544 (2019).
Pan, G., Liu, Y., Shang, L., Zhou, F. & Yang, S. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun. (Lond.) 41, 199–217 (2021).
Zhang, J. & Ma, L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 31, 653–662 (2012).
Li, J., Yu, H., Xi, M., Ma, D. & Lu, X. The SNAI1 3’UTR functions as a sponge for multiple migration-/invasion-related microRNAs. Tumour Biol. 36, 1067–1072 (2015).
Xiao, B., Shi, X. & Bai, J. miR-30a regulates the proliferation and invasion of breast cancer cells by targeting Snail. Oncol. Lett. 17, 406–413 (2019).
Liu, Y. N. et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene 32, 296–306 (2013).
Dou, R. et al. EMT-cancer cells-derived exosomal miR-27b-3p promotes circulating tumour cells-mediated metastasis by modulating vascular permeability in colorectal cancer. Clin. Transl. Med 11, e595 (2021).
Lu, N. et al. miRNA‑490‑3p promotes the metastatic progression of invasive ductal carcinoma. Oncol. Rep. 45, 706–716 (2021).
Li, Y., Long, X., Wang, J., Peng, J. & Shen, K. miRNA-128 modulates bone neoplasms cells proliferation and migration through the WNT/β-catenin and EMT signal pathways. J. Orthop. Surg. Res. 16, 71 (2021).
Wang, D. et al. MiRNA-155-5p inhibits epithelium-to-mesenchymal transition (EMT) by targeting GSK-3β during radiation-induced pulmonary fibrosis. Arch. Biochem. Biophys. 697, 108699 (2021).
Dagogo-Jack, I. & Shaw, A. T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94 (2018).
Fanelli, G. N., Naccarato, A. G. & Scatena, C. Recent Advances in Cancer Plasticity: Cellular Mechanisms, Surveillance Strategies, and Therapeutic Optimization. Front Oncol. 10, 569 (2020).
Morel, A. P. et al. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3, e2888 (2008).
Nimmakayala, R. K. et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene 40, 215–231 (2021).
Lei, X. et al. Cancer stem cells in colorectal cancer and the association with chemotherapy resistance. Med. Oncol. 38, 43 (2021).
Lam, K.-H. & Ma, S. Noncellular components in the liver cancer stem cell niche: Biology and potential clinical implications. Hepatology 21, 1–15 (2022).
Quintana, E. et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 18, 510–523 (2010).
Boiko, A. D. et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 466, 133–137 (2010).
Charles, N. et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 6, 141–152 (2010).
Nakano, M. et al. Dedifferentiation process driven by TGF-beta signaling enhances stem cell properties in human colorectal cancer. Oncogene 38, 780–793 (2019).
de Sousa e Melo, F. et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 543, 676–680 (2017).
Oxnard, G. R. et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 34, 3375–3382 (2016).
Chabon, J. J. et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 7, 11815 (2016).
Suda, K. et al. Heterogeneity in resistance mechanisms causes shorter duration of epidermal growth factor receptor kinase inhibitor treatment in lung cancer. Lung Cancer 91, 36–40 (2016).
Le Magnen, C., Shen, M. M. & Abate-Shen, C. Lineage Plasticity in Cancer Progression and Treatment. Annu Rev. Cancer Biol. 2, 271–289 (2018).
Easwaran, H. et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res 22, 837–849 (2012).
Xiao, Y. & Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharm. Ther. 221, 107753 (2021).
Anderson, N. M. & Simon, M. C. The tumor microenvironment. Curr. Biol. 30, R921–r925 (2020).
Hanahan, D. & Coussens, L. M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322 (2012).
Baghban, R. et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 18, 59 (2020).
Lequeux, A. et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 458, 13–20 (2019).
Paolicchi, E. et al. Targeting hypoxic response for cancer therapy. Oncotarget 7, 13464–13478 (2016).
Bae, H. J., Liu, S., Jin, P. & Stroncek, D. Mesenchymal stromal cell plasticity and the tumor microenvironment. Emerg. Top. Life Sci. 1, 487–492 (2017).
Wang, X., Zhang, H. & Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2, 141–160 (2019).
Reynolds, T. Y., Rockwell, S. & Glazer, P. M. Genetic instability induced by the tumor microenvironment. Cancer Res 56, 5754–5757 (1996).
Zhang, A., Miao, K., Sun, H. & Deng, C. X. Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. Int. J. Biol. Sci. 18, 3019–3033 (2022).
Rambow, F. et al. Toward minimal residual disease-directed therapy in melanoma. Cell 174, 843–855. e819 (2018).
Ramirez, M. et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. commun. 7, 1–8 (2016).
Tan, C.-S., Gilligan, D. & Pacey, S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 16, e447–e459 (2015).
Winder, M. & Virós, A. Mechanisms of drug resistance in melanoma. Handb. Exp. Pharm. 249, 91–108 (2018).
Sharma, S. V. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010).
Ravindran Menon, D. et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 34, 4448–4459 (2015).
Liau, B. B. et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. cell stem cell 20, 233–246. e237 (2017).
Kruidenier, L. et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488, 404–408 (2012).
Heinemann, B. et al. Inhibition of demethylases by GSK-J1/J4. Nature 514, E1–E2 (2014).
Johansson, C. et al. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat. Chem. Biol. 12, 539–545 (2016).
Yang, G. J. et al. Selective inhibition of lysine‐specific demethylase 5A (KDM5A) using a rhodium (III) complex for triple‐negative breast cancer therapy. Angew. Chem. Int Ed. Engl. 57, 13091–13095 (2018).
Liu, J. et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl Acad. Sci. U. S. A. 110, 20224–20229 (2013).
Liang, X., Lin, A., Wang, Q., Zhang, J. & Luo, P. Cell plasticity in patients with NSCLC: The controversial origins of transformed SCLC. Biomed. Pharmacother. 149, 112909 (2022).
Marcoux, N. et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J. Clin. Oncol. 37, 278 (2019).
Hangauer, M. J. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Yoo, S.-E. et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic. Biol. Med. 52, 1820–1827 (2012).
Eaton, J. K. et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 16, 497–506 (2020).
Santamaria, P. G., Moreno-Bueno, G., Portillo, F. & Cano, A. EMT: Present and future in clinical oncology. Mol. Oncol. 11, 718–738 (2017).
Voon, D. C., Huang, R. Y., Jackson, R. A. & Thiery, J. P. The EMT spectrum and therapeutic opportunities. Mol. Oncol. 11, 878–891 (2017).
Baram, T., Rubinstein-Achiasaf, L., Ben-Yaakov, H. & Ben-Baruch, A. Inflammation-driven breast tumor cell plasticity: stemness/EMT, therapy resistance and dormancy. Front. Oncol. 10, 614468 (2021).
Hasan, T. et al. Interleukin-8/CXCR2 signaling regulates therapy-induced plasticity and enhances tumorigenicity in glioblastoma. Cell Death Dis. 10, 1–17 (2019).
Fousek, K., Horn, L. A. & Palena, C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol. Ther. 219, 107692 (2021).
Xiao, P. et al. Neurotensin/IL-8 pathway orchestrates local inflammatory response and tumor invasion by inducing M2 polarization of Tumor-Associated macrophages and epithelial-mesenchymal transition of hepatocellular carcinoma cells. Oncoimmunology 7, e1440166 (2018).
Kahraman, D. C., Kahraman, T. & Cetin-Atalay, R. Targeting PI3K/Akt/mTOR pathway identifies differential expression and functional role of IL8 in liver cancer stem cell enrichment. Mol. Cancer Ther. 18, 2146–2157 (2019).
Dominguez, C., McCampbell, K. K., David, J. M. & Palena, C. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI insight 2, e94296 (2017).
Fernando, R. I. et al. IL-8 signaling is involved in resistance of lung carcinoma cells to erlotinib. Oncotarget 7, 42031 (2016).
Devapatla, B., Sharma, A. & Woo, S. CXCR2 inhibition combined with sorafenib improved antitumor and antiangiogenic response in preclinical models of ovarian cancer. PLoS One 10, e0139237 (2015).
Xu, J., Lamouille, S. & Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res 19, 156–172 (2009).
Grauel, A. L. et al. TGFβ-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat. commun. 11, 1–17 (2020).
Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. 13, 616–630 (2012).
Akhurst, R. J. & Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Disco. 11, 790–811 (2012).
de Gramont, A., Faivre, S. & Raymond, E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology 6, e1257453 (2017).
Pattabiraman, D. R. et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 351, aad3680 (2016).
Yingling, J. M. et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget 9, 6659 (2018).
Melisi, D. et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 119, 1208–1214 (2018).
Horn, L. A. et al. Remodeling the tumor microenvironment via blockade of LAIR-1 and TGF-β signaling enables PD-L1–mediated tumor eradication. J. Clin. Investig. 132, e155148 (2022).
Xing, Y. et al. Therapeutic efficacy and mechanism of CD73-TGFβ dual-blockade in a mouse model of triple-negative breast cancer. Acta Pharmacol. Sin. 43, 2410–2418 (2022).
Gan, L. et al. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark. Res. 6, 1–10 (2018).
Gallardo, A. et al. EZH2 endorses cell plasticity to carcinoma cells facilitating mesenchymal to epithelial transition and tumour colonization. Oncogene 41, 3611–3624 (2022).
Dardenne, E. et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 30, 563–577 (2016).
Knutson, S. K. et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 13, 842–854 (2014).
Vaswani, R. G. et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1, 2-dihydropyridin-3-yl) methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl) piperidin-4-yl) ethyl)-1 H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J. Med. Chem. 59, 9928–9941 (2016).
Gagliardi, M. et al. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 10, 902 (2019).
Igarashi, K., Nishizawa, H., Saiki, Y. & Matsumoto, M. The transcription factor BACH1 at the crossroads of cancer biology: From epithelial-mesenchymal transition to ferroptosis. J. Biol. Chem. 297, 101032 (2021).
Xie, J. et al. Targeting cancer cell plasticity by HDAC inhibition to reverse EBV-induced dedifferentiation in nasopharyngeal carcinoma. Signal Transduct. Target Ther. 6, 333 (2021).
Liu, Y. et al. Yap-Sox9 signaling determines hepatocyte plasticity and lineage-specific hepatocarcinogenesis. J. Hepatol. 76, 652–664 (2022).
Pei, J. et al. Targeting lysosomal degradation pathways: new strategies and techniques for drug discovery. J. Med. Chem. 64, 3493–3507 (2021).
Sievers, Q. L. et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362, eaat0572 (2018).
Kong, F.-E. et al. Targeting tumor lineage plasticity in hepatocellular carcinoma using an anti-CLDN6 antibody-drug conjugate. Sci. Transl. Med. 13, eabb6282 (2021).
Huang, F. et al. Inhibiting the MNK1/2-eIF4E axis impairs melanoma phenotype switching and potentiates antitumor immune responses. J. Clin. Investig. 131, e140752 (2021).
Chae, Y. K. et al. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 8, 2918 (2018).
Lou, Y. et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin. Cancer Res 22, 3630–3642 (2016).
Wu, T. et al. PD-L1-Mediated Immunosuppression in Oral Squamous Cell Carcinoma: Relationship With Macrophage Infiltration and Epithelial to Mesenchymal Transition Markers. Front. Immunol. 12, 693881 (2021).
Guo, Y. et al. Zeb1 induces immune checkpoints to form an immunosuppressive envelope around invading cancer cells. Sci. Adv. 7, eabd7455 (2021).
Zhao, K. et al. TIGIT blockade enhances tumor response to radiotherapy via a CD103 + dendritic cell-dependent mechanism. Cancer Immunol. Immunother. 72, 193–209 (2023).
Mahmoudian, R. A. et al. Correlation between the immune checkpoints and EMT genes proposes potential prognostic and therapeutic targets in ESCC. J. Mol. Histol. 52, 597–609 (2021).
Chen, X. et al. eEF2K promotes PD-L1 stabilization through inactivating GSK3β in melanoma. J. Immunother. Cancer 10, e004026 (2022).
Wu, Y. et al. Effect of ISM1 on the Immune Microenvironment and Epithelial-Mesenchymal Transition in Colorectal Cancer. Front Cell Dev. Biol. 9, 681240 (2021).
Dongre, A. et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res 77, 3982–3989 (2017).
Wu, Q. et al. MAGE-C3 promotes cancer metastasis by inducing epithelial-mesenchymal transition and immunosuppression in esophageal squamous cell carcinoma. Cancer Commun. (Lond.) 41, 1354–1372 (2021).
Kim, S. et al. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum. Pathol. 58, 7–14 (2016).
Cui, M. et al. Development and Validation of a Tumor Mutation Burden-Related Immune Prognostic Signature for Ovarian Cancers. Front Genet 12, 688207 (2021).
Cao, L. et al. Exploring Immune-Related Prognostic Signatures in the Tumor Microenvironment of Colon Cancer. Front Genet 13, 801484 (2022).
Song, J. et al. Pan-cancer analysis reveals RIPK2 predicts prognosis and promotes immune therapy resistance via triggering cytotoxic T lymphocytes dysfunction. Mol. Med. 28, 47 (2022).
Yang, C. Y. et al. Association between programmed death-ligand 1 expression, immune microenvironments, and clinical outcomes in epidermal growth factor receptor mutant lung adenocarcinoma patients treated with tyrosine kinase inhibitors. Eur. J. Cancer 124, 110–122 (2020).
Isomoto, K. et al. Impact of EGFR-TKI Treatment on the Tumor Immune Microenvironment in EGFR Mutation-Positive Non-Small Cell Lung Cancer. Clin. Cancer Res 26, 2037–2046 (2020).
Haratani, K. et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann. Oncol. 28, 1532–1539 (2017).
Jia, Y. et al. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: Implications for combination therapies. Int. J. Cancer 145, 1432–1444 (2019).
Tian, T. et al. Front-Line ICI-Based Combination Therapy Post-TKI Resistance May Improve Survival in NSCLC Patients With EGFR Mutation. Front. Oncol. 11, 739090 (2021).
Chen, Y. et al. Immunotherapy-based combination strategies for treatment of EGFR-TKI-resistant non-small-cell lung cancer. Future Oncol. 18, 1757–1775 (2022).
Jiang, T. et al. Toripalimab plus chemotherapy as second-line treatment in previously EGFR-TKI treated patients with EGFR-mutant-advanced NSCLC: a multicenter phase-II trial. Signal Transduct. Target Ther. 6, 355 (2021).
Liu, S. et al. Patients With Short PFS to EGFR-TKIs Predicted Better Response to Subsequent Anti-PD-1/PD-L1 Based Immunotherapy in EGFR Common Mutation NSCLC. Front. Oncol. 11, 639947 (2021).
Greten, T. F., Lai, C. W., Li, G. & Staveley-O’Carroll, K. F. Targeted and Immune-Based Therapies for Hepatocellular Carcinoma. Gastroenterology 156, 510–524 (2019).
Shemesh, C. S. et al. Atezolizumab and Bevacizumab in Patients with Unresectable Hepatocellular Carcinoma: Pharmacokinetic and Safety Assessments Based on Hepatic Impairment Status and Geographic Region. Liver Cancer 10, 485–499 (2021).
Qin, S. et al. Atezolizumab plus Bevacizumab versus Sorafenib in the Chinese Subpopulation with Unresectable Hepatocellular Carcinoma: Phase 3 Randomized, Open-Label IMbrave150 Study. Liver Cancer 10, 296–308 (2021).
Rizzo, A., Ricci, A. D., Gadaleta-Caldarola, G. & Brandi, G. First-line immune checkpoint inhibitor-based combinations in unresectable hepatocellular carcinoma: current management and future challenges. Expert Rev. Gastroenterol. Hepatol. 15, 1245–1251 (2021).
Cheng, A. L. et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 76, 862–873 (2022).
Tang, Z. et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res 27, 4898–4909 (2021).
Siewe, N. & Friedman, A. Combination therapy for mCRPC with immune checkpoint inhibitors, ADT and vaccine: A mathematical model. PLoS One 17, e0262453 (2022).
Guan, X. et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature 606, 791–796 (2022).
Wang, Y., Ma, X., Wei, Y., Ma, D. & Gong, P. Effect of platinum-based chemotherapy on EGFR gene mutation status in lung adenocarcinoma. Med. (Baltim.) 97, e9602 (2018).
Leonetti, A. et al. Molecular basis and rationale for combining immune checkpoint inhibitors with chemotherapy in non-small cell lung cancer. Drug Resist Updat 46, 100644 (2019).
Borghaei, H. et al. Pembrolizumab plus chemotherapy versus chemotherapy alone in patients with advanced non-small cell lung cancer without tumor PD-L1 expression: A pooled analysis of 3 randomized controlled trials. Cancer 126, 4867–4877 (2020).
Zhang, J. et al. MA11.06 A PII Study of Toripalimab, a PD-1 mAb, in Combination with Chemotherapy in EGFR+ Advanced NSCLC Patients Failed to Prior EGFR TKI Therapies. J. Thorac. Oncol. 14, s292 (2019).
Reck, M. et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med 7, 387–401 (2019).
Janjigian, Y. Y. et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet 398, 27–40 (2021).
Janjigian, Y. Y. et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 600, 727–730 (2021).
Miles, D. et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 32, 994–1004 (2021).
Schmid, P. et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 379, 2108–2121 (2018).
Zhang, Y. et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 39, 1578–1593 e1578 (2021).
Lu, Q. et al. Bioengineered Platelets Combining Chemotherapy and Immunotherapy for Postsurgical Melanoma Treatment: Internal Core-Loaded Doxorubicin and External Surface-Anchored Anti-PD-L1 Antibody Backpacks. Nano Lett. 22, 3141–3150 (2022).
Ahn, M. J., Sun, J. M., Lee, S. H., Ahn, J. S. & Park, K. EGFR TKI combination with immunotherapy in non-small cell lung cancer. Expert Opin. Drug Saf. 16, 465–469 (2017).
Yang, J. C. et al. Osimertinib Plus Durvalumab versus Osimertinib Monotherapy in EGFR T790M-Positive NSCLC following Previous EGFR TKI Therapy: CAURAL Brief Report. J. Thorac. Oncol. 14, 933–939 (2019).
Oxnard, G. R. et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 31, 507–516 (2020).
Oshima, Y., Tanimoto, T., Yuji, K. & Tojo, A. EGFR-TKI-Associated Interstitial Pneumonitis in Nivolumab-Treated Patients With Non-Small Cell Lung Cancer. JAMA Oncol. 4, 1112–1115 (2018).
Jia, Y. et al. Impact of EGFR-TKIs combined with PD-L1 antibody on the lung tissue of EGFR-driven tumor-bearing mice. Lung Cancer 137, 85–93 (2019).
Schoenfeld, A. J. et al. Severe immune-related adverse events are common with sequential PD-(L)1 blockade and osimertinib. Ann. Oncol. 30, 839–844 (2019).
Ribas, A., Hodi, F. S., Callahan, M., Konto, C. & Wolchok, J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 368, 1365–1366 (2013).
Yu, Y. Multi-target combinatory strategy to overcome tumor immune escape. Front. Med. 16, 208–215 (2022).
Wolchok, J. D. et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 377, 1345–1356 (2017).
Kariolis, M. S. et al. Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J. Clin. Invest 127, 183–198 (2017).
Leconet, W. et al. Therapeutic Activity of Anti-AXL Antibody against Triple-Negative Breast Cancer Patient-Derived Xenografts and Metastasis. Clin. Cancer Res 23, 2806–2816 (2017).
Ghiso, E. et al. YAP-Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 19, 1012–1021 (2017).
Yu, M. et al. YAP1 contributes to NSCLC invasion and migration by promoting Slug transcription via the transcription co-factor TEAD. Cell Death Dis. 9, 464 (2018).
Nyakas, M. et al. AXL inhibition improves BRAF-targeted treatment in melanoma. Sci. Rep. 12, 5076 (2022).
Scherschinski, L. et al. Regulation of the Receptor Tyrosine Kinase AXL in Response to Therapy and Its Role in Therapy Resistance in Glioblastoma. Int. J. Mol. Sci. 23, 982 (2022).
Shen, Y., Chen, X., He, J., Liao, D. & Zu, X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 198, 99–111 (2018).
Myers, S. H., Brunton, V. G. & Unciti-Broceta, A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 59, 3593–3608 (2016).
Wang, J. et al. Ginsenoside Rg3 sensitizes hypoxic lung cancer cells to cisplatin via blocking of NF-κB mediated epithelial-mesenchymal transition and stemness. Cancer Lett. 415, 73–85 (2018).
Wang, C. et al. Tumor-derived secretory clusterin induces epithelial-mesenchymal transition and facilitates hepatocellular carcinoma metastasis. Int. J. Biochem. Cell Biol. 44, 2308–2320 (2012).
Praharaj, P. P., Patra, S., Panigrahi, D. P., Patra, S. K. & Bhutia, S. K. Clusterin as modulator of carcinogenesis: A potential avenue for targeted cancer therapy. Biochim Biophys. Acta Rev. Cancer 1875, 188500 (2021).
Shahbaz, S. et al. CD71+VISTA+ erythroid cells promote the development and function of regulatory T cells through TGF-β. PLoS Biol. 16, e2006649 (2018).
Yingling, J. M. et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-beta receptor type I inhibitor. Oncotarget 9, 6659–6677 (2018).
Song, B. et al. Targeting FOXA1-mediated repression of TGF-beta signaling suppresses castration-resistant prostate cancer progression. J. Clin. Invest 129, 569–582 (2019).
Kelley, R. K. et al. A Phase 2 Study of Galunisertib (TGF-beta1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 10, e00056 (2019).
Harding, J. J. et al. Phase 1b study of galunisertib and ramucirumab in patients with advanced hepatocellular carcinoma. Cancer Med 10, 3059–3067 (2021).
Wick, A. et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Invest. N. Drugs 38, 1570–1579 (2020).
Melisi, D. et al. Safety and activity of the TGFbeta receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 9, e002068 (2021).
Tauriello, D. V. F. et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543 (2018).
Lan, Y. et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci. Transl. Med. 10, eaan5488 (2018).
Knudson, K. M. et al. M7824, a novel bifunctional anti-PD-L1/TGFβ Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology 7, e1426519 (2018).
Ravi, R. et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat. Commun. 9, 741 (2018).
Strauss, J. et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with human papillomavirus-associated malignancies. J. Immunother. Cancer 8, e001395 (2020).
Tan, B. et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Patients with Esophageal Adenocarcinoma: Results from a Phase 1 Cohort. Target. Oncol. 16, 435–446 (2021).
Yoo, C. et al. Phase I study of bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with pretreated biliary tract cancer. J. Immunother. Cancer 8, e000564 (2020).
Ma, F. et al. Autocrine Canonical Wnt Signaling Primes Noncanonical Signaling through ROR1 in Metastatic Castration-Resistant Prostate Cancer. Cancer Res 82, 1518–1533 (2022).
Moore, K. N. et al. A phase 1b dose escalation study of ipafricept (OMP54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 154, 294–301 (2019).
Dotan, E. et al. Phase Ib Study of Wnt Inhibitor Ipafricept with Gemcitabine and nab-paclitaxel in Patients with Previously Untreated Stage IV Pancreatic Cancer. Clin. Cancer Res 26, 5348–5357 (2020).
Kaur, A. et al. WNT inhibition creates a BRCA-like state in Wnt-addicted cancer. EMBO Mol. Med. 13, e13349 (2021).
Le, P. N. et al. Wnt signaling dynamics in head and neck squamous cell cancer tumor-stroma interactions. Mol. Carcinog. 58, 398–410 (2019).
Sun, L. et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 4, e126853 (2019).
Greene, S. et al. Inhibition of MDSC Trafficking with SX-682, a CXCR1/2 Inhibitor, Enhances NK-Cell Immunotherapy in Head and Neck Cancer Models. Clin. Cancer Res 26, 1420–1431 (2020).
Kim, M. J. et al. Reduced HIF-1α Stability Induced by 6-Gingerol Inhibits Lung Cancer Growth through the Induction of Cell Death. Molecules 27, 2106 (2022).
Tirpe, A. A., Gulei, D., Ciortea, S. M., Crivii, C. & Berindan-Neagoe, I. Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes. Int. J. Mol. Sci. 20, 6140 (2019).
Qin, Y. et al. Salidroside improves the hypoxic tumor microenvironment and reverses the drug resistance of platinum drugs via HIF-1α signaling pathway. EBioMedicine 38, 25–36 (2018).
Minoura, K., Abe, K., Nam, H., Nishikawa, H. & Shimamura, T. A mixture-of-experts deep generative model for integrated analysis of single-cell multiomics data. Cell Rep. Methods 1, 100071 (2021).
Longo, S. K., Guo, M. G., Ji, A. L. & Khavari, P. A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 22, 627–644 (2021).
Song, W. M. et al. Network models of primary melanoma microenvironments identify key melanoma regulators underlying prognosis. Nat. Commun. 12, 1214 (2021).
Acknowledgements
We thank Figdraw (www.figdraw.com) for providing us the tools for the figure presentation. We appreciate the assistance received in data processing and consultation from Intanx Life Co. Ltd. (Shanghai). We also thank KRGENE (Shanghai KR Pharmtech, Inc., Ltd.) and Fountanx Biomedical (Shanghai) Co., Ltd. for valuable comments and suggestions on the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (12271467,81774089, 82004110); the Jiangsu Province Key Research and Development Program (BE2020758, BE2019637); the Xuzhou Medical Outstanding Talents (Xuzhou Health Education Research [2017] No.22); the Jiangsu Medical Innovation Team (CXTDA2017048); the Key Projects of Xuzhou Science and Technology Plan (KC19075); the Xuzhou Clinical Medicine Expert Team Project (2018TD004); the Xuzhou Medical Key Talents Project (XWRCHT20220055); and the Xuzhou Medical Reserve Talents Project (XWRCHT20220009, XWRCHT20220012).
Author information
Authors and Affiliations
Contributions
Conception and design of the research: Z.-D.S. Drafting the manuscript: K.P., Z.-X.W. and Y.D. Production of figures in the article: L.H., J.-X.Q. and W.W. Manuscript revision for important intellectual content: Z.-S.C. and C.-H.H. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shi, ZD., Pang, K., Wu, ZX. et al. Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies. Sig Transduct Target Ther 8, 113 (2023). https://doi.org/10.1038/s41392-023-01383-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01383-x
This article is cited by
-
Targeting triple negative breast cancer stem cells using nanocarriers
Discover Nano (2024)
-
Targeting of REST with rationally-designed small molecule compounds exhibits synergetic therapeutic potential in human glioblastoma cells
BMC Biology (2024)
-
Unveiling the mechanisms and challenges of cancer drug resistance
Cell Communication and Signaling (2024)
-
Signaling, cancer cell plasticity, and intratumor heterogeneity
Cell Communication and Signaling (2024)
-
Targeting cellular plasticity: esculetin-driven reversion of stem cell-like characteristics and EMT phenotype in transforming cells with sequential p53/p73 knockdowns
BMC Cancer (2024)
