
Abstract
To date, the coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has determined 399,600,607 cases and 5,757,562 deaths worldwide. COVID-19 is a serious threat to human health globally. The World Health Organization (WHO) has declared COVID-19 pandemic a major public health emergency. Vaccination is the most effective and economical intervention for controlling the spread of epidemics, and consequently saving lives and protecting the health of the population. Various techniques have been employed in the development of COVID-19 vaccines. Among these, the COVID-19 messenger RNA (mRNA) vaccine has been drawing increasing attention owing to its great application prospects and advantages, which include short development cycle, easy industrialization, simple production process, flexibility to respond to new variants, and the capacity to induce better immune response. This review summarizes current knowledge on the structural characteristics, antigen design strategies, delivery systems, industrialization potential, quality control, latest clinical trials and real-world data of COVID-19 mRNA vaccines as well as mRNA technology. Current challenges and future directions in the development of preventive mRNA vaccines for major infectious diseases are also discussed.
Similar content being viewed by others

Introduction
Coronavirus disease 2019 (COVID-19) is an emerging disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).1,2,3 SARS-CoV-2 is an enveloped positive-sense single-stranded RNA (ssRNA) virus of the Betacoronavirus genus included in the Coronaviridae family. The full-length genome of SARS-CoV-2 isolate Wuhan-Hu-1 consists of 29,881 nucleotides (GenBank accession number: MN908947) with a methylated 5′-cap and a 3′-poly(A) tail, consisting of 9860 amino acids encoding 16 nonstructural proteins (nsp), 9 accessory proteins, and 4 structural proteins. The four structural proteins include spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins.4,5 As of February 9, 2022, COVID-19 has spread to 227 countries and regions worldwide,6 causing over 399 million confirmed cases and 5.75 million deaths.7 The COVID-19 pandemic is an unprecedented event which has caused a huge impact on human health and global public health security. Currently, no specific drug has been identified for COVID-19 prevention or treatment, and vaccination is the most economical and effective intervention to limit the spread of SARS-CoV-2.
To control the spread of the epidemic, governments worldwide have mobilized a considerable amount of manpower and material resources into research and development (R&D) efforts linked to the COVID-19 vaccine. Several approaches to COVID-19 vaccine development have been tested concurrently, including inactivated-virus,8,9,10,11 live attenuated,12,13,14 recombinant protein,15,16,17,18,19 adenovirus vector,20,21,22,23,24 influenza virus vector,25,26 mRNA27,28,29 and DNA vaccines.30,31,32,33,34 As a revolutionary innovation, the mRNA vaccine technology has played a unique role in controlling the COVID-19 pandemic.
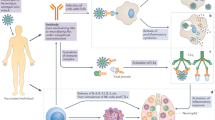
The fundamental mechanism underlying the mRNA vaccine technology is based on a vehicle that enables the delivery of a nucleic acid molecule encoding the antigen of interest into the target cell in the human host, thus allowing the host cell to fabricate the target protein and express the antigen to elicit the immune response. In this way, upon invasion by a pathogen carrying the antigen, the immune system of the host can quickly trigger humoral and cellular immune responses, thereby preventing the disease (Fig. 1). Three types of host cells can be transfected after administration of an mRNA vaccine intramuscularly, intracutaneously, or subcutaneously35: (1) non-immune cells (such as muscle cells and epidermal cells) at the injection site36,37,38; (2) immune cells found in the tissues at the injection site (such as dendritic cells and macrophages)39,40; (3) immune cells in peripheral lymphoid organs after the injected mRNA is transferred through the lymphatic system to adjacent lymph nodes or the spleen.39,41,42 Since mRNA is a negatively charged and unstable molecule, it is generally encapsulated in a delivery vehicle in order to enter the target cell. For instance, mRNA delivered by vaccine vehicles based on lipid nanoparticles (LNPs) enters cells exclusively by endocytosis, forming an endosome without destroying the cell membrane. After entering the cytoplasm, the endosome is directed immediately to lysosomes for degradation. Therefore, in order to ensure structural integrity and thus translation of injected mRNA, endosomal fusion with lysosomes and disruption must be evaded. Studies have shown that the ionizable lipids in LNPs play a role in mRNA release and endosomal escape. In the acidic environment inside endosomes, the headgroup of the ionizable lipid is protonated to a cationic state. After attracting and combining with the anionic headgroup of phospholipids in the endosomal membrane, the hydrophobic tail of cationic lipid and phospholipid expands, and the stable phospholipid bilayer structure is disrupted, which in turn allows mRNA to evade the endosome and reach the cytoplasmic compartment (Fig. 2).35,43,44,45,46 mRNA is then translated into proteins by ribosomes, used as an endogenous antigen, and degraded by the proteasome into antigenic peptides, which are presented to CD8+ cytotoxic T cells through the major histocompatibility complex (MHC) class I molecular pathway to activate cell-mediated immune responses, thereby constituting the key advantage of mRNA vaccines. In addition, translated proteins based on the information contained in the mRNA can be secreted into the extracellular environment, thereby entering the circulatory system in which they are uptaken by antigen-presenting cells (APCs). The antigenic peptide is presented to CD4+ T cells through MHC class II molecules as an exogenous antigen, which can elicit cellular immune response via the secretion of cytokines and activate B cells to produce antibodies and exert humoral immune effects.47 In addition, mRNA vaccines possess a self-adjuvant effect.48,49 ssRNA can be recognized by Toll-like receptor 7 (TLR7) and TLR8 in endosomes50,51 and activate the myeloid differentiation marker 88 (MyD88) pathway.52 Double-stranded (dsRNA) can be recognized by TLR3,53 retinoic-acid-inducible gene I protein (RIG-I),54 melanoma differentiation-associated gene 5 (MDA5)55,56 and other molecules, to cause downstream activation of TIR-domain-containing adapter-inducing interferon-β (TRIF) and mitochondrial antiviral signaling protein (MAVS) molecules,48,52 thereby mediating the production of type-I interferons (IFN-I) and pro-inflammatory cytokines57,58 as well as activating signaling pathways and several IFN-stimulated genes.59 In general, mRNA vaccines induce the production of antibodies, unique cellular immune responses, and self-adjuvant effects by the above-described mechanisms.

mRNA delivered in an mRNA vaccine enters cells by endocytosis and, after release from the endosome, is translated into protein by ribosomes. Translated proteins can then activate the immune system primarily in two ways: i) proteins are degraded by the proteasome into peptides subsequently presented as antigens on the cell surface by major histocompatibility complex (MHC) class I molecules which bind to the T cell receptor (TCR) to activate CD8+ T cells to kill infected cells thorugh the secretion of perforin and granzyme; ii) proteins secreted extracellularly are engulfed by antigen-presenting cells (APCs) and degraded into peptides subsequently presented on the cell surface by MHC class II molecules for recognition by CD4+ T cells, which can activate both the cellular immune responses by secreting cytokines and the humoral immune responses by co-activating B cells. In addition, single-stranded RNA and double-stranded RNA delivered in mRNA vaccines bind to Toll-like receptor (TLR) in the endosome to activate the antiviral innate immune responses via the production of type-I interferon (IFN-I) which results in the induction of several IFN-1-stimulated genes involved in antiviral innate immunity, in a mechanism known as the self-adjuvant effect of a sequence-engineered mRNA. This figure is created with BioRender.com

Endosomal escape of delivered mRNA is largely dependable on interactions between ionizable lipids and naturally occurring anionic phospholipids in the endosomal membrane.43 Prior to membrane fusion, ionizable lipids in lipid nanoparticles (LNPs) and anionic lipids in the endosomal membrane adopt a cylindrical conformation which is compatible with molecular packing in a bilayer phase. The acidic environment in endosomes facilitates protonation of ionizable lipids into cationic lipids. Cationic and anionic lipids generate ion pairs whose combined cross-sectional headgroup area is smaller than the total of individual headgroup areas before membrane fusion. Consequently, the ion pair adopts a conical shape which promotes the formation of inverted, non-bilayer phases, such as the hexagonal shape illustrated above. Thus, the formation of ion pairs between lipids promotes membrane fusion and disruption, allowing mRNA to escape from endosomes. This figure is created with BioRender.com
The successful development of mRNA vaccines is a result of years of research and groundwork. The mRNA molecule was first described by Brenner and colleagues in 1961,60 and due to the highly unstable nature of the mRNA molecule, it was not until 1969 that the first protein was produced in vitro from isolated mRNA.61 Dimitriadis and colleagues attempted to employ unilamellar liposome wrapping to deliver exogenous mRNA into human and murine cells in order to avoid mRNA degradation by nucleases.62,63 However, RNA is easily degraded and difficult to obtain in vitro, which greatly hinders the progress of research in RNA transfected cells. In 1984, Krieg and colleagues were the first to employ SP6 RNA polymerase to successfully transcribe and synthesize mRNA in vitro, establishing the foundation for subsequent in vitro mRNA studies.64,65,66,67,68,69,70,71,72,73,74 Subsequently, in 1987 Malone and colleagues employed cationic lipids to encapsulate mRNA for injection into eukaryotic cells, obtaining a highly efficient system for the expression of mRNA in vitro.75 In 1990, Wolff and colleagues injected for the first time into mouse quadriceps muscle mRNA resulted from in vitro transcription (IVT) which was successfully expressed, thus paving the way for mRNA therapeutic research.76 After immunizing mice with liposome-encapsulated mRNA encoding the influenza-virus nucleoprotein (NP), the production of anti-influenza cytotoxic T lymphocytes (CTLs) was induced in the host, thus marking a milestone in the development of the first mRNA vaccine.77 Later, Conry and colleagues tested the first mRNA tumor vaccine encoding the carcinoembryonic antigen (CEA) in mice, which broadened the perspectives for human anticancer research.78,79,80 However, due to the unsatisfactory stability and safety of mRNA vaccines, their use has been widely disregarded. In 2005, Karikó and colleagues found that mRNA synthesized using modified uridine could avoid recognition and degradation by the immune system, which greatly improved mRNA stability and immunogenicity in vivo, thereby inaugurating a new era in mRNA vaccine.81 After years of investigation, in August 2018 the first-ever RNA interference (siRNA) therapeutic drug, Onpattro ® (patisiran) (Alnylam Pharmaceuticals Inc., Cambridge, MA, USA), was approved by the U.S. Food and Drug Administration (FDA).82,83,84 mRNA vaccines for various infectious diseases, such as rabies, influenza, Ebola, Zika, and dengue virus, have entered the preclinical research or clinical trials in recent years.85,86,87,88,89,90,91,92,93,94,95,96,97,98,99 Since the beginning of the COVID-19 pandemic in 2019, mRNA vaccines have become a current research hotspot owing to their shorter R&D cycle, simple production process, and ability to induce strong immune responses.
The mRNA vaccine is the first batch of COVID-19 vaccine candidates in clinical trials. As of February 8, 2022, WHO reported 337 COVID-19 vaccine candidates currently under development, of which 47 are mRNA vaccines, and 23 among these have entered clinical trials.100 The mRNA vaccines Pfizer-BioNTech (BNT162b2), Moderna (mRNA-1273), and CureVac101,102,103 were the fastest vaccine development in medical history. The first two obtained emergency use authorization (EUA)104 from many regulatory agencies in the United States, the United Kingdom, Canada, and Hong Kong, China. On August 23, 2021, the Pfizer-BioNTech was the first COVID-19 vaccine officially approved for commercialization by the FDA,105 being also the first-ever approved on October 29, 2021 for use in children aged 5–11.106 Thus, the mRNA vaccine technology has the most promising application prospects for COVID-19. Thus, this review will cover the different types of COVID-19 mRNA vaccines, antigen design strategies, delivery vehicles, clinical trials, production process, and quality control, among other related topics.
Different Types of COVID-19 mRNA Vaccines
mRNA vaccines can be categorized as non-replicating mRNA, self-amplifying mRNA (saRNA) and circular RNA (circRNA) based on their genetic characteristics (Fig. 3).107 Non-replicating mRNA vaccines deliver exclusively genetic information coding for the target antigen, thus containing the 5′-cap, 5′ untranslated region (UTR), 3′ UTR, and 3′-poly(A) tail regions.108 saRNA vaccines can deliver genetic information encoding the target antigen and other genes, e.g., viral RNA polymerase, to enable mRNA to self-replicate.109,110 Based on saRNA technology, safe trans-amplifying RNA (taRNA) vaccines have been optimized and developed.111 In addition, circRNA has recently been developed for COVID-19 mRNA vaccines due to its natural high stability.112 Most COVID-19 mRNA vaccines currently in clinical trials or those already on the market are non-replicating mRNA vaccines. The advantages of non-replicating mRNA vaccines include the use of an RNA molecule of simple structure and shorter length. An optimized or modified mRNA can have greatly enhanced biological activity. Lastly, saRNA vaccines are currently in preclinical and clinical trials.

A The vaccine immunogen is encoded by a non-replicating RNA flanked by 5′ and 3′ UTRs (S protein). B Self-amplifying RNA (saRNA) encodes four nonstructural proteins (nsp 1–4) and a subgenomic promoter derived from the alphavirus genome. saRNA encodes a replicase and amplifies vaccine-encoding transcripts. C Trans-amplifying RNA (taRNA) uses two transcripts to enable self-amplification of replicase and the immunogen. D Circular RNA (circRNA) is circularized by the autocatalytic Group I ribozyme.223 The exon 2 is ligated upstream to exon 1, and a coding region is inserted between the exon-exon junction. During splicing, the 3′-OH of a guanosine nucleotide engages in a transesterification reaction at the 5′ splice site. The 5′ intron is excised, and the 3′-OH at the end of the intermediate engages in a second transesterification reaction at the 3′ splice site, resulting in the circularization of the immunogen mRNA. Upon entering the cell, the internal ribosome entry site (IRES) of circRNA initiates protein translation. The figures are created with BioRender.com
Conventional non-replicating mRNA
A non-replicating mRNA contains an open reading frame (ORF) encoding the gene coding for the target antigen flanked by 5′ and 3′ UTR. The 5′ end contains a 7-methylguanosine cap structure (5′-cap, m7G), whereas the 3′ end contains a poly(A) tail structure. This structure enhances stability of the delivered mRNA while improving accuracy and efficiency of mRNA translation (Fig. 4).113,114

mRNA molecules are synthesized in vitro with a 5′-cap 1 structure and chemically modified nucleotides as substitutes for natural nucleotides, which enhances stability and translation efficiency of mRNA as well as reduces innate immune response. This figure is created with BioRender.com
5′-cap
The 5′-cap structure prevents mRNA from degradation by exonucleases, thereby maintaining mRNA stability and enabling translation initiation.115 m7G is found at the 5′ end of mature mRNA in eukaryotic cells and connected to the first nucleotide of mRNA transcription by triphosphates to form an m7G cap structure (m7GpppNp).116 According to the degree of methylation, three main cap structures are possible: cap 0, cap 1, and cap 2. A cap 0 structure is the most elementary, namely m7GpppNp; however, an mRNA of cap 0 is likely to be recognized as exogenous RNA by the host, which could stimulate the innate immune response of the host and ultimately trigger inflammatory responses.117 A cap1 structure (m7GpppN1mp) has a methylated 2′-OH on the first nucleotide connecting the 5′ end of the mRNA to the cap.118 Since the cap1 structure has only been described to date in eukaryotic mRNAs, it can be used as a signature of self-RNA, thus reducing the activation of pattern recognition receptor (PRR) and consequently improving translation efficiency of mRNA in vivo.119 Lastly, cap2 (m7GpppN1mpN2mp) has a methylated 2′-OH on both the first and second nucleotides that connect the 5′ end of the mRNA to the cap, and methylation improves mRNA translation efficiency.115 At present, the cap1 structure is most commonly applied for capping mRNA vaccines.
Two types of capping methods are possible during IVT of mRNA (Fig. 5). The first method employs a capping enzyme RNA 5′-triphosphatase (RTPase) which hydrolyzes the 5′ γ-phosphate of RNA transcripts, with a transfer of guanosine monophosphate (GMP) to 5′-diphosphate RNA by guanylyltransferase (GTase), and the resulting 5′-end β-phosphate is combined with GMP to form GpppNp-RNA. Finally, the guanosine moiety is methylated by a cap-specific S-adenosylmethionine-(AdoMet)-dependent (guanine-N7) methyltransferase (N7MTase), forming a cap0 structure (m7GpppNp). The cap0 structure can be further modified to cap1 (m7GpppN1mp) by 2′-O-methyltransferase (2′-O-MTase).120 The vaccinia capping enzyme (VCE) integrates the enzymatic activity of RTPase, GTase, and G-N7 MTase, which can be capped to generate a cap0 structure, and then 2′-O-MTase can be used to generate a methylated cap1 structure, thus reaching a capping efficiency of 100%.52 Of note, it has been reported that the COVID-19 vaccine mRNA-1273 developed by Moderna employs the capping enzyme described above.28 The second capping method employs cap analogs (m7GpppG) during transcription of mRNA, involving T7, T3, or SP6 RNA polymerases to achieve mRNA co-transcriptional capping.121 Co-transcriptional capping is the most commonly used method in IVT for mRNA,115 but studies have found that cap analogs bind mRNA strands in reverse orientation.122 The reverse-capped mRNA cannot be recognized by the ribosome, resulting in reduced translation efficiency.123 To avoid reverse capping, a modified cap analog with methylation modification, namely anti-reverse cap analog (ARCA) (m7(3′-O-methyl)-GpppG), has emerged.124 Since the 3′-OH group in proximity to m7G is methylated, thus the ARCA cap analog can only bind the 5′ end of mRNA in forward orientation, which is recognized by the eukaryotic translation initiation factor 4E (eIF4E) to initiate ribosome recruitment and translation.74,125 However, the cap0 structure produced by ARCA capping requires additional methylation modification to yield a stable cap1 structure. Therefore, the ARCA capping approach results in inefficient capping and is not widely adopted. The current new generation of cap analogs is the CleanCap® cap analogs developed by TriLink BioTechnologies (San Diego, CA, USA),126 which can be co-transcribed with the target mRNA to obtain the cap1 structure, thus solving the issues of low efficiency and high enzyme costs of traditional capping methods. At present, there are several capping analogs such as CleanCap® Reagent AG (m7GpppA2′OMepG), CleanCap® Reagent AU (m7GpppA2′OMepU), and CleanCap® Reagent AG 3′OMe (m7G3′OMepppA2′OMepG),127 among which CleanCap® Reagent AG is commonly used for non-replicating mRNA,128 requiring that the T7 promoter sequence at the 5′ end of the DNA template must be followed by an AG start. In contrast, CleanCap® Reagent AU is a capping analog designed specifically for self-replicating RNA,129 and the start sequence at the 5′ end of the DNA template must be AU (Fig. 5). The use of CleanCap® cap analogs reduces the probability of reverse capped, uncapped, and cap0 intermediates, and the co-transcribed mRNA only possesses a cap1 structure, therefore capping rate can be 90% or higher.52,126 Currently, COVID-19 mRNA vaccines BNT162b1 and BNT162b2 developed by BioNTech employ TriLink Cap1 cap analog (m27,3′-O)Gppp(m2′-O)ApG for co-transcriptional capping.27,130

A Production of post-transcriptional modifications of mRNA with cap0 requires three enzymes: triphosphatase, guanylyltransferase, and N7-methyltransferase with S-adenosylmethionine (SAM) as the methyl donor. Subsequently, the cap0 is modified with 2′-O-ribose methyltransferase to generate the cap1 structure. B Cap analogs commonly used for in vitro transcription of mRNA are CleanCap® Reagent AG (TriLink) and CleanCap® Reagent AU (TriLink). The proposed mechanism of CleanCap co-transcriptional initiation involves the docking of AmG or AmU dimers onto the +1 and +2 positions in template nucleotides. Initiation occurs upon coupling of CleanCap with an nucleoside triphosphate (NTP) occupying the +3 position.157
In summary, cap structure of mRNA can both protect mRNA from destruction and facilitate its recognition by the host due to chemical modifications. Additionally, co-transcriptional capping can increase productivity of mRNA vaccines.
5′ and 3′ UTRs
mRNA contains 5′ and 3′ UTRs, whose functions are related, respectively, to regulating translation and maintaining mRNA stability.131 The 5′ UTR is mainly involved in translation of its downstream ORF sequence.132,133 The Kozak sequence is generally added after the 5′ UTR sequence to improve translation efficiency.134 Conversely, the function of the 3′ UTR is to maintain mRNA stability.135,136 Studies have shown that adenylate-uridylate-rich elements are involved in mRNA degradation. Degradation rate and translation life cycle can be adjusted by replacing adenylate-uridylate-rich sequences found in the 3′ UTR.137,138,139 At present, the 3′ UTR is mainly derived from hemoglobin subunit α (HBA) and subunit β (HBB) genes,140 but it can also be derived from albumin (ALB), heat-shock protein 70 (Hsp70), tyrosine hydroxylase (TH), and collagen alpha 1 (COL1A1) genes.141,142,143 In contrast, the 5′ UTR is mostly retrieved from genes such as globin, Hsp70, axon dynein heavy chain 2 (DNAH2), and hydroxysteroid dehydrogenase (3β-HSD).144,145 Design of proper 5′ and 3′ UTRs sequences is crucial for the success of mRNA vaccines. Many investigations have been conducted to screen and design the most effective 5′ and 3′ UTR sequences for mRNA vaccines, therefore UTR sequences are considered intellectual properties of vaccine manufacturers. Table 1 summarizes current UTR sequences of COVID-19 mRNA vaccines from different vaccine manufacturers.
Poly(A) tail
The poly(A) tail plays an important role in maintaining mRNA stability and translation efficiency.116 mRNA stability can be improved by inhibiting exonuclease-mediated mRNA degradation.52 The poly(A) tail can also bind to multiple poly(A)-binding proteins (PABPs) while working synergistically with 5′ m7G cap sequences to regulate translational efficiency.146,147 Polyadenylation of engineered mRNA can occur in two ways: i) by traditional enzymatic polyadenylation, adding the poly(A) tail to the 3′ end of mRNA, but which does not allow regulation of tail length148; and ii) by designing a fixed-length poly(A) sequence on a DNA template and transcribing the resulting length-controllable poly(A) tail.149
In mammalian cells, actively translated mRNAs generally contain 100–250 adenosine residues.117,150 A poly(A) tail of optimal length can improve translation efficiency and mRNA stability.150,151 Studies have shown that when poly(A) tail size increases to 120 bp, the expression level of the corresponding protein increases accordingly. However, when poly(A) tail size is greater than 120 bp, the expression level of the corresponding protein did not increase.152 In addition, other design strategies of poly(A) tails exist. For instance, BioNTech uses a segmented poly(A) tail whose two-tail structures are connected in tandem by a 10 bp UGC linker sequence (A30LA70).27,153 Current studies have shown that the segmented poly(A) tail extends mRNA half-life and improves translation efficiency compared to the long-chain poly(A) tail.154
Modified nucleosides
Naturally occurring modified nucleosides are found in mRNAs in humans.155 The host immune system can easily recognize unmodified mRNA or by-products formed during IVT of engineered mRNA as exogenous molecules.74 dsRNA can activate PRR such as TLR3,52,156 whereas ssRNA activates TLR7 and TLR8 to produce IFN-I, thus inducing inflammation in the host and interrupting mRNA translation.157,158 To avoid this, Karikó and colleagues found that adding modified nucleosides during IVT of mRNA can significantly reduce the host inflammatory response without affecting protein expression.81,159 Currently, the following modified nucleosides are available for mRNA modification: pseudouridine (ψ), N1-methylpseudouridine (m1ψ), 5-methoxyuridine (mo5U), 2-thiouridine (s2U), 5-methylcytidine (m5C) and N6-methyladenosine (m6A) (Fig. 4).115,160,161,162,163,164,165,166,167,168,169,170 Studies have found that replacing original nucleosides with m6A and s2U inhibits activation of TLR3, whereas activation of TLR7 and TLR8 is blocked when using ψ, 5-methyluridine (m5U), m6A, and s2U, thereby inhibiting the innate immune responses and improving protein translation efficiency.81,159,171 Kormann and colleagues152 replaced 25% of mRNA cytosine with m5C and 25% of uridine with s2U, which improved mRNA stability and increased protein translation in mice. However, replacing natural nucleosides in the right proportion might be challenging, which might hinder vaccine quality control and consistency. Currently, nucleoside-modified mRNA vaccines employ 100% chemically modified nucleosides replacing natural nucleosides, and m1ψ is often used to replace uridine during IVT to improve the safety and stability of mRNA vaccines.52,134,172
At present, mRNA vaccines that have entered clinical trials or been approved for commercialization include both nucleoside-modified and nucleoside-unmodified mRNA with sequence-optimized mRNA. Nucleoside-modified mRNA can reduce the activation of TLRs, retinoic acid-inducible gene I (RIG-I), protein kinase R (PKR), and 2′-5′-oligoadenylate synthetase (OAS). Additionally, nucleoside modification increases translation activity and resistance against RNase L-mediated degradation.54,81,173,174,175,176 Many nucleoside-modified mRNA COVID-19 vaccines, including BNT162b2, BNT162b1, and BNT162b3 developed by BioNTech27,177; mRNA-1273, TAK919, mRNA-1273.211, mRNA-1273.351, mRNA-1283 developed by Moderna28,178; ChulaCov19 developed by Chulalongkorn University; and PTX-COVID19-B developed by Providence Therapeutics, have been approved for commercialization or entered clinical trials.104 In addition, nucleoside-modified mRNA vaccines have been widely used in vaccine development for other viral agents such as Cytomegalovirus (CMV), Respiratory syncytial virus (RSV), Influenza A virus, Chikungunya virus, Zika virus, Dengue virus.92,98,179,180,181,182
In contrast, studies on mRNA vaccines with unmodified nucleosides have yielded inconsistent conclusions. Thess and colleagues showed that unmodified, GC-rich mRNA engineered with an optimized UTR sequence yielded more sustained antigen expression compared to nucleoside-modified mRNA.183 In contrast, Pardi and colleagues reported that protein levels after intradermal injection of m1ψ-modified mRNA in mice were 20 times higher compared to sequence-optimized but unmodified mRNA.184 Among unmodified-nucleoside COVID-19 vaccines currently in clinical trials are included CVnCoV developed by CureVac (clinical trial terminated), ARCoV developed by Abogen,185 BNT162a1 developed by BioNTech,130 and MRT5500 developed by Translate Bio (clinical trial terminated).104 A vaccine platform (RNActive®) was designed by CureVac186 combining the use of co-delivered RNA and protamine (a polycationic peptide) complex as adjuvant, which has been shown to effectively trigger innate immune responses and enhance vaccine immunogenicity.186,187,188,189 Using this technique, the Rabies vaccine CV7201 was developed by CureVac and is currently in phase I clinical trial.190
In summary, chemical modifications regulate the functional specificity of biological macromolecules, and to date, a total of 16 modifications have been found in eukaryotic mRNA. Both Moderna and BioNTech use pseudouridine modifications to ensure mRNA stability in their COVID-19 vaccine formulation.
saRNA
Engineered saRNA vaccines rely on the insertion of the gene encoding the target antigen into the genome of an RNA virus (mainly alphavirus) as well as the use of its replication machinery to amplify the delivered RNA, thereby increasing antigen expression.191,192,193 In terms of structures, in addition to the conventional elements of non-replicating mRNA, saRNA contains a long ORF after 5′ UTR encoding the four NSPs (nsP1, nsP2, nsP3, and nsP4) of alphavirus that functions as an mRNA capping enzyme, an NTPase/helicase/protease, a macrodomain, or an RNA-dependent RNA polymerase (RDRP), respectively. A subgenomic promoter can then be used to initiate transcription of the gene coding for the target antigen.194 Once in the cytoplasm of a host cell, saRNA undergoes translation by the endogenous ribosomal machinery, thereby enabling translation of nsP precursors to form an early replication complex. The positive-strand RNA is then used as a template to synthesize negative-strand RNA, which is the replication intermediate. With the cleavage of nsP precursors, a late replication complex is produced. Then, the negative-strand RNA of the replication intermediate is used as a template to synthesize a full-length positive-strand genomic RNA. At the same time, a subgenomic positive-strand RNA containing only information coding for the antigen is also synthesized (Fig. 3). As a result, one copy of saRNA produces multiple copies of RNA transcripts by the above-described mechanism to initiate self-amplification of antigen genes in the cell.35,195,196
The idea of using in vitro synthesized saRNA as a preventive vaccine was first proposed by Zhou and colleagues in 1994, using a modified Semliki Forest virus (SFV) replicon to express the nucleoprotein (NP) of the influenza virus.197 Subsequently, Fleeton and colleagues used the same SFV replicon to develop saRNA vaccines for influenza A virus, RSV, and louping ill virus (LIV). After direct intramuscular injection in mice, the naked saRNA could induce protective immune response.198 Decades later, Geall and colleagues were the first to use LNP to encapsulate a saRNA chimera composed of the Venezuelan equine encephalitis virus (VEEV) and Sindbis virus (SINV) replicons into a vaccine which was used to immunize mice, and immunogenicity was significantly improved compared with unencapsulated group.199 In recent years, several viral replicons have been used in saRNA vaccines, such as those of VEEV, classical swine fever virus (CSFV), tick-borne encephalitis virus (TBEV).96,200,201,202,203 In previous studies, the alphavirus genome have been screened and multiple superior mutations that could improve and optimize RNA replicons have been identified.204,205,206,207,208,209 Li and colleagues developed an in vitro evolution strategy, and six mutations (namely A1979G, G3936C, A4311G, A4758G, G4796T, G4944A) were identified in the nsP2 and nsP3 of the VEEV replicon, which were shown to promote expression of subgenomic RNA in cells.210 Moreover, saRNAs have yielded promising results in preclinical research for COVID-19 vaccines. Recently, two different saRNA vaccines developed independently by Arcturus Therapeutics and Imperial College London have shown favorable immune responses against SARS-CoV-2, and have entered clinical trials.211,212,213
The most advantageous aspect of saRNA vaccine is that it can be produced with ultra-low doses of saRNA. Compared with the dose of mRNA in the Moderna vaccine (100 μg) and that of Pfizer-BioNTech vaccine (30 μg), the amount of saRNA required for vaccine development is within a range of 0.1~10 μg.212 This ultra-low injection dose has several advantages214: (i) greater production potential, since the same amount of raw materials and the same equipment yield more vaccine production; (ii) reduced side effects considering the lower dosage; (iii) allows combination with other vaccines due to its lower dosage; (iv) intrinsic adjuvant effect; (v) high levels of antigen expression and long-term duration of immunity. saRNA vaccines have nonetheless certain shortcomings, including the risk of excessive activation of the inflammatory response, and the production of viral nsP produced by the alphavirus replicon that may interfere with normal signal transduction in target host cells.214,215 In addition, considering that the length of nsP1–4 sequence is approximately 7 kb, the full length of a saRNA sequence is usually above 9 kb, which might hinder cloning construction. Hence, the delivery vector employed in saRNA vaccines must allow for higher loading capacity and encapsulation efficiency.35
taRNA
taRNA is a self-amplified RNA composed of two separate RNA molecules (Fig. 3). To circumvent the problems caused by large and complex sequences of saRNA, the R&D team of the Imperial College London has developed a split replicon (splitzicon) system which enables encoding the alphavirus nsPs and the heterologous gene of interest (GOI) on separate RNA molecules whilst conserving the self-amplification properties of the replicon RNA.216 Blakney and colleagues216 used fluorescent reporter genes as encoding proteins and designed positive and negative splitzicons to identify structural components affecting self-amplification characteristics of VEEV replicons, thus providing a new strategy for developing saRNA vaccines based on alphavirus RNA replicons. In a recent study, Beissert and colleagues developed a novel bipartite vector system using taRNA,111 containing a transreplicon expressing hemagglutinin antigen (TR-HA) of influenza virus obtained by deleting the replicase gene in the amplified RNA of alphavirus together with an optimized non-replicating mRNA carrying a replicase gene. After application of the resulting vaccine in mice, it was shown that 0.05 μg of taRNA resulted in complete protection comparable to non-replicating mRNA vaccine or saRNA vaccine.
taRNAs usually yield safer vaccines compared to saRNA vaccines. The alphavirus replicon gene is divided into two different RNA molecules encoding vaccine antigens, which reduces the possibility of transfer of recombinant virus particles to host cells. In addition, taRNA technology has potential advantages in transfer capacity, versatility, and production scale-up, thus showing promising applications.217 At present, taRNA technology has only been applied in preclinical studies for influenza vaccines.111 COVID-19 vaccines based on taRNA technology have not been reported.
CircRNA
CircRNA is a highly stable single-stranded RNA with a covalently closed loop structure (Fig. 3), including a large category of non-coding RNAs generated by backsplicing in eukaryotic cells.218,219,220 In the 1970s, Sanger and colleagues discovered single-stranded circRNA viruses in higher plants.221 Later, circRNA was also identified in yeast mitochondria and hepatitis D virus.222 Despite the lack of essential elements for cap-dependent translation, circRNA can be translated by adding the IRES element or m6A modification incorporated to its 5′ UTR region.223,224 Unlike linear RNA, circRNA offers several advantages in vaccine development. The covalently closed loop structure of circRNA protects from exonuclease degradation, thus increasing circRNA half-life and stability. Moreover, previous studies have reported that cell transfection efficiency was maintained when circRNA was kept at room temperature for two weeks.112 In addition, unmodified circRNA has been shown to induce TLR/RIG-I-mediated innate immune response compared to unmodified linear mRNA.225,226
Recently, Liang and colleagues112 developed a circRNA vaccine against SARS-CoV-2 encoding a trimeric receptor-binding domain (RBD) of the spike protein of SARS-CoV-2, considering that in RBD the signal peptide sequence of human tissue plasminogen activator (tPA) was fused to the N-terminus to ensure antigen secretion, whereas the trimerization motif of bacteriophage T4 fibritin protein (foldon) was fused to the C-terminus to ensure the native conformation of the antigen protein trimer. In addition, the IRES element was inserted before the coding gene to initiate translation, and circRNA was produced using a group I ribozyme. Finally, LNP was used for encapsulation to obtain a circRNA vaccine. After immunizing mice with the obtained vaccine, long-lasting neutralizing antibodies and Th1-biased cellular immune responses were produced. Moreover, the vaccine also showed neutralizing activity against the Beta variant (B.1.351). Liang and colleagues227 further improved the circRNA vaccine by constructing multiple circRNAs based on several SARS-CoV-2 variants; the results revealed that circRNA prepared with sequences of Delta strains resulted in broad-spectrum protection and production of neutralizing antibodies against both Delta and Omicron. However, vaccines produced with circRNA prepared based on Omicron sequences provided a narrower protection, and produced neutralizing antibodies could protect only against Omicron but not against Delta. In addition, vaccination with circRNA prepared with the original SARS-CoV-2 strain sequence followed by a booster dose of the vaccine containing circRNA prepared with Delta sequences conferred good protection against Delta and Omicron. Since RBD is the main region inducing the production of neutralizing antibodies, it can be speculated that the future development of COVID-19 vaccines should focus on the Delta variant.
In summary, although considered a byproduct of the mRNA splicing process, circRNA has now emerged as an important new class of non-coding RNAs. With its highly stable properties without nucleotide modification, circRNA can potentially become a novel platform for vaccine and drug development.
Antigen design for COVID-19 mRNA vaccines
The trimeric S protein on the surface of SARS-CoV-2 plays a key role in mediating host cell invasion. Therefore, the S protein is considered the main antigen for vaccine design.228,229,230 The S protein is cleaved into S1 and S2 subunits during viral infection by the Furin enzyme and transmembrane serine protease 2 (TMPRSS2).231,232 The S1 subunit comprises the signal peptide (SP), RBD, N-terminal domain (NTD), C-terminal domain 1 (CTD1), and C-terminal domain 2 (CTD2), and primarily interacts with the cellular angiotensin-converting enzyme 2 (ACE2) receptor via RBD. The S2 subunit comprises the fusion peptide (FP), double heptad repeat (HR), central helix region (CH), connector domain (CD), transmembrane domain (TMD), and cytoplasmic tail (CT), and the S2 subunit is responsible for mediating the fusion between the virus and the host cell membrane233,234,235,236 (Fig. 6). Upon binding of RBD and the ACE2 receptor on the host cell membrane, the S protein undergoes a structural rearrangement that results in a postfusion conformation.236 Studies have found that the S protein prefusion conformation induces better immunogenicity and can be therefore considered an ideal target antigen.237 Most neutralizing antibodies are molded based on epitopes of S protein in prefusion conformation, which is covered once the S protein acquires the postfusion conformation, thus reducing the production of neutralizing antibodies.238

Representation of the SARS-CoV-2 reference genome showing structural, nonstructural, and accessory proteins, consisting of ORF1a, ORF1b, Spike protein (S), ORF3a, ORF3b, Envelope (E), Membrane (M), ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF14, Nucleocapsid (N) and ORF10.485 Spike and receptor-binding domain (RBD) proteins are mainly used as target antigens for the design and optimization of COVID-19 mRNA vaccines. This figure is created with BioRender.com
The ORF containing the coding sequence that is translated into protein in vivo is the most critical component of the mRNA vaccine. To improve the safety, efficacy, and stability of mRNA vaccines, researchers usually performed codon optimization172,239,240,241,242,243 on the antigen-coding sequence to enhance translation efficiency. Optimization of mRNA secondary structure244,245 and stability can be achieved by increasing the GC content of the coding sequence.246,247,248 Codon preference varies extensively in different organisms,249 therefore adjusting the balance between codon usage frequency and host tRNA availability can significantly improve translation efficiency and in vivo expression of the target antigen.250,251 Several online codon optimization tools are available,252,253,254,255,256,257,258,259,260,261,262,263,264,265,266 and optimization algorithms have been conceived for different research purposes.267 A few studies have indicated that optimization algorithms designed by BioNTech and Moderna may have shortcomings. In addition, since mRNA vaccines are often injected intramuscularly, a better immune response can be expected if codon optimization is adjusted for skeletal muscle preference.153
At present, two strategies are commonly used for designing COVID-19 S protein: 2P mutation and S1/S2 Cleavage site (Fig. 6). The 2 P mutation strategy is based on the findings of studies exploring the S protein in prefusion conformation in Middle East Respiratory Syndrome Coronavirus (MERS-CoV),268 SARS-CoV,269, and human coronavirus HKU1 (HCoV-HKU1).270 By adopting this strategy, two amino acids at the top of the helical position of the S2 subunit center are substituted with prolines (K986P and V987P), which was shown to improve stability of S protein in prefusion conformation effectively. The 2P mutation method is applicable to SARS-CoV-2271 and other β-coronavirus viruses,28 and BioNTech, Moderna, CureVac, and other developers have all adopted the 2P mutation strategy.27,28,185,272 The S1/S2 cleavage site strategy employs direct deletion of the sequence Q677TNSPRRARYSV687 in wild-type SARS-CoV-2 protein S to Q677TILRYSV683238 or mutation of amino acids (RRAR to GGSG)141 that ultimately prevent the S protein from cleavage in the host cell, thus maintaining its structural stability and inducing stronger immune responses. In addition to RiboBio mutation of the S1/S2 cleavage site (682–685: RRAR to GGSG),141 antigen design for the protein found in the COVID-19 recombinant vaccine developed by Novavax also introduced similar mutations (682-685: RRAR to QQAQ) to maintain the stability of S protein conformation.16,273,274,275
Certain research institutions have developed unique strategies for optimizing the S protein (Fig. 6), such as deleting TMD, CT, FP on the S2 subunit and mutating the S2′ cleavage site (K814A, R815N) to improve the conformational stability of prefusion S protein.141 In certain cases, an additional sequence can be inserted in the anterior segment of the ORF region to increase the expression of antigenic proteins.276 Moreover, since the S protein is trimeric, studies have shown that the trimeric motif of T4 bacteriophage fibritin introduced at the 3′ end of the coding region of the S protein or RBD protein can mimic the native structure of S protein and enhance antigen immunogenicity.141,277,278 Furthermore, previous studies revealed significant differences in protein expression levels of target antigens when different signal peptides are selected.93 In the COVID-19 recombinant vaccine developed by WESTVAC, expression of RBD protein was enhanced when the GP67 signal peptide was used.15 The S protein signal peptide MFVFLVLLPLVSSQCV has been used in COVID-19 mRNA vaccines by several developers, including BioNTech and Moderna. However, other signal peptides have also been used. For example, RiboBio employs the immunoglobulin heavy chain variable region (IGVH) signal peptide sequence (MDWIWRILFLVGAATGAHS) in COVID-19 mRNA vaccines to increase target protein expression. In brief, since mRNA vaccines involve sequence editing, structure, stability, and expression of the S protein may be modified to improve spatial conformation and thus vaccine-induced immune response (Table 2).
Delivery systems
Passage of mRNA through the phospholipid bilayer of the cell membrane is difficult due to its large molecular weight (104–106 Da), negative charge, and proneness to degradation by nucleases. Therefore, in recent years, various delivery vehicles have been developed for mRNA encapsulation, including LNPs, polyplexes and polymeric nanoparticles, lipopolyplexes (LPPs), and cationic polypeptides. Lipids and their derivatives are considered a new delivery system for mRNA vaccines and have been attracting much attention due to their low immunogenicity, biocompatibility, and high encapsulation rate. As an early version of LNPs first discovered in 1965,279 liposomes are the earliest nanomedicine delivery platform to pass from concept to clinical application successfully.280 The next generation of LNPs, which includes solid LNPs, nanostructured lipid carriers, and cationic lipid-nucleic acid complexes,281,282,283 possesses more complex internal structures, stability, and targeting capacity. In addition to vaccines, these substances can be used as a new drug delivery platform for anticancer and nucleic acid therapeutics.
LNPs
LNPs is a nano-scale vesicle which simulates the lipid structure of the cell membrane and can encapsulate mRNA in its cavity, being considered the most investigated mRNA vaccine delivery system. Currently, most COVID-19 mRNA vaccine candidates use LNPs as the delivery system. LNPs are composed of four components: ionizable lipids, helper phospholipids, cholesterol, and PEGylated lipids, among which, ionizable lipids are considered the key components. COVID-19 mRNA vaccines designed by different developers vary widely in structural design (Fig. 7).

LNPs are composed of four components: ionizable lipid, helper lipid, cholesterol, and PEGylated lipid. Binding with mRNA occurs by the ionizable lipid that occupies the central core of the LNP. PEGylated lipid is found on the surface of LNPs along with helper lipid forming the bilayer. Cholesterol, charged ionizable lipids, and neutral ionizable lipids are distributed throughout LNPs. The confirmed or the most likely chemical structure of ionizable lipids employed in COVID-19 mRNA vaccines developed by Moderna, BioNTech, CureVac, Arcturus, Imperial College London, and Chulalongkorn University.289 *Molar lipid ratio (%) of ionizable lipid: helper lipid: cholesterol: PEGylated lipid; **NA: Not applicable
The cationic lipid N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA) was first used by Malone and colleagues to transfect mRNA into cells.75 Although DOTMA has high delivery efficiency in vitro, it is quickly cleared in the host blood circulation and has pro-inflammatory and pro-apoptotic toxic effects.284 Accordingly, since ionizable lipids are positively charged in an acidic buffer environment, they can bind to negatively charged RNA and facilitate endosomal escape of mRNA after entering the host cell. Ionizable lipids are neutral at physiological pH, thus making them safer and more stable for use as delivery systems in vaccines.285 DLin-MC3-DMA is the ionizable lipid used in LNP formulation in Onpattro®286 the first-ever siRNA drug approved by the FDA. Moderna used DLin-MC3-DMA ionizable lipids to prepare mRNA vaccines for Zika virus and influenza on which preclinical and clinical studies were conducted.90,287,288 However, it was later found that the di-linoleic alkyl tail in DLin-MC3-DMA is prone to degradation, and repeated booster doses can potentially lead to cumulative toxicity.289 Based on these observations, Moderna has developed an ionizable lipid (namely Lipid H, SM-102), whose tail adopts larger branches which in turn increases potency, and whose introduced ester bonds increase biodegradability (Fig. 7).290,291 In contrast, BioNTech uses ALC-0315, whose chemical structure is similar to SM-102, as the ionizable lipid in the LNP formulation of their COVID-19 mRNA vaccine. (Fig. 7).289,292
Although ionizable lipids are essential components of LNPs, the other three components (i.e., helper phospholipids, cholesterol, and PEGylated lipids) play an important role in LNPs assembly and function. Helper phospholipids are amphiphilic lipids that support the lipid bilayer structure, help promote fusion with endosomal membranes, and determine the specificity of target organs.293,294 The choice of helper phospholipid for LNPs formulation is highly dependent on the length of the delivered RNA molecule. For instance, saturated helper lipids (such as 1,2-Dioctadecanoyl-sn-glycero-3-phophocholine —DSPC) are indicated for the transport of short siRNAs,295 whereas unsaturated lipids (e.g., 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine —DOPE) are more conducive to delivering longer mRNAs.296,297 However, DSPC is employed as helper phospholipid in the formulation of both Moderna and BioNTech COVID-19 mRNA vaccines (Fig. 7),27,298,299,300 which may be justified by the fact that DSPC performs better than DOPE when combined with ionizable lipids, as well as that DSPC is the only FDA-approved molecule for LNP formulation.104
As a naturally occurring lipid, cholesterol can modulate the bilayer structure of biological membranes in various ways by altering fluidity, thickness, compressibility, water penetration capacity, and intrinsic curvature.283,301 In LNPs formulations, cholesterol increases stability by filling gaps among LNPs molecules and aiding fusion with endosomal membranes, thereby promoting uptake of the vaccine complex.302 A previous study showed that LNPs made from oxidatively modified cholesterol can deliver mRNA to the liver microenvironment in a targeted manner.303
The PEGylated lipid is the least abundant component in LNPs formulation and is composed of hydrophilic polyethylene glycol (PEG) and a hydrophobic anchoring lipid [dimyristoyl Phosphoethanolamine (DMPE) or dimyristoyl glycerol (DMG)]. Both substances affect the size, permeability, and immunogenicity properties of LNPs. The main role of PEGylated lipid in LNPs is to reduce aggregation and non-specific uptake by immune cells.304,305,306 The molecular weight of PEG is typically 350–3000 Da,41 and the tail length of the anchoring lipid is typically 10−18 carbon,307 which are parameters that determine how extensively LNPs will circulate in the host as well as their uptake rate by immune cells. The greater the tail length of the anchoring lipid, the longer the half-life of the LNP complex in the host, and the lower the probability of being assimilated by macrophages in a non-specific manner.104 PEG2000-DMG is used in LNPs formulation of the FDA-approved siRNA drug OnpattroTM and Moderna COVID-19 mRNA vaccine (Fig. 7).308
Other delivery systems
Polymers are another widely used mRNA delivery system, and offer better physical stability than lipid carriers. Three main types of polymer-based delivery vehicles have been described previously: poly(ethylene imine) (PEI), poly(L-lysine) (PLL), and poly (amidoamine) (PAMAM), among which only PEI has been widely used as a delivery system in mRNA vaccines.96,309,310,311,312,313,314 Although the optimized chemical structure of PEI has higher gene transfection efficiency, it still induces strong cytotoxicity due to the high cationic charge density. Targeted modification of PEG chains can reduce cytotoxicity and significantly improve delivery efficiency in vivo and in vitro.315,316,317
In addition, another challenge with using polymer delivery vehicles is their biodegradability,318 which required the use of polyesters as mRNA carriers,316,319,320,321 among which are included poly(beta-amino esters) (PBAE), poly(amine-co-esters) (PACE), and poly(lactic acid) (PLA). Su and colleagues322 employed encapsulation of PBAE with lipids in which mRNA loaded on the surface of LNPs by electrostatic interaction. PBAE biocompatibility conferred by lipids facilitates cell entry. In addition, the pH-responsive and biodegradability of PBAE facilitate endosomal escape of delivered mRNAs and minimize cytotoxicity. Recently, Blakney and colleagues323 obtained a disulfide-linked poly(amidoamine) polymer (named pABOL), which can be used to generate polydisperse nanocomplexes of 100 nm diameter in size. In addition, in vivo experiments in mice showed that the delivery efficiency of pABOL is higher than that of PEI carriers. The pABOL system, which was developed by the Imperial College London R&D team, is considered a delivery system with the saRNA of SARS-CoV-2 in the vaccine formulation. However, its delivery efficiency was 1,000 times lower than that of LNP employed in the Acuitas vaccine formulation.289,324 Although certain characteristics of polymers, such as relatively low delivery efficiency and innate heterogeneity, limit its clinical application and industrial production, it has potential application prospects and areas for improvement.
Lipid shell-coated LPPs are a ternary complex containing a condensed mRNA core packaged in a lipid shell.325,326 LPPs have higher stability, low cytotoxicity, cell delivery and endosomal escape efficiency.327,328,329 Shen and colleagues326 developed a PbAE-based LPP platform that can efficiently deliver mRNA; in this delivery system, the PbAE-mRNA complex is encapsulated in a lipid shell which is mainly composed of 1,2dioleoyl-sn-glycero-3-ethylphosphocholine (EDOPC)/DOPE/1,2distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2k). The findings of this study revealed that, compared with naked PbAE-mRNA, cellular transfection efficiency was greatly improved when LPP-coated PbAE-mRNA was used. Moreover, this LPP-based mRNA vaccine exhibited intrinsic adjuvant activity, which stimulates dendritic cells (DCs) to secrete cytokines and inhibit tumor growth by activating the TLR7/8 signaling pathway, resulting in a significant antitumor activity. Yang and colleagues330 applied similar LPP technology to encapsulate mRNA in two steps using ionizable lipid, DOPE and PEG-lipid to generate COVID-19 mRNA vaccine with a core-shell structure, which showed significant protection in mice and non-human primates.
In addition to lipid and polymer carriers, peptides can also be used for mRNA delivery.331,332,333 Since some amino acids carry cationic or amphiphilic amino groups, they can electrostatically bind mRNA to form nanocomplexes. A commercial peptide, PepFect14, was shown to effectively deliver therapeutic mRNAs to ovarian tumor cells in mice.334 In addition, protamine was shown to activate TLR7 and TLR8 pathways, thereby showing potential as a delivery vehicle with adjuvant effect for vaccines or gene therapy.188 Based on this observation, the protamine-containing delivery platform developed by CureVac has been used in various vaccines and gene therapy for cancer treatment.85,335 Finally, cationic squalene emulsions can be applicable for mRNA delivery.336,337 These nanoemulsions are composed of a squalene-based core and a lipid shell. Squalene has an adjuvant effect, and cationic lipids on the surface of the lipid shell can bind to mRNA by electrostatic adsorption. The Lipid InOrganic Nanoparticles (LION) delivery vehicle developed by HDT Bio is composed of squalene, Span 60, Tween80, cationic lipid 1,2-dioleoyl-3-trimethylammonium propane (DOTAP), and superparamagnetic iron oxide (SPIO), and has been used to deliver self-replicating mRNA encoding the S protein of SARS-CoV-2. Preclinical results indicate that this delivery vehicle can improve vaccine stability, delivery efficiency and immunogenicity, thereby inducing strong neutralizing antibodies and T cell response in mice and non-human primates.338
Progress in clinical research on mRNA Vaccines
With the advent of mRNA delivery systems and nucleic acid modification technology, research on mRNA technology for cancer treatment as well as prevention of several infectious diseases has progressed rapidly. mRNA vaccines have shown good efficacy in the treatment of acute myelocytic leukemia (AML),339,340,341 non-small cell lung cancer (NSCLC),342,343 and melanoma344,345,346,347,348 (Table 3). The mRNA vaccine BNT111 developed by BioNTech’s FixVac platform to treat advanced melanoma has entered phase II clinical trials and was assigned the FDA Fast Track designation on November 19, 2021.349 The mRNA contained in the BNT111 vaccine encodes the four tumor-associated antigens (TAAs)—NY-ESO-1, MAGE-A3, tyrosinase, and TPTE—delivered in an RNA-lipoplex formulation. Previous studies have demonstrated that the use of BNT111 alone or in combination with PD-1 antibody can activate tumor antigen-specific CD4+ and CD8+ T cells and elicit durable immune responses.345,350,351,352 Additionally, the mRNA cancer vaccine CV9201 developed by CureVac encoding five NSCLC antigens has entered phase I/IIa clinical trials comprising 7 patients with locally advanced NSCLC and 39 patients with metastatic NSCLC. Specific immune responses against at least one antigen were detected in 63% of patients after treatment, and the frequency of activated IgD+ CD38hi B cells increased by more than two-fold in 60% of evaluated patients.343,353,354 Moderna’s mRNA personalized cancer vaccine mRNA-4157 is comprised of 34 unique neoantigen genes (encoded by tumor-specific mutated genes) combined in a single mRNA vaccine; this vaccine was proven safe and tolerable in combination with pembrolizumab in phase I clinical trials. Finally, the overall response rate (ORR) for the treatment of 10 cases of HPV-negative head and neck squamous cell carcinoma (HPV-HNSCC) with the mRNA-4157 vaccine was 50%, of which 2 cases achieved complete remission (CR).355,356,357,358
In addition to mRNA cancer vaccines, studies on mRNA vaccines to prevent infectious diseases have gradually expanded. Several mRNA vaccine candidates for viral agents other than SARS-CoV-2 have entered clinical trials (Table 4), including CMV,359 influenza virus,288,360 rabies virus,86,88 Zika virus,90,361,362,363,364 RSV,365,366 human metapneumovirus (hMPV).367 Currently, five mRNA vaccines for influenza virus encoding the HA antigen have entered clinical trials: mRNA-1851, mRNA-1440, and mRNA-1010368,369 from Moderna; MRT-5400 and MRT-5401 co-developed by Sanofi and Translate Bio.370 Current clinical trials showed that the mRNA-1440 vaccine against H10N8 and the mRNA-1851 vaccine against H7N9 influenza viruses were well tolerated and elicited robust humoral immune responses when tested separately (Table 4).360
In 2021, a rare and highly contagious SARS-CoV-2 variant emerged. SARS-CoV-2 was first identified in late 2019 and has constantly been evolving, as multiple new variants have emerged since then. To facilitate monitoring and investigation, WHO has divided the SARS-CoV-2 variants into three classes: variants of concern (VOCs), variants of interest (VOIs), and variants under monitoring (VUMs). For VOCs, 4 variants, Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), and Delta (B.1.617.2), are included.371 Each variant determined the rise of a new wave of COVID-19 infections, resulting in a massive spike in the number of deaths worldwide. On November 26, 2021, a new variant termed Omicron (B.1.1.529) was designated the fifth VOC by the WHO, immediately triggering a global health alert.372,373,374 To limit the spread of the current pandemic, governments from different countries have launched a special review task and approval of new drugs into clinical trials which showed promising application in COVID-19 vaccine production.375
BioNTech and Pfizer collaborated to develop five COVID-19 mRNA vaccine candidates at the beginning of the pandemic, which were based on nucleoside-modified mRNA (BNT162b2, BNT162b1, BNT162b3), non-modified mRNA (BNT162a1) and self-amplifying mRNA (BNT162c2). Till now, all of these candidates have entered clinical trials (Table 5).130,376,377 Among them, three different antigen types were designed in the nucleoside modified mRNA: transmembrane prefusion spike (BNT162b2), secreted spike RBD (BNT162b1), transmembrane spike RBD (BNT162b3), among which the first two were considered lead candidates; BNT162b2 encodes a full-length spike glycoprotein with two proline mutations in the S2 subunit, which is designed to maintain the protein in pre-fusion conformation; BNT162b1 encodes a secreted form of the trimeric spike protein RBD. In a phase I clinical trial, 195 subjects were divided into two groups based on the administered vaccine (BNT162b1 or BNT162b2); each group was further divided based on vaccination doses: individuals in the BNT162b1 group received either two doses with 10, 20, or 30 μg of the immunogen, or a single dose with 100 μg, or the placebo; individuals in the BNT162b2 group received either two doses with 10, 20, or 30 μg of the immunogen, or the placebo. Clinical trial data indicated that both mRNA vaccine candidates induced neutralizing antibodies at a comparable level. However, the vaccine developers eventually decide to proceed with BNT162b2 to phase II/III international clinical trials in view of its milder reactogenicity.277,278,378 BioNTech/Pfizer proceeded with BNT162b2 in global phase II/III clinical trials on July 27, 2020, which received EUA by the US government on December 11, 2020379,380 as well as the European Union conditional marketing authorization (CMA) on December 21, 2020.381,382 The BNT162b2 mRNA vaccine was then added to the WHO Emergency Use Listing (EUL) on December 31, 2020,383 and approved on May 10 and May 28, 2021 by the US government and the European Union, respectively, for administration in adolescents aged 12 to 15.384 The US FDA officially authorized BNT162b2 for vaccination of individuals aged 16 and older on August 23, 2021.105 The FDA granted BNT162b2 emergency approval for use in children and adolescents aged 5 to 11 on October 29, 2021.103,106
BNT162b2 is administered intramuscularly as a two-dose scheme of 30 μg of immunogen per dose, 21 days apart. The efficacy of the vaccine at preventing COVID-19 infection in individuals aged 16 was 95%, and over the six subsequent months, vaccine efficacy was 91.3%.385,386,387 Studies have shown that the protective effect of BNT162b2 against SARS-CoV-2 infection peaks after the second dose and then quickly declines, with the humoral immune response sharply decreasing.388,389,390,391 However, results from clinical trials with over 5,000 people who had received BioNTech/Pfizer booster injections showed that protection conferred by the vaccine was 95.6%.392,393,394 On September 22, 2021, the FDA approved the BNT162b2 vaccine for administration in high-risk populations as a booster,395 then expanded eligibility on November 19, 2021 as a single booster of both BNT162b2 and mRNA-1273 to individuals aged 18 and older after completion of the primary vaccination scheme with any FDA-authorized or previously approved COVID-19 vaccine.396 In a mass vaccination study which included 3,159,136 participants from Israel, vaccine effectiveness of BNT162b2 as a two-dose scheme was 94%.397 However, the vaccine was associated with increased risk of myocarditis in Israeli participants, reaching a rate of approximately 3 events per 100,000 persons.398,399,400 Another study indicated that the incidence of anaphylaxis after BNT162b2 vaccination in Japan was higher, which points towards considering race-related adverse effects401 whose underlying causes are still unknown. The PEG additive,402 which is also used in several cosmetics and pharmaceutical drugs, has been incriminated as a possible cause for anaphylaxis induced by the BNT162b2 mRNA vaccine. This is related to the fact 57% of the 37 people who presented anaphylaxis had a history of allergy, and four had a history of cosmetic allergy, suggesting a potential role for PEG in inducing anaphylaxis.
The SARS-CoV-2 variant B.1.617.2 was first identified in India in December 2020, being later designated the Delta variant, and became predominant in several countries.403 Real-world data from Qatar indicated that the BNT162b2 vaccine had only 51.9% effectiveness against the Delta variant, which was significantly lower compared to 75.0% and 89.5% effectiveness conferred by the vaccine against Beta and Alpha variants, respectively.102 As mentioned previously, protection conferred by BNT162b2 decreases significantly over time. The effectiveness of the BNT162b2 vaccine against the Beta variant was measured shortly after the population of Qatar had been vaccinated, whereas the effectiveness of BNT162b2 against the Delta variant was conducted several months after the second dose, which could be one of the reasons for low effectiveness of BNT162b2 against the Delta variant.404,405 More recently, and using a pseudovirus neutralization test (pVNT), neutralization titers induced after two doses of BNT162b2 were 160, 7, 24, and 73 GMTs for wild-type SARS-CoV-2, Omicron, Beta, and Delta variants, respectively, but improved to 368, 164, 279, and 413 GMTs, after one month following a booster vaccination. Comparable trends were observed in live virus neutralization testing. Thus vaccine booster with BNT162b2 may enhance neutralization of the Omicron variant.406 Similar findings suggest that vaccination with three doses of the mRNA vaccine BNT162b2 may protect against Omicron-mediated COVID-19.407,408
The mRNA-1273 developed by Moderna encodes the full-length prefusion spike protein of SARS-CoV-2 and is the second mRNA vaccine received EUA by the US government on December 18, 2020, and received Biologics License Application (BLA) on Janurary 31, 2022.409 Results of clinical trials have shown that mRNA-1273 is generally well tolerated and safe to use in adolescents and adults. No serious safety concerns have been identified so far, and most adverse events were mild or moderate; the most common adverse effect was pain at the vaccine injection site on both shots, whereas headache, fatigue, myalgia and chills were adverted after the second shot.410,411 In a phase III clinical trial involving 30,420 people, participants aged 18 or older were given two doses of 100 μg of mRNA-1273 with a 28-day interval, and vaccine efficacy was 94.1% (Table 5), with similar immune responses among adolescents aged 12 to 17.410,412,413,414 The mRNA-1273.351 developed based on SARS-CoV-2 Beta variant first identified in South Africa, together with the mRNA-1273.211 molecule containing both mRNA-1273.351 and mRNA-1273 has entered phase III clinical trials as a vaccine booster. Approximately six months after administration of the two injections of the mRNA-1273 vaccine, each group of twenty participants received a booster with the immunogen: 50 μg mRNA-1273, mRNA-1273.211 or mRNA-1273.351. The neutralization effect of the booster in each immunization group against SARS-CoV-2 variants Beta, Gamma and Delta reached a level comparable to that observed against the wild-type D614G strain. Among the three booster vaccines evaluated, the multivalent mRNA-1273.211 induced the largest geometric mean titers (GMT) for variants Beta, Gamma and Delta.415,416 Between December 28, 2020, and May 10, 2021, 256,037 people in Qatar received at least one dose of the mRNA-1273 vaccine, whereas 181,034 people received two doses. Real-world data showed that effectiveness of mRNA-1273 against Alpha and Beta variants was 100% and 96.4%, respectively,417,418 whereas effectiveness against Delta was slightly lower (73.1%).102 Other studies have shown that mRNA-1273 is still quite effective in congregate settings or higher-risk exposure such as prisons or hospitals in which the Delta variant was prevalent.419,420 Further real-world data on vaccine effectiveness for Omicron and Delta variants showed that administration of three doses of mRNA-1273 provided a high and durable protection against Delta infection (95.2%) but lower protection against Omicron (62.5%). However, none of the vaccinated individuals with three doses of mRNA-1273 were hospitalized, which indicates a promising alternative.421 Overall, despite the fact that vaccine effectiveness against SARS-CoV-2 decreases over time, vaccination with BNT162b2 and mRNA-1273 was still effective in preventing infection with Delta and other variants, reducing hospitalization and mortality, with mRNA-1273 showing a slightly superior performance compared to BNT162b2.420,422,423,424
Additionally, CureVac’s first-generation COVID-19 mRNA vaccine candidate (CVnCoV) is administered as a two-dose series with 12 μg, 28 days apart. Preliminary results from phase IIb/III clinical trials showed that the overall efficacy of the vaccine was 48.2% (Table 5), which failed to meet prespecified success criteria.425 In previously preclinical trials, CVnCoV was shown to induce high levels of neutralizing antibodies in rodents and non-human primates, outbalancing immune responses mediated by CD4+ and CD8+ T cells as well as showing better efficacy against SARS-CoV-2 D614G variant.426 Phase I clinical data showed that CVnCoV has acceptable tolerance and high immunogenicity.29,427 The low performance of CVnCoV in phase II clinical trials has been attributed to the rise of multiple SARS-CoV-2 variants. In phase II clinical trials, 228 COVID-19 cases were reported, from which 204 have been sampled for whole-genome sequencing, which showed that only 3% of the patients had been infected by wild-type SARS-CoV-2, whereas 14 variants have been identified in the remaining patients.425 The low vaccination dosage of CVnCoV and the use of unmodified nucleotides might have contributed to the performance of this vaccine in phase II clinical trials compared to the licensed COVID-19 vaccine.428 On October 12, 2021, the European Medicines Agency (EMA) announced the suspension of the rolling review of CVnCoV.429 On the same day, CureVac announced that they would abandon CVnCoV follow-up clinical studies to focus their research efforts on a second-generation mRNA vaccine candidate developed in collaboration with GSK.430
The COVID-19 vaccine PTX-COVID19-B produced by Providence Therapeutics in Canada has entered phase II clinical trial, proposing a two-dose scheme with 40 μg per injection, 28 days apart. PTX-COVID19-B was shown to induce high titers of neutralizing antibodies against wild-type SARS-CoV-2 and variants, including Alpha, Beta, and Delta, which were comparable to those elicited by the approved COVID-19 mRNA vaccines when assessed by the same neutralization assay.431,432 Moreover, Sanofi Pasteur collaborated with Translate Bio to develop the first-generation vaccine MRT5500, which encodes the full-length spike protein of SARS-CoV-2. Results of preclinical trials showed that MRT5500 induced a Th1-biased immune response in mice and non-human primates and prevented Th2-bias response which can induce vaccine-related enhanced respiratory disease (VAERD).433,434,435 Mid-term results of phase I clinical trial of MRT5500 showed that the seroconversion rate was 91% to 100%. However, considering that global supply of COVID-19 vaccines is sufficient, Sanofi decided to abandon MRT5500 follow-up studies.436,437 In addition, the mRNA vaccine ARCoV developed in China by Abogen has entered phase III clinical trials438; ARCoV is reportedly safe and well-tolerated at an amount of 15 μg, which induced high titers of neutralizing antibodies titers (approximately two-fold higher that those of patients which had recovered from COVID-19 infection).439 Moreover, mRNA vaccines of Stemirna and LIVERNA have also entered phase I and II clinical trials.440
Furthermore, the saRNA vaccine LNP-nCoVsaRNA developed by the Imperial College London showed seroconversion rate from 8% to 61% as determined by ELISA when 0.1–10.0 μg was administered per dose group, demonstrated inferior immunogenicity in humans compared to that observed in mice.212 In addition, the results of phase I/II clinical trials of the saRNA vaccine ARCT-021 developed by Arcturus Therapeutics indicated the production of robust anti-spike specific antibodies when 5.0–7.5 μg was administered per dose group, but it also failed to reach 100% seroconversion rate.441,442 saRNA vaccines have dosage advantages compared to the approved mRNA vaccines BNT162b2, mRNA-1273, and other non-replicating mRNA vaccines. However, current results of clinical trials suggest that immunogenicity profiles of saRNA vaccines may not be comparable to those obtained with non-self-replicating mRNA vaccines. This might likely reflect differences in exogenous RNA restriction by the innate immune sensing. Thus, incorporation of encoded modulators of human PRR or of a wider range of potential modifications may positively affect immunogenicity and efficacy of saRNA vaccines.443,444
Production and quality control of mRNA vaccines
Production of mRNA vaccines
Production of mRNA vaccines does not require culturing cells or viruses as in traditional vaccine production technology, relying instead on in vitro synthesis technology. Therefore, the production cycle is shorter and easy to scale up, hence offering the possibility of quick industrialization of vaccine production. From IVT of mRNA to preparation of mRNA-LNP complexes, the entire production cycle for an mRNA vaccine might last approximately 10 days. Considering also the time required for qualification and release, the product can be available on the market within 40 days. As a technology platform, mRNA vaccine technology is broadly compatible with any mRNA sequences and virtually all vaccines based on proteins can be produced using this technology. The production process mainly involves the following steps: target antigen sequence design; plasmid construction; establishing a three-level bacterial biobank; DNA template preparation; IVT of mRNA; mRNA purification; LNP formulation and encapsulation; mRNA-LNP complex dilution; mRNA-LNP complex concentration; sterile filtration and filling; and other minor final steps (Fig. 8).445 The production of mRNA vaccines is carried out under conditions that comply with current laws, regulations, and management guidelines preconized by governments and regulatory authorities in various countries. In this context, the five main elements (man, material, machine, method, and environment) must meet local and international requirements of good manufacturing practices and other standards.446,447,448,449,450

The design of an mRNA vaccine is conditioned to the definition of the antigen sequence of the target pathogen. By determining the target antigen and optimizing its coding sequence, the mRNA can be transcribed in vitro by RNA polymerase. The synthesized mRNA is purified by different processes and then mixed with a lipid phase using microfluidics and encapsulated into an mRNA-lipid nanoparticle (mRNA-LNP) complex. Subsequently, self-assembly of LNPs is completed by dilution and concentration by ultrafiltration. Finally, after sterile filtration, filling, and capping, the mRNA vaccine is obtained
The production of mRNA vaccines starts with the synthesis of the target antigen. After the antigen gene sequence is optimized and cloned into a plasmid, engineered bacteria are amplified and cultured to retrieve the desired gene sequences. Two strategies can be employed to obtain the linearized DNA template for IVT: using restriction enzymes to linearize the plasmid, or using PCR to amplify target DNA in preclinical studies or small-scale production. The purified and recovered DNA template is then used in IVT to obtain the corresponding mRNA. Using linear DNA as a template, NTPs are employed as precursors for the synthesis of the desired mRNA molecule by T7, SP6, or T3 RNA polymerase. In addition to the linearized DNA template and RNA polymerase, the IVT reaction also requires other components: ribonuclease inhibitor, pyrophosphatase, polymerase cofactor MgCl2, and a pH buffer containing polyamine and antioxidants.451 After a few hours of IVT, milligram quantities of the desired mRNA can be produced per milliliter of transcription reaction.452 Compared with the traditional production methods for inactivated, subunit, or viral vector vaccines, mRNA vaccine technology avoids complicated and time-consuming production steps, while also reducing the risk of contamination from cell sources.
In addition to the target product, the mRNA IVT reaction has many impurities, such as enzymes, NTPs, DNA, and abnormal transcription products. In a lab setting, treatment with DNase is often used to eliminate DNA from the obtained mRNA preparation, and lithium chloride precipitation is used to purify mRNA further.115 Nonetheless, these methods do not enable removal of abnormal transcription products, such as dsRNA and truncated RNA fragments. Abnormal transcription products can activate the host innate immune response, thereby causing inflammation and reducing the translation efficiency of delivered mRNA. Previous studies showed that the protein yield of mRNA purified by reverse phase HPLC increases by 10–1000 times.156,453 In addition to HPLC, magnetic beads, anion exchange, ultrafiltration, and dialysis can also be used as purification methods. Then, purified mRNA is dissolved in aqueous phase.
Purified mRNA is not suitable for clinical use unless it is protected by a delivery formulation. Most advanced mRNA vaccines employ LNPs as delivery systems. The four lipids (ionizable lipid, DSPC, cholesterol, and PEG-lipid) constituting LNPs are dissolved in ethanol, and each solution is prepared at a known concentration, then mixed at different molar ratios to prepare the mixed lipid phase. The target mRNA is then diluted in a buffer solution at pH ~4 to prepare the aqueous phase. With an appropriate lipid/mRNA ratio, the lipid solution is mixed with the mRNA aqueous phase in a microfluidic or T-junction channel to obtain lipid-encapsulated mRNA. The assembly principle underlying the mRNA-LNP preparation is that the ionizable lipid becomes protonated in the aqueous phase at pH 5.5. Due to its positive charge, it electrostatically binds to the negatively charged mRNA, thereby promoting vesicle formation and encapsulation of mRNA by hydrophobic forces. Once vesicles are formed, dilution and subsequent concentration by ultrafiltration are carried out to remove ethanol and replace the buffer to increase the pH of the preparation. At this time, the ionizable lipid adopts a more hydrophobic, uncharged condition, which drives vesicle fusion to promote encapsulation of mRNA, thereby forming a stable mRNA-LNP spherical complex.289 Finally, the obtained mRNA vaccine preparation is further submitted to final processing steps of sterile filtration and filling.
Quality control of mRNA vaccines
Although the mRNA vaccine is a new technology and its production relatively simple for scaling-up, most approaches employed during the process are sophisticated, thus quality control of mRNA vaccine production still represents a challenge.454 Safety, efficacy, and quality control of vaccine production are determined by measuring critical process parameters (CPPs) and intermediate critical quality attributes (IQAs). The management of LNPs encapsulation is directly related to the quality of the final mRNA vaccine, particularly involving target gene sequence design, raw materials, mRNA purity and integrity, and mRNA/lipid ratio. Quality control of mRNA vaccines should adhere to criteria preconized by laws and regulations of the producing countries. Quality control and quality management should be incorporated throughout the production process and life cycle of mRNA vaccines, thereby submitting the entire chain to stringent quality control monitoring.455,456,457,458,459,460,461,462,463,464 Quality control of COVID-19 mRNA vaccines focuses on raw materials (plasmids, biobanks, lipids, nucleotides, and enzymes used in the production process), semi-finished, and final product.445,465
Each country may have different regulamentations depending on whether the plasmid is used as a raw material for the production of mRNA vaccines. However, plasmid quality control must be under constant vigilance, i.e., ensuring the target gene sequence is unmodified, therefore the production process must be tightly controlled. Other biological features of plasmids, such as purity, proportion of supercoiled structures, and residual substance derived from plasmid extraction, should also be considered. In addition to strain quality control, ensuring that bacteria employed in transformations are indeed Escherichia coli, plasmid gene sequence, retention rate, copy number, and bacterial strain purity are parameters that should also be considered.
Lipid excipients are essential in mRNA vaccine production. Lipid functions should be assessed in addition to their source, physical and chemical features, such as appearance, type, purity, and residual solvents. Moreover, evaluation with pharmaceutical drugs should also be performed to ensure that novel lipid excipients are in compliance with applicable regulations. Furthermore, any animal (including human)-derived starting or raw material should be submitted to control to determine the source, quality control, and risk assessment, as well as should comply with WHO guidelines on transmissible spongiform encephalopathies concerning biological and pharmaceutical products.466
In addition, identification, purity, content of the mRNA stock solution must be considered. Process impurities in the drug substance include residual DNA, RNA polymerase, restriction endonuclease, DNA ligase, unincorporated nucleotides, caps, dsRNA, and misfolded RNA. Proper mRNA capping plays a critical role in mRNA transcription and translation efficiency, as well as in reducing inflammation response. As a result, determining mRNA capping efficiency is critical for quality control of mRNA vaccines, which is usually determined by liquid chromatography and mass spectrometry (LC-MS).467
Lipid-encapsulated mRNA forms the mRNA-LNP complex, which is a critical step in mRNA vaccine technology. A reproducible production process should be guaranteed by ensuring proper calibration and adjustment of encapsulating pump equipment and flow rate accuracy after a comprehensive and in-depth optimization. To assure size and homogeneity of LNPs, appropriate criteria should be stipulated, which in turn can increase vaccine stability and induce a better immune response. As a result, mRNA-LNP intermediate products must be scrutinized by measuring average particle size, particle size distribution, mRNA content, encapsulation efficiency, and sterility.
Collectively, it is paramount that internal quality control and release standards must be prepared. The mRNA vaccine should be stored and transported under ultra-low temperature conditions, due to the intrinsic instability of mRNA. Onpattro™, the first approved siRNA drug, also relies on an LNP-based delivery system,308 and the drug can be stored at 2–8 °C for three years. Studies showed that mRNA is highly susceptible to degradation by the ubiquitous RNases, therefore ultra-low temperature storage is recommended to limit RNase activity whilst ensuring mRNA stability effectively. In addition, ultra-low temperature preservation is more conducive to maintaining the original conformation of mRNA-LNP complexes.468 In fact, mRNA vaccines that have been licensed for clinical trials, commercialization, or emergency use must currently be stored and delivered at low temperatures. Although the current delivery system used in mRNA vaccines is based on LNPs, storage conditions and stability vary among vaccine manufacturers. For instance, the mRNA-1273 vaccine from Moderna maintains its effectiveness for six months at −20 °C, whereas the effectiveness of the BNT162b2 vaccine from BioNTech is maintained for six months when stored at −60 to −80 °C. Conversely, the CVnCoV vaccine from CureVac can be stored at −60 °C for three months. Therefore, existing COVID-19 mRNA vaccines undoubtedly require storage under ultra-low temperatures.469 As a result, appropriate verification of cold chain transportation and storage conditions should be taken into consideration in order to ensure the quality of the mRNA vaccine.470,471 A more recent storage approach involves employing freeze-drying to mRNA vaccine conservation, which was shown to effectively reduce storage requirements, being thus considered a promising technique to improve mRNA vaccine stability.472,473
Furthermore, for scientific and ethical standards, the international pharmacopeia and WHO urged the reduction of animal trials, and instead employing appropriate in vitro alternative approaches for evaluating mRNA safety and efficacy.474 At present, with the expansion of COVID-19 mRNA vaccine production and the incorporation of new production lines, changes in the production process have become inevitable. It is necessary to perform inter-batch quality comparisons based on relevant regulations and standards, including procedures and data requirements preconized in WHO guidelines.475 If quality comparison studies cannot fully attest product safety, effectiveness, and controllability, bridging studies in clinical trials are likely to offer a more reliable perspective.
Conclusion and future perspectives
After over 30 years of research, mRNA vaccines have become a promising technology platform for vaccine development. Prior to the emergence of COVID-19, mRNA technology was mostly used for developing novel cancer therapeutic drugs showing promising results. The COVID-19 pandemic has fostered the growth of mRNA vaccine platforms as a means to prevent and treat several infectious diseases, and a new generation of vaccines has progressively reached the public and gained increasing attention. At the moment, COVID-19 mRNA vaccines are playing a key role in limiting the spread of the current pandemic. mRNA vaccines, unlike traditional vaccines, may enable adjustment of antigen design and even allow combining sequences from several variants to respond to new mutations in the virus genome. In the future, the mRNA technology platform will enable preventing and managing infectious diseases as well as treating other disorders. Due to its advantages, such as a quick development cycle, no requirement for cell culture, and high immunogenicity, an mRNA vaccine has become the world’s first COVID-19 vaccine authorized by the FDA. However, the requirement for storage at ultra-low temperature conditions might represent a challenge in transportation and storage of mRNA vaccines. Therefore, stability of mRNA vaccines has to be further explored and optimized. Moreover, delivery of mRNA has evolved from its naked form to LNP-based delivery. Additional studies are being conducted to explore novel polymer materials to be used in mRNA delivery in the future. We believe that the safety and protection conferred by current mRNA vaccines are satisfactory, but their long-term protection will only be determined after additional clinical studies are performed.
References
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020).
Zhang, J.-j et al. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. ALLERGY 75, 1730–1741 (2020).
Hu, B., Guo, H., Zhou, P. & Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 19, 141–154 (2021).
Chen, L. et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg. Microbes Infect. 9, 313–319 (2020).
Zhu, G., Zhu, C., Zhu, Y. & Sun, F. Minireview of progress in the structural study of SARS-CoV-2 proteins. Curr. Res. Microb. Sci. 1, 53–61 (2020).
WHO Coronavirus (COVID-19) Dashboard. Situation by Region, Country, Territory & Area, https://covid19.who.int/table (2021).
WHO Coronavirus (COVID-19) Dashboard, https://covid19.who.int/ (2021).
Xia, S. et al. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBIBP-CorV: a randomised, double-blind, placebo-controlled, phase 1/2 trial. Lancet Infect. Dis. 21, 39–51 (2021).
Zhang, Y. et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18–59 years: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect. Dis. 21, 181–192 (2021).
Xia, S. et al. Effect of an inactivated vaccine against SARS-CoV-2 on safety and immunogenicity outcomes: interim analysis of 2 randomized clinical trials. JAMA 324, 951–960 (2020).
Ella, R. et al. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBV152: a double-blind, randomised, phase 1 trial. Lancet Infect. Dis. 21, 637–646 (2021).
Wang, Y. et al. Scalable live-attenuated SARS-CoV-2 vaccine candidate demonstrates preclinical safety and efficacy. Proc. Natl Acad. Sci. USA 118, e2102775118 (2021).
Trimpert, J. et al. Development of safe and highly protective live-attenuated SARS-CoV-2 vaccine candidates by genome recoding. Cell Rep. 36, 109493 (2021).
Seo, S. H. & Jang, Y. Cold-adapted live attenuated SARS-Cov-2 vaccine completely protects human ACE2 transgenic mice from SARS-Cov-2 infection. Vaccines 8, 584 (2020).
Yang, J. et al. A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity. Nature 586, 572–577 (2020).
Guebre-Xabier, M. et al. NVX-CoV2373 vaccine protects cynomolgus macaque upper and lower airways against SARS-CoV-2 challenge. Vaccine 38, 7892–7896 (2020).
Yang, S. et al. Safety and immunogenicity of a recombinant tandem-repeat dimeric RBD-based protein subunit vaccine (ZF2001) against COVID-19 in adults: two randomised, double-blind, placebo-controlled, phase 1 and 2 trials. Lancet Infect. Dis. 21, 1107–1119 (2021).
Dai, L. et al. A universal design of betacoronavirus vaccines against COVID-19, MERS, and SARS. Cell 182, 722–733. e711 (2020).
Wu, Y. et al. A recombinant spike protein subunit vaccine confers protective immunity against SARS-CoV-2 infection and transmission in hamsters. Sci. Transl. Med. 13, eabg1143 (2021).
Zhu, F.-C. et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. Lancet 395, 1845–1854 (2020).
Folegatti, P. M. et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 396, 467–478 (2020).
Logunov, D. Y. et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 397, 671–681 (2021).
Sadoff, J. et al. Interim results of a phase 1–2a trial of Ad26. COV2. S Covid-19 vaccine. N. Engl. J. Med. 384, 1824–1835 (2021).
Bos, R. et al. Ad26 vector-based COVID-19 vaccine encoding a prefusion-stabilized SARS-CoV-2 Spike immunogen induces potent humoral and cellular immune responses. npj Vaccines 5, 1–11 (2020).
Loes, A. N., Gentles, L. E., Greaney, A. J., Crawford, K. H. & Bloom, J. D. Attenuated influenza virions expressing the SARS-CoV-2 receptor-binding domain induce neutralizing antibodies in mice. Viruses 12, 987 (2020).
Chaparian, R. R. et al. Influenza viral particles harboring the SARS-CoV-2 spike RBD as a combination respiratory disease vaccine. BioRxiv, https://doi.org/10.1101/2021.04.30.441968 (2021).
Vogel, A. et al. A prefusion SARS-CoV-2 spike RNA vaccine is highly immunogenic and prevents lung infection in non-human primates. BioRxiv, https://doi.org/10.1101/2020.09.08.280818 (2020).
Corbett, K. S. et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 586, 567–571 (2020).
Kremsner, P. G. et al. Safety and immunogenicity of an mRNA-lipid nanoparticle vaccine candidate against SARS-CoV-2: A phase 1 randomized clinical trial. Wien. Klin. Wochenschr. 133, 931–941 (2021).
Smith, T. R. et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 11, 1–13 (2020).
Silveira, M. M., Moreira, G. M. S. G. & Mendonça, M., DNA vaccines against COVID-19: Perspectives and challenges. Life Sci., 118919 (2020).
Dey, A. et al. Immunogenic potential of DNA vaccine candidate, ZyCoV-D against SARS-CoV-2 in animal models. Vaccine 39, 4108–4116 (2021).
Yu, J. et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 369, 806–811 (2020).
Momin, T. et al. Safety and Immunogenicity of a DNA SARS-CoV-2 vaccine (ZyCoV-D): Results of an open-label, non-randomized phase I part of phase I/II clinical study by intradermal route in healthy subjects in India. EClinicalMedicine 38, 101020 (2021).
Kim, J., Eygeris, Y., Gupta, M. & Sahay, G. Self-assembled mRNA vaccines. Adv. Drug Deliv. Rev. 170, 83–112 (2021).
Pardi, N. et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Controlled Release 217, 345–351 (2015).
Lazzaro, S. et al. CD 8 T-cell priming upon mRNA vaccination is restricted to bone-marrow-derived antigen-presenting cells and may involve antigen transfer from myocytes. Immunology 146, 312–326 (2015).
Probst, J. et al. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. 14, 1175–1180 (2007).
Lindsay, K. E. et al. Visualization of early events in mRNA vaccine delivery in non-human primates via PET–CT and near-infrared imaging. Nat. Biomed. Eng. 3, 371–380 (2019).
Liang, F. et al. Efficient targeting and activation of antigen-presenting cells in vivo after modified mRNA vaccine administration in rhesus macaques. Mol. Ther. 25, 2635–2647 (2017).
Oberli, M. A. et al. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 17, 1326–1335 (2017).
Firdessa-Fite, R. & Creusot, R. J. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 16, 50–62 (2020).
Semple, S. C. et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 28, 172–176 (2010).
Varkouhi, A. K., Scholte, M., Storm, G. & Haisma, H. J. Endosomal escape pathways for delivery of biologicals. J. Control. Rel. 151, 220–228 (2011).
Wittrup, A. et al. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 33, 870–876 (2015).
Selby, L. I., Cortez-Jugo, C. M., Such, G. K. & Johnston, A. P. Nanoescapology: progress toward understanding the endosomal escape of polymeric nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 9, e1452 (2017).
Cagigi, A. & Loré, K. Immune responses induced by mRNA vaccination in mice, monkeys and humans. Vaccines 9, 61 (2021).
Xu, S., Yang, K., Li, R. & Zhang, L. Mrna vaccine era—Mechanisms, drug platform and clinical prospection. Int. J. Mol. Sci. 21, 6582 (2020).
Verbeke, R., Lentacker, I., De Smedt, S. C. & Dewitte, H. Three decades of messenger RNA vaccine development. Nano Today 28, 100766 (2019).
Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. & e Sousa, C. R. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004).
Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529 (2004).
Linares-Fernández, S., Lacroix, C., Exposito, J. Y. & Verrier, B. Tailoring mRNA vaccine to balance innate/adaptive immune response. Trends Mol. Med. 26, 311–323 (2019).
Alexopoulou, L., Holt, A. C., Medzhitov, R. & Flavell, R. A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 (2001).
Hornung, V. et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Kato, H. et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid–inducible gene-I and melanoma differentiation–associated gene 5. J. Exp. Med. 205, 1601–1610 (2008).
Brisse, M. & Ly, H. Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Front. Immunol. 10, 1586 (2019).
De Beuckelaer, A., Grooten, J. & De Koker, S. Type I interferons modulate CD8+ T cell immunity to mRNA vaccines. Trends Mol. Med. 23, 216–226 (2017).
Hervas-Stubbs, S. et al. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res 17, 2619–2627 (2011).
Devoldere, J., Dewitte, H., De Smedt, S. C. & Remaut, K. Evading innate immunity in nonviral mRNA delivery: don’t shoot the messenger. Drug Discov. Today 21, 11–25 (2016).
Brenner, S., Jacob, F. & Meselson, M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 190, 576–581 (1961).
Lockard, R. E. & Lingrel, J. B. The synthesis of mouse hemoglobin chains in a rabbit reticulocyte cell-free system programmed with mouse reticulocyte 9S RNA. Biochem. Biophys. Res. Commun. 37, 204–212 (1969).
Dimitriadis, G. J. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature 274, 923–924 (1978).
Ostro, M. J., Giacomoni, D., Lavelle, D., Paxton, W. & Dray, S. Evidence for translation of rabbit globin mRNA after liposomemediated insertion into a human cell line. Nature 274, 921–923 (1978).
Krieg, P. A. & Melton, D. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res 12, 7057–7070 (1984).
Krieg, P. A. & Melton, D. In vitro RNA synthesis with SP6 RNA polymerase. Methods Enzymol. 155, 397–415 (1987).
Jobling, S. A., Cuthbert, C. M., Rogers, S. G., Fraley, R. T. & Gehrke, L. In vitro transcription and translational efficiency of chimeric SP6 messenger RNAs devoid of 5′ vector nucleotides. Nucleic Acids Res 16, 4483–4498 (1988).
Melton, D. et al. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res 12, 7035–7056 (1984).
Stump, W. T. & Hall, K. B. SP6 RNA polymerase efficiently synthesizes RNA from short double-stranded DNA templates. Nucleic Acids Res 21, 5480–5484 (1993).
Edelmann, A., Kirchberger, J., Naumann, M. & Kopperschläger, G. Generation of catalytically active 6-phosphofructokinase from Saccharomyces cerevisiae in a cell-free system. FEBS J. 267, 4825–4830 (2010).
Melton, D. Injected anti-sense RNAs specifically block messenger RNA translation in vivo. Proc. Natl Acad. Sci. U.S.A. 82, 144–148 (1985).
Pakdel, F., Le Gac, F., Le Goff, P. & Valotaire, Y. Full-length sequence and in vitro expression of rainbow trout estrogen receptor cDNA. Mol. Cell. Endocrinol. 71, 195–204 (1990).
Gantt, J. S. & Key, J. L. Molecular cloning of a pea H1 histone cDNA. Eur. J. Biochem 166, 119–125 (1987).
Wawrousek, E. F., Nickerson, J. M. & Piatigorsky, J. Two δ-crystallin polypeptides are derived from a cloned δ1-crystallin cDNA. FEBS Lett. 205, 235–240 (1986).
Sahin, U., Karikó, K. & Türeci, Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Malone, R. W., Felgner, P. L. & Verma, I. M. Cationic liposome-mediated RNA transfection. Proc. Natl Acad. Sci. USA 86, 6077–6081 (1989).
Wolff, J. A. et al. Direct gene transfer into mouse muscle in vivo. Science 247, 1465–1468 (1990).
Martinon, F. et al. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 23, 1719–1722 (1993).
Conry, R. M. et al. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 55, 1397–1400 (1995).
Zhou, W.-Z. et al. RNA melanoma vaccine: induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Hum. Gene Ther. 10, 2719–2724 (1999).
Lattime, E. C., Mastrangelo, M. J., Bagasra, O., Li, W. & Berd, D. Expression of cytokine mRNA in human melanoma tissues. Cancer Immunol. Immunother. 41, 151–156 (1995).
Karikó, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Hoy, S. M. Patisiran: first global approval. Drugs 78, 1625–1631 (2018).
Adams, D. et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med 379, 11–21 (2018).
Kristen, A. V. et al. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 9, 5–23 (2019).
Schnee, M. et al. An mRNA vaccine encoding rabies virus glycoprotein induces protection against lethal infection in mice and correlates of protection in adult and newborn pigs. PLoS Negl. Trop. Dis. 10, e0004746 (2016).
Alberer, M. et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 390, 1511–1520 (2017).
Lutz, J. et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. npj Vaccines 2, 1–9 (2017).
Aldrich, C., Leroux–Roels, I., Huang, K. B., Bica, M. A. & Oostvogels, L. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: A phase 1 trial. Vaccine 39, 1310–1318 (2021).
Wong, S.-S. & Webby, R. J. An mRNA vaccine for influenza. Nat. Biotechnol. 30, 1202–1204 (2012).
Richner, J. M. et al. Modified mRNA vaccines protect against Zika virus infection. Cell 168, 1114–1125. e1110 (2017).
Roth, C. et al. A modified mRNA vaccine targeting immunodominant NS epitopes protects against dengue virus infection in HLA class I transgenic mice. Front. Immunol. 10, 1424 (2019).
Zhang, M., Sun, J., Li, M. & Jin, X. Modified mRNA-LNP vaccines confer protection against experimental DENV-2 infection in mice. Mol. Ther. Methods Clin. Dev. 18, 702–712 (2020).
Wollner, C. J. et al. A mRNA-LNP vaccine against Dengue Virus elicits robust, serotype-specific immunity. BioRxiv, https://doi.org/10.1101/2021.01.05.425517 (2021).
Petsch, B. et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 30, 1210–1216 (2012).
Hekele, A. et al. Rapidly produced SAM® vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2, 1–7 (2013).
Chahal, J. S. et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl Acad. Sci. U.S.A. 113, E4133–E4142 (2016).
Meyer, M. et al. Modified mRNA-based vaccines elicit robust immune responses and protect guinea pigs from Ebola virus disease. J. Infect. Dis. 217, 451–455 (2018).
Pardi, N. et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543, 248–251 (2017).
Pardi, N. et al. Characterization of HIV-1 nucleoside-modified mRNA vaccines in rabbits and rhesus macaques. Mol. Ther. Nucleic Acids 15, 36–47 (2019).
COVID-19 vaccine tracker and landscape, https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (2021).
Barda, N. et al. Effectiveness of a third dose of the BNT162b2 mRNA COVID-19 vaccine for preventing severe outcomes in Israel: an observational study. Lancet., https://doi.org/10.1016/S0140-6736(21)02249-2 (2021).
Tang, P. et al. BNT162b2 and mRNA-1273 COVID-19 vaccine effectiveness against the SARS-CoV-2 Delta variant in Qatar. Nat. Med. 27, 2136–2143 (2021).
Walter, E. B. et al. Evaluation of the BNT162b2 Covid-19 vaccine in children 5 to 11 years of age. N. Engl. J. Med 386, 35–46 (2021).
Chaudhary, N., Weissman, D. & Whitehead, K. A. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat. Rev. Drug Discov. 20, 817–838 (2021).
FDA Approves First COVID-19 Vaccine | FDA, https://www.fda.gov/news-events/press-announcements/fda-approves-first-covid-19-vaccine (2021).
FDA Authorizes Pfizer-BioNTech COVID-19 Vaccine for Emergency Use in Children 5 through 11 Years of Age, https://www.fda.gov/news-events/press-announcements/fda-authorizes-pfizer-biontech-covid-19-vaccine-emergency-use-children-5-through-11-years-age (2021).
Iavarone, C., O’hagan, D. T., Yu, D., Delahaye, N. F. & Ulmer, J. B. Mechanism of action of mRNA-based vaccines. Expert Rev. Vaccines 16, 871–881 (2017).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Blakney, A. K., Ip, S. & Geall, A. J. An update on self-amplifying mRNA vaccine development. Vaccines 9, 97 (2021).
Atasheva, S. et al. Enhancement of protein expression by alphavirus replicons by designing self-replicating subgenomic RNAs. Proc. Natl Acad. Sci. USA 111, 10708–10713 (2014).
Beissert, T. et al. A trans-amplifying RNA vaccine strategy for induction of potent protective immunity. Mol. Ther. 28, 119–128 (2020).
Qu, L. et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. BioRxiv, https://doi.org/10.1101/2021.03.16.435594 (2021).
Schlake, T., Thess, A., Fotin-Mleczek, M. & Kallen, K.-J. Developing mRNA-vaccine technologies. RNA Biol. 9, 1319–1330 (2012).
Youn, H. & Chung, J. K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin. Biol. Ther. 15, 1–12 (2015).
Kwon, H. et al. Emergence of synthetic mRNA: in vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 156, 172–193 (2018).
Gallie, D. R. The cap and poly (A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 5, 2108–2116 (1991).
To, K. K. & Cho, W. C. An overview of rational design of mRNA-based therapeutics and vaccines. Expert Opin. Drug Discov. 16, 1307–1317 (2021).
Wang, J. et al. Quantifying the RNA cap epitranscriptome reveals novel caps in cellular and viral RNA. Nucleic Acids Res 47, e130–e130 (2019).
Hyde, J. L. & Diamond, M. S. Innate immune restriction and antagonism of viral RNA lacking 2׳-O methylation. Virology 479, 66–74 (2015).
Ramanathan, A., Robb, G. B. & Chan, S.-H. mRNA capping: biological functions and applications. Nucleic Acids Res 44, 7511–7526 (2016).
Pardi, N., Hogan, M. J. & Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 65, 14–20 (2020).
Stepinski, J., Waddell, C., Stolarski, R., Darzynkiewicz, E. & Rhoads, R. E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl (3′-O-methyl) GpppG and 7-methyl (3′-deoxy) GpppG. RNA 7, 1486–1495 (2001).
Jemielity, J. et al. Novel “anti-reverse” cap analogs with superior translational properties. RNA 9, 1108–1122 (2003).
Kuhn, A. N. et al. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 17, 961–971 (2010).
Grudzien-Nogalska, E. et al. Synthesis of anti-reverse cap analogs (ARCAs) and their applications in mRNA translation and stability. Methods Enzymol. 431, 203–227 (2007).
Henderson, J. M. et al. Cap 1 messenger RNA synthesis with co-transcriptional CleanCap® analog by in vitro transcription. Curr. Protoc. 1, e39 (2021).
TriLink. Cap Analogs, https://www.trilinkbiotech.com/products-services/nucleoside-triphosphates-nucleotides/cap-analogs.html (2021).
TriLink. CleanCap®AG, https://www.trilinkbiotech.com/media/folio3/productattachments/product_insert/n7113_insert_v3.pdf (2020).
TriLink. CleanCap®AU, https://www.trilinkbiotech.com/media/folio3/productattachments/product_insert/cleancapau_n7114_productinsert.pdf (2020).
Sahin, U., Muik, A., Derhovanessian, E., Vogler, I. & Türeci, Z. Publisher Correction: COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature, 590, E17 (2021).
Zarghampoor, F., Azarpira, N., Khatami, S. R., Behzad-Behbahani, A. & Foroughmand, A. M. Improved translation efficiency of therapeutic mRNA. Gene 707, 231–238 (2019).
Balzer, Le,S., Onsager, I., Lorentzen, J. A. & Lale, R. Dual UTR-A novel 5′ untranslated region design for synthetic biology applications. Synth. Biol. 5, ysaa006 (2020).
Leppek, K., Das, R. & Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 19, 158–174 (2018).
Nance, K. D. & Meier, J. L. Modifications in an emergency: the role of N1-Methylpseudouridine in COVID-19 vaccines. ACS Cent. Sci. 7, 748–756 (2021).
Mayr, C. Regulation by 3′-untranslated regions. Annu. Rev. Genet. 51, 171–194 (2017).
Matoulkova, E., Michalova, E., Vojtesek, B. & Hrstka, R. The role of the 3’untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 9, 563–576 (2012).
Mayr, C. What are 3′ UTRs doing?. Cold Spring Harb. Perspect. Biol. 11, a034728 (2018).
Fotin-Mleczek, M. et al. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 34, 1–15 (2011).
Eberhardt, W., Doller, A., Akool, E.-S. & Pfeilschifter, J. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 114, 56–73 (2007).
Wadhwa, A., Aljabbari, A., Lokras, A., Foged, C. & Thakur, A. Opportunities and challenges in the delivery of mRNA-based vaccines. Pharmaceutics 12, 102 (2020).
Zhang, B. Vaccines and compositions based on S antigen protein of SARS-CoV-2. CN112480217A (2021).
Karikó, K., Muramatsu, H., Keller, J. M. & Weissman, D. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol. Ther. 20, 948–953 (2012).
Sepac, A. et al. Comparison of cardiomyogenic potential among human ESC and iPSC lines. Cell Transpl. 21, 2523–2530 (2012).
Carralot, J. P. et al. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell. Mol. Life Sci. 61, 2418–2424 (2004).
Weng, Y. et al. The challenge and prospect of mRNA therapeutics lan-dscape. Biotechnol. Adv. 40, 107534 (2020).
Goss, D. J. & Kleiman, F. E. Poly (A) binding proteins: are they all created equal? Wiley Interdiscip. Rev.: RNA 4, 167–179 (2013).
Wang, Z. & Kiledjian, M. The poly (A)-binding protein and an mRNA stability protein jointly regulate an endoribonuclease activity. Mol. Cell. Biol. 20, 6334–6341 (2000).
Weissman, D. mRNA transcript therapy. Expert Rev. Vaccines 14, 265–281 (2015).
Holtkamp, S. et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108, 4009–4017 (2006).
Tang, T. T. & Passmore, L. A. Recognition of poly (A) RNA through its intrinsic helical structure. Cold Spring Harb. Symp. Quant. Biol. 84, 21–30 (2019).
Yu, S. & Kim, V. N. A tale of non-canonical tails: gene regulation by post-transcriptional RNA tailing. Nat. Rev. Mol. Cell Biol. 21, 542–556 (2020).
Kormann, M. S. et al. Expression of therapeutic proteins after deliveryof chemically modified mRNA in mice. Nat. Biotechnol. 29, 154–157 (2011).
Xia, X. Detailed dissection and critical evaluation of the Pfizer/BioNTech and Moderna mRNA vaccines. Vaccines 9, 734 (2021).
Trepotec, Z., Geiger, J., Plank, C., Aneja, M. K. & Rudolph, C. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA 25, 507–518 (2019).
Fisher, A. J. & Beal, P. A. Structural basis for eukaryotic mRNA modification. Curr. Opin. Struct. Biol. 53, 59–68 (2018).
Kariko, K., Muramatsu, H., Ludwig, J. & Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res 39, e142–e142 (2011).
Vaidyanathan, S. et al. Uridine depletion and chemical modification increase Cas9 mRNA activity and reduce immunogenicity without HPLC purification. Mol. Ther. Nucleic Acids 12, 530–542 (2018).
Isaacs, A., Cox, R. & Rotem, Z. Foreign nucleic acids as the stimulus to make interferon. Lancet, 282, 113–116 (1963).
Karikó, K. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008).
Song, J. & Yi, C. Chemical Modifications to RNA: a New Layer of Gene Expression Regulation. ACS Chem. Biol. 12, 316–325 (2017).
Mauer, J., Luo, X., Blanjoie, A., Jiao, X. & Jaffrey, S. R. Reversible methylation of m(6)Am in the 5′ cap controls mRNA stability. Nature 541, 371–375 (2016).
Gilbert, W. V., Bell, T. A. & Schaening, C. Messenger RNA modifications: form, distribution, and function. Science 352, 1408–1412 (2016).
Roundtree, I. A., Evans, M. E., Pan, T. & He, C. Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017).
Desrosiers, R., Friderici, K. & Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl Acad. Sci. USA 71, 3971–3975 (1974).
Safra, M. et al. The m 1 A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 551, 251–255 (2017).
Carlile, T. M. et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146 (2014).
Dubin, D. T. & Taylor, R. H. The methylation state of poly A-containing-messenger RNA from cultured hamster cells. Nucleic Acids Res 2, 1653–1668 (1975).
Chu, J.-M. et al. Existence of internal N 7-methylguanosine modification in mRNA determined by differential enzyme treatment coupled with mass spectrometry analysis. ACS Chem. Biol. 13, 3243–3250 (2018).
Zhang, L.-S. et al. Transcriptome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA. Mol. Cell 74, 1304–1316. e1308 (2019).
Malbec, L. et al. Dynamic methylome of internal mRNA N 7-methylguanosine and its regulatory role in translation. Cell Res. 29, 927–941 (2019).
Yan, K. et al. Toll-like receptor 3 and RIG-I-like receptor activation induces innate antiviral responses in mouse ovarian granulosa cells. Mol. Cell. Endocrinol. 372, 73–85 (2013).
Mauger, D. M., Cabral, B. J., Presnyak, V., Su, S. V. & Mcfadyen, I. J. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl Acad. Sci. USA 116, 201908052 (2019).
Andries, O. et al. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Rel. 217, 337–344 (2015).
Anderson, B. R. et al. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res 38, 5884–5892 (2010).
Anderson, B. R. et al. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res 39, 9329–9338 (2011).
Svitkin, Y. V. et al. N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res 45, 6023–6036 (2017).
Sahin, U. et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH 1 T cell responses. Nature 586, 594–599 (2020).
Corbett, K. S. et al. Immune Correlates of Protection by mRNA-1273 Immunization against SARS-CoV-2 Infection in Nonhuman Primates. BioRxiv, https://doi.org/10.1101/2021.04.20.440647 (2021).
Nelson, C. S. et al. Human cytomegalovirus glycoprotein B nucleoside-modified mRNA vaccine elicits antibody responses with greater durability and breadth than MF59-adjuvanted gB protein immunization. J. Virol. 94, e00186–00120 (2020).
Espeseth, A. S. et al. Modified mRNA/lipid nanoparticle-based vaccines expressing respiratory syncytial virus F protein variants are immunogenic and protective in rodent models of RSV infection. npj Vaccines 5, 1–14 (2020).
Freyn, A. W. et al. A multi-targeting, nucleoside-modified mRNA influenza virus vaccine provides broad protection in mice. Mol. Ther. 28, 1569–1584 (2020).
Shaw, C. et al. Safety and immunogenicity of a mRNA-based chikungunya vaccine in a phase 1 dose-ranging trial. Int J. Infect. Dis. 79, 17 (2019).
Thess, A. et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 23, 1456–1464 (2015).
Pardi, N. et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med 215, 1571–1588 (2018).
Zhang, N. et al. A Thermostable mRNA Vaccine against COVID-19. Cell 182, 1271–1283 (2020).
Rauch, S., Lutz, J., Kowalczyk, A., Schlake, T. & Heidenreich, R. RNActive® technology: generation and testing of stable and immunogenic mRNA vaccines. Methods Mol. Biol. 1499, 89–107 (2017).
Edwards, D. K. et al. Adjuvant effects of a sequence-engineered mRNA vaccine: translational profiling demonstrates similar human and murine innate response. J. Transl. Med 15, 1–18 (2017).
Kallen, K.-J. et al. A novel, disruptive vaccination technology: self-adjuvanted RNActive® vaccines. Hum. Vaccine Immunother. 9, 2263–2276 (2013).
Kowalczyk, A. et al. Self-adjuvanted mRNA vaccines induce local innate immune responses that lead to a potent and boostable adaptive immunity. Vaccine 34, 3882–3893 (2016).
Armbruster, N., Jasny, E. & Petsch, B. Advances in RNA vaccines for preventive indications: a case study of a vaccine against rabies. Vaccines 7, 132 (2019).
Zhou, X., Berglund, P., Zhao, H., Liljeström, P. & Jondal, M. Generation of cytotoxic and humoral immune responses by nonreplicative recombinant Semliki Forest virus. Proc. Natl Acad. Sci. U.S.A. 92, 3009–3013 (1995).
Smerdou, C. & Liljeström, P. Non-viral amplification systems for gene transfer: vectors based on alphaviruses. Curr. Opin. Mol. Ther. 1, 244–251 (1999).
Schott, J. W., Morgan, M., Galla, M. & Schambach, A. Viral and synthetic RNA vector technologies and applications. Mol. Ther. 24, 1513–1527 (2016).
Abu Bakar, F. & Ng, L. F. Nonstructural proteins of alphavirus—potential targets for drug development. Viruses 10, 71 (2018).
Pepini, T. et al. Induction of an IFN-mediated antiviral response by a self-amplifying RNA vaccine: implications for vaccine design. J. Immunol. 198, 4012–4024 (2017).
Beal, J. et al. Model-driven engineering of gene expression from RNA replicons. ACS Synth. Biol. 4, 48–56 (2015).
Zhou, X. et al. Self-replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine 12, 1510–1514 (1994).
Fleeton, M. N. et al. Self-replicative RNA vaccines elicit protection against influenza A virus, respiratory syncytial virus, and a tickborne encephalitis virus. J. Infect. Dis. 183, 1395–1398 (2001).
Geall, A. J. et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl Acad. Sci. USA 109, 14604–14609 (2012).
McCullough, K. C. et al. Self-replicating replicon-RNA delivery to dendritic cells by chitosan-nanoparticles for translation in vitro and in vivo. Mol. Ther. Nucleic Acids 3, e173 (2014).
Englezou, P. C. et al. Self-amplifying replicon RNA delivery to dendritic cells by cationic lipids. Mol. Ther. Nucleic Acids 12, 118–134 (2018).
Aberle, J. H., Aberle, S. W., Kofler, R. M. & Mandl, C. W. Humoral and cellular immune response to RNA immunization with flavivirus replicons derived from tick-borne encephalitis virus. J. Virol. 79, 15107–15113 (2005).
Bloom, K., van den Berg, F. & Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 28, 117–129 (2021).
Frolov, I. et al. Selection of RNA replicons capable of persistent noncytopathic replication in mammalian cells. J. Virol. 73, 3854–3865 (1999).
Rose, N. F. et al. In vitro evolution of high-titer, virus-like vesicles containing a single structural protein. Proc. Natl Acad. Sci. U.S.A. 111, 16866–16871 (2014).
MacDonald, M. R., Machlin, E. S., Albin, O. R. & Levy, D. E. The zinc finger antiviral protein acts synergistically with an interferon-induced factor for maximal activity against alphaviruses. J. Virol. 81, 13509–13518 (2007).
Garmashova, N., Gorchakov, R., Frolova, E. & Frolov, I. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J. Virol. 80, 5686–5696 (2006).
Mayuri, Geders, T. W., Smith, J. L. & Kuhn, R. J. Role for conserved residues of Sindbis virus nonstructural protein 2 methyltransferase-like domain in regulation of minus-strand synthesis and development of cytopathic infection. J. Virol. 82, 7284–7297 (2008).
LaStarza, M. W., Lemm, J. A. & Rice, C. M. Genetic analysis of the nsP3 region of Sindbis virus: evidence for roles in minus-strand and subgenomic RNA synthesis. J. Virol. 68, 5781–5791 (1994).
Li, Y. et al. In vitro evolution of enhanced RNA replicons for immunotherapy. Sci. Rep. 9, 1–10 (2019).
McKay, P. F. et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 11, 1–7 (2020).
Pollock, K. M. et al. Safety and Immunogenicity of a Self-Amplifying RNA Vaccine Against COVID-19: COVAC1, a Phase I, Dose-Ranging Trial. EClinicalMedicine, https://doi.org/10.1016/j.eclinm.2021.101262 (2022).
de Alwis, R. et al. A single dose of self-transcribing and replicating RNA-based SARS-CoV-2 vaccine produces protective adaptive immunity in mice. Mol. Ther. 29, 1970–1983 (2021).
Maruggi, G., Zhang, C., Li, J., Ulmer, J. B. & Yu, D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol. Ther. 27, 757–772 (2019).
Jensen, S. & Thomsen, A. R. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 86, 2900–2910 (2012).
Blakney, A. K., McKay, P. F. & Shattock, R. J. Structural components for amplification of positive and negative strand VEEV splitzicons. Front. Mol. Biosci. 5, 71 (2018).
Fuller, D. H. & Berglund, P. Amplifying RNA vaccine development. N. Engl. J. Med 382, 2469–2471 (2020).
Zhang, X.-O. et al. Complementary sequence-mediated exon circularization. Cell 159, 134–147 (2014).
Kristensen, L. S. et al. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 20, 675–691 (2019).
Chen, L.-L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 17, 205–211 (2016).
Sanger, H. L., Klotz, G., Riesner, D., Gross, H. J. & Kleinschmidt, A. K. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl Acad. Sci. USA 73, 3852–3856 (1976).
Chen, Y. G. et al. N6-methyladenosine modification controls circular RNA immunity. Mol. Cell 76, 96–109. e109 (2019).
Wesselhoeft, R. A., Kowalski, P. S. & Anderson, D. G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. 9, 1–10 (2018).
Yang, Y. et al. Extensive translation of circular RNAs driven by N 6-methyladenosine. Cell Res 27, 626–641 (2017).
Wesselhoeft, R. A. et al. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol. Cell 74, 508–520. e504 (2019).
Chen, Y. G. et al. Sensing self and foreign circular RNAs by intron identity. Mol. Cell 67, 228–238. e225 (2017).
Qu, L. et al. Circular RNA Vaccines against SARS-CoV-2 and Emerging Variants. BioRxiv, https://doi.org/10.1101/2021.03.16.435594 (2022).
Yao, H. et al. Molecular architecture of the SARS-CoV-2 virus. Cell 183, 730–738. e713 (2020).
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020).
Hsieh, C.-L. et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369, 1501–1505 (2020).
Hoffmann, M., Kleine-Weber, H. & Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 78, 779–784. e775 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280. e278 (2020).
Wang, Q. et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904. e899 (2020).
Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020).
Singh, S. P., Pritam, M., Pandey, B. & Yadav, T. P. Microstructure, pathophysiology and potential therapeutics of COVID-19: A comprehensive review. J. Med. Virol. 93, 275–299 (2020).
Cai, Y. et al. Distinct conformational states of SARS-CoV-2 spike protein. Science 369, 1586–1592 (2020).
Yadav, T. et al. Recombinant vaccines for COVID-19. Hum. Vaccin Immunother. 16, 2905–2912 (2020).
Walls, A. C. et al. Structure, function and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181 (2020).
Hanson, G. & Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 19, 20–30 (2018).
Presnyak, V. et al. Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124 (2015).
Buhr, F. et al. Synonymous codons direct cotranslational folding toward different protein conformations. Mol. Cell 61, 341–351 (2016).
Yang, Y., Ma, X. & Huo, Y.-X. Application of codon optimization strategy in heterologous protein expression. Chin. J. Biotechnol. 35, 2227–2237 (2019).
Kudla, G., Murray, A. W., Tollervey, D. & Plotkin, J. B. Coding-sequence determinants of gene expression in Escherichia coli. Science 324, 255–258 (2009).
Zhong, C., Wei, P. & Zhang, Y. H. P. Enhancing functional expression of codon-optimized heterologous enzymes in Escherichia coli BL21(DE3) by selective introduction of synonymous rare codons. Biotechnol. Bioeng. 114, 1054–1064 (2016).
Zhang, H. et al. Lineardesign: Efficient algorithms for optimized mrna sequence design. arXiv preprint arXiv:2004.10177, https://arxiv.org/abs/2004.10177 (2021).
Keiichi, H., Tamotsu, N. & Satoshi, F. Codon usage is less optimized in eukaryotic gene segments encoding intrinsically disordered regions than in those encoding structural domains. Nucleic Acids Res 44, 10051–10061 (2016).
Courel, M. et al. GC content shapes mRNA storage and decay in human cells. Elife 8, e49708 (2019).
Kudla, G., Lipinski, L., Caffin, F., Helwak, A. & Zylicz, M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 4, e180 (2006).
Sharp, P. M. & Li, W. H. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucl. Acids Res 15, 1281–1295 (1987).
Wu, Q. et al. Translation affects mRNA stability in a codon-dependent manner in human cells. elife 8, e45396 (2019).
Gustafsson, C., Govindarajan, S. & Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 22, 346–353 (2004).
Chin, J. X., Chung, B. K.-S. & Lee, D.-Y. Codon Optimization OnLine (COOL): a web-based multi-objective optimization platform for synthetic gene design. Bioinformatics 30, 2210–2212 (2014).
Fu, H. et al. Codon optimization with deep learning to enhance protein expression. Sci. Rep. 10, 1–9 (2020).
Puigbo, P., Guzman, E., Romeu, A. & Garcia-Vallve, S. OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res 35, W126–W131 (2007).
Fath, S. et al. Multiparameter RNA and codon optimization: a standardized tool to assess and enhance autologous mammalian gene expression. PLoS One 6, e17596 (2011).
Raab, D., Graf, M., Notka, F., Schödl, T. & Wagner, R. The GeneOptimizer Algorithm: using a sliding window approach to cope with the vast sequence space in multiparameter DNA sequence optimization. Syst. Synth. Biol. 4, 215–225 (2010).
Graf, M., Deml, L. & Wagner, R. Codon-optimized genes that enable increased heterologous expression in mammalian cells and elicit efficient immune responses in mice after vaccination of naked DNA. Methods Mol. Med 94, 197–210 (2004).
Villalobos, A., Ness, J. E., Gustafsson, C., Minshull, J. & Govindarajan, S. Gene Designer: a synthetic biology tool for constructing artificial DNA segments. BMC Bioinf. 7, 1–8 (2006).
Wu, G. et al. SGDB: a database of synthetic genes re-designed for optimizing protein over-expression. Nucleic Acids Res. 35, D76–D79 (2007).
Puigbo, P., Romeu, A. & Garcia-Vallve, S. HEG-DB: a database of predicted highly expressed genes in prokaryotic complete genomes under translational selection. Nucleic Acids Res 36, D524–D527 (2007).
Daniel, E. et al. ATGme: Open-source web application for rare codon identification and custom DNA sequence optimization. BMC Bioinf. 16, 1–6 (2015).
Grote, A. et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–W531 (2005).
Puigbò, P., Bravo, I. G. & Garcia-Vallve, S. CAIcal: a combined set of tools to assess codon usage adaptation. Biol. Direct 3, 1–8 (2008).
VectorBuilder Codon Optimization, https://www.vectorbuilder.cn/tool/codon-optimization.html (2022).
GENEWIZ Codon Optimization, https://climsprod.genewiz.com.cn/Toolbox/CodonOptimization (2022).
GenScript Codon Optimization, https://www.genscript.com/tools/gensmart-codon-optimization (2022).
Şen, A., Kargar, K., Akgün, E. & Pınar, M. Ç. Codon optimization: a mathematical programing approach. Bioinformatics 36, 4012–4020 (2020).
Pallesen, J., Wang, N., Corbett, K. S., Wrapp, D. & Mclellan, J. S. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl Acad. Sci. USA 114, E7348–E7357 (2017).
Tortorici et al. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531, 114–117 (2016).
Kirchdoerfer, R. N. et al. Pre-fusion structure of a human coronavirus spike protein. Nature 531, 118–121 (2016).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Corbett, K. S., Flynn, B., Foulds, K. E., Francica, J. R. & Graham, B. S. Evaluation of the mRNA-1273 vaccine against SARS-COV-2 in nonhuman primates. N. Engl. J. Med. 383, 1544–1555 (2020).
Tian, J. H., Patel, N., Haupt, R., Zhou, H. & Smith, G. SARS-CoV-2 spike glycoprotein vaccine candidate NVX-CoV2373 immunogenicity in baboons and protection in mice. Nat. Commun. 12, 372 (2021).
Keech, C. et al. Phase 1–2 trial of a SARS-CoV-2 recombinant spike protein nanoparticle vaccine. N. Engl. J. Med. 383, 2320–2332 (2020).
Bangaru, S. et al. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science 370, 1089–1094 (2020).
Li, H. Vaccine agent for treating or preventing coronavirus diseases. CN113186203A (2021).
Walsh, E. E. et al. RNA-based COVID-19 vaccine BNT162b2 selected for a pivotal efficacy study. Medrxiv, https://doi.org/10.1101/2020.08.17.20176651 (2020).
Walsh, E. E. et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med 383, 2439–2450 (2020).
Bangham, A. D., Standish, M. M. & Watkins, J. C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 13, 238–IN227 (1965).
Working, P. & Dayan, A. Pharmacological-toxicological expert report CAELYXTM:(stealth® liposomal doxorubicin HCl). Hum. Exp. Toxicol. 15, 751–785 (1996).
Müller, R. H., Mäder, K. & Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery–a review of the state of the art. Eur. J. Pharm. Biopharm. 50, 161–177 (2000).
Müller, R. H., Radtke, M. & Wissing, S. A. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv. Drug Deliv. Rev. 54, S131–S155 (2002).
Tenchov, R., Bird, R., Curtze, A. E. & Zhou, Q. Lipid nanoparticles—from liposomes to mRNA vaccine delivery, a landscape of research diversity and advancement. ACS Nano 15, 16982–17015 (2021).
Cui, S. et al. Correlation of the cytotoxic effects of cationic lipids with their headgroups. Toxicol. Res. 7, 473–479 (2018).
Cullis, P. R. & Hope, M. J. Lipid nanoparticle systems for enabling gene therapies. Mol. Ther. 25, 1467–1475 (2017).
Jayaraman, M. et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int Ed. Engl. 124, 8657–8661 (2012).
Chen, S. et al. Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J. Control. Rel. 235, 236–244 (2016).
Bahl, K. et al. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol. Ther. 25, 1316–1327 (2017).
Buschmann, M. D. et al. Nanomaterial delivery systems for mRNA vaccines. Vaccines 9, 65 (2021).
Sabnis, S. et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 26, 1509–1519 (2018).
Hassett, K. J. et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids 15, 1–11 (2019).
Hope, M. J. Lipid nanoparticle formulations. National Center for Biotechnology Information. PubChem PatentSummary for WO-2018081480-A1, Lipid nanoparticle formulations. https://pubchem.ncbi.nlm.nih.gov/patent/WO-2018081480-A1. Accessed Mar. 16, 2022 (2018).
Cheng, X. & Lee, R. J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 99, 129–137 (2016).
Cheng, Q. et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 15, 313–320 (2020).
Lokugamage, M. P., Sago, C. D., Gan, Z., Krupczak, B. R. & Dahlman, J. E. Constrained nanoparticles deliver siRNA and sgRNA to T cells in vivo without targeting ligands. Adv. Mater. 31, 1902251 (2019).
Kauffman, K. J. et al. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 15, 7300–7306 (2015).
Ball, R. L., Hajj, K. A., Vizelman, J., Bajaj, P. & Whitehead, K. A. Lipid nanoparticle formulations for enhanced co-delivery of siRNA and mRNA. Nano Lett. 18, 3814–3822 (2018).
mRNA-1273-P301-Protocol, https://www.modernatx.com/sites/default/files/mRNA-1273-P301-Protocol.pdf (2020).
Corbett, K. S. et al. SARS-CoV-2 mRNA Vaccine Development Enabled by Prototype Pathogen Preparedness. BioRxiv, https://doi.org/10.1101/2020.06.11.145920 (2020).
Maier, M. A. et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 21, 1570–1578 (2013).
Patel, S. et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 11, 1–13 (2020).
Yang, S.-T., Kreutzberger, A. J., Lee, J., Kiessling, V. & Tamm, L. K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 199, 136–143 (2016).
Paunovska, K. et al. Nanoparticles containing oxidized cholesterol deliver mRNA to the liver microenvironment at clinically relevant doses. Adv. Mater. 31, 1807748 (2019).
Hashizaki, K. et al. Effects of poly (ethylene glycol)(PEG) concentration on the permeability of PEG-grafted liposomes. Chem. Pharm. Bull. 53, 27–31 (2005).
Hashizaki, K. et al. Effects of poly (ethylene glycol)(PEG) chain length of PEG-lipid on the permeability of liposomal bilayer membranes. Chem. Pharm. Bull. 51, 815–820 (2003).
Kanasty, R., Dorkin, J. R., Vegas, A. & Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 12, 967–977 (2013).
Zhu, X. et al. Surface De-PEGylation controls nanoparticle-mediated siRNA delivery in vitro and in vivo. Theranostics 7, 1990–2002 (2017).
Schoenmaker, L. et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int. J. Pharm. 601, 120586 (2021).
Zeng, C., Zhang, C., Walker, P. G. & Dong, Y. Formulation and delivery technologies for mRNA vaccines. Curr. Top. Microbiol. Immunol., https://doi.org/10.1007/82_2020_217 (2020).
Li, W. et al. West Nile virus infectious replicon particles generated using a packaging-restricted cell line is a safe reporter system. Sci. Rep. 7, 1–10 (2017).
Vogel, A. B. et al. Self-amplifying RNA vaccines give equivalent protection against influenza to mRNA vaccines but at much lower doses. Mol. Ther. 26, 446–455 (2018).
Moyo, N. et al. Efficient induction of T cells against conserved HIV-1 regions by mosaic vaccines delivered as self-amplifying mRNA. Mol. Ther. Methods Clin. Dev. 12, 32–46 (2019).
Akinc, A. et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 14, 1084–1087 (2019).
Leroux, J. C. Drug delivery: too much complexity, not enough reproducibility? Angew. Chem. Int Ed. Engl. 56, 15170–15171 (2017).
Park, T. G., Jeong, J. H. & Kim, S. W. Current status of polymeric gene delivery systems. Adv. Drug Deliv. Rev. 58, 467–486 (2006).
Wahane, A. et al. Role of lipid-based and polymer-based non-viral vectors in nucleic acid delivery for next-generation gene therapy. Molecules 25, 2866 (2020).
Miyazaki, T. et al. Polymeric nanocarriers with controlled chain flexibility boost mRNA delivery in vivo through enhanced structural fastening. Adv. Healthc. Mater. 9, 2000538 (2020).
Kowalski, P. S., Rudra, A., Miao, L. & Anderson, D. G. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther. 27, 710–728 (2019).
Jiang, Y. et al. Quantitating endosomal escape of a library of polymers for mRNA delivery. Nano Lett. 20, 1117–1123 (2020).
Jiang, Y. et al. A “top-down” approach to actuate poly (amine-co-ester) terpolymers for potent and safe mRNA delivery. Biomaterials 176, 122–130 (2018).
Coolen, A.-L. et al. Poly (lactic acid) nanoparticles and cell-penetrating peptide potentiate mRNA-based vaccine expression in dendritic cells triggering their activation. Biomaterials 195, 23–37 (2019).
Su, X., Fricke, J., Kavanagh, D. G. & Irvine, D. J. In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Mol. Pharm. 8, 774–787 (2011).
Blakney, A. K. et al. Big is beautiful: enhanced saRNA delivery and immunogenicity by a higher molecular weight, bioreducible, cationic polymer. ACS Nano 14, 5711–5727 (2020).
Lipids and lipid nanoparticle formulations for delivery of nucleic acids. US10221127B2.
Meng, C., Chen, Z., Li, G., Welte, T. & Shen, H. Nanoplatforms for mRNA Therapeutics. Adv. Ther. 4, 2000099 (2021).
Persano, S. et al. Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 125, 81–89 (2017).
Rezaee, M., Oskuee, R. K., Nassirli, H. & Malaekeh-Nikouei, B. Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J. Control. Rel. 236, 1–14 (2016).
Guevara, M. L., Jilesen, Z., Stojdl, D. & Persano, S. Codelivery of mRNA with α-Galactosylceramide using a new lipopolyplex formulation induces a strong antitumor response upon intravenous administration. ACS Omega 4, 13015–13026 (2019).
Kaczmarek, J. C. et al. Polymer–lipid nanoparticles for systemic delivery of mRNA to the lungs. Angew. Chem. Int Ed. Engl. 128, 14012–14016 (2016).
Yang, R. et al. A core-shell structured COVID-19 mRNA vaccine with favorable biodistribution pattern and promising immunity. Signal Transduct. Target. Ther. 6, 1–10 (2021).
Li, W., Nicol, F. & Szoka, F. C. Jr GALA: a designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv. Drug Deliv. Rev. 56, 967–985 (2004).
McCarthy, H. O. et al. Development and characterization of self-assembling nanoparticles using a bio-inspired amphipathic peptide for gene delivery. J. Controlled Release 189, 141–149 (2014).
Udhayakumar, V. K. et al. Arginine-rich peptide-based mRNA nanocomplexes efficiently instigate cytotoxic T cell immunity dependent on the amphipathic organization of the peptide. Adv. Healthc. Mater. 6, 1601412 (2017).
van den Brand, D. et al. Peptide-mediated delivery of therapeutic mRNA in ovarian cancer. Eur. J. Pharm. Biopharm. 141, 180–190 (2019).
Papachristofilou, A. et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J. ImmunoTher. Cancer 7, 1–14 (2019).
Brito, L. A. et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 22, 2118–2129 (2014).
Tsai, T. F. Fluad®-MF59®-adjuvanted influenza vaccine in older adults. Infect. Chemother. 45, 159–174 (2013).
Erasmus, J. H. et al. An Alphavirus-derived replicon RNA vaccine induces SARS-CoV-2 neutralizing antibody and T cell responses in mice and nonhuman primates. Sci. Transl. Med. 12, eabc9396 (2020).
Van Tendeloo, V. F. et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl Acad. Sci. USA 107, 13824–13829 (2010).
Anguille, S. et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 130, 1713–1721 (2017).
Khoury, H. J. et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 123, 3061–3072 (2017).
Sebastian, M. et al. Messenger RNA vaccination and B-cell responses in NSCLC patients. J. Clin. Oncol. 30, 2573–2573 (2012).
Sebastian, M. et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 68, 799–812 (2019).
Kranz, L. M. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016).
Sahin, U. et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 585, 107–112 (2020).
Wilgenhof, S., Corthals, J., Heirman, C., Neyns, B. & Thielemans, K. Clinical trials with MRNA electroporated dendritic cells for stage III/IV melanoma patients. J. ImmunoTher. Cancer 3, 1–1 (2015).
Sofie et al. Phase II study of autologous monocyte-derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in patients with pretreated advanced melanoma. J. Clin. Oncol. 34, 1330–1338 (2016).
Keersmaecker, B. D. et al. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: link between T-cell activation and clinical responses in advanced melanoma. J. ImmunoTher. Cancer 8, e000329 (2020).
BioNTech Receives FDA Fast Track Designation for its FixVac Candidate BNT111 in Advanced Melanoma, https://investors.biontech.de/news-releases/news-release-details/biontech-receives-fda-fast-track-designation-its-fixvac (2021).
Hou, X., Zaks, T., Langer, R. & Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 66, 1078–1094 (2021).
Tay, B. Q. et al. Evolution of cancer vaccines—challenges, achievements, and future directions. Vaccines 9, 535 (2021).
Jin, M.-Z. & Wang, X.-P. Immunogenic cell death-based cancer vaccines. Front. Immunol. 12, 697964 (2021).
Tang, X. et al. Therapeutic prospects of mRNA-based gene therapy for glioblastoma. Front. Oncol. 9, 1208 (2019).
Jarzebska, N. T., Mellett, M., Frei, J., Kuendig, T. M. & Pascolo, S. Protamine for RNA transfection: from heparin antagonist to RNA delivery. Pharmaceutics 13, 877 (2021).
Moderna Announces Clinical Updates on Personalized Cancer Vaccine Program, https://investors.modernatx.com/news-releases/news-release-details/moderna-announces-clinical-updates-personalized-cancer-vaccine (2020).
Burris, H. A. et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J. Clin. Oncol. 37, 2523 (2019).
Bauman, J. et al. 798 Safety, tolerability, and immunogenicity of mRNA-4157 in combination with pembrolizumab in subjects with unresectable solid tumors (KEYNOTE-603): an update. J. Immunother. Cancer 8, A846–A846 (2020).
Burris, H. A. III et al. A phase 1, open-label, multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in subjects with resected solid tumors and in combination with pembrolizumab in subjects with unresectable solid tumors (Keynote-603). J. Clin. Oncol. 37, 2523–2523 (2019).
Making Strides Toward CMV Prevention, https://www.modernatx.com/newsroom/our-blog-coding-region/making-strides-toward-cmv-prevention (2021).
Feldman, R. A. et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37, 3326–3334 (2019).
A Milestone Week at Moderna, https://www.modernatx.com/moderna-blog/welcome-to-the-moderna-blog (2017).
Moderna Data at IDWeek 2019, https://www.modernatx.com/moderna-blog/sharing-science-idweek-100719 (2019).
Jagger, B. W. et al. Protective efficacy of nucleic acid vaccines against transmission of zika virus during pregnancy in mice. J. Infect. Dis. 220, 1577–1588 (2019).
Moderna Receives FDA Fast Track Designation for Zika Vaccine mRNA-1893, https://www.businesswire.com/news/home/20190819005557/en/Moderna-Receives-FDA-Fast-Track-Designation-Zika/ (2019).
Aliprantis, A. O. et al. A phase 1, randomized, placebo-controlled study to evaluate the safety and immunogenicity of an mRNA-based RSV prefusion F protein vaccine in healthy younger and older adults. Hum. Vaccine Immunother. 17, 1248–1261 (2021).
Moderna Receives FDA Fast Track Designation for Respiratory Syncytial Virus (RSV) Vaccine (mRNA-1345), https://investors.modernatx.com/news-releases/news-release-details/moderna-receives-fda-fast-track-designation-respiratory (2021).
Moderna Announces Positive Interim Phase 1 data for first combination vaccine against the respiratory viruses hMPV and PIV3, https://www.businesswire.com/news/home/20190212005810/en/Moderna-Announces-Positive-Interim-Phase-1-Data (2019).
A seasonal flu investigational vaccine clinical trial for healthy adults, https://trials.modernatx.com/study/?id=mRNA-1010-P101 (2021).
Abbasi, J. Moderna’s mRNA vaccine for seasonal flu enters clinical trials. JAMA 326, 1365–1365 (2021).
Sanofi and Translate Bio Initiate Phase 1 Clinical Trial of mRNA Influenza Vaccine, https://translatebio.gcs-web.com/news-releases/news-release-details/sanofi-and-translate-bio-initiate-phase-1-clinical-trial-mrna (2021).
WHO. Tracking SARS-CoV-2 variants, https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (2022).
Graham, F. Daily briefing: Omicron coronavirus variant puts scientists on alert. Nature, https://doi.org/10.1038/d41586-021-03564-6 (2021).
Karim, S. S. A. & Karim, Q. A. Omicron SARS-CoV-2 variant: a new chapter in the COVID-19 pandemic. Lancet 398, 2126–2128 (2021).
WHO. B.1.1.529, https://cov-lineages.org/global_report_B.1.1.529.html (2022).
FDA. Coronavirus (COVID-19) | CBER-Regulated Biologics, https://www.fda.gov/vaccines-blood-biologics/industry-biologics/coronavirus-covid-19-cber-regulated-biologics (2021).
Mulligan, M. J. et al. Phase 1/2 study to describe the safety and immunogenicity of a COVID-19 RNA Vaccine Candidate (BNT162b1) in adults 18 to 55 years of age: interim report. medRxiv, https://doi.org/10.1101/2020.06.30.20142570 (2020).
Li, J. et al. Safety and immunogenicity of the SARS-CoV-2 BNT162b1 mRNA vaccine in younger and older Chinese adults: a randomized, placebo-controlled, double-blind phase 1 study. Nat. Med. 27, 1062–1070 (2021).
Mulligan, M. J. et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586, 589–593 (2020).
Lamb, Y. N. BNT162b2 mRNA COVID-19 Vaccine: First Approval. Drugs 81, 495-501, https://doi.org/10.1007/s40265-021-01480-7
FDA takes key action in fight against COVID-19 by issuing emergency use authorization for first COVID-19 Vaccine, https://www.fda.gov/news-events/press-announcements/fda-takes-key-action-fight-against-covid-19-issuing-emergency-use-authorization-first-covid-19 (2020).
Pfizer and BioNTech receive authorization in the European Union for COVID-19 vaccine, https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-receive-authorization-european-union (2020).
Cavaleri, M., Enzmann, H., Straus, S. & Cooke, E. The European Medicines Agency’s EU conditional marketing authorisations for COVID-19 vaccines. Lancet. (2021).
World Health Organisation. WHO issues its first emergency use validation for a COVID-19 vaccine and emphasizes need for equitable global access, https://www.who.int/news/item/31-12-2020-who-issues-its-first-emergency-use-validation-for-a-covid-19-vaccine-and-emphasizes-need-for-equitable-global-access (2020).
Coronavirus (COVID-19) Update: FDA Authorizes Pfizer-BioNTech COVID-19 Vaccine for Emergency Use in Adolescents in Another Important Action in Fight Against Pandemic, https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-pfizer-biontech-covid-19-vaccine-emergency-use (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Thomas, S. J. et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months. N. Engl. J. Med., NEJMoa2110345, https://doi.org/10.1056/NEJMoa2110345 (2021).
Tober-Lau, P. et al. Long-term immunogenicity of BNT162b2 vaccination in the elderly and in younger health care workers. medRxiv, https://doi.org/10.1101/2021.08.26.21262468 (2021).
Chemaitelly, H. et al. Waning of BNT162b2 vaccine protection against SARS-CoV-2 infection in Qatar. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2114114 (2021).
Levin, E. G. et al. Waning Immune humoral response to BNT162b2 Covid-19 vaccine over 6 months. N. Engl. J. Med. 385, e84 (2021).
Moustsen-Helms, I. R. et al. Vaccine effectiveness after 1st and 2nd dose of the BNT162b2 mRNA Covid-19 Vaccine in long-term care facility residents and healthcare workers–a Danish cohort study. MedRxiv, https://doi.org/10.1101/2021.03.08.21252200 (2021).
Levine-Tiefenbrun, M. et al. Viral loads of Delta-variant SARS-CoV2 breakthrough infections following vaccination and booster with the BNT162b2 vaccine. medRxiv, https://doi.org/10.1101/2021.08.29.21262798 (2021).
Pfizer. Pfizer and BioNTech announce Phase 3 trial data showing high efficacy of a booster dose of their COVID-19 vaccine, https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-announce-phase-3-trial-data-showing (2021).
Bar-On, Y. M. et al. Protection of BNT162b2 vaccine booster against Covid-19 in Israel. N. Engl. J. Med. 385, 1393–1400 (2021).
Romero-Ibarguengoitia, M. E. et al. Effect of the third dose of BNT162b2 vaccine in quantitative SARS-CoV-2 spike 1-2 IgG antibody titers in healthcare workers. medRxiv, https://doi.org/10.1101/2021.10.20.21265269 (2021).
FDA. FDA Authorizes Booster Dose of Pfizer-BioNTech COVID-19 Vaccine for Certain Populations, https://www.fda.gov/news-events/press-announcements/fda-authorizes-booster-dose-pfizer-biontech-covid-19-vaccine-certain-populations (2021).
FDA. Coronavirus (COVID-19) Update: FDA Expands Eligibility for COVID-19 Vaccine Boosters, https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-expands-eligibility-covid-19-vaccine-boosters (2021).
Dagan, N. et al. BNT162b2 mRNA Covid-19 vaccine in a nationwide mass vaccination setting. N. Engl. J. Med. 384, 1412–1423 (2021).
Barda, N. et al. Safety of the BNT162b2 mRNA Covid-19 vaccine in a nationwide setting. N. Engl. J. Med. 385, 1078–1090 (2021).
Mevorach, D. et al. Myocarditis after BNT162b2 mRNA vaccine against Covid-19 in Israel. N. Engl. J. Med. 385, 2140–2149 (2021).
Witberg, G. et al. Myocarditis after Covid-19 vaccination in a large health care organization. N. Engl. J. Med. 385, 2132–2139 (2021).
Hashimoto, T. et al. High anaphylaxis rates following vaccination with the Pfizer BNT162b2 mRNA vaccine against COVID-19 in Japanese healthcare workers: a secondary analysis of initial post-approval safety data. J. Travel Med. 28, taab090 (2021).
Bruusgaard-Mouritsen, M. A., Johansen, J. D. & Garvey, L. H. Clinical manifestations and impact on daily life of allergy to polyethylene glycol (PEG) in ten patients. Clin. Exp. Allergy 51, 463–470 (2021).
Lopez Bernal, J. et al. Effectiveness of Covid-19 vaccines against the B.1.617.2 (Delta) variant. N. Engl. J. Med. 385, 585–594 (2021).
Abu-Raddad, L. J. et al. Pfizer-BioNTech mRNA BNT162b2 Covid-19 vaccine protection against variants of concern after one versus two doses. J. Travel Med. 28, taab083 (2021).
Chemaitelly, H. & Abu-Raddad, L. J. Effectiveness of the BNT162b2 Covid-19 Vaccine against the B.1.1.7 and B.1.351 Variants. N. Engl. J. Med. 385, 187–189 (2021).
Muik, A. et al. Neutralization of SARS-CoV-2 Omicron by BNT162b2 mRNA vaccine-elicited human sera. Science, 375, 678–680 (2022).
Liu, L. et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature, https://doi.org/10.1038/s41586-021-04388-0 (2021).
Planas, D. et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature, https://doi.org/10.1038/s41586-021-04389-z (2021).
FDA. Coronavirus (COVID-19) update: FDA takes key action by approving second COVID-19 vaccine, https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-takes-key-action-approving-second-covid-19-vaccine (2022).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Moderna announces TeenCOVE study of its COVID-19 vaccine in adolescents meets primary endpoint and plans to submit data to regulators in early June, https://investors.modernatx.com/news-releases/news-release-details/moderna-announces-teencove-study-its-covid-19-vaccine (2021).
Ali, K. et al. Evaluation of mRNA-1273 SARS-CoV-2 vaccine in adolescents. N. Engl. J. Med. 385, 2241–2251 (2021).
Teo, S. P. Review of COVID-19 mRNA Vaccines: BNT162b2 and mRNA-1273. J. Pharm. Pract., 08971900211009650, https://doi.org/10.1177/08971900211009650 (2021).
Chu, L. et al. Immune memory response after a booster injection of mRNA-1273 for severe acute respiratory Syndrome Coronavirus-2 (SARS-CoV-2). medRxiv, https://doi.org/10.1101/2021.09.29.21264089 (2021).
Choi, A. et al. Safety and immunogenicity of SARS-CoV-2 variant mRNA vaccine boosters in healthy adults: an interim analysis. Nat. Med., https://doi.org/10.1038/s41591-021-01527-y (2021).
Wu, K. et al. Preliminary analysis of safety and immunogenicity of a SARS-CoV-2 variant vaccine booster. Medrxiv, https://doi.org/10.1101/2021.05.05.21256716 (2021).
Chemaitelly, H. et al. mRNA-1273 COVID-19 vaccine effectiveness against the B.1.1.7 and B.1.351 variants and severe COVID-19 disease in Qatar. Nat. Med. 27, 1614–1621 (2021).
Günl, F. et al. Shooting at a moving target—effectiveness and emerging challenges for SARS-CoV-2 vaccine development. Vaccines 9, 1052 (2021).
Chin, E. T. et al. Effectiveness of the mRNA-1273 vaccine during a SARS-CoV-2 Delta outbreak in a prison. N. Engl. J. Med., https://doi.org/10.1056/NEJMc2114089 (2021).
Paris, C. et al. Effectiveness of mRNA-BNT162b2, mRNA-1273, and ChAdOx1 nCoV-19 vaccines against COVID-19 in healthcare workers: an observational study using surveillance data. Clin. Microbiol. Infect. (2021).
Tseng, H. F. et al. Effectiveness of mRNA-1273 against SARS-CoV-2 omicron and delta variants. medRxiv, 2022.2001.2007.22268919, https://doi.org/10.1101/2022.01.07.22268919 (2022).
Cohen, K., Islam, P. N., Jarvis, M. & Sheils, N. E. Comparative efficacy over time of the mRNA-1273 (Moderna) vaccine and the BNT162b2 (Pfizer-BioNTech) vaccine. https://doi.org/10.21203/rs.3.rs-1071804/v1 (2021).
Self, W. H. et al. Comparative effectiveness of Moderna, Pfizer-BioNTech, and Janssen (Johnson & Johnson) vaccines in preventing COVID-19 hospitalizations among adults without immunocompromising conditions—United States, March–August 2021. MMWR Morb. Mortal. Wkly Rep. 70, 1337 (2021).
Wang, L., Davis, P. B., Kaelber, D. C., Volkow, N. D. & Xu, R. Comparison of mRNA-1273 and BNT162b2 vaccines on breakthrough SARS-CoV-2 infections, hospitalizations, and death during the delta-predominant period. JAMA, https://doi.org/10.1001/jama.2022.0210 (2022).
Kremsner, P. G. et al. Efficacy and safety of the CVnCoV SARS-CoV-2 mRNA vaccine candidate in ten countries in Europe and Latin America (HERALD): a randomised, observer-blinded, placebo-controlled, phase 2b/3 trial. Lancet Infect. Dis., https://doi.org/10.1016/s1473-3099(21)00677-0 (2021).
Rauch, S. et al. mRNA-based SARS-CoV-2 vaccine candidate CVnCoV induces high levels of virus-neutralising antibodies and mediates protection in rodents. npj Vaccines 6, 57 (2021).
Gebre, M. et al. Optimization of non-coding regions improves protective efficacy of an mRNA SARS-CoV-2 vaccine in nonhuman primates. BioRxiv, https://doi.org/10.1101/2021.08.13.456316 (2021).
Cohen, J. What went wrong with CureVac’s mRNA vaccine? Science 372, 1381 (2021).
EMA ends rolling review of CVnCoV COVID-19 vaccine following withdrawal by CureVac AG | European Medicines Agency, https://www.ema.europa.eu/en/news/ema-ends-rolling-review-cvncov-covid-19-vaccine-following-withdrawal-curevac-ag (2021).
CureVac to Shift Focus of COVID-19 Vaccine Development to Second-Generation mRNA Technology–CureVac, https://www.curevac.com/en/2021/10/12/curevac-to-shift-focus-of-covid-19-vaccine-development-to-second-generation-mrna-technology/ (2021).
Abe, K. T. et al. Neutralizing antibody responses to SARS-CoV-2 variants in vaccinated Ontario long-term care home residents and workers. medRxiv, https://doi.org/10.1101/2021.08.06.21261721 (2021).
Providence Therapeutics Reports PTX-COVID19-B, its mRNA Vaccine for COVID-19, Neutralizes SARS-CoV-2 and Variants of Concern, Including Delta, https://providencetherapeutics.com/article-details/providence-therapeutics-reports-ptx-covid19-b-its-mrna-vaccine-for-covid-19-neutralizes-sars-cov-2-and-variants-of-concern-including-delta.html (2021).
Graham, B. S. Rapid COVID-19 vaccine development. Science 368, 945–946 (2020).
Kalnin, K. V. et al. Immunogenicity and efficacy of mRNA COVID-19 vaccine MRT5500 in preclinical animal models. npj Vaccines 6, 61 (2021).
Kalnin, K. V. et al. Immunogenicity of novel mRNA COVID-19 vaccine MRT5500 in mice and non-human primates. BioRxiv, https://doi.org/10.1101/2020.10.14.337535 (2020).
Sanofi to focus its COVID-19 development efforts on the recombinant vaccine candidate, https://www.sanofi.com/en/media-room/press-releases/2021/2021-09-28-18-44-47-2304800 (2021).
Sanofi announces positive Phase 1/2 study interim results for its first mRNA-based vaccine candidate, https://www.sanofi.com/en/media-room/press-releases/2021/2021-09-28-08-00-00-2304069 (2021).
A Phase III Clinical Study of a SARS-CoV-2 Messenger Ribonucleic Acid (mRNA) vaccine candidate against COVID-19 in population aged 18 years and above, https://clinicaltrials.gov/ct2/show/NCT04847102 (2021).
Chen, G.-L. et al. Safety and immunogenicity of the SARS-CoV-2 ARCoV mRNA vaccine in Chinese adults: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Microbe., https://doi.org/10.1016/s2666-5247(21)00280-9 (2022).
Study of ameliorating effect of COVID-19 mRNA vaccine in individuals immunized with inactivated vaccine (COVID-19), https://clinicaltrials.gov/ct2/show/NCT04944381 (2021).
Arcturus therapeutics announces positive interim ARCT-021 (LUNAR-COV19) Phase 1/2 study results for both single shot and prime-boost regimens, and up to $220 million in additional financial commitments from Singapore | Arcturus Therapeutics, Inc, https://ir.arcturusrx.com/news-releases/news-release-details/arcturus-therapeutics-announces-positive-interim-arct-021-lunar (2020).
Low, J. G. et al. A phase 1/2 randomized, double-blinded, placebo controlled ascending dose trial to assess the safety, tolerability and immunogenicity of ARCT-021 in healthy adults. medRxiv, https://doi.org/10.1101/2021.07.01.21259831 (2021).
Hagai, T. et al. Gene expression variability across cells and species shapes innate immunity. Nature 563, 197–202 (2018).
Blakney, A. K. et al. Innate inhibiting proteins enhance expression and immunogenicity of self-amplifying RNA. Mol. Ther. 29, 1174–1185 (2021).
Center for Drug Evaluation. Guideline on the Chemistry, Manufacture and Control (CMC) of Prophylactic COVID-19 mRNA Vaccines (trial), https://www.nmpa.gov.cn/directory/web/nmpa/images/2020081423091712896.docx (2020).
National Medical Products Administration. Vaccine administration law of the People’s Republic of China, https://www.nmpa.gov.cn/xxgk/fgwj/flxzhfg/20190702121701506.html (2019).
National Medical Products Administration. Medicinal Product Administration Law of the People’s Republic of China, https://www.nmpa.gov.cn/xxgk/fgwj/flxzhfg/20190827083801685.html (2019).
National Medical Products Administration. Measures for the supervision and management of drug production, https://www.nmpa.gov.cn/directory/web/nmpa/xxgk/fgwj/bmgzh/20200330182901110.html (2020).
National Medical Products Administration. Measures for the administration of drug registration, https://www.nmpa.gov.cn/zhuanti/ypzhcglbf/ypzhcglbfzhcwj/20200330180501220.html (2020).
National Institutes for Food and Drug Control. Regulation on drug registration verification procedures and technical requirements, https://www.nifdc.org.cn/directory/web/nifdc/infoAttach/956958cb-bd3a-49dd-809d-32353a3b1a10.docx (2020).
Geall, A. J., Mandl, C. W. & Ulmer, J. B. RNA: the new revolution in nucleic acid vaccines. Semin. Immunol. 25, 152–159 (2013).
Baronti, L., Karlsson, H., Marušič, M. & Petzold, K. A guide to large-scale RNA sample preparation. Anal. Bioanal. Chem. 410, 3239–3252 (2018).
Ssr, A., Dmfp, A., Ama, A. & Mpcm, B. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine 39, 2190–2200 (2021).
Center for Drug Evaluation. Guiding principles for research and development technical of COVID-19 preventive vaccines (trial), https://www.nmpa.gov.cn/zhuanti/yqyjzxd/yqyjxd/20200814230916157.html (2020).
WHO good manufacturing practices: supplementary guidelines for the manufacture of pharmaceutical excipients, https://www.who.int/medicines/areas/quality_safety/quality_assurance/SupplementaryGMPPharmaceuticalExcipientsTRS885Annex5.pdf?ua=1 (1999).
Center For Drug Evaluation. Technical Guidelines for Pharmaceutical Research on SARS-CoV-2 mRNA Vaccines for Prevention (for Trial Implementation), https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=9efa70b504dc5c66965001181a49c30b (2021).
EMA. Quality aspects included in the product information for vaccines for human use, https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-aspects-included-product-information-vaccines-human-use-revision-1_en.pdf (2018).
EMA. Regulatory requirements for vaccines intended to provide protection against variant strain(s) of SARS-CoV-2, https://www.ema.europa.eu/en/regulatory-requirements-vaccines-intended-provide-protection-against-variant-strains-sars-cov-2 (2021).
EMA. Guideline on quality, non-clinical and clinical requirements for investigational advanced therapy medicinal products in clinical trials, https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-non-clinical-clinical-requirements-investigational-advanced-therapy_en.pdf (2019).
FDA. General principles for the development of vaccines to protect against global infectious diseases, https://www.fda.gov/media/82306/download (2011).
FDA. Emergency use authorization for vaccines to prevent COVID-19 guidance for industry, https://www.fda.gov/media/142749/download (2021).
FDA. Development and licensure of vaccines to prevent COVID-19 guidance for Industry, https://www.fda.gov/media/139638/download (2020).
FDA. Chemistry, Manufacturing, and Control (CMC) information for human gene therapy Investigational New Drug Applications (INDs) guidance for industry, https://www.fda.gov/media/113760/download (2020).
European Medicines Agency. Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products, https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-gene-therapy-medicinal-products_en.pdf (2018).
Poveda, C., Biter, A. B., Bottazzi, M. E. & Strych, U. Establishing preferred product characterization for the evaluation of RNA vaccine antigens. Vaccines 7, 131 (2019).
Guidelines on Transmissible Spongiform Encephalopathies in relation to biological and pharmaceutical products, https://apps.who.int/iris/handle/10665/68932 (2020).
Beverly, M., Dell, A., Parmar, P. & Houghton, L. Label-free analysis of mRNA capping efficiency using RNase H probes and LC-MS. Anal. Bioanal. Chem. 408, 5021–5030 (2016).
Mamontov, E., Daemen, L. L., Novak, E. & Stone, M. B. Melting and re-freezing leads to irreversible changes in the morphology and molecular-level dynamics of Pfizer-BioNTech COVID-19 Vaccine. Medicina 57, https://doi.org/10.3390/medicina57121343 (2021).
Uddin, M. N. & Roni, M. A. Challenges of storage and stability of mRNA-Based COVID-19 vaccines. Vaccines 9, https://doi.org/10.3390/vaccines9091033 (2021).
National Medical Products Administration. Regulation on vaccine storage and transportation, https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20171228165101753.html (2017).
National Medical Products Administration. Guideline on stability study techniques for biological products (trial), https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20150415120001189.html (2015).
Stitz, L. et al. A thermostable messenger RNA based vaccine against rabies. PLoS Negl. Trop. Dis. 11, e0006108 (2017).
Studies to address new formulations, https://www.pfizer.com/science/coronavirus/vaccine/studies-address-new-formulations (2022).
Lei, D. et al. Removal of the innocuity test from The International Pharmacopoeia and WHO recommendations for vaccines and biological products. Biologicals 66, 17–20 (2020).
Guideline on procedures and data requirements for changes to approved vaccines, In: WHO Expert Committee on Biological Standardization. Sixty-fifth Report. Geneva, World Health Organization, 2015, Annex 4 (WHO Technical Report Series, No. 993), https://www.who.int/biologicals/vaccines/Annex4_Guidelines_changes_to_approved_vaccines_eng.pdf?ua=1 (2015).
Jeong, D.-E. et al. Assemblies of putative SARS-CoV2-spike-encoding mRNA sequences for vaccines BNT-162b2 and mRNA-1273.docx, https://virological.org/t/assemblies-of-putative-sars-cov2-spike-encoding-mrna-sequences-for-vaccines-bnt-162b2-and-mrna-1273/663 (2021).
World Health Organization. Messenger RNA encoding the full-length SARS-CoV-2 Spike Glycoprotein, https://web.archive.org/web/20210105162941/https://mednet-communities.net/inn/db/media/docs/11889.doc (2020).
Roth, N. et al. CV2CoV, an enhanced mRNA-based SARS-CoV-2 vaccine candidate, supports higher protein expression and improved immunogenicity in rats. BioRxiv, https://doi.org/10.1101/2021.05.13.443734 (2021).
Peng, Y. mRNAs encoding SARS-CoV-2 virus antigen and vaccine and preparation method of vaccine. CN111218458A (2020).
Rauch, S., Gooch, K. E., Hall, Y., Salguero, F. J. & Carroll, M. W. mRNA vaccine CVnCoV protects non-human primates from SARS-CoV-2 challenge infection. BioRxiv, https://doi.org/10.1101/2020.12.23.424138 (2020).
Li, J. Novel coronavirus SARS-CoV-2 vaccine and preparation method thereof. CN111217917A (2020).
Yan, J. Novel coronavirus SARS-CoV-2mRNA vaccine and its preparation method and application. CN113151312A (2021).
FDA Takes additional action in fight against COVID-19 by issuing emergency use authorization for second COVID-19 Vaccine | FDA, https://www.fda.gov/news-events/press-announcements/fda-takes-additional-action-fight-against-covid-19-issuing-emergency-use-authorization-second-covid (2020).
Takeda announces approval of Moderna’s COVID-19 vaccine in Japan, https://www.takeda.com/newsroom/newsreleases/2021/takeda-announces-approval-of-modernas-covid-19-vaccine-in-japan/ (2021).
Naqvi, A., Fatima, K., Mohammad, T., Fatima, U. & Hassan, M. I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim Biophys. Acta Mol. Basis Dis. 1866, 165878 (2020).
Author information
Authors and Affiliations
Contributions
E. F., X. L. and M. L. wrote the manuscript. Y. L. and E. F. organized and designed the manuscript. B. Z., Z. Z. and L. S. revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, E., Liu, X., Li, M. et al. Advances in COVID-19 mRNA vaccine development. Sig Transduct Target Ther 7, 94 (2022). https://doi.org/10.1038/s41392-022-00950-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-00950-y
This article is cited by
-
Efficacy, immunogenicity and safety of respiratory syncytial virus prefusion F vaccine: systematic review and meta-analysis
BMC Public Health (2024)
-
Integrative single-cell analysis: dissecting CD8 + memory cell roles in LUAD and COVID-19 via eQTLs and Mendelian Randomization
Hereditas (2024)
-
Decoding epitranscriptomic regulation of viral infection: mapping of RNA N6-methyladenosine by advanced sequencing technologies
Cellular & Molecular Biology Letters (2024)
-
Exosome for mRNA delivery: strategies and therapeutic applications
Journal of Nanobiotechnology (2024)
-
mRNA vaccines in tumor targeted therapy: mechanism, clinical application, and development trends
Biomarker Research (2024)
