
Abstract
Monoclonal antibodies constitute a promising class of targeted anticancer agents that enhance natural immune system functions to suppress cancer cell activity and eliminate cancer cells. The successful application of IgG monoclonal antibodies has inspired the development of various types of therapeutic antibodies, such as antibody fragments, bispecific antibodies, and antibody derivatives (e.g., antibody–drug conjugates and immunocytokines). The miniaturization and multifunctionalization of antibodies are flexible and viable strategies for diagnosing or treating malignant tumors in a complex tumor environment. In this review, we summarize antibodies of various molecular types, antibody applications in cancer therapy, and details of clinical study advances. We also discuss the rationale and mechanism of action of various antibody formats, including antibody–drug conjugates, antibody–oligonucleotide conjugates, bispecific/multispecific antibodies, immunocytokines, antibody fragments, and scaffold proteins. With advances in modern biotechnology, well-designed novel antibodies are finally paving the way for successful treatments of various cancers, including precise tumor immunotherapy, in the clinic.
Similar content being viewed by others

Introduction
Over the past 30 years, therapeutic antibodies have revolutionized the field of targeted cancer therapy. Therapeutic application of monoclonal antibodies (mAbs) emerged after the hybridoma technique to produce mAbs was introduced by Kohler and Milstein in 1975.1 The antibody humanization technique pioneered by Greg Winter in 1988 further promoted the development of therapeutic mAbs for treating various cancers.2 To date, >100 mAbs have been approved by the US Food and Drug Administration (FDA) for the treatment of different human diseases, including cancer and autoimmune and chronic inflammatory diseases.3 mAbs can specifically bind to target antigens and induce cytotoxicity by exerting neutralizing or proapoptotic effects, as well as promote innate immune responses, such as antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP).4 Inspired by the successful application of immunoglobulin G (IgG) mAbs, other antibody formats (e.g., antibody fragments, bispecific antibodies (BsAbs), and non-IgG scaffold proteins) and antibody derivatives (e.g., antibody–drug conjugates (ADCs) and immunocytokines) have been successively accepted as alternative therapeutic agents for a broad range of cancers.5
The antitumor efficacy of an antibody can be remarkedly improved by linking highly a cytotoxic small molecule to the mAb, generating a novel type of antibody derivative, an ADC.6 ADCs can selectively deliver highly cytotoxic small-molecule drugs directly to targeted cancer cells and induce their apoptosis,7 which fulfills the requirements of a “magic bullet” as postulated by German physician and scientist Paul Ehrlich more than one century ago.8 The FDA has approved 10 ADCs for cancer treatment, and >80 ADCs are under clinical investigation.9 In addition to small-molecule drugs, antibodies can be conjugated to other types of molecules, such as oligonucleotides,10 radionuclides,11 and protein toxins.12
Harnessing the power of the human immune system is steadily gaining recognition for its importance in the treatment of cancer.13 BsAbs can simultaneously bind to two different antigens.14 The most widely used BsAb is a bispecific T cell engager (BiTE), with one arm targeting CD3 on T cells and the other recognizing target proteins on tumor cells, thereby activating the T cells to kill the tumor cells.15 One first-in-class BiTE, blinatumomab, which targets both CD19 and CD3, was approved by the FDA for the treatment of patients with relapsed and/or refractory B cell precursor acute lymphoblastic leukemia (R/R B-ALL) in 2014.16 In addition to their interaction with T cells, BsAbs have also been designed to engage other effector cells, such as natural killer (NK) cells17 and macrophages18 for cancer therapy. Antibody–cytokine fusion proteins (also named immunocytokines) represent another novel class of antibody-based immunotherapies.19 Cytokines constitute a broad and loosely defined class of relatively small proteins that regulate the immune response.20 The systemic administration of proinflammatory cytokines is often associated with severe off-target toxicity, particularly flu-like symptoms, which may limit the dose and prevent the escalation of dosages needed for developing therapeutically effective regimens.21 Similar to the ADC strategy, a strategy for using immunocytokines with antibodies or antibody fragments as vehicles has been used for the targeted delivery of immunomodulatory cytokines (such as interleukin (IL)-2, IL-12, and tumor necrosis factor (TNF)) to leverage the local tumor microenvironment (TME) and activate anticancer immune responses.22
IgG is the predominant antibody used in current antibody drugs, but in certain cases, the application of full-length antibody is limited in cancer treatment because these large antibodies such as poor penetration into solid tumors and because the Fc can mediate bystander activation of the immune system.23 Recent advantages in antibody engineering have facilitated the production of different types of antibody fragments (e.g., Fab, F(ab’)2), engineered antibodies (e.g., single-chain variable (scFv) fragments scFabs), and Ig domains (e.g., VHH).24 These fragments usually retain the antigen specificity of the full-length antibody and are expected to show better penetration into tumors and fewer Fc-related adverse effects. In addition, non-IgG scaffold proteins, such as affibodies, designed ankyrin repeat proteins (DARPins), and monobodies, represent promising classes of therapeutic and diagnostic molecules.25
mAbs usually recognize cell-surface antigens, whereas most cancer-associated proteins reside in intracellular compartments.26 T cell receptors (TCRs) can recognize certain small fragments of intracellular proteins by binding with the peptide-major histocompatibility complex (pMHC), which comprises a short peptide derived from intracellular proteins presented in the context of the MHC on the cell surface.27 An antibody that mimics the epitope-recognizing segment of a TCR, termed a TCR mimic (TCRm) antibody, and TCRms are being used to target proteins of interest inside tumor cells or other cells.28 A TCRm combines the pMHC-targeting ability of a TCR with the robustness of IgG mAbs, which is expected to improve druggability.29
In this review, we summarize the advances in the development of new therapeutic antibodies and their applications in cancer treatment.
Antibody conjugates
Antibody–drug conjugates
Design and structure of ADCs
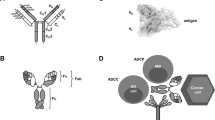
In recent years, the proposed use of ADCs has gradually gained steam, and they are rising stars in the tumor treatment field. An ADC comprises three main components: a mAb, cytotoxic payload, and linker. Upon binding with a target antigen on tumor cells, am ADC can deliver a cytotoxin payload into the targeted cell cytoplasm via receptor-mediated endocytosis, release the cytotoxic drug from the ADC during lysosomal degradation to destroy DNA or otherwise inhibit cell division and eventually kill tumor cells30 (Fig. 1c).

Representative therapeutic antibodies and their derivatives. a TCR-mimic antibody; b IgG antibody and antibody fragments; c antibody-drug conjugate (ADC) and its mechanism of action; d multifunctional antibodies, such as bispecific antibodies, immunocytokine (antibody-cytokine fusion protein)
Suitable drug targeting, which is highly tumor-specific and readily internalized by cancer cells, is a key factor that determines the druggability of an ADC.31 To minimize on-target/off‐target toxicity and open an acceptable therapeutic window for ADC applications, tumor-specific or overexpressed target antigens are preferable for ADC targeting and cytotoxic payload delivery. Ideal ADCs are rapidly and efficiently internalized via the clathrin-mediated pathway and are efficaciously trafficked to lysosomes, where they rapidly accumulate. Currently, more than 50 antigens have been used as targets for the preclinical or clinical development of ADCs; these antigens include human epidermal growth factor receptor 2 (HER2),32 trophoblast cell-surface antigen-2 (Trop-2),33 and B cell maturation antigen (BCMA).34
The cytotoxic payload of an ADC is a highly potent drug capable of efficient cell killing. Compared with the effect of conventional chemotherapy, these payloads showed higher toxic potency (from 100- to 1000-fold). Free ADC payloads cannot be effectively administered as chemotherapy agent candidates due to their extreme potency, but their toxicities can be minimized by directing the potency of the cytotoxic payload by conjugating it to a tumor-specific antibody. ADC payloads can be classified into two major types: (i) tubulin inhibitors inhibit tubulin polymerization and trigger cell cycle arrest in the G2/M phase and subsequent cell apoptosis; these inhibitors include monomethyl auristatin E (MMAE),35 monomethyl auristatin F (MMAF),36 and a derivative of maytansine 1 (DM1).32 (ii) DNA-damaging agents bind the minor groove in DNA, leading to cell death via DNA cleavage, DNA alkylation or interrupted DNA replication (these agents include calicheamicin,37 SN-38,33 DXd,38 and PBD39). Other small-molecule payloads, such as α‐amanitin (a selective RNA polymerase II inhibitor), are also under investigation.40
Linkers, which covalently conjugate the cytotoxic payloads to the antibody, are also essential components of ADCs. Ideal linkers are stable before they reach the targeted tumor site and are rationally designed for rapid liberation of payloads from an ADC upon entry into lysosomes. Based on the mechanism of payload release, linkers can be categorized into cleavable or noncleavable linkers. Cleavable linkers are designed to conditionally respond to the TME or intracellular environment, such as low pH (e.g., the acid-labile hydrazone-based linker in gemtuzumab ozogamicin (GO)41), proteolysis (e.g., the valine-citrulline linker in brentuximab vedotin (BV)42), or high-glutathione concentrations (e.g., the disulfide linker in the maytansinoid-based ADC mirvetuximab soravtansine43). On the other hand, noncleavable linkers (e.g., the thioether linker in ado-trastuzumab emtansine) rely on complete lysosomal degradation of the antibody for payload release.44 The chemical conjugation strategies of ADCs play a significant role in the therapeutic potential of ADCs. Usually, an ADC payload is conjugated to a surface lysine or cysteine residue of an antibody, resulting in the patterned distribution of ADCs with different drug-to-antibody ratios (DARs) during chromatographical separation. Different DARs, which may vary from zero to eight, indicate different ADC pharmacokinetics, efficacy, and safety profiles.7 Hence, site-specific conjugation approaches are being explored to generate homogeneous ADCs.45
Clinical results obtained with approved ADCs
To date, ten ADCs have been approved by the FDA for cancer treatment. An overview of these ADCs, including ADC design and indications, is presented in Table 1 and Fig. 2. A detailed discussion of these FDA-approved ADCs is presented below. ADCs at phase III clinical trial are summarized in Table 2.

Schematic representation of the FDA-approved antibody-drug conjugate (ADC)
Gemtuzumab ozogamicin
The FDA approved the first ADC drug—GO (Mylotarg®, Pfizer/Wyeth)—in 2000 for the treatment of patients aged >60 years with relapsed CD33-positive acute myeloid leukemia (AML).46 GO is a CD33-directed ADC consisting of recombinant humanized IgG4 antibody specific to CD33, a calicheamicin derivative, and an acid-labile hydrazone-based linker that covalently attaches the toxin to the antibody.37 However, in a postmarket clinical trial, GO failed to show improvement in complete response (CR), overall survival (OS), or disease-free survival; in contrast, an increase in treatment-related mortality was shown.47 Therefore, Pfizer withdrew the anti-CD33 ADC Mylotarg from the market in 2010.
In September 2017, on the basis of results from 3 clinical trials (ALFA-0701,48 AML-19,49 and MyloFrance-150), the FDA reapproved GO for patients newly diagnosed with CD33-positive ALL and for patients 2 years and older with relapsed or refractory CD33-positive AML. A phase III ALFA-0701 study compared standard front-line chemotherapy (daunorubicin and cytarabine) with and without a low fractionated dose of GO for patients with untreated de novo AML. The results showed that event-free survival (primary endpoint) was longer for patients in the GO group than for those in the chemotherapy-alone group (median 15.6 vs. 9.7 months; hazard ratio (HR), 0.58; 95% confidence interval (CI), 0.43–0.78; p = 0.0003). increased OS (median 34.0 vs. 19.2 months; HR 0.69; 95% CI, 0.49–0.98; p = 0.0368) and relapse-free survival (median 28.1 vs. 11.4 months; HR, 0.52; 95% CI, 0.36–0.75; p = 0.0003) were also observed. In the GO group, there was no increase in the risk of death from toxicity, although severe adverse events were more frequent. The randomized phase III AML-19 trial was performed to evaluate GO monotherapy and compare the results with that of the best supportive care (BSC) used for treating elderly patients with previously untreated AML. Patients assigned to the GO group exhibited a significantly longer OS than patients in the BSC group (median 4.9 months vs. 3.6 months; HR, 0.69; 95% CI, 0.53–0.90; p = 0.005). In a third trial, MyloFrance-1, a phase 2, single-arm, open-label study, included 57 patients with CD33-positive AML in their first relapse. Patients received GO at a dose of 3 mg/m2 on Days 1, 4, and 7. Fifteen (26%; 95% CI, 16–40%) patients achieved CR with a median relapse-free survival of 11.6 months. Both AML-19 and MyloFrance-1 showed the effectiveness and safety of GO administered as a single drug.
Ongoing studies are being performed to investigate the treatment of AML on the basis of various GO dosing schedules and in combination with chemotherapy.
Brentuximab vedotin
BV (Adcetris®, Seattle Genetics, Inc.) is an ADC composed of a chimeric anti-CD30 IgG1 antibody covalently linked to the microtubule-disrupting small-molecule MMAE via a cleavable valine-citrulline linker.51 In 2011, BV was approved by the FDA to treat patients with systemic anaplastic large-cell lymphoma (ALCL) and relapsed or refractory CD30-positive Hodgkin lymphoma (HL). Approval was based on two single-arm phase II trials.
ALCL is an aggressive subtype of T cell lymphoma characterized by uniform expression of CD30. In a single-arm phase II multicenter trial, patients with relapsed or refractory systemic ALCL after at least one prior therapy were treated with BV 1.8 mg/kg intravenously every 3 weeks. The trial showed an OR of 86% and a CR of 57%.52 For patients with relapsed or refractory HL (R/R HL), a phase II study showed that BV was associated with manageable toxicity and high efficacy. Of the 102 patients treated in the study, 76 patients (75%) achieved an objective response, 35 patients (34%) achieved complete remission, and 41 patients (40%) achieved partial remission. Grade 1 or 2 adverse effects were the most common; they included peripheral sensory neuropathy, nausea, fatigue, neutropenia, and diarrhea.53
Cutaneous T cell lymphomas (CTCLs) are rare non-HL subtypes that involve the skin and blood, lymph nodes, and other internal organs. In 2017, the FDA approved BV to treat adult patients with CD30+ CTCL, including patients with primary cutaneous ALCL (pcALCL) and mycosis fungoides who had been previously treated. Approval for the use of BV was based on the phase III ALCANZA trial, which showed a significant increase in an objective response lasting at least 4 months after BV treatment compared to the objective response to a physician’s choice of either methotrexate or bexarotene (56.3 vs. 12.5%, respectively; 95% CI, 29.1–58.4; p < 0.0001).54
BV is also effective when used in combination with chemotherapy. Based on results from the ECHELON-1 and ECHELON-2 trials, BV showed potent single-agent activity, and BV in combination with chemotherapy agents was shown to be more effective than chemotherapy alone.55,56 Hence, the FDA expanded approval of BV in combination with chemotherapy for the first-line treatment of stage III or IV classical HL or for previously untreated systemic ALCL or other CD30-expressing peripheral T cell lymphomas.
Currently, 78 active (phases I–III) clinical trials registered at ClinicalTrials.gov are being conducted to evaluate BV as a treatment for patients with hematological malignancies.
Ado-trastuzumab emtansine
HER2 is overexpressed in approximately 20% of breast cancer patients.57 In 2013, Ado-trastuzumab emtansine (also known as T-DM1 or Kadcyla®, Genentech, Inc.), a HER2-targeting ADC incorporating the anti-HER2 trastuzumab with the microtubule inhibitor DM1 (a maytansine derivative) via a stable thioether linker, was approved for the treatment of patients with HER2-positive metastatic breast cancer.58 The approval was based on a phase III trial (EMILIA).59 In the EMILIA trial, patients were randomly administered either T-DM1 (n = 495) or lapatinib plus capecitabine (n = 496). Median progression-free survival (PFS) (30.9 vs. 25.1 months; HR, 0.68; 95% CI, 0.55–0.85; p < 0.001) and median OS (9.6 vs. 6.4 months; HR, 0.65; 95% CI, 0.55–0.77; p < 0.001) were significantly better in the patients who received T-DM1 than in the patients who received lapatinib plus capecitabine.
In addition to breast cancer, T-DM1 has been studied in patients with other solid tumors, including lung, bladder, brain, and colorectal cancer.
Inotuzumab ozogamicin
The FDA licensed inotuzumab ozogamicin (BESPONSA®, Wyeth/Pfizer) for the treatment of patients with R/R B-ALL in 2017. Inotuzumab ozogamicin is an ADC composed of a recombinant humanized IgG4 mAb against CD22 covalently linked to the DNA-damaging agent calicheamicin via an acid-labile hydrazone-based linker.60 In a phase III trial (INO-VATE), the effects of inotuzumab ozogamicin treatment were compared with standard intensive chemotherapy in ALL patients, and the results showed that the CR was 81% for the inotuzumab ozogamicin group and 29% for the standard therapy group.61 Improvements in PFS (median, 5.0 months vs. 1.8 months; HR, 0.45; 95% CI, 0.34–0.61; p < 0.001) and OS (7.7 months vs. 6.7 months; HR, 0.77; 95% CI, 0.58–1.03; p = 0.04) were also observed.
Currently, 24 ongoing clinical trials (phases I-III registered at ClinicalTrials.gov) are being performed to test the effect of inotuzumab ozogamicin for treating ALL patients.
Polatuzumab vedotin-piiq
Polatuzumab vedotin-piiq (POLIVY®, Genentech, Inc.) is a CD79b-specific ADC that consists of a humanized anti-CD79b IgG1 antibody, MMAE, and a cleavable valine-citrulline linker that covalently conjugates MMAE to the polatuzumab antibody.62 In June 2019, the FDA granted accelerated approval to polatuzumab vedotin-piiq for adult patients with relapsed or refractory diffuse large B cell lymphoma (DLBCL), which is the most common type of non-Hodgkin lymphoma (NHL).63 Approval was based on a multicenter, open-label study (NCT02257567), and on the basis of the study design, polatuzumab vedotin-piiq was administered in combination with bendamustine (B) and rituximab (R).64 In this study, polatuzumab vedotin administered in combination with BR resulted in a significantly increased CR (40% vs. 17.5%; p = 0.026), median PFS (9.5 vs. 3.7 months; HR, 0.36; 95% CI, 0.21–0.63; p < 0.001) and median OS (12.4 months vs. 4.7 months; HR, 0.42;, 95% CI, 0.24–0.75; p = 0.002) compared with BR combination therapy without polatuzumab vedotin.
Twenty clinical trials have been recently established for studying polatuzumab vedotin-piiq for the treatment of patients with NHL or DLBCL.
Enfortumab vedotin
Enfortumab Vedotin (Padcev®, Astellas Pharma US, Inc.), also known as ASG-22ME, is a first-in-class ADC directed against Nectin-4 (Poliovirus receptor-related 4; PVRL4), which is highly expressed in urothelial carcinoma as well as breast, gastric, and lung cancers.65 Urothelial cancer, which typically occurs in the urinary system, is the most common type of bladder cancer.66 In December 2019, enfortumab vedotin (PADCEV, Astellas Pharma US, Inc.) was granted accelerated approval from the FDA for treating patients with locally advanced or metastatic urothelial cancer and who had been previously treated with anti-PD-1 (programmed cell death-1)//PD-L1 (programmed cell death-1 ligand 1) therapy or platinum-containing chemotherapy. The clinical basis for the FDA approval was the phase II single-arm trial EV-201.67 For patients with metastatic urothelial cancer, 55 (44%) of 125 patients showed an objective response, the median duration of the response was 7.6 months, and 15 (12%) patients showed a CR.
Enfortumab vedotin is currently being evaluated for its effect on urothelial cancers of various stages in eight ongoing clinical trials and for its effect on breast, lung, and other solid tumors in three other ongoing clinical studies, according to the registration information at ClinicalTrials.gov.
Trastuzumab deruxtecan
Trastuzumab deruxtecan (also known as DS-8201, ENHERTU®, AstraZeneca and Daiichi Sankyo) is an ADC composed of anti-HER2 trastuzumab, a cleavable tetrapeptide-based linker (GGFG), and a potent topoisomerase I inhibitor (an exatecan derivative, DXd) as the payload.68 On the basis of two key clinical trials, trastuzumab deruxtecan was granted accelerated approval by the FDA in 2019 for the treatment of patients with unresectable or metastatic breast cancer and who had been treated with at least two prior anti-HER2 regimens in the metastatic context. The phase I trial (NCT02564900) evaluated the safety and tolerability of trastuzumab deruxtecan in patients with HER2-positive advanced-stage breast cancer and with previously administered T-DM1 treatment. Trastuzumab deruxtecan showed a manageable safety profile and potent preliminary activity.69 In the second clinical trial (NCT03248492), the ORR was 60.9% (95% CI, from 53.4 to 68.0), and the median response duration was 14.8 months (95% CI, from 13.8 to 16.9). The median PFS was 16.4 months (95% CI, from 12.7 with an unreached maximum).70
Encouraged by the results of these clinical trials, approximately 29 clinical trials are currently aimed at evaluating trastuzumab deruxtecan as a treatment for patients with HER2-positive solid tumors, as reported at ClinicalTrials.gov.
Sacituzumab govitecan
Triple-negative breast cancer (TNBC) cells lack estrogen receptor, progesterone receptor or HER2 expression, limiting their response to hormonal therapy or HER2-targeted therapies.71 Patients with TNBC have limited treatment options. Sacituzumab govitecan (TRODELVY®, Immunomedics, Inc.) is an FDA-approved ADC that incorporates a humanized anti-Trop-2 mAb (hRS7), a cleavable linker, and the topoisomerase 1 inhibitor SN-38 as the payload.72 Compared to the payloads of other FDA-approved ADCs (pM), the potency of the SN-38 payload is moderate (at the single-digit nM level).73 Owing to the novel linker technology (a polar PEG-based linker), sacituzumab govitecan achieved a relatively high DAR (~7.6) in comparison with currently administered ADCs (DAR = ~4). Sacituzumab govitecan was approved by the FDA on an accelerated basis for refractory metastatic TNBC in April 2020 based on positive results from one clinical trial of 108 patients with TNBC (NCT01631552);74 in this clinical trial, the response rate was 33.3%, and the median duration of the response was 7.7 months. Sacituzumab govitecan is an important advancement for the treatment of patients, as it is the first approved ADC specifically targeted to metastatic TNBC.
Phase III trials of sacituzumab govitecan for treating patients with TNBC (NCT04595565), metastatic breast cancer (NCT04639986, NCT03901339) and other solid tumors (NCT04319198, NCT04527991) are ongoing.
Belantamab mafodotin
BCMA plays a central role in multiple myeloma (MM) pathogenesis in vivo and is overexpressed in MM cells. Therefore, BCMA has been shown to be a promising cell surface antigen for targeted therapies, such as CAR-T therapy, BsAbs, and ADCs, improving the landscape for patients with relapsed or refractory multiple myeloma (RRMM).75 Belantamab mafodotin (BLENREP®, GlaxoSmithKline) is a first-in-class anti-BCMA ADC that was granted accelerated approval by the FDA in August 2020 as a monotherapy treatment for adult patients with RRMM who had received at least four prior therapies; belantamab mafodotin includes an anti-CD38 mAb, the proteasome inhibitor MMAF and a non-cleavable maleimidocaproyl (mc) linker.76 The approval was based on results from a two-arm randomized open-label phase II study (DREAMM-2).77 In this trial, 97 patients were intravenously administered the recommended dose of 2.5 mg/kg belantamab mafodotin once every 3 weeks. The ORR was 31% (97.5% CI, 21%-43%), and 73% of the responders showed improvement that persisted for 6 months or longer. However, belantamab mafodotin can cause serious eye problems, including corneal changes, decreased vision, and/or blurred vision.
Clinical trials of belantamab mafodotin administered as a monotherapy and in combination with chemotherapy are ongoing for treating patients with MM.
Loncastuximab tesirine-lpyl
CD19 is widely expressed during various stages of B cell development and differentiation from pre-B cells to plasma cells, and it is an attractive target for treating B cell malignancies.78 Loncastuximab tesirine-lpyl (ZYNLONTA®, ADC Therapeutics SA) is a novel anti-CD19 ADC composed of a humanized anti-CD19 antibody, the alkylating agent pyrrolobenzodiazepine (PBD) dimer SG3199, and a cathepsin-cleavable valine-alanine linker.79 On April 23, 2021, loncastuximab tesirine-lpyl was granted accelerated approval by the FDA as a monotherapy treatment for adult patients with relapsed or refractory DLBCL who had received at least two prior systemic therapies.80 The approval was based on phase II multicenter open-label and single-arm LOTIS-2 trial (NCT03589469),81 in which a total of 145 patients were enrolled. Loncastuximab tesirine-lpyl showed an ORR of 48.3% (95% CI, 39.3–56.7), and the median duration of the response was 10.3 months (95% CI, from 6.9 to an inestimable value). PBD is approximately 50–100-fold more toxic than other cytotoxic ADC payloads,39 but loncastuximab tesirine-lpyl with PBD as the payload showed an acceptable safety profile. In the LOTIS-2 trial, the most common (≥10%) grade ≥3 adverse effects including laboratory-measured abnormalities were neutropenia (26.2%), thrombocytopenia (17.9%), an increased gamma-glutamyltransferase (17.2%) level, and anemia (10.3%).
Currently, loncastuximab tesirine-lpyl is under investigation for the treatment of various lymphomas as a single agent or in combination therapy in four active phase I–III clinical trials (ClinicalTrials.gov).
Antibody–small interfering RNA (siRNA) conjugates (ARCs)
As of January 2021, four siRNA-based drugs with lipid nanoparticle (LNP) or N-acetylgalactosamine (GalNAc) delivery systems had received regulatory approval from the FDA or European Medicines Agency (EMA).82 Encapsulated in LNPs or covalently conjugated to GalNAc ligands, siRNAs can be successfully delivered to the liver. Although RNA interference-based therapies are enabling progress in RNA delivery to the liver, low delivery efficiency and limited target organs (namely, the liver and eye) appear to be major obstacles for targeted siRNA therapeutics. Since antibodies show high specificity and affinity toward overexpressed antigens in certain cell types or tissues, they are drawing increasing attention as suitable vehicles for siRNA delivery. It has been sixteen years since antibodies were first used to mediate the in vivo delivery of siRNAs via cell-surface receptors.83 Through noncovalent interactions with antibody–fusion molecules or covalent linkages with lysine or cysteine residues, siRNAs can be coupled with antibodies for treating cancer (breast cancer,84 prostate cancer,85 colon cancer,86 and MM87) or other diseases (HIV83 and leukemia88). A schematic diagram of ARCs is shown in Fig. 3.

Schematic representation of antibody-siRNA conjugates (ARCs). a Antibody siRNA complex is constructed using electrostatic non-covalent interactions. b The THIOMAB-based ARC. The siRNA is chemically conjugated with the introduced cysteine of IgG antibody (THIOMAB, Genentech Inc) via thiol-maleimide reaction to achieve site-specific conjugation. c IgG-based ARC that incorporates cell-penetrating peptide in the linker to facilitate endosomal escape of siRNA. d Fab-based ARC. The siRNA or ASO is chemically conjugated to the C-terminus of Fab
In late 2015, using its THIOMAB platform, Genentech achieved site-specific, large-scale siRNA conjugation to antibodies.89 However, the results indicated that the entrapment of ARC in endosomes was a major limiting factor for its further application. Recently, Alnylam Pharmaceuticals, which had previously developed three siRNA drugs (i.e., patisiran, givosiran, and lumasiran) that were approved by the FDA, reported the simple generation of a structurally defined ARC (DVD-ARC) without introducing mutations or using enzymes.86 Moreover, the reported ARCs significantly downregulated the target mRNA and protein expression levels in tumor cells, further demonstrating the rationale of using antibodies as vehicles to specifically deliver siRNAs to nonliver tissues. In the late 2010s, Avidity Biosciences described an IgG-based antibody-siRNA conjugate strategy that achieved significant mRNA silencing in muscles and tumors, according to its published patent.90 For the development of antibody–oligonucleotide conjugates (AOCs), the Avidity AOC platform enables coupling of various types of oligonucleotides, including siRNAs and ASOs, to IgG antibodies. AOC 1001, a leading AOC drug candidate for treating adult patients with DM1, will enter a phase I/II clinical trial in 2021. This ARC comprises a mAb targeting transferrin receptor 1 (TfR1), a bridging motif (a bifunctional linker with or without a cell-penetrating peptide), and a rationally designed siRNAs to specifically target certain mRNAs. In addition to muscle disease, ARC applications have been expanded to other cell types, such as immune cells and heart cells, according to one manufacturer’s website (Avidity Bioscience). In addition to an IgG-based ARC strategy, using antigen-binding fragments (Fabs) as delivery vehicles has also been extensively explored.91 Due to the lack of a Fc domain, Fabs offer multiple powerful advantages over mAbs, including increased tissue penetration and tolerance, enhanced tissue penetration, and reduced risk of immune system activation.92 Dyne Therapeutics is pioneering the development of Fab-siRNA/ASO conjugates through its FORCE platform to treat rare muscle diseases, such as DM1, Duchene muscular dystrophy, and facioscapulohumeral muscular dystrophy. The FORCE platform targets TfR1, which is highly expressed on the surface of muscle cells,93 enabling efficient and targeted delivery of siRNAs or ASOs directly to skeletal, cardiac, and smooth muscle tissue.
However, the applications of ARCs in the cancer treatment field are still at an early stage, awaiting further exploration. There are several issues in using ARCs that remain unsolved. First, ARCs do not readily enter cells because the negative charge of the appended siRNA makes it difficult to overcome the thermodynamic barriers presented by the cell membrane.94 Second, similar to other delivery systems, endosomal escape is a major obstacle for intracellular delivery of siRNAs, leading to inefficient localization of siRNAs into the cytoplasmic RISC.95 Third, the inherent endocytic property of the targeted antigen determines the efficiency of siRNA delivery.90 For example, Fab-type ARCs have only been shown effective against the target antigen TfR1,91 a receptor prevalent in the endocytic pathway. Fourth, the lack of quantitative approaches for investigating the endosomal escape of siRNAs (released from ARCs) and the interaction of siRNAs with the RISC machinery further limits the rational design and optimization of ARCs.87 Finally, steric hindrance created during the conjugation of antibodies with a siRNA lowers conjugation efficiency. With the current technology of the ARC field, the ADC linker cannot simply be simply grafted into the conjugate, suggesting that extensive linker optimization is needed to generate certain ARCs. With the boom in ADC production, the strategic use of ARCs, which is similar to the rationale of ADC use, is expected to create new opportunities for the targeted interference of gene expression in multiple organs in vivo in the near future.
Multispecific antibodies
In the past three decades, multispecific antibodies have rapidly received tremendous attention as therapeutic agents to address unmet clinical needs.14,96 Multiple mediators contribute to the activation of cancer-related signaling pathways in the complex pathogenesis of cancer, which limits the efficacy of monospecific-based cancer treatment.97,98 A decrease in activated lymphocytes in the TME has been demonstrated to result in an unfavorable immune response.99,100 A broad variety of multispecific antibody formats has been developed to function through different mechanisms in cancer immunotherapy, including but not limited to (1) engaging T cells or other immune cells (e.g., NK cells) to specifically eliminate tumor cells, (2) bridging receptors to block or activate synergistic signaling pathways, and (3) targeting multiple tumor antigens or different antigen epitopes on tumor cells to increase tumor selectivity (Fig. 1d).14,101 Here we summarize the advances in the development of multispecific antibodies (mainly BsAbs) used for cancer treatment.
The formats of multispecific antibodies
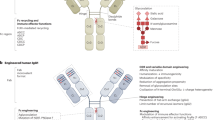
In the 1960s, the original concept of mixing univalent antibody fragments to generate multispecific antibodies was first described by Nisonoff and colleagues.102 Initially, BsAbs were generated by chemical conjugation of two antibody fragments and then by somatic cell fusion of two different hybridoma cell lines. The development of recombinant DNA technology and antibody engineering technology made it possible to assemble different antibody domains into various formats of multispecific antibodies with a desired orientation and stability. In summary, the formats of multispecific antibodies can be classified into two major categories: IgG-like antibody formats (with an Fc domain) and non-IgG-like antibody formats (without an Fc domain) (Fig. 4). The full-length IgG-like multispecific antibody contains an Fc domain that can bind with the neonatal Fc receptor FcRn, showing better pharmacokinetic characteristics than antibody fragments and exhibiting multiple antitumor mechanisms.103 Novel strategies such as Knobs-into-holes and CrossMab enable the correct pairing of different Ig heavy and light chains (Fig. 4). Compared with full-length IgG-like multispecific antibodies, non-IgG-like multispecific antibodies lack the Fc domain and thus have a lower molecular weight, which enables greater penetration efficiency in solid tumors. As a result, antibody formats without an Fc or albumin-binding domain have a relatively short serum half-life owing to rapid renal clearance. Non-IgG-like multispecific antibodies without an Fc domain can potentially be used to prevent nonspecific activation of the innate immune system as well as Fc-mediated ADCC or CDC, which is crucial for reducing immune cell engagement-related side effects.103 The well-known multispecific antibody formats include dual-variable-domain Ig, tetravalent IgG-like antibodies, multispecific scFvs, IgG-scFv fusion proteins, multispecific Fabs, tandem VHH domains, and others (Fig. 4).

Schematic overview of the IgG antibody structure and representative multispecific antibody formats at clinical stages. a The classical IgG antibody structure that consists of Fab and Fc regions. The single chain variable fragment (scFv) is a combination of the variable region of heavy chain (VH) and variable region of light chain (VL) domains. b Various multispecific antibody formats that are FDA-approved or under clinical studies. They are classified into the following categories: IgG-like constructs (with Fc region), non-IgG-like fragments (without Fc region). ScFv single-chain variable fragment, DVD-Ig dual-variable-domain immunoglobulin, BiTE bispecific T-cell engager, HLE-BiTE half-life-extended BiTE, TandAb tandem diabody, DART dual-affinity retargeting molecule, CODV cross-over dual variable region, TriKE trispecific NK-cell engager, TriTAC trispecific T-cell activation constructs
Mechanisms of action of different multispecific antibodies for cancer treatment
Multispecific antibodies are engineered to target distinct antigen epitopes simultaneously and thus may exhibit synergistic therapeutic efficacy. The modes of action of multispecific antibodies include but are not limited to redirecting the human immune system to fight cancer, regulating a receptor signaling pathway, combinational targeting of different tumor antigens to increase tumor selectivity or mitigate antigen loss-related relapse. With suitable target combinations and optimal format design, multispecific antibodies may achieve the desired therapeutic outcome and are feasible for large-scale production. In the near future, an extensive pipeline of multispecific antibodies may provide valuable candidates to meet the needs for clinical cancer treatment.
Targeting CD3 to engage T cells
Recruitable effector cells play critical roles in cancer immunotherapy. The early therapeutics BsAbs were T cell engagers, and their effects were first demonstrated in the mid-1980s.104,105 BiTEs are capable of binding CD3ε within the TCR complex and a selected tumor-associated antigen (TAA) on tumor cells simultaneously. Then, cytotoxic T cells are redirected to specifically kill the recognized tumor cells via T cell engagers.15,106 Catumaxomab (Removab®) was the first EMA-approved T cell-engaging BsAb for the treatment of malignant ascites; it was approved initially in 2009 and is currently used worldwide. As a trifunctional Triomab® family member, catumaxomab consists of an anti-CD3 rat IgG2b half-antibody and an anti-epithelial cell adhesion molecule (anti-EpCAM) mouse IgG2a isotype, with one arm binding to a T cell and the other arm binding to a tumor cell.107 Additionally, the binding to Fcγ receptors on accessory cells (e.g., macrophages, monocytes, NK cells, and dendritic cells) via the functional Fc region results in highly concerted antitumor immune responses.108 The efficacy and safety of catumaxomab were demonstrated in a pivotal phase II/III study, and these results were corroborated in phase I/II studies.107,109 However, catumaxomab was withdrawn from the market in 2017 for commercial reasons. Currently, another clinical trial of catumaxomab is being conducted in China for treating patients with recurrent or metastatic gastric cancer with peritoneal metastasis (NCT04222114, ClinicalTrials.gov).
Another advanced T cell-engaging BsAb format at the forefront of cancer clinical trials is BiTE® (Micromet, acquired by Amgen, Inc. in 2012). Blinatumomab (Blincyto; Amgen, Inc.) is composed of two scFvs targeting CD3ε on T cells and the B-lymphocyte antigen CD19. The striking clinical phase II trial results revealed a CR rate of 43% (81/189) in adult patients with R/R B-ALL after the first two cycles of continuous intravenous infusion of blinatumomab treatment110 (MT103–211, NCT01466179), which formed the basis for the accelerated approval granted by FDA in 2014 for the treatment of Philadelphia-chromosome negative (Ph−) R/R B-ALL in adults.16 Based on the encouraging clinical results of the TOWER trial (NCT02013167),106,111 Alcantara trial (NCT02000427),112 MT103–205 trial (NCT01471782)113 and BLAST trial (NCT01207388),114 the FDA approved the expansion of clinical indications for blinatumomab to patients with Philadelphia chromosome‐positive R/R B‐ALL and pediatric patients with R/R ALL or MRD-positive ALL.115
Recently, a chemotherapy-free first-line treatment strategy with blinatumomab and dasatinib (an ABL tyrosine kinase inhibitor (TKI)) showed high rates of molecular response and OS in 63 patients with Ph-positive ALL. Complete remission was observed in 98% of the patients, and at a median follow-up of 18 months, the OS was 95%, and the disease-free survival was 88%.116 Ongoing trials are evaluating the efficacy of blinatumomab administered in combination with chemotherapy, targeted therapies, or other immunotherapies, aiming to increase the OS rate and reduce side effects.116,117,118
The impressive clinical data of catumaxomab and blinatumomab sparked interest in the BsAb field and prompted researchers to develop a diverse variety of BsAb formats, leading to explosive growth in clinical trials. CD3-targeted BsAbs are rapidly becoming the most revolutionary strategic modalities in cancer immunotherapy, accounting for more than 50% of global BsAbs used in various clinical stages of cancer. Compared with IgG-like BsAbs, BiTEs show greater penetration into tumors owing to their small molecular weight (~55 kDa) and are relatively less immunogenic due to the lack of Fc domain-mediated effector toxicities.15 Researchers from Amgen have designed a series of BiTE®-based antibodies (anti-CD3 × tumor cell antigen) to validate their efficacy for use in cancer treatment; the drugs tested included AMG 330 (anti-CD3/CD33), AMG 427 (anti-CD3/FLT3, a half-life-extended BiTE® molecule in HLE-BiTE format, also known as a BiTE-Fc), AMG 673 (anti-CD3/CD33 in HLE-BiTE format), and AMG 701 (anti-CD3/BCMA in HLE-BiTE format) for use against hematological malignancies and AMG 160 (anti-CD3/a prostate-specific membrane antigen (PSMA) in HLE-BiTE format), AMG 596 (anti-CD3/epidermal growth factor receptor (EGFR) vIII), and AMG 757 (anti-CD3/DLL-3 in HLE-BiTE format) for use against solid malignancies; most of these drugs are still in the early stages of clinical development.
The recent spike of interest in antibody-mediated immune cell engagement for cancer treatment has driven many multifunctional antibodies into clinical studies. Approximately 30% of T cell-engaging BsAbs are in clinical trials for treating hematological malignancies, and the specific targets are mainly well-known TAAs, such as CD19, CD20, CD33, CD38, CD123, and BCMA.110,119,120,121,122,123 The application of T cell engaging BsAbs in solid tumors faces more-challenging hurdles, such as poor T cell infiltration, a complex immunosuppressive TME, and the higher likelihood of on-target off-tumor toxicities, raising concerns for the safety and efficacy of T cell engaging BsAbs. T cell-engaging BsAbs used for solid tumor treatment have been explored by targeting established tumor antigens, such as HER2, EpCAM, carcinoembryonic antigen (CEA), and PSMA.124,125,126,127 Many preclinical studies have demonstrated that T cell-engaging BsAbs can induce sufficient activation of T cells and subsequent killing of tumor cells in various mouse xenograft tumor models.128,129,130 To date, the most advanced T cell-engaging BsAbs that have entered clinical trials for investigation in patients are in phase I trials.103 In addition to the incorporation of extracellular antigens, some T cell-engaging BsAbs have incorporated TCR variable regions to recognize peptide/MHC I complexes that consist of MHC-I molecules and short peptides (8–9 aa) derived from intracellular proteins, and their binding is peptide-specific and MHC-restricted. Bispecific ImmTAC molecules (Immunocore, Inc.), fusion proteins composed of an affinity-enhanced TCR and an anti-CD3 scFv antibody, have provided potent and highly specific access to the vast landscape of intracellular targets via TCR-based tumor targeting. An exemplary ImmTAC molecule, tebentafusp (IMCgp100), which targets a gp100 peptide (a melanocyte differentiation antigen) presented by HLA-A02 on tumor cells, has shown promising clinical outcomes as a monotherapy agent for patients with metastatic uveal melanoma.131 The FDA has given a breakthrough therapy designation to tebentafusp (IMCgp100) for HLA-A2-positive, inoperable, or advanced uveal melanoma based on interim data obtained from the IMCgp100–202 Phase III trial (NCT03070392), which demonstrated the superiority of tebentafusp for extending survival in adults with newly diagnosed metastatic uveal melanoma. Tebentafusp has also been given a fast-track designation and orphan drug designation for use in uveal melanoma by the FDA and a Promising Innovative Medicine designation under the UK Early Access to Medicines Scheme for metastatic uveal melanoma. Currently, an IMCgp100–202 phase III clinical trial has met the predetermined criteria for statistical significance of the primary OS endpoint in tebentafusp treatment for patients with previously untreated metastatic uveal melanoma.
Targeting T cell costimulatory receptors
Costimulatory receptors on T cells, such as 4–1BB (CD137) and OX40 (CD134), can be leveraged for cancer immunotherapy. To reduce hepatotoxicity after systemic T cell costimulation via mAbs (e.g., urelumab), anti-TAA antibodies were fused to agonistic antibodies recognizing costimulatory receptors for tumor localization, TAA-dependent clustering, and activation of costimulatory receptors, such as the HER2/4–1BB BsAb.132 Several costimulatory receptors, including CD28 and members of the TNF receptor (TNFR) superfamily, such as 4–1BB and OX40, are promising targets for T cell-costimulation-related cancer immunotherapy.133,134 For instance, the 4–1BB/HER2 bispecific molecule PRS-343 induced T cell costimulation by HER2-dependent 4–1BB clustering and activation, increasing the infiltration and activation of lymphocytes in tumors and thus significantly suppressing tumor growth in HER2+ mouse xenograft tumor models.132 A phase I clinical trial of the first-in-class 4–1BB/HER2 bispecific molecule PRS-343 was initiated to evaluate its in vivo antitumor efficacy. Recently, Wu et al. described the application of a novel trispecific antibody format, termed a crossover dual variable (CODV) format, to simultaneously target CD38, CD3, and CD28 for treating multiple CD38-positive hematological malignancies, such as MM, AML, and chronic lymphocytic leukemia.135 Dual engagement of CD3 and CD28, representing the first and second signals required for optimal T cell proliferation, respectively, afforded efficient T cell activation and costimulation and boosted sustained T cell proliferation to kill CD38-positive MM cells in vitro and in vivo.135
Targeting CD16 to engage NK cells
NK cells have been identified as playing indispensable roles in the innate immune system, and growing evidence has demonstrated their ability to reduce tumor load and exert tumor immunosurveillance.136 CD16 (FcγRIII), mainly expressed on mature NK cells, is a low-affinity receptor that binds with the Fc domain of IgG antibodies to mediate ADCC. Bispecific and trispecific killer T cell engagers (BiKEs and TriKEs) are capable of exclusively triggering ADCC by crosslinking the CD16 receptor on NK cells together with antigen targets on cancer cells.137 AFM13 employs a tetravalent bispecific CD30/CD16A tandem diabody (TandAb®) with two binding sites for the CD16A isoform on NK cells and two binding sites for the CD30 antigen on lymphoma cells.138,139 Cytotoxicity assays demonstrated that AFM13 exhibited higher efficacy and potency with respect to the diabody and anti-CD30 IgG antibodies and showed inhibited lysis of CD30+ target cells.139 Early clinical studies suggested that AFM13 represents a well-tolerated, potent NK cell-engaging immunotherapy for treating CD30+ malignancies.140,141 A phase I dose-escalation study with patients with R/R HL revealed that the overall disease control rate of AFM13 was 61.5% in 26 evaluable patients, and the partial remission rate reached 11.5%.140 Another phase Ib study of AFM13 administered in combination with pembrolizumab in patients with R/R HL recently showed an 83% overall response rate for all recipients, and it showed good tolerability.141 In addition, a novel anti-CD16 × IL-15 × CD33 TriKE (GTB-3550/OXS-35504) construct designed by Vallera et al. incorporated an IL-15 cross-linker within scFvs recognizing CD16 on NK cells and CD33 on myeloid tumor cells.142,143 In vitro and in vivo studies revealed that GTB-3550 engaged NK cells to induce ADCC and eradicate CD33+ tumor cells efficiently, indicating the profound impact of IL-15 on maintaining the survival and proliferation of NK cells.142,143 A phase I/II clinical trial (ClinicalTrials.gov NCT03214666) for the treatment of CD33-positive leukemias such as AML, myelodysplastic syndrome, and other CD33+ hematopoietic malignancies is ongoing. The successful application of NK cell engagers in clinical stages of cancer strongly suggests that BsAb-mediated NK cell engagement is an alternative and viable strategy for leveraging immune responses to cancer. Notably, a higher proportion of NK cell infiltration within tumors has been demonstrated to be associated with better clinical outcomes in patients.144 Collectively, NK cell engagers hold promise in the near future as the most powerful agents for treating hematological malignancies and solid tumors.
Targeting checkpoint receptors on T cells
In an alternative strategy implemented following the improved clinical benefit observed in patients, the results from checkpoint blockade immunotherapy have shed light on manipulating the activation of immune cells in cancer treatment.145,146 Several studies have shown that the upregulated expression level of PD-L1 on advanced tumor cells inhibited the T cell response and facilitated cancer cell immune evasion, which limited the in vivo efficacy of cancer therapies.147,148 The utilization of therapeutic antibodies to target checkpoint molecules, including cytotoxic T lymphocyte antigen-4 (CTLA-4), PD-1, and PD-L1, unleashed pre-existing antitumor immune responses.149 Moreover, checkpoint blockades have been incorporated into BsAbs to achieve tumor-localized and TAA-dependent checkpoint blockage.150,151 For example, an anti-PD-1 × HER2 (human epidermal growth factor receptor 2) BsAb (IBI315, Innovent, Inc., China) was designed to bridge PD-1-expressing T cells to HER2-expressing tumor cells, block the PD-1/PD-L1 signaling pathway in a HER2-dependent manner, and inhibit activation of the HER2 signaling pathway.152 As a result, IBI315 combines targeted therapy with immunotherapy, thereby potentially enhancing antitumor activity via multiple mechanisms of action. Currently, a phase Ia/Ib clinical study is evaluating the safety, tolerability, and efficacy of IBI315 in patients with HER2-expressing advanced solid tumors (NCT04162327). Similarly, an anti-PD-1 × EGFR IgG scaffold-based BsAb has exhibited potent antitumor efficacy and enhanced T cell-based antitumor immunity through PD-L1 blockade in both in vitro and in vivo models.151 Simultaneous blockade of two immune checkpoints with synergic mechanisms of action has been used to maximize checkpoint blockade in the TME.153,154 A clinical trial showed that five-year outcomes of applying nivolumab (anti-PD-1) plus ipilimumab (anti-CTLA-4) to treat patients with advanced melanoma led to sustained long-term OS compared with treatment with ipilimumab alone,155 providing a rationale for concurrently targeting two immune checkpoints. Anti-PD-1 × CTLA-4 BsAbs, including AK104,156 MEDI5752,157 and MGD019,158 are expected to synergistically inhibit checkpoint signaling pathways in PD-1/CTLA-4 double-positive tumor-infiltrated lymphocytes (TILs) while enhancing PD-1 degradation and CTLA-4 inhibition in the TME.157
Moreover, to reverse acquired T cell-anergy-mediated resistance, the idea of targeting novel inhibitory receptors expressed on TILs, such as LAG-3 (lymphocyte-activation gene-3) and TIM-3 (T cell immunoglobulin and mucin-domain-containing molecule 3) with or without PD-1 and/or PD-L1, is also being explored in early clinical trials159 (NCT03219268, NCT03708328, and NCT03440437). A T cell-costimulation pathway can be combined with an immune checkpoint axis blockade to achieve sustained antitumor immune responses against advanced cancer.160,161 ICOS (inducible T cell costimulator, also known as CD278) and TNFR superfamily members such as 4–1BB and OX40 are promising targets of therapeutic BsAbs. The combinations of immune checkpoint blockade and costimulatory receptor activation, such as PD-L1 × 4–1BB (MCLA-145, FS222), CTLA-4 × OX40 (ATOR 1015), and PD-1 × ICOS (XmAb23104), are under preclinical and clinical investigation.162,163,164,165 To prevent potential toxic side effects due to unspecific overactivation, a monovalent trispecific antibody NM21–1480 comprising three scFvs (αPD-L1, α4–1BB and αHSA) fused in a single chain was designed to restrict 4–1BB signaling upon PD-L1 blockage.166 A similar immunomodulatory design strategy was used to construct GNC-038, a tetraspecific IgG-scFv-conjugated antibody (αCD19/CD3/4–1BB/PD-L1), now in phase I clinical trial (NCT04606433).
Recently, Cotton et al. showed that the fully recombinant BsAb AC-1, an antibody-based proteolysis-targeting chimera (PROTAC) termed AbTAC, simultaneously bound PD-L1 and the membrane-bound E3 ligase RNF43 to degrade PD-L1 by recruiting RNF43.167 This AbTAC efficiently recruited membrane-bound E3 ligase to degrade cell-surface PD-L1 in different cell lines with high PD-L1 expression levels (MDA-MB-231, HCC827, and T24 cells) via lysosomal degradation.167 AbTACs represent a new archetype of BsAbs for use in the targeted degradation of cell-surface proteins, further expanding the range of BsAb applications.
Targeting CD47 to enhance macrophage-mediated phagocytosis
CD47 is widely expressed on both healthy and cancer cells and transmits a “do not eat me” signal upon interaction with the signal-regulatory protein alpha (SIRPα) receptor on myeloid cells, including monocytes, macrophages, neutrophils, and a subset of dendritic cells.168 Blockade of the CD47-SIRPα axis by anti-CD47 mAbs augmented antitumor activity by unleashing macrophage-mediated phagocytosis; however, it also increased the possibility of damaging normal CD47-positive cells, such as erythrocytes.169,170 CD47-based BsAbs represent s viable strategy for protecting red blood cells while maximizing the potency of CD47 blockade. The introduction of a tumor-specific targeting arm into CD47-based BsAbs may potentially restrict CD47-SIRPα blockade to the tumor site. TG-1801 (also known as NI-1701, TG Therapeutics, Inc.) is a 1:1 IgG1 bispecific κλ antibody (CD19 × CD47) that combines malignant B cell targeting and selective CD47 blockade.171,172 Accordingly, TG-1801 specifically targeted double-positive tumor cells and showed antitumor efficacy through ADCP in vitro and in vivo.171 A phase I clinical trial for evaluating the safety and optimal dosage of TG-1801 (NCT03804996) is ongoing. Another anti-CD47-based BsAb, N1801 (TG Therapeutics, Inc.) was designed to target CD47 and mesothelin (MSLN), an antigen overexpressed in multiple solid tumors, and was demonstrated to efficiently kill MSLN+ tumor cells without generating an adverse hematological profile in a nonhuman primate study (AACR Annual Meeting 2019).173,174 The synergistic activation of innate and adaptive immune responses is also promising for tumor immunotherapy because it leads to the modulated activity of two highly potent effector cells, NK cells, and T cells. For example, the dual blockage of the PD-1/PD-L1 axis and CD47-SIRPα axis has been incorporated into single BsAbs, namely, IBI322 (PD-L1 × CD47) and HX009 (PD-1 × CD47), to simultaneously potentiate the local adaptive and innate immune responses against tumor cells and is being evaluated in phase I trials (NCT04338659 and NCT04097769 for IBI322 and HX009, respectively).175
Targeting receptor signaling pathways in tumor cells
Multispecific antibodies that recognize different receptors involved in signaling crosstalk can be utilized to obviate bypass signal transmission in cancer development.176,177,178 Receptor tyrosine kinases, such as EGFR, HER2, HER3, and MET, are enzyme-linked transmembrane receptors that consist of an extracellular ligand-binding domain, a transmembrane domain, and an intracellular protein–tyrosine kinase domain.179 Multiple studies have shown that phosphorylation and activation of c-Met signaling contribute to the resistance of EGFR TKIs in patients with non-small cell lung cancer (NSCLC).180 Recently, it was reported that the combination of osimertinib (EGFR TKI) plus savolitinib (MET TKI) showed an acceptable safety profile and encouraging antitumor efficacy in patients with advanced NSCLC.181 Therefore, dual inhibition of aberrant cell survival or proliferation-related signaling pathways has become a promising strategic focus for treating patients with MET-driven resistance to EGFR TKIs. Amivantamab/JNJ-61186372 is a dual-targeting EGFR × MET BsAb generated through controlled Fab arm exchange and can inhibit EGFR and MET signaling pathways while triggering Fc-mediated effector interactions.177 In various tumor models, amivantamab efficiently induced the downmodulation of EGFR and MET expression and exhibited immune-directed antitumor activity, translating clinically into robust antitumor responses in two exon 20ins patients.182 Notably, on May 21, 2021, the FDA granted accelerated approval to JNJ-61186372 (Rybrevant, Janssen Biotech, Inc.) for the treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion (exon 20ins) mutations. Another example is MCLA 128, an IgG1 BsAb specifically binding HER2 and HER3, which potently inhibits heregulin-driven signaling of HER2/HER3 and downstream PI3K/Akt signaling to suppress tumor cell survival and proliferation.183 In addition to those directed to breast cancer, clinical studies on MCLA 128 are ongoing in patients with metastatic pancreatic ductal adenocarcinoma or NSCLC that harbor fusion neuregulin 1 (NRG1) genes.
In addition to targeting extracellular membrane receptors, multifunctional antibodies can be employed to neutralize active mediators in the TME. Importantly, the inhibition of angiogenesis in tumor tissue, also known as tumor starvation, has been shown to be an effective way to deprive solid tumor cells of oxygen and nutrients needed for growth and metastasis.184,185 RO5520985/Vanucizumab, a BsAb in crossMab format, acts as a dual-target inhibitor of angiogenin-2 and vascular endothelial growth factor-A (VEGF-A).186 Preclinical studies have demonstrated that RO5520985 can induce potent antiangiogenic and tumor growth inhibition and antimetastatic efficacy in patient-derived tumor xenograft models.186 A first-in-human phase I study was performed to evaluate the safety and antitumor activity of RO5520985 as a monotherapy, and the results showed no disease progression in ten patients for ≥6 months and the induced toxicity was generally manageable.187 However, the McCAVE trial was terminated because the Vanucizumab/mFOLFOX-6 treatment did not improve the PFS of patients with previously untreated metastatic colorectal carcinoma.188 Thorough mechanistic studies and well-designed clinical trials with multispecific antibodies are needed to determine the principles underlying multiple-signaling pathway modulation in the TME.
Combinational targeting of TAAs
Tumor-specific targeting is important for therapeutic antibodies to minimize on-target off-tumor toxicity because TAAs are also expressed in noncancerous cells. To increase the specificity of tumor targeting, one appealing approach is to incorporate different antigen-binding modules into one multispecific antibody modality. The density, specificity, and stability of a target antigen on the surface of the tumor cell affect the duration of efficacious antitumor treatment with multispecific-antibody-engaged effector cells. Many receptor targets are subject to enzymatic cleavage that releases the soluble extracellular domain, e.g., CEA, HER2, CD19, and BCMA, thereby leading to variable and heterogeneous target receptor expression on tumor cells and even antigen escape.189 Multispecific antibodies with different targeting abilities are viable agents for limiting antigen escape and increasing the specificity of tumor targeting. For example, a dual-target single‐chain Fv antibody (CD123 × CD33) displayed more effective antitumor responses than a monotarget antibody in AML.190 Another example is DT2219ARL, a bispecific single-chain fusion protein that recognizes CD19 and CD22 and is fused with a diphtheria toxin (DT390) variant.191 Although CD19 is a validated target in B-ALL treatment, antigen loss-mediated relapse is a major limitation of potent CD19-targeted immunotherapies.192 However, CAR-T cells targeting CD22, which is also highly expressed on B cell malignant cells and are retained after CD19 loss, were found to be effective in killing CD22-positive B-ALL cells.193 A phase I trial of DT2219ARL for refractory B cell malignancy treatment demonstrated its safety, and the optimal dosage was determined.194 Complete remission or partial response (PR) in a 1-year timeframe was observed in 2 of 18 enrolled in a phase I/II trial (NCT02370160).
The strategy of combining targeting tumor antigens has also been applied to chimeric antigen receptor (CAR) T cell therapy and is a promising immunotherapy for several malignancies, such as lymphomas, leukemias, and MM.195,196,197 T cells obtained from a patient are engineered to express CARs in vitro. Recent advances have enabled the development of CAR T cells that target multiple antigens on the tumor cell surface, such as bispecific CARs and tandem CARs (Tan‑CARs), to overcome target antigen escape in patients with relapsed and refractory malignancies.197,198,199 A CAR is composed of an extracellular antigen-binding domain derived from a mAb (e.g., scFv, Fab, or VHH domain), a transmembrane domain, and an intracellular signaling domain.200,201 Bispecific CARs, with two distinct antibodies or antigen-binding domains (e.g., tandem VHH domains or scFvs) on one CAR T cell, can recognize different antigens or epitopes in one antigen during the targeting of tumor cells.202 For example, bispecific anti-CD20/CD19 (LV20.19) CAR T cells have shown low toxicity and high efficacy in the treatment of relapsed, refractory B cell malignancy (NCT03019055).203,204 Similarly, LCAR-B38M, a dual BCMA epitope-binding CAR T cell, demonstrated clinical efficacy and a manageable safety profile in patients with relapsed/refractory (R/R) MM.205
In a different strategy of cancer cell dual targeting, multispecific antibodies can be designed to recognize antigens on different tumor cells for the purpose of tumor-restricted activation with limited toxicity induction. For example, RG7386/RO6874813, a symmetric tetravalent 2:2 CrossMab BsAb that possesses a high affinity for fibroblast activation protein (FAP) on tumor stromal fibroblasts and low affinity for death receptor 5 (DR5), potently triggered death receptor-mediated apoptosis of tumor cells while sparing normal cells.206 This anti-FAP×DR5 BsAb demonstrated superior antitumor efficacy against FAP-expressing malignant cells in vitro as well as in patient-derived xenograft mouse models compared to that of conventional DR5 antibodies.206 A list of FDA-approved and clinical-stage multispecific antibodies is presented in Table 3.
Antibody–cytokine fusion proteins
Cytokines, such as ILs (e.g., IL-2), interferons (IFNs; e.g., IFN-γ), colony-stimulating factors (e.g., G-CSF), TNFs (e.g., TNF-α), and chemokines (e.g., CXCL3), are small proteins produced by immune cells or nonimmune cells and play vital roles in enhancing the efficacy of biological drugs. A complex network is formed by cytokines through their pleiotropic effects as well as complex autocrine and paracrine endocrine effects.207 At the same time, the high efficiency of cytokines drives the evolution of strict negative regulation mechanisms, which greatly weaken their biological functions.207 Moreover, dose-dependent side effects, unfavorable pharmacokinetics, poor drug tolerance, and high toxicity also limit their applications.19 With great progress in cancer research, the remarkable success of therapeutic antibodies in treating various cancer types has sparked interest in the development of novel immunotherapies administered alone or in combination.22 Fusion of cytokines to antibodies or antibody fragments may lead to more-specifically targeted TAAs, which may improve the efficacy, pharmacokinetics, and local concentration of cytokines and prevent systemic toxicity (Fig. 1d).208 A list of representative antibody–cytokine fusion proteins is presented in Table 4.
A variety of antibody–cytokine fusion proteins that can be classified into different categories based on their structures have been identified in recent decades (Fig. 5).208,209 An intact IgG, Fc, Fab or scFv can be fused with either monomers (e.g., IL-2 or IFN-α) or homomultimers of cytokines (e.g., IFN-γ or TNF).207 In addition, for some cytokines with two different polypeptide chains or heteromultimers (e.g., IL-12 and IL-27), different fusion strategies can be considered. Optimal combinations of antibodies or antibody fragments with cytokines may be acquired by considering various essential aspects (e.g., antibody type and fusion strategy) before applications are tested in clinical contexts. In principle, fusion with an IgG antibody or Fc domain conveys inherent advantages, such as binding to the neonatal Fc receptor (FcRn) and specific effector functions, to cytokines of interest to improve their pharmacokinetics, in vivo stability and in vivo efficacy. Furthermore, antibodies also provide a specific binding ability to target proteins to facilitate the efficient localization of cytokines to tumors in the form of immunocytokines. However, proinflammatory immunocytokines can trigger further cytokine production. In the clinic, the main side effects observed are hypotension, vomiting, flu-like symptoms, and nausea. Dose-dependent side effects and systemic toxicity can be reduced by reducing administration frequency.22 High-dose IL-2 (up to 800 million international units (IU)) administered for one week induces substantial toxicity,210 and therefore, IL-2-based immunocytokines are usually injected at much lower doses, such as 20–60 million IU, once a week for >6 months. It has been reported that the median half-life of an antibody–cytokine fusion protein, hu14.18-IL-2, which has been used for the treatment of refractory or recurrent neuroblastoma and melanoma, was only 3.1 h in clinical application.211 Therefore, the half-life of the fusion protein and its therapeutic effect were disappointing. Most likely, the accumulation of the fusion protein around a tumor might change the microenvironment, including densely packed activated immune cells, which may cause steric hindrance and block antigen presentation. In addition, the format and payload should be reconsidered during fusion protein development.19

Different antibody or antibody fragment-cytokine fusion proteins. a Cytokine fused to the N-terminus of the Fc domain; b Cytokine fused to the C-terminus of the Fc domain; c Cytokine fused to the C-terminus of the Fab; d Cytokine fused to the C-terminus of the scFv; e ScFv-cytokine-scFv fusion protein; f F(ab')-cytokine fusion protein; g IgG format immunocytokine without CH1 and CL; h Representative IgG format immunocykine-Bintrafusp alfa (M7824). A bifunctional antibody fusion protein composed of anti-PD-L1 human IgG1 and human TGF-βRII extracellular domain as the TGF-β trap, though a flexible glycine-serine linker fused to CH3-C terminus of IgG. Bintrafusp alfa blocks PD1-PDL1 as well as TGFR2-TGFβ signaling pathways to relieve immune suppression and remove the immune inhibition
IL-2 fusion protein
IL-2 is mainly produced by CD4+ T cells212 and can stimulate the immune system pleiotropically, including activation of NK cells, cytotoxic T cells, and monocytes. Human IL-2 displays biological activity by binding to the human IL-2 receptor (IL-2R), which consists of α, β, and γ subunits. Different subunits perform different functions. IL-2Rα (also known as p55 or CD25) binds to IL-2 with low affinity; IL-2Rβ (also known as p75 or CD122) mainly binds to Janus kinase 1 (JAK1) and is essential to the active signal transducer and activator of transcription 5 (STAT5); IL-2Rγ (also known as p64 or CD132) is essential for proliferative signaling, but the γ subunit alone does not show affinity for IL-2.213 Based on the function of IL-2, the development of fusion proteins that selectively favor the activation of IL-2Rβγ, but not IL-2Rα, may specifically activate T cells or NK cells to kill tumor cells while preventing side effects. OMCP-mutIL-2 is composed of a mutated form of IL-2 with a poor affinity for IL-2Rα and a cowpox virus encoding the NKG2D-binding protein (OMCP).214 This fusion protein (OMCP-MUTIL-2) activates IL-2 signaling effectively only in NKG2D cells, such as NK cells, not in IL-2Rα-positive cells. OMCP-MUTIL-2 has shown superior antitumor efficacy in several mouse xenograft models without inducing severe toxicity.
Since 1984, high-dose IL-2 has been used to treat malignant melanoma patients with tumor regression.215 In 1992 and 1998, the FDA approved IL-2 for the treatment of metastatic renal cell carcinoma and metastatic melanoma, respectively. However, high-dose side effects and vascular leakage syndrome, as well as a short serum half-life, were identified as major obstacles that hampered the therapeutic effect of IL-2.216 To address these issues, various antibody-IL-2 fusion proteins, such as IL-2 fused with an intact IgG, scFv, or a Fc domain, have been generated to produce higher local concentrations, reduce the effective dose and prolong the half-life of IL-2.
The immunocytokine NHS-IL-2-LT2 (EMD 521873), whose antibody moiety targets the DNA-histone complex in necrotic tumor cells, showed low toxicity when administered as a single agent in vivo.217 An IgG-IL-2 fusion protein enhanced the pharmacokinetic properties of IL-2 and increased its half-life from 7.8 to 11 h, similar to the effect of an IgG antibody (~150 kDa).218 A new IL-2 mutant (FSD13), with high efficacy and low toxicity, was identified by site-directed mutagenesis at IL-2 interaction sites and was obtained by replacing the proline residue at the 65th position with a lysine residue.219 The IL-2 mutant FSD13 was more potent than wild-type human IL-2 in activating CD4+ T cells, CD8+ T cells, and NK cells but was threefold weaker in inducing the conversion of CD4+ T cells into Treg cells.219 In addition, FSD13 significantly inhibited the growth and metastasis of melanoma without causing severe adverse side effects in the liver or lungs of a mouse xenograft model.219 The preclinical antitumor activity of the IL-2 mutant FSD13 was further investigated in combination with immune checkpoint blockers.219
IL-12 fusion protein
IL-12, composed of two subunits, p40 and p35, is a proinflammatory cytokine produced by antigen-presenting cells. IL-12 can induce the proliferation of NK and T cells and the production of IFN-γ, which results in the activation of T cells and the differentiation of Th1 cells.220 The co-inoculation of tumor cells with IL-12-secreting fibroblasts markedly inhibited tumor growth and neovascularization surrounding tumors in immunodeficient mice, suggesting a potent antitumor effect of IL-12.221 Furthermore, IL-12 showed cytotoxic potency against MHC-negative tumor cells, which was likely a result of the macrophage-mediated production of nitric oxide and activation of NK cells.222,223 IL-2 upregulated the expression of IFN-γ and IFN-γ-inducible protein 10 (IP-10) in endothelial cells. IL-12 was found to play a vital role in creating an inflammatory microenvironment during cancer treatment.224 However, in clinical trials, the local concentration of IL-12 at tumor sites was much lower than the overall administration dosage, inducing unfavorable immune responses and severe systemic toxicity.223,225
Murine IL-12 (mIL-12) was fused to scFv L19 of the human antibody L19, which recognizes the extra domain B (ED-B) domain of human fibronectin, an angiogenesis marker secreted by endothelial cells and tumor cells, to generate the protein L19-(mIL-12). The antitumor effect of fusion protein L19-(mIL-12) was enhanced in a syngeneic murine lung metastasis tumor model.226 A heterodimeric fusion protein including two scFv moieties, with one scFv fused to the N-terminus of the p35 subunit and another scFv fused to the C-terminus of the p40 subunit, showed tumor-targeted accumulation.227
Granulocyte-macrophage colony–stimulating factor (GM–CSF) fusion proteins
GM–CSF, a hemopoietic growth factor, plays an important role in the proliferation and differentiation of hematopoietic cells and is involved in the activation of neutrophils and macrophages as well as the expression of proinflammatory cytokines, adhesion molecules, and costimulatory molecules.228 GM–CSF is a monomeric glycoprotein cytokine composed of 127 amino acids forming a four-helix bundle.229 GM–CSF has been shown to be a pleiotropic host factor and to be involved in upregulating the expression of antigens,230,231 enhancing the production of adhesion molecules on granulocytes228 and monocytes and boosting IL-2-induced T cell growth in vitro.232 It has been demonstrated that GM–CSF induced tumor necrosis and reduced tumor size in mice vaccinated with irradiated GM–CSF-expressing tumor cells.233 In a clinical trial, systemic administration of GM–CSF in melanoma patients enhanced the immune response and greatly contributed to CTL-mediated tumor rejection in vivo.234 However, systemic administration of GM–CSF also induced dose-dependent side effects, such as myalgia, fever, fluid retention, and serosal effusion.235
GM–CSF was fused to the C-terminus of a scFv targeting the ED-B domain of fibronectin to generate GM–CSF-scFv fusion protein.236 Fusion of GM–CSF to the C-terminus of the ch17217 antibody, a rat/mouse chimeric anti-mouse transferrin receptor (TfR) antibody involved in iron uptake, maintained both the function of the antibody and cytokine.237 The ch17217-(GM–CSF) fusion protein eliminated hepatic metastases of neuroblastoma NXS2 cells and pulmonary metastases of CT26 colon carcinoma cells in syngeneic A/J and BALB/c mice.238 Another fusion protein, ch14.18-(GM–CSF), consisting of human chimeric antiganglioside GD2 antibody 14.18 and GM–CSF, maintained the ability to induce Fc domain-mediated ADCC and CDC activities.239 GM–CSF has also been fused to the N-terminus of a scFv fragment of an anti-idiotypic antibody mimicking the TAA CEA.240
IFN-γ fusion proteins
IFN-γ (a type II IFN) is composed of 143 amino acids and contains two N-glycosylation sites. IFN-γ is a head-to-tail dimer, in which one monomer aligns with the N-terminus of another monomer, with a molecular weight of ~100 kDa.241 The antitumor activity of IFN-γ depends on its function in the upregulation of class I and class II MHC molecules and the activation of monocytes/macrophages, CD8+ T cells, and NK cells. In this regard, IFN-γ is often used in combination with chemotherapy.242 In early research, an anti-TAG72 scFv-IFN-γ fusion protein exhibited the antigen-binding specificity of scFv moiety and cytokine activity in vitro.243 L19 scFv against the ED-B domain of fibronectin was fused to the N-terminus of a cysteine-free mutant of murine IFN-γ to generate an anti-ED-B-scFv-IFN-γ fusion protein.242 The fusion protein targeted blood vessels in solid tumors, and the targeting efficiency was strikingly increased in tumor‐bearing IFN‐γ receptor-knockout mice. scFv(L19)‐IFN‐γ displayed a strong antitumor effect in both subcutaneous and metastatic murine F9 teratocarcinomas. Immunocytokines are usually composed of mAbs against overexpressing antigens (e.g., fibronectin and FAP) in the TME and potent cytokines (e.g., IL-2, IL-12, and GM–CSF). Upon binding to the target antigen, the cytokines are released from immunocytokines directly into the TME, achieving high local concentrations and minimizing systemic side effects.244 The protagonists of immunocytokines are cytokines, which share mechanisms similar to that ADC payload release. In recent years, immunocytokines, also known as antibody–cytokine fusion proteins, have attracted increasing attention since the successful application of immune checkpoint antibodies, especially anti-PD1/PD-L1 antibodies. Ideally, in an exemplary immunocytokine context, an immune checkpoint antibody is expected to disable the immune escape mechanism, which is hijacked by tumor cells, and the cytokine payload locally and synergistically regulates the immune response.
At the 2018 American Society of Clinical Oncology conference, Merck KGaA announced an updated clinical study of Bintrafusp alfa (M7824, MSB0011359C), a bifunctional immunocytokine consisting of the anti-PD-L1 antibody Bavencio (avelumab) and the extracellular domain of transforming growth factor-β (TGFβ) type II receptor (TGFR-2).245 TGFβ was reported to attenuate the tumor response to PD-L1 blockade by contributing to the exclusion of T cells. Preclinical studies of Bintrafusp alfa revealed that the anti-PD-L1/TGFβ Trap fusion protein, not the anti-PD-L1 antibody, reversed the mesenchymalization of cancer cells, thereby sensitizing tumor cells to chemotherapy (NCT03631706). Furthermore, targeting both PD-L1 and TGF β in the TME is effective for inhibiting immune escape and “warming” cold tumors with limited infiltration of immune cells.245 Theoretically, these bifunctional fusion proteins show better clinical benefits than traditional PD1/PD-L1 mAbs. However, a clinical trial (NCT03631706) aimed at evaluating the efficacy of M7824 compared to that of Pabolizumab as a first-class treatment of advanced NSCLC with PD-L1 expression was terminated in January 2021 because it was unlikely to reach a common major endpoint, especially in terms of PFS, according to the collected data (Table 5). To date, antibodies have been fused not only with cytokines but also with other immune checkpoint targets; for example, Cinrebafusp alfa is the fusion of an anti-HER2 antibody with 4–1BB, an immune checkpoint protein, to generate the FAP-targeted 4–1BB agonist (FAP-4–1BBL). In China, I-Mab Biopharma developed the fusion protein efineptakin (also called TJ107), which is composed of an anti-PD-1 antibody and IL-7, and this protein was entered into a 2019 phase Ib clinical trial.
Cytokines can activate the immune system to achieve antitumor effects. Some cytokines, such as IL-2, have been authorized for the treatment of certain types of malignancies.246 However, dose-dependent side effects and systemic toxicity have limited the clinical applications of cytokines. Immunocytokines link cytokines to antibodies specific to TAAs, similar to BsAbs. Antibodies targeting TAAs or tumor-specific antigens (TSAs) can deliver cytokines to tumor sites where they can locally exert their antitumor effects.
Antibody fragments and non-Ig scaffold proteins
Fab
A Fab is a monovalent fragment consisting of one light chain and one heavy chain linked by a disulfide bond (Figs. 1b and 6a). Many in vivo experiments have shown that diffusion plays an important role in transporting molecules into tumor tissues.247 With a molecular weight of ~55 kDa, a Fab is much smaller than full-size IgG and thus shows greater penetration in solid tumors.248 For example, Lucentis, a VEGF-A-specific Fab fragment was more effective than the corresponding full-length mAb Avastin, in treating patients with the wet form of macular degeneration.24 Lacking the Fc region, a Fab can be produced in an economic expression system (e.g., yeast) and prevent Fc-related adverse effects in vivo. Predictably, a Fab has a shorter half-life due to the lack of FcRn-mediated recycling, similar to a scFv.249 The Fab half-life can be easily extended by conjugation to PEG or fusion with an albumin-binding protein.250 The FDA has approved certolizumab pegol (Cimzia®), a PEGylated and humanized anti-TNFα Fab, for the treatment of Crohn’s disease and rheumatoid arthritis.
Fab was considered the first therapeutic fragment antibody format, and eight Fab fragment antibodies were entered into clinical trials in the mid-1990s.251 To date, three Fabs have been approved by the FDA: abciximab (Reopro®), idarucizumab (Praxbind®), and ranibizumab (Lucentis®).250 No Fab has been approved for cancer treatment. Notably, naptumomab estafenatox (5T4FabV18-SEA/E-120 or ABR-217620), a fusion protein consisting of an anti-5T4 Fab and superantigen-staphylococcal enterotoxin A (SEA), was reported to successfully improve the OS of renal cell carcinoma patients in phase II/III trial.252,253
F(ab’)2
F(ab’)2, with a molecular weight of 110 kDa, is a bivalent fragment composed of two Fab segments linked together by a hinge region (Figs. 1b and 6b). They show better penetration than mAbs due to their smaller size.
The China FDA approved Metuximab-I131, a radioactive iodine [131I]-labeled anti-CD147 F(ab’)2 mAb, for the treatment of liver cancer. No F(ab’)2 has been approved by the FDA for cancer therapy.
Single-chain variable fragment (scFv)
The scFv was first described by Bird et al. in 1988.254 The scFv antibody consists of variable regions of heavy (VH) and light (VL) chains, which are joined together by a flexible peptide linker (e.g., GGGGS), and the biologically active form of scFv can be easily expressed in Escherichia coli, dramatically reducing production costs (Figs. 1b and 6c).255 The molecular weight of an scFv is ~25 kDa, which is much smaller than that of a full-length mAb, thus showing penetration in tumors. The use of an scFv can prevent Fc-related side effects resulting from ADCC or CDC.
However, the lack of the Fc region causes faster renal clearance (half-life ~5 h) and poor stability of the scFv, which tends to aggregate and show high immunogenicity.24,249 The multimerization of scFvs is a viable strategy to improve the pharmacokinetic properties and affinity of scFvs, such as diabodies (bivalent dimers, 55 kDa),256 triabodies (trivalent dimers, 80 kDa),257 tetrabodies (tetravalent dimers, 110 kDa),258 and minibodies (80 kDa).
Clinical trials of scFv antibodies have been conducted since the first scFv-focused clinical study performed in 1995.259 For example, several monovalent scFv fusion proteins are under development in clinical trials for cancer therapy, including L19-IL-2, rM28, D2C7-IT (in phase I), and Vicinium (in phase III). Multivalent or multispecific scFvs, such as BiTE, are discussed in the section on multispecific antibodies.
The leading scFv drug Vicinium (Sesen Biotech) is a humanized single-chain antibody fragment specific to the EpCAM antigen fused with Pseudomonas exotoxin A (ETA) (252–608 aa) toxin and was designed for treating bladder cancer. Upon binding with the EpCAM antigen on the surface of cancer cells, Vicinium can be internalized through the endocytic pathway and release the toxin ETA (252–608), thereby inducing cell apoptosis by irreversibly blocking protein synthesis.
VHH domain
In 1993, Hamers–Casterman discovered a special antibody in camelid that lacks a light chain and heavy chain CH1 domain, so it was called a heavy chain antibody (HCAb) (Figs. 1b and 6h).260 Later, HCAbs were found in Camelidae (bactrian and dromedary camels, alpacas, and llamas), as well as cartilaginous fish (e.g., sharks, rays, and skates).261 Interestingly, the variable domain of the heavy chain in a HCAb (also known as the VHH domain, a nanobody or single-domain antibody), despite a molecular weight (~15 kDa) one-tenth that of IgG (~150 kDa), retains high antigen-binding affinity, and is therefore the smallest naturally derived antigen-binding fragment.261 A crystal structure assay of the VHH domain revealed that its dimension is 4 nm × 2.5 nm × 3 nm.262
The homology discovered between the VHH and VH domains in the VH III human Ig family was greater than 80%.263 This means that the VHH sequence may induce relatively low immunogenicity when applied to cancer immunotherapy.264
VHH domains were demonstrated to be highly soluble and more stable than conventional antibodies and are capable of being stored for more than months at 4 or −20 °C without losing notable antigen-binding capacity.265 VHH domains can survive under very harsh conditions, for example, a wide pH range (range 3.0–9.0) and chemical (e.g., 6–8 M urea treatment) and thermal denaturing conditions (e.g., a VHH domain can maintain antigen-binding activity after prolonged incubation at 90 °C),265 making various routes, intravenous, oral, intraperitoneal or intratumor injection, feasible.266 Importantly, a VHH domains have a completely hydrophilic surface, which accounts for their superior stability and solubility compared to that of IgG VH domains, and VHHs show significantly less aggregation during production or multimerization (e.g., tandem VHH-based multispecific antibodies).265
In addition, the CDR3 loop in camelid VHH domains is usually longer (3–28 amino acids) than conventional VH domains in human IgG (8–15 amino acids).267 The long CDR3 of a VHH, which determines its recognition specificity, expands the potential interaction surface with a target antigen in the absence of a VL domain.265,268 Interestingly, the longer CDR3 in VHH domains can form a finger-like appendage that fits into a protein cleft, enabling the recognition of epitopes that are not accessible to the larger mAbs.267
The small size of the VHH domain results in fast renal clearance (half-life ~2 h),269 which is a major disadvantage for the application of VHH domains in cancer treatment but an advantage for obtaining an optimal signal-to-noise ratio during its application as an in vivo imaging agent.270 VHH domains have recently emerged as promising targeted imaging probes in combination with traditional imaging techniques, such as positron emission tomography (PET)271 and single-photon emission computed tomography (SPECT).272 The most noteworthy case of a VHH domain-based probe used for in vivo tumor imaging is the 68Ga-NOTA anti-HER2 VHH, which is a radiolabeled VHH domain currently in phase II clinical trial being evaluated for its ability to detect brain metastasis in breast cancer patients via PET/CT (NCT03331601).
Recently, the FDA and EMA approved the first VHH drug, caplacizumab (Ablynx Inc), a humanized anti-VWF VHH domain used for treating adults with acquired thrombotic thrombocytopenic purpura. A variety of VHH domains are being studied in the clinic to assess their effects for different purposes, including cancer treatment; for example, anti-EGFR VHH,36,273 anti-HER2 VHH,274 anti-VEGFR2 VHH,275,276 anti-c-Met VHH,6,277 and anti-chemokine receptor type 7 (CXCR7) VHH278 are currently being evaluated. Two VHH domains are in phase I clinical trials for cancer therapy; these VHH-based drugs are a biparatopic anti-CXCR4 VHH ALX-0651 (Ablynx, Inc.) for use in stem cell mobilization and novel agonistic tetravalent anti-DR5 VHH TAS266 (Novartis) used against advanced solid tumors. In its phase I clinical trial, TAS266 was found to show unexpected and significant but reversible hepatotoxicity, which can probably be attributed to enhanced DR5 clustering and activation of hepatocyte apoptosis.279 Tables 6 and 7 summarize the antibody fragments that have been approved for use by the FDA and those that have been entered into a clinical trial. The formats of presentative antibody fragments and their derivatives are summarized in Fig. 6.

Different formats of antibody fragments and their derivatives
Affibody
One key priority in current studies is the generation of homogeneous BsAbs to overcome hybrid pairing of heavy and light chains derived from two antibodies expressed in the same cell.280 Utilizing non-Ig scaffolds as building blocks is a viable approach to circumventing this problem.280 Non-IgG proteins include affibodies,281 anticalins,282 DARPins,283 monobodies,284 and so on.
Non-IgG protein scaffolds are typically composed of a single polypeptide chain folded into a structured core,280 which is much smaller than an antibody. The large size of an IgG antibody limits its penetration and infiltration in tumor tissues. The attractive characteristics of non-IgG proteins include but are not limited to their small size, high thermal stability, high solubility, the inclusion of a cysteine-free sequence and storage stability.285
Affibodies constitute a class of binding proteins based on the scaffold Z-domain, which is derived from domain B of staphylococcal protein A (SPA).281 An affibody is composed of a single polypeptide containing 58 amino acid residues with a molecular weight of approximately 6.5 kDa. The substitution of a glycine residue29 with an alanine residue in the B domain confers high stability and rapid folding to the Z-domain through the stabilization of helix 2.286 Therefore, affibodies are characterized by small size, high stability, and rapid and independent folding of the Z-domain structure.287 The combinatorial randomization of 13 amino acid residues in the Z-domain surface (helices 1 and 2), determined on the basis of the X-ray crystallography complex structure obtained for the homologous B-domain of SPA and human IgG, has led to the generation of affibody molecule libraries for antigen-binding screening.288 Affibodies have advantageous features for therapeutic applications, such as (i) easy production in E. coli or through solid-phase peptide synthesis due to their robust physical properties, including stability, fast folding, and ability to withstand a wide range of pH values and temperatures; (ii) small size resulting in rapid tissue penetration and efficient delivery of high molar doses compared to larger proteins; (iii) high affinity (with a KD of ~pM) for the target protein with nonspecific binding289; and (iv) site-specific conjugation facilitated by a unique C-terminal cysteine residue can be performed. Affibodies show the most promise as imaging probes due to their high tumor retention and tissue penetration, rapid blood clearance kinetics, low uptake by nontumorous organs, and the ability to undergo rapid, site-specific labeling with different radionuclides depending on the preferred modality.290 In vivo imaging studies using affibody proteins mainly focus on targeting HER2291, which is overexpressed in breast carcinomas and a validated target for antibody-based immunotherapy.
Affibody molecules, which is a rapidly growing class of non-IgG affinity ligands, show some advantages over antibodies. In addition to their potential in vivo imaging, affibodies can be applied in therapeutics. In 2020, a bispecific affibody (ABY-035) targeting subunit IL17A and human albumin for the extension of serum half-life was developed to treat patients with psoriatic arthritis (PsA), and the results from a phase I trial demonstrated that ABY-035 was safe and well tolerated (NCT03591887).
Another group of non-IgG scaffold proteins consists of anticalins, which are composed of a rigid β-barrel with four exposed loops that are engineered binding proteins based on the natural lipocalin fold.282 The central β-barrel supports four structurally variable loops that form a binding site.292 According to X-ray structural analysis, reshaping these loops can lead to the anticalin ability to recognize and tightly bind a wide range of molecules, from small molecules to peptides and proteins.292 The rigid and small structure of the non-IgG scaffold proteins enables flexible protein fusion and chemical conjugation with or incorporation into multifunctional molecules, such as multispecific antibodies and ADCs.280
A modified anticalin specifically binds to fibronectin ED-B,293 which can be used for the diagnosis of glioblastomas.294 ED-B-specific anticalins are located in the blood vessels of a glioblastoma, especially in the endothelial cells of glioblastoma origin.294 On the other hand, anticalins can be used to construct BsAbs for binding targets. For example, bispecific duocalin generated from two anticalins recognizing fluorescein and digoxigenin, respectively, has been successfully applied to fluorescence titration experiments.295
DARPins are scaffold proteins engineered on the basis of human ankyrin repeat proteins.283 DARPins are composed of 33 amino acid residues that form tightly packed repeats.296 Two antiparallel α-helices following a β-turn in each repeat form a structural unit.297 A right-handed solenoid structure with a continuous hydrophobic core constitutes four to six repeats in each ankyrin repeat domain.298 Currently, DARPins are applied in many fields, for example, as selective inhibitors of c-Jun N-terminal kinase 1 (JNK1) or JNK2.299 An EpCAM-specific soluble DARPin was produced as a fusion protein with Pseudomonas aeruginosa exotoxin A (ETA) in E. coli.300 This DARPin-ETA fusion protein was found to be highly toxic to EpCAM-positive tumor cells and exhibited strong antitumor efficacy in a mouse xenograft model.300
Monobodies are synthetic scaffold proteins based on the fibronectin type III (FN3) domain in human fibronectin.284 Monobodies can strongly bind to an epitope on the desired target301 to disrupt the biological function of the target molecule or protein.284 Monobodies have no cysteine residues with which to form intrachain disulfide bonds, and therefore, their functions are not influenced by the reducing environments of intracellular compartments.284 The SH2-kinase interaction is necessary for leukemogenesis, and intracellularly expressed monobodies targeting SH2-kinase can inhibit leukemia cell survival and oncogenic transformation.302,303 RAS and its mutants remain the most challenging drug targets.304 NS1, a monobody identified by the unbiased selection, binds to KRAS and HRAS by recognizing the uncharacterized α4/α5 region located on the opposite side of RAS.305 However, NS1 did not inhibit the GAP, GEF, or other downstream effectors of RAS.306 This evidence indicated that NS1 disrupted the formation of a signaling complex containing two RAS molecules and two RAF kinase molecules by inhibiting RAS self-association.284
The discovery of agents capable of disrupting protein–protein interactions is one of the major goals of the biopharmaceutical industry aimed at therapeutics development.307 These non-IgG scaffold proteins have been advanced in mechanistic studies and used to identify prospective therapeutic target drug-binding sites.
TCRm antibody
Most cancer-specific targets are intracellular proteins that are inaccessible to traditional mAbs.308 Intracellular proteins can be degraded and processed into peptides by the proteasome and ultimately presented on the cell surface in the context of MHC class I molecules for recognition by TCRs on CD8+ T cells.309 Although TCRs can bind target peptide-loaded MHC molecules specifically, their application as therapeutic agents has been limited by a low binding affinity for pMHC (1–100 µM)310 and poor yield during production in expression systems.311 It is feasible to stabilize TCRs computationally via rationally designed mutagenesis,312 but TCR affinity maturation and TCR expression remain challenging. An alternative strategy for targeting pMHC is the development of TCRm mAbs (Fig. 1a). Compared with TCR antibodies, TCRm antibodies show greater affinity (100–1000×) for pMHC and maintain all the advantages of IgG antibodies, such as stability, high yield, and a well-established production system.30 In addition, anti-TCRm mAbs can be generated efficiently via animal immunization and hybridoma technology or through in vitro display technologies, e.g., phage and yeast displays.313
In contrast to traditional mAbs, whose targets are mainly cell surface antigens (~10% of their total targets), TCRm mAbs provide a viable strategy to target the ~90% remaining intracellular antigens through pMHC.314 The targets of TCRm mAbs are classified into two types: TAAs and neoantigens, which are also known as tumor-specific antigens. Based on the origin of intracellular proteins, TAAs are classified into tumor-associated viral antigens and tumor-associated self-antigens. In some virus-induced cancers, viral proteins (e.g., CMV proteins315 and EBV proteins316) can be degraded and presented on the cell surface. They are referred to as tumor-associated viral antigens. Tumor-associated self-antigens are normal proteins that are abnormally expressed in tumor cells, including cancer-testis antigens, oncofetal antigens, differentiation antigens, and overexpressed antigens, e.g., Wilms tumor protein (WT1),317 glycoprotein (gp100), melanoma antigen (MAGE),318 melanoma-associated antigen recognized by T cells-1 (MART-1),319 and NY-ESO-1.320 On the other hand, neoantigens are results of tumor-specific somatic missense mutations, such as Kras G12V/D321 and p53.322 With respect to the high level of tumor specificity, the aforementioned pMHC antigens are emerging as conceptually ideal targets for targeted therapies prevent on-target off-tumor toxicity.323
The average low density of pMHC antigens appears to be the major hurdle for using TCRm mAbs in clinical application. Various strategies have been applied to enhance the potency of TCRm mAbs whose efficacy and affinity can be significantly affected by the low expression level of pMHC. One strategy is to enhance the Fc region functionalities, such as those that induce ADCC, CDC, and ADCP. ESK1 is a TCRm mAb that specifically recognizes the WT1 RMF/HLA-A*02:01 complex.324 To enhance the efficacy of ESK1, Nicholas Veomett et al. altered its Fc glycosylation to obtain a glycoengineered TCRm mAb, “ESKM.” As a result, ESKM exhibited potent ADCC activity at lower doses and superior in vivo efficacy than the parental antibody ESK1.325
The potency of TCRm mAbs can be enhanced by transforming TCRm mAbs into ADCs, BsAbs, or CARs. After equipping them with highly cytotoxic payloads, TCRm mAbs confer additional cytotoxicity, and thus, they can specifically deliver cytotoxic payloads into target tumor cells. In 2008, Klechevsky, E. et al. first fused TCRm antibodies targeting MART-126–35/HLA-A*02:01 and gp100280–288/HLA-A*02:01 with a truncated form of Pseudomonas exotoxin to generate a TCRm immunotoxin with antitumor activity.326 Our group developed EA1 HL-vcMMAE, a TCRm-ADC-targeting MART-126–35/HLA-A*02:01, for the treatment of metastatic melanomas, and we found that it showed potent in vivo efficacy in a mouse xenograft model.327 Recently, we generated a novel TCRm ADC 2A5-MMAE against the neoantigen Kras G12V/HLA-A*0201, and it showed specific antitumor activity both in vitro and in vivo.328 However, the potency of TCRm ADCs has been limited by extremely low levels of cell surface pMHC density compared with ADCs against normal targets. In another validated strategy, TCRm mAbs are applied as tumor-binding modules in the context of BsAbs to redirect and mediate T cells to specifically kill tumor cells. For example, the same group that discovered ESK1 engineered ESK1 into a BiTE.329 Despite the extremely low density of WT1/HLA-A*02:01 on the cell surface, ESK1-BiTE showed potent efficacy against multiple leukemias and solid tumors in vitro and in vivo. Interestingly, ESK1-BiTE also induced a robust secondary T cell response on the basis of its high specificity for HER2/Neu epitope 369 in an autologous in vitro setting. Thus, an epitope-spreading effect can potentially be another ESK1-BiTE mechanism of action. A TCRm can also be engineered into a TCRm CAR to redirect T cells.330 An ongoing early phase I clinical trial is aimed at evaluating GPA-TriMAR-T cells, which are TCRm-CAR-T cells targeting gp100209–217/HLA-A*0201, in patients with malignant melanoma (NCT03649529).
Drug combinations can improve therapeutic efficacy. FDA-approved TKIs are not effective in patients with Ph+ ALL. Since high expression levels of WT1 RMF/HLA-A*02:01 are found in Ph+ ALL, a TCRm mAb ESKM alone or in combination with TKIs was used in patients with Ph+ ALL. The results showed that ESKM alone is more effective than TKIs. Furthermore, the combination therapy with ESKM and TKIs showed superior efficacy than monotherapy with only TCRm ESKM or TKIs.331 Many small-molecule agents, such as proteasome inhibitors, histone deacetylase inhibitors, and MEK inhibitors, are capable of upregulating MHC expression and presentation, which facilitate pMHC-targeted tumor therapy.332 For example, our previous study showed that the MEK inhibitor trametinib augmented the antitumor efficacy of EA1 HL-vcMMAE both in vitro and in vivo by increasing MART-126–35 peptide presentation.328
In conclusion, TCRm mAbs, which combine the specificity of TCR recognition with the favorable properties of antibodies, have emerged as promising therapeutics, particularly in cancer treatment. Despite their potential for killing tumor cells specifically, clinical trials to evaluate TCRm mAbs are still pending. Future development of TCRm mAbs will focus on identifying druggable neoantigens and addressing challenges of TCRm mAbs application, including inadequate antigen presentation, lack of MHC internalization, and cross-reactivity with other epitopes.332
Conclusions and future perspectives
Recently, new formats of therapeutic antibodies have been described, and studies on traditional non-IgG immunoglobins, such as IgA and IgM, are undergoing a renaissance.333,334 Here we summarize the advances of new-format therapeutic antibodies in cancer therapy, including antibody conjugates (e.g., ADCs, AOCs, and radiolabeled antibodies), bispecific/multispecific antibodies, immunocytokines, antibody fragments (e.g., Fabs, scFvs, and VHH domains), and scaffold proteins. Miniaturization and multifunctionalization represent two major directions in antibody development. As described in this review, full-length antibodies have been transformed into fragments, e.g., Fabs, scFvs, and VHH domains, and small scaffold proteins (e.g., affibodies and DARPins) have been rationally designed to enhance tumor penetration and facilitate fast serum clearance, which are advantages for their applications in tumor diagnostic imaging. Radiolabeled VHH domains are promising in vivo imaging probes when combined with traditional imaging techniques (PEG/SPECT).335 Moreover, the small size and robustness of antibody fragments show superior local performance, for example, eye injection of ranibizumab (Lucentis, an anti-VEGF Fab) and the subcutaneous injection of sonelokinab (an anti-IL-17 A/F VHH).336,337
The multifunctionalization of therapeutic antibodies includes antibody derivatives (e.g., ADCs and AOCs) and antibody fusion proteins (e.g., multispecific antibodies and immunocytokines). ADCs and AOCs utilize payload-induced cytotoxicity and oligonucleotide functionality, respectively, in combination with the exquisite targeting ability of the antibody to show improved biodistribution profiles, attacking tumor cells via multiple mechanisms of action. An Fc-enhancing technique, such as P329G LALA mutation for ADCC enhancement and E430G for CDC enhancement, can further increase the potency of multifunctionalized antibodies in the treatment of solid tumors.338,339 Multispecific antibody-based strategies have been extensively explored to leverage the local TME in an antigen-dependent manner, such as T cell costimulation, engagement of innate and adaptive immune cells, simultaneous blockade of two immune checkpoints, and targeting multiple antigens to increase tumor selectivity. The poor infiltration of immune effector cells and complex immunosuppressive TME in solid tumors requires combination therapies consisting of multispecific antibodies with other immunomodulatory agents, for example, immune checkpoint blockers, personalized neoantigen vaccines, and oncolytic virus.161
Soluble TCRs, TCRm antibodies, and their derivatives (e.g., ImmTAC molecules) can recognize highly tumor-specific HLA-restricted peptides (e.g., p53 and KRAS), which are considered undruggable targets of therapeutic antibodies.340,341 The rapid development of second-generation sequencing, single-cell RNA sequencing, spatial omics, and integrated bioinformatics analysis enables an in-depth and comprehensive understanding of malignant tumor tissues and will lead to an era of antibody-based intracellular antigen targeting and precise tumor therapy.
References
Hafeez, U., Gan, H. K. & Scott, A. M. Monoclonal antibodies as immunomodulatory therapy against cancer and autoimmune diseases. Curr. Opin. Pharmacol. 41, 114–121 (2018).
Buss, N. A., Henderson, S. J., McFarlane, M., Shenton, J. M. & de Haan, L. Monoclonal antibody therapeutics: history and future. Curr. Opin. Pharmacol. 12, 615–622 (2012).
Kaplon, H. & Reichert, J. M. Antibodies to watch in 2021. MAbs 13, 1860476 (2021).
Golay, J. & Introna, M. Mechanism of action of therapeutic monoclonal antibodies: promises and pitfalls of in vitro and in vivo assays. Arch. Biochem. Biophys. 526, 146–153 (2012).
Weiner, G. J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15, 361–370 (2015).
Chau, C. H., Steeg, P. S. & Figg, W. D. Antibody-drug conjugates for cancer. Lancet 394, 793–804 (2019).
Birrer, M. J., Moore, K. N., Betella, I. & Bates, R. C. Antibody-drug conjugate-based therapeutics: state of the science. J. Natl Cancer Inst. 111, 538–549 (2019).
Strebhardt, K. & Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 8, 473–480 (2008).
Zhao, P. et al. Recent advances of antibody drug conjugates for clinical applications. Acta Pharm. Sin. B 10, 1589–1600 (2020).
Dovgan, I., Koniev, O., Kolodych, S. & Wagner, A. Antibody-oligonucleotide conjugates as therapeutic, imaging, and detection agents. Bioconjug. Chem. 30, 2483–2501 (2019).
Steiner, M. & Neri, D. Antibody-radionuclide conjugates for cancer therapy: historical considerations and new trends. Clin. Cancer Res. 17, 6406–6416 (2011).
Kreitman, R. J. et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 32, 1768–1777 (2018).
Yang, Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J. Clin. Investig. 125, 3335–3337 (2015).
Labrijn, A. F., Janmaat, M. L., Reichert, J. M. & Parren, P. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 18, 585–608 (2019).
Goebeler, M. E. & Bargou, R. C. T cell-engaging therapies - BiTEs and beyond. Nat. Rev. Clin. Oncol. 17, 418–434 (2020).
Przepiorka, D. et al. FDA approval: blinatumomab. Clin. Cancer Res. 21, 4035–4039 (2015).
Gleason, M. K. et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 123, 3016–3026 (2014).
Li, B. et al. CD89-mediated recruitment of macrophages via a bispecific antibody enhances anti-tumor efficacy. Oncoimmunology 7, e1380142 (2017).
Hutmacher, C. & Neri, D. Antibody-cytokine fusion proteins: Biopharmaceuticals with immunomodulatory properties for cancer therapy. Adv. Drug Deliv. Rev. 141, 67–91 (2019).
Waldmann, T. A. Cytokines in cancer immunotherapy. Cold Spring Harb. Perspect. Biol. 10, a028472 (2018).
Panelli, M. C. et al. Forecasting the cytokine storm following systemic interleukin (IL)-2 administration. J. Transl. Med. 2, 17 (2004).
Neri, D. Antibody-cytokine fusions: versatile products for the modulation of anticancer immunity. Cancer Immunol. Res. 7, 348–354 (2019).
Chames, P., Van Regenmortel, M., Weiss, E. & Baty, D. Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 157, 220–233 (2009).
Kholodenko, R. V., Kalinovsky, D. V., Doronin, I. I., Ponomarev, E. D. & Kholodenko, I. V. Antibody fragments as potential biopharmaceuticals for cancer therapy: success and limitations. Curr. Med. Chem. 26, 396–426 (2019).
Stahl, S. et al. Affibody molecules in biotechnological and medical applications. Trends Biotechnol. 35, 691–712 (2017).
Srivastava, P. K. Neoepitopes of cancers: looking back, looking ahead. Cancer Immunol. Res. 3, 969–977 (2015).
He, Q., Jiang, X., Zhou, X. & Weng, J. Targeting cancers through TCR-peptide/MHC interactions. J. Hematol. Oncol. 12, 139 (2019).
Jones, H. F., Molvi, Z., Klatt, M. G., Dao, T. & Scheinberg, D. A. Empirical and rational design of T cell receptor-based immunotherapies. Front. Immunol. 11, 585385 (2020).
Chang, A. Y. et al. A therapeutic T cell receptor mimic antibody targets tumor-associated PRAME peptide/HLA-I antigens. J. Clin. Investig. 127, 2705–2718 (2017).
Dubrovsky, L. et al. T cell receptor mimic antibodies for cancer therapy. Oncoimmunology 5, e1049803 (2016).
Hafeez, U., Parakh, S., Gan, H. K. & Scott, A. M. Antibody-drug conjugates for cancer therapy. Molecules 25, 4764 (2020).
Krop, I. & Winer, E. P. Trastuzumab emtansine: a novel antibody-drug conjugate for HER2-positive breast cancer. Clin. Cancer Res. 20, 15–20 (2014).
An ADC for triple-negative breast cancer. Cancer Discov. 6, OF8 (2016).
Tai, Y. T. et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 123, 3128–3138 (2014).
Okeley, N. M. et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin. Cancer Res. 16, 888–897 (2010).
Phillips, A. C. et al. Characterization of ABBV-221, a tumor-selective EGFR-targeting antibody drug conjugate. Mol. Cancer Ther. 17, 795–805 (2018).
Ricart, A. D. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 17, 6417–6427 (2011).
Xu, Z. et al. Novel HER2-targeting antibody-drug conjugates of trastuzumab beyond T-DM1 in breast cancer: trastuzumab deruxtecan (DS-8201a) and (Vic-)trastuzumab duocarmazine (SYD985). Eur. J. Med. Chem. 183, 111682 (2019).
Hartley, J. A. Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy. Expert Opin. Biol. Ther. 21, 931–943 (2021).
Moldenhauer, G. et al. Therapeutic potential of amanitin-conjugated anti-epithelial cell adhesion molecule monoclonal antibody against pancreatic carcinoma. J. Natl Cancer Inst. 104, 622–634 (2012).
van Der Velden, V. H. et al. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 97, 3197–3204 (2001).
Younes, A. et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 363, 1812–1821 (2010).
Moore, K. N. et al. Safety and activity of mirvetuximab soravtansine (IMGN853), a folate receptor alpha-targeting antibody-drug conjugate, in platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: a phase I expansion study. J. Clin. Oncol. 35, 1112–1118 (2017).
Isakoff, S. J. & Baselga, J. Trastuzumab-DM1: building a chemotherapy-free road in the treatment of human epidermal growth factor receptor 2-positive breast cancer. J. Clin. Oncol. 29, 351–354 (2011).
Tsuchikama, K. & An, Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 9, 33–46 (2018).
Bross, P. F. et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 7, 1490–1496 (2001).
Petersdorf, S. H. et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 121, 4854–4860 (2013).
Castaigne, S. et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 379, 1508–1516 (2012).
Amadori, S. et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J. Clin. Oncol. 34, 972–979 (2016).
Taksin, A. L. et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 21, 66–71 (2007).
van de Donk, N. W. & Dhimolea, E. Brentuximab vedotin. MAbs 4, 458–465 (2012).
Pro, B. et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J. Clin. Oncol. 30, 2190–2196 (2012).
Younes, A. et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 30, 2183–2189 (2012).
Prince, H. M. et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet 390, 555–566 (2017).
Horwitz, S. et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393, 229–240 (2019).
Connors, J. M. et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin’s lymphoma. N. Engl. J. Med. 378, 331–344 (2018).
Harbeck, N. & Gnant, M. Breast cancer. Lancet 389, 1134–1150 (2017).
Barok, M., Joensuu, H. & Isola, J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 16, 209 (2014).
Verma, S. et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 367, 1783–1791 (2012).
Lamb, Y. N. Inotuzumab ozogamicin: first global approval. Drugs 77, 1603–1610 (2017).
Kantarjian, H. M. et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 375, 740–753 (2016).
Deeks, E. D. Polatuzumab vedotin: first global approval. Drugs 79, 1467–1475 (2019).
Pasqualucci, L. & Dalla-Favera, R. Genetics of diffuse large B-cell lymphoma. Blood 131, 2307–2319 (2018).
Sehn, L. H. et al. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J. Clin. Oncol. 38, 155–165 (2020).
Challita-Eid, P. M. et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 76, 3003–3013 (2016).
Patel, V. G., Oh, W. K. & Galsky, M. D. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J. Clin. 70, 404–423 (2020).
Rosenberg, J. E. et al. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J. Clin. Oncol. 37, 2592–2600 (2019).
Ogitani, Y. et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 22, 5097–5108 (2016).
Tamura, K. et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: a dose-expansion, phase 1 study. Lancet Oncol. 20, 816–826 (2019).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. J. Med. 382, 610–621 (2020).
Bianchini, G., Balko, J. M., Mayer, I. A., Sanders, M. E. & Gianni, L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690 (2016).
Sahota, S. & Vahdat, L. T. Sacituzumab govitecan: an antibody-drug conjugate. Expert Opin. Biol. Ther. 17, 1027–1031 (2017).
Kawato, Y., Aonuma, M., Hirota, Y., Kuga, H. & Sato, K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 51, 4187–4191 (1991).
Bardia, A. et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N. Engl. J. Med 380, 741–751 (2019).
Shah, N., Chari, A., Scott, E., Mezzi, K. & Usmani, S. Z. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia 34, 985–1005 (2020).
Markham, A. Belantamab mafodotin: first approval. Drugs 80, 1607–1613 (2020).
Lonial, S. et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 21, 207–221 (2020).
Wang, K., Wei, G. & Liu, D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 1, 36 (2012).
Zammarchi, F. et al. ADCT-402, a PBD dimer-containing antibody drug conjugate targeting CD19-expressing malignancies. Blood 131, 1094–1105 (2018).
Mullard, A. FDA approves ADC Therapeutics’ loncastuximab tesirine, ushering in a new cytotoxic payload. Nat. Rev. Drug Discov. 20, 414 (2021).
Caimi, P. F. et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 22, 790–800 (2021).
Roberts, T. C., Langer, R. & Wood, M. J. A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 19, 673–694 (2020).
Song, E. et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 23, 709–717 (2005).
Yao, Y. D. et al. Targeted delivery of PLK1-siRNA by ScFv suppresses Her2+ breast cancer growth and metastasis. Sci. Transl. Med. 4, 130ra48 (2012).
Su, Y. et al. PSMA specific single chain antibody-mediated targeted knockdown of Notch1 inhibits human prostate cancer cell proliferation and tumor growth. Cancer Lett. 338, 282–291 (2013).
Baumer, N. et al. Antibody-coupled siRNA as an efficient method for in vivo mRNA knockdown. Nat. Protoc. 11, 22–36 (2016).
Nanna, A. R. et al. Generation and validation of structurally defined antibody-siRNA conjugates. Nucleic Acids Res. 48, 5281–5293 (2020).
Peer, D., Zhu, P., Carman, C. V., Lieberman, J. & Shimaoka, M. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proc. Natl Acad. Sci. USA 104, 4095–4100 (2007).
Cuellar, T. L. et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 43, 1189–1203 (2015).
Geall, A. J. et al. Nucleic acid-polypeptide compositions and uses thereof. US Patent Application 16/129,694 (2017).
Sugo, T. et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Controlled Release 237, 1–13 (2016).
Chames, P., Van Regenmortel, M., Weiss, E. & Baty, D. Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 157, 220–233 (2009).
Fava, R. A., Comeau, R. D. & Woodworth, R. C. Specific membrane receptors for diferric-transferrin in cultured rat skeletal myocytes and chick-embryo cardiac myocytes. Biosci. Rep. 1, 377–385 (1981).
Tushir-Singh, J. Antibody-siRNA conjugates: drugging the undruggable for anti-leukemic therapy. Expert Opin. Biol. Ther. 17, 325–338 (2017).
Dominska, M. & Dykxhoorn, D. M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 123, 1183–1189 (2010).
Carter, P. J. & Lazar, G. A. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 17, 197–223 (2018).
Jackman, D. et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 28, 357–360 (2010).
Wheeler, D. L. et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 27, 3944–3956 (2008).
Galon, J. et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964 (2006).
Zhang, L. et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 348, 203–213 (2003).
Li, H., Er Saw, P. & Song, E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell. Mol. Immunol. 17, 451–461 (2020).
Nisonoff, A. & Rivers, M. M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch. Biochem. Biophys. 93, 460–462 (1961).
Suurs, F. V., Lub-de Hooge, M. N., de Vries, E. G. E. & de Groot, D. J. A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 201, 103–119 (2019).
Perez, P., Hoffman, R. W., Shaw, S., Bluestone, J. A. & Segal, D. M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 316, 354–356 (1985).
Staerz, U. D., Kanagawa, O. & Bevan, M. J. Hybrid antibodies can target sites for attack by T cells. Nature 314, 628–631 (1985).
Yuraszeck, T., Kasichayanula, S. & Benjamin, J. E. Translation and clinical development of bispecific T-cell engaging antibodies for cancer treatment. Clin. Pharmacol. Ther. 101, 634–645 (2017).
Chelius, D. et al. Structural and functional characterization of the trifunctional antibody catumaxomab. MAbs 2, 309–319 (2010).
Zeidler, R. et al. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br. J. Cancer 83, 261–266 (2000).
Seimetz, D., Lindhofer, H. & Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 36, 458–467 (2010).
Topp, M. S. et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 16, 57–66 (2015).
Kantarjian, H. et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 376, 836–847 (2017).
Martinelli, G. et al. Complete hematologic and molecular response in adult patients with relapsed/refractory philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: results from a phase II, single-arm, multicenter study. J. Clin. Oncol. 35, 1795–1802 (2017).
von Stackelberg, A. et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J. Clin. Oncol. 34, 4381–4389 (2016).
Curran, E. & Stock, W. Taking a “BiTE out of ALL”: blinatumomab approval for MRD-positive ALL. Blood 133, 1715–1719 (2019).
Jen, E. Y. et al. FDA approval: blinatumomab for patients with b-cell precursor acute lymphoblastic leukemia in morphologic remission with minimal residual disease. Clin. Cancer Res. 25, 473–477 (2019).
Foa, R. et al. Dasatinib-blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N. Engl. J. Med. 383, 1613–1623 (2020).
King, A. C., Pappacena, J. J., Tallman, M. S., Park, J. H. & Geyer, M. B. Blinatumomab administered concurrently with oral tyrosine kinase inhibitor therapy is a well-tolerated consolidation strategy and eradicates measurable residual disease in adults with Philadelphia chromosome positive acute lymphoblastic leukemia. Leuk. Res. 79, 27–33 (2019).
Schwartz, M. S., Jeyakumar, D., Damon, L. E., Schiller, G. J. & Wieduwilt, M. J. A phase I/II study of blinatumomab in combination with pembrolizumab for adults with relapsed refractory B-lineage acute lymph oblastic leukemia: University of California Hematologic Malignancies Consortium Study 1504. J. Clin. Oncol. https://doi.org/10.1200/JCO.2019.37.15_suppl.TPS7064 (2019).
Engelberts, P. J. et al. DuoBody-CD3xCD20 induces potent T-cell-mediated killing of malignant B cells in preclinical models and provides opportunities for subcutaneous dosing. EBioMedicine 52, 102625 (2020).
Bannerji, R. et al. Odronextamab (REGN1979), a human CD20 x CD3 bispecific antibody, induces durable, complete responses in patients with highly refractory B-cell non-Hodgkin lymphoma, including patients refractory to CAR T therapy. Blood 136, 42–43 (2020).
Ravandi, F. et al. Complete responses in relapsed/refractory acute myeloid leukemia (AML) patients on a weekly dosing schedule of vibecotamab (XmAb14045), a CD123 x CD3 T cell-engaging bispecific antibody; initial results of a phase 1 study. Blood 136, 4–5 (2020).
Subklewe, M. Y. et al. Preliminary results from a phase 1 first-in-human study of AMG 673, a novel half-life extended (HLE) anti-CD33/CD3 BiTE (R) (bispecific T-cell engager) in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML). Blood 134, 833 (2019).
Garfall, A. L. et al. Updated phase 1 results of teclistamab, a B-cell maturation antigen (BCMA) x CD3 bispecific antibody, in relapsed and/or refractory multiple myeloma (RRMM). Blood 136, 27 (2020).
Bacac, M. et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin. Cancer Res. 22, 3286–3297 (2016).
Borlak, J., Langer, F., Spanel, R., Schondorfer, G. & Dittrich, C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcgamma receptors. Oncotarget 7, 28059–28074 (2016).
Lemon, B. et al. HPN424, a half-life extended, PSMA/CD3-specific TriTAC for the treatment of metastatic prostate cancer. Cancer Res. 78, 1773 (2018).
Lopez-Albaitero, A. et al. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody. Oncoimmunology 6, e1267891 (2017).
Laszlo, G. S. et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 123, 554–561 (2014).
Friedrich, M. et al. Regression of human prostate cancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol. Cancer Ther. 11, 2664–2673 (2012).
Pillarisetti, K. et al. Teclistamab is an active T cell-redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 4, 4538–4549 (2020).
Boudousquie, C. et al. Polyfunctional response by ImmTAC (IMCgp100) redirected CD8(+) and CD4(+) T cells. Immunology 152, 425–438 (2017).
Hinner, M. J. et al. Tumor-localized costimulatory T-cell engagement by the 4-1BB/HER2 bispecific antibody-anticalin fusion PRS-343. Clin. Cancer Res. 25, 5878–5889 (2019).
Chester, C., Sanmamed, M. F., Wang, J. & Melero, I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood 131, 49–57 (2018).
Sanmamed, M. F. et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin. Oncol. 42, 640–655 (2015).
Wu, L. et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 1, 86–98 (2020).
Shimasaki, N., Jain, A. & Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 19, 200–218 (2020).
Gleason, M. K. et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 11, 2674–2684 (2012).
Wu, J., Fu, J., Zhang, M. & Liu, D. AFM13: a first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J. Hematol. Oncol. 8, 96 (2015).
Reusch, U. et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs 6, 728–739 (2014).
Rothe, A. et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 125, 4024–4031 (2015).
Bartlett, N. L. et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 136, 2401–2409 (2020).
Vallera, D. A. et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin. Cancer Res. 22, 3440–3450 (2016).
Yun, H. D. et al. Trispecific killer engager CD16xIL15xCD33 potently induces NK cell activation and cytotoxicity against neoplastic mast cells. Blood Adv. 2, 1580–1584 (2018).
Habif, G., Crinier, A., Andre, P., Vivier, E. & Narni-Mancinelli, E. Targeting natural killer cells in solid tumors. Cell. Mol. Immunol. 16, 415–422 (2019).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Gong, J., Chehrazi-Raffle, A., Reddi, S. & Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J. Immunother. Cancer 6, 8 (2018).
Peng, S. et al. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol. Cancer 18, 165 (2019).
Kong, T. et al. CD44 promotes PD-L1 expression and its tumor-intrinsic function in breast and lung cancers. Cancer Res. 80, 444–457 (2020).
Postow, M. A., Callahan, M. K. & Wolchok, J. D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 33, 1974–1982 (2015).
Coward, J. et al. A phase I study of AK112, a bispecific antibody that targets PD-1 and VEGF co-expressing T cells, in patients with advanced solid tumors. J. Clin. Oncol. https://doi.org/10.1200/JCO.2020.38.15_suppl.TPS3155 (2020).
Li, L. et al. Tumor-targeting anti-EGFR x anti-PD1 bispecific antibody inhibits EGFR-overexpressing tumor growth by combining EGFR blockade and immune activation with direct tumor cell killing. Transl. Oncol. 14, 100916 (2021).
Liu, J., Song, N. & Yang, Y. Anti-PD-1/Anti-HER2 natural antibody structure-like bispecific antibody of heterodimeric form and preparation thereof. Patent WO2018090950; PCT/CN2017/111310 (2018).
Chae, Y. K. et al. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J. Immunother. Cancer 6, 39 (2018).
Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 38, 255 (2019).
Larkin, J. et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 381, 1535–1546 (2019).
Millward, M. et al. Safety and antitumor activity of AK104, a bispecific antibody targeting PD-1 and CTLA-4, in patients with mesothelioma which is relapsed or refractory to standard therapies. Ann. Oncol. 31, S705–S706 (2020).
Dovedi, S. J. et al. Design and efficacy of a monovalent bispecific PD-1/CTLA4 antibody that enhances CTLA4 blockade on PD-1(+) activated T cells. Cancer Discov. 11, 1100–1117 (2021).
Berezhnoy, A. et al. Development and preliminary clinical activity of PD-1-guided CTLA-4 blocking bispecific DART molecule. Cell Rep. Med. 1, 100163 (2020).
Kraman, M. et al. Dual blockade of PD-L1 and LAG-3 with FS118, a unique bispecific antibody, induces CD8+T-cell activation and modulates the tumor microenvironment to promote antitumor immune responses. Cancer Res. 78, 2719 (2018).
Redmond, W. L., Linch, S. N. & Kasiewicz, M. J. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol. Res. 2, 142–153 (2014).
Galon, J. & Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019).
Kvarnhammar, A. M. et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J. Immunother. Cancer 7, 103 (2019).
Lakins, M. A. et al. FS222, a CD137/PD-L1 tetravalent bispecific antibody, exhibits low toxicity and antitumor activity in colorectal cancer models. Clin. Cancer Res. 26, 4154–4167 (2020).
Moore, G. et al. Anti-PD1 x anti-ICOS bispecific antibody XmAb23104 brings together PD1 blockade and ICOS costimulation to promote human T cell activation and proliferation. J. Immunother. Cancer 5, P347 (2017).
Bol, K. et al. MCLA-145 (CD137XPD-L1): a potent CD137 agonist and immune checkpoint inhibitor that that does not show signs of peripheral toxicity. J. Immunother. Cancer 8, A487 (2020).
Snell, D. et al. Preclinical development and mechanism of action studies of NM21-1480, a PD-L1/4-1BB/HSA trispecific MATCH3 therapeutic clinical candidate. Cancer Res. https://doi.org/10.1158/1538-7445.AM2020-2276 (2020).
Cotton, A. D., Nguyen, D. P., Gramespacher, J. A., Seiple, I. B. & Wells, J. A. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J. Am. Chem. Soc. 143, 593–598 (2021).
Logtenberg, M. E. W., Scheeren, F. A. & Schumacher, T. N. The CD47-SIRP alpha immune checkpoint. Immunity 52, 742–752 (2020).
Ingram, J. R. et al. Localized CD47 blockade enhances immunotherapy for murine melanoma. Proc. Natl Acad. Sci. USA 114, 10184–10189 (2017).
Advani, R. et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med. 379, 1711–1721 (2018).
Dheilly, E. et al. Selective blockade of the ubiquitous checkpoint receptor CD47 is enabled by dual-targeting bispecific antibodies. Mol. Ther. 25, 523–533 (2017).
Fischer, N. et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat. Commun. 6, 6113 (2015).
Shang, L. et al. Abstract 546: Selectively targeting CD47 with bispecific antibody to efficiently eliminate mesothelin-positive solid tumors. Cancer Res. 79, 546 (2019).
Hatterer, E. et al. Targeting a membrane-proximal epitope on mesothelin increases the tumoricidal activity of a bispecific antibody blocking CD47 on mesothelin-positive tumors. MAbs 12, 1739408 (2020).
Wang, Y. et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol. Immunother. 70, 365–376 (2021).
Huang, S. et al. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 73, 824–833 (2013).
Moores, S. L. et al. A novel bispecific antibody targeting EGFR and cMet is effective against EGFR inhibitor-resistant lung tumors. Cancer Res. 76, 3942–3953 (2016).
McDonagh, C. F. et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol. Cancer Ther. 11, 582–593 (2012).
Hubbard, S. R. Structural analysis of receptor tyrosine kinases. Prog. Biophys. Mol. Biol. 71, 343–358 (1999).
Jo, M. et al. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem. 275, 8806–8811 (2000).
Sequist, L. V. et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 21, 373–386 (2020).
Yun, J. et al. Antitumor activity of amivantamab (JNJ-61186372), an EGFR-MET bispecific antibody, in diverse models of EGFR exon 20 insertion-driven NSCLC. Cancer Discov. 10, 1194–1209 (2020).
Geuijen, C. A. W. et al. Unbiased combinatorial screening identifies a bispecific IgG1 that potently inhibits HER3 signaling via HER2-guided ligand blockade. Cancer Cell 33, 922.e10–936.e10 (2018).
Kim, K. J. et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362, 841–844 (1993).
Jain, R. K. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622 (2014).
Kienast, Y. et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin. Cancer Res. 19, 6730–6740 (2013).
Hidalgo, M. et al. First-in-human phase I study of single-agent vanucizumab, a first-in-class bispecific anti-angiopoietin-2/anti-VEGF-A antibody, in adult patients with advanced solid tumors. Clin. Cancer Res. 24, 1536–1545 (2018).
Bendell, J. C. et al. The McCAVE trial: vanucizumab plus mFOLFOX-6 versus bevacizumab plus mFOLFOX-6 in patients with previously untreated metastatic colorectal carcinoma (mCRC). Oncologist 25, e451–e459 (2020).
Hamieh, M. et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 568, 112–116 (2019).
Kugler, M. et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br. J. Haematol. 150, 574–586 (2010).
Vallera, D. A. et al. A bispecific recombinant immunotoxin, DT2219, targeting human CD19 and CD22 receptors in a mouse xenograft model of B-cell leukemia/lymphoma. Clin. Cancer Res. 11, 3879–3888 (2005).
Gardner, R. et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127, 2406–2410 (2016).
Fry, T. J. et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 24, 20–28 (2018).
Bachanova, V. et al. Phase I study of a bispecific ligand-directed toxin targeting CD22 and CD19 (DT2219) for refractory B-cell malignancies. Clin. Cancer Res. 21, 1267–1272 (2015).
June, C. H., O’Connor, R. S., Kawalekar, O. U., Ghassemi, S. & Milone, M. C. CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018).
Mikkilineni, L. & Kochenderfer, J. N. CAR T cell therapies for patients with multiple myeloma. Nat. Rev. Clin. Oncol. 18, 71–84 (2021).
Shah, N. N., Maatman, T., Hari, P. & Johnson, B. Multi targeted CAR-T cell therapies for B-cell malignancies. Front. Oncol. 9, 146 (2019).
Shah, N. N. & Fry, T. J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 16, 372–385 (2019).
Zah, E., Lin, M. Y., Silva-Benedict, A., Jensen, M. C. & Chen, Y. Y. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res. 4, 498–508 (2016).
Sadelain, M., Brentjens, R. & Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 3, 388–398 (2013).
Guedan, S., Calderon, H., Posey, A. D. Jr. & Maus, M. V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 12, 145–156 (2019).
Grada, Z. et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2, e105 (2013).
Shah, N. N. et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat. Med. 26, 1569–1575 (2020).
Schneider, D. et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother. Cancer 5, 42 (2017).
Zhao, W. H. et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 11, 141 (2018).
Brunker, P. et al. RG7386, a novel tetravalent FAP-DR5 antibody, effectively triggers FAP-dependent, avidity-driven DR5 hyperclustering and tumor cell apoptosis. Mol. Cancer Ther. 15, 946–957 (2016).
Zhou, T. et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 583, 609–614 (2020).
Kontermann, R. E. Antibody-cytokine fusion proteins. Arch. Biochem. Biophys. 526, 194–205 (2012).
Muller, D. & Kontermann, R. E. Bispecific antibodies for cancer immunotherapy: current perspectives. BioDrugs 24, 89–98 (2010).
Smith, F. O. et al. Treatment of metastatic melanoma using interleukin-2 alone or in conjunction with vaccines. Clin. Cancer Res. 14, 5610–5618 (2008).
Osenga, K. L. et al. A phase I clinical trial of the hu14.18-IL2 (EMD 273063) as a treatment for children with refractory or recurrent neuroblastoma and melanoma: a study of the Children’s Oncology Group. Clin. Cancer Res. 12, 1750–1759 (2006).
Boyman, O. & Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 12, 180–190 (2012).
Mitra, S. & Leonard, W. J. Biology of IL-2 and its therapeutic modulation: mechanisms and strategies. J. Leukoc. Biol. 103, 643–655 (2018).
Ghasemi, R. et al. Selective targeting of IL-2 to NKG2D bearing cells for improved immunotherapy. Nat. Commun. 7, 12878 (2016).
Rosenberg, S. A. et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 313, 1485–1492 (1985).
Harvill, E. T. & Morrison, S. L. An IgG3-IL2 fusion protein activates complement, binds Fc gamma RI, generates LAK activity and shows enhanced binding to the high affinity IL-2R. Immunotechnology 1, 95–105 (1995).
Gillies, S. D. et al. A low-toxicity IL-2-based immunocytokine retains antitumor activity despite its high degree of IL-2 receptor selectivity. Clin. Cancer Res. 17, 3673–3685 (2011).
Gillessen, S. et al. A phase I dose-escalation study of the immunocytokine EMD 521873 (Selectikine) in patients with advanced solid tumours. Eur. J. Cancer 49, 35–44 (2013).
Chen, X. et al. A novel human IL-2 mutein with minimal systemic toxicity exerts greater antitumor efficacy than wild-type IL-2. Cell Death Dis. 9, 989 (2018).
Amemiya, K. et al. Interleukin-12 induces a Th1-like response to Burkholderia mallei and limited protection in BALB/c mice. Vaccine 24, 1413–1420 (2006).
Duda, D. G. et al. Direct in vitro evidence and in vivo analysis of the antiangiogenesis effects of interleukin 12. Cancer Res. 60, 1111–1116 (2000).
Mocellin, S., Wang, E. & Marincola, F. M. Cytokines and immune response in the tumor microenvironment. J. Immunother. 24, 392–407 (2001).
Valedkarimi, Z., Nasiri, H., Aghebati-Maleki, L. & Majidi, J. Antibody-cytokine fusion proteins for improving efficacy and safety of cancer therapy. Biomed. Pharmacother. 95, 731–742 (2017).
Xu, M. et al. Regulation of antitumor immune responses by the IL-12 family cytokines, IL-12, IL-23, and IL-27. Clin. Dev. Immunol. 2010, 832454 (2010).
Antony, G. K. & Dudek, A. Z. Interleukin 2 in cancer therapy. Curr. Med. Chem. 17, 3297–3302 (2010).
Halin, C. et al. Synergistic therapeutic effects of a tumor targeting antibody fragment, fused to interleukin 12 and to tumor necrosis factor alpha. Cancer Res. 63, 3202–3210 (2003).
Gafner, V., Trachsel, E. & Neri, D. An engineered antibody-interleukin-12 fusion protein with enhanced tumor vascular targeting properties. Int. J. Cancer 119, 2205–2212 (2006).
Clive, K. S. et al. Use of GM-CSF as an adjuvant with cancer vaccines: beneficial or detrimental? Expert Rev. Vaccines 9, 519–525 (2010).
Singhal, S. et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell 30, 120–135 (2016).
Fischer, H. G., Frosch, S., Reske, K. & Reske-Kunz, A. B. Granulocyte-macrophage colony-stimulating factor activates macrophages derived from bone marrow cultures to synthesis of MHC class II molecules and to augmented antigen presentation function. J. Immunol. 141, 3882–3888 (1988).
Steis, R. G. et al. Recombinant human granulocyte-macrophage colony-stimulating factor in patients with advanced malignancy: a phase Ib trial. J. Natl Cancer Inst. 82, 697–703 (1990).
Santoli, D., Clark, S. C., Kreider, B. L., Maslin, P. A. & Rovera, G. Amplification of IL-2-driven T cell proliferation by recombinant human IL-3 and granulocyte-macrophage colony-stimulating factor. J. Immunol. 141, 519–526 (1988).
Dranoff, G. et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl Acad. Sci. USA 90, 3539–3543 (1993).
Jager, E. et al. Granulocyte-macrophage-colony-stimulating factor enhances immune responses to melanoma-associated peptides in vivo. Int. J. Cancer 67, 54–62 (1996).
Ruef, C. & Coleman, D. L. Granulocyte-macrophage colony-stimulating factor: pleiotropic cytokine with potential clinical usefulness. Rev. Infect. Dis. 12, 41–62 (1990).
Kaspar, M., Trachsel, E. & Neri, D. The antibody-mediated targeted delivery of interleukin-15 and GM-CSF to the tumor neovasculature inhibits tumor growth and metastasis. Cancer Res. 67, 4940–4948 (2007).
Daniels, T. R., Delgado, T., Rodriguez, J. A., Helguera, G. & Penichet, M. L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 121, 144–158 (2006).
Dreier, T. et al. Recombinant immunocytokines targeting the mouse transferrin receptor: construction and biological activities. Bioconjug. Chem. 9, 482–489 (1998).
Batova, A., Kamps, A., Gillies, S. D., Reisfeld, R. A. & Yu, A. L. The Ch14.18-GM-CSF fusion protein is effective at mediating antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity in vitro. Clin. Cancer Res. 5, 4259–4263 (1999).
Schwegler, C., Dorn-Beineke, A., Nittka, S., Stocking, C. & Neumaier, M. Monoclonal anti-idiotype antibody 6G6.C4 fused to GM-CSF is capable of breaking tolerance to carcinoembryonic antigen (CEA) in CEA-transgenic mice. Cancer Res. 65, 1925–1933 (2005).
Windbichler, G. H. et al. Interferon-gamma in the first-line therapy of ovarian cancer: a randomized phase III trial. Br. J. Cancer 82, 1138–1144 (2000).
Ebbinghaus, C. et al. Engineered vascular-targeting antibody-interferon-gamma fusion protein for cancer therapy. Int. J. Cancer 116, 304–313 (2005).
Xiang, J., Qi, Y., Luo, X. & Liu, E. Recombinant bifunctional molecule FV/IFN-gamma possesses the anti-tumor FV as well as the gamma interferon activities. Cancer Biother. 8, 327–337 (1993).
Anupa Kudva, S. M. in Neuroblastoma 147–173 (American Cancer Society, 2019).
Lan, Y. et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci. Transl. Med. 10, eaan5488 (2018).
Neri, D. & Sondel, P. M. Immunocytokines for cancer treatment: past, present and future. Curr. Opin. Immunol. 40, 96–102 (2016).
Pluen, A. et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc. Natl Acad. Sci. USA 98, 4628–4633 (2001).
Chames, P., Van Regenmortel, M., Weiss, E. & Baty, D. Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 157, 220–233 (2009).
Cumber, A. J., Ward, E. S., Winter, G., Parnell, G. D. & Wawrzynczak, E. J. Comparative stabilities in vitro and in vivo of a recombinant mouse antibody FvCys fragment and a bisFvCys conjugate. J. Immunol. 149, 120–126 (1992).
Bates, A. & Power, C. A. David vs. Goliath: the structure, function, and clinical prospects of antibody fragments. Antibodies 8, 28 (2019).
Nelson, A. L., Dhimolea, E. & Reichert, J. M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 9, 767–774 (2010).
Elkord, E. et al. Immunological response and overall survival in a subset of advanced renal cell carcinoma patients from a randomized phase 2/3 study of naptumomab estafenatox plus IFN-α versus IFN-α. Oncotarget 6, 4428–4439 (2015).
Hawkins, R. E. et al. A randomized phase II/III study of naptumomab estafenatox + IFNalpha versus IFNalpha in renal cell carcinoma: final analysis with baseline biomarker subgroup and trend analysis. Clin. Cancer Res. 22, 3172–3181 (2016).
Bird, R. E. et al. Single-chain antigen-binding proteins. Science 242, 423–426 (1988).
Ahmad, Z. A. et al. scFv antibody: principles and clinical application. Clin. Dev. Immunol. 2012, 980250 (2012).
Holliger, P., Prospero, T. & Winter, G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc. Natl Acad. Sci. USA 90, 6444–6448 (1993).
Iliades, P., Kortt, A. A. & Hudson, P. J. Triabodies: single chain Fv fragments without a linker form trivalent trimers. FEBS Lett. 409, 437–441 (1997).
Todorovska, A. et al. Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J. Immunol. Methods 248, 47–66 (2001).
Nelson, A. L. & Reichert, J. M. Development trends for therapeutic antibody fragments. Nat. Biotechnol. 27, 331–337 (2009).
Hamers-Casterman, C. et al. Naturally occurring antibodies devoid of light chains. Nature 363, 446–448 (1993).
Kijanka, M., Dorresteijn, B., Oliveira, S. & van Bergen en Henegouwen, P. M. Nanobody-based cancer therapy of solid tumors. Nanomedicine 10, 161–174 (2015).
De Genst, E., Saerens, D., Muyldermans, S. & Conrath, K. Antibody repertoire development in camelids. Dev. Comp. Immunol. 30, 187–198 (2006).
Muyldermans, S., Cambillau, C. & Wyns, L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem. Sci. 26, 230–235 (2001).
Revets, H., De Baetselier, P. & Muyldermans, S. Nanobodies as novel agents for cancer therapy. Expert Opin. Biol. Ther. 5, 111–124 (2005).
Muyldermans, S. Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem 82, 775–797 (2013).
Jovcevska, I. & Muyldermans, S. The therapeutic potential of nanobodies. BioDrugs 34, 11–26 (2020).
Bannas, P., Hambach, J. & Koch-Nolte, F. Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front. Immunol. 8, 1603 (2017).
Wesolowski, J. et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 198, 157–174 (2009).
Harmsen, M. M. & De Haard, H. J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 77, 13–22 (2007).
Duranti, C. & Arcangeli, A. Ion channel targeting with antibodies and antibody fragments for cancer diagnosis. Antibodies 8, 33 (2019).
Vosjan, M. J. W. D. et al. Facile labelling of an anti-epidermal growth factor receptor Nanobody with 68Ga via a novel bifunctional desferal chelate for immuno-PET. Eur. J. Nucl. Med. Mol. Imaging 38, 753–763 (2011).
Huang, L. et al. SPECT imaging with 99mTc-labeled EGFR-specific nanobody for in vivo monitoring of EGFR expression. Mol. Imaging Biol. 10, 167–175 (2008).
Tijink, B. M. et al. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol. Cancer Ther. 7, 2288–2297 (2008).
Pruszynski, M. et al. Targeting breast carcinoma with radioiodinated anti-HER2 nanobody. Nucl. Med. Biol. 40, 52–59 (2013).
Kazemi-Lomedasht, F. et al. Inhibition of angiogenesis in human endothelial cell using VEGF specific nanobody. Mol. Immunol. 65, 58–67 (2015).
Behdani, M. et al. Generation and characterization of a functional Nanobody against the vascular endothelial growth factor receptor-2; angiogenesis cell receptor. Mol. Immunol. 50, 35–41 (2012).
Heukers, R. et al. Targeting hepatocyte growth factor receptor (Met) positive tumor cells using internalizing nanobody-decorated albumin nanoparticles. Biomaterials 35, 601–610 (2014).
Maussang, D. et al. Llama-derived single variable domains (nanobodies) directed against chemokine receptor CXCR7 reduce head and neck cancer cell growth in vivo. J. Biol. Chem. 288, 29562–29572 (2013).
Papadopoulos, K. P. et al. Unexpected hepatotoxicity in a phase I study of TAS266, a novel tetravalent agonistic nanobody (R) targeting the DR5 receptor. Cancer Chemother. Pharmacol. 75, 887–895 (2015).
Hober, S., Lindbo, S. & Nilvebrant, J. Bispecific applications of non-immunoglobulin scaffold binders. Methods 154, 143–152 (2019).
Nygren, P. A. Alternative binding proteins: affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 275, 2668–2676 (2008).
Beste, G., Schmidt, F. S., Stibora, T. & Skerra, A. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc. Natl Acad. Sci. USA 96, 1898–1903 (1999).
Binz, H. K. et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 22, 575–582 (2004).
Hantschel, O., Biancalana, M. & Koide, S. Monobodies as enabling tools for structural and mechanistic biology. Curr. Opin. Struct. Biol. 60, 167–174 (2020).
Feldwisch, J. & Tolmachev, V. Engineering of affibody molecules for therapy and diagnostics. Methods Mol. Biol. 899, 103–126 (2012).
Nilsson, B. et al. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1, 107–113 (1987).
Arora, P., Oas, T. G. & Myers, J. K. Fast and faster: a designed variant of the B-domain of protein A folds in 3 microsec. Protein Sci. 13, 847–853 (2004).
Nygren, P. A. & Skerra, A. Binding proteins from alternative scaffolds. J. Immunol. Methods 290, 3–28 (2004).
Orlova, A. et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 66, 4339–4348 (2006).
Tolmachev, V. Imaging of HER-2 overexpression in tumors for guiding therapy. Curr. Pharm. Des. 14, 2999–3019 (2008).
Wikman, M. et al. Selection and characterization of HER2/neu-binding affibody ligands. Protein Eng. Des. Sel. 17, 455–462 (2004).
Rothe, C. & Skerra, A. Anticalin((R)) proteins as therapeutic agents in human diseases. BioDrugs 32, 233–243 (2018).
Locher, R. et al. Abundant in vitro expression of the oncofetal ED-B-containing fibronectin translates into selective pharmacodelivery of (131)I-L19SIP in a prostate cancer patient. J. Cancer Res. Clin. Oncol. 140, 35–43 (2014).
Albrecht, V. et al. Anticalins directed against the fibronectin extra domain B as diagnostic tracers for glioblastomas. Int. J. Cancer 138, 1269–1280 (2016).
Schlehuber, S. & Skerra, A. Duocalins: engineered ligand-binding proteins with dual specificity derived from the lipocalin fold. Biol. Chem. 382, 1335–1342 (2001).
Walker, R. G., Willingham, A. T. & Zuker, C. S. A Drosophila mechanosensory transduction channel. Science 287, 2229–2234 (2000).
Pluckthun, A. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 55, 489–511 (2015).
Kobe, B. & Kajava, A. V. When protein folding is simplified to protein coiling: the continuum of solenoid protein structures. Trends Biochem. Sci. 25, 509–515 (2000).
Parizek, P. et al. Designed ankyrin repeat proteins (DARPins) as novel isoform-specific intracellular inhibitors of c-Jun N-terminal kinases. ACS Chem. Biol. 7, 1356–1366 (2012).
Martin-Killias, P., Stefan, N., Rothschild, S., Pluckthun, A. & Zangemeister-Wittke, U. A novel fusion toxin derived from an EpCAM-specific designed ankyrin repeat protein has potent antitumor activity. Clin. Cancer Res. 17, 100–110 (2011).
Sha, F., Salzman, G., Gupta, A. & Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 26, 910–924 (2017).
Grebien, F. et al. Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis. Cell 147, 306–319 (2011).
Wojcik, J. et al. Allosteric inhibition of Bcr-Abl kinase by high affinity monobody inhibitors directed to the Src homology 2 (SH2)-Kinase Interface. J. Biol. Chem. 291, 8836–8847 (2016).
Simanshu, D. K., Nissley, D. V. & McCormick, F. RAS proteins and their regulators in human disease. Cell 170, 17–33 (2017).
Khan, I., Spencer-Smith, R. & O’Bryan, J. P. Targeting the alpha4-alpha5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene 38, 2984–2993 (2019).
Spencer-Smith, R. et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 13, 62–68 (2017).
Diem, M. D. et al. Selection of high-affinity Centyrin FN3 domains from a simple library diversified at a combination of strand and loop positions. Protein Eng. Des. Sel. 27, 419–429 (2014).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Donaldson, J. G. & Williams, D. B. Intracellular assembly and trafficking of MHC class I molecules. Traffic 10, 1745–1752 (2009).
Rudolph, M. G., Stanfield, R. L. & Wilson, I. A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24, 419–466 (2006).
Gunnarsen, K. S. et al. Soluble T-cell receptor design influences functional yield in an E. coli chaperone-assisted expression system. PLoS ONE 13, e0195868 (2018).
Froning, K. et al. Computational stabilization of T cell receptors allows pairing with antibodies to form bispecifics. Nat. Commun. 11, 2330 (2020).
Hoydahl, L. S., Frick, R., Sandlie, I. & Loset, G. A. Targeting the MHC ligandome by use of TCR-like antibodies. Antibodies 8, 32 (2019).
Xu, Y., Salazar, G. T. A., Zhang, N. & An, Z. T-cell receptor mimic (TCRm) antibody therapeutics against intracellular proteins. Antibody Ther. 2, 22–32 (2019).
Makler, O., Oved, K., Netzer, N., Wolf, D. & Reiter, Y. Direct visualization of the dynamics of antigen presentation in human cells infected with cytomegalovirus revealed by antibodies mimicking TCR specificity. Eur. J. Immunol. 40, 1552–1565 (2010).
Sim, A. C. N. et al. Defining the expression hierarchy of latent T-cell epitopes in Epstein-Barr virus infection with TCR-like antibodies. Sci. Rep. 3, 3232 (2013).
Pinilla-Ibarz, J. et al. Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein. Leukemia 20, 2025–2033 (2006).
Liu, G. et al. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 64, 4980–4986 (2004).
van den Berg, J. H. et al. Case report of a fatal serious adverse event upon administration of T cells transduced with a MART-1-specific T-cell receptor. Mol. Ther. 23, 1541–1550 (2015).
Bethune, M. T. et al. Isolation and characterization of NY-ESO-1-specific T cell receptors restricted on various MHC molecules. Proc. Natl Acad. Sci. USA 115, E10702–E10711 (2018).
Wang, Q. J. et al. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res. 4, 204–214 (2016).
Malekzadeh, P. et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 129, 1109–1114 (2019).
Zamora, A. E., Crawford, J. C. & Thomas, P. G. Hitting the target: how T cells detect and eliminate tumors. J. Immunol. 200, 392–399 (2018).
Dao, T. et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci. Transl. Med. 5, 176ra133 (2013).
Veomett, N. et al. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin. Cancer Res. 20, 4036–4046 (2014).
Klechevsky, E. et al. Antitumor activity of immunotoxins with T-cell receptor-like specificity against human melanoma xenografts. Cancer Res. 68, 6360–6367 (2008).
Lai, J. et al. Elimination of melanoma by sortase A-generated TCR-like antibody-drug conjugates (TL-ADCs) targeting intracellular melanoma antigen MART-1. Biomaterials 178, 158–169 (2018).
Shen, Y. et al. TCR-mimic antibody-drug conjugates targeting intracellular tumor-specific mutant antigen KRAS G12V mutation. Asian J. Pharm. Sci. 15, 777–785 (2020).
Dao, T. et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol. 33, 1079–1086 (2015).
Zhang, G. et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci. Rep. 4, 3571 (2014).
Dubrovsky, L. et al. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood 123, 3296–3304 (2014).
Chang, A. Y. et al. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opin. Biol. Ther. 16, 979–987 (2016).
Breedveld, A. & van Egmond, M. IgA and FcalphaRI: pathological roles and therapeutic opportunities. Front. Immunol. 10, 553 (2019).
Fereidan-Esfahani, M., Nayfeh, T., Warrington, A., Howe, C. L. & Rodriguez, M. IgM natural autoantibodies in physiology and the treatment of disease. Methods Mol. Biol. 1904, 53–81 (2019).
Renard, E. et al. Site-specific dual-labeling of a VHH with a chelator and a photosensitizer for nuclear imaging and targeted photodynamic therapy of EGFR-positive tumors. Cancers 13, 428 (2021).
Stahl, A. et al. Ranibizumab versus laser therapy for the treatment of very low birthweight infants with retinopathy of prematurity (RAINBOW): an open-label randomised controlled trial. Lancet 394, 1551–1559 (2019).
Honma, M. & Hayashi, K. Psoriasis: recent progress in molecular-targeted therapies. J. Dermatol. 48, 761–777 (2021).
Herter, S. et al. GA101 P329GLALA, a variant of obinutuzumab with abolished ADCC, ADCP and CDC function but retained cell death induction, is as efficient as rituximab in B-cell depletion and antitumor activity. Haematologica 103, e78–e81 (2018).
Wang, X., Mathieu, M. & Brezski, R. J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 9, 63–73 (2018).
Hsiue, E. H. et al. Targeting a neoantigen derived from a common TP53 mutation. Science 371, 1009 (2021).
Douglass, J. et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 6, eabd5515 (2021).
Duell, J. et al. Functionally defective T cells after chemotherapy of B-cell malignancies can be activated by the tetravalent bispecific CD19/CD3 antibody AFM11. J. Immunother. 42, 180–188 (2019).
Zieger, N. et al. Treatment-free intervals mitigate T-cell exhaustion induced by continuous CD19xCD3-BiTE (R) construct stimulation in vitro. Blood 136, 44–45 (2020).
Djebli, N. et al. Population pharmacokinetics and exposure-response analyses for glofitamab in relapsed/refractory B-cell non-Hodgkin lymphoma (R/R NHL): confirmation of efficacy and CRS mitigation in patients with step-up dosing. Blood 136, 1–2 (2020).
Patel, K. et al. Preliminary safety and anti-tumor activity of XmAb13676, an anti-CD20 x anti-CD3 bispecific antibody, in patients with relapsed/refractory non-Hodgkin’s lymphoma and chronic lymphocytic leukemia. Blood 134, 4079 (2019).
Assouline, S. E. et al. Mosunetuzumab shows promising efficacy in patients with multiply relapsed follicular lymphoma: updated clinical experience from a phase I dose-escalation trial. Blood 136, 42–44 (2020).
Westervelt, P. et al. Phase 1 first-in-human trial of AMV564, a bivalent bispecific (2:2) CD33/CD3 T-cell engager, in patients with relapsed/refractory acute myeloid leukemia (AML). Blood 134, 834 (2019).
Doucey, M.-A. et al. ISB 1342: a first-in-class CD38 T cell engager for the treatment of relapsed refractory multiple myeloma. J. Clin. Oncol. 39, 8044 (2021).
Babich, A. et al. Immunoprofiling of normal human donor blood identified potential pharmacodynamic markers for JNJ-63709178 (CD123xCD3) duobody (R) antibody treatment. Blood 128, 5215 (2016).
Steinmetz, A. et al. CODV-Ig, a universal bispecific tetravalent and multifunctional immunoglobulin format for medical applications. MAbs 8, 867–878 (2016).
Topp, M. S. et al. Anti-B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J. Clin. Oncol. 38, 775–783 (2020).
Hagner, P. R. et al. Targeting B-cell maturation antigen (BCMA) with CC-93269, a 2+1 T cell engager, elicits significant apoptosis in diffuse large B-cell lymphoma preclinical models. Blood 134, 1580 (2019).
Lesokhin, A. M. et al. Preliminary safety, efficacy, pharmacokinetics, and pharmacodynamics of subcutaneously (SC) administered PF-06863135, a B-cell maturation antigen (BCMA)-CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma (RRMM). Blood 136, 8–9 (2020).
DiLillo, D. J. et al. A BCMAxCD3 bispecific T cell-engaging antibody demonstrates robust antitumor efficacy similar to that of anti-BCMA CAR T cells. Blood Adv. 5, 1291–1304 (2021).
De Santis, R. Anti-ErbB2 immunotherapeutics: struggling to make better antibodies for cancer therapy. MAbs 12, 1725346 (2020).
Yu, S. et al. A novel asymmetrical anti-HER2/CD3 bispecific antibody exhibits potent cytotoxicity for HER2-positive tumor cells. J. Exp. Clin. Cancer Res. 38, 355 (2019).
Back, J. et al. GBR1302: Effect of CD3-HER2, a bispecific T cell engager antibody, in trastuzumab-resistant cancers. J. Clin. Oncol. 36, 1 (2018).
Lehmann, S. et al. In vivo fluorescence imaging of the activity of CEA TCB, a novel T-cell bispecific antibody, reveals highly specific tumor targeting and fast induction of T-cell-mediated tumor killing. Clin. Cancer Res. 22, 4417–4427 (2016).
Moek, K. L. et al. (89)Zr-labeled bispecific T-cell engager AMG 211 PET shows AMG 211 accumulation in CD3-rich tissues and clear, heterogeneous tumor uptake. Clin. Cancer Res. 25, 3517–3527 (2019).
Deegen, P. et al. The PSMA-targeting half-life extended BiTE therapy AMG 160 has potent antitumor activity in preclinical models of metastatic castration-resistant prostate cancer. Clin. Cancer Res. 27, 2928–2937 (2021).
Hernandez-Hoyos, G. et al. MOR209/ES414, a novel bispecific antibody targeting PSMA for the treatment of metastatic castration-resistant prostate cancer. Mol. Cancer Ther. 15, 2155–2165 (2016).
Drake, C. G. (eds Jingsong Zhang Mark N. Stein Yuanfang Xu Frank A. Seebach Israel Lowy Kosalai Kal Mohan Glenn Kroog Elizabeth Miller Charles G. Drake et al.) A phase I/II study of REGN5678 (Anti-PSMAxCD28, a costimulatory bispecific antibody) with cemiplimab (anti-PD-1) in patients with metastatic castration-resistant prostate cancer. American Society of Clinical Oncology (2020).
Gaspar, M. et al. CD137/OX40 bispecific antibody induces potent antitumor activity that is dependent on target coengagement. Cancer Immunol. Res. 8, 781–793 (2020).
Reusch, U. et al. AFM24, a bispecific EGFR/CD16A innate cell engager with the potential to overcome resistance to current targeted treatments for EGFR-positive malignancies. Cancer Res. 80, 5659 (2020).
Shum, E. et al. Preliminary safety, pharmacokinetics/pharmacodynamics, and antitumor activity of Xmab20717, a Pd-1 X Ctla-4 bispecific antibody, in patients with advanced solid tumors. J. Immunother. Cancer 8, A247–A248 (2020).
Wang, J. et al. A phase 1, open-label study of MGD013, a bispecific DART (R) molecule binding PD-1 and LAG-3 in patients with relapsed or refractory diffuse large B-cell lymphoma. Blood 136, 21–22 (2020).
Laura, L. et al. RG7769 (PD1-TIM3), a novel heterodimeric avidity-driven T cell specific PD-1/TIM-3 bispecific antibody lacking Fc-mediated effector functions for dual checkpoint inhibition to reactivate dysfunctional T cells. Cancer Res. 80, 2270 (2020).
Zhao, H. Y. et al. The preliminary efficacy and safety data of KN046 in patients failed on prior immune checkpoint inhibitors therapy. J. Clin. Oncol. 38, 2 (2020).
Hellmann, M. D. et al. Safety and immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clin. Cancer Res. 27, 2773–2781 (2021).
Kotanides, H. et al. Bispecific targeting of PD-1 and PD-L1 enhances T-cell activation and antitumor immunity. Cancer Immunol. Res. 8, 1300–1310 (2020).
Qiao, Y. et al. Cancer immune therapy with PD-1-dependent CD137 co-stimulation provides localized tumour killing without systemic toxicity. Nat. Commun. 12, 6360 (2021).
Tian, W. et al. Abstract 545: Preclinical development of a bispecific antibody-trap selectively targeting CD47 and CD20 for the treatment of B cell lineage cancer. Cancer Res. 79, 545 (2019).
Wang, Y. et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol. Immunother. 70, 365–376 (2021).
Huang, Y., Zhang, F. & Xi, G. Anti-PD-1/CD47 bispecific antibody and application thereof. WO2019109357; PCT/CN2017/115323 (2019).
Vijayaraghavan, S. et al. Amivantamab (JNJ-61186372), an Fc enhanced EGFR/cMet bispecific antibody, induces receptor downmodulation and antitumor activity by monocyte/macrophage trogocytosis. Mol. Cancer Ther. 19, 2044–2056 (2020).
Roovers, R. et al. Preclinical evaluation of MCLA-158: a bispecific antibody targeting LGR5 and EGFR using patient-derived colon carcinoma organoids. Cancer Res. 77, 32 (2017).
MCLA-128 fights NRG1 fusion-positive cancers. Cancer Discov. 9, 1636 (2019).
Gong, J. F. et al. Preliminary safety, tolerability and efficacy results of Kn026 in combination with Kn046 in patients with Her2 aberrated solid tumors. J. Immunother. Cancer 8, A485–A486 (2020).
Huang, S. et al. Structural and functional characterization of MBS301, an afucosylated bispecific anti-HER2 antibody. MAbs 10, 864–875 (2018).
ZW25 effective in HER2-positive cancers. Cancer Discov. 9, 8 (2019).
Hamblett, K. J. et al. ZW49, a HER2 targeted biparatopic antibody drug conjugate for the treatment of HER2 expressing cancers. Cancer Res. 79, P6–17–13 (2019).
Kundranda, M. et al. Randomized, double-blind, placebo-controlled phase II study of istiratumab (MM-141) plus nab-paclitaxel and gemcitabine versus nab-paclitaxel and gemcitabine in front-line metastatic pancreatic cancer (CARRIE) (vol 31, pg 79, 2020). Ann. Oncol. 31, 1094–1094 (2020).
Girard, N. et al. Phase Ib study of BI 836880, a VEGF/Ang2-blocking nanobody (R), in combination with BI 754091, an anti-PD-1 antibody: Initial results in patients (pts) with solid tumours. Ann. Oncol. 31, S467–S467 (2020).
Li, Y. et al. ABT-165, a dual variable domain immunoglobulin (DVD-Ig) Targeting DLL4 and VEGF, demonstrates superior efficacy and favorable safety profiles in preclinical models. Mol. Cancer Ther. 17, 1039–1050 (2018).
Jimeno, A. et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Investig. N. Drugs 37, 461–472 (2019).
Acknowledgements
This study was supported by Joint Funds of the National Natural Science Foundation of China (Grant No. U20A20409), National Natural Science Foundation of China (Grant No. 82073750), Key research and development project of Zhejiang province (No. 2018C03022), the Fundamental Research Funds for the Central Universities (No. 2020QNA7005) and Zhejiang Province “Qianjiang Talent Plan”.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jin, S., Sun, Y., Liang, X. et al. Emerging new therapeutic antibody derivatives for cancer treatment. Sig Transduct Target Ther 7, 39 (2022). https://doi.org/10.1038/s41392-021-00868-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00868-x
This article is cited by
-
Therapeutic antibodies for the prevention and treatment of cancer
Journal of Biomedical Science (2024)
-
Nanocrystals: an emerging paradigm for cancer therapeutics
Future Journal of Pharmaceutical Sciences (2024)
-
The enhanced antitumor activity of bispecific antibody targeting PD-1/PD-L1 signaling
Cell Communication and Signaling (2024)
-
Repurposing an endogenous degradation domain for antibody-mediated disposal of cell-surface proteins
EMBO Reports (2024)
-
Unveiling the Biosimilar Paradox of Oncologists’ Perceptions and Hesitations in South Korea: A Web-Based Survey Study
BioDrugs (2024)
