
Abstract
Cancer development and its response to therapy are regulated by inflammation, which either promotes or suppresses tumor progression, potentially displaying opposing effects on therapeutic outcomes. Chronic inflammation facilitates tumor progression and treatment resistance, whereas induction of acute inflammatory reactions often stimulates the maturation of dendritic cells (DCs) and antigen presentation, leading to anti-tumor immune responses. In addition, multiple signaling pathways, such as nuclear factor kappa B (NF-kB), Janus kinase/signal transducers and activators of transcription (JAK-STAT), toll-like receptor (TLR) pathways, cGAS/STING, and mitogen-activated protein kinase (MAPK); inflammatory factors, including cytokines (e.g., interleukin (IL), interferon (IFN), and tumor necrosis factor (TNF)-α), chemokines (e.g., C-C motif chemokine ligands (CCLs) and C-X-C motif chemokine ligands (CXCLs)), growth factors (e.g., vascular endothelial growth factor (VEGF), transforming growth factor (TGF)-β), and inflammasome; as well as inflammatory metabolites including prostaglandins, leukotrienes, thromboxane, and specialized proresolving mediators (SPM), have been identified as pivotal regulators of the initiation and resolution of inflammation. Nowadays, local irradiation, recombinant cytokines, neutralizing antibodies, small-molecule inhibitors, DC vaccines, oncolytic viruses, TLR agonists, and SPM have been developed to specifically modulate inflammation in cancer therapy, with some of these factors already undergoing clinical trials. Herein, we discuss the initiation and resolution of inflammation, the crosstalk between tumor development and inflammatory processes. We also highlight potential targets for harnessing inflammation in the treatment of cancer.
Similar content being viewed by others

Introduction
Despite the employment in the clinical setting of a series of strategies for cancer treatment (e.g., surgery, chemotherapy, irradiation, and immunotherapy), cancer-related mortality remains one of the leading causes of death worldwide, accounting for 13% of all human deaths.1 Because cancer is considered a cell-intrinsic genetic disease, most treatment modalities are focused on killing tumor cells directly, with multidrug resistance of cancer cells being a crucial reason for the low efficacy of cancer therapy.2,3 Inflammation has been demonstrated closely associated with all stages of development and malignant progression of most types of cancer, as well as with the efficacy of anti-cancer therapies.4,5,6 In detail, chronic inflammation is involved in immunosuppression, thereby providing a preferred microenvironment for tumorigenesis, development, and metastasis.7 Besides, inflammatory responses can be induced by anti-cancer therapies.8,9 Acute inflammation contributes to cancer cell death by inducing an anti-tumor immune response, while therapy-elicited chronic inflammation promotes therapeutic resistance and cancer progression.
The correlation between inflammation and cancer was firstly suggested by Rudolf Virchow in the mid-19th century, based on observations that cancer originated in sites of chronic inflammation, and that inflammatory cells were abundant in tumor biopsies.10 Nowadays, cancer-related inflammation is considered as a key characteristic of cancer, with a well-established link between chronic inflammation and tumor development.11 In fact, chronic, dysregulated, persistent, and unresolved inflammation has been associated with an increased risk of malignancies, as well as the malignant progression of cancer in most types of cancer.4,5,12 Moreover, growing evidence have implied that the inflammatory tumor microenvironment (TME) is a key determinant for the therapeutic efficacy of conventional chemotherapy (e.g., radiotherapy and chemotherapy) and immunotherapy.2,6 However, acute inflammation induced by exogenous stimulators has been reported to enhance anti-tumor immunity by promoting the maturation and function of dendritic cells (DCs) and the initiation of effector T cells.13
Inflammation involving the innate and adaptive immune systems is known to be the protective immune response for maintaining tissue homeostasis by eliminating harmful stimuli, including damaged cells, irritants, pathogens, and sterile lesions.5,14 Unlike wound healing and infection, the inflammatory response during cancer development has been demonstrated to be non-resolving.14 Furthermore, tumor-extrinsic inflammation is known to be triggered by various factors, including autoimmune diseases, bacterial and viral infections, obesity, smoking, asbestos exposure, and excessive alcohol consumption, all of which have been reported to increase cancer risk and accelerate malignant progression. In contrast, cancer-intrinsic or cancer-elicited inflammation might be caused by cancer-initiating mutations and contribute to tumor progression via the recruitment and activation of inflammatory cells.15,16,17 Both extrinsic and intrinsic inflammation are known to result in immunosuppressive TME, thereby providing a preferred condition for tumor development. Once the inflammatory TME is established, inflammatory factors derived from tumor cells or interstitial cells would induce cell proliferation and prolong cell survival by initially activating oncogenes and subsequently inactivating tumor suppressor genes.15,16
Owing to the relationship between inflammation and tumor,13 harnessing inflammation appears to be an important approach for a more efficient anti-cancer treatment. The powerful chemopreventive effects of non-steroidal anti-inflammatory drugs (NSAIDs), particularly aspirin, have been demonstrated in numerous clinical studies.18,19,20 Administration of statins has also been reported to significantly reduce the risk of development of multiple types of cancer, including breast cancer, colorectal cancer (CRC), and hepatocellular carcinoma (HCC), by exerting anti-inflammatory effects.21,22,23 In addition, increasing the level of specialized proresolving lipid mediators (SPM, e.g., lipoxin A4 (LXA4) and resolvin D1 (RVD1)) and their synthetic pathways was also shown to significantly inhibit the tumor growth.24,25,26 Moreover, enhancing tumor immunity by blocking inhibitory checkpoints or using chimeric antigen receptor T-cell (CAR-T) immunotherapy has shown promising efficacy in certain cancer types.27,28 However, side-effects of these therapies, such as coagulopathy and “cytokine storm” have hindered their full application to cancer therapy,29,30 suggesting that reduction of these harmful immunotherapy-generated inflammation events would be beneficial for the outcome of patients with cancer.
In brief, tumor-related chronic inflammation has been shown to promote immunosuppression of the TME and the development of tumor.13 Thus, a better understanding of the relationship between the dysregulated inflammation and tumor progression would be conducive toward the development of new strategies for combating tumors, and would enhance the efficacy of immunotherapy, chemo- or radiotherapeutic approaches. In this review, we discuss the initiation and resolution of inflammation, crosstalk between tumor development and inflammatory processes, as well as cancer therapeutic approaches by modulating inflammation.
The initiation and resolution of inflammation
Inflammation is known to be a protective response of the host against infection and tissue damage, which can prevent the spread of pathogens or promote tissue repair.31,32 In the early or acute stages of inflammation, pathogen-associated molecular patterns (PAMPs) are recognized by tissue macrophages or mast cells, activating the secretion of pro-inflammatory cytokines, chemokines, vasoactive amines, and eicosanoids, thereby enhancing the immune response.33,34,35 These pro-inflammatory mediators are known to increase vascular permeability, leading to a massive influx of plasma containing antibodies and other soluble components.36 In addition, the injury site has been shown to release a variety of signaling molecules, including chemokines, cytokines, eicosanoids, and adhesion molecules, leading to the recruitment of neutrophils and monocytes.33,37 As the inflammatory response progresses, monocytes and lymphocytes accumulate in the inflammation sites to neutralize harmful substances. Subsequently, inflammatory cells undergo apoptosis and cleared by macrophages. In addition, SPM biosynthesis during the resolution of inflammation, have been reported to prevent the infiltration of neutrophils, reduce the secretion of pro-inflammatory mediators, stimulate macrophages to phagocytose apoptotic neutrophils, remove bacteria, and restore tissue homeostasis.38,39,40,41 At the final stage of the inflammatory cascade, the tissue repair process replaces the inflammatory process, alleviating the inflammatory response and re-establishing tissue homeostasis.39,40 Therefore, the inflammatory process involves different types of cells and mediators, which can regulate cell chemotaxis, migration, and proliferation in a highly-programmed manner.
Acute and chronic inflammation
Inflammation can be divided into two categories according to the length of the disease: acute and chronic inflammation. Acute inflammation is the initial response to harmful stimuli and persists for a couple of days or weeks. The majority of infiltrating inflammatory cells in acute inflammation are granulocytes.38,42,43 Chronic inflammation is characterized by the simultaneous occurrence of destruction and healing of tissues. The main infiltrating immune cells in chronic inflammation sites are macrophages and lymphocytes.44,45 If the pro-inflammatory stimulus is not eliminated during the acute inflammation process, it will lead to chronic inflammation, autoimmunity, tissue fibrosis, and necrosis. The persistence of inflammatory factors and damage to tissues are the key factors of chronic inflammation.46,47 Sustained acute inflammation without obvious symptoms are also known to be a cause of chronic inflammation, such as chronic cholecystitis and chronic pyelonephritis.48 Chronic inflammation has also been demonstrated to be induced by chronic intracellular viral infections, such as infection with Mycobacterium tuberculosis. These pathogens are less virulent but have been found to cause immune responses with no clinical manifestation of acute inflammation.49 Long-term exposure to nondegradable but potentially toxic substances, such as silicosis,50 or persistent immune response against self-tissues could cause autoimmune diseases, e.g., rheumatoid arthritis.41,51 Moreover, insufficient exercise, obesity, gut microbiota disorders, and an “inflammatory diet” (high in meat and fat, and low in fiber and ratio of omega-3/omega-6 fatty acids) are also known to be incentives of chronic inflammation.52,53,54 Chronic inflammation has been linked to many chronic diseases either directly or indirectly, such as atherosclerosis, myocardial infarction, chronic heart failure, Parkinson’s disease, Alzheimer’s disease, asthma, diabetes, psoriasis, osteoporosis, and cancer.55,56,57 Almost 20% of human cancers and infections have been related to chronic inflammation.6,58 Common risk factors associated with cancer development during chronic inflammation are known to include Helicobacter pylori infection in gastric cancer, hepatitis B or C infection in HCC, human papilloma virus (HPV) infection in cervical cancer, and so on.59,60,61
Inflammatory cells
Vascular endothelial cells
Vascular endothelial cells are known to play an important role in the inflammatory process. They are widely distributed in the inner side of the vascular cavity, forming a relatively stable barrier, separating the blood from the subcutaneous tissue. In the early stage of inflammation, they have been shown to regulate the permeability of blood vessels and affect the infiltration of inflammatory cells.62 During inflammation, leukocyte-synthesized and released TNF-α and IL-1 cytokines have been found to promote the pro-inflammatory phenotype of endothelial cells and fibroblasts through the activation of the TNFR/IL-1 pathway and NF-κB signaling.63,64 Activated endothelial cells express adhesion molecules, such as selectins and intercellular adhesion molecule (ICAM)−1, and secrete a large amount of chemokines.65 In addition, immobilization of CXC and CC chemokines on endothelial and matrix glycosaminoglycans was reported to create a chemotactic gradient, leading to the recruitment and extravasation of neutrophils and monocytes.66 More specifically, CXC chemokines, including CXCL8 (IL-8), macrophage inflammatory protein 2 (MIP-2, known also as CXCL2), complement C5a, leucine, and platelet-activating factor (PAF) have been reported to mediate the process of neutrophil infiltration.66,67
Neutrophils
Upon an inflammatory stimulus, numerous immune cells are recruited to the site of inflammation. Among these cells, neutrophils constitute the largest circulating leukocyte population in blood and are critical in defending against microbial pathogens infection.68 Their rapid recruitment to inflammatory sites is known to occur through a multistep adhesion cascade process.69 Initially, circulating neutrophils in circulating blood are “trapped” in blood vessels and migrate along the capillaries to the venule endothelium. This adhesion interaction is known to be mediated by members of the selectin family, such as P- and E-selectins expressed on the surface of endothelial cells. After traumatic stimulation, the surface of vascular endothelial cells rapidly express P-selectin, thus fulfilling the adherence of leukocytes to endothelial cells. The P-selectin glycoprotein ligand 1 (PSGL1) is commonly expressed in all lymphocytes, monocytes, eosinophils, and neutrophils. L-selectin expressed on neutrophils promote their attachment to the surface of the endothelium and sensing of inflammatory mediators, such as CXC chemokines and components of the complement cascade, leading to the activation of integrins.70 Subsequently, adhered neutrophils interact with endothelial transmembrane proteins, including platelet endothelial cell adhesion molecule (PECAM)−1, intercellular adhesion molecule (ICAM)−1, vascular endothelial (VE)-cadherin, and members of the junctional adhesion molecule (JAM) family to penetrate the vascular endothelium and migrate to the site of inflammation.71,72 Neutrophils display a wide range of roles during the inflammatory process, including phagocytosis of microorganisms, production of reactive oxygen species (ROS), secretion of proteases, and formation of neutrophil extracellular traps (NETs).73 These cells are crucial for the resolution of inflammation and reestablishment of tissue homeostasis.74 It has been found that wound healing is delayed in neutrophil depletion murine models,75 and depletion of neutrophils lead to the exacerbation of autoimmune diseases, such as ulcerative colitis,76 suggesting that these cells have pivotal roles during the inflammatory process.
Monocytes
The recruitment of monocytes and their differentiation into macrophages are essential for the onset, progression, and resolution of inflammation. During the onset of the inflammation process, the chemokine monocyte chemotactic protein (MCP)1/CCL2 was found to mediate the recruitment of pro-inflammatory monocytes expressing the chemokine receptor CCR2.77 As the inflammation progresses, the macrophage colony-stimulating factor (M-CSF), which can promote the differentiation of monocytes to macrophages, was significantly upregulated in the inflammation site.78,79 Macrophages have multiple functions and a plastic phenotype in responding to their inflammatory environment: M1 macrophages have a pro-inflammatory phenotype and produce pro-inflammatory factors, whereas M2 macrophages have immunosuppressive effects.80 These immunosuppressive macrophages express elevated 15-lipoxygenase (15-LOX) and transforming growth factor (TGF)-β, thus dampening leukocyte trafficking, promoting efferocytosis and wound repair.81 In addition, SPM were reported to upregulate microRNAs targeting inflammatory genes in macrophages, thereby downregulating the translation of inflammatory cytokines and chemokines.82
Mast cells
Mast cells are long-lived tissue-resident immune cells that play a protective role in limiting infections by microorganisms.83 They are maintained in constant numbers in healthy tissues, whereas their population increases dramatically during inflammation. Among various receptors, TLRs are the most studied pattern recognition receptors known to interact with a multitude of pathogen-associated molecular patterns from microorganisms or damaged cells. Mast cells have 9 types of TLRs84 and express various pro-inflammatory mediators upon activation. For instance, activation of TLR2 has been shown to induce the secretion of TNF, IL-6, IL-13, IL-4, and IL-5, while activation of TLR-4 elicits the expression of TNF, IL-6, IL-13, and IL-1β.85 Importantly, mast cells are known to reside in most tissues, especially located in epithelial barriers exposed to external environmental factors, such as the skin, airways, and gut tract. These locations particularly highlight the importance of mast cells in the initiation and propagation of immune responses.86 Moreover, activated mast cells have also been reported to release histamine and proteases, promoting the production of pro-inflammatory IL-1 family members, including IL-1, IL-6, and IL-33.87
T cells
T cells play a crucial role in antiviral responses through the production of cytokines.88 T cells are activated during inflammation, and differentiate into various T-cell subsets, including T-helper (Th)1, Th2, Th17, and regulatory T (Treg) cells, depending on the cytokines secreted around the inflammation loci. In particular, Th1 cells are derived following stimulation with interferon (IFN)-γ and TNF-α and secrete IFN-γ, TNF-α, and IL-2, whereas Th2 cells are derived in the presence of IL-4 or IL-10 and secrete IL-4, IL-5, IL-9, and IL-13. In addition, Th17 cells, which secrete IL-17, IL-23, and IL-22, are derived in the presence of TGF-β, IL-1β, and IL-6. In contrast, Treg cells are raised in the presence of TGF-β, and secrete immunosuppressive cytokines, including IL-10 and TGF-β. IL-17 is known to stimulate the production of inflammatory mediators, including TNF-α, IL-6, and IL-1β, whereas Treg cells have been shown to effectively regulate the resolution of inflammation. In addition, CD4+ T cells have been reported to promote the production of virus-specific antibodies by activating B cells, whereas CD8+ T cells produce IFN-γ and TNF-α and can kill viral-infected cells.89 T-helper cells are known to produce a variety of pro-inflammatory cytokines and chemokines by activating NF-κB signaling, recruiting lymphocytes and leukocytes to the site of inflammation, where all these immune cells express and secrete additional chemokines and cytokines amplifying the inflammatory process in response to viral infections.90
Dendritic cells
DCs are antigen-presenting cells that sense microbial and capture, process, and present antigens to lymphocytes.91 They stimulate the activation and proliferation of antigen-specific T and B lymphocytes to initiate the adaptive immune response.92,93 DC activation leads to the secretion of pro-inflammatory mediators which include antimicrobial mediators and chemokines, and recruit more immune cells to the site of infection. Also, DCs regulate T cells differentiation into distinct subsets such as Th1, Th2, Th17, and Treg cells.94,95
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) are immature myeloid cells involved in the regulation of acute and chronic inflammatory conditions such as autoimmune and infectious diseases.96 It is known that MDSCs can be recruited into inflamed tissues where they trigger the resolution of inflammation.96 Various studies show MDSCs suppress the activity of immune cells through different mechanisms involving the degradation of L-arginine, the production of ROS, and the secretion of anti-inflammatory cytokines like IL-10 and TGF-β.97 In addition, MDSCs can inhibit T cell activity by downregulating the pro-inflammatory cytokines, such as IL-12 and prostaglandin E2 (PGE2).98
Basophils and eosinophils
Although represent only about 0.5% of all leukocytes in human blood, basophils are important immune cells of both innate and acquired immunity.99,100 Basophils release a variety of pro-inflammatory mediators and cytokines such as IL-4, IL-13, IL-6, IL-9, CCL5, granulocyte-macrophage colony-stimulating factor (GM-CSF), MIP-1, and monocyte chemoattractant protein-1 (MCP-1/CCL2).101 It was demonstrated that basophils can be activated by IL-18 and IL-33.102 Upon stimulation they undergo degranulate, release and synthesize pro-inflammatory, vasodilative, chemotactic, and cytotoxic substances. These cells are crucial for allergy and inflammation. Eosinophils are other innate immune leukocytes and play important roles in host defense against parasitic, viral, fungal, and bacterial infections.103 Moreover, there is emerging evidence that eosinophils have an immune regulatory and homeostatic function. Eosinophils constitutively express 12/15-LOX which is a key enzyme for the synthesis of SPM, thereby promoting the resolution of inflammation.104
Natural killer and B cells
Natural killer (NK) and B cells are also involved in the inflammatory process. For instance, NK cells are important immunosurveillance cells that detect infected, transformed, or stressed cells with their activating receptors NKG2D and NKp46.104 Once activated, NK cells become cytotoxic and release lytic granules (perforin, granzymes) or induce death signals (e.g. TNF-related apoptosis-inducing ligand (TRAIL)/TRAL-R, Fas ligand (Fas-L)/Fas), thereby kill microorganisms.105 B cells are transformed into plasma cells and secrete antibodies to kill microorganisms, in a mechanism called antibody-dependent cell-mediated cytotoxicity (ADCC). Macrophages, B cells, and DCs are also known to activate T cells through antigen cross-presentation.106,107 However, the chemotactic mechanisms driving the recruitment of monocytes to repair tissues as the inflammation progresses, are not well understood.106 Still, the phenotype of monocytes at the site of inflammation has been demonstrated to be dynamically regulated by inflammatory cytokines and mediators. These pro- and anti-inflammatory factors were reported to lead to the production of subpopulations of macrophages with different functional characteristics that regulated the activity of fibroblast cells, matrix metabolism, angiogenesis, and promoted tissue repair processes.78,79
Pro- and anti-inflammatory factors
During the inflammatory response, an extremely complex regulatory network takes place, involving pro-inflammatory cytokines, pro-inflammatory cytokine-releasing cells, and pro-inflammatory cytokine target cells.108 In addition to pro-inflammatory cytokines, there exist many other inflammatory mediators, which are small molecule compounds closely related to the vascular response, nervous system response, and cell hyperplasia response.33 Various inflammatory factors are produced by specialized immune cells, especially tissue-resident macrophages and mast cells, or cells present in local tissues.33 Some inflammatory mediators (e.g., histamine and serotonin) are known to be expressed and stored in the granules of mast cells, basophils, and platelets.35 Whereas, other mediators are formed and circulate in the plasma as inactive precursors. The plasma concentration of these mediators has been demonstrated to increase significantly during acute inflammation due to the increased secretion of precursors.33,109 Inflammatory mediators can be divided into seven groups based on their biochemical properties: vasoactive amines, vasoactive peptides, cytokines, chemokines, fragments of complement components, lipid mediators, and proteolytic enzymes.
Vasoactive amines, including histamine and 5-hydroxytryptamine (5-HT), are mainly released by mast cells. Histamine synthesis occurs through the decarboxylation of the amino acid histidine by an enzyme called L-histidine decarboxylase (HDC), which has been found in mast cells, basophils, and gastric mucosal cells. Likewise, 5-HT is produced by the decarboxylation of tryptophan, and is stored in the granules of mast cells.110 Mast cells have been shown to release histamine and 5-HT when stimulated by physical factors, such as trauma, heat, immune response, and complements. These mediators have complex effects on the vascular system, including increased vascular permeability, vasodilation or vasoconstriction.
Vasoactive peptides, such as substance P, can be stored in secreted vesicles either in their active form or as inactive precursors (e.g., kinin, fibrinopeptide A/B, and fibrin degradation products) that can be processed by proteolytic enzymes.110 Substance P is released by sensory neurons, and has been reported to cause the degranulation of mast cells.111 Other vasoactive peptides are known to be produced by proteolysis of hageman factors, thrombin, or plasmin, and they have been found to cause vasodilation and increase vascular permeability directly or indirectly by inducing the release of histamine from mast cells.112 Hageman factors play a key role in these reactions, acting both as sensors of vascular damage and as inducers of inflammation.113
Cytokines are the major signaling molecules released by inflammatory cells and involved in multiple functions. They are classified into pro-inflammatory cytokines (IL-1, IL-6, IL-15, IL-17, IL-23, TNF-α, and IFN-γ) and anti-inflammatory cytokines (IL-4, IL-10, IL-13, and TGF-β).114 Among them, TNF-α, which is mainly produced by macrophages and mast cells, is one of the earliest and most important inflammatory mediators. TNF-α is known to have multiple roles in the inflammatory response, including the activation of inflammatory cytokines coded by the NF-κB signal pathway, adhesion molecules, gene expression of prostaglandin synthesis pathway enzymes (e.g., cyclooxygenase-2 (COX2)), induction of nitric oxide synthase (iNOS), leading to the activation of endothelium and white blood cells.15,115,116,117,118 It has also been reported to activate neutrophils and lymphocytes, increase the permeability of vascular endothelial cells, regulate the metabolic activities of other tissues, and promote the synthesis and release of other cytokines.119,120 Accordingly, IL-1 and IL-6 are well-known interleukins that participate in the production of ROS and reactive nitrogen species (RNS), and in the synthesis of inflammatory molecules, such as chemokines, integrins, and matrix metalloproteinase (MMP). Macrophages and T cells are the major cell sources of these interleukins. Both IL-1 and IL-6 bind to their respective IL-1R and IL-6R receptors, leading to the activation of NF-κB and JAKs-STAT pathways. Besides, IL-6 is also known to induce the differentiation of B cells for production of antibodies, and promotes the activation, proliferation, and differentiation of T cells.121,122
Chemokines are a family of small (generally 8–10 kDa) signaling peptides that have an important role in the recruitment of inflammatory cells during inflammation.123 They are divided into four families (C, CC, CXC, and CX3C) based on the spacing of their N-terminal cysteines.124 The major secreted chemokines during inflammation, which direct leukocyte migration and influence the activity of infiltrating immune cells, belong to the CC and CXC families. Chemokines have been shown to bind to their G protein-coupled cell-surface receptors (GPCRs) to exert their cellular effects, such as cell movement and activation.125,126,127 Chemokines are mainly released by innate immune cells including neutrophils, mast cells, and eosinophils.127,128 For instance, CCL2 is a chemokine important for the recruitment of monocytes in response to stimuli. Monocytes have been found to migrate to the inflammatory site following CCL2 gradients orchestrated by vascular endothelial cells in response to PAMPs.129,130 Likewise, CXCL12, also known as stromal cell-derived factor-1 (SDF-1), is another well-studied chemokine that contributes to tissue repair by mobilizing mesenchymal stem cells (MSCs) to injury sites trough binding to CXCR4.131 More specifically, binding of CXCL12 to CXCR4 has been reported to lead to the activation of G protein-coupled receptor (GPCR) downstream signaling, such as phosphatidylinositol-3-kinase (PI3K)/mechanistic target of rapamycin (mTOR) and mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling, thereby promoting tissue repair.132
The C3a, C4a, and C5a complement fragments are small soluble peptide fragments that play key roles in the regulation of inflammation.133 They are produced through several complement activation pathways and are known to promote the recruitment of granulocytes and monocytes, as well as induce the degranulation of mast cells.134,135 The activation of the complement system is triggered by either the classical, the lectin, or the alternative pathway.136 These pathways share the same proteolytic cascade processes, thereby promoting the cleavage of inactivated complement components into active peptide fragments.137 In this regard, the C3 and C5 complement components are cleaved into the C3a, C3b, C5a, and C5b fragments.138 In particular, C3a is known to be an anaphylatoxin with chemotactic activity and participates in the production of pro-inflammatory cytokines. Meanwhile, C3b produced by C3 convertases has been shown to function as a constituent of C5 convertases. The C5b fragment bind to target cells, allowing the assembly of the membrane attack complex (C5b-9 or MAC), thus leading to the lysis of target cells. In addition, the C3a and C5a complement fragments have been demonstrated to orchestrate inflammatory responses by binding to their C3aR and C5aR receptors, respectively.139,140 Once these complement fragments bind to their receptors, target cells show various responses, such as migration, antigen presentation, and the production of inflammatory mediators.141,142
Lipid mediators constitute one of the most important category of mediators of inflammation.143 Following the activation of cells by Ca2+ ions, cytoplasmic phospholipase A2 generates arachidonic (AA) and lysophosphatidic acid from phosphatidylcholine. Subsequently, AA is metabolized either by cyclooxygenases (COX1 and COX2) to produce prostaglandins and thromboxanes, or by lipoxygenases (LOXs) to produce leukotrienes and lipoxins.144,145,146 The PGE2 and prostacyclin I2 (PGI2) in turn cause vasodilatation, with PGE2 being also an effective stimulator of hyperalgesia and fever.144 In addition, AA-derived lipoxins and dietary omega-3 fatty acid-derived resolvins and protectins have been reported to inhibit inflammation and promote the resolution of inflammation, and tissue repair.146,147
Several proteolytic enzymes, including elastin and cathepsin, have multiple roles in inflammation, partly by degrading extracellular matrix (ECM) and basement membrane proteins.148,149,150 These proteases play an important role in many processes, including host defense, tissue remodeling, and leukocyte migration. Elastin is a dominant ECM protein in the lung, and plays a significant role in cardiovascular inflammation and calcification.151 Fragments of elastin have been shown to induce the differentiation of Th1 cells and enhance the release of IFN-γ from T cells.152 Cathepsins are lysosomal proteases composed of 11 members, including cathepsin B, C, F, H, K, L, O, S, V, W, and Z in humans,153,154 and have been reported to be involved in immune modulation through the proteolysis of the ECM and extracellular or membrane-bound proteins.155 For instance, cathepsin S is a cysteine protease involved in the cleavage of elastin and generation of bioactive elastin peptides.156 Cathepsin B (CatB) is known to have a role in the production of mature IL-1β and TNF-α,157 while cathepsin K (CatK) contributes to the activation of TLR9.158,159
The regulation of inflammatory mediators can occur at multiple levels, including transcription, mRNA translation, posttranslational modification, and mRNA degradation.160 The posttranscriptional regulation has been shown to play an important role in controlling the expression of these mediators for the normal and efficient initiation and resolution of inflammation. The mRNA of many inflammatory mediators has been shown to be unstable, partly because of the presence of AU-rich elements in their 3′-untranslated regions. Moreover, it has been found that binding of many RNA binding proteins to these AU-rich elements could lead to the regulation of the stability or translation of mRNA. For example, the infusion of E. coli in primates has been demonstrated to trigger the rapid release of TNF and other inflammatory cytokines, with their serum levels reaching a peak in 90 min and then quickly disappearing.161,162 The mRNA transcripts encoding these proteins contain regulatory elements that direct their degradation or translational inhibition. When these transcripts are induced, they synthesize proteins in a short time interval, and then are destroyed or silenced, thus preventing the overexpression of pro-inflammatory proteins.163,164,165
IFN-γ is another important pro-inflammatory factor in the inflammatory process that has been reported to both activate and inhibit the mRNA transcription of inflammatory genes.166 For example, in macrophages, IFN-γ was shown to induce the transcription of the pro-inflammatory gene ceruloplasmin,167 and also initiated the formation of IFN-γ-activated translation inhibitor (GAIT) complexes, which interact with the GAIT element located on the 3′-UTR of mRNAs encoding a variety of pro-inflammatory genes, including ceruloplasmin.168 Binding of the GAIT complex to the GAIT element in the mRNA inhibits the protein translation machinery and consecutively protein synthesis. As a result, ceruloplasmin has been found to be secreted by IFN-γ-activated macrophages for about 16 h, after which its levels are decreased.169 This fine adjustment of inflammatory mediators ensures the timely termination of the inflammatory process and returns the expression levels of pro-inflammatory factors and proteins to baseline levels.
Inflammation resolution
In order to prevent the progression from acute-resolving to persistent-chronic inflammation and allow organs to restore homeostasis, the inflammatory reaction must be actively resolved, to prevent further tissue damage.170,171 Historically, it was believed that the resolution of inflammation was a passive process involving the dilution of chemokine gradients over time, thus stopping the recruitment of circulating leukocytes to the site of injury.172 However, extensive work over the past few decades has revealed that the resolution of inflammation is a programmed active process, and deficiency in any of its components might lead to overactive, uncontrolled chronic inflammation. With the advancement of lipidomics and metabolomics, Serhan et al. showed that the resolution phase of inflammation is regulated by a class of enzymatically produced SPM.36,173 They also introduced the quantitative resolution indices (defined as follows: Tmax: time point when PMN infiltration to maximum; Ѱmax: PMN maximum number; T50: time point when PMNs reduction to half of Ѱmax; Ѱ50: 50% of Ѱmax; Ri: resolution interval, time interval from Tmax to T50; K50: the rate of PMN reduction from Tmax to T50), which indicated reduced PMN infiltration and shortened resolution interval after SPM biosynthesized.174
Upon inflammation initiation, the pro-inflammatory lipid mediators (LM) are produced, whereas during the resolution of inflammation, the SPM are abundantly biosynthesized, i.e., LM class switching occurs (Fig. 1). SPM have been shown to not only function as signals for the termination of the inflammatory response, but also promote macrophages to engulf dead cells to accelerate the resolution of inflammation. Removal of apoptotic neutrophils by macrophages is a prerequisite for macrophage efferocytosis, which has been reported to coincide with the biosynthesis of SPM, reducing the expression of pro-inflammatory lipid mediators and cytokines.

SPM biosynthesis and their roles in the resolution of inflammation. a SPM including lipoxins, E-series resolvins, D-series resolvins, protectins (neuroprotectin D1), and maresins are biosynthesized from arachidonic acid (AA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA). The main structures of these SPM and their receptors are depicted. b Anti-inflammatory lipid mediators (LM) class are produced to help restore tissue homeostasis during the resolution of inflammation
Lipoxins are a class of metabolite derivatives of AA via the lipoxygenase pathway. In the vascular cavity, leukocyte-derived 5-LOX is known to catalyzes the synthesis of leukotriene A4 (LTA4) which is then catalyzed by platelet-derived 12-LOX to produce LXA4 or LXB4.175 Lipoxins have also been found to be catalyzed by 15-LOX in epithelial cells, monocytes, and eosinophils to produce intermediate products, followed by their catalysis by 5-LOX in neutrophils to produce LXA4 or LXB4. Serhan et al. discovered that aspirin-mediated acetylation of COX2 inhibited the production of prostaglandin but led to the conversion of AA to 15(R)-hydroxyeicosatetraenoic acid (15(R)-HETE), a substrate used for the synthesis of 15-epi-lipoxins (AT-lipoxins). In addition, lipoxins have been reported to promote the resolution of inflammation through activating lipoxin receptor (ALX)/N-formyl peptide receptor (FPR)−2 receptors to antagonize pro-inflammatory mediators, resulting in decreased recruitment of leukocytes and deactivation of NF-κB, decreased production of superoxide, and diminished production of pro-inflammatory chemokines/cytokines.176,177
Resolvins are another series of important endogenous SPM. Depending on their source, they either contain E-series (RvE) derived from eicosapentaenoic acid (EPA), D-series (RvD) from docosahexaenoic acid (DHA) and aspirin-triggered resolvin D (AT-RvD1-RvD6), or Dp series (RvDn-3DPA) derived from docosapentaenoic acid (DPA).178,179,180 Resolvins are synthesized through interactions between the activities of aspirin-acetylated COX2 and LOX in endothelial cells and leukocytes. In particular, RvE1 is known to activate downstream pathways by binding to ERV1/ChemR23, leading to the inhibition of the NF-κB pathway in inflammatory cells.181 Meanwhile, RvD1 and RvD3 exert their bioactions through binding to ALX/FPR2 and DRV1/GPR32, respectively, whereas RvD2 and RvD5 activate their DRV2/GPR18 and DRV1/GPR32 receptors, respectively.178,182,183 Of interest, the activation of the RvE1-ERV1/ChemR23 axis has been shown to promote the apoptosis and macrophage-mediated phagocytosis of neutrophils, while reducing the production of pro-inflammatory cytokines.184,185,186 Recently, we also found that RvDP5 inhibited the infiltration of neutrophils and promoted the phagocytic function of macrophages through the ALX/FPR2 receptor.187
In addition to lipoxins and resolvins, additional families of SPM, namely protectins and maresins have also been identified. Protectin is derived from metabolites of DHA epoxidation. Because of its potent protective effect in central neurons, protectin D1 is also called neuroprotectin.174,188,189 The synthesis of protectins and maresins is known to be catalyzed by 15-LOX and 5-LOX.190 Protectin D1 has been shown to promote macrophage phagocytosis of apoptotic polymorphonuclear leukocytes (PMNs) and regulate the infiltration of leukocytes.191 In macrophages, DHA has been demonstrated to generate intermediate 14S-HDHA from the lipoxygenation of 12-LOX, subsequently generating maresins, including MaR1 and MaR2, through epoxidation or hydrolysis.192,193 Moreover, it has been shown that cytochrome oxidase could also convert DHA into 19,20-EDP, which could also be quickly converted by soluble epoxide hydrolase into the inactive 19,20-Di DHPA metabolite. Maresin 1 is the first member of the maresin family and has been found to restrict the infiltration of neutrophils, enhance the phagocytosis of apoptotic neutrophils and necrotic cells by macrophages, downregulate the production of pro-inflammatory mediators, inhibit the activation of NF-κB, and increase the content of regulatory T cells. Maresin 1 has also been demonstrated to increase the level of intracellular cyclic adenosine monophosphate to promote the resolution of inflammation; however, the receptors that interact with maresins and their mechanisms of action remain unclear.194,195
Recently, a group of peptide-conjugated SPM, such as protectin conjugates in tissue regeneration (PCTRs), maresin conjugates in tissue regeneration (MCTRs), and resolvin conjugates in tissue regeneration (RCTRs) have been discovered.196,197,198 These SPM are biosynthesized from DHA. PCTR1, an endogenous novel peptide-conjugated SPM that exerts anti-inflammatory and proresolving functions during infection have been recently discovered.199,200 More specifically, PCTR1 is produced from DHA in leukocytes and reduce pro-inflammatory factors in serum and improve the survival rate of mice during LPS-induced acute inflammation. In addition, PCTR1 reduced the levels of LPS-induced serum linoleic acid (LA), AA, and PGE2 via the activation of ALX.199 Moreover, PCTR1 promotes the conversion of LA to AA through the upregulation of LPS-inhibited fatty acid desaturase 1/2 (FADS1/2) and elongation of very long-chain fatty acids 2 (ELOVL2), and the inhibition of the expression of phospholipase A2 (PLA2) resulted in the increased intrahepatic content of AA. Similar to PCTRs, MCTRs act as anti-inflammatory and proresolving agents and contribute to host defense, organ protection, and pain modulation.201,202 MCTRs are produced by macrophages and participate in phagocytosis and tissue repair and regeneration.203 However, the signaling mechanisms underlying MCTRs functions have not yet been fully established. RCTRs are new chemical signals that play a role in inflammation resolution and tissue regeneration.204 RCTRs stimulate macrophage phagocytosis and efferocytosis of apoptotic PMNs, limiting PMN chemotaxis and infiltration, and exert anti-inflammatory and proresolving actions during resolution of inflammation.
The important protective actions of SPM in both acute inflammation (e.g., sepsis,199 lung injury,180 and ischemia-reperfusion injury200) and chronic inflammation (e.g., asthma205 and Alzheimer’s disease206) have been widely reported. These SPM are known to display protective effects through direct antimicrobial actions in host defense or indirectly by controlling the pathogen-mediated inflammation. For instance, the production of endogenous protectin D1 was shown to be increased during the infection of hosts with influenza viruses to directly inhibit the pathogenicity of influenza by interacting with the RNA replication machinery. Accordingly, insufficient upregulation of protectin D1 led to more efficient viral replication and host demise, whereas treatment of the host with exogenous protectin D1 could restore the inhibition of viral replication and improve host survival. Protectin D1 has pivotal roles in regulating the resolution of inflammation through limiting further recruitment of neutrophils, promoting macrophage clearance of apoptotic neutrophils and efferocytosis, accelerating tissue regeneration, and reducing pain and viral pathogenicity.
The coronavirus disease (COVID-19), an infection caused by a novel ssRNA betacoronavirus (SARS-CoV-2), has spread worldwide and already affected the population in more than 180 countries. Almost all patients with COVID-19 are clinically presented with fever, cough, and dyspnea.207,208 Moreover, infection with SARS-CoV-2 has been associated with systemic inflammation, and increased serum levels of inflammatory cytokines and chemokines, including IL-1, IL-7, IL-8, IL-9, IL-10, GM-CSF, and IFN-γ, which have been associated with disease severity and death.209,210,211
As mentioned above, SPM play an important antimicrobial protective role during infection and control pathogen-mediated inflammation, with deregulation of protectin D1 leading to viral replication and systemic inflammation. Thus, we speculated that SARS-CoV-2 might be able to suppress the production of SPM to facilitate its replication, and treatment with exogenous SPM, such as protectin D1 might inhibit its replication, prevent the subsequent cytokine storm, and improve survival rate.212 However, whether the production of SPM is changed during infection with SARS-CoV-2 remains unknown. In addition, whether the mechanism by which SARS-CoV-2 might suppress the production of endogenous SPM is to be evaluated. Further investigations of the functional role of SPM in patients with COVID-19 are imperative before SPM could be applied as potential agents.
Inflammation and immunity
The TLR, NOD-like receptor (NLR), and retinoic acid-inducible gene-like receptor (RLR) families are 3 major pathogen sensor families of innate immunity.213,214 The binding of pathogenic or endogenous dangerous factors to these receptors, including TLR and NLR is known to activate a variety of downstream intracellular signaling pathways, leading to the release of a plethora of pro-inflammatory mediators, including cytokines, chemokines, leukotrienes, and eicosanoids. Members of the TLR family can identify bacteria, viruses, fungi, and protozoa. The function of NLR is to detect bacteria, whereas the function of RLR is to sense viruses. These innate immune receptors are essential for the protection of the host from bacterial, viral, fungal, and protozoan infections, as well as in response to cellular stress. Despite the diversity of the TLR family, all members are known to be involved in the inflammatory response and the progression of certain inflammatory diseases, such as atherosclerosis.214 Eleven TLRs (TLR1~TLR11) have been identified in human cells, of which TLR1, TLR2, TLR4, TLR5, TLR6, TLR10, and TLR11 are expressed on the cell surface, whereas TLR3, TLR7, and TLR9 are expressed in the cytoplasm.214 Briefly, TLRs have 3 structural features: (1) an extracellular region composed of leucine; (2) a transmembrane region; (3) and a cytoplasmic region homologous to the IL-1 receptor, namely the Toll/interleukin-1 receptor (TIR), which is essential for the activation of its downstream signaling pathway.215,216 The first step after the activation of TLRs is their dimerization or synergy with other receptors, as well as their redistribution and aggregation on the cell surface. The downstream signaling pathways of TLRs include myeloid differentiation factor (MyD88), IL-1R-related protein kinase (IRAK), TRAF6, TAK1, TAB1, and TAB2. Studies have shown that there are 2 signaling pathways involved in the process of the transduction of the TLR signal, namely the MyD88-dependent and MyD88-independent pathways.217,218 Activation of TLR has been found to promote the effects of IRAK (IL-1RI-related protein kinase) 4 and IRAK1 through the recruitment of MyD88 adaptor molecules.219 More specifically, IRAK4 was reported to phosphorylate IRAK1, with IRAK1 further interacting with TRAF6 to form a complex, leading to the phosphorylation of TAK1 and TAB2. Then, TAK1 was shown to phosphorylate the inhibitory kappa B kinase (IKK) complex, leading to the activation of the NF-κB transcription factor and promoting the production of inflammatory cytokines, adhesion molecules, and prostaglandins.220 Both TLR3 and TLR4 were reported to interact with 2 TIR adaptor proteins, TIRAP and TRIF, independent of the MyD88 adaptor protein.221 Although TIRAP plays a role in the signaling pathways of TLR2 and TLR4, it does not participate in the signaling pathways of other TLRs. In contrast, TLR3 and TLR4 could be directly linked to TRIF, inducing the transduction of downstream factors without passing through MyD88.222
The NOD-like receptors are pattern recognition receptors in the cytoplasm. The structural features of NLRs are as follows: (1) the central nucleotide-binding oligomerization region (NACHT), which is very important for the oligomerization and activation of NLRs, is a structure shared by the NLR family; (2) the N-terminal effector binding region, that is, the N-terminal protein-protein interaction domain, such as caspase activation and recruitment domain (CARD); and (3) the C-terminal enrichment leucine-containing repeats (LRRs).223,224 The NLR family consists of 22 types of intracellular pattern recognition molecules, which are distributed in a variety of tissue cells, including monocytes, macrophages, T cells, B cells, dendritic-like cells of the small intestine, and Paneth cells. Human NLRs are divided into the following 5 categories: NLRA, NLRB, NLRC, NLRP, and NLRX.225 It has been shown that NOD1 and NOD2 recruit receptor-interacting protein (RIP)-2 through CARD-CARD interactions, thereby activating the NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways. The combination of the PYD-containing NLRP protein and CARD-containing apoptosis-associated speck-like (ASC) protein has been shown to cause the activation of caspase-1, promoting an inflammatory reaction.226 In addition, large amounts of NLR could form inflammasomes. An inflammasome is a multiprotein complex, including NLRs, the ASC intracellular adaptor protein, and caspase-1, which is known to regulate the processing and activation of IL-1β, IL-18, IL-33, and other pro-inflammatory cytokines, and participates in the activation of the innate immune system.227 As a result, a complex network is formed between NLR members and inflammatory factors to synergistically regulate the immune response and strengthen the inflammatory response and antimicrobial ability. Excessive activation of NLRP3 or gene mutations have been reported to cause severe inflammatory diseases, such as familial cold-induced autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and neonatal onset multisystem inflammatory disorder or chronic infantile neurologic cutaneous and articular syndrome (NOMID/CINCA).228
In addition to promoting the maturation and extracellular release of the IL-1β and IL-18 pro-inflammatory cytokines, the activation of the inflammasome could also induce pyroptosis.229 Pyrolysis, also known as cell inflammatory necrosis, is a kind of programmed cell necrosis, which is manifested by the continuous expansion of cells until the rupture of the cell membrane, which causes the release of cell contents and activates a strong inflammatory response.230,231 The cysteine protease caspase-1 is known to cut the linker between the amino and carboxyl ends of gasermin D (GSDMD), thereby regulating cell pyrolysis.232 Recent studies have shown that GSDMD is associated with familial mediterranean fever (FMF),233 neonatal multiple inflammatory disease,234 nonalcoholic steatohepatitis235, and multiple sclerosis in murine models.236 In addition, the NLR protein inflammasome-mediated inflammatory response has also been involved in the occurrence and development of certain tumors.237 For example, NLRP3 was involved in the inflammatory response caused by anti-tumor drugs.238,239 Therefore, these proteins could become targets for the future development of novel drugs and improved treatment approaches.
It is widely known that NF-κB is a family of key transcription factors participating in innate immunity and inflammation, and also involved in the occurrence and development of tumors.240 There are 5 proteins in this family in mammals, namely: RelA p65), RelB, c-Rel, NF-κB1 (p50), and NF-κB2 (p52). Their N-terminus has a highly conserved Rel homology region (RHR).115 The most common NF-κB dimer is the heterodimer composed of RelA and p50. The NF-κB signaling pathway is activated by extracellular signaling factors, including TNF-α, IL-1β, IL-2, IL-6, IL-8, IL-12, iNOS, COX2, chemokines, adhesion molecules, colony-stimulating factor, and many more.241,242 The activation of NF-κB results in the phosphorylation and degradation of inhibitors of NF-κB (IκBs), and the subsequent nuclear translocation of NF-κB and upregulation of numerous pro-inflammatory chemokines and cytokines, such as the IL-1, IL-6, IL-8, and PGE2, further promoting the inflammatory response.243,244,245,246 In addition, studies have shown that one of the important functions of NF-κB in tumor cells is to promote cell survival through the induction of the expression of antiapoptotic genes, such as BCL2, and promotion of the expression of the hypoxia-inducible factor-1α (HIF-1α).126,246 These cumulative evidence have linked innate immunity to inflammation and hypoxia. Studying the role of NF-κB in leukocytes infiltrated in the site of inflammation will further strengthen our understanding of the link between immunity and inflammation.
The activation of the NF-κB pathway has been demonstrated to be rapidly induced by viral and bacterial infections, necrotic cell components, and pro-inflammatory cytokines during immune responses.247 However, the NF-κB pathway is also known to accelerate cell proliferation, inhibit apoptosis, and promote cell migration and invasion. Notably though, in the TME, NF-κB is constitutively activated, promoting the expression of cytokines, chemokines, and growth factors.248 These results highlighted the important roles of NF-κB in inflammation and cancer progression.
Inflammation roles in cancer: promoting VERSUS inhibiting
As mentioned above, inflammation has been demonstrated to not only promote the immune response but also lead to immune surveillance. The innate and adaptive immunity involved in the inflammatory response were also shown to play an important role in cancer initiation, progression, and metastasis.5
The acute inflammatory response is the first line of defense against external infection or injury, promoting innate and adaptive immune responses. The innate immune system consists of evolutionary diversified hematopoietic cells, such as neutrophils, macrophages, DCs, mast cells, and so on.249 These cell populations are known to participate in the phagocytosis of pathogens, microorganisms, and necrotic substances, thereby mediating the resolution of inflammation. Moreover, as antigen-presenting cells, DCs and macrophages have also been shown to provide specific antigens to T cells for recognition and activation of the adaptive immune response.250 Therefore, acute inflammation could eliminate pathogens and protect the body from infections.
However, if the acute inflammatory reaction does not resolve in time, it could be transformed into chronic inflammation resulting in an immunosuppressive microenvironment with a large number of immunosuppressive cells (M2 macrophages, MDSCs, Treg cells, etc.) and cytokines.5,15 These changes have been reported to promote the activation of oncogenes, DNA and protein damage, release of ROS, and affect multiple signaling pathways including NF-κB, K-RAS, and P53, leading to chronic diseases including cancer.5 In addition, epigenetic alterations, such as DNA methylation, histone modification, chromatin remodeling, and noncoding RNA, play an important role in the transformation of inflammation into cancer as well as in the occurrence, development, invasion, metastasis, and drug resistance of cancer.247,251,252,253,254 Particularly worth mentioning is the histone lactylation in macrophages that might promote inflammatory resolution and tumor immune escape,251,255,256,257,258 but whether lactylation could modify other proteins and their effects on protein functions remain unknown. Moreover, lactic acids in the inflammatory microenvironment are known to play an important role in promoting the progression of inflammation and cancer via acting on immune cells (such as cytotoxic T cells (CTLs), DCs, and APCs),259,260,261 and immunosuppressive cells (such as M2-macrophages, MDSCs, and Treg cells).262,263,264
Meanwhile, gene mutations would lead to abnormal cellular proliferation, but immune cells could recognize specific antigens on these tumor cells, and stimulate immune response to clear them. Multiple inflammatory factors and signaling pathways, such as 5-LOX, COX-2, TGF-β, and VEGF are well-known molecules linking inflammation and chronic diseases.252 What’s more, the dysregulation of inflammatory molecules or factors is often caused by aberrant inflammatory pathways that including NF-κB, MAPK, JAK-STAT, and PI3K/AKT, etc (Fig. 2). For instance, more than 500 cancer-related genes are regulated by the NF-κB signaling pathway.247

Inflammatory signaling pathways involved in cancer development. Intracellular signaling pathways involved in inflammation and tumor development are activated via distinct receptors at the cell membrane. Subsequent downstream signaling events activate several well-characterized pathways: NF-κB, MAPK, JAK-STAT, and PI3K-AKT. These pathways regulate various inflammatory factors
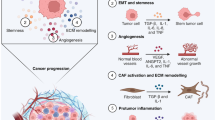
The immune system is known to broadly participate in cancer-related inflammation that could precede the development of malignancy or be induced by oncogenic changes, thus generating a pro-tumor inflammatory environment.9 In this section we retrospectively present the relationship of the innate and adaptive immune system during response to inflammation with tumor initiation and progression and discuss the outstanding questions that remain to be answered (Fig. 3).

The relationship between inflammation and cancer development. During acute inflammatory responses (left panel): after tumor antigen uptake or activation by TLR agonist, mature DCs can regulate anti-tumor immune responses by inducing inflammatory responses via multiple mechanisms, such as cross-presenting the tumor antigens and priming tumor-specific CD8+ T cells, polarizing immune cells toward tumor suppression (e.g., M1 polarization of TAMs), recruiting NK cells which can sustain T-cell responses. However, if the acute inflammatory reaction does not resolve in time, it subsequently transforms into chronic inflammation (right panel). In this microenvironment, cancer cells can not only hijack DCs to prevent TAA presentation, but also recruit a large number of immunosuppressive cells (e.g., MDSCs, Treg cells, Breg cells, M2-TAMs, N2-TANs, and Th2 cells) by secreting various cytokines, chemokines, and inflammatory mediators. In turn, these immunosuppressive cells provide a rich proangiogenic and pro-tumoral microenvironment, and prevent the innate immunity and T-cell anti-tumor immunity
Cancer-promoting inflammation
Inflammation has been recognized closely involved in cancer, substantially contributing to the development and progression of malignancies.253 Chronic inflammation driven by immune cells and molecular signaling pathways has been reported to lead to the susceptibility of the human body to various cancers. Evidence has shown that up to 25% of cancers are related to chronic inflammatory diseases; however, the exact mechanism underlying this connection remains unclear.254 Certain chronic inflammatory diseases have been recognized as precancerous lesions of tumors in clinical terms. For instance, the inflammatory bowel disease (IBD) is well known as a precancerous lesion of CRC. Clinical observations have shown that IBD might evolve into malignant tumors in the span of several years to decades. Furthermore, chemical induction of IBD is known to be a classical method for induction of CRC in mice.265,266 IBD-associated colon cancer shows different DNA methylation level compared with sporadic colon cancers.267 Besides, a research using the single-cell multiomics sequencing of human CRC has revealed that epigenetic inheritance plays an important regulatory role in the occurrence and development of CRC.268
Some viruses and bacteria-induced chronic inflammatory diseases have also been reported to contribute to carcinogenesis. For instance, infection with H. pylori was demonstrated to lead to gastritis and stomach cancer.269,270 IBD was not sufficient to induce CRC in the absence of intestinal microbiota or microbial products,271 and the use of a common antimicrobial additive could increase colonic inflammation and colitis-associated colon tumorigenesis in mice.272
Infection with HBV was also shown to induce chronic hepatitis, which could progress to primary HCC.4,273 Cervical carcinoma is known to be caused by infection with HPV.274 Besides, microbiota are known to directly or indirectly (via their metabolites, such as polysaccharide β-dextran, LPS, deoxycholic acid (DCA), short-chain fatty acid (SCFA), butyrate, and propionate) affect the differentiation and function of immune cells (e.g., M2 macrophages,275 neutrophils,276 Treg cells,277,278,279 T cells,280 and NKT cells281), potentially altering their effects on cancer. Therapies targeting gut microbiota showed significant improvement on immunotherapy efficacy.282
In addition, some chronic autoimmune diseases have also been associated with tumorigenesis.283 Moreover, in the chronic inflammatory microenvironment, a large number of immunosuppressive cells inhibit the killing function of T cells and lead to immune escape, thus promoting tumor formation. Evidence has suggested that chronic inflammatory stimuli increase the risk of cancer, promote tumor progression, and support metastatic spread.253 Thus, inflammatory cells and cytokines during chronic inflammation might act as tumor promoters affecting cell survival, proliferation, invasion, and angiogenesis. Here we focus on the role of the activated innate and adaptive immunity, as well as systemic inflammation in promoting cancer.
Cancer-promoting systemic inflammation
Systemic inflammation is a cardinal characteristics of malignancy, and it is well-established that patients with systemic inflammation always have poorer outcomes.284 It has been indicated that low-grade systemic inflammation related to obesity or depression promotes the progression of cancer by remodeling of the immune cell landscape.285,286
Obesity, characterized by chronic and low-grade systemic inflammation, increases the risk of many cancer types, such as breast cancer,287 CRC,288 liver cancer,289, and ovarian cancer,290 is associated with poor outcomes.291 Obesity-associated inflammation is always triggered by excessive nutrients, and is primarily localized in specialized metabolic tissues such as white adipose tissue.292 Tumor cell biology is directly affected by multiple cellular players present in the adipose tissue microenvironment, which have diverse morphologies and play opposing functions.293 White adipose tissues secrete a variety of inflammatory molecules that can potently fuel cancer, such as TNF-α, IL-6, IL-1β, and CCL2.294,295 These cytokines can establish chronic inflammatory environment by recruiting lymphocytes and macrophages. For example, cancer-associated adipocytes can facilitate radio-resistance by secreting IL-6.296 However, brown adipose tissue possesses a therapeutic potential role against cancer. The activation of the brown adipose metabolism can improve insulin resistance, reduce inflammation and increase the secretion of anti-inflammatory molecules, creating an anti-tumorigenic microenvironment.297 On the other hand, during the process of white adipocytes transdifferentiate into pink adipocytes in breast tissue, mammary epithelial secretory cells will lose the expression of peroxisome proliferator-activated receptor (PPAR)-γ, resulting in a pro-tumorigenic microenvironment.298 Given the key role of inflammation in obesity-associated cancers,299,300 anti-inflammatory therapies in obese patient populations may be beneficial to cancer prevention and treatment.
Chronic stress triggered by depression, anxiety, or loneliness/social isolation can also cause corresponding changes in immune function and inflammatory response, which are implicated in tumorigenesis and cancer development.286,301,302 First, chronic stress stimulates the classical neuroendocrine system, such as hypothalamic-pituitary-adrenal (HPA) axis, and the sympathetic nervous system (SNS), and causes a dysfunction of the prefrontal cortex and the hippocampus under stress. Subsequently, stress hormones produced during the activation of HPA axis and the SNS can facilitate tumorigenesis and cancer development through a variety of mechanisms, such as inducing DNA damage, accelerating p53 degradation, and regulating the TME. Chronic stress can also activate the inflammatory response, and the interaction between inflammatory cells and tumor cells to form the inflammatory TME, and promote tumorigenesis.303,304 Moreover, chronic stress can also selectively suppress the CTLs-mediated cellular immunity and interferon production, and dampen immune surveillance, thereby increase the risks of metastasis and decrease the effectiveness of anti-tumor therapy.305,306
In consideration of a long-term inflammatory response and the decline of the immune surveillance capabilities are implicated in tumorigenesis and cancer development,4 clinical management of systemic inflammation is essential for prevention and treatment of cancer.
Cancer-promoting inflammation in innate immunity
The innate immune response is the non-specific defense function that is formed during the development and evolution of lineage after birth.249,307 Innate immune cells including NK cells, macrophages, neutrophils, DCs, and innate lymphoid cells (ILCs), are known to be involved in the initial response to tissue injury and can promote or prevent tumor initiation and progression.4 Meanwhile, they have also been reported to facilitate cellular transformation and malignant development. Understanding the mechanism by which the innate immune system affects cancer formation and progression is crucial for developing strategies to treat cancer. In addition, other innate immune cells, such as mast cells, and MDSCs found in the TME are also involved in cancer promotion.5
Inflammation is often accompanied by the recruitment of fibroblasts and the induction of fibrosis. Cancer-associated fibroblast (CAFs) are responsible for the deposition of collagen and various ECM components in the TME, where they have been shown to facilitate cancer cell proliferation and angiogenesis.308,309 Moreover, CAFs are also known to have a critical immune function, as they produce numerous cytokines and chemokines, including osteopontin, CXCL1, CXCL2, CXCL12, CXCL13, IL-6, IL-1β, and CCL-5.310,311 It has been reported that during tumorigenesis, fibroblasts sense the alterations in tissue architecture caused by the increased proliferation of neighboring epithelial cells, and respond to these changes by producing pro-inflammatory mediators.312 In addition, CAFs have also been found to be activated during therapy-induced hypoxia, producing abundant TGF-β and numerous chemokines, including CXCL13.313 Subsequently, the CAF-secreted TGF-β inhibits the activation of NK cells and CTLs, and suppresses the differentiation of Treg cells and immunosuppressive plasmocytes.314,315 Besides, CAF-secreted CXCL13 was demonstrated to mediate the recruitment of B cells into androgen-deprived prostate cancer, resulting in hormone resistance.313,316 In breast cancer, the CAF-secreted CCL2 was shown to lead to the recruitment of macrophages to the TME.317 Furthermore, activated CAFs expressing the fibroblast activation protein-α (FAP) were also reported to attenuate anti-tumor immunity in established Lewis lung carcinoma mouse model.318
As tissue-resident sentinel cells, mast cells are first in the line of defense among innate immune cells responding to allergens, pathogens, or other pro-inflammatory and toxic agents.319 Upon activation, mast cells were found to not only rapidly release a series of biologically active mediators stored in their cytoplasmic granules, such as histamine, serotonin, TNF-α, proteoglycans, and various proteases, but could also release de novo synthesized lipid mediators (e.g., prostaglandins and leukotrienes), cytokines, chemokines, leukotrienes, and growth factors.320 In turn, many mast cell-released mediators, such as IL-1β, IL-6, TNF-α, PGE2, LTB4, and leukotriene D4 (LTD4), can attract or activate other immune, endothelial, epithelial, neuronal, and stromal cells. Accumulation of DCs has been observed in inflammatory diseases and multiple types of cancer, such as CRC, prostate cancer, pancreatic adenocarcinomas, esophagus squamous cell carcinomas (ESCC), non-small-cell lung cancer (NSCLC), and several types of hematologic neoplasms, such as non-Hodgson’s lymphoma, and follicular lymphoma.321,322,323 Furthermore, high density of mast cells was shown to be predictive of poor clinical outcome in CRC, lung cancer, and pancreatic cancer.324,325,326,327 Evidence have shown that mouse mast cells highly express programmed death ligand-1 (PD-L1) and PD-L2,328 indicating an additional mast cell-driven mechanism enhancing the pro-tumorigenic effect of the programmed death-1 (PD-1)/PD-L1 axis. Collectively, through shaping an inflammatory TME for immune escape, mast cells have been suggested to promote tumor development and progression. Mast cells were reported to promote the growth of endothelial cells and angiogenesis by either producing heparin or releasing lysozyme to dissolve the surrounding stromal tissue and then promote tumor growth and metastasis.319,320 Certain substances in the granular composition of mast cells could also promote collagen lysozyme produced by fibroblasts and tumor cells, and indirectly caused the disintegration of collagen, thus promoting tumor invasion and metastasis.329
Tumor-associated macrophages (TAMs), mainly M2-type macrophages are known to inhibit the killing function of T cells, and secret cytokines to maintain the immunosuppressive state in the TME, thus acting as a paradigm for tumor-promoting inflammation.330,331 In addition, M2-TAMs were found to regulate the distortion of adaptive responses, angiogenesis, cell proliferation, deposition, and remodeling of stromal cells in the TME.332 The functional reprogramming of TAMs was shown to be orchestrated by stimulations and signals from cancer cells, T and B cells. In general, the M2-like properties of TAMs resemble those of immune-tolerant macrophages, although there are multifarious phenotypes and signaling pathways in different tumors.332
Neutrophils act as the first line of defense of the body against infection and have been shown to respond to diverse inflammatory stimulation, with their persistent infiltration being a hallmark of chronic inflammation that contributes to tissue damage.333,334 They are regarded as “kamikaze” cells, sacrificing themselves while killing invading pathogens through the employment of multiple mechanisms: phagocytosis, secretion of ROS, hyperchlorous acid, and antimicrobial proteins (e.g., defensin, lysozyme, elastase, and cathepsin), or extrusion of DNA to generate NETs. In addition to playing tumor-promoting roles in the context of innate immune inflammation and tumor initiation, tumor-associated neutrophils (TANs) have also been reported to promote tumor progression by suppressing the function of the adaptive immune response in the TME.335,336,337 The increase of TANs has been found that negatively correlated with severe disease and poor outcome of patients in a broad variety of neoplasias.338 Similar to TAMs, TANs are also classified into N1 anti-tumor and N2 pro-tumor subsets, with neutrophil polarization influencing the role they play in the TME.308 N2 neutrophils were found to induce the switch of tumor angiogenesis during early tumor promotion, remodel the extracellular matrix of TME to promote tumor cell growth, regulate the biological behaviors of tumor cells, maintain the immunosuppressive state in the microenvironment by secreting various cytokines (e.g., iNOS, VEGF, Arg-1, CCL17, PGE2, and B/MMP9 gelatinase), and promote the invasion and metastasis of tumor cells at the later stage.339
Eosinophils, which are characterized by large secretory granules within their cytoplasm, are known to regulate the immune response through the presentation of antigens to T cells and release of immunomodulatory molecules.340 Responding to diverse stimuli, they have been reported to migrate to sites of inflammation, synthesizing and secreting several immunomodulatory molecules, including granule proteins that could potentially kill tumor cells.103 Alternatively, eosinophils could secrete both proangiogenic and matrix-remodeling soluble mediators that could facilitate tumor progression. Tumor-associated tissue eosinophilia (TATE) has been observed in many hematological and solid malignancies (e.g., colon, breast, colorectal, nasopharyngeal, oral, gastric, and head and neck cancers) with a generally good prognostic value,341 suggesting the involvement of eosinophils in the anti-tumor response. In these types of cancers, eosinophils were observed to display cytotoxicity via the secretion of granule proteins, TNF-α, and granzyme A,342 and shaped the TME via the induction of CD8+ T cells, promotion of vascular normalization, and shifting of TAMs into a pro-inflammatory (M1) phenotype.343 It was also found that IL-10- and IL-12-activated eosinophils suppressed the growth of prostate cancer cells in vitro and upregulated the expression of adhesion molecules, potentially limiting cancer metastasis.344 However, TATE has also been associated with poor prognosis in Hodgkin lymphoma and oral squamous cell carcinomas (OSCCs).345,346 Besides, tumor cell-derived thymic stromal lymphopoietin (TSLP) was shown to facilitate proliferation, increase the production of anti-inflammatory cytokines (e.g., IL-10, IL-4, IL-5, and IL-13), and decrease the expression of CD80 and CD86 in eosinophils, thus enhancing the proliferation of cervical cancer cells. Eosinophils were demonstrated to promote tumor metastasis and angiogenesis via the secretion of MMP9, VEGF, FGF, and PDGF, while polarize TAMs into a pro-tumor (M2) phenotype via the production of IL-4 and IL-13.347 Thus, the function of eosinophils might depend on the cellular composition of the TME in different cancer types.
The accumulation of MDSCs in peripheral tissues in cancer is well known along with their pro-tumor role in tumor progression. More specifically, MDSCs are known to produce Arg-1, iNOS, IL-10, TGF-β, and COX-2 to inhibit the proliferation and function of T cells.348 In addition to their immune suppressive function, MDSCs were shown to promote tumor progression by remodeling of the TME and facilitated tumor angiogenesis by producing cytokines, such as VEGF and FGF.349 In addition, MDSCs were observed to participate in the formation of premetastatic lesions, and metastasis by infiltrating primary tumors. They inhibited cellular senescence in spontaneous prostate cancer by antagonizing the IL-1α signaling pathway. Moreover, the recruitment of g-MDSCs promoted IBD and contributed to the initiation and development of CRC.350
Cancer-promoting inflammation in adaptive immunity
The adaptive immune response which occurs after the innate immune response, is a specific response of lymphocytes to antigen stimulation, followed by the immune memory effect.351 When antigen-presenting cells (APCs) present antigens to T cells, the T-cell receptor (TCR) recognizes the antigen and activates the secretion of tumor-killer molecules, such as IFN-γ and granzymes with the action of synergistic stimulatory molecules. Meanwhile, helper T cells secrete cytokines to activate B cells, which produce antibodies, mediating the ADCC.249,352 Generally, adaptive immune responses are known to inhibit tumorigenesis and progression. However, some types of T cells have been shown to mainly participate in adaptive immune responses, promoting tumor progression. In fact, Th2, Th17, and Treg cells have often been associated with tumor progression and unfavorable prognosis.249
T-helper 2 (Th2) cells are known to not only regulate protective type 2 immune responses to extracellular pathogens, such as helminthes, but also contribute to chronic inflammatory diseases including asthma, allergy, as well as cancer. Increasing evidence have demonstrated a crucial role of Th2 cells in orchestrating the progress and metastasis of tumors.353 In addition, Th2 cells and their cytokines were shown to construct an inflammatory TME involving M2-TAMs and promote tumor metastasis in breast cancer.354 For example, Th2 cells are known to produce IL-4, IL-5, and IL-13, and hence are able to regulate immunity. High levels of Th2 cell-derived cytokines were detected in tumor sites of patients with breast cancer, with the levels of IL-4 and the amount of tumor-infiltrating CD4+ T cells being positively correlated with tumor progression, as well as with metastasis to sentinel lymph nodes,15,355,356 highlighting the clinical relevance of Th2 cells in the pathogenesis of breast tumors. Through the secretion of IL-4, Th2 cells were also shown to regulate the polarization and function of M2 macrophages in the TME.
Th17 cells are a specific subset of T-helper lymphocytes characterized by the high production of IL-17. Th17 cells have been associated with tumor prognosis. More specifically, Th17 cells have been reported to promote tumor growth by inducing angiogenesis and exerting immunosuppressive functions. In contrast, Th17 cells were also demonstrated to recruit immune cells into tumors, activate effector CD8+ T cells, directly convert them toward the Th1 phenotype, and produce IFN-γ to kill tumor cells.357 Moreover, specific IL-17+ γδT-cell subsets were observed to play an unexpected role in driving tumor development and progression.358 They induce an immunosuppressive microenvironment and promote angiogenesis by producing various cytokines as regulatory Th17/Treg/Th2-like cells.358 Moreover, these pro-tumoral IL-17+γδ-T cells can suppress the maturation and function of DCs and subsequently inhibit the anti-tumor adaptive immunity by the PD-1/PD-L1 pathway.358,359,360
Studies have revealed that Treg cells could inhibit the maturation of DCs, as well as block the phagocytosis of tumor cells and the expansion of CTLs, which leads to immune surveillance and tumor progression.361 Treg cells were shown to promote the development and progression of tumors by inhibiting the anti-tumor immunity in TME. In particular, Treg cells were reported to lead to immune suppression by inhibiting co-stimulatory signals by CD80 and CD86 through the cytotoxic T-lymphocyte antigen-4 (CTLA-4), secreting inhibitory cytokines, and directly killing effector T cells.362 Treg cells have been shown to be chemoattracted to the TME by chemokines, such as chemokine receptors (CCR4)-CCL17/22 and CXCR3-CCL9/10/11, where they are activated to inhibit anti-tumor immune responses.363 Indeed, a high accumulation of Treg cells in various types of cancer is associated with poor survival.364
Regulatory B (Breg) cells represent a subset of B cells with immunosuppressive properties.365 According to the expression of tumor surface markers, the production of soluble factors, and the characteristics of promoting tumor growth, a variety of human and animal tumors of the Breg subtype have been identified. Although the phenotypic markers of different tumors have been reported to be different, the typical phenotypes of both human and mouse were shown to be concentrated in memory CD27+ and transitional CD38+ B cells, exhibiting the same phenotype as plasma cells (e.g., IgA+CD138+ and IgM+CD147+).366,367 In both human and mouse studies, Breg cells were observed to exhibit their specific immunosuppressive effects through the secretion of cytokines, such as IL-10 and TGF-β, or through the upregulation of immunomodulatory ligands, such as PD-L1 and CTLA-4, which could attenuate the response of T and NK cells and enhance the pro-tumor effect of Treg cells, MDSCs, and TAMs.368
Cancer-inhibiting inflammation
Although chronic inflammation might lead to tumorigenesis, most inflammatory cells are known to kill pathogens, promote tissue repair, and inhibit tumor growth. Both innate and adaptive immune responses inhibit tumor initiation and progression.369 More specifically, the immune system can recognize and destroy nascent tumor cells in a process called cancer immune surveillance, which plays an important role in cancer prevention.15 Recently, data obtained from a large number of studies in murine models and patients with cancer provided convincing evidence that specific innate and adaptive immune cell types, effector molecules, and pathways could sometimes work together as endogenous tumor suppression factors.362 However, in many cases, tumor-associated inflammation, mainly supported by innate immune cells, was reported to contribute to tumor growth. The initial innate activation is known to trigger the secretion of inflammatory, regenerative, and anti-inflammatory cytokines, subsequently activating an adaptive immune response to tumors.
Cancer-inhibiting inflammation in innate immunity
DCs are professional APCs linking the innate and adaptive immune system.370 Typically, DCs recognize a wide range of “danger signals” both from invading microbes and injured host cells through binding either PAMPs or damage-associated molecular patterns (DAMPs) to PRRs.371 For instance, the activation of TLRs in DCs has been shown to trigger a rapid inflammatory response to pathogens;372 with the presentation of tumor-associated antigens (TAA) by DCs being necessary for T-cell-mediated cancer immunity.373 Furthermore, DCs can also regulate immune responses by generating both central and peripheral tolerance and controlling inflammatory responses via multiple mechanisms, such as triggering apoptosis of autoreactive T cells and T-cell anergy, expanding Treg cells, and limiting other effector cell responses.374 Moreover, DCs were reported to control malignant development of colitis-associated colorectal cancer (CAC) through the production of IL-22BP, which neutralized the effect of IL-22,375 while IL-15-cultured DCs were shown to possess the capacity to enhance the anti-tumoral functions of γδT cells.376 However, it has been found that cancer cells could hijack DCs to promote chronic inflammation and prevent TAA presentation, thus accelerating tumor development. For example, TME-derived factors (e.g., IL-6 and M-CSF), as well as intracellular signaling proteins of DCs, including STAT transcription factors were demonstrated to switch the differentiation of monocytes to macrophages rather than DCs, preventing the priming of tumor-specific T cells.377,378 In addition, inflammatory DCs (infDCs), a subpopulation of DCs only forming in response to inflammatory stimuli, are critical to the anti-tumor immune response.379 In particular, infDCs were shown to migrate to lymphoid nodes and present antigens to naive CD4+ T cells to induce the differentiation of Th1,380 Th2,381 or Th17382 cells depending on the inflammatory environment. Therefore, inflammatory DCs appear not only during pathogenic inflammation, but also in experimental models of inflammatory diseases, such as in patients with rheumatoid arthritis or cancer.383 In summary, DCs have the potential to promote efficient anti-tumor immunity by recruiting and activating various immune cells. However, the TME is rich in immunosuppresive factors (e.g., VEGF, IL-6, PGE2, and IL-10) that suppress the immunostimulatory capacity of DCs and instead shift DCs into an anti-inflammatory phenotype.384 Nowadays, modulating the function of DCs to improve cancer immunotherapy is of particular research interest.384
TAMs have a dominant role as orchestrators of cancer-related inflammation. In nascent tumors, TAMs are known to display a pro-inflammatory phenotype (M1), eliminating some immunogenic tumor cells by promoting a Th1 response.385 Furthermore, M1-polarized macrophages are characterized by the high production of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6, IL-12, CXCL9, and CXCL10), and NOS and ROS intermediates, high expression of major histocompatability complex class II (MHC-II) and co-stimulatory molecules, efficient antigen presentation, but low expression of IL-10 and arginase.331,386,387 Through the secretion of pro-inflammatory cytokines and chemokines, such as IL-12, CXCL9, and CXCL10, M1-macrophages have been shown to drive the polarization and recruitment of Th1 cells, thereby amplifying a type 1 response, mediating phagocytosis of intracellular pathogens and tumor cells, and eliciting tissue-disruptive reactions.388 Several M1 stimuli, such as LPS and IFN-γ signals can polarize macrophages toward the M1 phenotype. For example, M1-like macrophages polarized with IFN-γ and exhibiting anti-tumor activity are usually characterized by high expression of HLA-DR (MHC-II),389 while activation of TLR9 by CpG, plus anti-IL-10 receptor antibodies were shown to redirect TAMs from an M2 to an M1 phenotype in vivo, leading to innate response debulking large tumors.390 Therefore, repolarization of macrophages from a pro-tumor phenotype (M2) to cytotoxic anti-tumor effectors (M1) is expected to improve the TME and contribute to anti-tumor immunotherapy.
Contrary to the N2 phenotype, tumor-inhibitory N1-TANs have been reported to produce multiple pro-inflammatory molecules (e.g., CCL2, CCL3, CXCL8, IL-6, TNF-α, and IFN-γ), and recruit other immune cells, including DCs and CD8+ T cells to the tumor sites.391 Moreover, N1-TANs can activate CD4+ and CD8+ T cells via the high expression of co-stimulatory molecules (e.g., CD86, ICAM-1, OX40L, and 4–1BBL), further potentiating anti-tumor immune responses.392 Furthermore, studies have shown that the strong anti-tumor properties of N1-TANs were also associated with the induction of ADCC, release of cytotoxic ROS, production of NETs, and their function as APCs.333,393,394
The roles of other innate lymphoid cells, such as ILC1, ILC2, and ILC3 subsets, in tumors are also of interest.395 The formal identification of ILCs increased our understanding of their tissue distribution and established the essential functions of ILCs in diverse physiological processes. These included the resistance to pathogens, regulation of autoimmune inflammation, tissue remodeling, as well as cancer and metabolic homeostasis.271 Briefly, ILCs are known to be major producers of cytokines in response to tissue damage and important regulators of the inflammatory response. However, their specific roles in cellular transformation and malignant progression remain largely unknown. Hence, unraveling the role of ILCs in cancer development and the interplay between ILCs and other immune cells would significantly improve the understanding of the mechanism by which the innate immune system tunes the inflammatory response in cancer. Notably, many ILC functions appear to be regulated by mechanisms distinct from those of other innate and adaptive immune cells.396 The ILC family subtypes are characterized primarily by their signature cytokine secretion profiles. For instance, ILC1 produces IFN-γ, whereas ILC2 produces IL-5 and IL-13, and ILC3 produces IL-17 and IL-22. Experimental evidences from murine models and patient-based studies have elucidated the effects of ILCs on the maintenance of tissue homeostasis and the associated consequences for health and disease.397
Natural killer cells are prototypes of innate lymphocytes with a strong cytolytic function, fulfilling their role in host defense against microbial infection and tumors.379 Based on their role in inhibiting microbial infections and tumor progression, NK cells are now considered to be an important component of the immune system. Moreover, conventional NK cells have been grouped as cytotoxic, IFN-γ-producing ILCs among the emerging population of ILCs.396 Upon activation, NK cells have been shown to induce target cell apoptosis via either the release of perforin and granzymes or the expression of Fas-L and TRAIL on their surface.398,399 Besides their cytotoxic capacity, NK cells can secrete various cytokines, chemokines, and growth factors, including IFN-γ, IL-13, TNF, FLT3L, CCL3, CCL4, CCL5, lymphotactin (XCL1), and GM-CSF.400 In addition, NK cells are presumed to bridge innate and adaptive immunity through the secretion of IFN-γ, which enhances the expression of MHC I on tumor cells and the expression of MHC-II on APCs, including monocytes, macrophages, and DCs.400 Impaired NK cells or NK cell deficiency have been associated with an increased incidence of various types of cancer.395,396 Thus, NK cells are considered to be key effectors in cancer immunosurveillance, transplantation rejection, as well as early viral immunity.400
Interestingly, NK-T cells, expressing both the TCR of T cells and natural killer cell receptor (NKR)-P1 receptors of NK cells, play an important role in innate immunity through the recognition of lipid antigens presented by CD1d.401 In addition, NK-T cells are known to not only secrete Th1- and Th2-related cytokines, but also have the same killing target cells as CD8+ killer T cells.401 The activation of NK-T cells has been shown to be usually accompanied by the activation of T cells, B cells, and NK cells, therefore having a great impact on the immune response after activation.395,402
Although in some cases, IL-17-producing γδ-T cells were demonstrated to promote tumor growth through the production of IL-17,403 they are generally considered as anti-tumor innate immune cells that provide IFN-γ-mediated protective responses in certain cases. Briefly, γδ-T cells recognize and directly kill tumor cells through TCR and natural killer cell receptors (NKR).404 The tumor cell killing of γδ-T cells was shown to be mediated by the TRAIL associated with TNF, Fas-L, or granulosa cell pathway (leading to the secretion of perforin and granzyme).405 Moreover, γδ-T cells could also destroy tumor cells through ADCC after treatment with tumor-specific antibodies.406
Anti-inflammatory metabolites produced by immune cells have also been reported to play an important role in orchestrating the resolution of inflammation. As mentioned, SPM, such as resolvins, protectins, lipoxins, and maresins, are a family of endogenous lipid mediators that exert proresolving and anti-inflammatory effects without suppressing the immune response.143 During the onset phase of inflammation, macrophages secrete SPM, promoting the resolution of inflammation, and increasing vascular permeability, thus enabling the infiltration of PMN into inflammatory tissues. Moreover, SPM have been found to stimulate self-limited innate responses, enhance innate microbial killing and clearance, hence avoiding the transition to chronic inflammation and inhibiting cancer progression.407 Accumulating evidence have shown that SPM not only possess anti-tumor ability, but also enhance the effects of other anti-tumor therapies. For instance, low dose of LXA4 could inhibit the proliferation and metastasis in OVC cells, Hela cells, as well as in papilloma.350,408,409 Besides, RvD1 and RvD2 were shown to prevent metastasis of A549 by reducing EMT induced by TGF-β1.410 Likewise, RvD1 was reported to induce caspase-3, thus increasing the apoptosis of pancreatic ductal adenocarcinoma cells in vitro.411 Furthermore, SPM are also known to display anti-tumor actions by targeting immune cells. For instance, an LXA4 isomer could inhibit the infiltration of neutrophils and reduced the production of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6, thereby inhibiting the severity of colitis in mice.412 In tumor-bearing mice, LXA4 significantly inhibited tumor growth by targeting IL-10-producing Breg cells;26 and selectively converted M2-TAMs to the M1 phenotype, triggering tumor cell apoptosis and thus attenuating tumor progression.26,412 Likewise, RvD1 was observed to enhance the killing function of NK cells in pancreatic ductal adenocarcinoma.411 Moreover, SPM have been reported to prevent tumorigenesis by targeting precancerous lesions. For instance, RvE1 could increase the survival rate and promote the regression of colitis.413 Similarly, the inhibitory effect of MaR1 in mice colitis was recently reported.414 In conclusion, SPM were demonstrated to play an important role in attenuating tumor-promoting inflammation, suppressing tumor development and progression, as well as enhancing anti-tumor immunity.
Cancer-inhibiting inflammation in adaptive immunity
Similar to APCs, macrophages and DCs also bridge innate and specific immunity.249,352,415 They can recognize tumor cell antigens and present them to the specialized members of the immune system to activate tumor-specific T cells for the killing and clearance of tumor cells. Adaptive immunity plays the most important role in the anti-tumor immune response.416 It has been noted that CD4+ Th1 cells, activated CD8+ T cells, and γδT cells are often involved in immune responses and have been associated with favorable prognosis in patients with lung cancer.417 In particular, CTLs are known to recognize the abnormal antigens of tumor cells, secrete granzyme and perforin to kill tumors, and express Fas-L, allowing them to bind with tumor cells to promote their apoptosis. Therapeutic reinvigoration with tumor-specific T cells has greatly improved the clinical outcome in many cancers. Nevertheless, many patients did not achieve a durable benefit. Recent evidence from studies in murine and human cancer have suggested that intratumoral T cells display a broad spectrum of dysfunctional states, shaped by the multifaceted suppressive signals occurring within the TME.418 However, this dysfunction of T cells in cancer might be utilized to develop personalized strategies to restore anti-tumor immunity. One such example is helper T cells that secrete cytokines, recruit, and activate CTLs to kill tumor cells.419
Th1 cells, a lineage of CD4+ effector T cells characterized by the secretion of IL-2, IFN-γ, TNF-α, and lymphotoxin are principally responsible for activating and regulating the development and persistence of CTLs. For example, Th1 cells have been shown to release IFN-γ, which stimulates the upregulation of molecules, such as LMP2, LMP7, MECL, PA28, and MHC class I in APCs, all of which contribute to increased antigen presentation to CTLs.420 Besides, Th1 cells are also known to recruit and activate inflammatory cells (macrophages, DCs, eosinophils, and NK cells) in the tumor, thereby enhancing their ability to eliminate intracellular microbes and to present antigens to CD8+ CTLs.421 In addition, Th1 cells were also found to directly destroy tumor cells via the release of cytokines, such as lymphotoxin, which activate death receptors on the surface of cancer cells.422 Furthermore, Th1 cells can directly interact with tumor cells through MHC class II molecules.423
B cells and humoral immunity have also been described to regulate anti-tumor immunity through other mechanisms, either by expressing cytokines, such as IL-10 or IL-35, inducing antibody-mediated cytotoxicity through NK cells, or by activating the C5a or C3a complement system components, which seem to either activate or suppress anti-tumor immunity in a context-dependent manner.424 However, the ways by which different B-cell types manifest their immunosuppressive effects remain poorly understood. Some studies have shown that B cells might play a pro-tumor role due to their immunosuppressive subtypes. For instance, tumor-infiltrating B-lymphocytes (TIBs) have been detected in all stages of lung cancer.425 The existence of TIBs has been reported to vary in different stages and histological subtypes, suggesting a critical role for B cells during lung cancer progression.426 Of interest, activated B cells were also found to be able to directly lyse tumor cells. Moreover, TIBs have been shown to possess cytotoxicity toward hepatoma cells through the secretion of granzyme B and TRAIL. In one such study, IFN-α- and TLR agonist-stimulated B cells produced functional TRAIL that was cytotoxic to melanoma cell lines.366 Abundant studies have assessed the function of TIL-B by immunohistochemical examination of CD20.427 Accordingly, 50.0% of these studies reported a positive prognostic effect for CD20+TIBs, whereas the rest showed neutral and negative effects. The prognostic significance of TIBs was basically reported to be consistent with that of CD3+ and CD8+ T cells, with the anti-tumor activity of T cells being generally shown to be more potent in the presence of TIB cells.428 Finally, accumulating evidence have supported a positive role for TIB in anti-tumor immunity,427,429,430 suggesting that enhancement of these responses should be considered in future cancer immunotherapies.
Therapy-elicited inflammation
Recently, anti-cancer therapy-induced inflammation has been recognized as a strong modulator of the TME. Several conventional classes of chemotherapeutic agents (e.g., anthracyclines and oxaliplatin) and radiation therapy can elicit immunogenic cell death (ICD) of tumor cells, and induce the secretion of DAMPs from dying cells.431,432,433 Subsequently, the ICD-induced DAMPs activate DC-mediated anti-tumor T-cell responses. In fact, the host immune response is indispensable for the therapeutic efficacy of these drugs.433
Chemotherapeutic drugs, also known as cytotoxic drugs, display anti-cancer effects by acting on key cellular biological events necessary for the proliferation and survival of cancer cells. Besides, chemotherapy can activate immune responses and enhance the activation of effector T cells, disrupting the immunosuppressive pathway of TME.434,435 However, chemotherapy resistance is one of the main factors that limit the therapeutic effect and affect the clinical outcome.2,436 Some chemotherapeutic drugs cause inflammation events, which play a pivotal role in tumor angiogenesis, metastasis, and failure of therapy.437 For instance, cisplatin is one of the most effective anti-cancer drugs used to treat a variety of solid tumors.438 Cisplatin-induced inflammation is mediated through multiple mechanisms including activating NF-κB, COX-2, and TNF-α.438,439 Furthermore, celecoxib, a specific COX2 inhibitor, increase the anti-tumor efficacy of cisplatin in cervix cancer cells, as well as in bladder cancer and gastric cancer.440,441,442 Paclitaxel induces apoptosis by stabilizing microtubules, thereby leading to cell arrest.442,443 In response to paclitaxel administration, a variety of inflammatory factors and signaling pathways, such as IL-1β, IL-8, IL-6, and NF-κB, can be activated.444,445,446,447
By using high doses of radiation to kill cancer cells and shrink tumors, radiotherapy is an important approach of cancer treatment. More than half of the cancer patients receive radiotherapy during their therapies.448 Radiation activates the interconnected network of cytokines, adhesion molecules, ROS/RNS and DAMPs, resulting in a self-amplified cascade, which generates pro-inflammatory TME, ultimately leads to tumor cell death.8,449 On the one hand, inflammation triggered by radiation feeds into adaptive antigen-specific immune responses and adds another dimension to the tumor-host crosstalk during radiotherapy, which can contribute to cancer cure.249,450 On the other hand, radiotherapy-induced chronic inflammation in the TME causes an increase in immunosuppressive populations, such as M2 macrophages, MDSCs, and Tregs.451 For instance, radiation can induce IL-6/STAT3 signaling pathway, which promotes tumor invasion and facilitates the survival of tumor cells after therapy, thereby conferring resistance to therapy.452,453 Moreover, silencing IL-6 by siRNA inhibits tumor recurrence after radiotherapy in prostate cancer and sensitizes tumor cells to radiation.454
Growing evidence suggests that TME is one of the major obstacles for cancer immunotherapy, where chronic inflammation plays a predominant role in tumor cell proliferation, angiogenesis, and immunosuppression.6,455 Furthermore, the side-effects of immune checkpoint blockade (ICB) and CAR-T therapies, such as coagulopathy and “cytokine storm” have limited their full application in cancer therapy,29,30,456 suggesting that reduction of these harmful immunotherapy-associated inflammation would be beneficial for the outcome of cancer patients. However, acute inflammation induced by other therapies can improve the effectiveness of immunotherapy. For example, activation of type 1 IFN response, such as recombinant IFN, CpG oligodeoxynucleotide, and 3′3′-Cgamp, can boost anti-cancer efficacy in synergy with immunotherapies.457 Besides, radiotherapy can trigger acute local inflammation, which sensitizing tumor cells to ICB therapy.458
Hence, acute inflammation triggered by some therapies can re-educate the pro-tumor TME toward an anti-tumor immune milieu. However, it is noteworthy that chronic death/injury-induced inflammation potentially promotes tumor progression and confers resistance to therapy,459 implying that therapy-elicited inflammation is a “double-edged sword” for cancer.
Understanding the means by which the immune system affects cancer development and progression has been one of the most challenging questions in anti-tumor therapy. Aforementioned, chronic inflammation shapes the TME, thereby promoting tumor development and progression, whereas acute inflammatory reactions can be used to improve the efficiency of anti-tumor therapies. Given its crucial role in cancer development, progression, and the anti-tumor effects of therapy, harnessing inflammation will open up new possibilities for long-lasting, multilayered tumor control.
Cancer therapy by targeting inflammation signaling pathways
Chronic inflammation is considered to be one of the characteristics of tumor initiation and progression,5 and therapy-induced chronic inflammation often endows residual cancer cells with resistance to subsequent courses of treatment (e.g., chemotherapy resistance and radiotherapy resistance).4 Anti-inflammatory drugs have been proven efficient for the prevention and treatment of tumors. However, the side-effects of ICB and CAR-T therapies, such as coagulopathy and the “cytokine storm” have limited their full application to cancer therapy,29,30 indicating that reduction of these pernicious inflammation reactions accompanying immunotherapy will improve therapeutic efficacy. Recently, a number of therapeutic strategies to limit inflammatory cells and their products have been successfully applied in clinical or preclinical tumor models. For instance, statins significantly reduced the risk of development of multiple types of cancer by exerting anti-inflammatory and other effects.21,22,23 Similarly, neutralization of IL-17A, IL-11, or IL-22 could inhibit colonic tumorigenesis at an early stage,460,461,462 while COXs inhibitors (e.g., celecoxib and aspirin) impaired tumor growth and metastasis.463
Nevertheless, not all inflammatory diseases or persistent infections are associated with increased cancer risk. Some cases of inflammation, such as allergic diseases that display a state of constant or recurring inflammation, might be even inversely correlated with cancer progression.464,465 In addition, although ICB has been reported to be clinically effective in presenting a durable response to treatment in some solid tumors, most patients with cancer did not respond to treatment for a variety of reasons. Infiltrated-inflamed tumors are considered “hot” tumors containing a high number of infiltrating cytotoxic lymphocytes expressing PD-1 that usually respond well to ICB.466 In contrast, infiltration-excluded tumors are characterized by the accumulation of CTLs along the border of the tumor mass and a lack of infiltrating CTLs into the tumor core. These tumors are generally considered “cold” tumors with poor sensitivity to ICB.467 Several promising strategies have been suggested to enhance the inflammatory infiltration that would contribute to the alteration of a cold into a “hot” tumor, thus rendering it sensitive to ICB.16 Herein, we discuss the targeted therapeutic approaches for the regulation of cancer-related inflammation (Fig. 4).

Harnessing the inflammation in cancer therapy. Several promising strategies for regulation of cancer-related inflammation have been suggested to improve anti-tumor response. On the one hand, inducing inflammation and modulating immune cell activation would overcome T-cell exclusion, turning “cold” tumors into “hot” tumors, for instance, local inflammation induced by irradiation or oncolytic viruses can promote innate immunity by activating nucleic-acid-sensing cytosolic receptors (cGAS–STING or RLRs) and subsequent type I IFN response. Besides, promoting DC maturation by cGAS–STING, TLR agonist, DC vaccine, or injection of GM-CSF can induce an acute inflammatory response and priming of T lymphocytes, which facilitate tumor regression. On the other hand, inhibiting chronic inflammation by anti-inflammatory drugs (e.g., aspirin) or accelerating inflammation resolution by proresolving mediators (SPM, e.g., lipoxins, resolvins, protectins, and maresins) also display an overall survival benefit for anti-cancer therapy. Furthermore, limitations of the infiltration and function of immunosuppressive cells (e.g., MDSCs, Treg cells, Breg cells, and M2-TAMs) by blocking inflammatory pathways is another way to restore immune surveillance and promote anti-tumor immunity. Green represents factors that enhancing these cancer-inhibiting inflammation, while red represents factors that blocking these cancer-promoting inflammation, would be beneficial to improve the effect of anti-tumor therapy
Non-specific agents targeting chronic inflammation
Non-steroidal anti-inflammatory drugs
NSAIDs are a family of agents that primarily inhibit the activity of COX enzymes and thereby suppress the synthesis of prostaglandins. Importantly, NSAIDs (including aspirin, celecoxib, and ibuprofen) use has been linked to reduced cancer risk and mortality.6 Aspirin, one of the most widely used anti-inflammatory drugs, has been identified as a broad-spectrum cancer-preventive agent based on multiple clinical and epidemiological studies.468,469,470 Besides, both preclinical and clinical studies have demonstrated promising results of the role of celecoxib in the treatment and prevention of cancer, and the best outcomes were observed in colon, breast, prostate, and head and neck cancers.471 Nevertheless, long-term administration of NSAIDs can result in side-effects including mucosal lesions, bleeding, peptic ulcer, and intestinal inflammation.20 Thus, the benefits of taking NASIDs for prevention and/or treatment of cancer must be assessed by risk versus benefit analyses.
Statins
Similar to aspirin, other agents with conventional anti-inflammatory activities have been repurposed for use in the prevention or/and treatment of cancer. Statins, a family of compounds that reduce blood cholesterol concentration by inhibiting the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, have a wide spectrum of activity as anti-cancer agents, including anti-angiogenic and anti-inflammatory actions in preclinical studies.472,473 However, a definitive benefit of statin use has not been confirmed in a randomized controlled trial setting to date. Several trials in patients with different tumor types designed to evaluate the benefit of statin are currently ongoing (NCT: 02161822, 01821404).
Corticosteroids
Corticosteroids, the most effective anti-inflammatory drugs for many chronic inflammatory diseases, are also shown to have anti-cancer activity.474 For example, pre-treatment with dexamethasone (DEX) improves the efficacy of chemotherapy in xenograft or syngeneic experimental tumor models of glioma, breast cancer, lung cancer, and CRC.475 Clinical trials have demonstrated that DEX in combination with carfilzomib and lenalidomide, obviously improved progression-free survival of patients with relapsed multiple myeloma.476,477
Natural anti-inflammatory products
By reducing the production of pro-inflammatory AA metabolites, omega-3 fatty acids reduce the production of eicosanoids that can activate AP-1 and NF-kB signaling and promote angiogenesis.474,478 Evidence has demonstrated that omega-3 fatty acids supplementation is associated with a reduced risk of CRC among individuals with low plasma levels of such fatty acids at baseline and in the African-American population.479
Several natural products such as polyphenols are able to modulate NF-κB, Wnt/β-catenin, PI3K/Akt, and MAPKs signaling and prevent the occurrence of inflammatory conditions.480 As a source of modulating agents to suppress chronic inflammation, dietary polyphenols may function as a chemopreventive agent against cancer, and improve the therapeutic effect of cancer.481 For example, curcumin (diferuloylmethane) is an active ingredient in plant turmeric spices, the anti-cancer activity of curcumin is related to its activity on inhibiting NF-κB, MAPK, PI3K/Akt/mTOR, Wnt/β-catenin, and JAK2/STAT3 signaling pathways.482,483 Besides, resveratrol is a natural polyphenol that provides a number of anti-aging health benefits including cardioprotection, and cancer prevention;484,485 while many factors need to be explored before resveratrol can be applied for human cancer prevention or treatment.
Reducing the therapy-elicited chronic inflammation
As discussed above, despite acute inflammation elicited-therapy contributes to destroy cancer cells during treatment, the therapy-elicited chronic inflammation (e.g., IL-1β, IL-6, IL-8, COX2/PGE2, NF-κB, and DAMP) plays a pivotal role in promoting therapeutic resistance and cancer progression.486,487 Scientists hypothesized that blocking chronic inflammation might enhance the therapeutic efficacy and benefit to cancer patients.16 For example, both chemotherapeutic drugs and radiation can induce IL-6 expression in tumor and stromal cells452,453 through the activation of NF-κB signaling, causing therapeutic resistance.488,489 These evidences suggest that blocking IL-6 or it’s downstream signaling pathways may provide therapeutic enhancement. Nowadays, several trials designed to evaluate the efficacy of Tocilizumab (human IL-6R-specific antibody) in chemotherapy are ongoing (Table 1).
Evidence demonstrates that many DAMPs are released from dying cells after chemo-/radiotherapy.490 In the TME, DAMPs can be ligands for TLRs expressed on immune cells and induce cytokines production and T-cell activation.491 However, DAMPs released from tumor cells can directly activate tumor-expressed TLRs that induce chemoresistance and metastasis.492,493 Furthermore, DAMP-induced chronic inflammation in the TME causes an increase in the infiltration of immunosuppressive cells, such as M2 macrophages, MDSCs, and Tregs.494,495,496 Therefore, regulation of DAMPs after chemo-/radiotherapy can reduce excessive inflammation to create an immunogenic TME.
In brief, combining educating inflammatory TME with anti-cancer therapies (including chemotherapy, radiotherapy, and immunotherapy) may provide more efficient strategy to inhibit tumor growth and improve patient survival.
Adjusting the inflammation in innate immunity
Inflammation is classically viewed as a feature of innate immunity, which differs from adaptive immunity owing to the receptors mediating its activation and its rapid onset. Once activated by PAMPs or DAMPs, MHC class I and II, and co-stimulatory molecules, as well as numerous inflammatory chemokines and cytokines are upregulated, attracting and priming T cells for activation through diverse antigen receptors.497,498 Given the crucial role of innate immune responses in immunity and inflammation,252,351 harnessing these responses opens up new possibilities for long-lasting, multilayered tumor control (Fig. 3).
Targeting mast cells
There have been compelling evidence that mast cells in the network of immune cells are involved in inflammatory disease and cancer.322,499 A number of studies have documented that mast cells potentially facilitate tumor progression via enhancing tumorigenesis, angiogenesis, and tissue remodeling, as well as via shaping an inflammatory microenvironment for immune escape.499,500,501,502,503 Today, several therapeutic strategies have been developed to inhibit tumor growth and improve the effect of immunotherapy by targeting mast cells. For instance, such strategies include the alteration of the numbers of mast cells, suppression of their activation, and prevention of the effects of inflammatory mediators.
Reducing the infiltrating numbers of mast cells is a promising treatment approach in inflammatory disease and cancer, in which their numbers are increased. The numbers of tumor-infiltrating mast cells have been reported to be reduced by the specific induction of apoptosis or by blocking the effects of factors that promote the recruitment of mast cell progenitors, their migration, differentiation, or survival.499 The stem cell factor (SCF-1), via the activation of the c-kit receptor (CD117) expressed on mast cells, has been identified as one of the most important factors for regulating the numbers of tissue mast cells under physiological conditions.504 Besides, several mediators of the recruitment of mast cell progenitors, such as CCR2, CCR3, CCR5, CCL2, CXCR2, IL-4, and CXCL12, were also considered to be important for the accumulation of mast cells in affected organs in murine models of disease.505,506 Blocking antibodies against these mediators have been used in various animal models to attempt to reduce the numbers of infiltrating mast cells in inflammatory diseases.507 Importantly, blocking these signals was also shown to display a potent anti-tumor effect.
Interestingly, mast cells are known to express a substantial number of activating and inhibitory receptors, which could be exploited to interfere with their activation.508 Recently, several drugs, are used in the clinic to treat mast cell-driven disorders, mostly allergic diseases. These include cromolyn sodium, nedocromil, lodoxamide, and antagonists for the histamine receptor H1, such as azelastine, ketotifen, olopatidine, bilastine, desloratadine, rupatadine, and epinastine.509 However, whether these drug have an anti-tumor effect remains unknown.
The degranulation of mast cells, the process of releasing inflammatory mediators from secretory granules, is a consistent feature of inflammatory lesions or tumors.509,510,511 Studies have shown that PI3K plays a crucial role in the biological functions of mast cells, including degranulation.512 Treatment with LY294002, a specific PI3K inhibitor, or inhibition of PI3K by overexpression of the Δp85 dominant-negative inhibitor was reported to lead to a significant decrease in the degranulation of mast cells via antigen-induced calcium (Ca2+) signals.513 Interference of the degranulation of mast cells was also shown to suppress tumor angiogenesis and progression in Myc-induced β-cell pancreatic cancer.514 Moreover, IL-10 inhibited the degranulation of mast cells by suppressing the expression and signaling of the IgE receptor of mast cells,515 while blocking IL-10 could hinder the antigen-induced recruitment of mast cell progenitors to the lungs of C57BL/6 mice.516 In addition, pharmacologic inhibition of the degranulation of mast cells using cromolyn was found to notably inhibit Myc-induced pancreatic islet tumors,514 experimental pancreatic and thyroid cancer,517,518,519 prostate cancer,520 and cholangiocarcinoma.521 Conclusively, blocking the degranulation of mast cells is considered as a promising approach for reducing inflammation and improving anti-tumor therapy.
Mast cells have been reported to play a pro-tumorigenic role in human bladder cancer through the stimulation of estrogen receptor β (ERβ).522 Accordingly, a selective EBβ antagonist inhibited mast cell-promoted tumor growth in a murine model of bladder cancer.522 Besides, mast cells were also shown to promote the proliferation of colon cancer in vivo, while injection of the Fcε-PE40 chimeric toxin, known to induce the apoptosis of mast cells, led to retrogradation of colon tumor progression in vivo.523
In addition, mast cells have been observed to display significant cyclooxygenase and lipoxygenase activities and to release inflammatory lipid metabolites of AA.524 In mice, the major cyclooxygenase products of mast cells are PGD2 and PGE2, while the primary lipooxygenase products are LTC4, LTD4, and LTE3.524 In particular, LTB4, a mast cell product of 5-lypoxygenase, is known to be a chemoattractant for mast cell progenitors.525 In general, these findings indicated that mast cells and their mediators deserve focused consideration as therapeutic targets in different types of cancer.
Activating DCs for uptake and presentation of tumor antigens
DCs are specific APCs that function as messengers between innate and adaptive immune responses.370 However, tumor cells have been shown to hijack DCs to promote chronic inflammation and prevent TAA presentation, thus accelerating tumor development.526,527 Nowadays, modulating the function of DCs to improve cancer immunotherapy is of particular interest.383
The maturation of DCs is essential for providing co-stimulatory signals to T cells. However, although the maturation of DCs occurs within TME, it is often insufficient to induce robust immunity.384 Bypassing suppressive pathways or directly activating DCs could trigger a T-cell response, and as such, therapeutic targeting of DCs holds translational potential in combinatorial approaches. The maturation of DCs is important for the initiation of Ag-specific T-cell responses. Nevertheless, the TME also contain a network of immunosuppressive factors (e.g., IL-6, M-CSF, PGE2, and VEGF) that could inhibit the infiltration of DCs and subdue their anti-tumor activity.527 Therefore, targeting these immunosuppressive pathways might improve the recruitment, infiltration, and anti-tumor activity of DCs in the TME. For instance, VEGF, correlated with poor prognosis in patients with different types of cancer, was found to inhibit the differentiation and maturation of DCs via the activation of VEGFR-1 and VEGFR-2.528,529 Moreover, blockade of the VEGF signaling significantly increased the proportion of mature DCs in patients with cancer; as a result, the inhibition of VEGF pathways has become an appreciated approach for the treatment of cancer.530 In addition, high levels of IL-6, known as a pro-inflammatory cytokine, were associated with a functional defect in DCs from patients with cancer.531 The IL-6-induced suppression of DCs could be intercepted by the AG490 JAK2/STAT3 inhibitor,487,532 indicating that the pro-inflammatory IL-6/JAK/STAT3 signaling pathway is a promising target for cancer immunotherapy. Moreover, due to the elevated levels of PGE2 and activity of COXs in patients with colon cancer and its correlation with tumor size and patient survival, PGE2 has been proposed as the principal prostanoid associated with CRC. Furthermore, PGE2 was reported to be responsible for the reduced differentiation of DCs from CD34+ precursors;533 and to mediate DC tolerance via the upregulation of the expression of indoleamine 2,3-deoxygenase (IDO1) in DCs, resulting in the differentiation of Treg cells and the inhibition of antigen-specific stimulatory potential of DCs.525 In addition, tumor-derived CSF-1 could inhibit the differentiation of hematopoietic progenitor CD34+ cells into DCs and induce the differentiation of cord blood monocytes to tolerogenic DCs.534 Evidence has shown that the regulation of DCs differentiation driven by CSF-1 was mediated by the PI3K-dependent pathway, delaying the activation of caspases in monocytes.535 Hence, removing or blocking immunosuppressive factors (e.g., IL-6, M-CSF, PGE2, IL-10, and VEGF) on DCs would promote anti-tumor efficiency by recruiting and promoting the maturation of DCs.
To launch a robust antigen-specific anti-tumor response, some immunostimulatory cytokines targeting the activation of DCs are applied in the development of therapeutic vaccines. First, GM-CSF was demonstrated to serve as a potent immune adjuvant inducing long-lasting anti-tumor immunity.536 Growing evidence have suggested that GM-CSF promotes the activation of DCs and enhances TAA presentation to T cells.537 In addition, studies revealed that treatment with GM-CSF and IL-4 in vitro could lead to the generation of bone marrow (BM)-derived DCs in mouse and monocyte-derived DCs from human peripheral blood mononuclear cells (PBMC); as a result, these findings have accelerated the clinical applications of GM-CSF.538 However, it was shown that in clinical trials administration of a high dose of GM-CSF resulted in immunosuppression, indicating a more complex role of GM-CSF in cancer immunotherapy.539,540 Second, the cytokine Fms-related tyrosine kinase 3 ligand (FLT3L), activating signaling through its FLT3 receptor expressed on DC precursors, was observed to be essential during the development of DCs.541 Culturing mouse BM precursors with recombinant FLT3L (rFLT3L) was sufficient to generate DCs in vitro.542 Furthermore, administration of rFLT3L in mice resulted in significant expansion of conventional DCs and plasmacytoid pre-DCs in vivo,543 whereas genetic ablation of FLT3L caused a marked decrease in these subsets.544,545
Typically, DCs can recognize a wide range of “danger signals” from both invading microbes and injured host cells through binding either PAMPs or DAMPs to specialized PRRs, such as TLRs, c-type lectin receptors, stimulator of interferon gene (STING), NOD-like receptors, and the RIG-I and MDA5 DNA/RNA receptors.370 Accumulating evidence has indicated that activating certain innate immune signaling pathways, especially TLRs, RLRs, and STING signaling pathways, might be a promising cancer immunotherapeutic approach.546 For example, the Coley toxin and Bacillus Calmette-Guerin TLR2 and TLR4 agonists have become common-used therapeutic agents against several cancers.547 The extract of larix leptolepis (ELL) was also found to potentially initiate TLR2 and TLR4 signaling in BM-derived DCs, inducing the activation of tumor-specific CTLs against cancers.548 According to these findings, combination therapy using TLR ligands and conventional radiation/chemotherapy, which displays a much higher growth-inhibitory effect compared with single application, might be a promising strategy for cancer treatment.549,550 Traditional therapy usually leads to the release of tumor antigens, which are subsequently phagocytosed and presented by macrophages and DCs. Stimulation of TLRs is known to further enhance the maturation of DCs, antigen presentation, and priming of tumor-specific CTLs, resulting in a more effective immunotherapy.551,552,553 Similarly, inducing the activation of melanoma differentiation-associated gene 5 (MDA5) using synthetic double-stranded RNA-poly (I:C) in OVC resulted in the increased expression of HLA-class I, release of cytokines (e.g., CXCL10, IL-6, and type I-IFN), and tumor cell apoptosis.554 This MDA5-mediated anti-cancer response was shown to require the DC-dependent phagocytosis of MDA5-activated tumor cells and subsequent generation of cytokines (CXCL10 and IFN-α), providing a pro-inflammatory milieu for boosting the cytolytic function and IFN-γ secretion of NK cells at the tumor site.555
Similar to the RLR signaling, activation of the STING pathway results in the production of IFNs for the induction of interferon-stimulated genes (ISGs), prompting cell death in an IFN-independent manner.556,557 Moreover, STING signaling has been shown to enhance the expression and secretion of inflammatory cytokines, such as IFNs, TNF-α, and IL-6.558 Thus, STING signaling could directly trigger tumor cell death and might provide a new cancer therapy option. Therefore, activation of RLRs could promote endogenous NK or CD8+ T-cell-mediated anti-tumor immune responses, and provide a promising approach for anti-cancer immunotherapy.
Radiotherapy is an effective treatment commonly used for various primary and metastatic cancers. In addition to directly destroying cancer cells, radiotherapy has been found to also induce a local inflammation with high levels of IFN-γ and ROS, whereas reduced levels of cytotoxic mediators and DAMPs.559 Furthermore, irradiation was demonstrated to trigger long-range effects through the induction of immunogenic cell death relying on the DC priming of CD8+ T cells.560,561 Moreover, irradiation-induced DNA damage is known to activate PRR, such as the cyclic GMP-AMP synthase cytosolic DNA sensor, which further activates STING, leading to a type I-IFN response by DCs, and contributing to anti-tumor immunity.562 In addition, canonical NF-κB signaling was reported to be necessary for the radiation therapy-induced anti-tumor immune responses.563 Combination of radiation with anti-PD1 treatment displayed an abscopal effect, with tumor regression in the nonirradiated secondary tumors.560 Through the induction of local inflammation and activation of DCs, radiotherapy might provide a nonpharmacological approach to improve the systemic response of immunotherapy.
Immunotherapy using DC-based vaccines could be an approved approach for boosting immune responses through the stimulation of tumor cell-killing T cells and induction of memory T cells for the prevention of cancer recurrence.564 These procedures would involve the isolation or in vitro generation and amplification of autologous DCs, followed by their ex vivo manipulation, including the induction of the maturation of DCs and tumor antigen loading, and finally their reinfusion into patients with cancer.565 In phase II studies, vaccination with DCs was shown to induce an immunologic response, increasing the number of TILs and providing overall survival benefit for at least a subpopulation of patients.566 Furthermore, injection of a DC vaccine comprising antigen-pulsed DCs induced antigen-specific immune responses in vivo, whereas soluble antigen alone failed to trigger immunity.567 Vaccination with cDCs loaded with tumor antigens was reported to synergize with the PD-1 blockade in vivo.568 Thus, strategies combining the application of DC vaccines with other therapies would be potential to improve anti-cancer efficacy.
Targeting tumor-associated macrophages
It is widely known that TAMs are the main infiltrating inflammatory cells in multiple tumors, contributing to an immunosuppressive environment.569 Abundant evidences have highlighted the correlation between high numbers of infiltrating TAMs with tumor progression and resistance to therapies.570 Therefore, TAMs are attractive targets for cancer therapies aiming to reduce cancer-promoting inflammation and TAM-orchestrated immune suppression.569 To date, several main strategies targeting TAMs have been used for the treatment of cancers and inflammatory diseases: reducing the accumulation of TAMs, direct depletion of TAMs, inducing polarization of M1 macrophages, and augmenting macrophage-mediated phagocytosis.
One strategy for targeting TAMs is to inhibit their recruitment or the infiltration of monocytes/macrophages into tumors. Tumor cell-derived CCL2 is known to be critical for the recruitment and infiltration of monocytes and TAMs in several tumor types, including ESCC, CRC, HCC, and breast cancer.571,572 Accumulating evidence have suggested that both CCL2 and its CCR2 receptor are implicated in both the inflammation and progression of tumors. Blockade of the CCL2/CCR2 pathway could effectively suppress the accumulation of TAMs in experimental tumor sites, and improve efficacy in combination with chemotherapy. For instance, blockade of the CCL2/CCR2 signaling inhibited the recruitment of inflammatory monocytes, infiltration and M2 polarization of TAMs, suppressed malignant growth and metastasis, reduced postsurgical recurrence, and enhanced survival in a mouse model of HCC.572 Similarly, CCR2-targeted therapy with PF-04136309 in combination with FOLFIRINOX displayed a benefit for patients with borderline resectable and locally advanced pancreatic cancer.573 In addition, treatment with bindarit, an anti-inflammatory indazolic derivative that can inhibit the synthesis of CCL2, displayed a potential inhibitory function in tumor progression and metastasis in prostate, melanoma, and breast cancers.574,575 Importantly, the anti-tumor effects of bindarit were revealed to be related to its ability of selective interference with the infiltration of TAMs and MDSCs. Taken together, these data indicated that the CCL2/CCR2 pathway might be a potential candidate for inhibiting the recruitment of TAMs into the inflammatory and immunosuppressive TME.
CCL5 is an inflammatory chemokine known to promote the migration of macrophages involved in the immune/inflammatory response.576 The CCL5/CCR5 axis has been implicated in tumor development or progression of multiple types of cancer (e.g., gastric cancer (GC), breast cancer, glioblastoma multiforme, and CRC), through the recruitment of TAMs and their polarization toward a M2-like phenotype.577,578,579,580 Maraviroc, a specific CCR5 antagonist, was demonstrated to reduce the infiltration of monocytes/macrophages in breast cancer, GC, glioblastoma, and advanced CRC.577,581,582 Therefore, the CCL5-CCR5 axis might be another chemokine pathway with potential for preventing the recruitment of macrophages.
In addition, CSF-1 has also been found to be involved in the differentiation and recruitment of TAMs to tumor milieus via the activation of CSF-1R.583 High expression of CSF1 and CSF1R have been correlated with poor prognosis in various cancer types, including breast cancer, GC, and OVC.584,585,586,587 Notably, chemo-radio and hormonal therapies were shown to exhibit the unwanted effect of upregulating the local expression of CSF-1.588,589 Emerging data have shown that blocking the CSF-1/CSF-1R signaling prevented the trafficking of TAMs, thereby achieving a meaningful clinical benefit for patients with cancer in clinical trials.590,591,592 For example, emactuzumab, a humanized monoclonal antibody (RG7155) that inhibits the activation of the CSF-1 receptor (CSF-1R), reduced the density of macrophages and increased the ratio of CD8+:CD4+ T cells in tumors.592 Besides, GW2580, a selective CSF-1R inhibitor, has been shown to reduce the infiltration of macrophages and the volume of ascites in a mouse model of ovarian cancer.589 Moreover, combining GW2580 with chemotherapy displayed synergistic results.593 GS-1101 is a specific inhibitor of the PI3Kp110δ kinase, whose activation is pivotal for the CSF-1-triggered infiltration of TAMs.594 Furthermore, GS-1101 inhibited the CSF-1-induced spreading and invasive capacity of TAMs.594 These findings suggested that targeting of the CSF-1/CSF-1R signaling could remove TAMs, leading to anti-tumor immune responses.
The selective elimination of TAMs in TME has also been explored for cancer therapy. One attractive strategy for depleting TAMs within the tumor milieu is the induction of their apoptosis. Several compounds (e.g., zoledronate, clodronate, and trabectedin) and bacterial pathogens have been demonstrated to trigger the apoptosis of macrophages. Bisphosphonates, which are primary agents for several bone diseases, have been used for depleting macrophages, as well as TAMs.595 Structurally, bisphosphonates are chemically stable derivatives of pyrophosphate, in which the P-O-P bond has been substituted by the P-C-P bond. Liposome-encapsulated bisphosphonates (e.g., zoledronic acid- or clodroenate-loaded liposomes) have been widely applied for the depletion of TAMs in murine tumor models, resulting in markedly reduced tumor growth and metastasis.596,597 Furthermore, treatment with bisphosphonates or their combination with other anti-tumor therapies markedly reduced the recurrence and overall mortality of breast and prostate cancer.595,598
As the first marine-derived anti-tumor agent approved for the treatment of soft tissue sarcoma and OVC, trabectedin was also found to suppress the accumulation of macrophages in TME and the production of inflammatory mediators from TAMs, such as IL-6, CCL2, CXCL8, angiopoietin 2, and VEGF.387,599,600 Moreover, as TRAIL receptors are more highly expressed in monocytes/macrophages than in neutrophils and lymphocytes, trabectedin was shown to specifically activate caspase-8-dependent apoptosis in monocytes/macrophages.387,601 In several types of cancer (e.g., breast cancer and sarcoma), trabectedin reduced the density of TAMs, and inhibited tumor growth in vivo.387,602,603 Recently, it was shown that the inhibitory effect of trabectedin on the accumulation of TAMs and production of inflammatory mediators from TAMs contributed to its anti-tumor activity, especially, in inflammation-associated cancers.
As a bacterial pathogen, Shigella flexneri is known to specifically induce apoptosis in macrophages. To evaluate the effectiveness of Shigella-induced depletion of macrophages in achieving tumor regression in vivo, Galmbacher et al. developed an attenuated strain of S. flexneri, termed M90TΔaroA.604 Interestingly, injection of M90TΔaroA led to caspase-1-dependent apoptosis of TAMs and a significant reduction in tumors of transgenic MMTV-HER-2 mice. Furthermore, depletion of TAMs was associated with complete tumor regression, suggesting that bacterial pathogens might serve as potential arms in the targeting of TAMs for attenuated inflammation and anti-tumor therapy.604
The polarization of macrophages has been shown to have a profound impact on inflammation and TME, with repolarization of macrophages from the pro-tumor phenotype (M2) into cytotoxic anti-tumor effectors (M1) improving the TME. Classically, M1 polarization of macrophages can be obtained by stimulation with pathogen-derived LPS alone or in combination with Th1 cytokines, such as IFN-γ and GM-CSF, whereas IL-10, TGF-β, and PGE2 contribute toward the M2 phenotype and have been associated with a suppression of inflammation and anti-tumor activities.605
Decades ago, the intravesical inoculation of the muramyl tripeptide phosphatidylethanolamine (MTP-PE), a synthetic analog of the muramyl dipeptide (MDP), was applied in the treatment of bladder carcinoma with the aim to activate cytotoxic macrophages against tumor cells.606 Besides, stimulation of the TLR signaling by LPS is known to trigger the translocation of NF-κB into the nucleus and the production of inflammatory cytokines, such as IL-1β, IL-6, IL-12, IL-23, and TNF-α.607
IFN-γ is a prototypic cytokine inducing the M1 polarization of macrophages, antimicrobial and anti-tumor effects, as well as the enhancement of antigen processing and presentation pathways.608 Upon exposure to IFN-γ, macrophages were shown to become primed for induction of the NF-kB pathway through direct and indirect mechanisms.609,610 Recently, recombinant IFN-γ displayed antiproliferative, antiangiogenic, and proapoptotic effects on cancer cells,611,612 as well as anti-inflammatory effects.613 Thus, IFN-γ has been adopted in the clinical management of a variety of malignancies, including bladder carcinoma, CRC, OVC, and adult T-cell leukemia.614,615,616 In general, IFN-γ has been expected to act as sine qua non in the clinical efficacy of anti-tumor immunotherapies.
More recently, an alternative approach has been explored to re-educate macrophages via the activation of the CD40 co-stimulatory molecule.617 Preclinical experiments have demonstrated that CP-870,893, an agonistic anti-CD40 antibody switches TAMs toward an M1-like phenotype and endowed them with antigen-presenting capabilities, leading to the reestablishment of immune surveillance and retardation in tumor growth.618 Moreover, in phase I trials, treatment with CP-870,893 in combination with gemcitabine showed a partial therapeutic benefit in patients with advanced pancreatic cancer.619 These findings suggested that the CD40 pathway could be therapeutically harnessed to reverse immune suppression by targeting TAMs involved in cancer-related inflammation.
The STAT3 transcriptional factor is a pivotal regulator of the inflammatory response of macrophages, as well as other immune cells.620 Evidence have demonstrated that tumor-secreted lactate induces the M2 polarization of macrophages through the activation of the ERK/STAT3 signaling pathway, thus promoting breast cancer progression.263 Likewise, activation of the IL-6/STAT3 pathway was reported to accelerate the progression of HCC via switching the M1/M2 polarization of macrophages.621 Blocking of the STAT3 signaling was reported to be part of the antiproliferative and anti-inflammatory properties of resveratrol.622,623 Furthermore, treatment with metformin suppressed the monocyte-to-macrophage differentiation via the AMP-activated protein kinase (AMPK)-mediated dephosphorylation of STAT3.624 These data suggested that the anti-inflammatory and anti-tumor properties of the STAT3 signaling inhibitors were related to the M1/M2 polarization of macrophages.
The scavenger receptor macrophage receptor with collagenous structure (MARCO) has been shown to be mainly expressed on macrophages or DCs, regulating inflammatory responses to bacterial pathogens though the recognition of PAMPs.625 Mounting evidence has recently indicated that MARCO also plays important roles in regulating the polarization of macrophages.625,626,627 For instance, MARCO is known to be an initial signaling receptor for asbestos, polarizing alveolar macrophages toward a profibrotic M2 phenotype.626 Besides, MARCO has been found to be also highly expressed on M2-TAMs with immunosuppressive gene signatures in the TME of both murine tumor models and human cancers. Blocking MARCO using antibodies led to reduced tumor growth and inhibition of metastasis, and a switch to the M1 phenotype of macrophages.626 Of note, MARCO could also be used as a viable target for reeducating TAMs.
Apart from depleting TAMs and suppressing the progression of glioma in preclinical murine models, BZL945 was demonstrated to induce TAMs toward a M1 phenotype, unlike other CSF-1R inhibitors.591 Administration of polyethylenimine-coated superparamagnetic iron oxide nanoparticles (EI-SPION) could inhibit the M2 polarization of macrophages by suppressing the phosphorylation of Src and cortactin.628 Moreover, by downregulating the placental growth factor (PlGF), the host-produced histidine-rich glycoprotein (HRG) skewed TAMs from a M2- to a M1-like phenotype, resulting in reduced tumor growth and metastasis, as well as improving chemotherapy.629
Macrophage-mediated phagocytosis is a pivotal process underlying the anti-tumor effect of activated macrophages.331 The signal regulatory protein α (SIRPα) expressed on (e.g., macrophages and DCs) was observed to trigger a cascade of events that inhibited phagocytosis when phagocytes interacted with CD47 highly expressed on cancer cells.630,631 Thereby CD47 appears to serve as a “don’t eat me” signal and a myeloid-specific immune checkpoint. Currently, antibodies that antagonize the CD47-SIRPα interaction were found to induce the antibody-dependent cellular phagocytosis (ADCP) of tumor cells, and the priming of tumor-specific T-cell responses. Several antibodies targeting the CD47-SIRPα interaction (e.g., Hu5F9-G4, TTI-621, CC-90002, and ALX-148) are being currently tested in clinical trials for both solid and hematologic malignancies.632,633 In addition, CD24 expressed in OVC or triple-negative breast cancer was reported to facilitate immune evasion through its interaction with the sialic-acid-binding Ig-like lectin 10 (Siglec-10) inhibitory receptor of TAMs.634 Blockade of the CD24-Siglec-10 interaction remarkably strengthened the phagocytosis of all CD24-expressing human tumors,634 highlighting that CD24 signaling might be a promising target for cancer immunotherapy.635 These findings suggested that targeting the “don’t eat me” signal could promote macrophage-mediated phagocytosis, leading to an anti-tumor innate immune response.
Targeting myeloid-derived suppressor cells
MDSCs represent a heterogeneous population of immature myeloid cells with anti-inflammatory effects and potent immunosuppressive activity that play a crucial role in TME.348 The accumulation and activation of MDSCs have been shown to correlate with tumor progression, metastasis, and recurrence of many types of tumors.636 Moreover, the efficacy of immunotherapy was negatively correlated with an increased density and activity of MDSCs.637,638 Hence, targeting MDSCs could become a promising approach to overcome tumor-mediated immunosuppression and enhance the efficiency of tumor immunotherapies. The modulation of MDSCs was achieved by facilitating their differentiation into mature myeloid cells, thus inhibiting their development, expansion, and function, as well as depleting their numbers.
As the immune suppressive phenotype of MDSCs is known to depend on their immature state,348 forcing their differentiation into mature myeloid cells (e.g., DCs or macrophages) would impair their suppressive function. It has been demonstrated that agents that can induce the differentiation of MDSCs include vitamin A and D, all-trans retinoic acid (ATRA), IL-12, the activation of TLR9, taxanes, beta-glucan particles, the inhibition of tumor-derived exosomes, and very small size proteoliposomes.639,640,641
Although the mechanism remains unclear, vitamins A and D have been demonstrated to promote the differentiation of MDSCs into mature cells. Compared with vitamin A-replete mice, vitamin A-deficient mice exhibited increased numbers of MDSCs in bone marrow, spleen, and peripheral blood.642 Similar results were also observed in tumor-bearing mice and patients with NSCLC.643,644 Furthermore, administration of high dose vitamin D reduced the numbers of immature myeloid cells, and increased the levels of IL-12 and IFN-γ, displaying an anti-tumor effect.645 ATRA, a vitamin A derivative, was reported to lead to the differentiation of MDSCs to DCs and macrophages by blocking the transduction of the retinoic acid signal in both patients with cancer and tumor-bearing mice.639,646 Mechanistically, administration of ATRA led to reduction in the levels of ROS in MDSCs by activating the ERK1/2 pathway.647 In addition, in vivo administration of ATRA notably reduced the number of MDSCs, whereas concomitantly boosting CD4+ and CD8+ T-cell-mediated tumor-specific immune responses. Depletion of MDSCs using ATRA in patients with small-cell lung cancer, dramatically enhanced the efficiency of a DC vaccine against p53 by increasing the levels of CD8+ T cells.648 In a phase I/II study, ATRA was demonstrated to significantly decrease the immunosuppressive function of MDSCs and increase the levels of CD8+ T cells in patients with melanoma receiving ipilimumab therapy compared with patients receiving ipilimumab alone.649
Taxanes (e.g., docetaxel and paclitaxel), a class of chemotherapeutic drugs, were also reported to facilitate the differentiation of MDSCs in mice and humans with cancer.650,651 Administration of docetaxel reduced the number of MDSCs, decreased their function, whereas increased the activity of CD8+ T cells in tumor-bearing mice.652 Low-dose paclitaxel promoted the differentiation of MDSCs toward DCs in vitro in a TLR4-independent manner.653 In a phase II study, women with HER-2 negative breast cancer treated with doxorubicin and cyclophosphamide followed with docetaxel displayed a complete response and lower levels of circulating MDSCs.654
Besides, treatment with β-glucan has been reported to lead to the maturation of M-MDSCs in vitro, and the suppression of the activity of M-MDSCs in tumor-bearing mice, thereby leading to delayed tumor progression.655 To date, there have been several ongoing or completed clinical trials evaluating the effects of β-glucans on cancer therapy.656 Moreover, tumor-derived exosomes, enriched in proteins and nucleic acids, were observed to prevent immature bone marrow myeloid cells from becoming mature DCs.657 Inhibiting the production of exosomes using dimethyl amiloride heightened the anti-tumor efficacy of cyclophosphamide in vivo.658 Furthermore, very small size proteoliposomes, such as nanosized particles formed from the outer membrane vesicles of Neisseria meningitidis and GM3 ganglioside, were found to induce the maturation of DCs and an anti-tumor response from CD8+ T cells in mice.659 These studies indicated that inducing the differentiation of MDSCs in patients with cancer might augment immunotherapeutic approaches.
The expansion and infiltration of MDSCs depend on several signaling pathways, including JAK/STAT, VEGF, CXCR2, and COX2/PGE2.660 First, STAT3 is the main transcription factor regulating the immunosuppressive activity of myeloid cells, and blockage of the STAT3 pathway by various inhibitors can decrease the number of G-MDSCs. For example, the herb derivative curcumin, which is a regulator of STAT3 signaling, was found to exhibit several pharmacologic effects (e.g., anti-inflammatory, antioxidant, and anti-cancer activities), as well as reduce the number of MDSCs at tumor sites and in the blood of tumor-bearing mice.661,662 The siRNA-inhibition of STAT3 eliminated the immunosuppressive effects of MDSCs on CD8+ T-cell functions by reducing the expression of Arg-1.663 However, small molecule inhibitors targeting STAT3 displayed limited efficacy and broad side-effects.664
Another strategy to prevent the expansion and infiltration of MDSCs was shown to be the blocking of local signaling in TME, for instance, by targeting VEGF. Although VEGF might promote tumor growth through several mechanisms, including angiogenesis and metastasis, evidence have indicated that the anti-tumor effect of the VEGF blockade was related to its inhibition of MDSCs. In a mouse model of metastatic renal carcinoma, neutralization of VEGF by bevacizumab reduced the CD11b+VEGFR1+ population of MDSCs in the peripheral blood.665 Clinical studies further confirmed that bevacizumab could decrease the number of G-MDSCs, whereas increase the numbers of mature circulating DCs in patients with metastatic CRC, as well as in patients with NSCLC and patients with RCC.666,667
The stem cell factor (SCF-1), which is highly expressed in tumor cells, has been shown to enhance the development and expansion of MDSCs.668 Decreasing the levels of SCF-1 in mice using tyrosine kinase inhibitors, such as sunitinib, pazopanib, and sorafenib was shown to inhibit the development of MDSCs in the bone marrow.669 Blocking CSF-1 by sunitinib agents also decreased the numbers of MDSCs in patients with RCC and RCC-bearing mice.670 Furthermore, blocking CSF-1 also increased anti-tumor responses, tumor shrinkage, and prolonged survival in murine models of colon carcinoma and lung cancer.494,671 Recently, there exist a number of ongoing clinical trials evaluating the levels of CSF-1 in tyrosine kinase therapy.
Semaphorin 4D (Sema4D) is a proangiogenic cytokine produced by several malignancies, known to induce the expansion of MDSCs from monocytes.672 Preclinical studies have demonstrated that antibody neutralization of Sema4D disrupted the expansion of MDSCs, which was associated with a shift in the cellular phenotypes and tumor-derived cytokines toward a pro-inflammatory and anti-tumor milieu.672 Treatment with humanized IgG4 mAb targeting Sema4D (VX15/2503) was shown to be well tolerated in patients with advanced solid tumors in a phase I trial.673 These results suggested that Sema4D might be a promising therapeutic target for the enhancement of the anti-tumorigenic inflammatory response in HNSCC and other epithelial malignancies.
The CXCR1 and CXCR2 chemokine receptors are known to recruit immune suppressive cells, such as MDSCs into the TME. Blocking the CXCR2 chemokine signaling using anti-CXCR2 mAb was reported to disrupt the CXCR2-mediated tumor trafficking of MDSCs, and improved the therapeutic efficacy of anti-PD1 therapy.674 Other approaches inhibiting the infiltration of MDSCs in patients include the inhibition of galectin-3 by GR-MD-02 and the inhibition of CCR2 by CCX872.675,676 In general, targeting cytokine or chemokines related to the expansion and infiltration of MDSCs has been shown to significantly improve the efficacy of anti-tumor immunotherapy.
In addition to tumor-derived cytokines or chemokines, the bioactive lipid PGE2 has been suggested to drive the expansion and infiltration of MDSCs in patients and experimental murine models.677 In response to doxorubicin, PGE2 is known to be secreted by tumor cells, increasing the recruitment of MDSCs into the TME.678 Moreover, it has been found that in CRC, the receptor-interacting protein kinase 3 (RIPK3) is downregulated in MDSCs in response to PGE2, causing an increase in the expression of Arg-1, as well as an NF-κB-mediated upregulation of COX2, thus creating an immunosuppressive positive feedback loop.679,680 Celecoxib, a specific COX2 inhibitor, prevented the expansion of MDSC subtypes and reduced the function of MDSCs in a murine model of mesothelioma, improving dendritic cell-based immunotherapy.681 In addition, targeting E-type prostanoid receptors (EPs) is another efficient approach for the suppression of the PGE2-mediated expansion of MDSCs. Combination of the E7046 EP4 antagonist682 with agents targeting Treg cells, was observed to increase the ratio of CD8+ CTLs to Tregs and induce the differentiation of MDSCs.683 Moreover, the COX2/PGE2 signaling has also been found to contribute to the immunosuppressive activity and PD-L1 upregulation of other myeloid cells (e.g., M2-like macrophages). It was further revealed that pharmacological blocking COX2 (celecoxib or aspirin) synergized with ICB therapy in preclinical models.684,685 Beyond that, the expansion and infiltration of MDSCs could also be stimulated by many soluble factors in TME, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), G-CSF, M-CSF, TGF-β, TNF-α, S100A9, S100A8, IL-1β, IL-6, and IL-10.
After infiltrating into TME, MDSCs utilize multiple mechanisms including oxidative stress and nutrient depletion to silence the function of effector cells.686 The modulation of oxidative stress was shown to be beneficial for controlling the MDSC-mediated immune suppression in tumor-bearing mice. For example, nitroaspirin, generated from the coupling of a NO-releasing moiety to the non-steroidal anti-inflammatory drug aspirin, has been demonstrated to possess properties, such as scavenging of ROS and inhibition of the release of pro-inflammatory cytokines, feedback inhibition of the activity of iNOS, and suppression of cancer cell proliferation.687 Oral administration of nitroaspirin restored immune responses in tumor-bearing mice, and increased the number and function of tumor antigen-specific T cells.688 In addition, inhibitors of phosphodiesterase-5 (PDE5) attenuated the activity of Arg-1 and iNOS and weakened the suppressive activities of MDSCs in tumor-bearing mice, resulting in suppressed tumor growth with greater infiltration and activation of T cells.688 Recently, a trial of tadalafil, another PDE5 inhibitor, displayed positive effects on the activation of CD8+ and CD4+ T cells, and improved the clinical outcome in patients with metastatic melanoma.689
Targeting the critical molecules in the signaling pathways shared by tumors and MDSCs, such as AMPK, HIF-1α, and PI3K, is also being actively investigated. Metformin has been reported to have a broad impact on AMPKα and HIF-1α, which are critical for the expression of CD39 and CD73 in MDSCs.690 Moreover, metformin can directly reduce the CD73/CD39-mediated suppressive functions of MDSCs. Importantly, metformin improved the functionality of CD8+ T cells and increased the overall survival in patients with OVC.691 Pharmacological targeting the highly expressed PI3Kγ on MDSCs using IPI-549 has been shown to be beneficial when in conjunction with the administration of immune checkpoint blocking antibodies.692 The efficacy of targeting PI3Kγ in promoting CTLs is currently being evaluated in a phase I clinical trial.693 PI3K-δ is an identified critical signaling molecule in Treg cells, and duvelisib, the dual inhibitor of PI3K-δ and PI3K-γ, has been shown to promote the efficacy of immune checkpoint antibodies in a preclinical study.694 Duvelisib showed a synergistic anti-tumor effect when combined with treatment with anti-PD-1 and anti-OX40 in an A20 B-cell lymphoma model; this effect was associated with a remarkable reduction in Treg cells, M2 macrophages, and MDSCs.694
Chemotherapeutic agents are known to employ a variety of mechanisms to rapidly destroy dividing cells, including MDSCs. In addition to apoptosis of tumor cells, some cytotoxic anti-cancer agents, including paclitaxel, doxorubicin, trabectedin, gemcitabine, docetaxel, and 5-Fu, have been reported to activate the immune system through the depletion of MDSCs.678,695,696 Hence, antibodies targeting MDSC-specific markers would serve as an effective approach for the depletion of MDSCs. For instance, anti-Gr-1 mAb enhanced the function of CD8+ T cells and delayed tumor progression in tumor-bearing mice, while the efficacy of the anti-Gr-1 mAb in the depletion of MDSCs was potently biased toward young mice.697 Moreover, in a clinical trial, DS-8273a, a mAb against the R2 TRAIL-death receptor, was shown to selectively deplete MDSCs without affecting mature myeloid cells and lymphoid cells in patients with advanced solid tumors.698 Employing a competitive peptide phage display platform, a new peptide-Fc fusion recombinant protein specifically targeting MDSCs was successfully identified.699 Intravenous injection of this peptide completely eliminated both G-MDSCs and M-MDSCs from the blood, spleen, and tumors in vivo, and inhibited tumor xenograft growth compared with controls and treatment using Gr-1 mAb.699 Therefore, therapeutic approaches targeting MDSCs would shed new lights on cancer immunotherapy, especially, inflammation-related cancer.
Targeting tumor-associated neutrophils
In cancer, neutrophils make up a central component of the immune cells that infiltrate tumor tissues. It has been shown that accumulation of TANs is correlated with the progression and poor outcome of patients with cancer, especially in HCC, ICC, HNC, NSCLC, and RCC.700 Similar to TAMs, TANs are also classified into N1 anti-tumor and N2 pro-tumor subsets, with neutrophil polarization influencing the role they play in the TME.335 Tumor-associated N2 neutrophils, characterized by high expression of CXCR4, VEGF, and B/MMP9 gelatinase, are known to exert pro-tumoral activities. Neutrophil-targeting agents are being currently developed for the treatment of inflammatory or autoimmune diseases.701,702 Growing evidence have suggested that neutrophils might regulate the innate and adaptive immune system during tumor evolution.339,703 Therefore, TANs might serve as a promising target for anti-cancer therapies.
The prominent immunosuppressive TGF-β cytokine within the TME has been reported to induce a population of TANs with a pro-tumor N2 phenotype, suppressing the cytotoxicity of neutrophils and restricting their entry into the tumor.335,704 TGF-β is abundant both at primary and metastatic tumors, and neutrophil cytotoxicity is not evident in these sites, whereas the pro-tumor functions are manifested. In murine tumor models, blocking the TGF-β signaling using the SM16 TGF-β receptor inhibitor or anti-TGF-β antibodies enhanced the recruitment of cytotoxic N1-neutrophils into tumor sites and activated CD8+ T cells, resulting in the recession of tumor growth.335 Beyond that, recent studies have demonstrated that inhibition of TGF-β could serve as a promising strategy for the induction of the infiltration of CD8+ T cells, improving immunotherapy.705,706 In phase I/II trials, administration of GC1008 (fresolimumab, a human anti-TGFβ) showed preliminary evidence of anti-tumor activity and acceptable safety in patients with advanced malignant melanoma, RCC, pleural mesothelioma, as well as breast cancer.707,708,709 Likewise, the combination of galunisertib, a small-molecule selective inhibitor of the TGFβ receptor I, and sorafenib showed acceptable safety and a prolonged overall survival outcome in patients with HCC.710 Additional clinical trials have demonstrated that galunisertib enhanced the anti-tumor immunity of other agents, such as gemcitabine, nivolumab (anti-PD1), and durvalumab (anti-PD-L1) in multiple types of metastatic cancers, including HCC, breast, and pancreatic cancer.711,712
Type I-IFNs, which were initially identified as having antiviral functions, have been found to possess a N1-promoting effect that opposes that of TGF-β.713 Through the activation of various immune cells including neutrophils, T cells, NK cells, DCs, and macrophages, type I-IFNs showed potent anti-tumor function and inflammation regulation activities.457 For instance, the deficiency of type I-IFN was shown to lead to a higher metastasis load with a massive accumulation of N2-TANs, which was characterized by the high expression of prometastatic proteins (e.g., S100A8, S100A9, VEGF, and MMP9), in the lungs of a premetastatic murine model.714,715 In contrast, compared with untreated patients, the numbers of neutrophils in type I-IFN-treated patients were significantly decreased.715 In addition to suppressing the expression of proangiogenic factors, such as VEGF and MMP9, IFN-β enhanced the recruitment of neutrophils and their life span in tumor sites, thereby inhibiting tumor growth.716 Thus, enhancing the activity of IFNs at the TME could promote the anti-tumor cytotoxicity of neutrophils and might be considered as an additional strategy in anti-tumor immunotherapy.
G-CSF is a potent growth factor stimulating the biogenesis of neutrophils from progenitors and their trafficking from the BM to blood circulation.334,717 More importantly, blockade of G-CSF has been reported to not only lead to a decrease in the numbers of neutrophils, but to also induce a phenotype alteration characterized by the reduced levels of ROS and increased expression of Rb1. Therefore, the tumor-derived G-CSF is responsible for both the development and the activity of TANs in TME, providing a promising target for restricting immunosuppressive TANs. A number of studies have demonstrated the potential beneficial effect of inhibiting G-CSF in improving the anti-cancer therapeutic efficacy. For example, anti-G-CSF treatment could also induce anti-tumor immunity through the activation of NK cells, IL12-producing macrophages, as well as CD4+ and CD8+ T cells in a CRC mouse model.708
The CXCR2 axis is known to enhance the efflux of neutrophils from BM to blood, and the TME, thereby promoting tumor progression and metastasis.718 Inhibition of the CXCR2 signaling by knocking-down CXCL3, a critical ligand for CXCR2, or treating mice with SX-682 reduced the recruitment of TANs and enhanced the response to anti-PD1 therapy.719 In addition, the CXCL12/CXCR4 axis has also been shown to be a key retention signal for bone marrow neutrophils.712 Blockade of CXCR4 signaling using plerixafor suppressed the infiltration of neutrophils into the TME and improved the antiangiogenic therapy in a CRC mouse model.720 Increasing studies have shown the potential of the CXCR4 blockade in reducing immunosuppression and facilitating the immunotherapy response.721,722,723,724
The hepatocyte growth factor receptor (HGF-R) has emerged as an important regulator of neutrophils in tumor immunity and the response to immunotherapy. Importantly, high levels of HGF in blood have been shown to correlate with increased numbers of neutrophils and poor responses to ICB therapy in patients with metastatic melanoma.725 The inhibition of immunosuppressive TANs using cabozantinib, an inhibitor targeting HGF-R, VEGFR2, RET, and AXL, sensitized immunotherapy in a syngeneic murine model of CRC.726,727 Moreover, cabozantinib could lead to the infiltration of anti-tumor CXCR4+ neutrophils via the upregulation of neutrophil chemotactic factors, such as CXCL12 and high mobility group box 1 (HMGB1),728 suggesting that the HGF-R blockade could convert neutrophils from a tumor-promoting N2 to a tumor-inhibiting N1 phenotype.
Activating NK cells
Recent studies have highlighted the fact that patients with low cytotoxic activity of NK cells have a higher incidence of cancer, indicating that NK cells interfere with tumorigenesis.729 NK cells show cytotoxicity against diverse tumor cell types, but in the TME, Treg cells, M2 macrophages, and MDSCs can inhibit the activation and anti-tumor activity of NK cells through a series of mechanisms, such as the secretion of immunosuppressive products or interfering with the complex receptor array.730 For example, activated platelets can directly inhibit NK cells, while cytokines and metabolites, including TGF-β, adenosine, PGE2, IDO, and others, can directly suppress the maturation, proliferation, and function of NK cells.400,730 Some clinical responses to activating T-cell cytotoxicity immunotherapy, antibody-based, and tyrosine kinase inhibitor-based immunotherapy were shown to positively correlate with the activation of NK cells.731,732 Generally, overcoming the immunosuppressive TME to restore the function of NK cells is a potential therapeutic option for cancer treatment.
NK cells express a broad variety of activating and inhibitory receptors, which can be targeted by antibodies and soluble ligands to enhance the activity of NK cells.733 During infection, the activation of NK cells has been shown to be triggered by multiple pro-inflammatory cytokines, such as IL-2, IL-12, IL-15, IL-18, type I-IFN (IFN-α and IFN-β), and IFN-γ. IL-15 plays an essential role in the regulation of the development and activation of NK cells.734 In patients with non-Hodgkin’s lymphomas, high concentrations of serum IL-15 following autologous peripheral blood hematopoietic stem cell transplantation (APHSCT) were associated with better survival.735 In addition, systemic administration of recombinant IL-15 could stimulate the activity of NK cells. In a phase I clinical trial of patients with metastatic malignancies, administration of recombinant IL-15 induced the proliferation of NK cells and substantially increased their numbers.736 In addition, following haploidentical stem cell transplantation, the IL-15-stimulated infusion of NK cells was shown to induce a clinical response in 4 of 6 patients with pediatric solid refractory tumors.737 Furthermore, exposure to IL-2 stimulated signaling from activating receptors of NK cells. A phase I clinical trial evaluating rituximab combined with IL-2 against B-cell non-Hodgkin’s lymphoma, revealed that addition of IL-2 to rituximab therapy was safe and resulted in NK cell accumulation and ADCC activity that correlated with the better responses.738 Type I IFN could also preactivate NK cells by activating their receptors. Cyclic GMP-AMP, a second-messenger was reported to activate the STING adaptor protein, stimulating the production of IFN-β, and resulting in the priming of NK cells for cytotoxicity.739 Furthermore, oncolytic viruses are known to be able to trigger the recruitment of immune cells and induce anti-cancer responses by activating NK cells and T cells, thus selectively killing cancer cells.740,741 In preclinical models, localized therapy using the oncolytic Newcastle disease virus (NDV) induced inflammatory immune infiltrates in distant tumors, making them susceptible to ICB immunotherapy through the activation of NK and T cells.742
Disruption of immunosuppression is another strategy to elicit NK cells in the TME. The inhibitory factors of solid tumors are known to be composed by a complex composition of immunosuppressive molecules, such as TGF-β, IL-10, IDO, PGE2, VEGF, iNOS, and ROS, produced by regulatory immune cells, such as Treg cells, MDSCs, and M2-TAMs, as well as by tumor cells themselves.730 These factors generate a chronic inflammatory and immunosuppressive TME, accelerating tumor progression. Besides, the PD-1 and CTLA-4 immune checkpoints, lymphocyte activation gene 3 protein (LAG3), and T-cell immunoglobulin mucin-3 (TIM3) are expressed in some types of NK cells, with their ligands potentially taking part in dampening NK anti-tumor responses; as a result, blockade of interaction with checkpoint inhibitors boosts the activity of NK cells.743,744,745
Killer cell immunoglobulin-like receptors (KIRs) are the most polymorphic among inhibitory receptors that bind the human leukocyte antigen (HLA) class I receptors. As such, blockage of KIRs using an anti-KIR blocking antibody (lirilumab) is currently tested in clinical trials.746 It has been reported that inhibition of the TAM tyrosine kinase receptors (e.g., Tyro3, Axl, and Mer) in NK cells can enhance the antimetastatic potential of NK cells in murine models.747 In humans, CD96 is known to be mainly expressed in NK cells, CD8+, and CD4+ T cells. Accordingly, Cd96 (−/−) mice displayed hyperinflammatory responses to LPS and resistance to 3′-methylcholanthrene (MCA)-induced carcinogenesis and lung metastases. Importantly, blockade of Cd96 using an anti-Cd96 antibody or gene knockout improved the function of NK cells in mice.748 After treatment with trastuzumab, NK cells were reported to be activated, expressing the co-stimulatory CD137 receptor in the peripheral blood of women with HER2-expressing breast cancer. An agonistic mAb specific for CD137 enhanced the trastuzumab-mediated cytokine secretion of NK cells and NK cell-mediated cytotoxicity.749,750 In addition, natural killer group protein (NKG)- 2A is an intracytoplasmic tyrosine-based inhibitory motifs (ITIMs)-bearing receptor expressed on both NK and T cells. Blocking the expression of the NKG2A inhibitory receptor enhanced the anti-tumor immunity mediated by NK and CD8+ T cells.751
It should be noted that NK cells often become anergic in TME, due to the chronic stimulation by putative self-ligands, with the stimulation not being mitigated by inhibitory receptors engaged with self-MHC.752 Similar to T cells, NK cells can also be reactivated ex vivo to be applied for adoptive cell transfer therapy.733 First, NK cells are transduced with cytokine-encoding genes to promote their expansion after infusion. For instance, when injected into immunodeficient mice, NK cells expressing IL-15 were demonstrated to expand well and infiltrate in multiple tissues; this expansion could be further heightened by treatment with IL-2. Second, a chimeric receptor composed of NKG2D, linked to CD3ζ could directly provide activation signals; so adding the DAP10 adaptor molecule to the vector construct could promote the expression of NKG2D and transduce the activation signals.379 NK cells endowed with the additional NKG2D-CD3ζ-DAP10 stimulus displayed a notably greater cytotoxic effect against multiple cancer cells both in vitro and in vivo, whereas toxicity toward nontransformed cells remained low.753 In addition, expression of anti-CD19 CAR in NK cells specifically and dramatically augmented the NK cell-mediated killing of leukemic cells.754 Given the successful anti-cancer therapeutic approaches involving the blockade of T-cell-directed immune checkpoints, these new combination strategies could also affect the anti-tumor functions of NK cells.
Targeting eosinophils
Given that TATE is associated with improved prognosis of some cancer types, promotion of the effector function of eosinophils cold serve as a potential strategy against some tumors. Several studies on preclinical murine tumor models have reported the eosinophil-targeting anti-tumor therapy. For example, eosinophils have been frequently observed in patients following immunotherapy with IL-2,755 IL-4,756 GM-CSF,757 or tumor vaccination.757 More specifically, a high count of eosinophils in the blood was associated with the responsiveness of patients to immunotherapy, in particular to ICB.758,759
Recruitment of eosinophils from the peripheral blood into tumor sites is known to be regulated by various mechanisms, including chemokines (CCR3 and CCR1 ligands), Th2 cell-derived cytokines (IL-5, IL-4), immunomodulators (GM-CSF, IL-4, and IL-2), as well as danger signal molecules (HMGB1 and IL-33).338,760 For instance, IL-5, which is known to be the most specific cytokine to eosinophils, was shown to be responsible for their selective expansion and survival.338 Besides, chemokine eotaxin-overexpressing HCC cells were observed to activate eosinophil-mediated anti-tumor immunity in the presence of IL-5.761 Furthermore, increased tumor-associated eosinophilia were observed in tumors of patients with bladder cancer following treatment with IL-2.755 IL-4 exhibited potent anti-tumor activity when present at the tumor site, accompanied by an inflammatory infiltrate comprised predominantly of eosinophils and macrophages; treatment with IL-4 stimulated systemic eosinophilia, as well as increased the levels of serum and urine major basic protein (MBP), which is an eosinophil granule protein.756 Moreover, administration of DAMPs or alarmins displayed anti-tumor immunity though the recruitment and activation of eosinophils into tumor sites.762,763
The anti-tumor responses of eosinophils are associated with the degranulation of eosinophils.764 For instance, IL-2 immunotherapy has been applied to treat both melanoma and RCC. The anti-tumor effect of systemic IL-2 therapy was also reported to correlate with the degranulation of eosinophils, which could have relied on antibody-dependent mechanisms.765,766 Consistent with IL-2, the administration of recombinant IL-4 led to the degranulation of eosinophils in a dose-dependent manner in patients with cancer.767 In addition, treatment with recombinant IL-25 was shown to induce eosinophilia, which was correlated with tumor suppression.768 Taken together, for some types of cancer, chemokines promoting the recruitment or degranulation of eosinophils appear to be a hopeful approach for improving the efficacy of immunotherapy.
Modulating the inflammation in acquired immunity
Activated adaptive immune cells, including T and B lymphocytes, are known to further amplify the initial inflammatory response.769 More specifically, Th1 cells activate macrophages both through cell-to-cell contact and secretion of IFN-γ,770 while Th2 cells activate eosinophils through the release of cytokines, and B cells secrete antibodies that activate the complement cascade, as well as phagocytes, NK cells, and mast cells through Fc receptors.351,771,772 However, certain adaptive immune cells, especially Treg cells and Breg cells have been found to be able to turn off the inflammatory response.773,774 Thus, activating acquired immunity is a promising new target for cancer immunotherapy and inflammation control.
Activating cytotoxic T cells
During the inflammation process, activated CD8+ T cells produce IFN-γ, TNF-α, and granzymes, which destroy target cells.775 Clinical evidence have shown that the number of TILs, particularly CD8+ T cells, is a positive prognostic marker of multiple solid tumors.776 However, the effector functions of CD8+ T cells have been shown to be gradually lost in TME during chronic inflammation, a condition named T-cell exhaustion.418 T-cell exhaustion, in which reduced and dysfunctional effector T cells lead to immune escape, is one of the mechanisms employed by pathogens or tumor cells to get rid of the control of immunologic surveillance.418 Currently, exogenous reactivation, or priming of CTLs has been demonstrated to overcome the chronic inflammatory TME, leading to successful immunotherapy strategies against cancer.
It is well known that cGAS–STING-mediated DNA sensing in cancer cells or phagocytes (e.g., DCs, macrophages) is crucial for detecting cytosolic DNA, inducing a type I-IFN response for augmenting anti-tumor immunity, as well as host defense against pathogens.777 It has been reported that once activated by ligands, STING aggregates in a perinuclear region and activates the TBK1 kinase, which in turn phosphorylates IRF3, directly launching the transcription of type I IFN genes. Evidence has shown that the STING agonist DMXAA induces a cooperation between T lymphocytes and myeloid cells, resulting in tumor regression in vivo.778
Besides, co-stimulatory and co-inhibitory receptors play a crucial role in T-cell biology, as they are known to determine the functional outcome of TCR signaling. Accumulative evidence have suggested that CD40 plays an intrinsic role in the co-stimulation of T cells.779 CD40 has been shown to activate multiple signaling pathways, including Ras, PI3K, and protein kinase C, resulting in NF-κB-dependent induction of cytotoxic mediators (e.g., granzyme and perforin), and boosting of CD8+ T cells.780 CD28, another major co-stimulatory molecule for the priming of T cells, was reported to recruit adaptors, such as PI3K, growth factor receptor-bound protein 2 (GRB2), and LCK, resulting in the activation of nuclear factor of activated T cells (NFAT), activator protein (AP)-1, and NF-κB.781 The CD28 signaling was further shown to amplify TCR signaling, including the expression of IL-2 and B-cell lymphoma (Bcl)-2, modulation of metabolism, and epigenetic changes.781,782 Nowadays, a substantial amount of drugs targeting co-stimulatory molecules are in clinical trials against cancer, including members of the TNF receptor superfamily, OX40 (CD134), CD27, and 4-1BB (CD137).783
One of the primary characteristics of exhaustion is the co-expression of high levels of a series of inhibitory receptors, including PD-1, CTLA-4, CD152, LAG-3, Tim-3, CD244/2B4, CD160, and TIGIT. Importantly, ICB has been found to be able to induce durable responses among multiple types of cancer both in patients and murine model.784 Two inhibitory molecules, the cytotoxic CTLA-4 and PD-1, have attracted much attention, because blockage of the CTLA-4 or PD-1 signaling has prominently improved the survival of patients with metastatic solid cancers.785 Furthermore, ICB alone or combinations with other immunotherapies, such as adoptive cell therapy and DC vaccination, has displayed some survival benefit for patients with advanced cancer.785
In the perspective of anti-tumor immunotherapy, modulating the actions of cytokines is an attractive strategy to control exhausted CD8+ T cells. For example, administration of IL-2 has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of metastatic RCC and melanoma.786 Moreover, establishing the effective combination of cytokine-targeted therapy and ICB is of great interest. Administration of IL-2 during chronic viral infection was demonstrated to exhibit striking synergistic effects with PD-1 blockade, thus enhancing virus-specific CD8+ T cells.787 The synergy of exogenous IL-2, PD-1 blockade, and a powerful T-cell vaccine combination therapy has been confirmed in a cancer model.788 Besides, blockade of IL-10R significantly improved the efficacy of anti-PD-L1 treatment, resulting in enhancing the function and viral clearance of T cells.789
Adoptive T-cell therapy includes the use of gene-modified T cells expressing novel TCR or CAR receptors that recognize tumor cells and carry out potent anti-tumor functions.790 CAR-T-cell therapy has shown tremendous success in B-cell malignancies, leading to the approval of this approach for certain types of leukemia and lymphomas by FDA.791 For instance, CD19-specific CAR-T cells (Kymriah and Yescarta) have yielded remarkable clinical trial results in the treatment of certain types of B-cell leukemia and lymphomas.792,793,794 However, the efficacy of CAR-T cells in solid tumors is limited to date, partly due to the lack of trafficking of CAR-T cells to the tumor site, insufficient activation of the transferred T cells, and the immunosuppressive TME in solid tumors.795
However, CAR-T-cell therapy can trigger a severe inflammatory storm, known as inflammatory cytokine release syndrome (CRS), which can lead directly to death. On a troubling note, the better therapeutic effects of CAR-T cells have been associated with a stronger CRS.29,30 T cells kill tumor cells partially by releasing perforin and granzymes. Nowadays, researchers have found that CAR-T cells attack leukemia cells, leading to the swelling and bursting of tumor cells. Subsequently, the lysis of tumor cells has been shown to activate macrophages, which release a large amount of inflammatory factors (e.g., IL-6 and IL-1), inducing CRS. Meanwhile, NK-T cells cause the apoptosis of tumor cells, which are more pyknotic.796 In order to prevent the CRS caused by CAR-T therapy, several approaches can be adopted. For instance: (1) shifting the module of death of tumor cells from pyroptosis to apoptosis, thereby controlling the occurrence of CRS; (2) eliminating the ATP-released pyroptosis of tumor cells; (3) suppressing the activation of macrophages. Collectively, restoring the function of exhausted CD8+ T cells is a potential strategy for achieving improved therapeutic benefits of anti-tumor immunotherapy.
Targeting Treg cells
Treg cells are usually identified as a specialized subset of CD4+ T cells functioning in the establishment and maintenance of immunosuppression, such as promoting the resolution of inflammation, suppressing aberrant immune responses against self-antigens, and limiting anti-tumor immune responses. Treg cells are known to suppress pro-inflammatory responses through the secretion of cytokines, such as IL-2, IL-10, IL-35, and TGF-β.797 Besides, Treg cells are able to regulate not only T cells but also B cells, NK cells, DCs, and macrophages via humoral and cell–cell contact mechanisms. For example, Treg cells have been reported to facilitate the conversion of DCs to a tolerogenic state through the expression of cytotoxic CTLA-4, and inhibit the proliferation of effector T cells (Teff) cells through the production of inhibitory molecules, such as tryptophan and adenosine, reducing the IL-2-dependent activation of CD8+ T cells and NK cells.797 Clinically, accumulation of infiltrating Treg cells in tumor has often been associated with poor prognosis of patients with cancer.798 Accumulating evidence suggesting that depletion of Treg cells or modulation of the function of Treg cells was able to evoke and enhance anti-tumor immune responses. Various molecules relatively specific to Treg cells are good candidates for the depletion or functional modulation of Tregs, such as immune checkpoint molecules (e.g., CTLA-4, GITR, LAG3, and PD-1), CCR4, and metabolites (e.g., PGE2, tryptophan, and adenosine) that have been targeted by Abs or small molecules.
As immune checkpoint molecules are known to be highly expressed in Treg cells, they could thus be targeted to control the function of Treg cells. One of the recent breakthroughs in cancer immunotherapy was the clinical use of anti-CTLA-4 antibodies (ipilimumab and tremelimumab), which were shown to induce tumor regression and improve the survival of patients with metastatic melanoma.799,800,801,802,803,804,805,806,807 Although targeting CTLA-4 was initially designed to reactivate Teff cells, CTLA-4 is also highly expressed on Treg cells, with anti-CTLA-4 antibodies inducing the depletion of Treg cells in the TME through the activation of ADCC.806,807 In addition, anti-CTLA-4 antibodies have been reported to exhibit complimentary activity with therapies targeting anti-PD-1 (nivolumab), another checkpoint inhibitor expressed on Treg cells, with their combined use being more beneficial than the use of either antibody alone.808,809,810 Furthermore, other molecules expressed by Treg cells with immunosuppressive activity, such as TIGIT, LAG3, and TIM3, are being currently considered and tested in clinical trials.811,812,813 Importantly, targeting TIM3 might be more advantageous than CTLA-4 and PD-1 blockage, because the expression of TIM3 is restricted to intratumoral T cells, and hence its inhibition is less likely to be associated with adverse autoimmune-like toxicities.814,815,816 Indeed, TIM3-deficient mice did not display autoimmune disorders.817
GITR, a member of TNFRs providing co-stimulatory signaling to activate T cells, is known to be highly expressed in intratumoral Treg cells. An agonistic antibody targeting GITR was shown to attenuate the activity of Treg cells, reduce their numbers, decrease their stability in tumors, and promote tumor regression in mice,818 particularly when combined with treatment with CTLA-4 or PD-1 inhibitors.819,820,821 Similar to CTLA-4 or PD-1 blockade, GITR ligation also improved the functions of Teff cells.822 In addition, OX40, another TNF receptor family member, showed similar patterns of expression and function to GITR. Treatment with anti-OX40 mAb impaired the function of Treg cells and enhanced Teff cell responses, resulting in increased anti-tumor immunity and improved tumor-free survival.823 Thus, TNFR2 is a potential target for the development of Treg cell-based immunomodulatory therapies.
Low-dose cyclophosphamide has been shown to deplete Treg cells by inhibiting their proliferation and inducing apoptosis, and to attenuate their function by suppressing the expression of FOXP3 and GITR.824 In addition, TKIs (sunitinib, sorafenib, and imatinib) have been reported to prevent the expansion and function of intratumoral Treg cells.825,826,827 Although these approaches have been shown to inhibit the proliferation and function of Treg cells, they are not specific to tumor-infiltrating Treg cells. Moreover, antibodies targeting CD25 (e.g., daclizumab, basiliximab, and LMB-2), have been applied for the depletion of Treg cells by inducing ADCC and complement-mediated cytotoxicity.797 Preclinical and clinical studies using a combination of anti-CD25 antibodies (DAB389IL-2) and DC vaccines displayed beneficial effects in patients with RCC.828 Furthermore, it was reported that in the treatment of relapses of leukaemia in patients who did not develop graft-versus-host disease (GVHD) during the first transplantation, depletion of Treg cells from hematopoietic stem cell transplantation (HSCT) showed better outcomes.829 However, global depletion of Treg cells has been shown to have variable efficacy and could potentially induce systemic complications.
Indoleamine 2,3-dioxygenase (IDO), an enzyme with 2 isoforms (IDO1 and IDO2), converts tryptophan to kynurenine, resulting in the exhaustion of tryptophan, a molecule that is crucial to the proliferation and differentiation of T cells. Accordingly, lack of tryptophan and overproduction of kynurenine has been shown to not only diminish the proliferation and survival of T cells, but also shift the differentiation of T cells into Treg cells.830 Interestingly, evidence have shown that combined treatment using an IDO inhibitor and a tumor vaccine induce the upregulation of IL-6 in pDCs and in situ, resulting in the conversion of a majority of Tregs to Th17-like cells, with marked enhancement in the activation and anti-tumor efficacy of Teff cells. These findings suggested that the combined application of IDO inhibitors and tumor vaccines could be an alternative strategy for the reduction of Treg cells by converting them to Teff-like cells.831
CCR4 is predominantly expressed by effector Treg cells, rather than naive Treg and Th2 cells,832 with the migration and infiltration of Treg cells into solid tumors appearing to be dependent on CCR4 ligands (e.g., CCL17 and CCL22).833,834 Blocking the interactions between CCL22 and CCR4 by anti-CCL22 antibody reduced the accumulation of intratumoral Treg cells and suppressed tumor growth in mice.834 Clinical trials conducted using a humanized anti-CCR4 antibody (mogamulizumab, KW-0761) displayed a depletion of intratumoral Treg cells and anti-tumor activity with minimal or moderate toxicity.832,835 Similarly, in a mouse model with orthotopically implanted human OVC cells overexpressing CCL28 (the ligand for CCR10), intraperitoneal injection of an anti-CCR10 immunotoxin led to complete depletion of intratumoral Treg cells, reducing tumor growth.836
The effects of anti-inflammatory cytokines (e.g., TGF-β, IL-10, and IL-35) secreted by Treg cells in the TME can be blocked by using neutralizing antibodies. For instance, blockade of TGF-β expressed by Treg cells improved the anti-tumor immune response against melanoma, and suppressed the metastasis of pancreatic tumors in mice.837,838 Treatment with anti-IL-35 revealed the ability of suppressing tumor growth in multiple murine models of cancer.839 Therefore, depletion or dysfunction of intratumoral Treg cells, together with the augmentation of the tumor-killing activity of Teff cells, would make cancer immunotherapy more effective with less adverse effects.
Activating Th1 cells
The malfunction of Th1 cells has been observed in the peripheral blood of multiple types of cancer, involving them in tumorigenesis and tumor progression.840 Furthermore, a high density of tumor-infiltrating Th1 cells is considered to be a beneficial prognostic marker in several types of solid cancers, including OVC, CRC, NSCLC, and breast cancer.841,842 Clinical trials have demonstrated that inflammation driven by tumor-specific Th1 cells prevented the progression of malignancies,843 providing a strong rationale to develop anti-tumor Th1 immunity-activating immunotherapy.
An efficient strategy to stimulate the Th1 response is using a cancer vaccine. In phase I/II trials of patients with NSCLC, a human telomerase reverse transcriptase (hTERT)-derived helper peptide vaccine (GV1001) induced CD4+ T cells displaying a Th1 cytokine profile, and stimulated T-cell responses in >50% of subjects, without exhibiting clinically important toxicity.844,845 These results indicated a positive correlation between GV1001-specific Th1 responses and prolonged survival. Recently, universal cancer peptides (UCP), novel anti-tumor Th1-inducer peptides derived from hTERT, were demonstrated to induce a spontaneous CD4 T-cell response in 38% of patients with metastatic NSCLC, with the high-avidity UCP-specific CD4 T cells being Th1 polarized.846 In addition, tumor cell loaded type-1 polarized DCs induced the activation of antigen-specific Th1-type CD4+ T cells, resulting in a significant reduction in tumor growth.847
Application of Th1 cytokines has been suggested to reinforce the anti-tumor effects of immunotherapy. Th1-type cytokines, including IL-1, IL-2, IL-12, and GM-CSF are known potent stimulators of the differentiation of Th1 cells and Th1-based anti-tumor response.848 Although many preclinical studies have demonstrated the anti-tumor effects of Th1 cytokines, their clinical efficacy remains limited. To date, most studies on cytokine immunotherapy have focused on the mechanism by which to augment the Th1 response. To this end, many studies have combined cytokine-based therapy with other therapies to reverse immunosuppression in TME.849 For instance, IL-12 was found to promote the differentiation of Th1 cells and activation of NK cells, as well as increase the production of IFN-γ. More specifically, IL-12 induced Th1 responses against cancers and improved the anti-tumor efficacy of cancer vaccines, DC vaccines, and other cytokines (IL-18 and IL-15).849 Besides, IL-12 gene-modified DCs showed positive clinical response in patients with stage IV melanoma.850
Many immune adjuvants are known to display potent capability toward enhancing the production of Th1 cytokines and amplifying Th-immunity in response to cancer vaccines. Currently, several important Th1 adjuvants have been used for the activation of Th1 immunity, such as Bacillus Calmette-Guérin (BCG), heat-shock proteins (HSPs), TLR9 agonists, and unmethylated cytosine phophateguanosine oligodeoxynucleotides (CpG-ODN).848
Evidence has shown that PGE2 shifts the balance away from Th1 responses toward Th2 responses.851 Overproduction of PGE2 has been observed in multiple Th2-associated diseases, including atopic dermatitis and asthma.852 Moreover, inhibition of prostaglandin synthesis using COX2 inhibitors was reported to cause an augmentation of the Th1 response,853 suggesting that reducing the production of PGE2 would induce the Th1 response, improving the efficacy of anti-tumor immunotherapy. Therefore, developing strategies focusing on the activation of the Th1 immunity response would contribute to successful anti-tumor immunotherapy.
Targeting Th2 cells
Th2 cells are regulated by the innate immune system through the use of IL-25, IL-33, and TSLP cytokines. Preclinical breast cancer models have demonstrated their important role in cancer development and metastasis. IL-33, an important member of the IL-1 family was shown to play a pivotal role in regulating immune responses through interactions with its suppression of tumorigenicity 2 (ST2) receptor.854 Once activated, the IL-33/ST2 pathway has been demonstrated to regulate Th2 immune responses in autoimmune and inflammatory conditions.855 In addition, IL-25, another Th2 promoting cytokine, was found to be highly expressed in human and murine breast cancer.856 Blockade of IL-25 by an antagonistic antibody decreased Th2 and M2 macrophages in the primary TME and inhibited tumor metastasis.856 Thus, restraining the activation of Th2 cells or reducing the production of type 2 effector cytokines would benefit cancer immunotherapy.
Targeting Th17 cells
The IL-17-secreting CD4+ T cells have been defined as Th17 cells, and constitute ~1% of CD4+ T cells in the peripheral blood of healthy donors.857 Through the secretion of IL-17, IL-17F, and IL-22, Th17 cells play key roles in many human diseases including inflammation, autoimmune diseases, and cancer. Importantly, Th17 cells have been reported in many types of human cancers, impacting the prognosis of patients. For example, high levels of Th17 cells have been associated with improved prognosis of patients with OSCCs,11 and salivary gland tumors.858 Patients with melanoma, early-stage OVC, and malignant pleural effusions exhibiting increased numbers of Th17 cells were reported to have better survival.859 As experimental and clinical studies have demonstrated that targeting IL-17 has achieved great efficacy in autoimmune diseases, such as psoriasis,860 it is predictable that manipulation of Th17-cell biology would be a promising therapeutic modality for the treatment of Th17-affected cancers.
In CRC, however, high levels of IL-17 were observed to refrain Th1-armed anti-tumor immunity, in part by attracting myeloid cells into tumors. Deletion or blockade of IL-17 suppressed the tumor-promoting inflammation, reactivated tumor immunosurveillance, and reduced the frequency of tumorigenesis in lung cancer models.460,861 Accordingly, patients with CRC could be benefited by cancer immunotherapy using the anti-IL-17 approach as adjuvant therapies, which would contribute to the inhibition of both IL-17-mediated tumor promotion and T-cell exclusion.
Furthermore, the development and function of Th17 cells have been shown to be regulated by innate system-derived pro-inflammatory cytokines, such as IL-6, IL-23, and IL-1. In particular, IL-23 promotes inflammatory responses, such as the upregulation of the MMP9 matrix metalloprotease, and increases angiogenesis, but reduces the infiltration of CD8 T cells.862,863 As a result, transplanted tumors were observed to be growth-restricted in IL-23R-deficient mice.864 Besides, IL-6 has been shown to stimulate the production of Th2 type cytokines and upregulate the expression of VEGF and NRP-1 in pancreatic cancer cells.865 Agents targeting IL-6, IL-6 receptor, or JAKs have already received U.S. FDA approval for the treatment of inflammatory conditions or myeloproliferative neoplasms. Moreover, to reduce the adverse effects of CAR-T cells, combinations with anti-IL-6/IL-6R signaling strategies are being currently evaluated in patients with hematopoietic malignancies and solid tumors.487,866
Activating B cells
As the central compose of humoral immunity, B-lymphocytes are known to function in the production of antibodies, presentation of antigens, and secretion of inflammatory cytokines.867 TIBs can be detected in various solid tumors.868,869 Evidence have suggested that in some cancers TIBs inhibit tumor progression by secreting antibodies and cytokines, promoting T-cell response, and directly destroying tumor cells.427,870 Nowadays, several strategies have been developed to fully unleash the anti-tumor potential of B cells.
In order to activate cytotoxic T cells against tumors, B-cell-based cancer vaccines have been designed for the stimulation of B cells. In this context, the use of CD40 stimulation has been widely studied. The ligation of CD40 with CD40L was reported to stimulate the expression of co-stimulatory molecules and cytokines, with CD40-activated B cells increasing the potential to facilitate the activation of naive and memory T cells.871,872 In addition, it has been shown that these CD40-activated B cells were resistant to the immunosuppressive TME,873 and could reach secondary lymphoid organs after being injected in vivo, where they could efficiently activate T cells.874 The CD40-activated B cells have been tested and validated in preclinical models of human papillomavirus 16 (HPV16) E6- and E7-expressing TC-1 tumors,875 B16-F10 melanoma, E.G7 lymphoma,872 4T1 breast tumor metastasis,876 and spontaneous non-Hodgkin’s lymphoma.871
CpG-ODN, a TLR9 ligand, can also be used to activate B cells. In a mouse model of B16-F10-derived lung metastases, injection of CpG-activated B cells was reported to cause a regression of metastases and a less immunosuppressive TME.877 The GIFT4, a fusion between GM-CSF and IL-4 cytokines, which were found to unexpectedly cluster the respective receptors on B cells, resulted in the activation of the JAK/STAT pathway in B cells.878 This clustering could promote the proliferation of B cells and their differentiation from naive B cells toward activate helper B cells. Subsequently, these activated B cells were observed to act as APCs, secreting cytokines and expressing co-stimulatory markers, resulting in the activation of T cells. Administration of GIFT4 to melanoma-bearing mice caused an efficient regression of tumors. Furthermore, tumors were found to be resistant to GIFT4 in B-cell-deficient mice, suggesting that the anti-tumor effect of GIFT4 was B-cell-dependent.878
In addition, tumor-derived autophagosomes enriched in defective ribosomal products (DRibbles) were shown to be captured and internalized by B cells in vivo.879 These DRibbles contain tumor-specific antigens and activate B cells with increased expression of MHC class I and II molecules, CD86, and CD40. Then, these activated B cells can present DRibbles-derived antigens to stimulate the anti-tumor T-cell response. In lymphoma- or HCC-bearing mice, the combined injection of DRibbles and DRibble-loaded B cells led to the control of tumor growth.879,880
Targeting Breg cells
In human cancer, the frequencies of regulatory B (Breg) cells have been shown to usually increase with tumor progression, and to be enriched in tumors compared with peripheral blood or adjacent normal tissues. Breg cells are known to mediate inflammation and maintain homeostasis mainly via the secretion of anti-inflammatory cytokines, such as IL-10, TGF-β, and IL-35,881 directly killing effector cells by expressing Fas-L.882 Through the secretion of TGF-β, Breg cells were reported to promote the transformation of effector CD4+ T cells to active Tregs, which in turn suppressed the proliferation of T cells and facilitated tumor metastasis.883 Moreover, IL-21 was shown to induce granzyme B (GrB)+ Breg cells contributing to tumor escape from an efficient anti-tumor immune response.884 In addition, Breg cells were observed to dampen immune responses through the inhibition of the differentiation of DCs and the proliferation of Th1 and Th17 cells.885 These results suggested that Breg cells play an important role in regulating inflammation and the immunosuppressive TME, which prevent the anti-tumor immune process.
LXA4, an endogenous eicosanoid derived from AA, has been highlighted in the regulation of inflammation.886 Evidence have shown that LXA4 repressed the generation of Breg cells by dephosphorylating both STAT3 and ERK, resulting in impaired tumor growth.26 Targeting Breg cells by LXA4 decreased the number of Treg cells in tumor tissues, as well as enhanced the activities of cytotoxic T cells.26 These findings revealed that targeting Breg cells through the administration of LXA4 could have potential clinical applications. Interestingly, another metabolite from this pathway was observed to display the opposing effect: LTB4 could trigger the conversion of naive B cells into Breg cells. Furthermore, MK886, a 5-lipoxygenase activating protein (FLAP) inhibitor and antagonist of PPARα, could inhibit the generation of Breg cells, suggesting the crucial role of the 5-LOX/FLAP pathway in the differentiation and suppressive function of Breg cells.887
The phytoalexin resveratrol, which is known to block the phosphorylation of STAT3, was demonstrated to cause a decreased proportion of Breg cells in 4T1-bearing mice, inhibiting the formation of lung metastases.887 Likewise, total glucosides of paeony (TGP) extracted from plant were found to exert anti-inflammatory and immunomodulatory activities. In a rat model of diethylnitrosamine (DEN)-induced HCC, treatment with TGP resulted in a reduction of nodules and improvement of survival through a reduction in the numbers of Breg cells.888
Furthermore, tumor-infiltrating Breg cells with increased expression of PD-L1 and TGF-β suppressed the proliferation of CD4+ T cells, CD8+ T cells, and NK cells. Monoclonal antibodies targeting TGF-β or PD-L1 notably suppressed tumor growth and reduced the number of Breg cells in mice.887 Treatment with ibrutinib was shown to improve the immunosuppressive TME of chronic lymphocytic leukemia (CLL) via the STAT3-mediated suppression of Breg cells and the PD-1/PD-L1 pathway.889 Taken together, selectively targeting Breg cells in the TME appears to be a hopeful strategy for tumor immunotherapy.
Nowadays, numerous trials designed to evaluate the efficacy of inflammation modulators in cancer prevention and treatment are ongoing. Some ongoing or completed clinical trials with agents targeting cancer-associated inflammation signaling pathways are listed in Table 1. On the one hand, monotherapy with anti-inflammatory agents has been proved limited efficacy in cancer treatment, for example, monotherapy with agents targeting IL-6 only showed moderate activity against solid tumors in non-stratified patients (NCT00841191, NCT01531998). On the other hand, modulating inflammation can boost anti-cancer efficacy in synergy with chemotherapy or immunotherapies, for example, compared with placebo, adjuvant therapy with the celecoxib can benefit disease free survival (DFS) of two years and overall survival (OS) in primary breast cancer patients received endocrine treatment according to local practice (NCT02429427).
Hence, based on the relationship between inflammation and cancer, targeting inflammatory cells or inflammatory factors would contribute to the achievement of better outcomes for cancer therapeutics. The acute inflammation induced by some therapies (e.g., recombination IFN, TLRs activator, STING activator) can redirect the pro-tumor TME toward an anti-tumor immune milieu, which can enhance efficiency of anti-cancer therapies (e.g., chemotherapy, radiotherapy and immunotherapy). Moreover, a number of therapeutic strategies to limit chronic inflammation (incuding systermic and local inflammation) have been successfully applied in clinical or preclinical tumor models to prevent tumorigenesis, or sensitize tumor to anti-cancer therapies including chemotherapy, radiotherapy, and immunotherapy.
Conclusion and remarks
Acute inflammation is the initial response to harmful stimuli, with the persistence of inflammatory factors potentially inducing chronic inflammation.46 Innate immune cells (endothelial cells, neutrophils, macrophages, mast cells, NK cells, and DCs) and adaptive immune cells (T cells and B cells), as well as pro-inflammatory factors (vasoactive amines, vasoactive peptides, complement fragments, and some cytokines, such as IL-1, IL-6, IL-15, IL-17, IL-23, TNF-α, and IFN-γ) are important for the initiation of inflammation. Besides, chemokines (CCL2, CXCL12) are necessary for the recruitment of inflammatory cells in the inflammatory area. However, anti-inflammatory cells (M2 macrophages, Th2, Tregs, and MDSCs), some cytokines (IL-4, IL-10, IL-13, and TGF-β) and SPM (LTA4, LXA4, LXB4, lipoxins, RvE, RvD, MaR1, MaR2, DHPA, PCTR1, and protectin D1) are involved in the resolution of inflammation. The effect of inflammation on most cancers is two-edged, with cancer also affecting the process of inflammation. Normally, the immune system recognizes and removes the pathogens and tumor cells, thus inhibiting tumor growth.369 However, during chronic inflammation, inflammatory cells and cytokines might act as tumor promoters affecting cell survival, proliferation, invasion, and angiogenesis.276
Based on the close relationship between inflammation and tumor, targeting inflammation is an important way for improving anti-cancer treatment. There are two aspects in targeting inflammation for cancer treatment. Activating anti-cancer immunity cells (e.g., DCs, NK cells, NKT cells, CTLs, Th1 cells, and B cells) can improve the cancer-killing ability of the immune system. Concomitantly, inhibiting procancer immune cells (e.g., mast cells, TAMs, MDSCs, TANs, eosinophils, Th2 cells, Th17 cells, Treg cells, and Breg cells) or converting their polarization to anti-tumor type by targeting key signal pathways can impede the immunosuppressive effect and the progression of cancer. For example, ablating unfolded protein response mediator PERK in MDSC can reverse their pro-tumor role and elicit anti-tumor T cells.890 Besides, the intestinal microbiome also played an important role in inflammation and cancer, especially between IBD and CRC.271,272 Microbiota are known to directly or indirectly (via their metabolites, such as polysaccharide β-dextran, LPS, deoxycholic acid (DCA), short-chain fatty acid (SCFA), butyrate, and propionate) affect the differentiation and function of immune cells (e.g., M2-TAMs, TANs, Treg cells, DCs, and CD8+IFN-γ+ T cells), potentially altering their effects on tumors.275,276,277,279,280,891,892 Thus, intestinal microbiota are promising targets for the treatment of inflammation-associated cancer and fecal microbiota transplant (FMT) is becoming an effective method to improve the intestinal microbiome.893
Although the inhibition of inflammation targeting the innate and adaptive immunity has offered remarkable achievements in the clinical field of cancer therapy, several obstacles and challenges remain exist. Cancer therapy-induced inflammation often endows residual cancer cells with resistance to subsequent courses of treatment, enhancing cancer progression.894 For example, the Fc side of the checkpoint antibody could cause ADCC and CDC.895 Besides, ipilimumab (anti-CTLA-4) was shown to promote colitis and hypophysitis,896,897 while anti-PD-1 therapy improved the incidence of thyroiditis, pneumonia, and diabetes.897,898,899 Moreover, CAR-T therapy could cause a “cytokine storm”, with insufficient persistence of CAR-T cells leading to high recurrence rates of cancer.900 Therefore, combination of anti-inflammatory strategies with cancer treatment have improved the anti-cancer effect in some clinical cases and in vivo experiments, such as COX-2 inhibitors,901 NSAIDs,902,903,904 LOX Inhibitors,905 and statins.906 Besides, precision medicine, also be called personalized medicine, should also be taken into account in different inflammatory responses of cancer patients during the anti-tumor process and adopt personalized therapeutic strategies targeting inflammation.
As we underlined the several mechanisms of the interaction between inflammation and cancer, the essence of inflammation-targeting cancer therapy is to promote cancer-inhibiting inflammation and inhibit cancer-promoting inflammation, while the biggest difficulty of treatment is to maintain the balance of inflammation. Except for the targets mentioned above, there are numerous molecules involved in the regulation of inflammation and cancer, like intestinal microbiota and their metabolites. Besides, anti-tumor therapies targeting inflammation should be incorporated into precision therapy. However, applying these theories to clinical cancer therapy is still a long way off. In addition, a large number of studies would continue to contribute to the reinforcement of the theoretical basis of inflammation-targeting cancer treatment, constantly updating the field.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Vasan, N., Baselga, J. & Hyman, D. M. A view on drug resistance in cancer. Nature 575, 299–309 (2019).
Tang, W. et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct. Target Ther. 5, 87 (2020).
Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, inflammation, and cancer. Cell 140, 883–899 (2010).
Coussens, L. M. & Werb, Z. Inflammation and cancer. Nature 420, 860–867 (2002).
Crusz, S. M. & Balkwill, F. R. Inflammation and cancer: advances and new agents. Nat. Rev. Clin. Oncol. 12, 584–596 (2015).
Shacter, E. & Weitzman, S. A. Chronic inflammation and cancer. Oncology 16, 217–226 (2002). 229; discussion 230-212.
Schaue, D. et al. Radiation and inflammation. Semin Radiat. Oncol. 25, 4–10 (2015).
Guthrie, G. J. et al. The systemic inflammation-based neutrophil-lymphocyte ratio: experience in patients with cancer. Crit. Rev. Oncol. Hematol. 88, 218–230 (2013).
Balkwill, F. & Mantovani, A. Inflammation and cancer: back to Virchow? Lancet 357, 539–545 (2001).
Punt, S. et al. A beneficial tumor microenvironment in oropharyngeal squamous cell carcinoma is characterized by a high T cell and low IL-17(+) cell frequency. Cancer Immunol. Immunother. 65, 393–403 (2016).
Elinav, E. et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 13, 759–771 (2013).
Ma, Y. et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38, 729–741 (2013).
Nathan, C. & Ding, A. Nonresolving inflammation. Cell 140, 871–882 (2010).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Ritter, B. & Greten, F. R. Modulating inflammation for cancer therapy. J. Exp. Med. 216, 1234–1243 (2019).
Diakos, C. I., Charles, K. A., McMillan, D. C. & Clarke, S. J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 15, e493–e503 (2014).
Cuzick, J. et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 10, 501–507 (2009).
Rothwell, P. M. et al. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 377, 31–41 (2011).
Cuzick, J. et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann. Oncol. 26, 47–57 (2015).
Bonovas, S., Filioussi, K., Tsavaris, N. & Sitaras, N. M. Use of statins and breast cancer: a meta-analysis of seven randomized clinical trials and nine observational studies. J. Clin. Oncol. 23, 8606–8612 (2005).
Maisonneuve, P. & Lowenfels, A. B. Statins and the risk of colorectal cancer. N. Engl. J. Med. 353, 952–954 (2005). author reply 952-954.
Bonovas, S., Nikolopoulos, G. & Sitaras, N. M. Statins and reduced risk of hepatocellular carcinoma in patients with hepatitis C virus infection: further evidence is warranted. J. Clin. Oncol. 31, 4160 (2013).
Hao, H. et al. Lipoxin A4 and its analog suppress hepatocellular carcinoma via remodeling tumor microenvironment. Cancer Lett. 309, 85–94 (2011).
Li, Y. et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene 30, 3887–3899 (2011).
Wang, Z. et al. Lipid mediator lipoxin A4 inhibits tumor growth by targeting IL-10-producing regulatory B (Breg) cells. Cancer Lett. 364, 118–124 (2015).
Mulder, W. J. M. & Gnjatic, S. Cancer immunotherapy: from local to global. Nat. Nanotechnol. 12, 840–841 (2017).
Gun, S. Y., Lee, S. W. L., Sieow, J. L. & Wong, S. C. Targeting immune cells for cancer therapy. Redox Biol. 25, 101174 (2019).
Davila, M. L. et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 6, 224ra225 (2014).
Bonifant, C. L., Jackson, H. J., Brentjens, R. J. & Curran, K. J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 3, 16011 (2016).
Dorward, D. A., Lucas, C. D., Rossi, A. G., Haslett, C. & Dhaliwal, K. Imaging inflammation: molecular strategies to visualize key components of the inflammatory cascade, from initiation to resolution. Pharm. Ther. 135, 182–199 (2012).
Sun, S. C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 17, 545–558 (2017).
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Kono, H. & Rock, K. L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289 (2008).
Nathan, C. Points of control in inflammation. Nature 420, 846–852 (2002).
Headland, S. E. & Norling, L. V. The resolution of inflammation: Principles and challenges. Semin. Immunol. 27, 149–160 (2015).
Wright, H. L., Moots, R. J., Bucknall, R. C. & Edwards, S. W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 49, 1618–1631 (2010).
Savill, J. S. et al. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest. 83, 865–875 (1989).
Rossi, A. G. et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 12, 1056–1064 (2006).
Gilroy, D. W., Lawrence, T., Perretti, M. & Rossi, A. G. Inflammatory resolution: new opportunities for drug discovery. Nat. Rev. Drug Disco. 3, 401–416 (2004).
Serhan, C. N. The resolution of inflammation: the devil in the flask and in the details. Faseb J. 25, 1441–1448 (2011).
Xu, H. et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830 (2003).
Kumar, R., Clermont, G., Vodovotz, Y. & Chow, C. C. The dynamics of acute inflammation. J. Theor. Biol. 230, 145–155 (2004).
Serhan, C. N. Controlling the resolution of acute inflammation: a new genus of dual anti-inflammatory and proresolving mediators. J. Periodontol. 79, 1520–1526 (2008).
Ploeger, H. E., Takken, T., de Greef, M. H. & Timmons, B. W. The effects of acute and chronic exercise on inflammatory markers in children and adults with a chronic inflammatory disease: a systematic review. Exerc. Immunol. Rev. 15, 6–41 (2009).
Boyle, M. et al. Chronic inflammation and angiogenic signaling axis impairs differentiation of dental-pulp stem cells. PLoS ONE 9, e113419 (2014).
Feehan, K. T. & Gilroy, D. W. Is resolution the end of inflammation? Trends Mol. Med. 25, 198–214 (2019).
Nasef, N. A., Mehta, S. & Ferguson, L. R. Susceptibility to chronic inflammation: an update. Arch. Toxicol. 91, 1131–1141 (2017).
Snell, L. M., McGaha, T. L. & Brooks, D. G. Type I interferon in chronic virus infection and cancer. Trends Immunol. 38, 542–557 (2017).
Pollard, K. M. Silica, silicosis, and autoimmunity. Front Immunol. 7, 97 (2016).
Monteiro, R. & Azevedo, I. Chronic inflammation in obesity and the metabolic syndrome. Mediators Inflamm. 2010, 289645 (2010).
Rogovskii, V. Modulation of inflammation-induced tolerance in cancer. Front. Immunol. 11, 1180 (2020).
DiNicolantonio, J. J. & O’Keefe, J. The importance of maintaining a low omega-6/omega-3 ratio for reducing the risk of inflammatory cytokine storms. Mo. Med. 117, 539–542 (2020).
Kozhieva, M. K. et al. Gut human microbiota and multiple sclerosis. Zh. Nevrol. Psikhiatr Im. S S Korsakova 117, 11–19 (2017).
Bozkurt, B., Mann, D. L. & Deswal, A. Biomarkers of inflammation in heart failure. Heart Fail Rev. 15, 331–341 (2010).
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C. & Gage, F. H. Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934 (2010).
Kawanishi, S., Ohnishi, S., Ma, N., Hiraku, Y. & Murata, M. Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. Int. J. Mol. Sci 18, 1808 (2017).
Eiro, N. & Vizoso, F. J. Inflammation and cancer. World J. Gastrointest. Surg. 4, 62–72 (2012).
Rogovskii, V. S. The linkage between inflammation and immune tolerance: interfering with inflammation in cancer. Curr. Cancer Drug Targets 17, 325–332 (2017).
Parkin, D. M., Hammerl, L., Ferlay, J. & Kantelhardt, E. J. Cancer in Africa 2018: the role of infections. Int. J. Cancer 146, 2089–2103 (2020).
Hashimoto, M. et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu. Rev. Med 69, 301–318 (2018).
Pober, J. S. & Cotran, R. S. The role of endothelial cells in inflammation. Transplantation 50, 537–544 (1990).
Steinke, J. W. & Borish, L. 3. Cytokines and chemokines. J. Allergy Clin. Immunol. 117, S441–S445 (2006).
Pober, J. S. & Sessa, W. C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 7, 803–815 (2007).
Butcher, E. C. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell 67, 1033–1036 (1991).
Locati, M. & Murphy, P. M. Chemokines and chemokine receptors: biology and clinical relevance in inflammation and AIDS. Annu Rev. Med 50, 425–440 (1999).
Kobayashi, Y. Neutrophil infiltration and chemokines. Crit. Rev. Immunol. 26, 307–316 (2006).
Kolaczkowska, E. & Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175 (2013).
Williams, M. R., Azcutia, V., Newton, G., Alcaide, P. & Luscinskas, F. W. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 32, 461–469 (2011).
Simpson, P. J. et al. Reduction of experimental canine myocardial reperfusion injury by a monoclonal antibody (anti-Mo1, anti-CD11b) that inhibits leukocyte adhesion. J. Clin. Invest. 81, 624–629 (1988).
Woodfin, A. et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 12, 761–769 (2011).
Corada, M. et al. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc. Natl Acad. Sci. USA 102, 10634–10639 (2005).
Nathan, C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6, 173–182 (2006).
Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 371, 531–539 (2018).
Harty, M. W. et al. Neutrophil depletion blocks early collagen degradation in repairing cholestatic rat livers. Am. J. Pathol. 176, 1271–1281 (2010).
Kuhl, A. A. et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology 133, 1882–1892 (2007).
Dewald, O. et al. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 96, 881–889 (2005).
Frantz, S. et al. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 27, 871–881 (2013).
Zhao, Y., Zou, W., Du, J. & Zhao, Y. The origins and homeostasis of monocytes and tissue-resident macrophages in physiological situation. J. Cell Physiol. 233, 6425–6439 (2018).
Bystrom, J. et al. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood 112, 4117–4127 (2008).
Stables, M. J. et al. Transcriptomic analyses of murine resolution-phase macrophages. Blood 118, e192–e208 (2011).
Fredman, G. & Serhan, C. N. Specialized proresolving mediator targets for RvE1 and RvD1 in peripheral blood and mechanisms of resolution. Biochem. J. 437, 185–197 (2011).
Galli, S. J., Nakae, S. & Tsai, M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 6, 135–142 (2005).
Yu, Y., Blokhuis, B. R., Garssen, J. & Redegeld, F. A. Non-IgE mediated mast cell activation. Eur. J. Pharm. 778, 33–43 (2016).
Costanza, M., Colombo, M. P. & Pedotti, R. Mast cells in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. Int. J. Mol. Sci. 13, 15107–15125 (2012).
Bischoff, S. C. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat. Rev. Immunol. 7, 93–104 (2007).
Dudeck, A. et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 34, 973–984 (2011).
Cecere, T. E., Todd, S. M. & Leroith, T. Regulatory T cells in arterivirus and coronavirus infections: do they protect against disease or enhance it? Viruses 4, 833–846 (2012).
Li, G. et al. Coronavirus infections and immune responses. J. Med. Virol. 92, 424–432 (2020).
Zabetakis, I., Lordan, R., Norton, C. & Tsoupras, A. COVID-19: the inflammation link and the role of nutrition in potential mitigation. Nutrients 12, 1466 (2020).
Kelly, B. & O’Neill, L. A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 25, 771–784 (2015).
Steinbach, E. C. & Plevy, S. E. The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm. Bowel Dis. 20, 166–175 (2014).
Martinek, J., Wu, T. C., Cadena, D., Banchereau, J. & Palucka, K. Interplay between dendritic cells and cancer cells. Int. Rev. Cell Mol. Biol. 348, 179–215 (2019).
Schuster, S., Hurrell, B. & Tacchini-Cottier, F. Crosstalk between neutrophils and dendritic cells: a context-dependent process. J. Leukoc. Biol. 94, 671–675 (2013).
Angeli, V. & Randolph, G. J. Inflammation, lymphatic function, and dendritic cell migration. Lymphat Res. Biol. 4, 217–228 (2006).
Salminen, A., Kaarniranta, K. & Kauppinen, A. The role of myeloid-derived suppressor cells (MDSC) in the inflammaging process. Ageing Res. Rev. 48, 1–10 (2018).
Goh, C. C. et al. Hepatitis C virus-induced myeloid-derived suppressor cells suppress NK cell IFN-gamma production by altering cellular metabolism via arginase-1. J. Immunol. 196, 2283–2292 (2016).
Huang, S. et al. EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res 79, 2009–2020 (2019).
Gleich, G. J. The functions of eosinophils. Ann. Inst. Pasteur Immunol. 137D, 136–141 (1986).
Knol, E. F. & Olszewski, M. Basophils and mast cells: underdog in immune regulation? Immunol. Lett. 138, 28–31 (2011).
Gilmartin, L., Tarleton, C. A., Schuyler, M., Wilson, B. S. & Oliver, J. M. A comparison of inflammatory mediators released by basophils of asthmatic and control subjects in response to high-affinity IgE receptor aggregation. Int. Arch. Allergy Immunol. 145, 182–192 (2008).
Silver, M. R. et al. IL-33 synergizes with IgE-dependent and IgE-independent agents to promote mast cell and basophil activation. Inflamm. Res 59, 207–218 (2010).
Gleich, G. J., Adolphson, C. R. & Leiferman, K. M. The biology of the eosinophilic leukocyte. Annu. Rev. Med 44, 85–101 (1993).
Strandmark, J. et al. Eosinophils are required to suppress Th2 responses in Peyer’s patches during intestinal infection by nematodes. Mucosal Immunol. 10, 661–672 (2017).
Tosello-Trampont, A., Surette, F. A., Ewald, S. E. & Hahn, Y. S. Immunoregulatory role of NK Cells in tissue inflammation and regeneration. Front. Immunol. 8, 301 (2017).
Nahrendorf, M. et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037–3047 (2007).
Hofmann, U. et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125, 1652–1663 (2012).
Wu, Y., Antony, S., Meitzler, J. L. & Doroshow, J. H. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 345, 164–173 (2014).
Back, M., Yurdagul, A. Jr, Tabas, I., Oorni, K. & Kovanen, P. T. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406 (2019).
Ding, N. et al. Mast cells are important regulator of acupoint sensitization via the secretion of tryptase, 5-hydroxytryptamine, and histamine. PLoS ONE 13, e0194022 (2018).
Shi, G. P., Bot, I. & Kovanen, P. T. Reply: The complexity of substance P-mediated mast cell activation. Nat. Rev. Cardiol. 14, 124 (2017).
Oschatz, C. et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity 34, 258–268 (2011).
Stoll, G. & Nieswandt, B. Thrombo-inflammation in acute ischaemic stroke - implications for treatment. Nat. Rev. Neurol. 15, 473–481 (2019).
Nissinen, L. & Kahari, V. M. Matrix metalloproteinases in inflammation. Biochim. Biophys. Acta 1840, 2571–2580 (2014).
Courtois, G. & Gilmore, T. D. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene 25, 6831–6843 (2006).
Rius, J. et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453, 807–811 (2008).
Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 441, 431–436 (2006).
Koehne, C. H. & Dubois, R. N. COX-2 inhibition and colorectal cancer. Semin. Oncol. 31, 12–21 (2004).
Szlosarek, P. W. & Balkwill, F. R. Tumour necrosis factor alpha: a potential target for the therapy of solid tumours. Lancet Oncol. 4, 565–573 (2003).
Uceyler, N. et al. Tumor necrosis factor-alpha links heat and inflammation with Fabry pain. Mol. Genet. Metab. 127, 200–206 (2019).
Jordan, S. C. et al. Interleukin-6, a cytokine critical to mediation of inflammation, autoimmunity and allograft rejection: therapeutic implications of IL-6 receptor blockade. Transplantation 101, 32–44 (2017).
Nguyen, D. P., Li, J. & Tewari, A. K. Inflammation and prostate cancer: the role of interleukin 6 (IL-6). BJU Int. 113, 986–992 (2014).
Lacy, P. Editorial: secretion of cytokines and chemokines by innate immune cells. Front. Immunol. 6, 190 (2015).
Rani, A., Dasgupta, P. & Murphy, J. J. Prostate cancer: the role of inflammation and chemokines. Am. J. Pathol. 189, 2119–2137 (2019).
Borroni, E. M., Savino, B., Bonecchi, R. & Locati, M. Chemokines sound the alarmin: The role of atypical chemokine in inflammation and cancer. Semin. Immunol. 38, 63–71 (2018).
Proost, P. et al. Chemokine isoforms and processing in inflammation and immunity. J. Autoimmun. 85, 45–57 (2017).
Lazennec, G. & Richmond, A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol. Med. 16, 133–144 (2010).
Chen, K. et al. Chemokines in homeostasis and diseases. Cell Mol. Immunol. 15, 324–334 (2018).
Yoshimura, T. The production of monocyte chemoattractant protein-1 (MCP-1)/CCL2 in tumor microenvironments. Cytokine 98, 71–78 (2017).
Yoshimura, T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol. Immunol. 15, 335–345 (2018).
Marquez-Curtis, L. A. & Janowska-Wieczorek, A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed. Res. Int. 2013, 561098 (2013).
Daniel, S. K., Seo, Y. D. & Pillarisetty, V. G. The CXCL12-CXCR4/CXCR7 axis as a mechanism of immune resistance in gastrointestinal malignancies. Semin. Cancer Biol. 65, 176–188 (2020).
Klos, A., Wende, E., Wareham, K. J. & Monk, P. N. International union of basic and clinical pharmacology [corrected]. LXXXVII. complement peptide C5a, C4a, and C3a receptors. Pharm. Rev. 65, 500–543 (2013).
Martin, M. & Blom, A. M. Complement in removal of the dead - balancing inflammation. Immunol. Rev. 274, 218–232 (2016).
Orsini, F., De Blasio, D., Zangari, R., Zanier, E. R. & De Simoni, M. G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell Neurosci. 8, 380 (2014).
Koelman, D. L. H., Brouwer, M. C. & van de Beek, D. Targeting the complement system in bacterial meningitis. Brain 142, 3325–3337 (2019).
Erdei, A., Andrasfalvy, M., Peterfy, H., Toth, G. & Pecht, I. Regulation of mast cell activation by complement-derived peptides. Immunol. Lett. 92, 39–42 (2004).
Gasque, P. Complement: a unique innate immune sensor for danger signals. Mol. Immunol. 41, 1089–1098 (2004).
Cain, S. A. & Monk, P. N. The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-Arg(74). J. Biol. Chem. 277, 7165–7169 (2002).
Janatova, J. C3, C5 components and C3a, C4a, and C5a fragments of the complement system. Methods Enzymol. 162, 579–625 (1988).
Lopez-Lera, A. et al. Complement as a diagnostic tool in immunopathology. Semin Cell Dev. Biol. 85, 86–97 (2019).
Morgan, B. P. & Harris, C. L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug Disco. 14, 857–877 (2015).
Basil, M. C. & Levy, B. D. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 16, 51–67 (2016).
Williams, K. I. & Higgs, G. A. Eicosanoids and inflammation. J. Pathol. 156, 101–110 (1988).
Serhan, C. N., Chiang, N. & Dalli, J. The resolution code of acute inflammation: novel pro-resolving lipid mediators in resolution. Semin. Immunol. 27, 200–215 (2015).
Serhan, C. N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Ess. Fat. Acids 73, 141–162 (2005).
Serhan, C. N., Chiang, N., Dalli, J. & Levy, B. D. Lipid mediators in the resolution of inflammation. Cold Spring Harb. Perspect. Biol. 7, a016311 (2014).
Withana, N. P. et al. Dual-modality activity-based probes as molecular imaging agents for vascular inflammation. J. Nucl. Med. 57, 1583–1590 (2016).
Zayani, Y. et al. Inflammations mediators and circulating levels of matrix metalloproteinases: biomarkers of diabetes in tunisians metabolic syndrome patients. Cytokine 86, 47–52 (2016).
Alameddine, H. S. & Morgan, J. E. Matrix metalloproteinases and tissue inhibitor of metalloproteinases in inflammation and fibrosis of skeletal muscles. J. Neuromuscul. Dis. 3, 455–473 (2016).
Baranek, T. et al. Elastin receptor (spliced galactosidase) occupancy by elastin peptides counteracts proinflammatory cytokine expression in lipopolysaccharide-stimulated human monocytes through NF-kappaB down-regulation. J. Immunol. 179, 6184–6192 (2007).
Maeno, T. et al. CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J. Immunol. 178, 8090–8096 (2007).
Campden, R. I. & Zhang, Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 670, 32–42 (2019).
Stoka, V., Turk, V. & Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 32, 22–37 (2016).
van Kasteren, S. I. & Overkleeft, H. S. Endo-lysosomal proteases in antigen presentation. Curr. Opin. Chem. Biol. 23, 8–15 (2014).
Ahmad, S. & Siddiqi, M. I. Insights from molecular modeling into the selective inhibition of cathepsin S by its inhibitor. J. Mol. Model 23, 92 (2017).
Tang, Y. et al. Cathepsin B inhibition ameliorates the non-alcoholic steatohepatitis through suppressing caspase-1 activation. J. Physiol. Biochem. 74, 503–510 (2018).
Pan, W. et al. Inhibition of Ctsk alleviates periodontitis and comorbid rheumatoid arthritis via downregulation of the TLR9 signalling pathway. J. Clin. Periodontol. 46, 286–296 (2019).
Asagiri, M. et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science 319, 624–627 (2008).
Anderson, P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat. Rev. Immunol. 10, 24–35 (2010).
Anel, R. & Kumar, A. Human endotoxemia and human sepsis: limits to the model. Crit. Care 9, 151–152 (2005).
Hesse, D. G. et al. Cytokine appearance in human endotoxemia and primate bacteremia. Surg. Gynecol. Obstet. 166, 147–153 (1988).
Mazumder, B., Sampath, P. & Fox, P. L. Regulation of macrophage ceruloplasmin gene expression: one paradigm of 3’-UTR-mediated translational control. Mol. Cells 20, 167–172 (2005).
Mazumder, B. et al. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell 115, 187–198 (2003).
Sampath, P. et al. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell 119, 195–208 (2004).
Muhl, H. & Pfeilschifter, J. Anti-inflammatory properties of pro-inflammatory interferon-gamma. Int. Immunopharmacol. 3, 1247–1255 (2003).
Mazumder, B., Mukhopadhyay, C. K., Prok, A., Cathcart, M. K. & Fox, P. L. Induction of ceruloplasmin synthesis by IFN-gamma in human monocytic cells. J. Immunol. 159, 1938–1944 (1997).
Sampath, P., Mazumder, B., Seshadri, V. & Fox, P. L. Transcript-selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3’ untranslated region. Mol. Cell Biol. 23, 1509–1519 (2003).
Mukhopadhyay, R., Jia, J., Arif, A., Ray, P. S. & Fox, P. L. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci. 34, 324–331 (2009).
Serhan, C. N. et al. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204 (2000).
Serhan, C. N. et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037 (2002).
Levy, B. D., Clish, C. B., Schmidt, B., Gronert, K. & Serhan, C. N. Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2, 612–619 (2001).
Serhan, C. N. & Chiang, N. Novel endogenous small molecules as the checkpoint controllers in inflammation and resolution: entree for resoleomics. Rheum. Dis. Clin. North Am. 30, 69–95 (2004).
Schwab, J. M., Chiang, N., Arita, M. & Serhan, C. N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874 (2007).
O’Meara, S. J., Rodgers, K. & Godson, C. Lipoxins: update and impact of endogenous pro-resolution lipid mediators. Rev. Physiol. Biochem. Pharm. 160, 47–70 (2008).
Serhan, C. N. Lipoxins and aspirin-triggered 15-epi-lipoxin biosynthesis: an update and role in anti-inflammation and pro-resolution. Prostaglandins Other Lipid Mediat 68-69, 433–455 (2002).
Serhan, C. N., Hamberg, M. & Samuelsson, B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl Acad. Sci. USA 81, 5335–5339 (1984).
Serhan, C. N., Arita, M., Hong, S. & Gotlinger, K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids 39, 1125–1132 (2004).
Serhan, C. N., Gotlinger, K., Hong, S. & Arita, M. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: an overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 73, 155–172 (2004).
Eickmeier, O. et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol. 6, 256–266 (2013).
Samson, M. et al. ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. Eur. J. Immunol. 28, 1689–1700 (1998).
Payrits, M. et al. Resolvin D1 and D2 inhibit transient receptor potential Vanilloid 1 and Ankyrin 1 ion channel activation on sensory neurons via lipid raft modification. Int. J. Mol. Sci. 21, 5019 (2020).
Chiang, N., Dalli, J., Colas, R. A. & Serhan, C. N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med 212, 1203–1217 (2015).
Back, M. & Hansson, G. K. Omega-3 fatty acids, cardiovascular risk, and the resolution of inflammation. FASEB J. 33, 1536–1539 (2019).
Lopez-Vicario, C. et al. Association of a variant in the gene encoding for ERV1/ChemR23 with reduced inflammation in visceral adipose tissue from morbidly obese individuals. Sci. Rep. 7, 15724 (2017).
Laguna-Fernandez, A. et al. ERV1/ChemR23 signaling protects against atherosclerosis by modifying oxidized low-density lipoprotein uptake and phagocytosis in macrophages. Circulation 138, 1693–1705 (2018).
Zhou, Y. et al. Fibrinogen-like protein 2 controls sepsis catabasis by interacting with resolvin Dp5. Sci. Adv. 5, eaax0629 (2019).
Marcheselli, V. L. et al. Neuroprotectin D1/protectin D1 stereoselective and specific binding with human retinal pigment epithelial cells and neutrophils. Prostaglandins Leukot. Ess. Fat. Acids 82, 27–34 (2010).
Kohli, P. & Levy, B. D. Resolvins and protectins: mediating solutions to inflammation. Br. J. Pharm. 158, 960–971 (2009).
Serhan, C. N. et al. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J. Immunol. 176, 1848–1859 (2006).
Serhan, C. N., Dalli, J., Colas, R. A., Winkler, J. W. & Chiang, N. Protectins and maresins: new pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta 1851, 397–413 (2015).
Chiurchiu, V. et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci. Transl. Med. 8, 353ra111 (2016).
Chatterjee, A. et al. The pro-resolving lipid mediator maresin 1 (MaR1) attenuates inflammatory signaling pathways in vascular smooth muscle and endothelial cells. PLoS ONE 9, e113480 (2014).
Hong, S. et al. Maresin-like lipid mediators are produced by leukocytes and platelets and rescue reparative function of diabetes-impaired macrophages. Chem. Biol. 21, 1318–1329 (2014).
Dalli, J. et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J. 27, 2573–2583 (2013).
Liening, S., Romp, E., Werz, O., Scriba, G. K. E. & Garscha, U. Liquid chromatography-coupled mass spectrometry analysis of glutathione conjugates of oxygenated polyunsaturated fatty acids. Prostaglandins Other Lipid Mediat. 144, 106350 (2019).
Liu, Y. J. et al. PCTR1 ameliorates lipopolysaccharide-induced acute inflammation and multiple organ damage via regulation of linoleic acid metabolism by promoting FADS1/FASDS2/ELOV2 expression and reducing PLA2 expression. Lab. Invest. 100, 904–915 (2020).
Ramon, S. et al. The protectin PCTR1 is produced by human M2 macrophages and enhances resolution of infectious inflammation. Am. J. Pathol. 186, 962–973 (2016).
Zhang, Q., Zhu, B. & Li, Y. Resolution of cancer-promoting inflammation: a new approach for anticancer therapy. Front. Immunol. 8, 71 (2017).
Xian, W. et al. The pro-resolving lipid mediator maresin 1 protects against cerebral ischemia/reperfusion injury by attenuating the pro-inflammatory response. Biochem. Biophys. Res. Commun. 472, 175–181 (2016).
Hwang, S. M., Chung, G., Kim, Y. H. & Park, C. K. The role of maresins in inflammatory pain: function of macrophages in wound regeneration. Int. J. Mol. Sci. 20, 5849 (2019).
Serhan, C. N., Chiang, N. & Dalli, J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol. Asp. Med. 64, 1–17 (2018).
Chiang, N. et al. New maresin conjugates in tissue regeneration pathway counters leukotriene D4-stimulated vascular responses. FASEB J. 32, 4043–4052 (2018).
de la Rosa, X. et al. Identification and complete stereochemical assignments of the new resolvin conjugates in tissue regeneration in human tissues that stimulate proresolving phagocyte functions and tissue regeneration. Am. J. Pathol. 188, 950–966 (2018).
Flesher, R. P., Herbert, C. & Kumar, R. K. Resolvin E1 promotes resolution of inflammation in a mouse model of an acute exacerbation of allergic asthma. Clin. Sci. 126, 805–814 (2014).
Wang, X. et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement. 11, 40–50 (2015). e41-42.
Yang, L. et al. Immune characteristics of severe and critical COVID-19 patients. Signal Transduct. Target Ther. 5, 179 (2020).
Xu, Z. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8, 420–422 (2020).
Mehta, P. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034 (2020).
Rokni, M., Hamblin, M. R. & Rezaei, N. Cytokines and COVID-19: friends or foes? Hum. Vaccin Immunother. 16, 2363–2365 (2020).
Otsuka, R. & Seino, K. I. Macrophage activation syndrome and COVID-19. Inflamm. Regen. 40, 19 (2020).
Das, U. N. Can bioactive lipids inactivate coronavirus (COVID-19)? Arch. Med. Res. 51, 282–286 (2020).
Fekete, T. et al. Regulatory NLRs control the RLR-mediated Type I interferon and inflammatory responses in human dendritic cells. Front. Immunol. 9, 2314 (2018).
Si, Y. et al. Posttranslational modification control of inflammatory signaling. Adv. Exp. Med. Biol. 1024, 37–61 (2017).
Istomin, A. Y. & Godzik, A. Understanding diversity of human innate immunity receptors: analysis of surface features of leucine-rich repeat domains in NLRs and TLRs. BMC Immunol. 10, 48 (2009).
Dolasia, K., Bisht, M. K., Pradhan, G., Udgata, A. & Mukhopadhyay, S. TLRs/NLRs: shaping the landscape of host immunity. Int. Rev. Immunol. 37, 3–19 (2018).
Broad, A., Kirby, J. A., Jones, D. E., Applied, I. & Transplantation Research, G. Toll-like receptor interactions: tolerance of MyD88-dependent cytokines but enhancement of MyD88-independent interferon-beta production. Immunology 120, 103–111 (2007).
Wieland, C. W. et al. The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable haemophilus influenzae from the mouse lung. J. Immunol. 175, 6042–6049 (2005).
Triantafilou, M. et al. Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J. Biol. Chem. 281, 31002–31011 (2006).
Bagchi, A. et al. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J. Immunol. 178, 1164–1171 (2007).
Tamassia, N. et al. The MyD88-independent pathway is not mobilized in human neutrophils stimulated via TLR4. J. Immunol. 178, 7344–7356 (2007).
Karnati, H. K. et al. TLR-4 signalling pathway: MyD88 independent pathway up-regulation in chicken breeds upon LPS treatment. Vet. Res. Commun. 39, 73–78 (2015).
Inohara, N. & Nunez, G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 3, 371–382 (2003).
Chamaillard, M., Girardin, S. E., Viala, J. & Philpott, D. J. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell Microbiol. 5, 581–592 (2003).
Ting, J. P. et al. The NLR gene family: a standard nomenclature. Immunity 28, 285–287 (2008).
Wagner, R. N., Proell, M., Kufer, T. A. & Schwarzenbacher, R. Evaluation of Nod-like receptor (NLR) effector domain interactions. PLoS ONE 4, e4931 (2009).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 (2010).
Davis, B. K., Wen, H. & Ting, J. P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735 (2011).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Man, S. M., Karki, R. & Kanneganti, T. D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 277, 61–75 (2017).
Xue, Y., Enosi Tuipulotu, D., Tan, W. H., Kay, C. & Man, S. M. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 40, 1035–1052 (2019).
Feng, S., Fox, D. & Man, S. M. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J. Mol. Biol. 430, 3068–3080 (2018).
Kanneganti, A. et al. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J. Exp. Med. 215, 1519–1529 (2018).
Xiao, J. et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLoS Biol. 16, e3000047 (2018).
Xu, B. et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 68, 773–782 (2018).
McKenzie, B. A. et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl Acad. Sci. USA 115, E6065–E6074 (2018).
Hu, B. et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl Acad. Sci. USA 107, 21635–21640 (2010).
Feng, X. et al. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res 36, 81 (2017).
Chen, L. et al. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell Mol. Life Sci. 75, 2045–2058 (2018).
Pikarsky, E. et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431, 461–466 (2004).
Carbia-Nagashima, A. et al. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell 131, 309–323 (2007).
Herrington, F. D., Carmody, R. J. & Goodyear, C. S. Modulation of NF-kappaB signaling as a therapeutic target in autoimmunity. J. Biomol. Screen 21, 223–242 (2016).
Peng, C., Ouyang, Y., Lu, N. & Li, N. The NF-kappaB signaling pathway, the microbiota, and gastrointestinal tumorigenesis: recent advances. Front. Immunol. 11, 1387 (2020).
Park, Y. R. et al. NF-kappaB signaling is key in the wound healing processes of silk fibroin. Acta Biomater. 67, 183–195 (2018).
McDaniel, D. K., Eden, K., Ringel, V. M. & Allen, I. C. Emerging roles for noncanonical NF-kappaB signaling in the modulation of inflammatory bowel disease pathobiology. Inflamm. Bowel Dis. 22, 2265–2279 (2016).
Mizukami, Y. et al. Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat. Med. 11, 992–997 (2005).
Taniguchi, K. & Karin, M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol. 18, 309–324 (2018).
Tanaka, T. Introduction for inflammation and cancer. Semin. Immunopathol. 35, 121–122 (2013).
Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
Corrales, L., Matson, V., Flood, B., Spranger, S. & Gajewski, T. F. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 27, 96–108 (2017).
Dawson, M. A. & Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 (2012).
Gupta, S. C., Kunnumakkara, A. B., Aggarwal, S. & Aggarwal, B. B. Inflammation, a double-edge sword for cancer and other age-related diseases. Front. Immunol. 9, 2160 (2018).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Murata, M. Inflammation and cancer. Environ. Health Prev. Med. 23, 50 (2018).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Ell, B. & Kang, Y. Transcriptional control of cancer metastasis. Trends Cell Biol. 23, 603–611 (2013).
Easwaran, H., Tsai, H. C. & Baylin, S. B. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 54, 716–727 (2014).
Iliopoulos, D., Hirsch, H. A. & Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706 (2009).
Chiarugi, P. & Cirri, P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 380, 272–280 (2016).
Fischer, K. et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819 (2007).
Gottfried, E. et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107, 2013–2021 (2006).
Bader, J. E., Voss, K. & Rathmell, J. C. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell 78, 1019–1033 (2020).
Mu, X. et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 17, 428–438 (2018).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
Jess, T. et al. Decreasing risk of colorectal cancer in patients with inflammatory bowel disease over 30 years. Gastroenterology 143, 375–381 (2012).
Keller, D. S., Windsor, A., Cohen, R. & Chand, M. Colorectal cancer in inflammatory bowel disease: review of the evidence. Tech. Coloproctol. 23, 3–13 (2019).
Pekow, J. et al. IBD-associated colon cancers differ in DNA methylation and gene expression profiles compared with sporadic colon cancers. J. Crohns Colitis 13, 884–893 (2019).
Bian, S. et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 362, 1060–1063 (2018).
Gottlieb, M. & Nakitende, D. Comparison of tamsulosin, nifedipine, and placebo for ureteric colic. CJEM 19, 156–158 (2017).
Loor, A. & Dumitrascu, D. L. Helicobacter pylori infection, gastric cancer and gastropanel. Rom. J. Intern. Med. 54, 151–156 (2016).
Bain, C. C. & Mowat, A. M. Macrophages in intestinal homeostasis and inflammation. Immunol. Rev. 260, 102–117 (2014).
Yang, H. et al. A common antimicrobial additive increases colonic inflammation and colitis-associated colon tumorigenesis in mice. Sci. Transl. Med. 10, eaan4116 (2018).
Chen, Y. & Tian, Z. HBV-induced immune imbalance in the development of HCC. Front. Immunol. 10, 2048 (2019).
Schiffman, M., Castle, P. E., Jeronimo, J., Rodriguez, A. C. & Wacholder, S. Human papillomavirus and cervical cancer. Lancet 370, 890–907 (2007).
Li, R. et al. Gut microbiota-stimulated cathepsin K secretion mediates TLR4-dependent M2 macrophage polarization and promotes tumor metastasis in colorectal cancer. Cell Death Differ. 26, 2447–2463 (2019).
Kalafati, L. et al. Innate immune training of granulopoiesis promotes anti-tumor activity. Cell 183, 771–785 e712 (2020).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013).
Arpaia, N. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013).
Routy, B. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018).
Ma, C. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931 (2018).
Elkrief, A., Derosa, L., Zitvogel, L., Kroemer, G. & Routy, B. The intimate relationship between gut microbiota and cancer immunotherapy. Gut Microbes 10, 424–428 (2019).
Jin, S. et al. Maresin 1 improves the Treg/Th17 imbalance in rheumatoid arthritis through miR-21. Ann. Rheum. Dis. 77, 1644–1652 (2018).
Laird, B. J. et al. The systemic inflammatory response and its relationship to pain and other symptoms in advanced cancer. Oncologist 18, 1050–1055 (2013).
Avgerinos, K. I., Spyrou, N., Mantzoros, C. S. & Dalamaga, M. Obesity and cancer risk: emerging biological mechanisms and perspectives. Metabolism 92, 121–135 (2019).
Reiche, E. M., Nunes, S. O. & Morimoto, H. K. Stress, depression, the immune system, and cancer. Lancet Oncol. 5, 617–625 (2004).
Chan, D. S. M. et al. Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Ann. Oncol. 25, 1901–1914 (2014).
Larsson, S. C. & Wolk, A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am. J. Clin. Nutr. 86, 556–565 (2007).
Larsson, S. C. & Wolk, A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br. J. Cancer 97, 1005–1008 (2007).
Olsen, C. M. et al. Obesity and the risk of epithelial ovarian cancer: a systematic review and meta-analysis. Eur. J. Cancer 43, 690–709 (2007).
Calle, E. E., Rodriguez, C., Walker-Thurmond, K. & Thun, M. J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med 348, 1625–1638 (2003).
Saltiel, A. R. & Olefsky, J. M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 127, 1–4 (2017).
Correa, L. H., Heyn, G. S. & Magalhaes, K. G. The impact of the adipose organ plasticity on inflammation and cancer progression. Cells 8, 662 (2019).
Rogers, N. H., Perfield, J. W. 2nd, Strissel, K. J., Obin, M. S. & Greenberg, A. S. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology 150, 2161–2168 (2009).
Howe, L. R., Subbaramaiah, K., Hudis, C. A. & Dannenberg, A. J. Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin. Cancer Res. 19, 6074–6083 (2013).
Bochet, L. et al. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem. Biophys. Res. Commun. 411, 102–106 (2011).
Lee, Y. H., Jung, Y. S. & Choi, D. Recent advance in brown adipose physiology and its therapeutic potential. Exp. Mol. Med. 46, e78 (2014).
Apostoli, A. J. et al. Loss of PPARgamma expression in mammary secretory epithelial cells creates a pro-breast tumorigenic environment. Int. J. Cancer 134, 1055–1066 (2014).
Kolb, R., Sutterwala, F. S. & Zhang, W. Obesity and cancer: inflammation bridges the two. Curr. Opin. Pharm. 29, 77–89 (2016).
Deng, T., Lyon, C. J., Bergin, S., Caligiuri, M. A. & Hsueh, W. A. Obesity, inflammation, and cancer. Annu. Rev. Pathol. 11, 421–449 (2016).
Stein, M. Stress, depression, and the immune system. J. Clin. Psychiatry 50, 35–40 (1989).
Chrousos, G. P. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 332, 1351–1362 (1995).
Miller, E. S. et al. Chronic stress induces persistent low-grade inflammation. Am. J. Surg. 218, 677–683 (2019).
Gao, X. et al. Chronic stress promotes colitis by disturbing the gut microbiota and triggering immune system response. Proc. Natl Acad. Sci. USA 115, E2960–E2969 (2018).
Curtin, N. M., Boyle, N. T., Mills, K. H. & Connor, T. J. Psychological stress suppresses innate IFN-gamma production via glucocorticoid receptor activation: reversal by the anxiolytic chlordiazepoxide. Brain Behav. Immun. 23, 535–547 (2009).
Yang, H. et al. Stress-glucocorticoid-TSC22D3 axis compromises therapy-induced antitumor immunity. Nat. Med. 25, 1428–1441 (2019).
Gonzalez, H., Hagerling, C. & Werb, Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284 (2018).
Orimo, A. et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348 (2005).
Bhowmick, N. A., Neilson, E. G. & Moses, H. L. Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337 (2004).
Ozdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014).
Chang, H. Y. et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2, E7 (2004).
Wong, V. W. et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med. 18, 148–152 (2011).
Ammirante, M., Shalapour, S., Kang, Y., Jamieson, C. A. & Karin, M. Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc. Natl Acad. Sci. USA 111, 14776–14781 (2014).
Shalapour, S. et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 521, 94–98 (2015).
Yang, L., Pang, Y. & Moses, H. L. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 31, 220–227 (2010).
Ammirante, M., Luo, J. L., Grivennikov, S., Nedospasov, S. & Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 464, 302–305 (2010).
Ksiazkiewicz, M. et al. Importance of CCL2-CCR2A/2B signaling for monocyte migration into spheroids of breast cancer-derived fibroblasts. Immunobiology 215, 737–747 (2010).
Kraman, M. et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330, 827–830 (2010).
Galli, S. J. et al. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu. Rev. Immunol. 23, 749–786 (2005).
Bulfone-Paus, S., Nilsson, G., Draber, P., Blank, U. & Levi-Schaffer, F. Positive and negative signals in mast cell activation. Trends Immunol. 38, 657–667 (2017).
Aller, M. A., Arias, J. I. & Arias, J. Pathological axes of wound repair: gastrulation revisited. Theor. Biol. Med. Model 7, 37 (2010).
Khazaie, K. et al. The significant role of mast cells in cancer. Cancer Metastasis Rev. 30, 45–60 (2011).
Kischer, C. W., Bunce, H. 3rd & Shetlah, M. R. Mast cell analyses in hypertrophic scars, hypertrophic scars treated with pressure and mature scars. J. Invest. Dermatol. 70, 355–357 (1978).
Carlini, M. J. et al. Mast cell phenotypes and microvessels in non-small cell lung cancer and its prognostic significance. Hum. Pathol. 41, 697–705 (2010).
Strouch, M. J. et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin. Cancer Res. 16, 2257–2265 (2010).
Kuronita, T. et al. A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J. Cell Sci. 115, 4117–4131 (2002).
Gulubova, M. & Vlaykova, T. Prognostic significance of mast cell number and microvascular density for the survival of patients with primary colorectal cancer. J. Gastroenterol. Hepatol. 24, 1265–1275 (2009).
Nakae, S. et al. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J. Immunol. 176, 2238–2248 (2006).
Roche, W. R. Mast cells and tumour angiogenesis: the tumor-mediated release of an endothelial growth factor from mast cells. Int. J. Cancer 36, 721–728 (1985).
Salmaninejad, A. et al. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol. 42, 591–608 (2019).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Kim, J. & Bae, J. S. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm. 2016, 6058147 (2016).
Giese, M. A., Hind, L. E. & Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 133, 2159–2167 (2019).
Hidalgo, A., Chilvers, E. R., Summers, C. & Koenderman, L. The neutrophil life cycle. Trends Immunol. 40, 584–597 (2019).
Fridlender, Z. G. et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16, 183–194 (2009).
Nicolas-Avila, J. A., Adrover, J. M. & Hidalgo, A. Neutrophils in homeostasis, immunity, and cancer. Immunity 46, 15–28 (2017).
Castanheira, F. V. S. & Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 133, 2178–2185 (2019).
Shaul, M. E. & Fridlender, Z. G. Cancer-related circulating and tumor-associated neutrophils - subtypes, sources and function. FEBS J. 285, 4316–4342 (2018).
Ocana, A., Nieto-Jimenez, C., Pandiella, A. & Templeton, A. J. Neutrophils in cancer: prognostic role and therapeutic strategies. Mol. Cancer 16, 137 (2017).
Sastre, B., Rodrigo-Munoz, J. M., Garcia-Sanchez, D. A. & Canas, J. A. & Del Pozo, V. Eosinophils: old players in a new game. J. Investig. Allergol. Clin. Immunol. 28, 289–304 (2018).
Gatault, S., Legrand, F., Delbeke, M., Loiseau, S. & Capron, M. Involvement of eosinophils in the anti-tumor response. Cancer Immunol. Immunother. 61, 1527–1534 (2012).
Legrand, F. et al. Human eosinophils exert TNF-alpha and granzyme A-mediated tumoricidal activity toward colon carcinoma cells. J. Immunol. 185, 7443–7451 (2010).
Carretero, R. et al. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat. Immunol. 16, 609–617 (2015).
Furbert-Harris, P. M. et al. Activated eosinophils upregulate the metastasis suppressor molecule E-cadherin on prostate tumor cells. Cell Mol. Biol. (Noisy-le.-Gd.) 49, 1009–1016 (2003).
Alaarg, A. et al. Applying nanomedicine in maladaptive inflammation and angiogenesis. Adv. Drug Deliv. Rev. 119, 143–158 (2017).
von Wasielewski, R. et al. Tissue eosinophilia correlates strongly with poor prognosis in nodular sclerosing Hodgkin’s disease, allowing for known prognostic factors. Blood 95, 1207–1213 (2000).
Reichman, H., Karo-Atar, D. & Munitz, A. Emerging roles for eosinophils in the tumor microenvironment. Trends Cancer 2, 664–675 (2016).
Gabrilovich, D. I. & Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174 (2009).
Kumar, V., Patel, S., Tcyganov, E. & Gabrilovich, D. I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 37, 208–220 (2016).
Wang, C. et al. IFN-gamma-mediated downregulation of LXA4 is necessary for the maintenance of nonresolving inflammation and papilloma persistence. Cancer Res. 73, 1742–1751 (2013).
Iwasaki, A. & Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 16, 343–353 (2015).
Vesely, M. D., Kershaw, M. H., Schreiber, R. D. & Smyth, M. J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 29, 235–271 (2011).
Coussens, L. M., Zitvogel, L. & Palucka, A. K. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 339, 286–291 (2013).
Dong, C. Helper T cells and cancer-associated inflammation: a new direction for immunotherapy? J. Interferon Cytokine Res. 37, 383–385 (2017).
Pedroza-Gonzalez, A. et al. Thymic stromal lymphopoietin fosters human breast tumor growth by promoting type 2 inflammation. J. Exp. Med. 208, 479–490 (2011).
Kohrt, H. E. et al. Profile of immune cells in axillary lymph nodes predicts disease-free survival in breast cancer. PLoS Med .2, e284 (2005).
Guery, L. & Hugues, S. Th17 cell plasticity and functions in cancer immunity. Biomed. Res. Int. 2015, 314620 (2015).
Fleming, C., Morrissey, S., Cai, Y. & Yan, J. gammadelta T cells: unexpected regulators of cancer development and progression. Trends Cancer 3, 561–570 (2017).
Ye, J. et al. Tumor-derived gammadelta regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J. Immunol. 190, 2403–2414 (2013).
Fleming, C. et al. Microbiota-activated CD103(+) DCs stemming from microbiota adaptation specifically drive gammadeltaT17 proliferation and activation. Microbiome 5, 46 (2017).
Knochelmann, H. M. et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol. Immunol. 15, 458–469 (2018).
Ohue, Y. & Nishikawa, H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 110, 2080–2089 (2019).
Bromley, S. K., Mempel, T. R. & Luster, A. D. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat. Immunol. 9, 970–980 (2008).
van Herk, E. H. & Te Velde, A. A. Treg subsets in inflammatory bowel disease and colorectal carcinoma: characteristics, role, and therapeutic targets. J. Gastroenterol. Hepatol. 31, 1393–1404 (2016).
Alhabbab, R. Y. et al. Regulatory B cells: Development, phenotypes, functions, and role in transplantation. Immunol. Rev. 292, 164–179 (2019).
Schwartz, M., Zhang, Y. & Rosenblatt, J. D. B cell regulation of the anti-tumor response and role in carcinogenesis. J. Immunother. Cancer 4, 40 (2016).
Mauri, C. & Menon, M. The expanding family of regulatory B cells. Int. Immunol. 27, 479–486 (2015).
Zhang, Y., Gallastegui, N. & Rosenblatt, J. D. Regulatory B cells in anti-tumor immunity. Int. Immunol. 27, 521–530 (2015).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011).
Schlitzer, A., McGovern, N. & Ginhoux, F. Dendritic cells and monocyte-derived cells: Two complementary and integrated functional systems. Semin. Cell Dev. Biol. 41, 9–22 (2015).
Durai, V. & Murphy, K. M. Functions of murine dendritic cells. Immunity 45, 719–736 (2016).
Shi, M., Chen, X., Ye, K., Yao, Y. & Li, Y. Application potential of toll-like receptors in cancer immunotherapy: systematic review. Med. 95, e3951 (2016).
Chrisikos, T. T. et al. Molecular regulation of dendritic cell development and function in homeostasis, inflammation, and cancer. Mol. Immunol. 110, 24–39 (2019).
Schraml, B. U. & Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 32, 13–20 (2015).
Martin, J. C. et al. Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal Immunol. 7, 101–113 (2014).
Van Acker, H. H. et al. Interleukin-15-cultured dendritic cells enhance anti-tumor gamma delta T cell functions through IL-15 secretion. Front. Immunol. 9, 658 (2018).
Chomarat, P., Banchereau, J., Davoust, J. & Palucka, A. K. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat. Immunol. 1, 510–514 (2000).
Chomarat, P., Dantin, C., Bennett, L., Banchereau, J. & Palucka, A. K. TNF skews monocyte differentiation from macrophages to dendritic cells. J. Immunol. 171, 2262–2269 (2003).
Morvan, M. G. & Lanier, L. L. NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer 16, 7–19 (2016).
Leon, B., Lopez-Bravo, M. & Ardavin, C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity 26, 519–531 (2007).
Fei, M. et al. TNF-alpha from inflammatory dendritic cells (DCs) regulates lung IL-17A/IL-5 levels and neutrophilia versus eosinophilia during persistent fungal infection. Proc. Natl Acad. Sci. USA 108, 5360–5365 (2011).
Hammad, H. et al. Inflammatory dendritic cells-not basophils-are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J. Exp. Med. 207, 2097–2111 (2010).
Qian, C. & Cao, X. Dendritic cells in the regulation of immunity and inflammation. Semin. Immunol. 35, 3–11 (2018).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020).
Lewis, C. E. & Pollard, J. W. Distinct role of macrophages in different tumor microenvironments. Cancer Res 66, 605–612 (2006).
Cook, J. & Hagemann, T. Tumour-associated macrophages and cancer. Curr. Opin. Pharm. 13, 595–601 (2013).
Germano, G. et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 23, 249–262 (2013).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Ino, Y. et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 108, 914–923 (2013).
Guiducci, C., Vicari, A. P., Sangaletti, S., Trinchieri, G. & Colombo, M. P. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 65, 3437–3446 (2005).
Scapini, P. et al. Neutrophils produce biologically active macrophage inflammatory protein-3alpha (MIP-3alpha)/CCL20 and MIP-3beta/CCL19. Eur. J. Immunol. 31, 1981–1988 (2001).
Eruslanov, E. B. et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Invest. 124, 5466–5480 (2014).
Wu, L., Saxena, S., Awaji, M. & Singh, R. K. Tumor-associated neutrophils in cancer: going pro. Cancers 11, 564 (2019).
Amulic, B., Cazalet, C., Hayes, G. L., Metzler, K. D. & Zychlinsky, A. Neutrophil function: from mechanisms to disease. Annu. Rev. Immunol. 30, 459–489 (2012).
Chiossone, L., Dumas, P. Y., Vienne, M. & Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 18, 671–688 (2018).
Artis, D. & Spits, H. The biology of innate lymphoid cells. Nature 517, 293–301 (2015).
Vacca, P. et al. Human natural killer cells and other innate lymphoid cells in cancer: Friends or foes? Immunol. Lett. 201, 14–19 (2018).
Smyth, M. J. et al. NKG2D function protects the host from tumor initiation. J. Exp. Med. 202, 583–588 (2005).
Smyth, M. J. et al. Activation of NK cell cytotoxicity. Mol. Immunol. 42, 501–510 (2005).
Vivier, E., Tomasello, E., Baratin, M., Walzer, T. & Ugolini, S. Functions of natural killer cells. Nat. Immunol. 9, 503–510 (2008).
Krijgsman, D., Hokland, M. & Kuppen, P. J. K. The role of natural killer T cells in cancer—a phenotypical and functional approach. Front. Immunol. 9, 367 (2018).
Bae, E. A., Seo, H., Kim, I. K., Jeon, I. & Kang, C. Y. Roles of NKT cells in cancer immunotherapy. Arch. Pharm. Res. 42, 543–548 (2019).
Rei, M., Pennington, D. J. & Silva-Santos, B. The emerging protumor role of gammadelta T lymphocytes: implications for cancer immunotherapy. Cancer Res. 75, 798–802 (2015).
Correia, D. V., Lopes, A. & Silva-Santos, B. Tumor cell recognition by gammadelta T lymphocytes: T-cell receptor vs. NK-cell receptors. Oncoimmunology 2, e22892 (2013).
Silva-Santos, B., Serre, K. & Norell, H. gammadelta T cells in cancer. Nat. Rev. Immunol. 15, 683–691 (2015).
Gertner-Dardenne, J. et al. Bromohydrin pyrophosphate enhances antibody-dependent cell-mediated cytotoxicity induced by therapeutic antibodies. Blood 113, 4875–4884 (2009).
Buckley, C. D., Gilroy, D. W. & Serhan, C. N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 40, 315–327 (2014).
Zhang, Q. et al. The SP1-12LOX axis promotes chemoresistance and metastasis of ovarian cancer. Mol. Med. 26, 39 (2020).
Hao, H. et al. Lipoxin A4 suppresses lipopolysaccharide-induced hela cell proliferation and migration via NF-kappaB pathway. Inflammation 38, 400–408 (2015).
Lee, H. J., Park, M. K., Lee, E. J. & Lee, C. H. Resolvin D1 inhibits TGF-beta1-induced epithelial mesenchymal transition of A549 lung cancer cells via lipoxin A4 receptor/formyl peptide receptor 2 and GPR32. Int. J. Biochem Cell Biol. 45, 2801–2807 (2013).
Halder, R. C. et al. Curcuminoids and omega-3 fatty acids with anti-oxidants potentiate cytotoxicity of natural killer cells against pancreatic ductal adenocarcinoma cells and inhibit interferon gamma production. Front. Physiol. 6, 129 (2015).
Gewirtz, A. T. et al. Lipoxin a4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. J. Immunol. 168, 5260–5267 (2002).
Arita, M. et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201, 713–722 (2005).
Marcon, R. et al. Maresin 1, a proresolving lipid mediator derived from omega-3 polyunsaturated fatty acids, exerts protective actions in murine models of colitis. J. Immunol. 191, 4288–4298 (2013).
Cronkite, D. A. & Strutt, T. M. The regulation of inflammation by innate and adaptive lymphocytes. J. Immunol. Res. 2018, 1467538 (2018).
Yang, Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J. Clin. Invest. 125, 3335–3337 (2015).
Cheng, M. & Hu, S. Lung-resident gammadelta T cells and their roles in lung diseases. Immunology 151, 375–384 (2017).
Thommen, D. S. & Schumacher, T. N. T cell dysfunction in cancer. Cancer Cell 33, 547–562 (2018).
Crespo, J., Sun, H., Welling, T. H., Tian, Z. & Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 25, 214–221 (2013).
Fruh, K. & Yang, Y. Antigen presentation by MHC class I and its regulation by interferon gamma. Curr. Opin. Immunol. 11, 76–81 (1999).
Hirahara, K. et al. Mechanisms underlying helper T-cell plasticity: implications for immune-mediated disease. J. Allergy Clin. Immunol. 131, 1276–1287 (2013).
Knutson, K. L. & Disis, M. L. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 54, 721–728 (2005).
Topalian, S. L. et al. Melanoma-specific CD4+ T lymphocytes recognize human melanoma antigens processed and presented by Epstein-Barr virus-transformed B cells. Int. J. Cancer 58, 69–79 (1994).
Sarvaria, A., Madrigal, J. A. & Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell Mol. Immunol. 14, 662–674 (2017).
Dieu-Nosjean, M. C., Goc, J., Giraldo, N. A., Sautes-Fridman, C. & Fridman, W. H. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 35, 571–580 (2014).
Gottlin, E. B. et al. The association of intratumoral germinal centers with early-stage non-small cell lung cancer. J. Thorac. Oncol. 6, 1687–1690 (2011).
Wang, S. S. et al. Tumor-infiltrating B cells: their role and application in anti-tumor immunity in lung cancer. Cell Mol. Immunol. 16, 6–18 (2019).
Wouters, M. C. A. & Nelson, B. H. Prognostic significance of tumor-infiltrating B cells and plasma cells in human cancer. Clin. Cancer Res. 24, 6125–6135 (2018).
Kurebayashi, Y. et al. Comprehensive immune profiling of lung adenocarcinomas reveals four immunosubtypes with plasma cell subtype a negative indicator. Cancer Immunol. Res. 4, 234–247 (2016).
Banat, G. A. et al. Immune and inflammatory cell composition of human lung cancer stroma. PLoS ONE 10, e0139073 (2015).
Barker, H. E., Paget, J. T., Khan, A. A. & Harrington, K. J. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425 (2015).
Krysko, D. V. et al. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860–875 (2012).
Kroemer, G., Galluzzi, L., Kepp, O. & Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72 (2013).
Hirata, E. & Sahai, E. Tumor microenvironment and differential responses to therapy. Cold Spring Harb. Perspect. Med. 7, a026781 (2017).
Li, H., Zhou, L., Zhou, J., Li, Q. & Ji, Q. Underlying mechanisms and drug intervention strategies for the tumour microenvironment. J. Exp. Clin. Cancer Res. 40, 97 (2021).
Rivera, G. & Wakelee, H. A. Resistance to therapy. Cancer Treat. Res. 170, 183–202 (2016).
Karagiannis, G. S., Condeelis, J. S. & Oktay, M. H. Chemotherapy-induced metastasis: molecular mechanisms, clinical manifestations, therapeutic interventions. Cancer Res. 79, 4567–4576 (2019).
Yao, X., Panichpisal, K., Kurtzman, N. & Nugent, K. Cisplatin nephrotoxicity: a review. Am. J. Med. Sci. 334, 115–124 (2007).
Grabosch, S. et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 38, 2380–2393 (2019).
Kashiwagi, E. et al. Prostaglandin receptors induce urothelial tumourigenesis as well as bladder cancer progression and cisplatin resistance presumably via modulating PTEN expression. Br. J. Cancer 118, 213–223 (2018).
Xu, H. B., Shen, F. M. & Lv, Q. Z. Celecoxib enhanced the cytotoxic effect of cisplatin in chemo-resistant gastric cancer xenograft mouse models through a cyclooxygenase-2-dependent manner. Eur. J. Pharm. 776, 1–8 (2016).
Robledo-Cadena, D. X. et al. Non-steroidal anti-inflammatory drugs increase cisplatin, paclitaxel, and doxorubicin efficacy against human cervix cancer cells. Pharmaceuticals 13, 463 (2020).
Rowinsky, E. K. The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu. Rev. Med. 48, 353–374 (1997).
Volk, L. D. et al. Nab-paclitaxel efficacy in the orthotopic model of human breast cancer is significantly enhanced by concurrent anti-vascular endothelial growth factor A therapy. Neoplasia 10, 613–623 (2008).
Pusztai, L. et al. Changes in plasma levels of inflammatory cytokines in response to paclitaxel chemotherapy. Cytokine 25, 94–102 (2004).
Lee, L. F., Haskill, J. S., Mukaida, N., Matsushima, K. & Ting, J. P. Identification of tumor-specific paclitaxel (Taxol)-responsive regulatory elements in the interleukin-8 promoter. Mol. Cell Biol. 17, 5097–5105 (1997).
White, C. M., Martin, B. K., Lee, L. F., Haskill, J. S. & Ting, J. P. Effects of paclitaxel on cytokine synthesis by unprimed human monocytes, T lymphocytes, and breast cancer cells. Cancer Immunol. Immunother. 46, 104–112 (1998).
Jaffray, D. A. Image-guided radiotherapy: from current concept to future perspectives. Nat. Rev. Clin. Oncol. 9, 688–699 (2012).
Schaue, D., Kachikwu, E. L. & McBride, W. H. Cytokines in radiobiological responses: a review. Radiat. Res. 178, 505–523 (2012).
Yang, X. et al. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 25, 37–48 (2014).
McLaughlin, M. et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 20, 203–217 (2020).
Wu, C. T., Chen, M. F., Chen, W. C. & Hsieh, C. C. The role of IL-6 in the radiation response of prostate cancer. Radiat. Oncol. 8, 159 (2013).
Chen, M. F. et al. IL-6 expression predicts treatment response and outcome in squamous cell carcinoma of the esophagus. Mol. Cancer 12, 26 (2013).
Culig, Z. & Puhr, M. Interleukin-6: a multifunctional targetable cytokine in human prostate cancer. Mol. Cell Endocrinol. 360, 52–58 (2012).
Hou, J., Karin, M. & Sun, B. Targeting cancer-promoting inflammation - have anti-inflammatory therapies come of age? Nat. Rev. Clin. Oncol. 18, 261–279 (2021).
Braaten, T. J. et al. Immune checkpoint inhibitor-induced inflammatory arthritis persists after immunotherapy cessation. Ann. Rheum. Dis. 79, 332–338 (2020).
Zitvogel, L., Galluzzi, L., Kepp, O., Smyth, M. J. & Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 15, 405–414 (2015).
Spiotto, M., Fu, Y. X. & Weichselbaum, R. R. The intersection of radiotherapy and immunotherapy: mechanisms and clinical implications. Sci. Immunol. 1, eaag1266 (2016).
Kuraishy, A., Karin, M. & Grivennikov, S. I. Tumor promotion via injury- and death-induced inflammation. Immunity 35, 467–477 (2011).
Chen, J. et al. IL-17 inhibits CXCL9/10-mediated recruitment of CD8(+) cytotoxic T cells and regulatory T cells to colorectal tumors. J. Immunother. Cancer 7, 324 (2019).
Yang, W. et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 11, 4457 (2020).
Lauko, A., Bayik, D. & Lathia, J. D. IL-11 drives postsurgical hepatocellular carcinoma recurrence. EBioMedicine 47, 18–19 (2019).
Schneider, C. & Pozzi, A. Cyclooxygenases and lipoxygenases in cancer. Cancer Metastasis Rev. 30, 277–294 (2011).
Kozlowska, R., Bozek, A. & Jarzab, J. Association between cancer and allergies. Allergy Asthma Clin. Immunol. 12, 39 (2016).
Turner, M. C., Chen, Y., Krewski, D. & Ghadirian, P. An overview of the association between allergy and cancer. Int. J. Cancer 118, 3124–3132 (2006).
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017).
Gajewski, T. F. The next hurdle in cancer immunotherapy: overcoming the non-T-Cell-inflamed tumor microenvironment. Semin. Oncol. 42, 663–671 (2015).
Salinas, C. A. et al. Use of aspirin and other nonsteroidal antiinflammatory medications in relation to prostate cancer risk. Am. J. Epidemiol. 172, 578–590 (2010).
Algra, A. M. & Rothwell, P. M. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 13, 518–527 (2012).
Rothwell, P. M. et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 379, 1602–1612 (2012).
Yona, D. & Arber, N. Coxibs and cancer prevention. J. Cardiovasc Pharm. 47(Suppl 1.), S76–S81 (2006).
Boudreau, D. M., Yu, O. & Johnson, J. Statin use and cancer risk: a comprehensive review. Expert Opin. Drug Saf. 9, 603–621 (2010).
Dulak, J. & Jozkowicz, A. Anti-angiogenic and anti-inflammatory effects of statins: relevance to anti-cancer therapy. Curr. Cancer Drug Targets 5, 579–594 (2005).
Dinarello, C. A. Anti-inflammatory agents: present and future. Cell 140, 935–950 (2010).
Wang, H., Li, M., Rinehart, J. J. & Zhang, R. Pretreatment with dexamethasone increases antitumor activity of carboplatin and gemcitabine in mice bearing human cancer xenografts: in vivo activity, pharmacokinetics, and clinical implications for cancer chemotherapy. Clin. Cancer Res. 10, 1633–1644 (2004).
Chari, A. et al. Daratumumab plus carfilzomib and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood 134, 421–431 (2019).
Dimopoulos, M. et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet 396, 186–197 (2020).
Sears, D. D. et al. 12/15-lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS ONE 4, e7250 (2009).
Song, M. et al. Effect of supplementation with marine omega-3 fatty acid on risk of colorectal adenomas and serrated polyps in the US general population: a prespecified ancillary study of a randomized clinical trial. JAMA Oncol. 6, 108–115 (2019).
Jantan, I. et al. Dietary polyphenols suppress chronic inflammation by modulation of multiple inflammation-associated cell signaling pathways. J. Nutr. Biochem. 93, 108634 (2021).
Guo, W., Kong, E. & Meydani, M. Dietary polyphenols, inflammation, and cancer. Nutr. Cancer 61, 807–810 (2009).
Banik, U., Parasuraman, S., Adhikary, A. K. & Othman, N. H. Curcumin: the spicy modulator of breast carcinogenesis. J. Exp. Clin. Cancer Res. 36, 98 (2017).
Arshad, L., Haque, M. A., Abbas Bukhari, S. N. & Jantan, I. An overview of structure-activity relationship studies of curcumin analogs as antioxidant and anti-inflammatory agents. Future Med. Chem. 9, 605–626 (2017).
Rauf, A. et al. Resveratrol as an anti-cancer agent: a review. Crit. Rev. Food Sci. Nutr. 58, 1428–1447 (2018).
Jiang, Z. et al. Resveratrol and cancer treatment: updates. Ann. N. Y Acad. Sci. 1403, 59–69 (2017).
Jang, G. Y. et al. Interactions between tumor-derived proteins and Toll-like receptors. Exp. Mol. Med. 52, 1926–1935 (2020).
Johnson, D. E., O’Keefe, R. A. & Grandis, J. R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248 (2018).
Yoon, S. et al. NF-kappaB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene 31, 3467–3481 (2012).
Wolf, J., Rose-John, S. & Garbers, C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine 70, 11–20 (2014).
Patidar, A. et al. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine 104, 114–123 (2018).
Fang, H. et al. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell Mol. Immunol. 11, 150–159 (2014).
Park, H. D. et al. Pancreatic adenocarcinoma upregulated factor promotes metastasis by regulating TLR/CXCR4 activation. Oncogene 30, 201–211 (2011).
Kelly, M. G. et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 66, 3859–3868 (2006).
Finke, J. et al. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int. Immunopharmacol. 11, 856–861 (2011).
Mantovani, A., Sozzani, S., Locati, M., Allavena, P. & Sica, A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 (2002).
Nishikawa, H. & Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 127, 759–767 (2010).
Medzhitov, R. & Janeway, C. A. Jr. Innate immune induction of the adaptive immune response. Cold Spring Harb. Symp. Quant. Biol. 64, 429–435 (1999).
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054–1066 (2018).
Siebenhaar, F., Redegeld, F. A., Bischoff, S. C., Gibbs, B. F. & Maurer, M. Mast cells as drivers of disease and therapeutic targets. Trends Immunol. 39, 151–162 (2018).
Nonomura, N. et al. Decreased number of mast cells infiltrating into needle biopsy specimens leads to a better prognosis of prostate cancer. Br. J. Cancer 97, 952–956 (2007).
Nielsen, H. J. et al. Independent prognostic value of eosinophil and mast cell infiltration in colorectal cancer tissue. J. Pathol. 189, 487–495 (1999).
Welsh, T. J. et al. Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J. Clin. Oncol. 23, 8959–8967 (2005).
Molin, D. et al. Mast cell infiltration correlates with poor prognosis in Hodgkin’s lymphoma. Br. J. Haematol. 119, 122–124 (2002).
Maurer, M. et al. The c-kit ligand, stem cell factor, can enhance innate immunity through effects on mast cells. J. Exp. Med. 188, 2343–2348 (1998).
Godot, V. et al. H4 histamine receptor mediates optimal migration of mast cell precursors to CXCL12. J. Allergy Clin. Immunol. 120, 827–834 (2007).
Dahlin, J. S. & Hallgren, J. Mast cell progenitors: origin, development and migration to tissues. Mol. Immunol. 63, 9–17 (2015).
Reber, L. L. & Frossard, N. Targeting mast cells in inflammatory diseases. Pharm. Ther. 142, 416–435 (2014).
Kalesnikoff, J. & Galli, S. J. New developments in mast cell biology. Nat. Immunol. 9, 1215–1223 (2008).
Zhang, T., Finn, D. F., Barlow, J. W. & Walsh, J. J. Mast cell stabilisers. Eur. J. Pharm. 778, 158–168 (2016).
Lv, Y. P. et al. Degranulation of mast cells induced by gastric cancer-derived adrenomedullin prompts gastric cancer progression. Cell Death Dis. 9, 1034 (2018).
Dvorak, A. M. Ultrastructural studies of human basophils and mast cells. J. Histochem. Cytochem. 53, 1043–1070 (2005).
Kim, M. S., Radinger, M. & Gilfillan, A. M. The multiple roles of phosphoinositide 3-kinase in mast cell biology. Trends Immunol. 29, 493–501 (2008).
Ching, T. T., Hsu, A. L., Johnson, A. J. & Chen, C. S. Phosphoinositide 3-kinase facilitates antigen-stimulated Ca(2+) influx in RBL-2H3 mast cells via a phosphatidylinositol 3,4,5-trisphosphate-sensitive Ca(2+) entry mechanism. J. Biol. Chem. 276, 14814–14820 (2001).
Soucek, L. et al. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 13, 1211–1218 (2007).
Kennedy Norton, S. et al. IL-10 suppresses mast cell IgE receptor expression and signaling in vitro and in vivo. J. Immunol. 180, 2848–2854 (2008).
Jones, T. G., Finkelman, F. D., Austen, K. F. & Gurish, M. F. T regulatory cells control antigen-induced recruitment of mast cell progenitors to the lungs of C57BL/6 mice. J. Immunol. 185, 1804–1811 (2010).
Soucek, L. et al. Modeling pharmacological inhibition of mast cell degranulation as a therapy for insulinoma. Neoplasia 13, 1093–1100 (2011).
Ma, Y., Hwang, R. F., Logsdon, C. D. & Ullrich, S. E. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 73, 3927–3937 (2013).
Melillo, R. M. et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene 29, 6203–6215 (2010).
Pittoni, P. et al. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res. 71, 5987–5997 (2011).
Johnson, C. et al. Inhibition of mast cell-derived histamine decreases human cholangiocarcinoma growth and differentiation via c-Kit/stem cell factor-dependent signaling. Am. J. Pathol. 186, 123–133 (2016).
Rao, Q. et al. Recruited mast cells in the tumor microenvironment enhance bladder cancer metastasis via modulation of ERbeta/CCL2/CCR2 EMT/MMP9 signals. Oncotarget 7, 7842–7855 (2016).
Wang, S. et al. Mast cell targeted chimeric toxin can be developed as an adjunctive therapy in colon cancer treatment. Toxins 8, 71 (2016).
Yano, H. et al. Mast cell infiltration around gastric cancer cells correlates with tumor angiogenesis and metastasis. Gastric Cancer 2, 26–32 (1999).
Trabanelli, S. et al. PGE2-induced IDO1 inhibits the capacity of fully mature DCs to elicit an in vitro antileukemic immune response. J. Immunol. Res. 2015, 253191 (2015).
Gardner, A. & Ruffell, B. Dendritic cells and cancer immunity. Trends Immunol. 37, 855–865 (2016).
Zong, J., Keskinov, A. A., Shurin, G. V. & Shurin, M. R. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 65, 821–833 (2016).
Toi, M., Matsumoto, T. & Bando, H. Vascular endothelial growth factor: its prognostic, predictive, and therapeutic implications. Lancet Oncol. 2, 667–673 (2001).
Gabrilovich, D. I. et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 2, 1096–1103 (1996).
Fricke, I. et al. Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin. Cancer Res. 13, 4840–4848 (2007).
Ratta, M. et al. Dendritic cells are functionally defective in multiple myeloma: the role of interleukin-6. Blood 100, 230–237 (2002).
Oosterhoff, D. et al. Tumor-mediated inhibition of human dendritic cell differentiation and function is consistently counteracted by combined p38 MAPK and STAT3 inhibition. Oncoimmunology 1, 649–658 (2012).
Sombroek, C. C. et al. Prostanoids play a major role in the primary tumor-induced inhibition of dendritic cell differentiation. J. Immunol. 168, 4333–4343 (2002).
Bazzoni, G. & Rasia, M. Effects of an amphipathic drug on the rheological properties of the cell membrane. Blood Cells Mol. Dis. 24, 552–559 (1998).
Lo, A. S. et al. Modulation of dendritic cell differentiation by colony-stimulating factor-1: role of phosphatidylinositol 3’-kinase and delayed caspase activation. J. Leukoc. Biol. 82, 1446–1454 (2007).
Dranoff, G. et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl Acad. Sci. USA 90, 3539–3543 (1993).
Mach, N. et al. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 60, 3239–3246 (2000).
Sallusto, F. & Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179, 1109–1118 (1994).
Morales, J. K., Kmieciak, M., Knutson, K. L., Bear, H. D. & Manjili, M. H. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res. Treat. 123, 39–49 (2010).
Parmiani, G. et al. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann. Oncol. 18, 226–232 (2007).
Lyman, S. D. et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell 75, 1157–1167 (1993).
Naik, S. H. et al. Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J. Immunol. 174, 6592–6597 (2005).
Karsunky, H., Merad, M., Cozzio, A., Weissman, I. L. & Manz, M. G. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J. Exp. Med. 198, 305–313 (2003).
McKenna, H. J. et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 95, 3489–3497 (2000).
Ginhoux, F. et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 206, 3115–3130 (2009).
Li, K., Qu, S., Chen, X., Wu, Q. & Shi, M. Promising targets for cancer immunotherapy: TLRs, RLRs, and STING-mediated innate immune pathways. Int. J. Mol. Sci. 18, 404 (2017).
Galluzzi, L. et al. Trial Watch: experimental Toll-like receptor agonists for cancer therapy. Oncoimmunology 1, 699–716 (2012).
Koizumi, S. et al. Extracts of Larix Leptolepis effectively augments the generation of tumor antigen-specific cytotoxic T lymphocytes via activation of dendritic cells in TLR-2 and TLR-4-dependent manner. Cell Immunol. 276, 153–161 (2012).
Jeung, H. C. et al. Phase III trial of adjuvant 5-fluorouracil and adriamycin versus 5-fluorouracil, adriamycin, and polyadenylic-polyuridylic acid (poly A:U) for locally advanced gastric cancer after curative surgery: final results of 15-year follow-up. Ann. Oncol. 19, 520–526 (2008).
Dewan, M. Z. et al. Synergy of topical toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clin. Cancer Res. 18, 6668–6678 (2012).
Tel, J. et al. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type I IFN secretion. J. Immunol. 191, 5005–5012 (2013).
Scholch, S., Rauber, C., Weitz, J., Koch, M. & Huber, P. E. TLR activation and ionizing radiation induce strong immune responses against multiple tumor entities. Oncoimmunology 4, e1042201 (2015).
Van De Voort, T. J., Felder, M. A., Yang, R. K., Sondel, P. M. & Rakhmilevich, A. L. Intratumoral delivery of low doses of anti-CD40 mAb combined with monophosphoryl lipid a induces local and systemic antitumor effects in immunocompetent and T cell-deficient mice. J. Immunother. 36, 29–40 (2013).
Kubler, K. et al. Immunogenic cell death of human ovarian cancer cells induced by cytosolic poly(I:C) leads to myeloid cell maturation and activates NK cells. Eur. J. Immunol. 41, 3028–3039 (2011).
Petrasek, J. et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl Acad. Sci. USA 110, 16544–16549 (2013).
Sze, A. et al. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 14, 422–434 (2013).
Tang, C. H. et al. Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 76, 2137–2152 (2016).
Ablasser, A. & Chen, Z. J. cGAS in action: expanding roles in immunity and inflammation. Science 363, eaat8657 (2019).
Liu, Y. et al. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J. Hematol. Oncol. 11, 104 (2018).
Galluzzi, L., Buque, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Rodriguez-Ruiz, M. E. et al. Abscopal effects of radiotherapy are enhanced by combined immunostimulatory mAbs and are dependent on CD8 T cells and crosspriming. Cancer Res. 76, 5994–6005 (2016).
Deng, L. et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852 (2014).
Hou, Y. et al. Non-canonical NF-kappaB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity 49, 490–503 e494 (2018).
Gelao, L. et al. Dendritic cell-based vaccines: clinical applications in breast cancer. Immunotherapy 6, 349–360 (2014).
Sabado, R. L., Meseck, M. & Bhardwaj, N. Dendritic cell vaccines. Methods Mol. Biol. 1403, 763–777 (2016).
Cheever, M. A. & Higano, C. S. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 17, 3520–3526 (2011).
Dhodapkar, M. V. et al. Rapid generation of broad T-cell immunity in humans after a single injection of mature dendritic cells. J. Clin. Invest. 104, 173–180 (1999).
Wculek, S. K. et al. Effective cancer immunotherapy by natural mouse conventional type-1 dendritic cells bearing dead tumor antigen. J. Immunother. Cancer 7, 100 (2019).
Xiang, X., Wang, J., Lu, D. & Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target Ther. 6, 75 (2021).
Haydar, D. et al. Azithromycin POlarizes Macrophages to an M2 phenotype via inhibition of the STAT1 and NF-kappaB signaling pathways. J. Immunol. 203, 1021–1030 (2019).
Svensson, S. et al. CCL2 and CCL5 are novel therapeutic targets for estrogen-dependent breast cancer. Clin. Cancer Res. 21, 3794–3805 (2015).
Li, X. et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 66, 157–167 (2017).
Nywening, T. M. et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 17, 651–662 (2016).
Zollo, M. et al. Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin. Exp. Metastasis 29, 585–601 (2012).
Gazzaniga, S. et al. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Invest. Dermatol. 127, 2031–2041 (2007).
Aldinucci, D. & Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014, 292376 (2014).
Wang, H. C. et al. Tumor-associated macrophages promote epigenetic silencing of gelsolin through DNA methyltransferase 1 in gastric cancer cells. Cancer Immunol. Res. 5, 885–897 (2017).
Aldinucci, D. & Casagrande, N. Inhibition of the CCL5/CCR5 axis against the progression of gastric cancer. Int. J. Mol. Sci. 19, 1477 (2018).
Walens, A. et al. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. Elife 8, e43653 (2019).
Suarez-Carmona, M. et al. CCR5 status and metastatic progression in colorectal cancer. Oncoimmunology 8, e1626193 (2019).
Frankenberger, C. et al. Metastasis suppressors regulate the tumor microenvironment by blocking recruitment of prometastatic tumor-associated macrophages. Cancer Res. 75, 4063–4073 (2015).
Nie, Y. et al. Breast phyllodes tumors recruit and repolarize rumor-associated macrophages via secreting CCL5 to promote malignant progression, which can be inhibited by CCR5 inhibition therapy. Clin. Cancer Res. 25, 3873–3886 (2019).
Guilliams, M., Thierry, G. R., Bonnardel, J. & Bajenoff, M. Establishment and maintenance of the macrophage niche. Immunity 52, 434–451 (2020).
Chockalingam, S. & Ghosh, S. S. Macrophage colony-stimulating factor and cancer: a review. Tumour Biol. 35, 10635–10644 (2014).
Chambers, S. K. Role of CSF-1 in progression of epithelial ovarian cancer. Future Oncol. 5, 1429–1440 (2009).
Okugawa, Y. et al. Colony-stimulating factor-1 and colony-stimulating factor-1 receptor co-expression is associated with disease progression in gastric cancer. Int. J. Oncol. 53, 737–749 (2018).
Richardsen, E., Uglehus, R. D., Johnsen, S. H. & Busund, L. T. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res .35, 865–874 (2015).
DeNardo, D. G. et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Disco. 1, 54–67 (2011).
Escamilla, J. et al. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 75, 950–962 (2015).
Cannarile, M. A. et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 5, 53 (2017).
Pyonteck, S. M. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272 (2013).
Ries, C. H. et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25, 846–859 (2014).
Weizman, N. et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 33, 3812–3819 (2014).
Mouchemore, K. A. et al. Specific inhibition of PI3K p110delta inhibits CSF-1-induced macrophage spreading and invasive capacity. FEBS J. 280, 5228–5236 (2013).
Goldvaser, H. & Amir, E. Role of bisphosphonates in breast cancer therapy. Curr. Treat. Options Oncol. 20, 26 (2019).
Bader, J. E. et al. Macrophage depletion using clodronate liposomes decreases tumorigenesis and alters gut microbiota in the AOM/DSS mouse model of colon cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 314, G22–G31 (2018).
Tang, X. et al. Targeted delivery of zoledronic acid through the sialic acid-siglec axis for killing and reversal of M2 phenotypic tumor-associated macrophages—a promising cancer immunotherapy. Int. J. Pharm. 590, 119929 (2020).
Macherey, S. et al. Bisphosphonates for advanced prostate cancer. Cochrane Database Syst. Rev. 12, CD006250 (2017).
Cuevas, C. & Francesch, A. Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 26, 322–337 (2009).
Allavena, P. et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. 65, 2964–2971 (2005).
Liguori, M. et al. Functional TRAIL receptors in monocytes and tumor-associated macrophages: a possible targeting pathway in the tumor microenvironment. Oncotarget 7, 41662–41676 (2016).
D’Incalci, M. & Zambelli, A. Trabectedin for the treatment of breast cancer. Expert Opin. Investig. Drugs 25, 105–115 (2016).
D’Incalci, M., Badri, N., Galmarini, C. M. & Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 111, 646–650 (2014).
Galmbacher, K. et al. Shigella mediated depletion of macrophages in a murine breast cancer model is associated with tumor regression. PLoS ONE 5, e9572 (2010).
Martinez, F. O., Sica, A., Mantovani, A. & Locati, M. Macrophage activation and polarization. Front. Biosci. 13, 453–461 (2008).
Liebes, L. et al. Modulation of monocyte functions by muramyl tripeptide phosphatidylethanolamine in a phase II study in patients with metastatic melanoma. J. Natl Cancer Inst. 84, 694–699 (1992).
Bode, J. G., Ehlting, C. & Haussinger, D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 24, 1185–1194 (2012).
Hu, X., Chakravarty, S. D. & Ivashkiv, L. B. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 226, 41–56 (2008).
Kelsey, S. M., Hazel, D., Murrell, C. & Newland, A. C. GM-CSF for peripheral blood stem cell harvest in myeloma. Br. J. Haematol. 92, 505 (1996).
Schroder, K., Hertzog, P. J., Ravasi, T. & Hume, D. A. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189 (2004).
Lee, S. & Margolin, K. Cytokines in cancer immunotherapy. Cancers 3, 3856–3893 (2011).
Conlon, K. C., Miljkovic, M. D. & Waldmann, T. A. Cytokines in the treatment of cancer. J. Interferon Cytokine Res. 39, 6–21 (2019).
Sheikh, S. Z. et al. Cutting edge: IFN-gamma is a negative regulator of IL-23 in murine macrophages and experimental colitis. J. Immunol. 184, 4069–4073 (2010).
Pujade-Lauraine, E. et al. Intraperitoneal recombinant interferon gamma in ovarian cancer patients with residual disease at second-look laparotomy. J. Clin. Oncol. 14, 343–350 (1996).
Miller, C. H., Maher, S. G. & Young, H. A. Clinical Use of Interferon-gamma. Ann. N. Y Acad. Sci. 1182, 69–79 (2009).
Alberts, D. S. et al. Randomized phase 3 trial of interferon gamma-1b plus standard carboplatin/paclitaxel versus carboplatin/paclitaxel alone for first-line treatment of advanced ovarian and primary peritoneal carcinomas: results from a prospectively designed analysis of progression-free survival. Gynecol. Oncol. 109, 174–181 (2008).
Vonderheide, R. H. CD40 agonist antibodies in cancer immunotherapy. Annu. Rev. Med. 71, 47–58 (2020).
Beatty, G. L. et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616 (2011).
Beatty, G. L. et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 19, 6286–6295 (2013).
Yu, H., Pardoll, D. & Jove, R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 (2009).
Yin, Z. et al. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J. Cell Biochem. 119, 9419–9432 (2018).
Sun, J. et al. Suppression of TLR4 activation by resveratrol is associated with STAT3 and Akt inhibition in oxidized low-density lipoprotein-activated platelets. Eur. J. Pharm. 836, 1–10 (2018).
Wang, H., Feng, H. & Zhang, Y. Resveratrol inhibits hypoxia-induced glioma cell migration and invasion by the p-STAT3/miR-34a axis. Neoplasma 63, 532–539 (2016).
Vasamsetti, S. B. et al. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes 64, 2028–2041 (2015).
Arredouani, M. S. Is the scavenger receptor MARCO a new immune checkpoint? Oncoimmunology 3, e955709 (2014).
Georgoudaki, A. M. et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 15, 2000–2011 (2016).
La Fleur, L. et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int. J. Cancer 143, 1741–1752 (2018).
Mulens-Arias, V. et al. Polyethylenimine-coated superparamagnetic iron oxide nanoparticles impair in vitro and in vivo angiogenesis. Nanomedicine 21, 102063 (2019).
Rolny, C. et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 19, 31–44 (2011).
Willingham, S. B. et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl Acad. Sci. USA 109, 6662–6667 (2012).
Logtenberg, M. E. W. et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPalpha axis and a target for cancer immunotherapy. Nat. Med. 25, 612–619 (2019).
Russ, A. et al. Blocking “don’t eat me” signal of CD47-SIRPalpha in hematological malignancies, an in-depth review. Blood Rev. 32, 480–489 (2018).
Schurch, C. M. et al. The “don’t eat me” signal CD47 is a novel diagnostic biomarker and potential therapeutic target for diffuse malignant mesothelioma. Oncoimmunology 7, e1373235 (2017).
Barkal, A. A. et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396 (2019).
Bradley, C. A. CD24 - a novel ‘don’t eat me’ signal. Nat. Rev. Cancer 19, 541 (2019).
Montero, A. J., Diaz-Montero, C. M., Kyriakopoulos, C. E., Bronte, V. & Mandruzzato, S. Myeloid-derived suppressor cells in cancer patients: a clinical perspective. J. Immunother. 35, 107–115 (2012).
Limagne, E. et al. Accumulation of MDSC and Th17 Cells in patients with etastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 76, 5241–5252 (2016).
Hansen, G. L., Gaudernack, G., Brunsvig, P. F., Cvancarova, M. & Kyte, J. A. Immunological factors influencing clinical outcome in lung cancer patients after telomerase peptide vaccination. Cancer Immunol. Immunother. 64, 1609–1621 (2015).
Bauer, R. et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res. 78, 3220–3232 (2018).
Fleet, J. C., Burcham, G. N., Calvert, R. D., Elzey, B. D. & Ratliff, T. L. 1alpha, 25 Dihydroxyvitamin D (1,25(OH)2D) inhibits the T cell suppressive function of myeloid derived suppressor cells (MDSC). J. Steroid Biochem. Mol. Biol. 198, 105557 (2020).
Roda, J. M., Parihar, R. & Carson, W. E. 3rd CpG-containing oligodeoxynucleotides act through TLR9 to enhance the NK cell cytokine response to antibody-coated tumor cells. J. Immunol. 175, 1619–1627 (2005).
Kuwata, T. et al. Vitamin A deficiency in mice causes a systemic expansion of myeloid cells. Blood 95, 3349–3356 (2000).
Kulbersh, J. S., Day, T. A., Gillespie, M. B. & Young, M. R. 1alpha,25-Dihydroxyvitamin D(3) to skew intratumoral levels of immune inhibitory CD34(+) progenitor cells into dendritic cells. Otolaryngol. Head. Neck Surg. 140, 235–240 (2009).
Young, M. R., Wright, M. A., Vellody, K. & Lathers, D. M. Skewed differentiation of bone marrow CD34+ cells of tumor bearers from dendritic toward monocytic cells, and the redirection of differentiation toward dendritic cells by 1alpha,25-dihydroxyvitamin D3. Int. J. Immunopharmacol. 21, 675–688 (1999).
Lathers, D. M., Clark, J. I., Achille, N. J. & Young, M. R. Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3. Cancer Immunol. Immunother. 53, 422–430 (2004).
Mirza, N. et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 66, 9299–9307 (2006).
Nefedova, Y. et al. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 67, 11021–11028 (2007).
Iclozan, C., Antonia, S., Chiappori, A., Chen, D. T. & Gabrilovich, D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. 62, 909–918 (2013).
Tobin, R. P. et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int. Immunopharmacol. 63, 282–291 (2018).
Young, M. R. & Lathers, D.M. Combination docetaxel plus vitamin D(3) as an immune therapy in animals bearing squamous cell carcinomas. Otolaryngol. Head. Neck Surg. 133, 611–618 (2005).
Tsavaris, N., Kosmas, C., Vadiaka, M., Kanelopoulos, P. & Boulamatsis, D. Immune changes in patients with advanced breast cancer undergoing chemotherapy with taxanes. Br. J. Cancer 87, 21–27 (2002).
Kodumudi, K. N. et al. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin. Cancer Res. 16, 4583–4594 (2010).
Michels, T. et al. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J. Immunotoxicol. 9, 292–300 (2012).
Montero, A. J. et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Res. Treat. 132, 215–223 (2012).
Tian, J. et al. beta-Glucan enhances antitumor immune responses by regulating differentiation and function of monocytic myeloid-derived suppressor cells. Eur. J. Immunol. 43, 1220–1230 (2013).
Chan, G. C., Chan, W. K. & Sze, D. M. The effects of beta-glucan on human immune and cancer cells. J. Hematol. Oncol. 2, 25 (2009).
Xiang, X. et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 124, 2621–2633 (2009).
Chalmin, F. et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Invest. 120, 457–471 (2010).
Fernandez, A. et al. Inhibition of tumor-induced myeloid-derived suppressor cell function by a nanoparticulated adjuvant. J. Immunol. 186, 264–274 (2011).
Liu, Y. et al. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol. Immunother. 67, 1181–1195 (2018).
Tu, S. P. et al. Curcumin induces the differentiation of myeloid-derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth. Cancer Prev. Res. 5, 205–215 (2012).
Deguchi, A. Curcumin targets in inflammation and cancer. Endocr. Metab. Immune Disord. Drug Targets 15, 88–96 (2015).
Hossain, D. M. et al. TLR9-targeted STAT3 silencing abrogates immunosuppressive activity of myeloid-derived suppressor cells from prostate cancer patients. Clin. Cancer Res. 21, 3771–3782 (2015).
Kortylewski, M. & Moreira, D. Myeloid cells as a target for oligonucleotide therapeutics: turning obstacles into opportunities. Cancer Immunol. Immunother. 66, 979–988 (2017).
Kusmartsev, S. et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J. Immunol. 181, 346–353 (2008).
Koinis, F. et al. Effect of first-line treatment on myeloid-derived suppressor cells’ subpopulations in the peripheral blood of patients with non-small cell lung cancer. J. Thorac. Oncol. 11, 1263–1272 (2016).
Limagne, E. et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 76, 5241–5252 (2016).
Albeituni, S. H., Ding, C. & Yan, J. Hampering immune suppressors: therapeutic targeting of myeloid-derived suppressor cells in cancer. Cancer J. 19, 490–501 (2013).
Pan, P. Y. et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood 111, 219–228 (2008).
Ko, J. S. et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 15, 2148–2157 (2009).
Panka, D. J., Liu, Q., Geissler, A. K. & Mier, J. W. Effects of HDM2 antagonism on sunitinib resistance, p53 activation, SDF-1 induction, and tumor infiltration by CD11b+/Gr-1+ myeloid derived suppressor cells. Mol. Cancer 12, 17 (2013).
Evans, E. E. et al. Antibody blockade of semaphorin 4D promotes immune infiltration into tumor and enhances response to other immunomodulatory therapies. Cancer Immunol. Res. 3, 689–701 (2015).
Patnaik, A. et al. Safety, pharmacokinetics, and pharmacodynamics of a humanized anti-semaphorin 4D antibody, in a first-in-human study of patients with advanced solid tumors. Clin. Cancer Res. 22, 827–836 (2016).
Highfill, S. L. et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 6, 237ra267 (2014).
Flores-Toro, J. A. et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl Acad. Sci. USA 117, 1129–1138 (2020).
Wang, T. et al. Galectin-3 contributes to cisplatin-induced myeloid derived suppressor cells (MDSCs) recruitment in Lewis lung cancer-bearing mice. Mol. Biol. Rep. 41, 4069–4076 (2014).
Obermajer, N., Muthuswamy, R., Lesnock, J., Edwards, R. P. & Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 118, 5498–5505 (2011).
Rong, Y. et al. Doxorubicin resistant cancer cells activate myeloid-derived suppressor cells by releasing PGE2. Sci. Rep. 6, 23824 (2016).
Takahashi, R. et al. Microsomal prostaglandin E synthase-1 promotes lung metastasis via SDF-1/CXCR4-mediated recruitment of CD11b(+)Gr1(+)MDSCs from bone marrow. Biomed. Pharmacother. 121, 109581 (2020).
Yan, G. et al. A RIPK3-PGE2 circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res 78, 5586–5599 (2018).
Veltman, J. D. et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 10, 464 (2010).
Hong, D. S. et al. First-in-human phase I study of immunomodulatory E7046, an antagonist of PGE2-receptor E-type 4 (EP4), in patients with advanced cancers. J. Immunother. Cancer 8, e000222 (2020).
Albu, D. I. et al. EP4 antagonism by E7046 diminishes myeloid immunosuppression and synergizes with Treg-reducing IL-2-diphtheria toxin fusion protein in restoring anti-tumor immunity. Oncoimmunology 6, e1338239 (2017).
Zelenay, S. et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, 1257–1270 (2015).
Prima, V., Kaliberova, L. N., Kaliberov, S., Curiel, D. T. & Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl Acad. Sci. USA 114, 1117–1122 (2017).
Gabrilovich, D. I., Ostrand-Rosenberg, S. & Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268 (2012).
De Santo, C. et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc. Natl Acad. Sci. USA 102, 4185–4190 (2005).
Serafini, P. et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 203, 2691–2702 (2006).
Hassel, J. C. et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic melanoma (TaMe). Oncoimmunology 6, e1326440 (2017).
Li, J. et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 6, e1320011 (2017).
Li, L. et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 78, 1779–1791 (2018).
De Henau, O. et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 539, 443–447 (2016).
Zhang, X., Shen, L., Liu, Q., Hou, L. & Huang, L. Inhibiting PI3 kinase-gamma in both myeloid and plasma cells remodels the suppressive tumor microenvironment in desmoplastic tumors. J. Control Release 309, 173–180 (2019).
Patel, K., Danilov, A. V. & Pagel, J. M. Duvelisib for CLL/SLL and follicular non-Hodgkin lymphoma. Blood 134, 1573–1577 (2019).
Suzuki, E., Kapoor, V., Jassar, A. S., Kaiser, L. R. & Albelda, S. M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 11, 6713–6721 (2005).
Vincent, J. et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 70, 3052–3061 (2010).
Hurez, V. et al. Mitigating age-related immune dysfunction heightens the efficacy of tumor immunotherapy in aged mice. Cancer Res. 72, 2089–2099 (2012).
Dominguez, G. A. et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin. Cancer Res. 23, 2942–2950 (2017).
Qin, H. et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat. Med. 20, 676–681 (2014).
Shen, M. et al. Tumor-associated neutrophils as a new prognostic factor in cancer: a systematic review and meta-analysis. PLoS ONE 9, e98259 (2014).
Granot, Z. Neutrophils as a therapeutic target in cancer. Front Immunol. 10, 1710 (2019).
Zhang, Y., Guoqiang, L., Sun, M. & Lu, X. Targeting and exploitation of tumor-associated neutrophils to enhance immunotherapy and drug delivery for cancer treatment. Cancer Biol. Med. 17, 32–43 (2020).
Shaul, M. E. & Fridlender, Z. G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 16, 601–620 (2019).
Haider, C. et al. Transforming growth factor-beta and axl induce CXCL5 and neutrophil recruitment in hepatocellular carcinoma. Hepatology 69, 222–236 (2019).
Tauriello, D. V. F. et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543 (2018).
Mariathasan, S. et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018).
Stevenson, J. P. et al. Immunological effects of the TGFbeta-blocking antibody GC1008 in malignant pleural mesothelioma patients. Oncoimmunology 2, e26218 (2013).
Morris, J. C. et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 9, e90353 (2014).
Formenti, S. C. et al. Focal irradiation and systemic TGFbeta blockade in metastatic breast cancer. Clin. Cancer Res. 24, 2493–2504 (2018).
Kelley, R. K. et al. A phase 2 study of galunisertib (TGF-beta1 receptor type I inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin. Transl. Gastroenterol. 10, e00056 (2019).
Melisi, D. et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 119, 1208–1214 (2018).
Holmgaard, R. B. et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 6, 47 (2018).
Glennon-Alty, L., Moots, R. J., Edwards, S. W. & Wright, H. L. Type I interferon regulates cytokine-delayed neutrophil apoptosis, reactive oxygen species production and chemokine expression. Clin. Exp. Immunol. 203, 151–159 (2021).
Wu, C. F. et al. The lack of type I interferon induces neutrophil-mediated pre-metastatic niche formation in the mouse lung. Int. J. Cancer 137, 837–847 (2015).
Andzinski, L. et al. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 138, 1982–1993 (2016).
Nauseef, W. M. How human neutrophils kill and degrade microbes: an integrated view. Immunol. Rev. 219, 88–102 (2007).
Semerad, C. L., Liu, F., Gregory, A. D., Stumpf, K. & Link, D. C. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity 17, 413–423 (2002).
Cheng, Y., Ma, X. L., Wei, Y. Q. & Wei, X. W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 1871, 289–312 (2019).
Liao, W. et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 35, 559–572 e557 (2019).
Jung, K. et al. Targeting CXCR4-dependent immunosuppressive Ly6C(low) monocytes improves antiangiogenic therapy in colorectal cancer. Proc. Natl Acad. Sci. USA 114, 10455–10460 (2017).
Zeng, Y. et al. Dual blockade of CXCL12-CXCR4 and PD-1-PD-L1 pathways prolongs survival of ovarian tumor-bearing mice by prevention of immunosuppression in the tumor microenvironment. FASEB J. 33, 6596–6608 (2019).
Xue, L. J., Mao, X. B., Ren, L. L. & Chu, X. Y. Inhibition of CXCL12/CXCR4 axis as a potential targeted therapy of advanced gastric carcinoma. Cancer Med. 6, 1424–1436 (2017).
Righi, E. et al. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res. 71, 5522–5534 (2011).
Gil, M. et al. CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells. J. Immunol. 193, 5327–5337 (2014).
Glodde, N. et al. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity 47, 789–802 e789 (2017).
Kurzrock, R. et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J. Clin. Oncol. 29, 2660–2666 (2011).
Kwilas, A. R., Ardiani, A., Donahue, R. N., Aftab, D. T. & Hodge, J. W. Dual effects of a targeted small-molecule inhibitor (cabozantinib) on immune-mediated killing of tumor cells and immune tumor microenvironment permissiveness when combined with a cancer vaccine. J. Transl. Med. 12, 294 (2014).
Patnaik, A. et al. Cabozantinib eradicates advanced murine prostate cancer by activating antitumor innate immunity. Cancer Disco. 7, 750–765 (2017).
Imai, K., Matsuyama, S., Miyake, S., Suga, K. & Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet 356, 1795–1799 (2000).
Vitale, M., Cantoni, C., Pietra, G., Mingari, M. C. & Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 44, 1582–1592 (2014).
Rusakiewicz, S. et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 73, 3499–3510 (2013).
Veeramani, S. et al. Rituximab infusion induces NK activation in lymphoma patients with the high-affinity CD16 polymorphism. Blood 118, 3347–3349 (2011).
Shimasaki, N., Jain, A. & Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Disco. 19, 200–218 (2020).
Huntington, N. D. The unconventional expression of IL-15 and its role in NK cell homeostasis. Immunol. Cell Biol. 92, 210–213 (2014).
Porrata, L. F. et al. Interleukin-15 affects patient survival through natural killer cell recovery after autologous hematopoietic stem cell transplantation for non-Hodgkin lymphomas. Clin. Dev. Immunol. 2010, 914945 (2010).
Conlon, K. C. et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 33, 74–82 (2015).
Perez-Martinez, A. et al. A phase I/II trial of interleukin-15-stimulated natural killer cell infusion after haplo-identical stem cell transplantation for pediatric refractory solid tumors. Cytotherapy 17, 1594–1603 (2015).
Gluck, W. L. et al. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-hodgkin’s lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clin. Cancer Res. 10, 2253–2264 (2004).
Marcus, A. et al. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity 49, 754–763 e754 (2018).
Breitbach, C. J. et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 477, 99–102 (2011).
Alvarez-Breckenridge, C. A. et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat. Med. 18, 1827–1834 (2012).
Zamarin, D. et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 6, 226ra232 (2014).
Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Invest. 128, 4654–4668 (2018).
Ohs, I. et al. Restoration of natural killer cell antimetastatic activity by IL12 and checkpoint blockade. Cancer Res. 77, 7059–7071 (2017).
Concha-Benavente, F. et al. PD-L1 mediates dysfunction in activated PD-1(+) NK Cells in head and neck cancer patients. Cancer Immunol. Res. 6, 1548–1560 (2018).
Kohrt, H. E. et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 123, 678–686 (2014).
Paolino, M. et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 507, 508–512 (2014).
Chan, C. J. et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 15, 431–438 (2014).
Kohrt, H. E. et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J. Clin. Invest. 122, 1066–1075 (2012).
Kohrt, H. E. et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood 117, 2423–2432 (2011).
Andre, P. et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175, 1731–1743 e1713 (2018).
Elliott, J. M. & Yokoyama, W. M. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 32, 364–372 (2011).
Chang, Y. H. et al. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 73, 1777–1786 (2013).
Imai, C., Iwamoto, S. & Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 106, 376–383 (2005).
Huland, E. & Huland, H. Tumor-associated eosinophilia in interleukin-2-treated patients: evidence of toxic eosinophil degranulation on bladder cancer cells. J. Cancer Res. Clin. Oncol. 118, 463–467 (1992).
Tepper, R. I., Coffman, R. L. & Leder, P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science 257, 548–551 (1992).
Schaefer, J. T. et al. Dynamic changes in cellular infiltrates with repeated cutaneous vaccination: a histologic and immunophenotypic analysis. J. Transl. Med. 8, 79 (2010).
Gebhardt, C. et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with Ipilimumab. Clin. Cancer Res. 21, 5453–5459 (2015).
Weide, B. et al. Baseline biomarkers for outcome of melanoma patients treated with Pembrolizumab. Clin. Cancer Res. 22, 5487–5496 (2016).
Davis, B. P. & Rothenberg, M. E. Eosinophils and cancer. Cancer Immunol. Res. 2, 1–8 (2014).
Kataoka, S., Konishi, Y., Nishio, Y., Fujikawa-Adachi, K. & Tominaga, A. Antitumor activity of eosinophils activated by IL-5 and eotaxin against hepatocellular carcinoma. DNA Cell Biol. 23, 549–560 (2004).
Rothenberg, M. E. & Hogan, S. P. The eosinophil. Annu. Rev. Immunol. 24, 147–174 (2006).
Rosenberg, H. F., Dyer, K. D. & Foster, P. S. Eosinophils: changing perspectives in health and disease. Nat. Rev. Immunol. 13, 9–22 (2013).
Caruso, R. A. et al. Ultrastructural descriptions of heterotypic aggregation between eosinophils and tumor cells in human gastric carcinomas. Ultrastruct. Pathol. 35, 145–149 (2011).
Simon, H. U. et al. Interleukin-2 primes eosinophil degranulation in hypereosinophilia and Wells’ syndrome. Eur. J. Immunol. 33, 834–839 (2003).
Rivoltini, L. et al. In vitro anti-tumor activity of eosinophils from cancer patients treated with subcutaneous administration of interleukin 2. Role of interleukin 5. Int. J. Cancer 54, 8–15 (1993).
Sosman, J. A. et al. Evidence for eosinophil activation in cancer patients receiving recombinant interleukin-4: effects of interleukin-4 alone and following interleukin-2 administration. Clin. Cancer Res. 1, 805–812 (1995).
Benatar, T. et al. IL-17E, a proinflammatory cytokine, has antitumor efficacy against several tumor types in vivo. Cancer Immunol. Immunother. 59, 805–817 (2010).
Steinman, R. M. Decisions about dendritic cells: past, present, and future. Annu. Rev. Immunol. 30, 1–22 (2012).
Monney, L. et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 (2002).
Carroll, M. C. & Isenman, D. E. Regulation of humoral immunity by complement. Immunity 37, 199–207 (2012).
Wernersson, S. & Pejler, G. Mast cell secretory granules: armed for battle. Nat. Rev. Immunol. 14, 478–494 (2014).
Chaudhry, A. & Rudensky, A. Y. Control of inflammation by integration of environmental cues by regulatory T cells. J. Clin. Invest. 123, 939–944 (2013).
Cai, X., Zhang, L. & Wei, W. Regulatory B cells in inflammatory diseases and tumor. Int. Immunopharmacol. 67, 281–286 (2019).
Volaric, I., Vicic, M. & Prpic-Massari, L. The role of CD8+ T-cells and their cytokines in the pathogenesis of psoriasis. Acta Dermatovenerol. Croat. 27, 159–162 (2019).
Fernandez-Poma, S. M. et al. Expansion of tumor-infiltrating CD8(+) T cells expressing PD-1 improves the efficacy of adoptive T-cell therapy. Cancer Res. 77, 3672–3684 (2017).
Woo, S. R., Corrales, L. & Gajewski, T. F. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 36, 250–256 (2015).
Weiss, J. M. et al. The STING agonist DMXAA triggers a cooperation between T lymphocytes and myeloid cells that leads to tumor regression. Oncoimmunology 6, e1346765 (2017).
Ara, A., Ahmed, K. A. & Xiang, J. Multiple effects of CD40-CD40L axis in immunity against infection and cancer. Immunotargets Ther. 7, 55–61 (2018).
Martin, S., Pahari, S., Sudan, R. & Saha, B. CD40 signaling in CD8+CD40+ T cells turns on contra-T regulatory cell functions. J. Immunol. 184, 5510–5518 (2010).
Esensten, J. H., Helou, Y. A., Chopra, G., Weiss, A. & Bluestone, J. A. CD28 costimulation: from mechanism to therapy. Immunity 44, 973–988 (2016).
Frauwirth, K. A. et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 (2002).
Jeong, S. & Park, S. H. Co-stimulatory receptors in cancers and their implications for cancer immunotherapy. Immune Netw. 20, e3 (2020).
Wei, S. C., Duffy, C. R. & Allison, J. P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Disco. 8, 1069–1086 (2018).
Wieder, T., Eigentler, T., Brenner, E. & Rocken, M. Immune checkpoint blockade therapy. J. Allergy Clin. Immunol. 142, 1403–1414 (2018).
Jiang, T., Zhou, C. & Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 5, e1163462 (2016).
West, E. E. et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Invest. 123, 2604–2615 (2013).
Moynihan, K. D. et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 22, 1402–1410 (2016).
Brooks, D. G. et al. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc. Natl Acad. Sci. USA 105, 20428–20433 (2008).
Labanieh, L., Majzner, R. G. & Mackall, C. L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2, 377–391 (2018).
Cruz-Ramos, M. & Garcia-Foncillas, J. CAR-T cell and personalized medicine. Adv. Exp. Med. Biol. 1168, 131–145 (2019).
Heng, G. et al. Sustained therapeutic efficacy of humanized anti-CD19 chimeric antigen receptor T Cells in relapsed/refractory acute lymphoblastic leukemia. Clin. Cancer Res. 26, 1606–1615 (2020).
Maude, S. L. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014).
Park, J. H. et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 378, 449–459 (2018).
Mardiana, S., Solomon, B. J., Darcy, P. K. & Beavis, P. A. Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Sci. Transl. Med. 11, eaaw2293 (2019).
Liu, Y. et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 5, eaax7969 (2020).
Sharabi, A. et al. Regulatory T cells in the treatment of disease. Nat. Rev. Drug Disco. 17, 823–844 (2018).
Shang, B., Liu, Y., Jiang, S. J. & Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci. Rep. 5, 15179 (2015).
Ribas, A. et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 31, 616–622 (2013).
Quirk, S. K., Shure, A. K. & Agrawal, D. K. Immune-mediated adverse events of anticytotoxic T lymphocyte-associated antigen 4 antibody therapy in metastatic melanoma. Transl. Res. 166, 412–424 (2015).
Khunger, A., Rytlewski, J. A., Fields, P., Yusko, E. C. & Tarhini, A. A. The impact of CTLA-4 blockade and interferon-alpha on clonality of T-cell repertoire in the tumor microenvironment and peripheral blood of metastatic melanoma patients. Oncoimmunology 8, e1652538 (2019).
Menjak, I. B., Elias, E. S., Jain, S., Lawrie, D. & Petrella, T. M. Evaluation of a multidisciplinary immunotherapy toxicity monitoring program for patients receiving Ipilimumab for metastatic melanoma. JCO Oncol. Pract. https://doi.org/10.1200/OP.20.00845 (2021).
Dimitriou, F., Long, G. V. & Menzies, A. M. Novel adjuvant options for cutaneous melanoma. Ann. Oncol. 32, 854–865 (2021).
Slingluff, C. L. et al. A phase 1 study of NY-ESO-1 vaccine + anti-CTLA4 antibody Ipilimumab (IPI) in patients with unresectable or metastatic melanoma. Oncoimmunology 10, 1898105 (2021).
Kanikarla Marie, P. et al. Pilot clinical trial of perioperative durvalumab and tremelimumab in the treatment of resectable colorectal cancer liver metastases. Clin. Cancer Res. 27, 3039–3049 (2021).
Sobhani, N. et al. CTLA-4 in regulatory T cells for cancer immunotherapy. Cancers 13, 1440 (2021).
Zappasodi, R. et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 591, 652–658 (2021).
Zhang, L. et al. Association of survival and immune-related adverse events with anti-PD-1/PD-L1 and anti-CTLA-4 inhibitors, alone or their combination for the treatment of cancer: a systematic review and meta-analysis of 13 clinical trials. Front. Oncol. 11, 575457 (2021).
Patel, S. P. et al. A phase II basket trial of dual anti-CTLA-4 and anti-PD-1 blockade in rare tumors (DART) SWOG S1609: high-grade neuroendocrine neoplasm cohort. Cancer https://doi.org/10.1002/cncr.33591 (2021).
Wong, S. K., Beckermann, K. E., Johnson, D. B. & Das, S. Combining anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4) and -programmed cell death protein 1 (PD-1) agents for cancer immunotherapy. Expert. Opin. Biol. Ther. https://doi.org/10.1080/14712598.2021.1921140 (2021).
Solinas, C., De Silva, P., Bron, D., Willard-Gallo, K. & Sangiolo, D. Significance of TIM3 expression in cancer: from biology to the clinic. Semin Oncol. 46, 372–379 (2019).
Qi, Y. et al. Research progress concerning dual blockade of lymphocyte-activation gene 3 and programmed death-1/programmed death-1 ligand-1 blockade in cancer immunotherapy: preclinical and clinical evidence of this potentially more effective immunotherapy strategy. Front. Immunol. 11, 563258 (2020).
Chauvin, J. M. & Zarour, H. M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 8, e000957 (2020).
Sakuishi, K. et al. TIM3(+)FOXP3(+) regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology 2, e23849 (2013).
Anderson, A. C. Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol. Res. 2, 393–398 (2014).
Ngiow, S. F. et al. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 71, 3540–3551 (2011).
Sabatos, C. A. et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 4, 1102–1110 (2003).
Schaer, D. A., Murphy, J. T. & Wolchok, J. D. Modulation of GITR for cancer immunotherapy. Curr. Opin. Immunol. 24, 217–224 (2012).
Lu, L. et al. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J. Transl. Med. 12, 36 (2014).
Ko, K. et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J. Exp. Med. 202, 885–891 (2005).
Wang, B. et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8(+) T cell dysfunction and maintain memory phenotype. Sci. Immunol. 3, eaat7061 (2018).
Shimizu, J., Yamazaki, S., Takahashi, T., Ishida, Y. & Sakaguchi, S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 3, 135–142 (2002).
Curti, B. D. et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 73, 7189–7198 (2013).
Lutsiak, M. E. et al. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 105, 2862–2868 (2005).
Whiteside, T. L. The role of regulatory T cells in cancer immunology. Immunotargets Ther. 4, 159–171 (2015).
Desar, I. M. et al. Sorafenib reduces the percentage of tumour infiltrating regulatory T cells in renal cell carcinoma patients. Int. J. Cancer 129, 507–512 (2011).
Adotevi, O. et al. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J. Immunother. 33, 991–998 (2010).
Dannull, J. et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J. Clin. Invest. 115, 3623–3633 (2005).
Maury, S. et al. Lymphodepletion followed by infusion of suicide gene-transduced donor lymphocytes to safely enhance their antitumor effect: a phase I/II study. Leukemia 28, 2406–2410 (2014).
Fallarino, F. et al. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 4, 1206–1212 (2003).
Sharma, M. D. et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 113, 6102–6111 (2009).
Sugiyama, D. et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl Acad. Sci. USA 110, 17945–17950 (2013).
Faget, J. et al. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res. 71, 6143–6152 (2011).
Curiel, T. J. et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–949 (2004).
Ogura, M. et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J. Clin. Oncol. 32, 1157–1163 (2014).
Facciabene, A. et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475, 226–230 (2011).
Okubo, Y., Mera, T., Wang, L. & Faustman, D. L. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci. Rep. 3, 3153 (2013).
Soares, K. C. et al. TGF-beta blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget 6, 43005–43015 (2015).
Turnis, M. E. et al. Interleukin-35 limits anti-tumor immunity. Immunity 44, 316–329 (2016).
Pellegrini, P. et al. Disregulation in TH1 and TH2 subsets of CD4+ T cells in peripheral blood of colorectal cancer patients and involvement in cancer establishment and progression. Cancer Immunol. Immunother. 42, 1–8 (1996).
Fridman, W. H. et al. The immune microenvironment of human tumors: general significance and clinical impact. Cancer Microenviron. 6, 117–122 (2013).
Zhang, Z. et al. Infiltration of dendritic cells and T lymphocytes predicts favorable outcome in epithelial ovarian cancer. Cancer Gene Ther. 22, 198–206 (2015).
Haabeth, O. A. et al. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat. Commun. 2, 240 (2011).
Brunsvig, P. F. et al. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 55, 1553–1564 (2006).
Kyte, J. A. Cancer vaccination with telomerase peptide GV1001. Expert Opin. Investig. Drugs 18, 687–694 (2009).
Godet, Y. et al. Analysis of spontaneous tumor-specific CD4 T-cell immunity in lung cancer using promiscuous HLA-DR telomerase-derived epitopes: potential synergistic effect with chemotherapy response. Clin. Cancer Res. 18, 2943–2953 (2012).
Hokey, D. A., Larregina, A. T., Erdos, G., Watkins, S. C. & Falo, L. D. Jr. Tumor cell loaded type-1 polarized dendritic cells induce Th1-mediated tumor immunity. Cancer Res 65, 10059–10067 (2005).
Xu, H. M. Th1 cytokine-based immunotherapy for cancer. Hepatobiliary Pancreat. Dis. Int. 13, 482–494 (2014).
Coughlin, C. M. et al. Interleukin-12 and interleukin-18 synergistically induce murine tumor regression which involves inhibition of angiogenesis. J. Clin. Invest. 101, 1441–1452 (1998).
Carreno, B. M. et al. IL-12p70-producing patient DC vaccine elicits Tc1-polarized immunity. J. Clin. Invest. 123, 3383–3394 (2013).
Kaisar, M. M. M. et al. Dectin-1/2-induced autocrine PGE2 signaling licenses dendritic cells to prime Th2 responses. PLoS Biol. 16, e2005504 (2018).
Kapsenberg, M. L., Hilkens, C. M., Wierenga, E. A. & Kalinski, P. The paradigm of type 1 and type 2 antigen-presenting cells. Implic. Atopic Allergy Clin. Exp. Allergy 29, 33–36 (1999).
Padol, I. T. & Hunt, R. H. Association of myocardial infarctions with COX-2 inhibition may be related to immunomodulation towards a Th1 response resulting in atheromatous plaque instability: an evidence-based interpretation. Rheumatology 49, 837–843 (2010).
Qian, Y. & Zhang, M. The functional roles of IL-33/ST2 axis in ocular diseases. Mediators Inflamm. 2020, 5230716 (2020).
Gao, X. et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J. Immunol. 194, 438–445 (2015).
Jiang, Z. et al. IL-25 blockade inhibits metastasis in breast cancer. Protein Cell 8, 191–201 (2017).
Kryczek, I. et al. Cutting edge: opposite effects of IL-1 and IL-2 on the regulation of IL-17+ T cell pool IL-1 subverts IL-2-mediated suppression. J. Immunol. 179, 1423–1426 (2007).
Haghshenas, M. R., Khademi, B., Faghih, Z., Ghaderi, A. & Erfani, N. Immune regulatory cells and IL17-producing lymphocytes in patients with benign and malignant salivary gland tumors. Immunol. Lett. 164, 109–116 (2015).
Kryczek, I. et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 114, 1141–1149 (2009).
Lai, Y. & Dong, C. Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int. Immunol. 28, 181–188 (2016).
Chang, S. H. et al. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl Acad. Sci. USA 111, 5664–5669 (2014).
Feng, S., Chen, X. M., Wang, J. F. & Xu, X. Q. Th17 cells associated cytokines and cancer. Eur. Rev. Med. Pharm. Sci. 20, 4032–4040 (2016).
Gorczynski, R. M. IL-17 signaling in the tumor microenvironment. Adv. Exp. Med. Biol. 1240, 47–58 (2020).
Langowski, J. L. et al. IL-23 promotes tumour incidence and growth. Nature 442, 461–465 (2006).
van Duijneveldt, G., Griffin, M. D. W. & Putoczki, T. L. Emerging roles for the IL-6 family of cytokines in pancreatic cancer. Clin. Sci. 134, 2091–2115 (2020).
Feurino, L. W. et al. IL-6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol. Ther. 6, 1096–1100 (2007).
Tsou, P., Katayama, H., Ostrin, E. J. & Hanash, S. M. The emerging role of B cells in tumor immunity. Cancer Res. 76, 5597–5601 (2016).
Burger, J. A. & Wiestner, A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat. Rev. Cancer 18, 148–167 (2018).
Largeot, A., Pagano, G., Gonder, S., Moussay, E. & Paggetti, J. The b-side of cancer immunity: the underrated tune. Cells 8, 449 (2019).
Yuen, G. J., Demissie, E. & Pillai, S. B lymphocytes and cancer: a love-hate relationship. Trends Cancer 2, 747–757 (2016).
Sorenmo, K. U. et al. CD40-activated B cell cancer vaccine improves second clinical remission and survival in privately owned dogs with non-Hodgkin’s lymphoma. PLoS ONE 6, e24167 (2011).
Wennhold, K. et al. CD40-activated B cells induce anti-tumor immunity in vivo. Oncotarget 8, 27740–27753 (2017).
Shimabukuro-Vornhagen, A. et al. The immunosuppressive factors IL-10, TGF-beta, and VEGF do not affect the antigen-presenting function of CD40-activated B cells. J. Exp. Clin. Cancer Res. 31, 47 (2012).
Gonzalez, N. K. et al. In vitro and in vivo imaging of initial B-T-cell interactions in the setting of B-cell based cancer immunotherapy. Oncoimmunology 4, e1038684 (2015).
Rossetti, R. A. M. et al. B lymphocytes can be activated to act as antigen presenting cells to promote anti-tumor responses. PLoS ONE 13, e0199034 (2018).
Li, Q. et al. Adoptive transfer of tumor reactive B cells confers host T-cell immunity and tumor regression. Clin. Cancer Res. 17, 4987–4995 (2011).
Sorrentino, R. et al. B cells contribute to the antitumor activity of CpG-oligodeoxynucleotide in a mouse model of metastatic lung carcinoma. Am. J. Respir. Crit. Care Med. 183, 1369–1379 (2011).
Deng, J. et al. Engineered fusokine GIFT4 licenses the ability of B cells to trigger a tumoricidal T-cell response. Cancer Res. 74, 4133–4144 (2014).
Li, W. et al. Tumor-derived autophagosomes (DRibbles) induce B cell activation in a TLR2-MyD88 dependent manner. PLoS ONE 8, e53564 (2013).
Ren, H. et al. Therapeutic antitumor efficacy of B cells loaded with tumor-derived autophagasomes vaccine (DRibbles). J. Immunother. 37, 383–393 (2014).
Rosser, E. C., Blair, P. A. & Mauri, C. Cellular targets of regulatory B cell-mediated suppression. Mol. Immunol. 62, 296–304 (2014).
Lundy, S. K. & Klinker, M. W. Characterization and activity of Fas ligand producing CD5(+) B cells. Methods Mol. Biol. 1190, 81–102 (2014).
Olkhanud, P. B. et al. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res. 71, 3505–3515 (2011).
Lindner, S. et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res. 73, 2468–2479 (2013).
Lampropoulou, V. et al. TLR-activated B cells suppress T cell-mediated autoimmunity. J. Immunol. 180, 4763–4773 (2008).
Serhan, C. N., Chiang, N. & Van Dyke, T. E. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361 (2008).
Zhang, Y. et al. Mammary-tumor-educated B cells acquire LAP/TGF-beta and PD-L1 expression and suppress anti-tumor immune responses. Int. Immunol. 28, 423–433 (2016).
Song, S. S. et al. Protective effects of total glucosides of paeony on N-nitrosodiethylamine-induced hepatocellular carcinoma in rats via down-regulation of regulatory B cells. Immunol. Invest. 44, 521–535 (2015).
Kondo, K. et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia 32, 960–970 (2018).
Mohamed, E. et al. The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity 52, 668–682 e667 (2020).
Malhi, H. & Camilleri, M. Modulating bile acid pathways and TGR5 receptors for treating liver and GI diseases. Curr. Opin. Pharm. 37, 80–86 (2017).
Schroeder, B. O. & Backhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 22, 1079–1089 (2016).
Wargo, J. A. Modulating gut microbes. Science 369, 1302–1303 (2020).
Shalapour, S. & Karin, M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J. Clin. Invest. 125, 3347–3355 (2015).
Liu, L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J. Pharm. Sci. 104, 1866–1884 (2015).
Dillard, T., Yedinak, C. G., Alumkal, J. & Fleseriu, M. Anti-CTLA-4 antibody therapy associated autoimmune hypophysitis: serious immune related adverse events across a spectrum of cancer subtypes. Pituitary 13, 29–38 (2010).
Boutros, C. et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 13, 473–486 (2016).
Osorio, J. C. et al. Antibody-mediated thyroid dysfunction during T-cell checkpoint blockade in patients with non-small-cell lung cancer. Ann. Oncol. 28, 583–589 (2017).
Gauci, M. L. et al. Autoimmune diabetes induced by PD-1 inhibitor-retrospective analysis and pathogenesis: a case report and literature review. Cancer Immunol. Immunother. 66, 1399–1410 (2017).
Shah, N. N. & Fry, T. J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 16, 372–385 (2019).
Schonthal, A. H., Chen, T. C., Hofman, F. M., Louie, S. G. & Petasis, N. A. Celecoxib analogs that lack COX-2 inhibitory function: preclinical development of novel anticancer drugs. Expert Opin. Investig. Drugs 17, 197–208 (2008).
Kashfi, K. et al. Nitric oxide-donating nonsteroidal anti-inflammatory drugs inhibit the growth of various cultured human cancer cells: evidence of a tissue type-independent effect. J. Pharm. Exp. Ther. 303, 1273–1282 (2002).
Rigas, B. & Williams, J. L. NO-donating NSAIDs and cancer: an overview with a note on whether NO is required for their action. Nitric Oxide 19, 199–204 (2008).
Zheng, L., Lv, W., Zhou, Y., Lin, X. & Yao, J. Progress on the mechanism for aspirin’s anti-tumor effects. Curr. Drug Targets 22, 105–111 (2021).
Ding, X. Z., Iversen, P., Cluck, M. W., Knezetic, J. A. & Adrian, T. E. Lipoxygenase inhibitors abolish proliferation of human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 261, 218–223 (1999).
Katz, M. S. Therapy insight: potential of statins for cancer chemoprevention and therapy. Nat. Clin. Pr. Oncol. 2, 82–89 (2005).
Acknowledgements
The authors thank the editors and anonymous reviewers for their efforts on improving this manuscript during the review process. This work was supported by the Major International (Regional) Joint Research Program of the National Natural Science Foundation of China (No. 81920108027), Chongqing Outstanding Youth Science Foundation, Chongqing Talent Innovation Leader Project, and Funding for Chongqing University Innovation Research Group.
Author information
Authors and Affiliations
Contributions
Y.L. and Y.W. conceived and organized the manuscript. H.Z., L.W., G.Y., Y.C., and M.Z. wrote the manuscript, prepared the figures, table, and contributed to the discussion. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, H., Wu, L., Yan, G. et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Sig Transduct Target Ther 6, 263 (2021). https://doi.org/10.1038/s41392-021-00658-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00658-5
This article is cited by
-
Pan-immune-inflammation value and body mass index to predict survival in diffuse large B-cell lymphoma
The Egyptian Journal of Internal Medicine (2024)
-
The opposing effect of acute and chronic Toxoplasma gondii infection on tumor development
Parasites & Vectors (2024)
-
Tobacco smoke condensate-induced senescence in endothelial cells was ameliorated by colchicine treatment via suppression of NF-κB and MAPKs P38 and ERK pathways activation
Cell Communication and Signaling (2024)
-
Pan-cancer analysis of CDKN2A alterations identifies a subset of gastric cancer with a cold tumor immune microenvironment
Human Genomics (2024)
-
Relationships between nine neuropsychiatric disorders and cervical cancer: insights from genetics, causality and shared gene expression patterns
BMC Women's Health (2024)
