
Abstract
Colorectal cancer (CRC) is among the most lethal and prevalent malignancies in the world and was responsible for nearly 881,000 cancer-related deaths in 2018. Surgery and chemotherapy have long been the first choices for cancer patients. However, the prognosis of CRC has never been satisfying, especially for patients with metastatic lesions. Targeted therapy is a new optional approach that has successfully prolonged overall survival for CRC patients. Following successes with the anti-EGFR (epidermal growth factor receptor) agent cetuximab and the anti-angiogenesis agent bevacizumab, new agents blocking different critical pathways as well as immune checkpoints are emerging at an unprecedented rate. Guidelines worldwide are currently updating the recommended targeted drugs on the basis of the increasing number of high-quality clinical trials. This review provides an overview of existing CRC-targeted agents and their underlying mechanisms, as well as a discussion of their limitations and future trends.
Similar content being viewed by others
Introduction
Current treatment for colorectal cancer
Colorectal cancer (CRC) ranks as the second most lethal cancer and the third most prevalent malignant tumor worldwide. In 2018, 1.8 million new CRC cases arose, and 881,000 deaths were reported, which accounted for nearly 10% of new cancer cases and deaths worldwide,1 and the number of new cases may increase to nearly 2.5 million in 2035.2 According to statistics in the USA, the death rate declined by ~50% in 2016 (13.7 per 10,000 patients) compared with that in 1970 (29.2 per 10,000 patients) because of the rapid development of screening methods and improved treatment methods. However, this trend seems to be observed only in highly developed countries.2 Meanwhile, the 5-year survival rate for CRC is ~64%, but drops to 12% for metastatic CRC, and further investigation is still required to develop effective approaches for medical intervention.3
Given the advances in primary and adjuvant treatments, the survival time in CRC has been improving. Typically, the ideal CRC treatment is to achieve complete removal of the tumor and metastases, which mostly requires surgical intervention.4 However, despite the emergence of numerous screening programs to reduce CRC incidence, nearly a quarter of CRCs are diagnosed at an advanced stage with metastases, and 20% of the remaining cases may develop metachronous metastases, which result in difficulties in curative surgical control and subsequent tumor-related deaths.5,6,7,8 For those patients with unresectable lesions or who are intolerant to surgery, the goal is maximum shrinkage of the tumor and suppression of further tumor spread and growth, and radiotherapy and chemotherapy are the leading strategies for controlling disease in such patients. Of note, in some cases, chemotherapy or radiotherapy might be applied before or after surgery as neoadjuvant or adjuvant treatment to maximally reduce and stabilize the tumor.9,10,11,12
Chemotherapy
Current chemotherapy includes both single-agent therapy, which is mainly fluoropyrimidine (5-FU)-based, and multiple-agent regimens containing one or several drugs, including oxaliplatin (OX), irinotecan (IRI), and capecitabine (CAP or XELODA or XEL). Although studies have argued that first-line single-agent therapy is not inferior to combined regimens in terms of overall survival (OS),13,14 the combined therapy regimens FOLFOX (5-FU+OX), FOXFIRI (5-FU+IRI), XELOX or CAPOX (CAP+OX), and CAPIRI (CAP+OX) remain the mainstream approaches in first-line treatment, while patients with poor performance or at low risk of deterioration are recommended to receive single-agent therapy. When choosing additive agents, efficacy appears to be similar, and only adverse events may differ among different regimens.12,15,16,17 Emerging evidence does not support stronger efficacy in the multiple-agent regimen FOLOXIRI (5-FU+OX+IRI), which is infrequently applied because of its potential increased toxicity.18,19 Nonetheless, data from research performed in recent decades show that using chemotherapy in patients with CRC, especially those with metastases, has pushed their OS time to almost 20 months, resulting in chemotherapy becoming the backbone of CRC treatment.15,20,21 However, chemotherapy is associated with certain limitations, such as existing systemic toxicity, unsatisfying response rate, unpredictable innate and acquired resistance, and low tumor-specific selectivity. Therefore, massive investments have been pledged to develop new approaches to refine or even replace existing CRC chemotherapy.
Targeted therapy
The idea of molecular targeted therapy has a relatively long history. The concept of a chemical that specifically targets a microorganism was first proposed in the early 1900s and expanded to cancer treatment in 1988,22 and this concept was renewed and has flourished in the past 20 years.23
Targeted therapies can work on cancerous cells by directly inhibiting cell proliferation, differentiation, and migration. The tumor microenvironment, including local blood vessels and immune cells, might also be altered by targeted drugs to impede tumor growth and enact stronger immune surveillance and attack. Small molecules, such as monoclonal antibodies, are major players in targeted therapies.24,25,26 Small molecules are a group of molecules with a molecular weight <900 Da that might penetrate into cells, mostly working within cells to inactivate selected enzymes, thereby interfering with tumor cell growth and even triggering apoptosis. Cyclin-dependent kinases, proteasomes, and poly ADP-ribose polymerase make up most of the molecular targets. Carfilzomib for multiple myeloma, ribociclib for metastatic breast cancer, and rucaparib for BRCA-positive ovarian cancer are a few examples.23 For targets outside cells, such as cell surface receptors or membrane-bound sites, monoclonal antibodies or therapeutic antibodies can recognize and bind them to directly regulate downstream cell cycle progression and cell death. In addition, certain monoclonal antibodies work on cells other than cancer cells, such as immune cells, which helps to manipulate the immune system to attack human cancer.
Landscape of current CRC-targeted therapy
The first targeted agent for CRC approved by the Food and Drug Administration (FDA) was cetuximab in 2004, followed by bevacizumab in the same year, and emerging FDA-approved targeted drugs for CRC have been brought to market successively since then, with more on the way (Fig. 1). Numerous agents have been developed and brought into preclinical and clinical trials. The list of recommended CRC-targeted agents from guidelines such as those from the National Comprehensive Cancer Network (NCCN) is being updated quickly, given the unprecedented speed of the emergence of large trials (Fig. 2).

VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor; EGFR: epidermal growth factor receptor

CRC: colorectal cancer; VEGF/VEGFR: vascular endothelial growth factor/vascular endothelial growth factor receptor; EGF/EGFR: epidermal growth factor/epidermal growth factor receptor; HGF: hepatocyte growth factor; c-MET: mesenchymal–epithelial transition factor; IGF/IGF-1R: insulin-like growth factor/ insulin-like growth factor 1 receptor; TGF: transforming growth factor
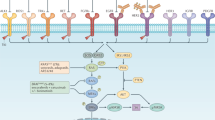
Various pathways mediating the initiation, progression, and migration of CRC, such as Wnt/β-catenin, Notch, Hedgehog, and TGF-β (transforming growth factor-β)/SMAD, as well as those capable of activating signaling cascades, such as phosphatidylinositol 3-kinase (PI3K)/AKT or RAS/rapidly accelerated fibrosarcoma (RAF), contain ideal sites for targeted therapy (Fig. 2).27,28 Given the complex downstream signaling and difficulties in completely inhibiting specific biological interactions, not all existing CRC-related pathways can be successfully interfered with, and current data cover only a few pathways in which experimentally identified targeted agents can be proved to be efficient in clinical studies, and a large group of targeted drugs remain in preclinical status or in phase I trials.
The EGFR-related pathway
Activities of the pathway
EGFR (epidermal growth factor receptor) belongs to the ErbB (erythroblastosis oncogene B)/HER (human epidermal growth factor receptor) family, which consists of four members: ErbB1 (EGFR/HER1), ErbB2 (Neu/HER2), ErbB3 (HER3), and ErbB4 (HER4).29,30 The ErbB receptors were first considered to be related to carcinogenesis almost 30 years ago and are quite unique among the multitude of receptor tyrosine kinases. The impaired kinase activity of HER3/ErbB3 and the absence of a direct HER2/ErbB2 ligand mean that these transmembrane glycoprotein can only be activated after homo- or heterodimerization with HER2, HER3, or HER4 through specific binding, mainly by EGF or TGF-α. Once activated, various downstream intracellular signaling pathways, including the RAS/RAF/MEK/ERK, PI3K/AKT, and JAK/STAT3 (Janus kinase/signal transducer and activator of transcription 3) pathways, are triggered to regulate cell growth, survival, and migration.31,32,33
Aberrant expression levels of EGFR and HER have been identified in a group of cancers, including glioma; melanoma; medulloblastoma; gastrointestinal tumors such as esophageal, colorectal, and gastric cancers; and cancers in the lung, breast, bladder, prostate, pancreas, and ovary.29 Overexpression of EGFR has been observed in 15–30% of breast cancers, 60% of NSCLCs (non-small-cell lung cancer), and 25–77% of CRCs, which might also indicate poor prognosis.34,35,36 HER2 overexpression occurs in ~20–30% of breast and ovarian cancers,33,37 in 3.8–36.6% of gastric cancers38 and in 1.3–47.7% of CRCs.39 HER3 showed higher expression in 83% of gastrointestinal tumors and 20% of breast, ovarian, and bladder cancers than in normal tissues;33,40 however, it was prohibited from becoming a drug target given difficulties in finding its ligand. HER4 remains controversial because both cancer-promoting and cancer-suppressing effects have been found.41,42 Therefore, substantial efforts are being made to develop mainstream targeted drugs for HER1 and HER2 while facing potential drug resistance caused by mutations of HER1 and HER2. For instance, mutations of EGFR and HER2 were found in 15–30% of NSCLC samples43,44 and in 1.6% of HER2-positive breast cancer cases.45
The typical ErbB receptor consists of a ligand-binding domain outside the cell, a transmembrane domain, and an intracellular domain with distinct tyrosine residues in the C-terminal region where subsequent phosphorylation may take place upon activation.46
Activation of EGFR triggers various downstream signaling pathways that mediate cellular proliferation or metabolism, playing vital roles in cancer initiation and progression. Activated EGFR initiates plasma recruitment of SOSs (son of sevenless homologs) to achieve RAS-RAF activation, which leads to phosphorylation of mitogen-activated protein kinase (MAPK or MEK) and activation of extracellular signal-related kinase (ERK), which might then translocate inside the nucleus to regulate the expression of transcription factors such as c-FOS and ELK1.47,48,49,50 It is worth mentioning that during RAS-RAF activation, the serine/threonine protein kinase in the RAF family, BRAF (BRAF proto-oncogene, serine/threonine kinase), plays vital roles in the RAS/RAF/MEK pathway. In contrast to RAS mutations, BRAF mutations, mostly comprising the V600E alteration, were found in 5–10% of metastatic CRC cases and cause activation of downstream MAPK regardless of RAS status.51,52,53,54
Activation of PI3K by RAS or direct activation by EGFR transforms the second messenger phosphatidylinositol-bisphosphate into phosphatidylinositol-trisphosphate (PIP3) through phosphorylation. PIP3 interacts with the SH3 domain of serine/threonine kinase PKB (also called AKT) recruited to the cell membrane. AKT plays significant roles in cell growth and apoptosis and works as a vital mediator in the ErbB-related pathway.55,56,57 In addition, the ErbB2-3 heterodimer is the strongest activator of the PI3K/AKT pathway among all the ErbB-dimer family members. Cancer and diabetes are closely related to poorly regulated AKT activity.58,59 AKT regulates cell cycle entry and survival via phosphorylation of forkhead box O, BCL2-associated agonist of cell death, and glycogen synthase kinase 3 (GSK-3), thereby preventing cellular apoptosis through mammalian target of rapamycin activation.57,58,59
Another vital protein that influences various cellular biological functions, such as cell motility, growth, differentiation, and membrane ruffle formation, which are mainly activated by EGFR, is phospholipase C-γ1 (PLC-γ1).60,61,62 This 145 kDa protein has one SH3 domain, two pleckstrin homology domains, and two SH2 domains that might interact with EGFR, eventually increasing enzyme activity to produce inositol-triphosphate (IP3) and diacylglycerol (DAG) from hydrolysis of phosphatidylinositol-bisphosphate.63,64,65,66,67 IP3 and DAG promote the release of intracellular Ca2+ and activate protein kinase C to promote carcinogenesis.68,69 Moreover, recent studies stated that the SH3 domain of PLC-γ1 might be of great importance in the interaction with EGFR. EGF mediates PLC-γ1 binding to AKT, altering its activity through the SH3 domain.70 In addition, PI3K enhancer, a nuclear GTPase that activates nuclear PI3K activity,71 dynamin-1, and Racl, which might enhance EGF-induced cell proliferation and migration, are regulated by the SH3 domain of PLC-γ1, acting as guanine nucleotide exchange factors.70,72
EGFR might directly bind to and phosphorylate STATs to enable them to dimerize and transfer into the nucleus, where they mediate cell growth, differentiation, and apoptosis by regulating related gene transcription.73,74 In addition, a nonreceptor tyrosine kinase (c-Src) acts on EGFR in an indirect way to govern STATs, which exerts a crucial effect. Overexpression of c-Src and EGFR occurs in many cancer cases, suggesting a close interaction between them and their potential contributions to tumor proliferation. The Src family is a group of nonreceptor tyrosine kinases that overlap with the STAT and PI3K pathways. SRCs enhance EGFR signaling through c-SRC-dependent phosphorylation and c-SRC-EGFR complex formation.75,76
Targeting EGFR and EGFR-related pathways
Methods to target the EGFR pathway typically comprise anti-EGFR monoclonal antibodies and tyrosine kinase inhibitors (TKIs) aimed at intracellular kinases (Tables 1 and 2).
Cetuximab and panitumumab
In 1995, the first monoclonal antibody targeted to EGFR with convincing preclinical data was announced. Named cetuximab, it is a chimeric immunoglobulin G (IgG) antibody that induces EGFR internalization and degradation once bound to the external domain of EGFR.77 Cetuximab showed great potential in progression-free survival (PFS) improvement in patients with low response to single-agent IRI therapy, according to the BOND trial, which contributed to the FDA approval of cetuximab for metastatic CRC in 2004.78 Moreover, a subsequent study also confirmed that cetuximab treatment prolonged OS and PFS in patients with CRCs when previous treatment with fluoropyrimidine, IRI and OX failed or was contraindicated.79 Combinations of cetuximab with other existing chemotherapies also displayed promising results. The phase III CRYSTAL trial found that cetuximab plus the FOLFIRI regimen had better progression control (8.9 vs. 8 months, hazard ratio (HR) 0.85; p = 0.048) than FOLFIRI alone, although the OS was not significantly different (HR, 0.93; p = 0.31).80 Interestingly, in different studies investigating cetuximab combined with FOLFOX in metastatic patients with CRC,81,82,83 no significant PFS or OS improvement was identified given that the doses in FOLFOX might have differed between studies because of the impact of the crossover design, but this lack of improvements in PFS and OS has also now been ascribed to CRC molecular heterogeneity.84 Maintaining cetuximab alone after a FOLFOX plus cetuximab regimen was not inferior to maintaining combination therapy in terms of PFS, with fewer adverse reactions noted.85 Escalating to the maximal dose of cetuximab based on the intensity of skin rash in the EVEREST trial suggested that an overall response might be achieved but without OS improvement.86
Murine-human chimeric antibodies might cause immunogenic reactions; therefore, the fully humanized antibody panitumumab has been developed, which does not trigger antibody-dependent cell-mediated cytotoxicity like cetuximab does87 and showed a lower risk of hypersensitivity reactions (0.6–3.0% for panitumumab and 3.5–7.5% for cetuximab).88 The efficacy of panitumumab against CRC was evaluated in the PRIME trial when FOLFOX plus panitumumab was compared with FOLFOX alone, and the combination regimen achieved a better PFS (10 vs. 8.6 months, HR 0.80, p = 0.01) and OS than FOLFOX alone (23.9 vs. 19.7 months, HR = 0.88, p = 0.17), with further demonstrated significance in the updated survival analysis (HR = 0.83, p = 0.003) in patients with metastatic CRC.89,90
Maintenance with panitumumab and 5-FU/LV after panitumumab plus FOLFOX showed numerical improvement in PFS and OS compared with single-agent panitumumab in the retrospective analysis of the PRIME and PEAK trials.91 The toxicity of this combination did not increase, which was confirmed in the VALENTINO trial, in which maintaining single-agent panitumumab appeared to have shorter PFS (HR = 1.55, p = 0.011) than treatment with panitumumab combined with 5-FU/LV.92
Cetuximab and panitumumab are both FDA-approved agents for the first-line treatment of CRC. No inferiority or superiority was identified in the phase III ASPECCT study between these two drugs. Cetuximab resulted in an OS of 10.0 months, and the OS was 10.4 months for panitumumab (HR 0.97, p < 0.0007 for noninferiority), in which no obvious adverse events were noted other than the incidence of grade 3 or 4 hypomagnesemia (3% for cetuximab and 7% for panitumumab).93 This also indicated that antibody-dependent cell-mediated cytotoxicity was not a major mechanism for these agents. However, in terms of quality-adjusted life-years, panitumumab seemed to be more economically efficient than cetuximab.93
For second-line treatment of CRC or beyond, anti-EGFR agents might be low priority because in several studies cetuximab and panitumumab have been demonstrated to fail to reach statistically better PFS or OS for patients with CRC.94,95,96 In fact, only one study97 reported that panitumumab significantly prolonged PFS (8 vs. 7.3 weeks, HR = 0.54, p < 0.001) compared with best supportive care in patients with chemorefractory CRC with an acceptable rate of adverse events. In general, anti-EGFR agents are among the least attractive choices in second-line treatment, especially compared with anti-vascular endothelial growth factor (VEGF) agents, which will be discussed in a subsequent section.
Notably, subgroup analysis has indicated that both of these anti-EGFR agents are robustly beneficial to those patients with RAS-wild-type tumors in the CRYSTAL, PRIME, and TAILOR trials,80,90,98 even though negative outcomes were experienced in patients with RAS mutations (KRAS and NRAS exon 2, 3, and 4 mutations). Interestingly, left-sided CRC tends to be more enriched for EGFR expression than right-sided CRC, in which MSI or BRAF mutations are predominantly activated.99 This sidedness leads to different clinical outcomes, such that worse OS and PFS have been observed in right-sided CRC than in left-sided CRC regardless of the choice of chemotherapy regimen or targeted agent.100,101 This biological factor has also been validated in anti-EGFR agent trials: in terms of RR, PFS, and OS within RAS-wild-type patients, those with left-sided tumors were expected to have better clinical outcomes than those with right-sided cancers.80,90,98 As demonstrated above, BRAF mutations are independent from RAS mutations and are closely related to a low anti-EGFR response, and both the NCCN and ESMO guidelines recommend using cetuximab and panitumumab in confirmed BRAF-wild-type and RAS-wild-type patients.102
BRAF inhibitors
A higher incidence of mutated BRAF is found in melanoma than in CRC. The efficiency of BRAF inhibitors in BRAF-V600E-mutated melanoma prompted the development of a similar approach in CRC. A few studies investigated blocking BRAF or BRAF/MEK using vemurafenib or dabrafenib or using selective BRAF inhibitors and trametinib; however, a selective MEK inhibitor failed to improve the PFS or OS of patients with metastatic CRC, even though downstream MAPK activity was inhibited after drug administration. Some scholars have suggested that BRAF/MEK blockade might trigger feedback reactivation of EGFR, which would bypass activating MAPK via RAS.103,104,105 Preclinical research indicated that a combination of BRAF inhibitors and an upstream-pathway inhibitor might be superior to BRAF inhibition alone in terms of tumor growth control in BRAF-mutated CRC xenograft models.104,106,107 Subsequent studies focused on the combined use of BRAF inhibitors and EGFR inhibitors.108,109 Promising survival outcomes and response rates were observed in trials using vemurafenib combined with IRI and cetuximab for patients with BRAF-mutant CRC.110,111 In a phase II trial using encorafenib (a BRAF inhibitor) plus cetuximab, with or without alpelisib (ALP), the PFS and OS were improved compared with those seen in historical data.112 A triplet regimen consisting of dabrafenib, trametinib, and panitumumab achieved a better response rate than the doublet regimens (21% vs. 10% for dabrafenib + panitumumab or 0% for trametinib + panitumumab) in patients with BRAF-V600E-mutated CRCs.109 Similar results were reported for the ongoing BEACON trial, in which a triplet regimen of encorafenib, binimetinib (a MEK inhibitor), and cetuximab was well tolerated and exceeded previous efficacy outcomes for BRAF inhibitors.113 New evidence emerged suggesting that a triple regimen of encorafenib, binimetinib, and cetuximab offered significantly better survival benefit for patients with BRAF-mutated metastatic CRC than that achieved historically with a comparable rate of adverse events (OS: 9 vs. 5.4 months, HR = 0.52, p < 0.001; RR: 26% vs. 2%, p < 0.01).113 Second- or third-line regimens treating BRAF-V600E-mutated mCRC now may include anti-EGFR agents combined with vemurafenib + IRI or dabrafenib + trametinib or encorafenib + binimitinib, as recommended by the NCCN.
HER2 inhibitor
As discussed above, HER2 acts similarly to EGFR because it shares many downstream pathways, such as RAS/RAF/MEK and PI3K/AKT, and overexpression of HER provides one explanation for anti-EGFR resistance.114,115,116 Unlike the rate in breast cancer or gastric cancer, the rate of HER2 overexpression is relatively low (2–3%) and is independent of RAS or RAF mutation in patients with CRC.117,118,119 Preclinical studies revealed that HER2 amplification might compensate for EGFR blockade, and combined targeting of HER2 and EGFR inhibited tumor cell proliferation, producing an effect that was stronger than that achieved using either single agent alone.115,118,120 Several clinical trials have been developed to determine whether targeted agents against HER2-positive CRC (determined by immunohistochemistry (IHC), fluorescence in situ hybridization (FISH), or chromogenic in situ hybridization) can be as effective as those against breast cancer or gastric cancer. A few of these studies using a single HER2-targeted agent, with or without chemotherapy, were terminated early because of a low patient response rate or insignificant patient survival benefits.121,122 By contrast, dual-targeted HER2 therapy was found to be promising in preclinical research.118,123 In the phase II MyPathway trial, doublet treatment with trastuzumab, a classic HER2 inhibitor, and pertuzumab, a HER2 dimerization inhibitor, both of which are FDA approved in HER-positive breast cancer treatment, helped patients with HER2-amplified metastatic CRC to gain an overall response rate of 32%, a PFS of 2.9 months and an OS of 11.5 months, which may be even better in patients with RAS-wild-type CRCs (PFS: 5.3 months and OS: 14 months OS).124 Another dual anti-HER2 agent combination of trastuzumab and lapatinib (a TKI targeting both EGFR and HER2) against metastatic CRC was studied in the phase II HERACLES trial and reached an overall response rate of 30%, a PFS of 21 weeks and an OS of 46 weeks.125 In addition, this combination was capable of overcoming resistance to pertuzumab and trastuzumab doublet treatment.126 Given the low rate of HER2 overexpression and difficulties in identifying suitable dual-HER2 regimens, the HERACLES trial took great pains to find a potentially effective doublet regimen consisting of lapatinib;125 thus, targeting HER2 might act as a backup regimen for patients with RAS-wild-type HER2-positive CRC. Notably, left-sided colon tumors tend to overexpress HER2 more than those on the right side. Thus, anti-HER2 therapy might offer a new choice for anti-EGFR-resistant CRC.125
EGFR resistance
Accumulating evidence shows that even patients with RAS-wild-type CRC might not benefit from EGFR-targeted therapy, which suggests that identifying certain factors predicting low anti-EGFR therapy response and introducing other agents or strategies to overcome resistance would be beneficial. Some of these factors are innate or intrinsic, some are acquired after anti-EGFR treatment, and some may occur in both situations.
RAS mutations
RAS mutations are found in nearly half of patients with CRC, most of whom also harbor KRAS or NRAS mutations (36% for KRAS and 3% for NRAS).127 However, data showed that not all KRAS-mutated patients developed EGFR resistance: 85–90% of patients had mutations in KRAS codons 12 and 13 (exon 2), which largely indicate EGFR therapy resistance.128,129,130 For other sites, such as KRAS G13D, the connection with drug resistance is uncertain.131,132 Moreover, even patients with wild-type KRAS exon 2 might have other RAS mutations in sites such as KRAS exons 3 and 4 and NRAS exons 2, 3, and 4, which are related to negative benefits from cetuximab or panitumumab treatment.84,133
PI3K mutations and PTEN loss
PI3K (encoding phosphatidylinositol-4,5-bisphosphate 3-kinase) mutations occur mostly in exons 9 and 20; mutations in exon 9 or exon 20 are found in 10–18% of patients with metastatic CRC and lead to constitutive activation of the downstream pathway to reverse EGFR-blocking effects in patients with CRC (response rate of 0% vs. 36.8% in mutated vs. nonmutated patients).134,135 PTEN (phosphatase and tensin homolog) is a suppressor in the PI3K/AKT pathway, the loss of which resulted in long-term tumor growth via activated PI3K/AKT and was found in 20–40% of patients with metastatic CRC.136 Theoretically, PTEN loss might be associated with EGFR blockade resistance; however, data from clinical studies remain contradictory.137,138 Given the low occurrence rate of these mutations in CRC, large trials are required for better confirmation.
EGFR alterations
Mutations in EGFR or low expression of EGFR or AREG (amphiregulin)/EREG (epiregulin), key ligands in the EGFR-specific autocrine loop, cause loss of target for anti-EGFR therapy, representing one of the major ways by which EGFR resistance develops in NSCLC and CRC. Although high EGFR levels might correlate slightly with stronger efficacy of anti-EGFR therapy, patients with low EGFR gene expression may benefit less from EGFR blockers than patients with high EGFR gene expression.139,140 Clinical studies also found that low AREG/EREG levels identified a low cetuximab response rate and vice versa.141,142 EGFR mutated sites vary, and the uncommon ones are linked to worse prognosis.136 For the common mutations, the T790M mutation is considered to be a primary alteration inducing EGFR TKI resistance, which is frequently observed in patients with NSCLC.143 For patients with CRCs, the EGFR S492R mutation in the extracellular domain of EGFR may be found in those receiving cetuximab and was responsible for their low drug response; however, they may still respond to panitumumab.144 New agents are being developed to maximize the affinity for mutated EGFR, such as Sym044145,146 and MM151,147 which might simultaneously target several different sites of the EGFR extracellular domain to overcome resistance to cetuximab or panitumumab, and both Sym044 and MM151 are in preclinical studies and clinical trials.
Compensative activation of alternative pathways, such as IGF-1R (insulin-like growth factor 1 receptor), JAK/STAT, c-MET, VEGF, and HER2, is responsible for acquired anti-EGFR resistance. Similar to EGFR, IGF-1R is bound by IGF1 or 2 and may activate RAS/RAF and PI3K/AKT signaling. Increased IGF-1R activation was noted in patients with CRC receiving cetuximab and was associated with a significantly lower response rate than that seen in patients without IGF-1R activation148 (22% vs. 65%, p = 0.002). This effect has also been observed in patients with breast cancer;149 thus, introducing an IGF-1R inhibitor combined with an EGFR blocker might be a practical solution. A phase III trial, combining the IGF-1R inhibitor dalotuzumab with cetuximab, showed numerically superior PFS and OS improvement in patients with CRC with IGF-1R-positive tumors,150 although preclinical studies did not support noticeable benefits from anti-IGF-1R treatment,151 which implied that more steps are needed for IGF-1R targeting. Persistent JAK/STAT activation might also be vital for EGFR-targeted resistance, although the increased level of STAT3 phosphorylation seen in in vivo and in vitro studies was related to gefitinib resistance, which could be overcome by silencing STAT3 in CRC cells.152,153
Bypass amplification and activation
c-MET and VEGF amplification and activation are discussed in the following parts of this review.
Another technique to develop novel anti-EGFR agents is to enclose conventional EGFR blockers within other agents such as nanoparticles, liposomes, and other protein-based drug delivery systems, which have shown promising tumor affinity and drug efficacy in several preclinical studies.154
The VEGF/VEFGR pathway
About the pathway
Angiogenesis, a physiological process by which new vessels form or reform from existing vessels, plays a vital role in tumor initiation, growth, and metastasis. Angiogenesis is also under complex regulation involving various proangiogenic and antiangiogenic factors, such as VEGF, fibroblast growth factors (FGFs), TGF-α, TGF-β, platelet-derived endothelial cell growth factor (PDGF), and angiopoietins produced from cancer or stromal cells.155,156,157 The relationship between neo-vessels and carcinogenesis remained theoretical until the identification of VEGF-A (also known as VEGF) and the production of its monoclonal antibody inhibitor, which finally demonstrated the tumor-promoting effect of angiogenesis.158 The VEGF family consists of five members (VEGF-A, -B, -C, and -D and placental growth factor (PIGF)), which may bind to endothelial cells via tyrosine kinase VEGF receptors. Vascular endothelial growth factor receptors (VEGFRs) are divided into three types, VEGFR-1, -2, and -3, along with the non-tyrosine kinase coreceptors neuropilin-1 (NP-1) and NP-2. The VEGF family may also interact with other proteins, such as integrins,157,159,160,161,162 to regulate angiogenesis, for example, by guiding the migration of endothelial cells.163 Among the complicated and diverse interactions between VEGF and VEGFR, VEGF-A, VEGF-B, and PIGF contribute predominantly to angiogenesis, while VEGF-C and VEGF-D tend to regulate lymphangiogenesis. VEGF-A and VEGF-B mainly bind to VEGFR-1 and VEGFR-2, which are mostly expressed on vascular endothelial cells and on some nonendothelial cells.164 VEGFR-3 is bound by VEGF-C and VEGF-D with greatest affinity and is expressed on endothelial lymphatic cells.165
VEGFR-1 is a 180 kDa member of the receptor tyrosine kinase family expressed on many kinds of cells, including epithelial cells, inflammatory cells, and cancer cells. VEGFR-1 has high affinity for VEGF-1 and relatively low affinity for VEGF-2 and PIGF. Interestingly, VEGFR-1 seems to make little contribution to cell proliferation during vascular formation. Instead, it regulates cell differentiation and migration, especially for epithelial cells,163,166 and promotes differentiation of epithelial cells during early vascular construction.166 In addition, activation of VEGFR-1 under pathological conditions in inflammatory cells mediates the activation of several downstream pathways, such as PI3K/AKT/MAPK/ERK, leading to upregulation of inflammatory cytokine production (TNF-α and some interleukins (IL-1β, IL-6, and IL-8)) and inflammatory cell migration. The detailed function of VEGFR-1 is not fully understood; however, it is believed to be a regulatory factor in angiogenesis. VEGFR-1 favors VEGF-A over VEGFR-2, and the interaction of PIGF with VEGFR-1 might allow VEGF-A to bind to VEGFR-2. Therefore, VEGFR-1 works as a decoy regulator to control the amount of free VEGF-A available to activate VEGFR-2 when angiogenic effects appear to be mediated by VEGF-A/VEGFR-2.164,167,168
In contrast to VEGFR-1, VEGFR-2 is actively involved in vascular formation. It has a molecular mass of 200–230 kDa and is mostly expressed on blood and lymphatic epithelial cells.166 VEGFR-2 mainly interacts with VEGF-A, and activated VEGFR-2 leads to phosphorylation of tyrosine residues and activation of various pathways, including the PLCγ and RAS/RAF/ERK/MAPK pathways, by which epithelial cell growth is promoted, and the PI3K/AKT pathway, by which cell apoptosis may be avoided.156,157,161,163,166 Moreover, adhesion molecules such as cadherins and β-catenin, which are activated by the PI3K and MAPK pathways, may further interact with VEGFR-2, causing deterioration of intercellular junction stability and epithelial cell cytoskeleton reorganization, thus elevating vascular permeability. Vascular permeability is also enhanced by epithelial cell production of endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) via AKT protein kinase activation.169 The above observations indicate the proangiogenic effect of VEGFR-2 in physiological and pathological conditions. Activated VEGFR-2 contributes to the differentiation, proliferation, migration, and apoptosis resistance of epithelial cells, thereby increasing vascular tubulogenesis and permeability, which is very important for cancer angiogenesis and progression.
VEGFR-3, activated by VEGF-C and VEGF-D, contributes relatively independently to lymphatic vessel formation.170,171 Activated VEGFR-3 mediates the differentiation, migration, proliferation and survival of lymphatic endothelial cells by activating the RAS/MAPK/ERK pathway and the PI3K–AKT/PKB pathway.169,170,171 Although the VEGFR-3 expression level in tumor cells remains controversial, high levels of VEGF-C and VEGF-D have been observed in tumors with lymphatic metastasis, which is considered a potential explanation for cancer migration through lymphatic vessels.172
VEGF levels and VEGFR activity are elevated in patients with CRC and other cancers and are considered to be related to poor prognosis.173,174,175,176 Some tumor cells produce VEGF and express VEGFR, suggesting that VEGF works as both an autocrine factor and an endocrine factor in this situation. Increased VEGF levels were observed in very early stages of colorectal neoplasia, e.g., adenoma, and were even higher in later stages of cancer, especially in the metastatic stage.177,178 VEGF regulation is complex in CRC. Mutated K-RAS and p53, expression of COX-2, and hypoxia inducible factor 1 (HIF-1) induced by hypoxia from high tumor cell density might all contribute to VEGF-VEGFR activity alteration, resulting in cancer growth and migration.178,179,180,181 The proangiogenic effects of VEGF-VEGFR are important both in local sites supporting tumor progression and migration and in metastatic sites for neovascularization to support cancer survival and growth; therefore, anti-VEGF/VEGFR therapy might be developed to target both steps in tumor metastasis.
Targeting angiogenesis
Bevacizumab: the milestone
The landmark trials based on antiangiogenic therapy for CRC were initiated in 2004, comprising the phase II and III AVF2107 trials, which confirmed the superiority of chemotherapy (IRI, 5-FU, and leucovorin) plus bevacizumab over chemotherapy plus placebo.182 Bevacizumab is a humanized IgG monoclonal antibody targeted to VEGF-A that, according to the AVF2107 trial, improves both PFS and OS in metastatic CRC (RR: 44% vs. 34.8%; OS: 20.3 vs. 15.6 months; HR: 0.66, p < 0.001; PFS: 10.6 vs. 6.2 months; HR: 0.54; p < 0.001). Therefore, the FDA-approved bevacizumab as the first VEGF-targeted agent for metastatic CRC, even though several trials investigating bevacizumab plus monotherapy or FOLFOX/FOXFIRI showed only a partial significant improvement in either OS or PFS.182,183,184,185,186,187 Using bevacizumab may lead to 10% more grade 3–5 adverse events, such as hypertension or leukopenia,188 while it remained relatively safe and effective when treating elderly patients with CRC (age over 70 years old) in the phase III AVEX trial.185 Further investigation found that both patients with KRAS mutations and those with a wild-type genotype may benefit from bevacizumab.189,190,191 Both left- and right-sided colon tumors respond well to bevacizumab.191 Two independent trials stated no difference in terms of efficacy against metastatic CRC between FOLFOX and FOLFIRI combined with bevacizumab.192,193 Yet interestingly, a bevacizumab-containing regimen seemed to have better efficacy with the triplet FOLFOXIRI regimen than FOLFIRI alone (PFS: 12.3 vs. 9.7 months; HR: 0.77; p = 0.006; OS: 29.8 vs. 25.8 months; HR: 0.80; p = 0.03), although the latter doublet regimen had fewer adverse reactions according to the TRIBE trial.187
In addition to first-line application of bevacizumab, various trials have validated its efficacy in the second-line setting. Longer PFS (7.3 vs. 4.7 months, HR = 0.61, p < 0.001) and OS (12.9 vs. 10.8 months, HR = 0.75, p = 0.0011), as well as a better response rate (22.7% vs. 8.6%, p = 0.0001), were seen in the E3200 trial with a combination of FOLFOX and bevacizumab than with FOLFOX alone for patients with CRC who progressed after FOLFOX therapy.194 Similar numerical differences were also noted in the comparison with bevacizumab alone. Even so, continuation on bevacizumab for those who progressed after first-line chemotherapy was still helpful for PFS (5.7 vs. 4.1 months, HR = 0.68, p < 0.001) and OS (11.2 vs. 9.9 months, HR = 0.81, p = 0.0062) improvement compared with standard chemotherapy alone in the phase III ML18147 trial.195
In terms of maintenance, that is, bevacizumab after first-line chemotherapy in stable CRC, a series of trials demonstrated that anti-VEGF agents might be quite attractive. The prospective and observational BRiTE study indicated that bevacizumab continuation dramatically improved the OS of patients with CRC (31.8 vs. 19.9 months, HR = 0.48, p < 0.001) in comparison with no maintenance.196 Continuation of CAP and bevacizumab significantly prolonged the progression time in patients after first-line XELOX plus bevacizumab compared with observation (11.7 vs. 8.5 months, HR = 0.67, p < 0.0001)186 regardless of RAS/BRAF mutation status and mismatch repair (MMR) status.197
Trends of longer OS (23.2 vs. 20.0 months, HR = 1.05, p = 0.65 in the MACRO trial and 25.4 vs. 23.8 months, HR = 0.83, p = 0.2 in the SAKK (Swiss Group for Clinical Cancer Research) trial) have been observed for maintenance bevacizumab plus XELOX over bevacizumab alone in the MACRO trial198 and for maintaining single-agent bevacizumab therapy compared with no treatment in the SAKK trial.199 No inferiority has been found for maintenance of bevacizumab alone over bevacizumab plus 5-FU or continuation of bevacizumab plus CAP over bevacizumab plus XELOX.200,201
Emerging anti-VEGFR agents
Until now, only bevacizumab has been FDA approved as a first- and second-line VEGF-targeted agent for CRC, although various novel agents are emerging, and some of them have been approved for second-line treatment of CRC.
Aflibercept is a VEGFR-1 and VEGFR-2 extracellular domain recombinant fusion protein that acts as a ligand trap targeting VEGF-A, VEGF-B, and PIGF. Aflibercept has a stronger affinity for VEGF-A than bevacizumab.202 The single-agent benefit of aflibercept seems to be limited,202 while chemo-combinations showed great potential according to the phase III VALOUR trial, in which the addition of aflibercept after OX or bevacizumab in metastatic CRC patients receiving FOXFIRI gained a better response (19.8% vs. 11.1%) as well as a longer PFS (6.9 vs. 4.7 months, HR = 0.76; p < 0.001) and OS (13.5 vs. 12.1 months, HR = 0.82; p = 0.0032) than FOXFIRI plus placebo.203 However, in terms of the first-line setting, as in the phase II AFFIRM trial, the combination of aflibercept with FOLFOX did not result in noticeable benefits in PFS or response rate, but did result in increased adverse event rates. Therefore, aflibercept should remain a second-line recommended CRC agent.204
Ramucirumab, a fully humanized monoclonal VEGFR-2-targeted IgG antibody, is another FDA-approved drug for second-line treatment of metastatic CRC based on the phase III RAISE trial. In this second-line-setting trial, a combination of ramucirumab and FOLFIRI significantly prolonged PFS (5.7 vs. 4.5 months; HR = 0.79, p < .0005) and OS (13.3 vs. 11.7 months, HR = 0.84, p = 0.022) compared with FOLFIRI-placebo.205 Similar to the findings with aflibercept, a phase II trial showed that the FOLFOX regimen may not benefit from addition to ramucirumab in terms of PFS.206
TKIs have become an appealing choice for patients with anti-EGFR-resistant NSCLC, while in patients with CRC, very few drugs have proven to be effective. Regorafenib, a TKI with multiple targets, such as VEGFR, PDGFR (platelet-derived growth factor receptor), FGFR (fibroblast growth factor receptor), and BRAF, was approved by the FDA to treat metastatic CRC. A first-line study concerning regorafenib plus FOLFOX in CRC found no improvement in the response rate compared with FOLFOX plus placebo.207 However, for refractory metastatic CRC treatment, in the phase III CORRECT trial,208 better median OS (6.4 vs. 5.0 months, HR = 0.77, p = 0.0052) and PFS (1.9 vs. 1.7 months, HR = 0.49, p < 0.0001) were achieved using regorafenib than using placebo, which has also been validated in an Asian population in the CONCUR trial (PFS: 3.2 vs. 1.7 months, HR = 0.31, p < 0.0001; OS: 8.8 vs. 6.3 months, HR = 0.55, p = 0.0002).209
Other agents are being developed quickly. The phase III FRESCO trial supported fruquintinib, a TKI with the ability to block VEGFR-1, VEGFR-2, and VEGFR-3, as a recommended choice for chemotherapy against refractory metastatic CRC. In this Chinese-based study, OS (9.3 vs. 6.6 months, HR = 0.65, p < 0.001) and PFS (3.7 vs. 1.8 months, HR = 0.26, p < 0.001) were significantly prolonged with fruquintinib compared with placebo,210 which led to approval of by the China Food and Drug Administration (CFDA) also known as NMPA (National Medical Products Administration). Famitinib is another TKI targeting the c-KIT receptor, VEGFR-2, and VEGFR-3, PDGFR, and RET that is being investigated in an ongoing phase II study, which has so far shown an improved PFS (2.8 vs. 1.5 months, HR = 0.58, p = 0.0034) and disease control rate (57.58% vs. 30.91%, p = 0.0023) for Famitinib, with results concerning OS waiting to be reported.211
New TKIs expressing remarkable antitumor effects in preclinical studies have produced unsatisfying OS and RR values in recent reports; however, PFS may be prolonged by drugs such as the VEGFR-2- and FGFR-targeted brivanib212 and cediranib, a TKI targeted to all three VEGFRs and PDGFR that failed to present efficacy towards CRC control in the phase II and III HORIZON study,213,214 as did nintedanib, a TKI with the ability to block all VEGFRs, FGFR1-3, PDGFR-α, and PDGFR-β, according to the phase III LUME-Colon 1 trial.215 Other on-market TKIs, such as the gastrointestinal stromal tumor (GIST)-targeted imatinib and sunitinib and the squamous cell carcinoma-targeted erlotinib and gefitinib, have no indication or supporting data for treating CRC. The major agents for antiangiogenic therapy under clinical investigation in CRC are summarized in Tables 3 and 4.
Resistance to antiangiogenic therapy
Resistance to anti-VEGF has been observed in various cancer types, including CRC, which may be explained by compensatory activation of other signaling pathways and alternative excretion of angiogenesis-related proteins.
The fact that PIGF is upregulated and overexpressed in CRC cases that are resistant to antiangiogenic therapies216 suggests that PIGF is a crucial factor in overcoming anti-VEGF resistance, which might explain why aflibercept performed better than bevacizumab in xenograft models.217
The angiopoietin/TIE (tyrosine kinase with Ig-like and EGF-like domains) signaling RTK pathway contributes to vascular formation and stabilization by mediating downstream the RAS/RAF and PI3K/AKT pathways, which may be negatively regulated by angiopoietin-2. Abnormally increased levels of angiopoietin-2 have been noticed in a wide range of cancers, including CRC, and are associated with resistance to bevacizumab.218 Targeting both VEGF and angiopoietin-2 in preclinical studies helped control proliferation and progression in cancers that were resistant to VEGF-targeted therapies.219,220,221 The VEGF-A and angiopoietin-2 cotargeting agent vanucizumab, which inhibited growth in a CRC xenograft model,222 has passed through a phase I study with acceptable safety and encouraging anticancer effects.223
The FGF/FGFR pathway is important in both normal and cancer tissues for cell growth, survival, and migration. Upregulation of the FGF/FGFR pathway has also been observed in anti-VEGF-resistant cases.224,225,226 Dual blockade of FGF/FGFR and VEGF/VEGFR in preclinical studies displayed positive effects against tumor cells, while in clinical trials, agents such as nintedanib and the FGF-VEGF dual blocker dovitinib failed to benefit anti-VEGF-refractory patients.215,227
Compensatory activation of the c-MET pathway is the mechanism most related to the loss of anti-VEGF agent effectiveness.228 Single-agent c-MET inhibition might be helpful, as we shall discuss in the following section. However, CRC-based evidence for c-MET and VEGF dual targeting remains rare, and a study on NSCLC stated no better effect by combined blocking.229
A number of studies found factors such as a high level of TGF-β,230,231 upregulation of IL-1,231 downregulation of MIF (macrophage migration inhibitory factor),232 and overexpression of PDGFR233 in a wide range of VEGF-blockade-resistant cancers, implying possible connections to antiangiogenic therapeutic resistance; however, a lack of adequate data on silencing these factors in clinical cases has limited their further confirmation for CRC therapy.
Anti-EGFR or antiangiogenic therapies?
Both anti-EGFR and antiangiogenic therapies have demonstrated decent effects against metastatic CRC; however, which one is the preferred first-line choice for a more precise and personalized targeted agent strategy has been a matter of intense debate. The first head-to-head comparison study was the phase III FIRE-3 trial, which compared bevacizumab and cetuximab in a combined regimen with FOLFIRI. No obvious difference was discovered in the response rate or PFS for both arms, yet OS was prolonged in the cetuximab arm (28.7 vs. 25 months, HR = 0.77, p = 0.017).189 Similar results were observed in a recent phase III trial investigating these two agents plus FOLFOX/FOLFIRI therapy, which reported few differences in the response rate, PFS, and OS between the two groups.191 The PEAK trial, focusing on panitumumab and bevacizumab with FOLFOX, stated that the response rate and PFS seemed alike, and a slightly longer OS for panitumumab than bevacizumab (34.2 vs. 24.3 months, HR = 0.77, p = 0.017) was noted.190 Further analysis in subgroups emphasized the importance of an individualized strategy. RAS mutation status might influence the efficacy of anti-EGFR therapy, but not that of anti-VEGF therapy; therefore, subgroup studies concerning gene information have been carried out. Cetuximab appeared to be the better choice for RAS-wild-type patients in the FIRE-3 post hoc analysis trial, given the increased rate of objective response (72.0% vs. 56.1%, p = 0.0029) and early tumor shrinkage (68.2% vs. 49.1%, p = 0.0005) that were achieved in the cetuximab arm in these patients.234 A recommendation that anti-EGFR over anti-VEGF is favored in RAS-wild-type patients has also been proposed via a meta-analysis that included the FIRE-3, CALGB, and PEAK trials.235 In addition, sidedness has been a critical factor that has marked impact on prognosis.236 Left-sided tumors responded more to cetuximab than to bevacizumab (38.3 vs. 28 months, HR = 0.63, p = 0.02), while those on the right side of the colon tended to behave oppositely (8.3 vs. 23 months, HR = 1.44, p = 0.28) in the FIRE-3 trial,237 which corresponded with subgroup findings from the CALGB study.236 Analysis of panitumumab supported the same side- and genetic-related trends.238 Even when BRAF-mutated cases are removed, right-sided cancer might still benefit little from anti-EGFR therapy.238 For second-line treatment, switching a bevacizumab maintenance strategy to cetuximab or panitumumab239 made no difference in patients with progressed RAS-wild-type CRC according to two independent phase II trials.
Given these data, patients with RAS-wild-type metastatic CRC with tumors on the left side of the colon are recommended to start first-line treatment with chemotherapy combined with anti-EGFR drugs, and anti-VEGF agents should be considered as an alternative choice at all times.
The HGF/c-MET pathway
About the pathway
Hepatocyte growth factor (HGF) and the receptor tyrosine kinase known as mesenchymal–epithelial transition factor (c-MET or MET) encoded by the MET proto-oncogene play vital roles in tumor proliferation, survival, metastasis, and acquired drug resistance.240,241,242,243,244 This signaling pathway was first discovered from TPR-MET fusion genes (translocated promoter region locus on chromosome 1 and MET sequence on chromosome 7) of a human osteosarcoma cell line in the 1980s, when HGF was also named scatter factor because it was initially isolated from rat platelets responsible for epithelial dispersal in organ healing and regeneration.245,246,247,248
HGF is secreted mostly from mesenchymal tissues and is currently the only known ligand for MET. Its tissue and serum expression levels are related to poor prognosis of patients with different malignant tumors, such as breast,249 esophageal,250 and gastric cancers,251 and especially CRC. Patients with advanced CRC have elevated serum HGF at diagnosis and decreased levels after cancer resection.252,253
MET is a member of the surface transmembrane receptor family expressed in both normal and malignant epithelial and endothelial cells, as well as in neural cells, hematopoietic cells, and hepatocytes.254,255 Overexpression of MET has been found in various carcinomas, such as hepatocellular carcinoma, lung cancer, breast cancer, thyroid cancer, kidney cancer, gastric cancer, and CRC.256,257,258,259,260,261,262 Increased mRNA and protein levels of MET were reported in CRC tissues, and its connection to tumor progression and metastasis was demonstrated in several studies.263,264,265
Activation of MET signaling starts from HGF binding to the MET receptor on the membrane, triggering the formation of an intracellular multifunctional docking site from two tyrosine residues, which bind to subsequent substrates. The activated HGF/MET pathway initiates various downstream signal transduction pathways, including the MAPK/ERK, PI3K/AKT, and STAT/JAK pathways and the nuclear factor-κB complex, to regulate hematopoiesis, organ regeneration, and wound healing.244,252,254,255,266 Gene amplification, overexpression, and mutation and ligand-dependent autocrine or paracrine signaling loops are commonly found in aberrant HGF/MET axes in oncogenic situations.267 Interestingly, MET mutations and amplifications are rarely discovered in patients with CRC, with rates of 2–5% and 0.5–2%, respectively.119,268 However, as mentioned above, overexpression of HGF/c-MET mRNA and protein was observed in over 70% and 50% of CRC tissue samples, respectively.269,270,271 The amplification of HGF-MET paracrine and autocrine loops was first identified by Boccaccio et al.272 Subsequent studies supported the theory that overactivated MET promotes enhanced HGF transcription and expression, thus contributing to subsequent MET expression to form a loop that can be further augmented through paracrine HGF produced by reactive stromal cells in the tumor microenvironment or under certain situations, such as hypoxia or inflammation.273,274,275,276,277
In addition to self-regulation, the HGF/MET signaling pathway might also be modulated by other factors. Plexin B family members have been reported to have structural similarity with MET. Activated plexin B1 might transactivate MET to modulate cancer growth and migration. However, the role of plexin B1 remains controversial because in various cancers, both tumor-promoting and tumor-suppressing effects have been observed.278,279,280 A recent study found that a newly identified gene, MACC1 (metastasis-associated in colon cancer 1), has high potential to be a key regulator of MET expression and further influence CRC progression and metastasis.281 Elevated levels of MACC1 expression were found in both local and metastatic malignant tissues. Accumulating data revealed HGF-induced MACC1 translocation from the plasma into nuclei, and MACC1 binding to the MET promoter contributed to enhanced MET transcription. MACC1 research provides new evidence for a positive loop of MET expression in CRC.
Another major method of regulating signaling activity relies on crosstalk between the MET pathway and other RTKs, especially EGFR. Overexpression of both MET and EGFR is commonly found in the same malignant tumor, such as CRC.282 Compensatory activity regains of MET or EGFR after targeted treatment of either of them has been observed in various studies, strongly implying the existence of crosstalk between MET and EGFR. As such, MET was the first factor to be identified as responsible for EGFR inhibitor resistance, even in the absence of known resistance-related mutations.240,283,284,285,286 Blocking either aberrant MET or aberrant EGFR leaves little restraint on downstream ERK or PI3K activation, while resistance abrogation was observed using combined therapy targeting both receptors in vivo and in vitro.283,286,287 Mutual regulation between MET and EGFR has several possible mechanisms. Downstream products of EGFR might induce phosphorylation of MET, whereas altered c-MET-induced protein may also lead to EGFR phosphorylation.282,287 Activated MET and EGFR might form different heterodimers, resulting in various tumor biological behaviors, such as cell growth and survival for MET-EGFR and MET-HER3 or migration for MET-HER2.288 In addition, MET activation has been observed occasionally in VEGF-targeted therapy, resulting in VEGF resistance; however, the underlying mechanism remains unclear.289,290
Targeting the HGF/c-MET pathway
Accumulating knowledge of the close relationship between cancer and the HGF-MET pathway identifies it as a highly promising site for targeted therapy. Various ways of blocking HGF-MET via newly developed monoclonal antibodies or small molecules with different pharmacological mechanisms have emerged. For HGF, drugs are aimed at either blocking HGF activation and production or interfering with the binding of HGF to MET receptors. In the latter case, agents either competitively bind to MET receptors (MET antagonists) or inhibit intracellular tyrosine kinase activity (MET TKIs). To date, no severe adverse events have been reported for these agents, although some patients complained about fatigue, poor appetite, allergic reactions, edema, skin rash, and neutropenia.252,266 There are several current clinical trials of HGF/c-MET-targeted agents in the context of CRC therapy (Table 5).
HGF inhibitors
HGF production relies on the maturation of its precursor (pro-HGF), mediated by an endogenous inhibitor protein family called HGF activator inhibitors (HAIs).291,292 High levels of HAI-1 have been observed in patients with benign lesions compared with those with prostate cancer, and the level of HAI-2 was decreased in highly invasive and progressed prostate cancer cells.293,294 The HAI protein family seems to be an attractive target to control HGF activation; however, no artificial compound or analog has been made so far, and thus, clinical testing is a long way off, although experimental data imply a potential role in an antimetastatic strategy for CRC.295,296
Instead of blocking HGF activation, neutralizing HGF to impede its ability to bind to receptors to interfere with whole pathway appears to be more practical. A few monoclonal antibodies have been synthesized and introduced in several clinical trials. Rilotumumab, a humanized IgG monoclonal antibody, has been investigated in phase I and II trials. In those patients with gastric or gastroesophageal cancer, a prolonged median PFS (6.8 vs. 4.4 months; HR = 0.46, 95% confidence interval (CI): 0.25–0.85) and OS (10.6 vs. 5.7 months; HR = 0.46, 95% CI: 0.24–0.87) were achieved in patients with MET overexpression using rilotumumab plus CAP compared with those in the placebo plus CAP arm.297 Further phase III studies (RILOMET-1 and RILOMET-2)261,298 in patients with untreated or advanced-stage gastric or gastroesophageal cancer were halted early because of a rapid increase in disease-related deaths. These trials highlighted the importance of stratification. Current trials commonly apply methods such as IHC or FISH to determine the existence of MET overexpression, and a further scoring system according to the percentage of tumor cells with high staining intensity is used to stratify MET-positive/high and MET-negative/low patients, although the criteria differ by small degrees.
For patients with CRC, a randomized phase Ib/II trial299 concerning rilotumumab or ganitumab vs. panitumumab in patients with KRAS-wild-type metastatic CRC showed no significant benefit with the combined use of rilotumumab and panitumumab in terms of median OS (13.8 vs. 13.7 months, p = 0.71) in patients with MET-high disease compared with MET-low disease.
Ficlatuzumab is a humanized IgG monoclonal antibody that has been investigated in a phase I trial for advanced solid tumors and liver metastases.300 TAK-701, another humanized anti-HGF monoclonal antibody, was combined with gefitinib to help overcome EGFR resistance in lung cancer and is also undergoing a phase I trial.301,302,303
MET antagonists
Agents that compete with HGF for binding to MET result in abnormal dimerization and degradation of MET. Various antibodies have been developed, including onartuzumab, DN-30, and ABT-700. Onartuzumab, a murine-derived monoclonal antibody with high specificity for the MET semaphorin domain,304 has been evaluated in several trials in patients with solid tumors, such as NSCLC, glioblastoma, gastroesophageal cancer, gastric cancer, and CRC.305,306,307,308 Improved median OS and PFS were observed in MET-positive lung cancer patients treated with erlotinib in a phase II trial; however, no such efficacy was reported in a phase III trial.306 Similarly, in gastric or gastroesophageal cancer, no significant improvement in PFS or OS was observed using onartuzumab plus mFOLFOX6 vs. placebo plus mFOLFOX6.305 In the case of metastatic CRC, no significant differences were identified in PFS between MET-positive and MET-negative patients using onartuzumab combined with mFOLFOX6 + bevacizumab or placebo.308
There are a few novel MET antibodies that function promisingly in cancer control yet lack supporting data in CRC. The antibody DN-30 binds to the IPT (Ig-like, plexins, transcription factors) domain of MET and shows a promising ability to inhibit the proliferation of MET-positive gastric cancer and metastatic melanoma in vitro and in vivo.309 ABT-700 is a humanized antibody that could induce gastric and liver tumor regression in preclinical cancer models with MET amplification and passed a phase I trial in several solid tumors with favorable safety and tolerability.310,311,312 Emibetuzumab, a humanized antibody targeting MET, has been used in phase I and II trials for NSCLC and gastric cancer.313,314,315,316 Other anti-MET agents, such as ABBV-399, YYB-101, and ARGX-111, are either undergoing or just past phase I trials for a range of solid tumors.
MET TKIs
A number of drugs functioning as selective or nonselective TKIs have been developed and brought to clinical trials. To some extent, their similar RTK structure to MET guarantees their pharmacological effects. Selective agents include tivantinib (ARQ 197), savolitinib (AZD 6094; volitinib), AMG 337, and capmatinib (INC 280), which target the MET kinase domain, and nonselective agents include crizotinib (PF-02341066), cabozantinib (XL-184), tepotinib (EMD-1214063), foretinib (GSK1363089), glesatinib (MGCD-265), golvatinib (E-7050), and merestinib (LY-2801653).
Tivantinib is an oral small-molecule allosteric RTK inhibitor that selectively keeps MET in the inactive state. In vivo and in vitro experiments confirmed its ability to impair the growth of various cancers.317,318,319 Tivantinib has been investigated as an independent drug or combined with sorafenib in phase II and III trials in patients with liver cancer and combined with erlotinib in phase III trials in patients with NSCLC. Data showed that patients with liver cancer with MET overexpression had a better OS than those without MET overexpression in a phase II trial, but the significance was lost in a phase III trial, while the use of tivantinib contributed little to the prognosis of NSCLC.320,321 For patients with metastatic CRC, a phase I/II trial enrolling patients to receive tivantinib or placebo plus cetuximab and IRI found no PFS improvement,322,323 and a phase II trial concerning tivantinib or placebo plus cetuximab did not meet its primary end point because of a low response rate.324 To date, various MET-specific TKIs have shown dramatic effects in treating several malignancies, such as NSCLC or gastric cancer, and have been used in phase I clinical trials. However, clinical trials on CRC are insufficient to evaluate the efficacy of tivantinib and other selective TKIs.
AMG 337, an oral ATP-competitive TKI specific to MET, caused a strong response in patients with MET-amplified upper gastrointestinal tract cancer in phase I and II trials.325,326 Savolitinib, a selective MET inhibitor, displayed marked antitumour potential under experimental conditions and appeared to be effective against renal cell cancer327,328 and is being investigated in a metastatic CRC phase I trial. Capmatinib, another selective MET inhibitor, has been demonstrated as a good supplementary agent to gefitinib in patients with EGFR-mutant, MET-amplified NSCLC.329
Nonselective TKIs targeting multiple factors, including the TKs of MET, may have a wider range of applications.
Crizotinib targets anaplastic lymphoma kinase (ALK) and the TKs of MET, and the RON (macrophage-stimulating 1 receptor) and ROS (ROS proto-oncogene 1, receptor tyrosine kinase) receptors and is an FDA-approved treatment for ALK-rearranged NSCLC.330 Crizotinib has attractive efficacy in prolonging the survival of patients with NSCLC.331 Although there is a lack of clinical evidence for crizotinib in CRC, a series of phase I and II trials are in progress. The use of crizotinib might enhance the response to radiation therapy in KRAS-mutant CRC cell lines, and a combination of crizotinib with mitomycin C seemed to have a synergistic effect against CRC in preclinical results, which showed promise for future anti-CRC treatments.332
Cabozantinib, specific for a wide range of TKs, such as MET, RET, and VEGFR-2, is also an FDA-approved drug for metastatic medullary thyroid cancer and renal carcinoma, with antitumor effects in liver cancer, as assessed through phase II and phase III trials.333,334,335 Similarly, an exciting antitumor performance of cabozantinib was observed in research based on CRC xenograft models and cell lines, while the results from CRC-focused clinical trials are awaited.289,336,337
Merestinib, an inhibitor of MET, AXL (Axl receptor tyrosine kinase), TEK (tunica interna endothelial cell kinase), ROS1, DDR (discoidin domain receptor tyrosine kinase), MKNK (MAP kinase-interacting serine/threonine protein kinase), and FLT3 (Fms-related tyrosine kinase 3), has just finished its first human phase I trial for various advanced cancers, including CRC,338 to determine a suitable dose for the phase II trial.
TKIs are being upgraded rapidly from generation to generation. Although highlighted in preclinical studies or phase I/II trials for solid tumors such as gastric cancer or NSCLC,339,340,341,342,343 new agents such as tepotinib, foretinib, glesatinib, golvatinib, and sitravatinib are only a few steps from clinical investigation for CRC.
As described above, owing to the crosstalk between HGF/MET and other major pathways targeted by most targeted agents, targeting HGF/MET could help overcome resistance against EGFR or VEGFR inhibitors. Combined inhibition of MET and EGFR showed improved PFS in patients with NSCLC with MET overexpression; however, the study was halted prematurely because of an increased rate of interstitial lung disease.344 Similar antitumor effects have been noted in renal cancer xenografts treated with therapies targeted at both VEGF and MET.345
Although clinical evidence of HGF/MET-targeted drug resistance has not been presented, a preclinical study observed acquired resistance to HGF/MET inhibitors in patients with gastric cancer.346 Under laboratory conditions, suppression of HGF/MET might induce subsequent MET mutations (Y1230 mutation in the MET loop) and compensatory activation of alternative pathways, such as EGFR or the RAS/RAF/MEK pathway, which might inevitably cause secondary resistance to anti-HGF or anti-MET therapies.277
Immune checkpoint inhibitor therapy
About the pathway
In addition to methods directly blocking pathways that contribute to tumor growth and spread, accumulating data suggest that targeting other pathways to enhance immunorecognition and the response against cancer cells might be effective. Malignancies harboring various genetic and epigenetic alterations may be identified and obliviated by the host immune system via the expression of abnormal antigens. The detection process comprises several steps, including T cells binding to major histocompatibility complex (MHC) molecules held by antigen-presenting cells (APCs), followed by the next step involving secondary signals mediated via costimulatory or inhibitory receptors that play a vital role in the activation and tolerance of T cells.347,348 This double-check mechanism is important both in physiological conditions to avoid an excessive immune response and in pathological conditions so that abnormal cells may be attacked in a flexible manner.349
Immune escape, which refers to cancer cells escaping from host immunorecognition and response, has been frequently identified in various cancers.350 Secretion of immunosuppressive factors, such as TGF-β and IL-6, recruitment of immunosuppressive cells, such as regulatory cells, or loss of immunogenicity via downregulation of MHC-1 might all contribute to immune escape.351,352,353,354,355 One more major explanation is tumor-related T cell inactivation and exhaustion via activation of coinhibitory receptors, the so-called immune checkpoint receptors, on the surface of T cells,356 which include programmed death-1 (PD-1), or CD279, and cytotoxic T lymphocyte antigen 4 (CTLA-4). PD-1, with its ligands PD-L1 (CD274) and PD-L2 (CD 273), is a peripheral immune checkpoint on tumor, stromal, and immune cells, while CTLA-4 is a central checkpoint targeting B7-1/B7-2 or CD80/CD86 on APCs.348,351,357 Engaged CTLA-4 downregulates IL-2 secretion and competitively binds to B7-1/B7-2 to diminish the CD28-derived stimulatory effect of T cells, and activated PD-1 leads to inhibition of downstream pathways, such as the PI3K/AKT pathway, resulting in both the abrogation of T cell proliferation and eventual immune anergy.358,359 Like other mutation-enriched cancers, metastatic CRC lesions express higher levels of PD-L1 than primary lesions,360 laying the foundation for interfering with the host immune response.
Immune cells, including B cells and T cells, infiltrating into the CRC microenvironment were found to be highly related to alterations in the adaptive immune response and antitumor outcomes.361,362 Among them, CD4+ T regulatory (Treg) cells, which suppress the activation and function of other immune cells, were found to infiltrate into CRC tissues and related lymph nodes in vitro and in vivo.363,364 Indeed, in paired patient samples from CRC tissues and normal mucosa, increased numbers of Treg cells were observed in the CRC tissues.365 These PD-1-expressing Treg cells are considered crucial in the immune tolerance and homeostasis of CRC. Moreover, MHC-1-loss-detecting NK cells, which are effective in killing tumors that use downregulation of MHC-1 as a method of immune escape, were found to be dysfunctional in CRC, also partially because of the PD-1/PD-L1 interaction.366,367 Preclinical studies found T cell activation and inactivation of tumor cells when blocking the PD-1/PD-L1 interaction or CTLA-4.368,369,370 In addition, checkpoint inhibitors displayed promising effects against different advanced cancers, such as melanoma, renal cell cancer, and NSCLC, in recent phase III trials.371,372,373,374,375,376 These data provide the basis for the development of immune checkpoint-targeting therapy against CRC.
Immune checkpoint blockade agents
Immune checkpoint targeted therapy aims to enhance immune surveillance and suppression against cancer by blocking the tumor’s attempt to escape from T cell detection.377,378 Currently, checkpoint inhibitors have been investigated in various solid tumors with promising responses. The first FDA-approved therapy was a CTLA-4 inhibitor, ipilimumab, for melanoma,379,380,381 followed by the PD-1 inhibitor nivolumab, which displayed significant effects with tolerable adverse events in a phase I trial and went through subsequent phase II and III trials. Another PD-1 inhibitor, pembrolizumab, was trialed for several advanced tumors, such as renal cell cancer, melanoma, and NSCLC, which led to its approval by the FDA for treating these malignancies.371,372,373,374,375,376,382
In metastatic CRC, although a significant response was noticed in some phase I trials,383 further studies found that only a small proportion of patients with CRC responded to immune checkpoint therapy.384 This subgroup of patients had a high tumor mutational burden and their malignant lesions showed high levels of microsatellite instability (MSI-H) or MMR deficiency (dMMR).385,386,387 Tumor mutational burden has been observed to be associated with the immune checkpoint response rate in melanoma and NSCLC;388,389 however, the underlying mechanism remains unclear. One hypothesis argued that tumor-related neoantigens derived from mutations favored attention from immune cells and thus attracted increased T cell infiltration.390,391,392
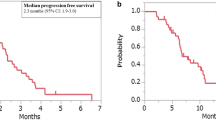
Pembrolizumab and nivolumab
The first PD-1 blocker to display good efficacy against MMR-deficient CRC was pembrolizumab, a humanized IgG4 antibody that started a new chapter in CRC immunotherapy when the FDA approved it for metastatic CRC treatment in 2017. The KEYNOTE-016 study found that patients with dMMR CRC might respond to pembrolizumab treatment (response rate of 40% and a 20-week PFS of 78%), while none of those with pMMR responded to the drug (response rate of 0% and a 20-week PFS of 11%).387 Another phase I trial also identified the antitumor activity of pembrolizumab only in patients with MSI-H CRC.393 Thus, the KEYNOTE-164 study investigated pembrolizumab in patients with MSI-H metastatic CRC in the second-line setting, which showed an objective response rate of 33% and PFS and OS values of 2.3 and 31.4 months, respectively.394,395 Further updated analysis of the data from KEYNOTE-016 demonstrated an objective response rate of 52%, a 2-year PFS of 59%, and an OS of 72%.396 Combined therapy with pembrolizumab and ipilimumab showed comparable efficacy in melanoma patients, but there is insufficient evidence in CRC.397
Nivolumab, another humanized monoclonal IgG4-based PD-1 antibody, gained FDA approval for dMMR or MSI-H metastatic CRC in 2017 based on the CheckMate-142 trial, in which 51 out of 74 patients enrolled achieved disease control for at least 12 weeks, with an objective overall response rate of 31.1%, regardless of the tumor PD-L1 level, and 1-year PFS and OS values of 50.4% and 73.4%, respectively.382 Further studies argued that a combination of nivolumab and the CTLA-4 inhibitor ipilimumab surpassed single-agent immune checkpoint blocking with an acceptable rate of adverse events.398,399,400 Combined therapy with nivolumab and ipilimumab helped patients with dMMR or MSI-H CRC who had previously received chemotherapy to reach a PFS of 71% and an OS of 85% at 1 year; 80% of them maintained disease control over 12 weeks. For first-line use of this doublet regimen, a new study assessed its efficacy and safety for patients with dMMR or MSI-H CRC.399 According to this trial of 45 patients, the 1-year PFS and OS values for the combined regimen were 77% and 83%, respectively, with an ORR of 60% and a disease control rate of 84%. This evidence gained the nivolumab and ipilimumab-containing doublet regimen FDA approval as a therapy for patients with chemotherapy-refractory metastatic CRC (Table 6).
Other agents
Other novel PD-1/PD-L1 inhibitors are under investigation (Table 7), including atezolizumab, avelumab, and durvalumab, which are IgG monoclonal antibodies targeting PD-L1 (BP 545758), some of which have been approved for the treatment of NSCLC and urothelial cancer. These agents have been through phase I trials for various solid tumors, including CRC, with manageable safety profiles and are undergoing further exploration. New immune checkpoint targets are being investigated under experimental conditions and are being evaluated for their safety profiles in phase I trials, such as tumor-overexpressed TIM-3 (T cell Ig and mucin domain-containing protein 3), T cell Ig, and ITIM domains (TIGIT), and T cell-derived LAG-3 (lymphocyte activation gene 3), which both contribute to T cell exhaustion and promote CRC progression.401,402
Overcoming resistance to immunotherapy
Given the unsatisfactory results for immune checkpoint blockade therapy observed in patients with MMR proficient (pMMR) or microsatellite stable (MSS) CRC, who constitute the major proportion of patients with CRC, it is unfortunate that the underlying mechanism has not been clearly determined. Investigators have tried to overcome the resistance of pMMR or MSS CRC to immune checkpoint inhibitors on the basis of several hypotheses related to reduced tumor-specific antigen expression, antigen presentation defects, altered immunosuppressive pathways (e.g., activation of MAPK and loss of PTEN), and alternative activation of other immune checkpoint signaling pathways, immune regulatory cells, and cytokines.403 Strategies to improve pMMR or MSS CRC immune checkpoint inhibitor responses are being developed, such as combined therapy with various approaches including radiotherapy, bispecific antibody therapy, other immune checkpoint modulators, and other targeted agents.377
Radiotherapy might lead to upregulated expression of tumor-specific neoantigens through cell damage and increase membrane MHC class I expression, thus activating the host immune response.404 Radiotherapy combinations showed antitumor effects with well-tolerated adverse events in melanoma treatment, and this strategy seemed to benefit only the combined use of PD-1 and CTLA-4 blockers because the effects of a single agent might be overcome by upregulation of the other signaling pathway.405 For MSS CRC, several studies are ongoing that introduce radiotherapy into a doublet regimen with immune checkpoint-blocking therapy (NCT03104439, NCT03007407, and NCT02888743).
Another method to enhance T cell surveillance is the use of a bispecific antibody such as the CEA-TCB antibodies RG7802 or RO6958688, which bind to CEA on tumor cells and CD3 on T cells, thereby helping T cells infiltrate and identify tumor cells. Preclinical studies and a phase I trial showed that the CEA-TCB antibody plus atezolizumab had anti-MSS CRC potential with acceptable toxicity.406,407,408,409 Among various combinations of immune checkpoint inhibitors and other targeted agents, MEK blockers seem to have attracted increased attention because MEK blockade is linked to an increased T cell response via upregulation of PD-L1 expression.410 Trials have been conducted for combined blocking of MEK and immune checkpoints inspired by the phase I and III studies focused on atezolizumab and cobimetinib (a MEK inhibitor), which found that this regimen was well tolerated yet offered no significant survival improvement over single drugs such as regorafenib or TAS-102 in patients with MSS CRC.411,412 Strategies that interfere with other pathways, such as VEGF/VEGFR blockade with PD-1/PD-L1 inhibition, are being investigated in a wide range of trials, and a phase I study has verified the safety of the combined method.413
Biomarkers for treatment surveillance
Considering that efficacy varies for immune checkpoint therapy and overdosing might lead to unwanted adverse events, identifying biomarkers for potentially sensitive patients and predicting their response becomes a vital task.
The PD-L1 expression level appeared to be a persuasive marker because PD-L1-positive lesions were more vulnerable to PD-1 inhibition therapy than PD-L1-negative lesions, yet clinical survival data did not show a significant relationship.414,415,416,417 The predictive role of PD-L1 expression in CRC is considered to be limited because in pMMR CRC, no obvious trend was observed between PD-L1 expression levels and drug efficacy.382,387
A high mutational burden correlates with elevated levels of neoantigens. It is not just dMMR and MSI-H tumors that harbor high mutational burdens. MSS and pMMR lesions might present with an ultramutated phenotype, such as the DNA polymerase epsilon (POLE) mutations that are found in ~1–2% of pMMR CRCs, which cause increased immunogenicity and upregulation of immune checkpoint genes such as PD-1/PD-L1 and CTLA-4, resulting in similar clinical responses to those seen in dMMR tumors.418,419,420 Only a few cases of POLE mutations indicating the efficacy of PD-1 blockade therapy have been reported; therefore, immune checkpoint modulator-based phase II or II trials enrolling larger groups of patients with POLE-mutated CRC have been initiated (NCT03150706, NCT03435107, and NCT03827044). Currently, whether genetic alterations contribute to high mutational burdens, other than MMR- and POLE-related alterations, remains uncertain, and whether the cost of testing the mutational load via methods like next-generation sequencing is justified might also be a matter of debate.421
In addition to immunorecognition, T cell infiltration is also fundamental for PD-1-blocking therapy; thus, the immunoscore based on the calculation of two lymphocyte populations (CD3/CD45-CD8 or CD8/CD45 populations) in the center and invasive margins of the tumor has been attractive for predicting the drug response and has acted as a reliable CRC classifier and recurrence estimator; however, it is waiting for further clinical validation.422,423,424
Some other factors indicating cytotoxic T cell activity, such as the level of granzymes, perforins, and IFN-γ, remain in the theoretical phase,403 and genetic PD-1 resistance-predicting markers such as β-2-microglobulin (B2M), JAK1, and JAK2, which have been validated in other tumors, such as melanoma, have not been confirmed to have similar effects in CRC, although the absence of B2M might correlate with a better clinical outcome.425,426
Adjuvant and neoadjuvant therapy with targeted agents
Targeted agents in adjuvant therapy
Fluoropyrimidine-based chemotherapy after curative surgery for CRC might help to minimize tumor recurrence and prolong survival. Adjuvant chemotherapy benefited patients with stage II cancer to a lesser degree than it benefited those who had tumors in stage III, suggesting that it might achieve better disease-free survival (DFS) and OS, as the former group typically had better prognosis, with a 5-year survival rate of almost 80% according to the randomized QUASAR study and the MOSAIC and NSABP C07 studies.427,428,429,430 MSI-H stage II CRC appeared to not respond to adjuvant chemotherapy.431 High-risk stage II tumors, such as those at stage T4, those with lymphovascular or perineural invasion, and those with perforation or obstruction; MSS tumors; and tumors in stage III were recommended to be treated with adjuvant FOLFOX or CAPOX therapy, which may reduce the 10–22% death rate.102,432 Notably, extra drug-derived toxicity should be taken into consideration433 in adjuvant therapy; thus, additive agents such as targeted drugs have been used to attempt to improve adjuvant treatment. Therefore, several subsequent trials have been initiated to refine current adjuvant therapy with previously proven targeted agents.
The phase III NSABP-C08 trial,434 AVANT trial,435 and QUASAR 2 trial436 aimed to introduce bevacizumab into either CAP, XELOX, or FOLFOX adjuvant regimens; however, none of them showed significantly prolonged DFS for patients with CRC, and only an increased rate of treatment-related adverse events was reported. A similar conclusion was drawn that no survival benefit could be gained from adjuvant application of cetuximab with the classic regimens FOLFOX or FOLFIRI, and for subgroup analysis of RAS- and BRAF-wild-type patients, an adjusted HR of 0.76 (p = 0.07) was noted, implying that a further larger randomized controlled study may be required.437,438,439 Moreover, the cell surface glycoprotein 17-1A antibody edrecolomab, which was once of high potential interest for use against CRC, also failed to improve adjuvant therapy.440,441 Based on these data, targeted agents have not been included in current adjuvant therapy; however, trials concerning immune checkpoint inhibitors are ongoing. Specifically, atezolizumab plus adjuvant FOLFOX for patients with dMMR or MSI-H CRC (ATOMIC trial: NCT02912559) and avelumab plus an adjuvant 5-FU-based regimen in patients with MSI-H or POLE-mutant CRC (POLEM trial: NCT03827044) are being studied.442,443
Targeted agents in neoadjuvant therapy
Typically, neoadjuvant therapy should be applied for colon cancer with potential resectable metastases and rectal cancer of stages II to IV to minimize local recurrence. Routine approaches include radiation, chemotherapy, or combined chemoradiation. Reaching a pathological complete response (pCR), which indicates complete tumor remission, has always been the main goal of neoadjuvant therapy for rectal cancer, enabling possible sphincter-saving curative surgery with a reduced recurrence rate of 5–6%;444,445,446,447,448 however, current statistics showed an unsatisfying pCR rate of less than 15%.448
Targeted agents are considered to be novel weapons to enhance neoadjuvant efficacy despite being contradicted in other cancers;449,450,451,452 thus, various studies have been conducted to introduce targeted drugs into neoadjuvant therapy for CRC. For KRAS-wild-type patients with resectable colorectal liver metastases, although a few phase II trials stated potential efficacy for anti-EGFR agents,453,454,455 preoperative and postoperative use of cetuximab plus chemotherapy did not help and resulted in a shorter PFS (14.1 vs. 20.5 months, HR = 1.48, p = 0.03) than that seen with chemotherapy alone according to the EPOC trial.456 However, in another randomized controlled trial enrolling patients with KRAS-wild-type CRC with liver metastases, preoperative chemotherapy with cetuximab might have contributed to a better rate of R0 resection of the metastases (25.7% vs. 7.4%) and longer survival (3-year OS rate: 41% vs. 18%, p = 0.013; median survival time: 46.4 vs. 25.7 months, p = 0.013)454 over single-agent chemotherapy, suggesting that preoperative use of cetuximab is controversial for these patients. While perioperative administration of bevacizumab and chemotherapy for patients with liver metastatic CRC might be tolerable and helpful in prolonging survival, several phase II trials showed that recurrence was unavoidable, which lacks further validation.457,458,459,460
As for locally advanced rectal cancer, abundant phase II studies have been conducted for the neoadjuvant use of anti-EGFR or anti-VEGF therapy, with pooled estimated pCR values of 27% and 14% being reported for bevacizumab- and cetuximab-relevant regimens, respectively, via a meta-analysis of 32 previous studies.448 Although newer trials echoed the results from meta-analysis,461 to elucidate whether the targeted agents are truly effective in the neoadjuvant setting still requires larger population-based head-to-head, time-to-event data. It is worth noting that the efficacy of immune checkpoint modulators for the neoadjuvant treatment of CRC has been investigated in various ongoing trials, offering more potential choices in the future (NCT03926338, NCT03299660, NCT03102047, NCT04130854, NCT03854799, and NCT02754856).
Interaction with the gut microbiota
The gut microbiota is closely related to carcinogenesis and tumor progression of CRC.462,463,464,465,466,467 Novel approaches for CRC screening and surveillance might be achieved through single or multiple microbial markers. Alteration of and intervention in the microbiome might also contribute to CRC treatment and further assist in predicting and monitoring treatment response and adverse events. Although most findings have been preliminary and await clinical validation, the gut microbiota represents one of most promising approaches for personized CRC therapy.
Current chemotherapy for CRC has been shown to potentially be mediated by the gut microbiota, which might respond to cytotoxic agents via alterations of their diversity, location, and metabolism.466,468,469,470 Specific types of organisms in the gut microbiota have been found to play a vital role in resistance to 5-FU and OX therapy by mediating autophagy.471 In addition, other gut residents might aggravate chemotherapy-related adverse reactions via microbial metabolism of chemotherapy agents such as IRI.472 There is limited evidence showing that the gut microbiota might interfere with targeted therapy; however, host–microbiome interactions in experimental settings might suggest some underlying clues for the microbial-mediated efficacy of targeted agents. Bile acids are among the major microbial metabolic products, which are also relevant to the initiation and progression of CRC.473 The crosstalk between the gut microbiota and the host intestinal cells is considered to be mediated by the progression of primary-secondary bile acid transformation, which mainly regulates the growth of colon epithelial cells via EGFR and nuclear farnesoid X receptor signaling.474 Thus, more investigation is needed to confirm that microbial interventions, such as the use of probiotics, could reduce intestinal inflammation through EGFR regulation. No further evidence has been presented stating that EGFR-targeted therapy might be interfered with by the gut microbiota. Similar conclusions can be drawn for anti-VEGF therapy, which may be even more related to the gut microbiota. An angiogenesis-mediated function for the gut microbiota was reported previously, by which the cancer microenvironment was formed and shaped.475,476 Probiotics might help to control local inflammation by downregulating the VEGF/VEGFR pathway in the liver and intestinal cells.477,478 Antibiotic use, which might dramatically reduce gut microbial diversity and density, was found to be related to poor survival in patients with metastatic CRC who had received bevacizumab therapy.479 However, the influence of the microbiota on anti-VEGF agents is still a matter of debate because of the controversial reported results480,481 in renal cell cancer-based studies stating an unclear role of antibiotics in VEGF-blockade therapy.
Interestingly, the gut microbiota turned out to be an indispensable factor for immune checkpoint blockade and affected responses and adverse reactions.482,483,484,485,486 Several strains of bacteria were related to the drug response,487,488,489,490,491,492 and further experiments showed that colonization by a combination of 11 strains of bacteria in germ-free mice might have enhanced the efficacy of immune checkpoint modulators, partially because the new bacterial infection led to stronger immune-protective infiltration and response from CD8+ T cells, as well as increased numbers of CD4+ T cells and CD103+/MHC class Ia-expressing dendritic cells.493 Antibiotic exposure was associated with reduced clinical activity in response to immunotherapy with a decreased PFS and OS in NSCLC (PFS: 1.9 vs. 3.8 months, HR = 1.5, p = 0.03; OS: 7.9 vs. 24.6 months, HR = 4.4, p < 0.01) and renal cancer (PFS: 1.9 vs. 7.4 months, HR = 3.1, p < 0.01; OS: 17.3 vs. 30.6 months, HR = 3.5, p = 0.03),489 findings that have been restated by recent studies that showed a dramatically reduced immunotherapeutic benefit (2 vs. 26 months, HR = 7.4) once antibiotics were given for several cancer types.494,495 Similarly, the efficacy of a PD-1 inhibitor in melanoma patients correlated with the gut microbiota, and reconstitution of the microbiota from PD-1 therapy-responding patients might result in an enhanced T cell response and improved PD-1-blocking effect, whereas transplantations from nonresponding patients had a negative outcome.490,491,492 Further metagenomics analysis identified Akkermansia muciniphila as the key bacteria that ameliorates PD-1 blockade, but this effect might be overcome by the administration of antibiotics.496,497 In addition, the gut microbiota might predict immunotherapy-related colitis because enriched levels of Firmicutes indicated a more frequent occurrence of ipilimumab-induced colitis.488 Currently, a few trials are in progress investigating whether immunotherapy can be modified by fecal microbiota transplantation (NCT04130763, NCT04116775, and NCT03341143). However, much remains to be learned about the gut microbiota in immune response regulation, and elucidating the underlying mechanism requires further investigation. Moreover, a lack of effective methods to precisely control the abundance or constitution of specific strains or groups of the gut microbiota limits the current opportunities for intervention.
Other pathways
The development of new targeted agents based on pathways other than previously known ones appears to be rather slow. A few clinical trials concerning drugs aimed at targets such as IGF-1R, Wnt, Notch, Hedgehog, human death receptor 5 and TGF-β have been initiated, yet no attractive results have emerged so far. For example, the γ-secretase inhibitor RO4929097 in Notch blockade therapy and the Hedgehog pathway inhibitor vismodegib displayed little effect in phase II trials.498,499 The limited progress of anti-TGF-β and anti-Wnt therapy against CRC has also been reviewed.500,501 Agents such as COX-2 inhibitors were found to be helpful in CRC prevention in terms of Wnt inhibition; however, the development of other agents that might enhance chemotherapy sensitivity, yet direct CRC-control-targeted drugs with high affinity to single targets, still lags behind. The existence of crossover between these pathways has also made blockade therapy inefficient, and other obstacles, such as difficulties in selecting patients who will respond well, identifying outcome-monitor markers, and efficiently blocking specific targets, have appeared; however, these have not halted investigations into novel agents. Table 8 summarizes those agents under clinical investigation for various targets.
Future directions and conclusions
Human genomic, transcriptional, proteomic, and epigenetic details have never been as accessible as they have in the past few decades, owing to evolving sequencing technologies. Alterations in cell differentiation, proliferation, and survival resulting from genetic profile changes contribute to cancer initiation and development. On the basis of identifying these heterogeneities, treatments targeted to specific enzymes, growth factor receptors, and signal transducers make personalized cancer therapy possible, such that many oncogenic cellular processes can be efficiently interfered with, which holds the promise of precise cancer eradication and better patient care.
After decades of development, great efforts have been devoted to updating CRC-targeted drugs for better patient compliance, fewer adverse events and more individualized treatment strategies. Current targeted agents for CRC and the NCCN-recommended strategy are summarized in Figs. 3 and 4. To date, there is no universal regimen that can easily treat every patient with equal efficacy, and our knowledge about CRC has also been advancing, resulting in the identification of novel targets.

HGF: hepatocyte growth factor; c-MET: mesenchymal–epithelial transition factor; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor; EGFR: epidermal growth factor receptor; EGF: epidermal growth factor; HER2: human epidermal growth factor 2; CTLA-4: cytotoxic T lymphocyte-associated antigen 4; PD-1: programmed death-1; PD-L1: programmed death ligand 1; PI3K: phosphoinositide 3-kinase; AKT: protein kinase B, also known as PKB; mTOR: mammalian target of rapamycin; MEK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase. *These agents have not been recommended by the NCCN. **This agent has been approved by the National Medical Products Administration of China (NMPA), but not by the United States of America Food and Drug Administration (FDA)

mCRC: metastatic colorectal cancer; EGFR: epidermal growth factor receptor; VEGF: vascular endothelial growth factor; PD-1/L1: programmed death-1/programmed death ligand 1; dMMR: deficient mismatch repair; pMMR: proficient mismatch repair; HER2: human epidermal growth factor 2; BSC: best supportive care; WT: wild type; mut: mutated; amp: amplified. *The NCCN recommends initial administration of PD-1/PD-L1 therapy only in patients in poor functional status
CRC classification changes swiftly because of the rapidly developing pathological and immunological findings that might increase existing knowledge of cancer biological characteristics. The genetic heterogenicity of CRC has been identified, together with a comprehensive understanding of the different molecular pathways and genetic profiles involved. Updated classification of CRC led to the announcement of the Consensus Molecular Subtype (CMS) classification, which took both tumor pathological characteristics and gene expression into account:502 CMS1, enriched for inflammatory or immune genes; CMS2, canonical; CMS3, metabolic; and CMS4, mesenchymal. Although it is still in development, the CMS system might indicate the prognosis of CRC and help guide drug development and application.503,504,505 Data showed that the immune desert CMS2 and CMS3 subtypes responded preferably to anti-EGFR or anti-VEGF therapy, and patients in all four CMS classifications might have different mechanisms of immune evasion, which will enable tailored targeted therapy.504,506,507 There has been a changing trend from a clonal perspective to a clonal-stromal-immune perspective when multiple genetic profiles in genomics, transcriptomics, and immunity are considered before making decisions on comprehensive personized targeted therapy.504
Although targeted therapy is associated with prolonged survival, there are several drawbacks: (1) The cost–benefit balance is questionable when current chemotherapy is much less expensive than extra targeted regimens, especially for those who may need multiple targeted agents. This is crucial because both producing targeted agents and testing necessary genetic markers remain costly, and the best result is longer survival and not full recovery. (2) Targeted therapy might cause extra adverse events. Level 3 or level 4 adverse events can be observed occasionally, and their incidence might increase when two or more targeted agents are combined. (3) Considering existing crossover and bypass mechanisms between pathways (Fig. 5), current drug resistance cannot be avoided, and acquired resistance adds further complications because the optimal solution might involve more treatment-related expense and toxicity. (4) Efficacy differs dramatically among people, leading to increased burdens associated with patient selection and surveillance. These issues may form a part of future directions for targeted drug development.

VEGFR: vascular endothelial growth factor receptor; EGFR: epidermal growth factor receptor; c-MET: mesenchymal–epithelial transition factor; IGF-1R: insulin-like growth factor 1 receptor; TGF-β: transforming growth factor-β; RON-R: recepteur d’Origine nantais; PDGFR: platelet-derived growth factor receptor; *with multiple isoforms
In general, we not only are encouraged by the fact that patients with CRC are living longer with plentiful choices of targeted treatments, of which one or more could ultimately be beneficial, but also expect even more individualized treatments to be developed that promote even longer survival, have fewer adverse reactions and have the potential for full recovery.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Dekker, E., Tanis, P. J., Vleugels, J. L. A., Kasi, P. M. & Wallace, M. B. Colorectal cancer. Lancet 394, 1467–1480 (2019).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 (2019).
Kuipers, E. J. et al. Colorectal cancer. Nat. Rev. Dis. Prim. 1, 15065 (2015).
Keum, N. & Giovannucci, E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 16, 713–732 (2019).
Sanchez-Gundin, J., Fernandez-Carballido, A. M., Martinez-Valdivieso, L., Barreda-Hernandez, D. & Torres-Suarez, A. I. New trends in the therapeutic approach to metastatic colorectal cancer. Int J. Med. Sci. 15, 659–665 (2018).
Wolf, A. M. D. et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J. Clin. 68, 250–281 (2018).
van der Stok, E. P., Spaander, M. C. W., Grunhagen, D. J., Verhoef, C. & Kuipers, E. J. Surveillance after curative treatment for colorectal cancer. Nat. Rev. Clin. Oncol. 14, 297–315 (2017).
Messersmith, W. A. NCCN guidelines updates: management of metastatic colorectal cancer. J. Natl Compr. Cancer Netw. 17, 599–601 (2019).
Brown, K. G. M., Solomon, M. J., Mahon, K. & O’Shannassy, S. Management of colorectal cancer. BMJ 366, l4561 (2019).
Labianca, R. et al. Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 24(Suppl. 6), vi64–vi72 (2013).
Van Cutsem, E., Cervantes, A., Nordlinger, B. & Arnold, D. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 25(Suppl. 3), iii1–iii9 (2014).
Seymour, M. T. et al. Different strategies of sequential and combination chemotherapy for patients with poor prognosis advanced colorectal cancer (MRC FOCUS): a randomised controlled trial. Lancet 370, 143–152 (2007).
Koopman, M. et al. Sequential versus combination chemotherapy with capecitabine, irinotecan, and oxaliplatin in advanced colorectal cancer (CAIRO): a phase III randomised controlled trial. Lancet 370, 135–142 (2007).
Colucci, G. et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J. Clin. Oncol. 23, 4866–4875 (2005).
Tournigand, C. et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J. Clin. Oncol. 22, 229–237 (2004).
Vera, R. et al. Current controversies in the management of metastatic colorectal cancer. Cancer Chemother. Pharm. 76, 659–677 (2015).
Falcone, A. et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: the Gruppo Oncologico Nord Ovest. J. Clin. Oncol. 25, 1670–1676 (2007).
Souglakos, J. et al. FOLFOXIRI (folinic acid, 5-fluorouracil, oxaliplatin and irinotecan) vs FOLFIRI (folinic acid, 5-fluorouracil and irinotecan) as first-line treatment in metastatic colorectal cancer (MCC): a multicentre randomised phase III trial from the Hellenic Oncology Research Group (HORG). Br. J. Cancer 94, 798–805 (2006).
Cassidy, J. et al. XELOX (capecitabine plus oxaliplatin): active first-line therapy for patients with metastatic colorectal cancer. J. Clin. Oncol. 22, 2084–2091 (2004).
Goldberg, R. M. et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol. 22, 23–30 (2004).
Brodsky, F. M. Monoclonal antibodies as magic bullets. Pharm. Res. 5, 1–9 (1988).
Lee, Y. T., Tan, Y. J. & Oon, C. E. Molecular targeted therapy: treating cancer with specificity. Eur. J. Pharm. 834, 188–196 (2018).
Oh, D. Y. & Bang, Y. J. HER2-targeted therapies—a role beyond breast cancer. Nat. Rev. Clin. Oncol. 17, 33–48 (2020).
Ferguson, F. M. & Gray, N. S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov. 17, 353–377 (2018).
Tariman, J. D. Changes in cancer treatment: Mabs, Mibs, Mids, Nabs, and Nibs. Nurs. Clin. N. Am. 52, 65–81 (2017).
Tiwari, A., Saraf, S., Verma, A., Panda, P. K. & Jain, S. K. Novel targeting approaches and signaling pathways of colorectal cancer: An insight. World J. Gastroenterol. 24, 4428–4435 (2018).
Krishnamurthy, N. & Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: update on effectors and inhibitors. Cancer Treat. Rev. 62, 50–60 (2018).
Arteaga, C. L. & Engelman, J. A. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25, 282–303 (2014).
Tebbutt, N., Pedersen, M. W. & Johns, T. G. Targeting the ERBB family in cancer: couples therapy. Nat. Rev. Cancer 13, 663–673 (2013).
Roskoski, R. Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharm. Res. 139, 395–411 (2019).
Vecchione, L., Jacobs, B., Normanno, N., Ciardiello, F. & Tejpar, S. EGFR-targeted therapy. Exp. Cell Res. 317, 2765–2771 (2011).
Wang, Z. ErbB receptors and cancer. Methods Mol. Biol. 1652, 3–35 (2017).
Hsu, J. L. & Hung, M. C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 35, 575–588 (2016).
Rotow, J. & Bivona, T. G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 17, 637–658 (2017).
Roskoski, R. Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharm. Res. 79, 34–74 (2014).
Hayes, D. F. HER2 and breast cancer—a phenomenal success story. N. Engl. J. Med. 381, 1284–1286 (2019).
Boku, N. HER2-positive gastric cancer. Gastric Cancer 17, 1–12 (2014).
Takegawa, N. & Yonesaka, K. HER2 as an emerging oncotarget for colorectal cancer treatment after failure of anti-epidermal growth factor receptor therapy. Clin. Colorectal Cancer 16, 247–251 (2017).
Kiavue, N. et al. ERBB3 mutations in cancer: biological aspects, prevalence and therapeutics. Oncogene https://doi.org/10.1038/s41388-019-1001-5 (2019).
Veikkolainen, V. et al. Function of ERBB4 is determined by alternative splicing. Cell Cycle 10, 2647–2657 (2011).
Junttila, T. T. et al. Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 65, 1384–1393 (2005).
Saintigny, P. & Burger, J. A. Recent advances in non-small cell lung cancer biology and clinical management. Discov. Med. 13, 287–297 (2012).
Wang, X., Goldstein, D., Crowe, P. J. & Yang, J. L. Next-generation EGFR/HER tyrosine kinase inhibitors for the treatment of patients with non-small-cell lung cancer harboring EGFR mutations: a review of the evidence. OncoTargets Ther. 9, 5461–5473 (2016).
Bose, R. et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 3, 224–237 (2013).
Ishibashi, K. et al. Nuclear ErbB4 signaling through H3K9me3 is antagonized by EGFR-activated c-Src. J. Cell Sci. 126, 625–637 (2013).
Pearson, G. et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22, 153–183 (2001).
Wu, P., Wee, P., Jiang, J., Chen, X. & Wang, Z. Differential regulation of transcription factors by location-specific EGF receptor signaling via a spatio-temporal interplay of ERK activation. PLoS ONE 7, e41354 (2012).
Kim, E. K. & Choi, E. J. Compromised MAPK signaling in human diseases: an update. Arch. Toxicol. 89, 867–882 (2015).
Mizukami, T., Izawa, N., Nakajima, T. E. & Sunakawa, Y. Targeting EGFR and RAS/RAF signaling in the treatment of metastatic colorectal cancer: from current treatment strategies to future perspectives. Drugs 79, 633–645 (2019).
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002).
Samatar, A. A. & Poulikakos, P. I. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov. 13, 928–942 (2014).
Taieb, J., Lapeyre-Prost, A., Laurent Puig, P. & Zaanan, A. Exploring the best treatment options for BRAF-mutant metastatic colon cancer. Br. J. Cancer 121, 434–442 (2019).
Ducreux, M. et al. Molecular targeted therapy of BRAF-mutant colorectal cancer. Ther. Adv. Med. Oncol. 11, 1758835919856494 (2019).
Okano, J., Gaslightwala, I., Birnbaum, M. J., Rustgi, A. K. & Nakagawa, H. Akt/protein kinase B isoforms are differentially regulated by epidermal growth factor stimulation. J. Biol. Chem. 275, 30934–30942 (2000).
Elmenier, F. M., Lasheen, D. S. & Abouzid, K. A. M. Phosphatidylinositol 3 kinase (PI3K) inhibitors as new weapon to combat cancer. Eur. J. Med. Chem. 183, 111718 (2019).
Alzahrani, A. S. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin. Cancer Biol. 59, 125–132 (2019).
Tang, F., Wang, Y., Hemmings, B. A., Ruegg, C. & Xue, G. PKB/Akt-dependent regulation of inflammation in cancer. Semin. Cancer Biol. 48, 62–69 (2018).
Manning, B. D. & Toker, A. AKT/PKB signaling: navigating the network. Cell 169, 381–405 (2017).
Weisheit, S., Schafer, C., Mertens, C., Berndt, A. & Liebmann, C. PKCepsilon acts as negative allosteric modulator of EGF receptor signalling. Cell Signal. 23, 436–448 (2011).
Koss, H., Bunney, T. D., Behjati, S. & Katan, M. Dysfunction of phospholipase Cgamma in immune disorders and cancer. Trends Biochem. Sci. 39, 603–611 (2014).
Choi, J. H. et al. Phospholipase C-gamma1 is a guanine nucleotide exchange factor for dynamin-1 and enhances dynamin-1-dependent epidermal growth factor receptor endocytosis. J. Cell Sci. 117, 3785–3795 (2004).
Wahl, M. I., Daniel, T. O. & Carpenter, G. Antiphosphotyrosine recovery of phospholipase C activity after EGF treatment of A-431 cells. Science 241, 968–970 (1988).
Margolis, B. et al. EGF induces tyrosine phosphorylation of phospholipase C-II: a potential mechanism for EGF receptor signaling. Cell 57, 1101–1107 (1989).
Meisenhelder, J., Suh, P. G., Rhee, S. G. & Hunter, T. Phospholipase C-gamma is a substrate for the PDGF and EGF receptor protein-tyrosine kinases in vivo and in vitro. Cell 57, 1109–1122 (1989).
Anderson, D. et al. Binding of SH2 domains of phospholipase C gamma 1, GAP, and Src to activated growth factor receptors. Science 250, 979–982 (1990).
Margolis, B. et al. Effect of phospholipase C-gamma overexpression on PDGF-induced second messengers and mitogenesis. Science 248, 607–610 (1990).
Nappi, A. et al. Metastatic colorectal cancer: role of target therapies and future perspectives. Curr. Cancer Drug Targets 18, 421–429 (2018).
Dai, L. et al. DAG/PKCdelta and IP3/Ca(2)(+)/CaMK IIbeta operate in parallel to each other in PLCgamma1-driven cell proliferation and migration of human gastric adenocarcinoma cells, through Akt/mTOR/S6 pathway. Int. J. Mol. Sci. 16, 28510–28522 (2015).
Wang, Y., Wu, J. & Wang, Z. Akt binds to and phosphorylates phospholipase C-gamma1 in response to epidermal growth factor. Mol. Biol. Cell 17, 2267–2277 (2006).
Ye, K. et al. Phospholipase C gamma 1 is a physiological guanine nucleotide exchange factor for the nuclear GTPase PIKE. Nature 415, 541–544 (2002).
Li, S., Wang, Q., Wang, Y., Chen, X. & Wang, Z. PLC-gamma1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol. Endocrinol. 23, 901–913 (2009).
Geiger, J. L., Grandis, J. R. & Bauman, J. E. The STAT3 pathway as a therapeutic target in head and neck cancer: barriers and innovations. Oral Oncol. 56, 84–92 (2016).
Khanna, P., Chua, P. J., Bay, B. H. & Baeg, G. H. The JAK/STAT signaling cascade in gastric carcinoma (Review). Int. J. Oncol. 47, 1617–1626 (2015).
Chen, Z. et al. EGFR family and Src family kinase interactions: mechanics matters? Curr. Opin. Cell Biol. 51, 97–102 (2018).
Egloff, A. M. & Grandis, J. R. Targeting epidermal growth factor receptor and SRC pathways in head and neck cancer. Semin. Oncol. 35, 286–297 (2008).
Mendelsohn, J., Prewett, M., Rockwell, P. & Goldstein, N. I. CCR 20th anniversary commentary: a chimeric antibody, C225, inhibits EGFR activation and tumor growth. Clin. Cancer Res. 21, 227–229 (2015).
Cunningham, D. et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 351, 337–345 (2004).
Jonker, D. J. et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 357, 2040–2048 (2007).
Van Cutsem, E. et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 360, 1408–1417 (2009).
Tabernero, J. et al. Phase II trial of cetuximab in combination with fluorouracil, leucovorin, and oxaliplatin in the first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 25, 5225–5232 (2007).
Maughan, T. S. et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet 377, 2103–2114 (2011).
Tveit, K. M. et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J. Clin. Oncol. 30, 1755–1762 (2012).
Van Cutsem, E. et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 33, 692–700 (2015).
Aranda, E. et al. First-line mFOLFOX plus cetuximab followed by mFOLFOX plus cetuximab or single-agent cetuximab as maintenance therapy in patients with metastatic colorectal cancer: phase II randomised MACRO2 TTD study. Eur. J. Cancer 101, 263–272 (2018).
Van Cutsem, E. et al. Intrapatient cetuximab dose escalation in metastatic colorectal cancer according to the grade of early skin reactions: the randomized EVEREST study. J. Clin. Oncol. 30, 2861–2868 (2012).
Yarom, N. & Jonker, D. J. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov. Med. 11, 95–105 (2011).
Fakih, M. & Vincent, M. Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr. Oncol. 17(Suppl. 1), S18–S30 (2010).
Douillard, J. Y. et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J. Clin. Oncol. 28, 4697–4705 (2010).
Douillard, J. Y. et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann. Oncol. 25, 1346–1355 (2014).
Modest, D. P. et al. Panitumumab-based maintenance after oxaliplatin discontinuation in metastatic colorectal cancer: A retrospective analysis of two randomised trials. Int. J. Cancer 145, 576–585 (2019).
Pietrantonio, F. et al. First-line FOLFOX plus panitumumab (Pan) followed by 5FU/LV plus Pan or single-agent Pan as maintenance therapy in patients with RAS wild-type metastatic colorectal cancer (mCRC): The VALENTINO study. J. Clin. Oncol. 36, 3505–3505 (2018).
Price, T. J. et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 15, 569–579 (2014).
Sobrero, A. F. et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 2311–2319 (2008).
Cohn, A. L. et al. An open-label, single-arm, phase 2 trial of panitumumab plus FOLFIRI as second-line therapy in patients with metastatic colorectal cancer. Clin. Colorectal Cancer 10, 171–177 (2011).
Seymour, M. T. et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 14, 749–759 (2013).
Van Cutsem, E. et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J. Clin. Oncol. 25, 1658–1664 (2007).
Qin, S. et al. Efficacy and tolerability of first-line cetuximab plus leucovorin, fluorouracil, and oxaliplatin (FOLFOX-4) versus FOLFOX-4 in patients with RAS wild-type metastatic colorectal cancer: the Open-Label, Randomized, Phase III TAILOR Trial. J. Clin. Oncol. 36, 3031–3039 (2018).
Missiaglia, E. et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 25, 1995–2001 (2014).
Holch, J. W., Ricard, I., Stintzing, S., Modest, D. P. & Heinemann, V. The relevance of primary tumour location in patients with metastatic colorectal cancer: A meta-analysis of first-line clinical trials. Eur. J. Cancer 70, 87–98 (2017).
Creasy, J. M. et al. The impact of primary tumor location on long-term survival in patients undergoing hepatic resection for metastatic colon cancer. Ann. Surg. Oncol. 25, 431–438 (2018).
Van Cutsem, E. et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 27, 1386–1422 (2016).
Lito, P. et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 22, 668–682 (2012).
Corcoran, R. B. et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2, 227–235 (2012).
Corcoran, R. B. et al. Efficacy and circulating tumor DNA (ctDNA) analysis of the BRAF inhibitor dabrafenib (D), MEK inhibitor trametinib (T), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E–mutated (BRAFm) metastatic colorectal cancer (mCRC). Ann. Oncol. 27, vi150 (2016).
Prahallad, A. et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103 (2012).
Yang, H. et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 72, 779–789 (2012).
van Geel, R. et al. A phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov. 7, 610–619 (2017).
Corcoran, R. B. et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAF(V600E)-mutant colorectal cancer. Cancer Discov. 8, 428–443 (2018).
Hong, D. S. et al. Phase IB study of vemurafenib in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discov. 6, 1352–1365 (2016).
Kopetz, S. et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG 1406). J. Clin. Oncol. 35, 520–520 (2017).
Tabernero, J. et al. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J. Clin. Oncol. 34, 3544–3544 (2016).
Kopetz, S. et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N. Engl J. Med. https://doi.org/10.1056/NEJMoa1908075 (2019).
H, B., M, K. & CP, P. Colorectal cancer. Lancet 383, 1490–1502 (2014).
Yonesaka, K. et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 3, 99ra86 (2011).
Mohan, S. et al. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet. 10, e1004271 (2014).
Richman, S. D. et al. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J. Pathol. 238, 562–570 (2016).
Bertotti, A. et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 1, 508–523 (2011).
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Luca, T. et al. In vitro combined treatment with cetuximab and trastuzumab inhibits growth of colon cancer cells. Cell Prolif. 47, 435–447 (2014).
Bekaii-Saab, T. S. et al. A phase I trial of paclitaxel and trastuzumab in combination with interleukin-12 in patients with HER2/neu-expressing malignancies. Mol. Cancer Ther. 8, 2983–2991 (2009).
Ramanathan, R. K. et al. Low overexpression of HER-2/neu in advanced colorectal cancer limits the usefulness of trastuzumab (Herceptin) and irinotecan as therapy. A phase II trial. Cancer Invest. 22, 858–865 (2004).
Leto, S. M. et al. Sustained inhibition of HER3 and EGFR is necessary to induce regression of HER2-amplified gastrointestinal carcinomas. Clin. Cancer Res. 21, 5519–5531 (2015).
Meric-Bernstam, F. et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 20, 518–530 (2019).
Sartore-Bianchi, A. et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 17, 738–746 (2016).
Fakih, M. G. Trastuzumab plus pertuzumab resistance does not preclude response to lapatinib plus trastuzumab in HER2-amplified colorectal cancer. Oncologist 23, 474–477 (2018).
Wilson, C. Y. & Tolias, P. Recent advances in cancer drug discovery targeting RAS. Drug Discov. Today 21, 1915–1919 (2016).
Misale, S., Di Nicolantonio, F., Sartore-Bianchi, A., Siena, S. & Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov. 4, 1269–1280 (2014).
Van Cutsem, E. et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 29, 2011–2019 (2011).
Amado, R. G. et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 1626–1634 (2008).
De Roock, W. et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 304, 1812–1820 (2010).
Peeters, M. et al. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J. Clin. Oncol. 31, 759–765 (2013).
Douillard, J. Y. et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 369, 1023–1034 (2013).
Mao, C., Yang, Z. Y., Hu, X. F., Chen, Q. & Tang, J. L. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann. Oncol. 23, 1518–1525 (2012).
De Roock, W. et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 11, 753–762 (2010).
Siena, S., Sartore-Bianchi, A., Di Nicolantonio, F., Balfour, J. & Bardelli, A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J. Natl Cancer Inst. 101, 1308–1324 (2009).
Laurent-Puig, P. et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin. Oncol. 27, 5924–5930 (2009).
Nagata, Y. et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6, 117–127 (2004).
Moroni, M. et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 6, 279–286 (2005).
Sartore-Bianchi, A. et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J. Clin. Oncol. 25, 3238–3245 (2007).
Jacobs, B. et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J. Clin. Oncol. 27, 5068–5074 (2009).
Khambata-Ford, S. et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 25, 3230–3237 (2007).
Yu, H. A. et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 19, 2240–2247 (2013).
Montagut, C. et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med 18, 221–223 (2012).
Dienstmann, R. et al. Safety and activity of the first-in-class Sym004 Anti-EGFR antibody mixture in patients with refractory colorectal cancer. Cancer Discov. 5, 598–609 (2015).
Montagut, C. et al. Efficacy of Sym004 in patients with metastatic colorectal cancer with acquired resistance to anti-EGFR Therapy and molecularly selected by circulating tumor DNA analyses: a Phase 2 Randomized Clinical Trial. JAMA Oncol. 4, e175245 (2018).
Arena, S. et al. MM-151 overcomes acquired resistance to cetuximab and panitumumab in colorectal cancers harboring EGFR extracellular domain mutations. Sci. Transl. Med. 8, 324ra314 (2016).
Scartozzi, M. et al. Analysis of HER-3, insulin growth factor-1, nuclear factor-kB and epidermal growth factor receptor gene copy number in the prediction of clinical outcome for K-RAS wild-type colorectal cancer patients receiving irinotecan-cetuximab. Ann. Oncol. 23, 1706–1712 (2012).
Yang, Y. & Yee, D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 17, 251–261 (2012).
Sclafani, F. et al. A randomized phase II/III study of dalotuzumab in combination with cetuximab and irinotecan in chemorefractory, KRAS wild-type, metastatic colorectal cancer. J. Natl Cancer Inst. 107, djv258 (2015).
Reidy, D. L. et al. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J. Clin. Oncol. 28, 4240–4246 (2010).
Yar Saglam, A. S., Alp, E., Elmazoglu, Z. & Menevse, S. Treatment with cucurbitacin B alone and in combination with gefitinib induces cell cycle inhibition and apoptosis via EGFR and JAK/STAT pathway in human colorectal cancer cell lines. Hum. Exp. Toxicol. 35, 526–543 (2016).
Li, Q. et al. Nuclear PKM2 contributes to gefitinib resistance via upregulation of STAT3 activation in colorectal cancer. Sci. Rep. 5, 16082 (2015).
Mahmood, M. Q., Shukla, S. D., Dua, K. & Shastri, M. D. The role of epidermal growth factor receptor in the management of gastrointestinal carcinomas: present status and future perspectives. Curr. Pharm. Des. 23, 2314–2320 (2017).
Saharinen, P., Eklund, L., Pulkki, K., Bono, P. & Alitalo, K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol. Med 17, 347–362 (2011).
Goel, H. L. & Mercurio, A. M. VEGF targets the tumour cell. Nat. Rev. Cancer 13, 871–882 (2013).
Ferrara, N., Gerber, H. P. & LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 9, 669–676 (2003).
Folkman, J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182–1186 (1971).
Falcon, B. L., Chintharlapalli, S., Uhlik, M. T. & Pytowski, B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharm. Ther. 164, 204–225 (2016).
Karaman, S., Leppanen, V. M. & Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 145, dev151019 (2018).
Koch, S. & Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2, a006502 (2012).
Peng, K., Bai, Y., Zhu, Q., Hu, B. & Xu, Y. Targeting VEGF-neuropilin interactions: a promising antitumor strategy. Drug Discov. Today 24, 656–664 (2019).
Olsson, A. K., Dimberg, A., Kreuger, J. & Claesson-Welsh, L. VEGF receptor signalling—in control of vascular function. Nat. Rev. Mol. Cell. Biol. 7, 359–371 (2006).
Lee, Y. J. et al. Differential effects of VEGFR-1 and VEGFR-2 inhibition on tumor metastases based on host organ environment. Cancer Res. 70, 8357–8367 (2010).
Vaahtomeri, K., Karaman, S., Makinen, T. & Alitalo, K. Lymphangiogenesis guidance by paracrine and pericellular factors. Genes Dev. 31, 1615–1634 (2017).
Takahashi, H. & Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. (Lond.) 109, 227–241 (2005).
Tjwa, M., Luttun, A., Autiero, M. & Carmeliet, P. VEGF and PlGF: two pleiotropic growth factors with distinct roles in development and homeostasis. Cell Tissue Res. 314, 5–14 (2003).
Fischer, C., Mazzone, M., Jonckx, B. & Carmeliet, P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat. Rev. Cancer 8, 942–956 (2008).
Cebe-Suarez, S., Zehnder-Fjallman, A. & Ballmer-Hofer, K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell Mol. Life Sci. 63, 601–615 (2006).
Garnier, L., Gkountidi, A. O. & Hugues, S. Tumor-associated lymphatic vessel features and immunomodulatory functions. Front. Immunol. 10, 720 (2019).
Secker, G. A. & Harvey, N. L. VEGFR signaling during lymphatic vascular development: from progenitor cells to functional vessels. Dev. Dyn. 244, 323–331 (2015).
Shibuya, M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2, 1097–1105 (2011).
Lopez, A., Harada, K., Vasilakopoulou, M., Shanbhag, N. & Ajani, J. A. Targeting angiogenesis in colorectal carcinoma. Drugs 79, 63–74 (2019).
Stitzlein, L., Rao, P. & Dudley, R. Emerging oral VEGF inhibitors for the treatment of renal cell carcinoma. Expert Opin. Investig. Drugs 28, 121–130 (2019).
Sammarco, G. et al. Mast cells, angiogenesis and lymphangiogenesis in human gastric cancer. Int. J. Mol. Sci. 20, 2106 (2019).
Kurzrock, R. & Stewart, D. J. Exploring the benefit/risk associated with antiangiogenic agents for the treatment of non-small cell lung cancer patients. Clin. Cancer Res. 23, 1137–1148 (2017).
Seeber, A., Gunsilius, E., Gastl, G. & Pircher, A. Anti-angiogenics: their value in colorectal cancer therapy. Oncol. Res. Treat. 41, 188–193 (2018).
Guba, M., Seeliger, H., Kleespies, A., Jauch, K. W. & Bruns, C. Vascular endothelial growth factor in colorectal cancer. Int. J. Colorectal Dis. 19, 510–517 (2004).
Jain, R. K. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622 (2014).
Amelio, I. & Melino, G. The p53 family and the hypoxia-inducible factors (HIFs): determinants of cancer progression. Trends Biochem. Sci. 40, 425–434 (2015).
Tarnawski, A. S., Ahluwalia, A. & Jones, M. K. Angiogenesis in gastric mucosa: an important component of gastric erosion and ulcer healing and its impairment in aging. J. Gastroenterol. Hepatol. 29(Suppl. 4), 112–123 (2014).
Hurwitz, H. et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 350, 2335–2342 (2004).
Saltz, L. B. et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J. Clin. Oncol. 26, 2013–2019 (2008).
Passardi, A. et al. Effectiveness of bevacizumab added to standard chemotherapy in metastatic colorectal cancer: final results for first-line treatment from the ITACa randomized clinical trial. Ann. Oncol. 26, 1201–1207 (2015).
Cunningham, D. et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): an open-label, randomised phase 3 trial. Lancet Oncol. 14, 1077–1085 (2013).
Simkens, L. H. et al. Maintenance treatment with capecitabine and bevacizumab in metastatic colorectal cancer (CAIRO3): a phase 3 randomised controlled trial of the Dutch Colorectal Cancer Group. Lancet 385, 1843–1852 (2015).
Cremolini, C. et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 16, 1306–1315 (2015).
Hurwitz, H. I. et al. Efficacy and safety of bevacizumab in metastatic colorectal cancer: pooled analysis from seven randomized controlled trials. Oncologist 18, 1004–1012 (2013).
Heinemann, V. et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 15, 1065–1075 (2014).
Schwartzberg, L. S. et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J. Clin. Oncol. 32, 2240–2247 (2014).
Venook, A. P. et al. Effect of first-line chemotherapy combined with cetuximab or bevacizumab on overall survival in patients with KRAS wild-type advanced or metastatic colorectal cancer: a Randomized Clinical Trial. JAMA 317, 2392–2401 (2017).
Parikh, A. R. et al. MAVERICC, a randomized, biomarker-stratified, phase II study of mFOLFOX6-bevacizumab versus FOLFIRI-bevacizumab as first-line chemotherapy in metastatic colorectal cancer. Clin. Cancer Res. 25, 2988–2995 (2019).
Yamazaki, K. et al. Randomized phase III study of bevacizumab plus FOLFIRI and bevacizumab plus mFOLFOX6 as first-line treatment for patients with metastatic colorectal cancer (WJOG4407G). Ann. Oncol. 27, 1539–1546 (2016).
Giantonio, B. J. et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 25, 1539–1544 (2007).
Bennouna, J. et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 14, 29–37 (2013).
Grothey, A. et al. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE). J. Clin. Oncol. 26, 5326–5334 (2008).
Goey, K. K. H. et al. Maintenance treatment with capecitabine and bevacizumab versus observation in metastatic colorectal cancer: updated results and molecular subgroup analyses of the phase 3 CAIRO3 study. Ann. Oncol. 28, 2128–2134 (2017).
Diaz-Rubio, E. et al. First-line XELOX plus bevacizumab followed by XELOX plus bevacizumab or single-agent bevacizumab as maintenance therapy in patients with metastatic colorectal cancer: the phase III MACRO TTD study. Oncologist 17, 15–25 (2012).
Koeberle, D. et al. Bevacizumab continuation versus no continuation after first-line chemotherapy plus bevacizumab in patients with metastatic colorectal cancer: a randomized phase III non-inferiority trial (SAKK 41/06). Ann. Oncol. 26, 709–714 (2015).
Yalcin, S. et al. Bevacizumab + capecitabine as maintenance therapy after initial bevacizumab + XELOX treatment in previously untreated patients with metastatic colorectal cancer: phase III ‘Stop and Go’ study results—a Turkish Oncology Group Trial. Oncology 85, 328–335 (2013).
Hegewisch-Becker, S. et al. Maintenance strategies after first-line oxaliplatin plus fluoropyrimidine plus bevacizumab for patients with metastatic colorectal cancer (AIO 0207): a randomised, non-inferiority, open-label, phase 3 trial. Lancet Oncol. 16, 1355–1369 (2015).
Tang, P. A. et al. Phase II clinical and pharmacokinetic study of aflibercept in patients with previously treated metastatic colorectal cancer. Clin. Cancer Res. 18, 6023–6031 (2012).
Van Cutsem, E. et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 30, 3499–3506 (2012).
Folprecht, G. et al. Oxaliplatin and 5-FU/folinic acid (modified FOLFOX6) with or without aflibercept in first-line treatment of patients with metastatic colorectal cancer: the AFFIRM study. Ann. Oncol. 27, 1273–1279 (2016).
Tabernero, J. et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 16, 499–508 (2015).
Moore, M. et al. Randomized phase II study of modified FOLFOX-6 in combination with ramucirumab or icrucumab as second-line therapy in patients with metastatic colorectal cancer after disease progression on first-line irinotecan-based therapy. Ann. Oncol. 27, 2216–2224 (2016).
Argiles, G. et al. Regorafenib plus modified FOLFOX6 as first-line treatment of metastatic colorectal cancer: a phase II trial. Eur. J. Cancer 51, 942–949 (2015).
Grothey, A. et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381, 303–312 (2013).
Li, J. et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 16, 619–629 (2015).
Li, J. et al. Effect of fruquintinib vs placebo on overall survival in patients with previously treated metastatic colorectal cancer: the FRESCO Randomized Clinical Trial. JAMA 319, 2486–2496 (2018).
Xu, R. H. et al. Famitinib versus placebo in the treatment of refractory metastatic colorectal cancer: a multicenter, randomized, double-blinded, placebo-controlled, phase II clinical trial. Chin. J. Cancer 36, 97 (2017).
Siu, L. L. et al. Phase III randomized, placebo-controlled study of cetuximab plus brivanib alaninate versus cetuximab plus placebo in patients with metastatic, chemotherapy-refractory, wild-type K-RAS colorectal carcinoma: the NCIC Clinical Trials Group and AGITG CO.20 Trial. J. Clin. Oncol. 31, 2477–2484 (2013).
Schmoll, H. J. et al. Cediranib with mFOLFOX6 versus bevacizumab with mFOLFOX6 as first-line treatment for patients with advanced colorectal cancer: a double-blind, randomized phase III study (HORIZON III). J. Clin. Oncol. 30, 3588–3595 (2012).
Hoff, P. M. et al. Cediranib plus FOLFOX/CAPOX versus placebo plus FOLFOX/CAPOX in patients with previously untreated metastatic colorectal cancer: a randomized, double-blind, phase III study (HORIZON II). J. Clin. Oncol. 30, 3596–3603 (2012).
Van Cutsem, E. et al. Nintedanib for the treatment of patients with refractory metastatic colorectal cancer (LUME-Colon 1): a phase III, international, randomized, placebo-controlled study. Ann. Oncol. 29, 1955–1963 (2018).
Kopetz, S. et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J. Clin. Oncol. 28, 453–459 (2010).
Chiron, M. et al. Differential antitumor activity of aflibercept and bevacizumab in patient-derived xenograft models of colorectal cancer. Mol. Cancer Ther. 13, 1636–1644 (2014).
Goede, V. et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br. J. Cancer 103, 1407–1414 (2010).
Rigamonti, N. et al. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 8, 696–706 (2014).
Scholz, A. et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 8, 39–57 (2016).
Kienast, Y. et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin. Cancer Res. 19, 6730–6740 (2013).
Mueller, T., Freystein, J., Lucas, H. & Schmoll, H. J. Efficacy of a bispecific antibody co-targeting VEGFA and Ang-2 in combination with chemotherapy in a chemoresistant colorectal carcinoma xenograft model. Molecules 24, 2865 (2019).
Hidalgo, M. et al. First-in-human phase I study of single-agent vanucizumab, a first-in-class bispecific anti-angiopoietin-2/anti-VEGF-A antibody, in adult patients with advanced solid tumors. Clin. Cancer Res. 24, 1536–1545 (2018).
Mitsuhashi, A. et al. Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nat. Commun. 6, 8792 (2015).
Incio, J. et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci. Transl. Med. 10 (2018).
Goto, H. & Nishioka, Y. Fibrocytes: a novel stromal cells to regulate resistance to anti-angiogenic therapy and cancer progression. Int. J. Mol. Sci. 19, 98 (2017).
Semrad, T. J. et al. Phase II study of dovitinib in patients progressing on anti-vascular endothelial growth factor therapy. Cancer Treat. Res. Commun. 10, 21–26 (2017).
Jahangiri, A. et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin. Cancer Res. 19, 1773–1783 (2013).
Wakelee, H. et al. Efficacy and safety of onartuzumab in combination with first-line bevacizumab- or pemetrexed-based chemotherapy regimens in advanced non-squamous non-small-cell lung cancer. Clin. Lung Cancer 18, 50–59 (2017).
Prete, A. et al. Pericytes elicit resistance to vemurafenib and sorafenib therapy in thyroid carcinoma via the TSP-1/TGFbeta1 axis. Clin. Cancer Res. 24, 6078–6097 (2018).
Carbone, C. et al. Combined inhibition of IL1, CXCR1/2, and TGFbeta signaling pathways modulates in-vivo resistance to anti-VEGF treatment. Anticancer Drugs 27, 29–40 (2016).
Castro, B. A. et al. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene 36, 3749–3759 (2017).
Liu, T. et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 9, 3439 (2018).
Stintzing, S. et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3): a post-hoc analysis of tumour dynamics in the final RAS wild-type subgroup of this randomised open-label phase 3 trial. Lancet Oncol. 17, 1426–1434 (2016).
Khattak, M. A., Martin, H., Davidson, A. & Phillips, M. Role of first-line anti-epidermal growth factor receptor therapy compared with anti-vascular endothelial growth factor therapy in advanced colorectal cancer: a meta-analysis of randomized clinical trials. Clin. Colorectal Cancer 14, 81–90 (2015).
Arnold, D. et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann. Oncol. 28, 1713–1729 (2017).
Tejpar, S. et al. Prognostic and predictive relevance of primary tumor location in patients with RAS wild-type metastatic colorectal cancer: retrospective analyses of the CRYSTAL and FIRE-3 Trials. JAMA Oncol. 3, 194–201 (2017).
Boeckx, N. et al. Primary tumor sidedness has an impact on prognosis and treatment outcome in metastatic colorectal cancer: results from two randomized first-line panitumumab studies. Ann. Oncol. 28, 1862–1868 (2017).
Hecht, J. R. et al. SPIRITT: a randomized, multicenter, phase II study of panitumumab with FOLFIRI and bevacizumab with FOLFIRI as second-line treatment in patients with unresectable wild type KRAS metastatic colorectal cancer. Clin. Colorectal Cancer 14, 72–80 (2015).
Wang, Q., Yang, S., Wang, K. & Sun, S. Y. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J. Hematol. Oncol. 12, 63 (2019).
Cheng, F. & Guo, D. MET in glioma: signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 38, 270 (2019).
Demkova, L. & Kucerova, L. Role of the HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol. Cancer 17, 26 (2018).
Lam, B. Q., Dai, L. & Qin, Z. The role of HGF/c-MET signaling pathway in lymphoma. J. Hematol. Oncol. 9, 135 (2016).
Bradley, C. A. et al. Targeting c-MET in gastrointestinal tumours: rationale, opportunities and challenges. Nat. Rev. Clin. Oncol. 14, 562–576 (2017).
Cooper, C. S. et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 311, 29–33 (1984).
Nakamura, T. et al. Molecular cloning and expression of human hepatocyte growth factor. Nature 342, 440–443 (1989).
Stoker, M., Gherardi, E., Perryman, M. & Gray, J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 327, 239–242 (1987).
Schmidt, C. et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 373, 699–702 (1995).
Xing, F. et al. Activation of the c-Met pathway mobilizes an inflammatory network in the brain microenvironment to promote brain metastasis of breast cancer. Cancer Res. 76, 4970–4980 (2016).
Ozawa, Y. et al. c-Met in esophageal squamous cell carcinoma: an independent prognostic factor and potential therapeutic target. BMC Cancer 15, 451 (2015).
Anestis, A., Zoi, I. & Karamouzis, M. V. Current advances of targeting HGF/c-Met pathway in gastric cancer. Ann. Transl. Med. 6, 247 (2018).
Safaie, Q. E. et al. The c-Met receptor: implication for targeted therapies in colorectal cancer. Tumour Biol. 39, 1010428317699118 (2017).
Otte, J. M. et al. Functional expression of HGF and its receptor in human colorectal cancer. Digestion 61, 237–246 (2000).
Matsumoto, K., Umitsu, M., De Silva, D. M., Roy, A. & Bottaro, D. P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 108, 296–307 (2017).
Bahrami, A. et al. C-Met as a potential target for the treatment of gastrointestinal cancer: current status and future perspectives. J. Cell Physiol. 232, 2657–2673 (2017).
Bouattour, M. et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 67, 1132–1149 (2018).
Drilon, A., Cappuzzo, F., Ou, S. I. & Camidge, D. R. Targeting MET in lung cancer: will expectations finally be MET? J. Thorac. Oncol. 12, 15–26 (2017).
Blumenschein, G. R. Jr., Mills, G. B. & Gonzalez-Angulo, A. M. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J. Clin. Oncol. 30, 3287–3296 (2012).
Cabanillas, M. E. et al. Cabozantinib as salvage therapy for patients with tyrosine kinase inhibitor-refractory differentiated thyroid cancer: results of a multicenter phase II International Thyroid Oncology Group Trial. J. Clin. Oncol. 35, 3315–3321 (2017).
Linehan, W. M. et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 374, 135–145 (2016).
Catenacci, D. V. T. et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 18, 1467–1482 (2017).
Sartore-Bianchi, A., Loupakis, F., Argiles, G. & Prager, G. W. Challenging chemoresistant metastatic colorectal cancer: therapeutic strategies from the clinic and from the laboratory. Ann. Oncol. 27, 1456–1466 (2016).
Gao, H., Guan, M., Sun, Z. & Bai, C. High c-Met expression is a negative prognostic marker for colorectal cancer: a meta-analysis. Tumour Biol. 36, 515–520 (2015).
Luo, H. Y. & Xu, R. H. Predictive and prognostic biomarkers with therapeutic targets in advanced colorectal cancer. World J. Gastroenterol. 20, 3858–3874 (2014).
Baldus, S. E., Kort, E. J., Schirmacher, P., Dienes, H. P. & Resau, J. H. Quantification of MET and hepatocyte growth factor/scatter factor expression in colorectal adenomas, carcinomas and non-neoplastic epithelia by quantitative laser scanning microscopy. Int. J. Oncol. 31, 199–204 (2007).
Mo, H. N. & Liu, P. Targeting MET in cancer therapy. Chronic Dis. Transl. Med. 3, 148–153 (2017).
Kentsis, A. et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 18, 1118–1122 (2012).
Lennerz, J. K. et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 29, 4803–4810 (2011).
Kammula, U. S. et al. Molecular co-expression of the c-Met oncogene and hepatocyte growth factor in primary colon cancer predicts tumor stage and clinical outcome. Cancer Lett. 248, 219–228 (2007).
El-Deiry, W. S. et al. Molecular profiling of 6,892 colorectal cancer samples suggests different possible treatment options specific to metastatic sites. Cancer Biol. Ther. 16, 1726–1737 (2015).
Di Renzo, M. F. et al. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin. Cancer Res. 1, 147–154 (1995).
Boccaccio, C., Gaudino, G., Gambarotta, G., Galimi, F. & Comoglio, P. M. Hepatocyte growth factor (HGF) receptor expression is inducible and is part of the delayed-early response to HGF. J. Biol. Chem. 269, 12846–12851 (1994).
Stellrecht, C. M. & Gandhi, V. MET receptor tyrosine kinase as a therapeutic anticancer target. Cancer Lett. 280, 1–14 (2009).
Comoglio, P. M., Giordano, S. & Trusolino, L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 7, 504–516 (2008).
Pennacchietti, S. et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 3, 347–361 (2003).
Bigatto, V. et al. TNF-alpha promotes invasive growth through the MET signaling pathway. Mol. Oncol. 9, 377–388 (2015).
Qi, J. et al. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res. 71, 1081–1091 (2011).
Peacock, J. W. et al. SEMA3C drives cancer growth by transactivating multiple receptor tyrosine kinases via Plexin B1. EMBO Mol. Med. 10, 219–238 (2018).
Worzfeld, T. et al. ErbB-2 signals through Plexin-B1 to promote breast cancer metastasis. J. Clin. Invest. 122, 1296–1305 (2012).
Malik, M. F., Ye, L. & Jiang, W. G. The Plexin-B family and its role in cancer progression. Histol. Histopathol. 29, 151–165 (2014).
Stein, U. et al. MACC1, a newly identified key regulator of HGF-MET signaling, predicts colon cancer metastasis. Nat. Med. 15, 59–67 (2009).
Parseghian, C. M., Napolitano, S., Loree, J. M. & Kopetz, S. Mechanisms of innate and acquired resistance to anti-EGFR therapy: a review of current knowledge with a focus on rechallenge therapies. Clin. Cancer Res. 25, 6899–6908 (2019).
Boccaccio, C., Luraghi, P. & Comoglio, P. M. MET-mediated resistance to EGFR inhibitors: an old liaison rooted in colorectal cancer stem cells. Cancer Res. 74, 3647–3651 (2014).
Murtuza, A. et al. Novel third-generation EGFR tyrosine kinase inhibitors and strategies to overcome therapeutic resistance in lung cancer. Cancer Res. 79, 689–698 (2019).
Luraghi, P. et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. 74, 1857–1869 (2014).
Van Emburgh, B. O., Sartore-Bianchi, A., Di Nicolantonio, F., Siena, S. & Bardelli, A. Acquired resistance to EGFR-targeted therapies in colorectal cancer. Mol. Oncol. 8, 1084–1094 (2014).
Viticchie, G. & Muller, P. A. J. c-Met and other cell surface molecules: interaction, activation and functional consequences. Biomedicines 3, 46–70 (2015).
Tanizaki, J., Okamoto, I., Sakai, K. & Nakagawa, K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br. J. Cancer 105, 807–813 (2011).
Song, E. K. et al. Potent antitumor activity of cabozantinib, a c-MET and VEGFR2 inhibitor, in a colorectal cancer patient-derived tumor explant model. Int. J. Cancer 136, 1967–1975 (2015).
Nandagopal, L., Sonpavde, G. P. & Agarwal, N. Investigational MET inhibitors to treat renal cell carcinoma. Expert Opin. Investig. Drugs 28, 851–860 (2019).
Reid, J. C. et al. In vitro evidence that KLK14 regulates the components of the HGF/Met axis, pro-HGF and HGF-activator inhibitor 1A and 1B. Biol. Chem. 397, 1299–1305 (2016).
Ye, F. et al. 3-Cl-AHPC inhibits pro-HGF maturation by inducing matriptase/HAI-1 complex formation. J. Cell Mol. Med. 23, 155–166 (2019).
Hu, C. et al. Expression of hepatocyte growth factor activator inhibitor-1 (HAI-1) gene in prostate cancer: clinical and biological significance. J. Buon 19, 215–220 (2014).
Tsai, C. H. et al. HAI-2 suppresses the invasive growth and metastasis of prostate cancer through regulation of matriptase. Oncogene 33, 4643–4652 (2014).
Garajova, I., Giovannetti, E., Biasco, G. & Peters, G. J. c-Met as a target for personalized therapy. Transl. Oncogenom. 7, 13–31 (2015).
Ishikawa, T., Kimura, Y., Hirano, H. & Higashi, S. Matrix metalloproteinase-7 induces homotypic tumor cell aggregation via proteolytic cleavage of the membrane-bound Kunitz-type inhibitor HAI-1. J. Biol. Chem. 292, 20769–20784 (2017).
Iveson, T. et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 15, 1007–1018 (2014).
Doi, T. et al. A phase 3, multicenter, randomized, double-blind, placebo-controlled study of rilotumumab in combination with cisplatin and capecitabine (CX) as first-line therapy for Asian patients (pts) with advanced MET-positive gastric or gastroesophageal junction (G/GEJ) adenocarcinoma: The RILOMET-2 trial. J. Clin. Oncol. 33, TPS226–TPS226 (2015).
Van Cutsem, E. et al. Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin. Cancer Res. 20, 4240–4250 (2014).
Tabernero, J. et al. A pharmacodynamic/pharmacokinetic study of ficlatuzumab in patients with advanced solid tumors and liver metastases. Clin. Cancer Res. 20, 2793–2804 (2014).
Jones, S. F. et al. Safety, tolerability, and pharmacokinetics of TAK-701, a humanized anti-hepatocyte growth factor (HGF) monoclonal antibody, in patients with advanced nonhematologic malignancies: First-in-human phase I dose-escalation study. J. Clin. Oncol. 28, 3081–3081 (2010).
Hori, A. et al. Monotherapeutic and combination antitumor activities of TAK-701, a humanized anti-hepatocyte growth factor neutralizing antibody, against multiple types of cancer. Cancer Res. 69, 1244 (2009).
Okamoto, W. et al. TAK-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation. Mol. Cancer Ther. 9, 2785–2792 (2010).
Merchant, M. et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc. Natl Acad. Sci. USA 110, E2987–E2996 (2013).
Shah, M. A. et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-negative, MET-positive gastroesophageal adenocarcinoma: the METGastric Randomized Clinical Trial. JAMA Oncol. 3, 620–627 (2017).
Spigel, D. R. et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: METLung. J. Clin. Oncol. 35, 412–420 (2017).
Cloughesy, T. et al. Randomized, double-blind, placebo-controlled, multicenter phase II study of onartuzumab plus bevacizumab versus placebo plus bevacizumab in patients with recurrent glioblastoma: efficacy, safety, and hepatocyte growth factor and O(6)-methylguanine-DNA methyltransferase biomarker analyses. J. Clin. Oncol. 35, 343–351 (2017).
Bendell, J. C. et al. A phase II randomized trial (GO27827) of first-line FOLFOX plus bevacizumab with or without the MET inhibitor onartuzumab in patients with metastatic colorectal cancer. Oncologist 22, 264–271 (2017).
Pacchiana, G. et al. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J. Biol. Chem. 285, 36149–36157 (2010).
Strickler, J. H. et al. First-in-human phase I, dose-escalation and -expansion study of telisotuzumab vedotin, an antibody–drug conjugate targeting c-Met, in patients with advanced solid tumors. J. Clin. Oncol. 36, 3298–3306 (2018).
Wang, J. et al. ABBV-399, a c-Met antibody-drug conjugate that targets both MET-amplified and c-Met-overexpressing tumors, irrespective of MET pathway dependence. Clin. Cancer Res. 23, 992–1000 (2017).
Wang, J. et al. Anti-c-Met monoclonal antibody ABT-700 breaks oncogene addiction in tumors with MET amplification. BMC Cancer 16, 105 (2016).
Scagliotti, G. et al. A randomized-controlled phase 2 study of the MET antibody emibetuzumab in combination with erlotinib as first-line treatment for EGFR mutation-positive NSCLC patients. J. Thorac. Oncol. 15, 80–90 (2020).
Rosen, L. S. et al. A first-in-human phase I study of a bivalent MET antibody, emibetuzumab (LY2875358), as monotherapy and in combination with erlotinib in advanced cancer. Clin. Cancer Res. 23, 1910–1919 (2017).
Harding, J. J. et al. A phase Ib/II study of ramucirumab in combination with emibetuzumab in patients with advanced cancer. Clin. Cancer Res. 25, 5202–5211 (2019).
Sakai, D. et al. A non-randomized, open-label, single-arm, Phase 2 study of emibetuzumab in Asian patients with MET diagnostic positive, advanced gastric cancer. Cancer Chemother. Pharm. 80, 1197–1207 (2017).
Li, H. et al. MET inhibitors promote liver tumor evasion of the immune response by stabilizing PDL1. Gastroenterology 156, 1849–1861.e1813 (2019).
Zaman, S. et al. Targeting the pro-survival protein MET with tivantinib (ARQ 197) inhibits growth of multiple myeloma cells. Neoplasia 17, 289–300 (2015).
Remsing Rix, L. L. et al. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem. Biol. 9, 353–358 (2014).
L, R. et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): a final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 19, 682–693 (2018).
Scagliotti, G. V. et al. Tivantinib in combination with erlotinib versus erlotinib alone for EGFR-mutant NSCLC: an exploratory analysis of the phase 3 MARQUEE study. J. Thorac. Oncol. 13, 849–854 (2018).
Eng, C. et al. A randomized, placebo-controlled, phase 1/2 study of tivantinib (ARQ 197) in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with wild-type KRAS who have received first-line systemic therapy. Int. J. Cancer 139, 177–186 (2016).
Scagliotti, G. et al. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J. Clin. Oncol. 33, 2667–2674 (2015).
Rimassa, L. et al. Phase II study of tivantinib and cetuximab in patients with KRAS wild-type metastatic colorectal cancer with acquired resistance to EGFR inhibitors and emergence of MET overexpression: lesson learned for future trials with EGFR/MET dual inhibition. Clin. Colorectal Cancer 18, 125–132.e122 (2019).
Van Cutsem, E. et al. A multicenter phase II study of AMG 337 in patients with -amplified gastric/gastroesophageal junction/esophageal adenocarcinoma and other -amplified solid tumors. Clin. Cancer Res. 25, 2414–2423 (2019).
Hong, D. S. et al. Phase I study of AMG 337, a highly selective small-molecule MET inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 25, 2403–2413 (2019).
Collie, G. W. et al. Structural and molecular insight into resistance mechanisms of first generation cMET inhibitors. ACS Med. Chem. Lett. 10, 1322–1327 (2019).
Gan, H. K. et al. First-in-human phase I study of the selective MET inhibitor, savolitinib, in patients with advanced solid tumors: safety, pharmacokinetics, and antitumor activity. Clin. Cancer Res. 25, 4924–4932 (2019).
Yl, W. et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J. Clin. Oncol. 36, 3101–3109 (2018).
Roskoski, R. Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharm. Res. 144, 19–50 (2019).
Arbour, K. C. & Riely, G. J. Systemic therapy for locally advanced and metastatic non-small cell lung cancer: a review. JAMA 322, 764–774 (2019).
Lev, A. et al. Preclinical rationale for combination of crizotinib with mitomycin C for the treatment of advanced colorectal cancer. Cancer Biol. Ther. 18, 694–704 (2017).
Choueiri, T. K. et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 17, 917–927 (2016).
Abou-Alfa, G. K. et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 379, 54–63 (2018).
Cabanillas, M. E., Ryder, M. & Jimenez, C. Targeted therapy for advanced thyroid cancer: kinase inhibitors and beyond. Endocr. Rev. 40, 1573–1604 (2019).
Scott, A. J. et al. Cabozantinib exhibits potent antitumor activity in colorectal cancer patient-derived tumor xenograft models via autophagy and signaling mechanisms. Mol. Cancer Ther. 17, 2112–2122 (2018).
Yang, S. et al. Cabozantinib induces PUMA-dependent apoptosis in colon cancer cells via AKT/GSK-3β/NF-κB signaling pathway. Cancer Gene Ther. https://doi.org/10.1038/s41417-019-0098-6 (2019).
He, A. R. et al. First-in-human phase I study of merestinib, an oral multikinase inhibitor, in patients with advanced cancer. Oncologist 24, e930–e942 (2019).
Shah, M. A. et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS ONE 8, e54014 (2013).
Engstrom, L. D. et al. Glesatinib exhibits antitumor activity in lung cancer models and patients harboring exon 14 mutations and overcomes mutation-mediated resistance to type I MET inhibitors in nonclinical models. Clin. Cancer Res. 23, 6661–6672 (2017).
Molife, L. R. et al. A phase I, dose-escalation study of the multitargeted receptor tyrosine kinase inhibitor, golvatinib, in patients with advanced solid tumors. Clin. Cancer Res. 20, 6284–6294 (2014).
Friese-Hamim, M., Bladt, F., Locatelli, G., Stammberger, U. & Blaukat, A. The selective c-Met inhibitor tepotinib can overcome epidermal growth factor receptor inhibitor resistance mediated by aberrant c-Met activation in NSCLC models. Am. J. Cancer Res. 7, 962–972 (2017).
Dolan, M. et al. Enhanced efficacy of sitravatinib in metastatic models of antiangiogenic therapy resistance. PLoS ONE 14, e0220101 (2019).
Yoshioka, H. et al. A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-Met inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study). Ann. Oncol. 26, 2066–2072 (2015).
Ciamporcero, E. et al. Combination strategy targeting VEGF and HGF/c-met in human renal cell carcinoma models. Mol. Cancer Ther. 14, 101–110 (2015).
Kim, D. C. et al. Resistance to the c-Met inhibitor KRC-108 induces the epithelial transition of gastric cancer cells. Oncol. Lett. 11, 991–997 (2016).
Sharpe, A. H. & Freeman, G. J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2, 116–126 (2002).
Jelinek, T., Mihalyova, J., Kascak, M., Duras, J. & Hajek, R. PD-1/PD-L1 inhibitors in haematological malignancies: update 2017. Immunology 152, 357–371 (2017).
Chen, L. & Han, X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J. Clin. Invest. 125, 3384–3391 (2015).
Markman, J. L. & Shiao, S. L. Impact of the immune system and immunotherapy in colorectal cancer. J. Gastrointest. Oncol. 6, 208–223 (2015).
Wang, J., Reiss, K. A., Khatri, R., Jaffee, E. & Laheru, D. Immune Therapy in GI malignancies: a review. J. Clin. Oncol. 33, 1745–1753 (2015).
Kalyan, A., Kircher, S., Shah, H., Mulcahy, M. & Benson, A. Updates on immunotherapy for colorectal cancer. J. Gastrointest. Oncol. 9, 160–169 (2018).
Havel, J. J., Chowell, D. & Chan, T. A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 19, 133–150 (2019).
Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, inflammation, and cancer. Cell 140, 883–899 (2010).
Ishihara, J. et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci. Transl. Med. 11, eaau3259 (2019).
Pauken, K. E. & Wherry, E. J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 36, 265–276 (2015).
Gianchecchi, E., Delfino, D. V. & Fierabracci, A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun. Rev. 12, 1091–1100 (2013).
Seidel, J. A., Otsuka, A. & Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front. Oncol. 8, 86 (2018).
Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 38, 255 (2019).
Wang, H. B. et al. Rise of PD-L1 expression during metastasis of colorectal cancer: Implications for immunotherapy. J. Dig. Dis. 18, 574–581 (2017).
Berntsson, J., Nodin, B., Eberhard, J., Micke, P. & Jirstrom, K. Prognostic impact of tumour-infiltrating B cells and plasma cells in colorectal cancer. Int. J. Cancer 139, 1129–1139 (2016).
Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
Zhang, X., Kelaria, S., Kerstetter, J. & Wang, J. The functional and prognostic implications of regulatory T cells in colorectal carcinoma. J. Gastrointest. Oncol. 6, 307–313 (2015).
Facciabene, A., Motz, G. T. & Coukos, G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 72, 2162–2171 (2012).
Fujimoto, H. et al. Deregulated mucosal immune surveillance through gut-associated regulatory T cells and PD-1(+) T cells in human colorectal cancer. J. Immunol. 200, 3291–3303 (2018).
Morvan, M. G. & Lanier, L. L. NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer 16, 7–19 (2016).
Liu, Y. et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 36, 6143–6153 (2017).
Juneja, V. R. et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 214, 895–904 (2017).
Topalian, S. L., Drake, C. G. & Pardoll, D. M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 24, 207–212 (2012).
Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016).
Motzer, R. J. et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1803–1813 (2015).
Robert, C. et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330 (2015).
Brahmer, J. et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 373, 123–135 (2015).
Borghaei, H. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
Ganesh, K. et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 16, 361–375 (2019).
Tintelnot, J. & Stein, A. Immunotherapy in colorectal cancer: available clinical evidence, challenges and novel approaches. World J. Gastroenterol. 25, 3920–3928 (2019).
Hoos, A. et al. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin. Oncol. 37, 533–546 (2010).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Wolchok, J. D. et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 369, 122–133 (2013).
Overman, M. J. et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 18, 1182–1191 (2017).
Passardi, A., Canale, M., Valgiusti, M. & Ulivi, P. Immune checkpoints as a target for colorectal cancer treatment. Int. J. Mol. Sci. 18, 1324 (2017).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Giannakis, M. et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 15, 857–865 (2016).
Llosa, N. J. et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 5, 43–51 (2015).
Le, D. T. et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014).
Ready, N. et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J. Clin. Oncol. 37, 992–1000 (2019).
Mouw, K. W., Goldberg, M. S., Konstantinopoulos, P. A. & D’Andrea, A. D. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 7, 675–693 (2017).
McGranahan, N. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
O’Neil, B. H. et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS ONE 12, e0189848 (2017).
Le, D. T. et al. KEYNOTE-164: pembrolizumab for patients with advanced microsatellite instability high (MSI-H) colorectal cancer. J. Clin. Oncol. 36, 3514–3514 (2018).
Le, D. T. et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J. Clin. Oncol. https://doi.org/10.1200/jco.19.02107 (2019).
Le, D. T. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Long, G. V. et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 18, 1202–1210 (2017).
Overman, M. J. et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 36, 773–779 (2018).
Lenz, H.-J. J. et al. LBA18_PRDurable clinical benefit with nivolumab (NIVO) plus low-dose ipilimumab (IPI) as first-line therapy in microsatellite instability-high/mismatch repair deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC). Ann. Oncol. 29, https://doi.org/10.1093/annonc/mdy424.019 (2018).
Morse, M. A. et al. Safety of nivolumab plus low-dose ipilimumab in previously treated microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer. Oncologist 24, 1453–1461 (2019).
Zhou, E. et al. Up-regulation of Tim-3 is associated with poor prognosis of patients with colon cancer. Int. J. Clin. Exp. Pathol. 8, 8018–8027 (2015).
Yu, X. et al. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. MABS 11, 1139–1148 (2019).
Lee, J. J. & Chu, E. Recent advances in the clinical development of immune checkpoint blockade therapy for mismatch repair proficient (pMMR)/non-MSI-H metastatic colorectal cancer. Clin. Colorectal Cancer 17, 258–273 (2018).
Wu, X. et al. Application of PD-1 Blockade in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 17, 661–674 (2019).
Twyman-Saint Victor, C. et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377 (2015).
Segal, N. H. et al. 403P Phase I studies of the novel carcinoembryonic antigen T-cell bispecific (CEA-CD3 TCB) antibody as a single agent and in combination with atezolizumab: preliminary efficacy and safety in patients (pts) with metastatic colorectal cancer (mCRC). Ann. Oncol. 28, https://doi.org/10.1093/annonc/mdx367.036 (2017).
Tabernero, J. et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 35, 3002–3002 (2017).
Bacac, M. et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin. Cancer Res. 22, 3286–3297 (2016).
Claus, C. et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 11, https://doi.org/10.1126/scitranslmed.aav5989 (2019).
Liu, L. et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 21, 1639–1651 (2015).
MD, H. et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann. Oncol. 30, 1134–1142 (2019).
Eng, C. et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 20, 849–861 (2019).
Callahan, M. K. et al. Phase 1 study to evaluate the safety and tolerability of MEDI4736 (durvalumab, DUR) + tremelimumab (TRE) in patients with advanced solid tumors. J. Clin. Oncol. 35, 3069–3069 (2017).
Droeser, R. A. et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur. J. Cancer 49, 2233–2242 (2013).
Mlecnik, B. et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J. Clin. Oncol. 29, 610–618 (2011).
Yu, H. et al. Correlation of PD-L1 expression with tumor mutation burden and gene signatures for prognosis in early-stage squamous cell lung carcinoma. J. Thorac. Oncol. 14, 25–36 (2019).
Yarchoan, M. et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 4, https://doi.org/10.1172/jci.insight.126908 (2019).
Mehnert, J. M. et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J. Clin. Invest. 126, 2334–2340 (2016).
Gong, J., Wang, C., Lee, P. P., Chu, P. & Fakih, M. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J. Natl Compr. Canc Netw. 15, 142–147 (2017).
Domingo, E. et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 1, 207–216 (2016).
Ngeow, J. & Eng, C. Mismatch repair deficiency in colorectal cancers: is somatic genomic testing the grab-bag for all answers? J. Clin. Oncol. 34, 2085–2087 (2016).
Pages, F. et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391, 2128–2139 (2018).
Mlecnik, B. et al. Comprehensive Intrametastatic Immune Quantification and Major Impact of Immunoscore on Survival. J Natl Cancer Inst 110, https://doi.org/10.1093/jnci/djx123 (2018).
Angell, H. K., Bruni, D., Barrett, J. C., Herbst, R. & Galon, J. The immunoscore: colon cancer and beyond. Clin Cancer Res. https://doi.org/10.1158/1078-0432.Ccr-18-1851 (2019).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016).
Koelzer, V. H., Baker, K., Kassahn, D., Baumhoer, D. & Zlobec, I. Prognostic impact of beta-2-microglobulin expression in colorectal cancers stratified by mismatch repair status. J. Clin. Pathol. 65, 996–1002 (2012).
Gray, R. et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 370, 2020–2029 (2007).
International Multicentre Pooled Analysis of Colon Cancer Trials (IMPACT) investigators. Efficacy of adjuvant fluorouracil and folinic acid in colon cancer. Lancet 345, 939–944 (1995).
Smith, R. E., Colangelo, L., Wieand, H. S., Begovic, M. & Wolmark, N. Randomized trial of adjuvant therapy in colon carcinoma: 10-year results of NSABP protocol C-01. J. Natl Cancer Inst. 96, 1128–1132 (2004).
Taieb, J., Andre, T. & Auclin, E. Refining adjuvant therapy for non-metastatic colon cancer, new standards and perspectives. Cancer Treat. Rev. 75, 1–11 (2019).
Sargent, D. J. et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 28, 3219–3226 (2010).
Schmoll, H. J. et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J. Clin. Oncol. 33, 3733–3740 (2015).
Andre, T. et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: updated 10-year survival and outcomes according to BRAF mutation and mismatch repair status of the MOSAIC study. J. Clin. Oncol. 33, 4176–4187 (2015).
Allegra, C. J. et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C-08. J. Clin. Oncol. 29, 11–16 (2011).
de Gramont, A. et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol. 13, 1225–1233 (2012).
Kerr, R. S. et al. Adjuvant capecitabine plus bevacizumab versus capecitabine alone in patients with colorectal cancer (QUASAR 2): an open-label, randomised phase 3 trial. Lancet Oncol. 17, 1543–1557 (2016).
Huang, J. et al. Comparison of FOLFIRI with or without cetuximab in patients with resected stage III colon cancer; NCCTG (Alliance) intergroup trial N0147. Clin. Colorectal Cancer 13, 100–109 (2014).
Alberts, S. R. et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 307, 1383–1393 (2012).
Taieb, J. et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol. 15, 862–873 (2014).
Fields, A. L. et al. Adjuvant therapy with the monoclonal antibody Edrecolomab plus fluorouracil-based therapy does not improve overall survival of patients with stage III colon cancer. J. Clin. Oncol. 27, 1941–1947 (2009).
Punt, C. J. et al. Edrecolomab alone or in combination with fluorouracil and folinic acid in the adjuvant treatment of stage III colon cancer: a randomised study. Lancet 360, 671–677 (2002).
Sinicrope, F. A. et al. Randomized trial of FOLFOX alone or combined with atezolizumab as adjuvant therapy for patients with stage III colon cancer and deficient DNA mismatch repair or microsatellite instability (ATOMIC, Alliance A021502). J. Clin. Oncol. 35, TPS3630–TPS3630 (2017).
Lau, D. et al. POLEM: Avelumab plus fluoropyrimidine-based chemotherapy as adjuvant treatment for stage III dMMR or POLE exonuclease domain mutant colon cancer—a phase III randomized study. J. Clin. Oncol. 37, TPS3615–TPS3615 (2019).
Qin, Q. & Wang, L. Neoadjuvant therapy and subsequent treatment in rectal cancer: balance between oncological and functional outcomes. J. Anus Rectum Colon 2, 47–58 (2018).
van Gijn, W. et al. Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer: 12-year follow-up of the multicentre, randomised controlled TME trial. Lancet Oncol. 12, 575–582 (2011).
Sauer, R. et al. Preoperative versus postoperative chemoradiotherapy for rectal cancer. N. Engl. J. Med. 351, 1731–1740 (2004).
Collette, L. et al. Patients with curative resection of cT3-4 rectal cancer after preoperative radiotherapy or radiochemotherapy: does anybody benefit from adjuvant fluorouracil-based chemotherapy? A trial of the European Organisation for Research and Treatment of Cancer Radiation Oncology Group. J. Clin. Oncol. 25, 4379–4386 (2007).
Zhong, X. et al. The efficacy of adding targeted agents to neoadjuvant therapy for locally advanced rectal cancer patients: a meta-analysis. Cancer Med. 7, 565–582 (2018).
Forster, T. et al. Cetuximab in pancreatic cancer therapy: a systematic review and meta-analysis. Oncology 1–8. https://doi.org/10.1159/000502844 (2019).
Kaneko, M. et al. Neoadjuvant imatinib therapy in rectal gastrointestinal stromal tumors. Surg. Today 49, 460–466 (2019).
Ruhstaller, T. et al. Neoadjuvant chemotherapy followed by chemoradiation and surgery with and without cetuximab in patients with resectable esophageal cancer: a randomized, open-label, phase III trial (SAKK 75/08). Ann. Oncol. 29, 1386–1393 (2018).
Brenner, B. et al. The addition of cetuximab to preoperative chemoradiotherapy for locally advanced esophageal squamous cell carcinoma is associated with high rate of long term survival: mature results from a prospective phase Ib/II trial. Radiother. Oncol. 134, 74–80 (2019).
Folprecht, G. et al. Survival of patients with initially unresectable colorectal liver metastases treated with FOLFOX/cetuximab or FOLFIRI/cetuximab in a multidisciplinary concept (CELIM study). Ann. Oncol. 25, 1018–1025 (2014).
Ye, L. C. et al. Randomized controlled trial of cetuximab plus chemotherapy for patients with KRAS wild-type unresectable colorectal liver-limited metastases. J. Clin. Oncol. 31, 1931–1938 (2013).
Carrato, A. et al. First-line panitumumab plus FOLFOX4 or FOLFIRI in colorectal cancer with multiple or unresectable liver metastases: a randomised, phase II trial (PLANET-TTD). Eur. J. Cancer 81, 191–202 (2017).
Primrose, J. et al. Systemic chemotherapy with or without cetuximab in patients with resectable colorectal liver metastasis: the New EPOC randomised controlled trial. Lancet Oncol. 15, 601–611 (2014).
Bisschop, C. et al. Short-course radiotherapy followed by neoadjuvant bevacizumab, capecitabine, and oxaliplatin and subsequent radical treatment in primary stage IV rectal cancer: long-term results of a phase II study. Ann. Surg. Oncol. 24, 2632–2638 (2017).
Chen, H. H. et al. Neoadjuvant therapy of bevacizumab in combination with oxaliplatin and capecitabine (XELOX) for patients with metastatic colorectal cancer with unresectable liver metastases: a phase II, open-label, single-arm, noncomparative trial. Asia Pac. J. Clin. Oncol. 14, 61–68 (2018).
Pietrantonio, F. et al. Perioperative bevacizumab-based triplet chemotherapy in patients with potentially resectable colorectal cancer liver metastases. Clin. Colorectal Cancer 18, 34–43.e36 (2019).
Yasuno, M. et al. mFOLFOX6 plus bevacizumab to treat liver-only metastases of colorectal cancer that are unsuitable for upfront resection (TRICC0808): a multicenter phase II trial comprising the final analysis for survival. Int. J. Clin. Oncol. 24, 516–525 (2019).
Hasegawa, S. et al. A multicenter phase 2 study on the feasibility and efficacy of neoadjuvant chemotherapy without radiotherapy for locally advanced rectal cancer. Ann. Surg. Oncol. 24, 3587–3595 (2017).
Louis, P., Hold, G. L. & Flint, H. J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 12, 661–672 (2014).
Litvak, Y., Byndloss, M. X. & Baumler, A. J. Colonocyte metabolism shapes the gut microbiota. Science 362, https://doi.org/10.1126/science.aat9076 (2018).
Tilg, H., Adolph, T. E., Gerner, R. R. & Moschen, A. R. The intestinal microbiota in colorectal cancer. Cancer Cell 33, 954–964 (2018).
Ma, W. et al. Gut microbiota shapes the efficiency of cancer therapy. Front Microbiol. 10, 1050 (2019).
Wong, S. H. & Yu, J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 16, 690–704 (2019).
Xie, Y. H. et al. Fecal Clostridium symbiosum for noninvasive detection of early and advanced colorectal cancer: test and validation studies. EBioMedicine 25, 32–40 (2017).
Garcia-Gonzalez, A. P. et al. Bacterial metabolism affects the C. elegans response to cancer chemotherapeutics. Cell 169, 431–441.e438 (2017).
Daillere, R. et al. Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 45, 931–943 (2016).
Alexander, J. L. et al. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat. Rev. Gastroenterol. Hepatol. 14, 356–365 (2017).
Yu, T. et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170, 548–563.e516 (2017).
Wallace, B. D. et al. Structure and inhibition of microbiome beta-glucuronidases essential to the alleviation of cancer drug toxicity. Chem. Biol. 22, 1238–1249 (2015).
Jia, W., Xie, G. & Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 15, 111–128 (2018).
Dossa, A. Y. et al. Bile acids regulate intestinal cell proliferation by modulating EGFR and FXR signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G81–G92 (2016).
Schirbel, A. et al. Pro-angiogenic activity of TLRs and NLRs: a novel link between gut microbiota and intestinal angiogenesis. Gastroenterology 144, 613–623.e619 (2013).
Suh, S. H. et al. Gut microbiota regulates lacteal integrity by inducing VEGF-C in intestinal villus macrophages. EMBO Rep. 20, https://doi.org/10.15252/embr.201846927 (2019).
Chen, X. et al. Probiotic yeast inhibits VEGFR signaling and angiogenesis in intestinal inflammation. PLoS ONE 8, e64227 (2013).
Ding, L. et al. Lactobacillus rhamnosus GG ameliorates liver injury and hypoxic hepatitis in rat model of CLP-induced sepsis. Dig. Dis. Sci. 64, 2867–2877 (2019).
Lu, L. et al. Association of antibiotic exposure with the mortality in metastatic colorectal cancer patients treated with bevacizumab-containing chemotherapy: a hospital-based retrospective cohort study. PLoS ONE 14, e0221964 (2019).
Hahn, A. W. et al. Targeting Bacteroides in stool microbiome and response to treatment with first-line VEGF tyrosine kinase inhibitors in metastatic renal-cell carcinoma. Clin. Genitourin. Cancer 16, 365–368 (2018).
Lalani, A. A. et al. Effect of antibiotic use on outcomes with systemic therapies in metastatic renal cell carcinoma. Eur. Urol. Oncol. https://doi.org/10.1016/j.euo.2019.09.001 (2019).
Zitvogel, L., Ma, Y., Raoult, D., Kroemer, G. & Gajewski, T. F. The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 359, 1366–1370 (2018).
Fessler, J., Matson, V. & Gajewski, T. F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 7, 108 (2019).
Elkrief, A., Derosa, L., Zitvogel, L., Kroemer, G. & Routy, B. The intimate relationship between gut microbiota and cancer immunotherapy. Gut Microbes 10, 424–428 (2019).
Sivan, A. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089 (2015).
Anderson, R., Theron, A. J. & Rapoport, B. L. Immunopathogenesis of immune checkpoint inhibitor-related adverse events: roles of the intestinal microbiome and Th17 cells. Front. Immunol. 10, 2254 (2019).
Vetizou, M. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084 (2015).
Chaput, N. et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 28, 1368–1379 (2017).
Derosa, L. et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 29, 1437–1444 (2018).
Elkrief, A. et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology 8, e1568812 (2019).
Gopalakrishnan, V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Matson, V. et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108 (2018).
Tanoue, T. et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605 (2019).
Zhao, S. et al. Antibiotics are associated with attenuated efficacy of anti-PD-1/PD-L1 therapies in Chinese patients with advanced non-small cell lung cancer. Lung Cancer 130, 10–17 (2019).
Pinato, D. J. et al. Association of prior antibiotic treatment with survival and response to immune checkpoint inhibitor therapy in patients with cancer. JAMA Oncol. https://doi.org/10.1001/jamaoncol.2019.2785 (2019).
Routy, B. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018).
Ansaldo, E. et al. Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science 364, 1179–1184 (2019).
Strosberg, J. R. et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur. J. Cancer 48, 997–1003 (2012).
Berlin, J. et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res. 19, 258–267 (2013).
Villalba, M., Evans, S. R., Vidal-Vanaclocha, F. & Calvo, A. Role of TGF-beta in metastatic colon cancer: it is finally time for targeted therapy. Cell Tissue Res. 370, 29–39 (2017).
Ghosh, N., Hossain, U., Mandal, A. & Sil, P. C. The Wnt signaling pathway: a potential therapeutic target against cancer. Ann. NY Acad. Sci. 1443, 54–74 (2019).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Lenz, H. J. et al. Impact of consensus molecular subtype on survival in patients with metastatic colorectal cancer: results from CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 37, 1876–1885 (2019).
Dienstmann, R. et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 17, 79–92 (2017).
Mooi, J. K. et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 29, 2240–2246 (2018).
Fontana, E., Eason, K., Cervantes, A., Salazar, R. & Sadanandam, A. Context matters-consensus molecular subtypes of colorectal cancer as biomarkers for clinical trials. Ann. Oncol. 30, 520–527 (2019).
Roelands, J. et al. Immunogenomic classification of colorectal cancer and therapeutic implications. Int. J. Mol. Sci. 18, https://doi.org/10.3390/ijms18102229 (2017).
Peeters, M. et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J. Clin. Oncol. 28, 4706–4713 (2010).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81421001, 81530072, 81830081 and 81772506). We thank Dr. Yao Zhang for her verification of all table data.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xie, YH., Chen, YX. & Fang, JY. Comprehensive review of targeted therapy for colorectal cancer. Sig Transduct Target Ther 5, 22 (2020). https://doi.org/10.1038/s41392-020-0116-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-0116-z
This article is cited by
-
Circulating vitamin D level before initiating chemotherapy impacts on the time-to-outcome in metastatic colorectal cancer patients: systematic review and meta-analysis
Journal of Translational Medicine (2024)
-
Regulation of metastatic potential by drug repurposing and mitochondrial targeting in colorectal cancer cells
BMC Cancer (2024)
-
Targeted RT study: results on early toxicity of targeted therapies and radiotherapy
Radiation Oncology (2024)
-
Inorganic nanoparticle-based treatment approaches for colorectal cancer: recent advancements and challenges
Journal of Nanobiotechnology (2024)
-
Acoustic waves and smart biomimetic nanoparticles: combination treatment from 2D to 3D colorectal cancer models
Cancer Nanotechnology (2024)
