
Abstract
Since cancer stem cells (CSCs) were first identified in leukemia in 1994, they have been considered promising therapeutic targets for cancer therapy. These cells have self-renewal capacity and differentiation potential and contribute to multiple tumor malignancies, such as recurrence, metastasis, heterogeneity, multidrug resistance, and radiation resistance. The biological activities of CSCs are regulated by several pluripotent transcription factors, such as OCT4, Sox2, Nanog, KLF4, and MYC. In addition, many intracellular signaling pathways, such as Wnt, NF-κB (nuclear factor-κB), Notch, Hedgehog, JAK-STAT (Janus kinase/signal transducers and activators of transcription), PI3K/AKT/mTOR (phosphoinositide 3-kinase/AKT/mammalian target of rapamycin), TGF (transforming growth factor)/SMAD, and PPAR (peroxisome proliferator-activated receptor), as well as extracellular factors, such as vascular niches, hypoxia, tumor-associated macrophages, cancer-associated fibroblasts, cancer-associated mesenchymal stem cells, extracellular matrix, and exosomes, have been shown to be very important regulators of CSCs. Molecules, vaccines, antibodies, and CAR-T (chimeric antigen receptor T cell) cells have been developed to specifically target CSCs, and some of these factors are already undergoing clinical trials. This review summarizes the characterization and identification of CSCs, depicts major factors and pathways that regulate CSC development, and discusses potential targeted therapy for CSCs.
Similar content being viewed by others

Introduction
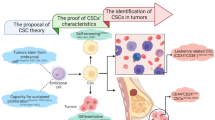
Cancers are chronologic diseases that seriously threaten human life. Many strategies have been developed for cancer treatment, including surgery, radiotherapy, chemotherapy, and targeted therapy. Because of all these treatments, the incidence rate of cancer has been stable in women and has declined slightly in men in the past decade (2006–2015), and the cancer death rate (2007–2016) also declined.1 However, traditional cancer treatment methods are effective only for some malignant tumors.2 The main reasons for the failure of cancer treatment are metastasis, recurrence, heterogeneity, resistance to chemotherapy and radiotherapy, and avoidance of immunological surveillance.3 All these failures could be explained by the characteristics of cancer stem cells (CSCs).4 CSCs can cause cancer relapse, metastasis, multidrug resistance, and radiation resistance through their ability to arrest in the G0 phase, giving rise to new tumors.5 Therefore, CSCs could be considered the most promising targets for cancer treatment.
CSCs were first identified in leukemia and then isolated via CD34+ and CD38− surface marker expression in the 1990s.6,7 CSCs expressing different surface markers, such as CD133, nestin, and CD44, have been subsequently found in many nonsolid and solid tumors, and these cells also form the bulk of the tumor.8,9 CSCs can generate tumors via the self-renewal and differentiation into multiple cellular subtypes.10 The activities of CSCs are controlled by many intracellular and extracellular factors, and these factors can be used as drug targets for cancer treatment.11 To understand the nature of CSCs, we summarized their characteristics, methods for identification and isolation, regulation and current research on targeting CSCs for cancer therapy both in basic research and clinical studies.
The concept of CSCs
Biological characteristics of CSCs
With the deepening of tumor biology research, clinical diagnosis and cancer treatment have significantly improved in recent years. However, the high recurrence rate and high mortality rate are still unresolved and are closely related to the biological characteristics of CSCs. With further understanding of CSC characteristics, research on tumor biology has entered a new era. Therefore, understanding the biological properties of CSCs is of great significance in the diagnosis and treatment of tumors.
CSCs have a strong self-renewal ability, which is the direct cause of tumorigenesis.12 CSCs can symmetrically divide into two CSCs or into one CSC and one daughter cell.13 CSCs expand in a symmetrical splitting manner to excessively increase cell growth, ultimately leading to tumor formation.14 CSCs isolated from original tumor tissue that were transplanted into severe combined immunodeficiency disease (SCID) mice then formed new tumors.15 CSCs and normal stem cells also share some of the same regulatory signaling pathways, such as the Wnt/β-catenin,16 Sonic Hedgehog (Hh),17 and Notch pathways, which are involved in the self-renewal process.18 In addition, other signaling molecules, such as PTEN and the polycomb family, also play important roles in the regulation of CSC growth.19 The regulation of CSC self-renewal is the key link to understanding tumorigenesis. These studies will provide a clear target for cancer treatment.
In addition to their self-renewal ability, CSCs also have the ability to differentiate into different cell types. Bonnet and Dick7 demonstrated in 1997 that CD34+/CD38− leukemia stem cells (LSCs) have the ability to differentiate and proliferate in SCID mice. Brain CSCs isolated from patients are positive for the markers CD133 and nestin, which are the same markers as those of normal neuronal stem cells, but some cells lack surface markers for differentiation.20 Generally, various signaling pathways regulate the self-renewal and differentiation of normal stem cells to promote their proliferation and differentiation in a relatively balanced manner. Once the regulatory balance is destroyed, uncontrolled CSCs ultimately lead to tumorigenesis.21 CSCs also transdifferentiate into other multilineage cells to regulate tumorigenesis.22 Bussolati et al.23 found that renal CSCs differentiated into vascular endothelial cells (ECs) in the bulk of tumors formed in SCID mice after injection of human renal CSCs. Additionally, CSCs that differentiate into vascular ECs and promote angiogenesis have been found in a variety of cancers, such as glioblastoma24 and liver cancer.25
Metastasis refers to the process by which cancer cells travel from the primary site through lymphatic vessels, blood vessels, or the body cavity.26 Since stromal cells (such as granulocytes and macrophages) secrete signaling molecules in the tumor microenvironment (TME), these cells stimulate epithelial–mesenchymal transformation (EMT) to promote the invasion of tumor cells,27 which induce differentiated human mammary epithelial cells to form mammary glands.28 Activation of the RAS/MAPK (mitogen-activated protein kinase) signaling pathway transforms nontumorigenic CD44−/CD24+ breast cancer cells into tumorigenic CD44+/CD24− breast cancer cells.29 A study showed that CSCs are closely related to EMT, and EMT is likely to be the basis for tumor invasion and metastasis. In addition, CD133+/CXCR4+ pancreatic cancer cells30 and CD44+/α2βhi1/CD133+ prostate cancer cells31 are also tumorigenic. Therefore, these studies indicate that CSCs play a crucial role in tumor metastasis and development.
Furthermore, understanding the mechanism of CSC drug resistance is vital for cancer treatment and preventing recurrence.32 CSCs efficiently express ATP-binding cassette (ABC) transporters (including MDR1 (ABCB1), MRP1 (ABCC1), and (ABCG2)), which are multidrug resistance proteins, and these proteins protect leukemia and some solid tumor cells from drug damage and induce drug resistance.33 According to previous studies, aldehyde dehydrogenase (ALDH), a marker in many CSCs,34 eliminates oxidative stress and enhances resistance to chemotherapeutic drugs, such as oxazolidine, taxanes, and platinum drugs.35 ALDH also removes free radicals induced by radiation and stimulates resistance to radiation.35 Inducing DNA damage and apoptosis through chemotherapy and radiotherapy are commonly used cancer treatments. However, CSCs can effectively protect cancer cells from apoptosis by activating DNA repair abilities.36
It is currently believed that CSCs are the key "seeds" for tumor initiation and development, metastasis, and recurrence.37 CSCs have evolved and are highly heterogeneous.38 Breast CSCs have different expression patterns of surface biomarkers, such as CD44+, CD24−, SP, and ALDH+.29,34,39 CD271− or CD271+ melanoma stem cells can form tumors in SCID mice.40 The heterogeneity of CSCs has also been found in other cancers, including glioblastoma,41 prostate cancer,42 and lung cancer.43 The heterogeneity of CSCs is so complex that more effective biomarkers are needed to identify CSCs or distinguish the heterogeneity of CSCs.
Isolation and identification of CSCs
It is known that the proportion of CSCs in tumor tissues is very low and generally accounts for only 0.01–2% of the total tumor mass. In addition, CSCs and normal stem cells also share similar transcription factors and signaling pathways. Therefore, it is more challenging to isolate and identify CSCs. However, an increasing number of techniques and means have emerged.
CSCs have been identified through different biomarkers in human cancers (Table 1). CSCs can be separated by combining specific biomarkers that are mostly located on the cell surface.3 The primary separation techniques are fluorescence-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS).44,45 Since Dick JE first screened CSCs from leukemia by using FACS technology,7 FACS has become the most widely used technique for cell separation. It can perform multibiomarker sorting at one time and has high purity and strong specificity. MACS is a MACS technique. MACS separation is relatively simple, but the technique is cumbersome. Therefore, this method requires high activity of CSCs.44,46 These two methods are effective in separating CSCs from large numbers of cells.
Additionally, there are other ways to separate CSCs from tumors. In 1996, Dr. Goodell observed that after adding Hoechst 33342 to a culture of bone marrow cells, a few cells did not accumulate dyes, and he claimed that these few cells were side population (SP) cells. Therefore, SP cells can be separated by fluorescence screening after the outflow of Hoechst 33342. Recently, SP cells have been identified in various normal tissues and tumor cells. SP cells have high homology, self-renewal and multidirectional differentiation potential.47,48 Some reports have shown that ABCG2 is highly expressed in SP cells.47,49 ABCG2 is highly related to the drug resistance of CSCs and is used as a phenotypic marker for CSCs,50,51 including ovarian cancer,52 AML,53 breast cancer,54 lung cancer,55 nasopharyngeal carcinoma,56 and hepatocellular carcinoma (HCC).57 Montanaro et al.58 explored the optimal concentration of Hoechst 33342 to reduce the toxic effect. The SP sorting method has universal applicability in the separation and identification of CSCs, especially CSCs with unknown cell surface markers, and is an effective method for CSC research.
The colony-forming ability of CSCs is also used for separation and identification.59 After digestion of the tumor tissues into single cells, low-density cell culture can be conducted in serum-free medium containing epithelial growth factor (EGF) and basic fibroblast growth factor (FGF).60 Under this condition, a single CSC will form a cell colony or sphere. Taylor et al.61 successfully isolated CSCs from a variety of neurological tumors by using this colony formation assay. However, the cell purification rate is low, and the CSC specificity is poor in this assay. The in vivo limited dilution assay (LDA) can be used for assessing CSC activity. After low-density transplantation of immune-deficient mice with the limiting dilution method, CSCs can be identified by ELDA software analysis, and this method is affected by cell density and the microenvironment in mice.62
Traditional chemotherapeutic drugs mainly affect cancer cells, but CSCs are mostly arrested in the G0 phase and are relatively static, thus evading the killing effect of chemotherapeutic drugs.63 Hence, the drug-resistant characteristics of CSCs can be used to isolate and identify CSCs.64 Previous studies have shown that radiotherapy combined with hypoxic culture can also be used to enrich CSCs.65 In addition, the separation of CSCs can also be accomplished by physical methods. Hepatoma stem cells can be isolated from rat liver cancer tissue by Percoll density gradient centrifugation; a cell fraction with a high nuclear-to-cytoplasmic ratio is obtained.66 Recently, Rahimi et al.67 used the miR-302 host gene promoter to overexpress neomycin in cancer cells and selected and collected neomycin-resistant CSCs.
Factors regulating CSCs
CSCs can originate from at least four cell types, including normal stem cells, directed group progenitor cells, mature cells, and the fusion of stem cells and other mutant cells.68 Therefore, transformed CSCs from normal cells require multiple gene mutations, epigenetic changes, uncontrolled signaling pathways, and continuous regulation of the microenvironment. It is currently believed that there are many similarities between CSCs and embryonic stem (ES) cells, especially regarding their ability to grow indefinitely and self-renew, signaling pathways and some transcription factors. In addition, CSCs exist in the supporting microenvironment, which is vital for their survival. Moreover, the complex interaction between CSCs and their microenvironment can further regulate CSC growth. This section will discuss the effects of transcription factors, signaling pathways, and the microenvironment on CSC survival, apoptosis, and metastasis.
Major transcription factors in CSCs
Generally, stem cells have at least two common characteristics: the ability to self-renew and the potential to differentiate into one or more specialized cell types.69 Somatic cells can be reprogrammed to become induced pluripotent stem cells by transient ectopic overexpression of the transcription factors Oct4, Sox2, Nanog, KLF4, and MYC.70,71,72 In addition, there are some similarities between CSCs and ES cells. It is reasonable that some embryonic transcription factors can be re-expressed or reactivated in CSCs.69 Therefore, these transcription factors play a very important role in the regulation of CSC growth.
Oct4, a homeodomain transcription factor of the Pit-Oct-Unc family, is recognized as one of the most important transcription factors.73 Recently, Oct4 has emerged as a master regulator that controls pluripotency, self-renewal, and maintenance of stem cells.74 Some studies have reported that Oct4 is highly expressed in CSCs.70,73 High expression of Oct4 is positively correlated with glioma grades75 and promotes self-renewal, chemoresistance, and tumorigenicity of HCC stem cells.76 High expression of Oct4 is also observed in breast CSC-like cells (CD44+/CD24−).77 Cisplatin, etoposide, adriamycin, and paclitaxel γ-irradiation upregulate the expression of Oct4 in lung cancer cells, and CD133+ cells are more resistant to drug treatments than CD133− cells.78 Data also show that Oct4 expression is associated with poor clinical outcome in hormone receptor-positive breast cancer.79 Knockdown of Oct4 also reduces the stemness of germ cell tumors.80 Hence, these studies have proven that Oct4 is a pluripotent factor in CSCs.
Sox2 belongs to the family of high-mobility group transcription factors and plays a significant function in the early development and maintenance of undifferentiated ESCs. It is also one of the key transcription factors in CSCs. Rodriguez-Pinilla et al.81 found that increased expression of Sox2 in basal-like breast cancer may help to characterize poorly differentiated/stem cell phenotypes.82 Hagerstrand et al.82 also found that a high level of Sox2 can induce xenograft glioma. Further studies showed that knockout of Sox2 inhibits glioblastoma cell proliferation and tumorigenicity, which suggests that Sox2 is the basis for maintaining the self-renewal ability of tumor-initiating cells (TICs).83 Sox2 also maintains the self-renewal of TICs in osteosarcomas, and downregulation of Sox2 drastically decreases its transformative characteristics and tumorigenesis ability in vitro. Furthermore, osteosarcoma cells that lose Sox2 cannot form osteospheres and differentiate into mature osteoblasts any longer.84 Sox2 is found in invasive cutaneous squamous cell carcinoma (SCC) and promotes the metastasis of cancer cells.85 These studies suggest that Sox2 promotes self-renewal and tumorigenesis and inhibits differentiation in CSCs.
Nanog, a differentiated homeobox (HOX) domain protein that was first discovered in ESCs, has typical self-renewal and multipotent transcriptional regulatory functions.86 Although Nanog is silenced in normal somatic cells, abnormal expression has been reported in human cancers, such as breast cancer, cervical cancer, brain cancer, colon cancer, head and neck cancer, lung cancer, and gastric cancer.86,87,88,89,90 Compared to levels in benign tissues, Nanog messenger RNA (mRNA) is elevated in malignant tumors. In a number of patients with colorectal cancer (n = 175), high Nanog protein is associated with lymph node positivity and Dukes grade.91 Similarly, overexpression of Nanog in colorectal CSCs promotes colony formation and tumorigenicity in vivo.92 In addition, gastric cancer patients with high Nanog levels have a lower 5-year survival rate.88 The expression level of Nanog is increased in HCC cell lines and primary tumors and is associated with advanced diseases (tumor node metastasis (TNM) stage III/IV).93 Through the study of prostatic cell lines, xenografts and primary tumors, it was found that Nanog short hairpin RNA inhibits the formation of primary prostate cancer cells (PCA) spheres, clonal growth, and tumorigenesis.94 In 43 cases of pancreatic cancer tissue microarray analysis, Kaplan–Meier analysis showed that high expression of Nanog (and Oct4) predicted worse prognosis and was negatively correlated with patient survival.95 These studies indicate that Nanog plays an important role in regulating the self-renewal and proliferation of CSCs.
KLF4 is expressed in many tissues and plays an important role in many different physiological processes. As a bifunctional transcription factor, KLF4 activates or inhibits transcription according to different target genes and utilizing different mechanisms. KLF4 can play an oncogenic or anticancer role, depending on the type of cancer involved. For example, KLF4 is an anticancer factor in the intestinal epithelium and gastric epithelium.96 The expression of KLF4 is downregulated with hypermethylation and loss of heterozygosity in colorectal CSCs and gastric CSCs.97 Downregulation of KLF4 is also found in other cancers, such as non-small-cell lung carcinoma,98 liver cancer,99 leukemia,100 anaplastic meningioma,101 bladder cancer,102 and esophageal cancer.103 Although these data clearly demonstrate that KLF4 plays an anticancer role in those cancers, KLF4 may also be an oncogene, which was demonstrated for the first time in nearly a decade.104 Overexpression of KLF4 in transformed rat renal epithelial cells induces tumorigenesis of laryngeal SCC.105 In addition, depletion of KLF4 inhibits melanoma xenograft growth in vivo.106 High expression of KLF4, an oncogene in human breast CSCs, is correlated with an aggressive phenotype in canine mammary tumors.107 These studies suggest that KLF4 has different functions in different CSCs.
MYC has three family members (C-Myc, N-Myc, and L-Myc, which are encoded by the proto-oncogene family and are essential transcription factors in the DNA-binding proteins of the basic helix–loop–helix (bHLH) superfamily). MYC regulates a large number of protein-coding and noncoding genes and coordinates various biological processes in stem cells, such as cell metabolism, self-renewal, differentiation, and growth.108,109 Although the MYC gene is one of the most commonly activated oncogenes that is involved in the pathogenesis of human cancer, overexpression of MYC alone is surprisingly unable to induce the transformation of normal cells into tumor cells. The overexpression of MYC in normal human cells may be ineffective or highly destructive, resulting in stagnation of proliferation, aging, or apoptosis.110 MYC is usually deregulated in human cancers, plays an important role in maintaining the number of invasive CSCs,111 and is also one of the most effective oncogenes for detecting the cell transformation phenotype in vitro and in vivo. Previous studies have shown that deletion of the tumor suppressor gene p53 and MYC synergizes to induce hepatocyte proliferation and tumorigenesis.112 In addition to p53 deletion, overexpression of Bcl-2 and Bmi-1 and loss of p19ARF also assist MYC in regulating the survival and proliferation of CSCs.113 The expression of the three members of the MYC family is different in different tumors, such as C-MYC in leukemia and tongue SCC stem cells114,115 and N-MYC in small-cell lung cancer, prostate cancer, neuroblastoma, and medulloblastoma.116,117 L-MYC is expressed in hematopoietic malignancies.118 In addition, inactivation of MYC results in HCC stem cells differentiating into hepatocytes and biliary duct cells to form bile duct structures, which might be associated with the loss of the tumor marker α-fetoprotein and increased expression of cytokeratin 8, hepatocyte markers, carcinoembryonic antigen, and the liver stem cell marker cytokeratin 19.119 Studies have also shown that MYC is highly expressed in glioblastoma multiforme stem cells and induces cell proliferation and invasion and inhibits apoptosis.111 Increased copy number of the MYC gene in human and mouse prostate CSCs has also been found.120 These studies indicate that MYC induces tumorigenesis with the help of other factors.
Major signaling pathways in CSCs
Many signaling pathways that contribute to the survival, proliferation, self-renewal, and differentiation properties of normal stem cells are abnormally activated or repressed in tumorigenesis or CSCs. Many endogenous or exogenous genes and microRNAs regulate these complex pathways. These signaling pathways can also induce downstream gene expression, such as cytokines, growth factors, apoptosis genes, antiapoptotic genes, proliferation genes, and metastasis genes in CSCs. These signaling pathways are not a single regulator but interwoven networks of signaling mediators to regulate CSC growth. Therefore, this section will describe how signaling pathways regulate CSC growth.
Wnt signaling pathway in CSCs
Wnts include large protein ligands that affect diverse processes, such as the generation of cell polarity, and cells fate.121 The Wnt pathway is highly complex and evolutionarily conserved and includes 19 Wnt ligands and more than 15 receptors.122 The Wnt signaling pathway can be divided into canonical Wnt signaling (through the FZD-LRP5/6 receptor complex, leading to derepression of β-catenin) and noncanonical Wnt signaling (through FZD receptors and/or ROR1/ROR2/RYK coreceptors, activating PCP, RTK, or Ca2+ signaling cascades).123 In canonical Wnt signaling, in the absence of Wnt ligands (inactive Wnt signaling state, Fig. 1, left), β-catenin is phosphorylated by glycogen synthase kinase 3β (GSK3β), which leads to β-catenin degradation via β-TrCP200 ubiquitination and inhibits translocation of β-catenin from the cytoplasm to the nucleus.124 In contrast, in the presence of Wnt ligands (e.g., Wnt3a and Wnt1), the ligands combine with Fzd receptors and LRP coreceptors (active Wnt signaling, Fig. 1, right). LRP receptors are phosphorylated by GSK3β and CK1α.125 β-Catenin is released from the Axin complex to enter the nucleus. In addition, β-catenin combines with LEF/TCF and enhances the recruitment of histone-modifying coactivators, such as BCL9, Pygo, CBP/p300, and BRG1, to activate transcription. Noncanonical Wnt signaling does not involve β-catenin. During Wnt/PCP signaling, Dvl is activated through binding of Wnt ligands and the ROR-Frizzled receptor.126 Dvl inhibits the binding of the small GTPase Rho and the cytoplasmic protein DAAM1.127 The small GTPases Rac1 and Rho together trigger ROCK (Rho kinase) and JNK (c-Jun N-terminal kinase). This results in cytoskeletal rearrangement and/or transcriptional responses.128 Wnt/Ca2+ signaling is activated by G protein-triggered phospholipase C activity, which results in intracellular calcium flux and downstream calcium-dependent cytoskeletal and/or transcriptional responses.129,130

Wnt/β-catenin pathway in cancer stem cells. The canonical Wnt/β-catenin pathway regulates the pluripotency of CSCs and determines the differentiation fate of CSCs. In the absence of Wnt signaling, β-catenin is bound to the Axin complex, which contains APC and GSK3β, and is phosphorylated, leading to ubiquitination and proteasomal degradation through the β-Trcp pathway. However, the complex (TAZ/YAP), the long noncoding RNA TIC1 and proteins (TRAP1 and TIAM1) regulate the β-Trcp pathway. In the presence of Wnt signaling, the binding of LRP5/6 and Fzd inhibits the activity of the Axin complex and the phosphorylation of β-catenin, which makes β-catenin enter the nucleus, and then bind to TEF/TCF to form a complex, which then recruits cofactors to initiate downstream gene expression. Some proteins (DKK2 (Dickkopf-related protein 2), DACT1, CDH11, GECG, PKM2, EZH2, CD44v6, MYC, and TERT), microRNAs (miR-1246, miR-9, miR-92a, miR-544a, and miR-483-5p), and long noncoding RNAs (lncR-β-catm and lncR-TCF7) regulate the activation of the Wnt/β-catenin pathway in CSCs
Aberrant Wnt signaling is found in many cancers, such as invasive ductal breast carcinomas,131 colorectal cancer,132 papillary thyroid cancer,133 esophageal cancer,134 and colorectal cancer.135 The activation of Wnt signaling is different in different tumors. Some Wnt activation is caused by mutations in Wnt components, such as Axin mutation in gastrointestinal cancers,136 APC mutation in colorectal cancer,137 and β-catenin mutation in gastric cancer and liver cancer.138,139 GSK3 genes are critical for β-catenin regulation; therefore, many researchers expect the occurrence of GSK3 mutations, but GSK3 mutations are not correlated with cancer occurrence. In addition, some genes (pyruvate kinase isozyme M2 (PKM2) in breast cancer140 and telomerase reverse transcriptase (TERT) in prostate cancer141) and microRNAs (miR-164a in colorectal cancer142 and miR-582-3p in non-small-cell lung cancer143) inhibit the activity of APC, Axin, and GSK3β to promote the accumulation of β-catenin in the cytoplasm.
Stem cell signaling pathways and transcriptional circuits are related to the alteration or reactivation of signaling pathways.144 Tumor dormancy is a lag phenomenon in tumor growth. Dormancy may occur during primary tumor formation or in the diffusion of some of the constituent tumor cells. However, primary tumor dormancy and metastatic dormancy seem to be different processes.145 In some cases, cells in the TME produce cytokines, such as Wnt proteins, secreted inhibitors of bone morphogenetic protein (BMP), and Delta, which activate the signaling pathway to maintain the self-renewal ability of CSCs.146 Activation of Wnt induce the transformation of dormant CSCs into active CSCs to promote cell cycle progression through β-catenin, increasing the expression of downstream cyclin D1 and MYC, and MYC also promotes the expression of the polycomb repressor complex 1 component Bmi-1 and induces the combination E2F with cyclin E.147 The extracellular matrix (ECM) protein tenascin C often exists in the gap of stem cells, which supports the cell cycle in breast cancer cells by increasing Wnt signals.148 In addition, aberrant Wnt signaling has also been observed in the self-renewal of CSCs (Fig. 1). Many reports have proven that numerous proto-oncogenes stimulate this process through the Wnt signaling pathway.135 PKM2 catalyzes the last step of glycolysis and plays an essential role in the proliferation of breast CSCs by associating with increased β-catenin levels at regions “−410 to 180 and −2250 to 2000”.140,145,149 Enhancer of zeste homolog 2 (EZH2), a key component of the polycomb PRC2 complex, promotes self-renewal of CSCs by activating β-catenin.150 Moreover, TERT, an RNA-dependent DNA polymerase, acts as a cofactor and forms a complex with β-catenin to activate Wnt downstream targets in prostate CSCs.141 Capillary morphogenesis gene 2 increases the expression of nuclear β-catenin to regulate the self-renewal and tumorigenicity of gastric CSCs,151 and SMYD3, which is located downstream of the Wnt pathway, has a similar effect.152 In addition, long noncoding RNAs and microRNAs also promote self-renewal of CSCs through the Wnt signaling pathway. LncTCF7 recruits the SWI/SNF complex to regulate the expression of the TCF7 promoter in liver CSCs.153 Lnc-β-Catm associates with the methyltransferase EZH2 to suppress the ubiquitination of β-catenin and promote its stability,154 and LncTIC1 interacts with β-catenin and maintains its stability, activating Wnt/β-catenin signaling.155 MicroRNA-1246, miR-19, and miR-92a suppress the expression of AXIN and GSK3β in CSCs.156 MicroRNA-544a downregulates GSK3β in lung CSCs.157 MicroRNA-483-5p upregulates the expression of β-catenin in gastric CSCs.158 In addition, there are still many genes, microRNAs, and noncoding RNAs in CSCs’ self-renewal through the Wnt signaling pathway.
Wnt signaling also plays an important role in the dedifferentiation of CSCs. HOXA5, which is a member of the HOX family, induces the differentiation of colorectal CSCs. However, Wnt indirectly suppresses indirectly via MYC, which is an important direct target of β-catenin/TCF in the intestine.159 PMP22, an integral membrane glycoprotein in myelin in the peripheral nervous system, induces the differentiation of gastric CSCs, but its mRNA level declines with activation of the Wnt/β-catenin pathway.160 Moreover, TRAP1, a component of the HSP90 (heat-shock protein 90) chaperone family, inhibits the differentiation of colorectal carcinoma stem cells by modulating β-catenin ubiquitination and phosphorylation.161 Lgr5, a member of the G protein-coupled receptor (GPCR) family of proteins, is located downstream of the Wnt signaling pathway and restrains the differentiation of esophageal SCC stem cells.162
Wnt signaling also plays an important role in regulating CSC apoptosis. Dickkopf-related protein 2 induces G0/G1 arrest and cell apoptosis by suppressing β-catenin activity in breast CSCs.163 DACT1, a homolog of Dapper that is located at chromosomal region 14q23.1, promotes apoptosis in breast CSCs by antagonizing the Wnt/β-catenin signaling pathway.164 Cadherin-11, a proapoptotic tumor suppressor, reduces the level of active phospho-β-catenin (ser552) to induce apoptosis in colorectal CSCs.165 Epigallocatechin-3-gallate increases apoptosis by degrading β-catenin in lung CSCs.166 The small-molecule inhibitor CWP232228 antagonizes the binding of β-catenin to TCF in the nucleus to induce apoptosis in liver CSCs.167 In addition, temozolomide combined with miR-125b significantly induces apoptosis by targeting the Wnt/β-catenin signaling pathway in glioma stem cells.168
Wnt/β-catenin signaling has been implicated in CSC-mediated metastasis.169 In the cytomembrane, Frizzled8 promotes bone metastasis in prostate CSCs.170 The leucine-rich repeat containing GPCR4 (LGR4, or GPR48), together with its family members LGR5/6, binds to R-spondins 1–4 and leads to Wnt3A potentiation, activating Wnt signaling in breast CSCs.171,172 Increased levels of CD44v6 mRNA in human pancreatic CSCs, lung CSCs, and colon CSCs promote migration and metastasis through the activation of β-catenin.173,174,175 In the cytoplasm, TAZ/YAP interacts directly with β-catenin and restricts β-catenin degradation,176 but TIAM1 antagonizes TAZ/YAP accumulation and translocation from the cytoplasm to the nucleus.177 Moreover, CDH11 inhibits the migration and invasion of colorectal CSCs by inhibiting Wnt/β-catenin and AKT/RhoA signaling.165 Wnt signaling decreases the expression of HOXA5 to promote CSC metastasis.159 These data suggest that amplified Wnt signaling is important for self-renewal, dedifferentiation, apoptosis inhibition, and metastasis of CSCs.
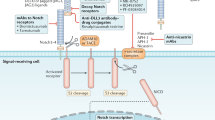
Notch signaling pathway in CSCs
The Notch signaling pathway consists of the Notch receptor, Notch ligand (DSL protein), CSL (CBF-1, suppressor of hairless, Lag), DNA-binding protein, other effectors, and Notch regulatory molecules. In 1917, studies discovered the Notch gene in a mutant Drosophila. Mammals have four Notch receptors (Notch1–4) and five Notch ligands (Delta-like 1, 3, and 4, Jagged 1, and Jagged 2).178 Notch and DSL ligands are transmembrane proteins that mediate communication between neighboring cells. Under physiological conditions, the ligand binds to a Notch receptor that is expressed on neighboring cells in a juxtacrine manner, thereby triggering proteolytic cleavage of the intracellular domain (ICD) of Notch and its translocation into the nucleus to bind to the transcription factor CSL, forming the NICD/CSL transcriptional activation complex, which activates target genes of the bHLH transcription inhibitor family, such as HES, HEY, and HERP.179,180
The Notch pathway regulates cancer cells in many tumors, such as glioblastoma, leukemia, and those of the breast, pancreas, colon, and lung, among others.181 Different tumors and tumor subtypes express different Notch ligands and receptors. Therefore, Notch is known to function as both an oncogene and a suppressive gene. As an oncogene, Notch is overexpressed in gastric cancer,182 breast cancer,183 colon cancer,184 and pancreatic cancer. In contrast, Notch expression is downregulated in prostate cancer,185 skin cancer,186 non-small-cell lung cancer,187 liver cancer,188 and some breast cancers.189 Whether Notch acts as an oncogene or a tumor suppressor gene is determined by the microenvironment.190 Moreover, post-translational modifications of Notch receptors change their affinity for ligands and their intracellular half-lives.191
Many studies on the Notch pathway in CSCs have shown that activation of Notch promotes cell survival, self-renewal, and metastasis and inhibits apoptosis. Aberrant Notch signaling (Notch1 and Notch4) promotes self-renewal and metastasis of breast and HCC stem cells.192,193 However, microRNA-34a downregulates Notch1.194 Similarly, abundant Delta-like ligand 4 (DLL4) also promotes tumor angiogenesis and metastasis in gastric CSCs.195 Delta-like 1 activation of Notch1 signaling requires the assistance of the actin-related protein 2/3 complex to maintain the stem cell phenotype of glioma-initiating cells.196 Additionally, some intracellular genes also regulate the Notch signaling pathway. For example, MAP17 (DD96, PDZKIP1), a nonglycosylated membrane-associated protein, is located on the plasma membrane and the Golgi apparatus. MAP17 interacts with NUMB through the PDZ-binding domain to activate the Notch pathway in cervical CSCs.197 Inducible nitric oxide synthase promotes the self-renewal capacity of CD24+CD133+ liver CSCs through TACE/ADAM17 activation to regulate Notch1 signaling.198 Moreover, tumor necrosis factor-α (TNFα) enhances the CSC-like phenotype by activating Notch1 signaling in oral SCC cells.199 Overexpression of PER3 decreases the expression of Notch1 and Jagged 1 in colorectal CSCs.200 In addition, KLF4 and BMP4 also increase Notch1 and Jagged 1 in breast CSCs to regulate cell migration and invasion.201,202 BRCA1 is a key regulator of breast cancer cell differentiation; however, it is localized to a conserved intronic enhancer region within the Notch ligand Jagged 1 gene to maintain the stemness of breast CSCs.203 Similarly, increased Gli3 also promotes cell proliferation and invasion in oral SCC by increasing Notch2.204 Hypoxia/hypoxia-inducible factor (HIF)-induced migration and invasion is a well-known phenomenon that has been reported in numerous CSCs.205 Notch1 can induce the migration and invasion of ovarian CSCs in the absence of hypoxia.206 Hypoxia-induced Jagged 2 activation enhances cell invasion of breast CSCs207 and lung CSCs.208 Moreover, HIF-1α/2α regulates self-renewal and maintenance of glioblastoma stem cells.209 In addition, increased miR-200b-3p decreases Notch signaling to promote pancreatic CSCs to become asymmetric.210 MiR-26a directly targets Jagged 1 to inhibit osteosarcoma CSC proliferation.211 These studies indicate that Notch plays an important role in regulating the self-renewal, growth, and metastasis of CSCs.
Hh signaling pathway in CSCs
The Hh signaling pathway consists of ligands and receptors. The Hh signaling network is very complex, including extracellular Hh ligands, the transmembrane protein receptor PTCH, the transmembrane protein SMO, intermediate transduction molecules, and the downstream molecule GLI.212 The components of the Hh signaling pathway play different roles. The membrane protein SMO plays a positive regulatory role, while the transmembrane protein PTCH plays a negative regulatory role. PTCH has two subtypes, PTCH1 and PTCH2,213 and there is 73% homology between the two subtypes. GLI, an effector protein, has three subtypes, Gli1, Gli2, and Gli3, in vertebrates,214 and these effector proteins have different functions. Gli1 strongly activates transcription, while Gli3 inhibits transcription.215 Gli2 has dual functions of activating and inhibiting transcription but mainly functions as a transcriptional activator.216,217 Numerous studies have confirmed that Hh signaling is involved in embryonic development and the formation of the nervous system, skeleton, limbs, lung, heart, and gut.218 As an extracellular signaling pathway, in the presence of ligand signals, Hh ligands bind to PTCH receptors on target cell membranes and initiate a series of intracellular signal transduction processes.219 When there is no ligand signal, the transmembrane receptor PTCH on the target cell membrane binds to SMO and inhibits SMO activity, which prevents signaling.220 When the Hh ligand is present, it binds to PTCH, which changes the spatial conformation of PTCH, removing the inhibition of SMO activating the transcription factor GLI and inducing it to enter the cell nucleus, where GLI regulates cell growth, proliferation, and differentiation.221
Studies have confirmed that abnormal activation of the Hh signaling pathway can be found in human cancers,222 such as breast cancer,223 lung cancer,224 bladder cancer,225 pancreatic cancer,226 chondrosarcoma,227 rhabdomyosarcoma,228 neuroblastoma,229 medulloblastoma,230 and gastric cancer.231 However, activation of Hh signaling is different in different tumors. Gorlin syndrome (basal cell nevus syndrome), an autosomal dominant condition, is associated with germline loss of the PTCH1 gene. This condition is very common in basal cell carcinoma, rhabdomyosarcoma, and medulloblastoma.232,233 Other Hh pathway components are also mutated in human cancers, such as Gli1 and Gli3 mutations in pancreatic adenocarcinoma, Gli1 gene amplification in glioblastoma, and SUFU (suppressor of fused) mutations in medulloblastoma.234,235 In addition, other genes also regulate the Hh signaling pathway. Speckle-type POZ protein, an E3 ubiquitin ligase adaptor, inhibits Hh signaling by accelerating Gli2 degradation in gastric cancer.236
Hh signaling plays distinct functions in different types of cancer.237 During tumor development, Hh signaling has three major roles: driving tumor development, promoting tumor growth, and regulating residual cancer cells after therapy. Based on these functions, the aberrant Hh pathway plays a causal role in CSCs238,239 (Fig. 2). The expression level of Hh signaling components is relatively high in CSCs. For example, Hh signaling promotes the maintenance, proliferation, self-renewal, and tumorigenicity of lung adenocarcinoma stem cells.240 In CD133+ glioma stem cells, SMO, GLI, and PTCH promote cell proliferation, self-renewal, migration, and invasion. The expression of Gli1, PTCH1, and PTCH2 is regulated by histone deacetylase 6.241 USP48 activates Gli-dependent transcription by stabilizing the Gli1 protein in glioma stem cells.242 The protein kinase CK2α enhances Gli1 expression and its transcriptional activity in lung CSCs.243 WIP1 (PPM1D), a nuclear Ser/Thr phosphatase, also enhances the function of Gli1 by increasing its transcriptional activity, protein stability, and nuclear localization in breast CSCs and medulloblastomas.244,245 F-box and leucine-rich repeat protein 17 mediates the release of Gli1 from SUFU for proper Hh signal transduction in medulloblastoma stem cells.246 Moreover, retinoic acid receptor α2 (RARα2) upregulates the expression of SMO and Gli1 in CD138+ multiple myeloma stem cells.247 PRKCI, which is regulated by miR-219 in tongue SCC,248 has a similar function as RARα2 in maintaining a stem-like phenotype in lung SCC cells.249 Interleukin-27 (IL-27) and IL-6 activate Hh signaling in CD133+ non-small-cell lung CSCs.250 During self-renewal and maintenance of stemness of BCMab1+CD44+ bladder CSCs, glycotransferase GALNT1-mediated glycosylation significantly activates Sonic Hh signaling by upregulating Gli1.251

Hedgehog signaling pathway in cancer stem cells. The Hedgehog pathway plays a key role in stem maintenance, self-renewal, and regeneration of CSCs. The secreted Hh protein acts in a concentration- and time-dependent manner to initiate a series of cell responses, such as cell survival, proliferation, and differentiation. After receiving the Shh signal, the transmembrane protein receptor PTCH relieves the inhibition of the transmembrane protein SMO, which induces Gli1/2 to detach from SUFU and enter the nucleus to regulate downstream gene transcription. During activation of the Hh pathway, some proteins (IL-6, IL-27, Fbxl17 (F-box and leucine-rich repeat protein 17), PPKCI, RARα2, RUXN3, SCUBE2, HDAC6 (histone deacetylase 6), USP48, CK2α, WIP1, GALNT1, VASH2 (Vasohibin 2), BCL6, FOXC1 (forkhead box C1), and p65), microRNAs (miR-324-5p, miR-122, and miR-326), and the long noncoding RNA HDAC2 are involved in the Hedgehog pathway to affect CSC growth
Furthermore, p63, a master regulator of normal epithelial stem cell maintenance, regulates the expression of Shh, Gli2, and PTCH1 by directly binding to their gene regulatory regions, which eventually contributes to the activation of Hh signaling in mammary CSCs.252 The N-terminal domain of forkhead box C1 binds directly to an internal region (amino acids (aa) 898–1168) of Gli2 to enhance transcriptional activation of Gli2 and determines the stem cell phenotype in breast CSCs.253 Through recruitment of the deubiquitinating enzyme ATXN3, tetraspanin-8 interacts with PTCH1 and inhibits the degradation of the SHH/PTCH1 complex. In addition, long noncoding microRNAs also activate Hh signaling. For example, lncHDAC2 promotes the self-renewal of liver CSCs by recruiting the NuRD complex onto the promoter of the PTCH1 gene to suppress its expression.254 In addition, the TME is crucial for the survival of CSCs. Consequently, breast CSCs secrete Shh, which upregulates cancer-associated fibroblasts (CAFs). Subsequently, CAFs secrete factors that promote the expansion and self-renewal of breast CSCs.255 Hh signaling also promotes self-renewal and metastasis of CSCs by upregulating the expression of related downstream markers of CSCs, such as Bmi-1, Wnt2, ALDH1, CD44, CCND1, Twist1, C-MYC, Nanog, Oct4, PDGFRα (platelet-derived factor receptor-α), Snail, Jagged 1, and C-MET.231,247,256,257,258,259,260,261,262,263,264
Some proto-oncogenes and suppressor genes also directly or indirectly regulate Hh signaling in the proliferation and migration of CSCs. The signal peptide CUB EGF-like domain-containing protein 2 (SCUBE2), a member of the SCUBE family of proteins, inhibits cell proliferation and migration in glioma stem cells by downregulating Hh signaling.265 BCL6, a transcriptional repressor and lymphoma oncoprotein, directly represses the Sonic Hh effectors Gli1 and Gli2 in medulloblastoma stem cells.266 The transcription factor RUNX3 suppresses metastasis and the stemness of colorectal CSCs by promoting ubiquitination of Gli1 at the intracellular level.267 Vasohibin 2 suppresses Smo, Gli1, and Gli2 expression in pancreatic CSCs.268 β-Catenin stably increases its physical interaction with Gli1, resulting in Gli1 degradation in medulloblastoma stem cells.269 In addition, microRNAs also target Hh signaling components to regulate CSC proliferation. For example, miR-324-5p significantly decreases SMO and Gli1 in myeloma stem cells.270 Mir-326 directly downregulates SMO and Gli2 in medulloblastoma stem cells.271 MiR-326 downregulates SMO in glioma stem cells.272 Mir-122 targets Shh and Gli1 in lung CSCs.273 These data demonstrate that amplified Hh signaling is important for the self-renewal, growth, and metastasis of CSCs.
NF-κB signaling pathway in CSCs
Nuclear factor-κB (NF-κB), a rapidly inducible transcription factor,274 consists of five different proteins (p65, RelB, c-Rel, NF-κB1, and NF-κB2). The main physiological function of NF-κB is the p50-p65 dimer.275,276,277 The primary mode of NF-κB regulation occurs at the level of subcellular localization. In the activation stage, transcription factor complexes must translocate from the cytoplasm to the nucleus.278 The activity of the complexes is regulated by two major pathways (canonical NF-κB signaling and noncanonical NF-κB signaling). In the canonical NF-κB activation pathway, activation occurs through the binding of ligands, such as bacterial cell components, IL-1β, TNF-α, or lipopolysaccharides, to their respective receptors, such as Toll-like receptors, TNF receptor (TNFR), IL-1 receptor (IL-1R), and antigen receptors.279 Stimulation of these receptors leads to the phosphorylation and activation of IκB kinase (IKK) proteins, subsequently initiating the phosphorylation of IκB proteins.276 The alternative pathway of NF-κB activation is termed the noncanonical pathway. The noncanonical pathway receptor originates from different classes, such as CD40, receptor activator for NF-κB, B cell activation factor, TNFR2 and Fn14, and lymphotoxin β-receptor.280 This pathway leads to activation of NF-κB by inducing the kinase (NIK), which then phosphorylates and predominantly activates IKK1. The activity of the latter enzyme induces the phosphorylation of p100 to generate p52.281
The NF-κB pathway plays an important role in regulating immune and inflammatory responses. In addition, the NF-κB pathway is involved in cellular survival, proliferation, and differentiation.276 The process of tumor development and progression produces cytokines, growth, and angiogenic factors and proteases to activate NF‐κB signaling.282 Inflammation has been recognized as a hallmark of cancer.283 Overactivation of NF-κB signaling has been reported in gastrointestinal, genitourinary, gynecological, and head and neck cancers, breast tumors, multiple myeloma, and blood cancers.278,284,285,286 However, direct or altered molecular mutations in NF-κB have rarely been reported in human cancers.287 Based on recent studies, NF-κB regulates many genes and is implicated in cell survival, proliferation, metastasis, and tumorigenesis of cancer.288 NF-κB activation also directly or indirectly enhances the expression of key angiogenesis factors and adhesion molecules, such as IL-8, vascular endothelial growth factor (VEGF), and growth-regulated oncogene 1.289
The NF-κB pathway has an essential connection regulating inflammation, self-renewal, or maintenance and metastasis of CSCs (Fig. 3). CD44+ cells promote self-renewal, metastasis, and maintenance of ovarian CSCs by increasing the expression of RelA, RelB, and IKKα and mediating nuclear activation of p50/RelA (p50/p65) dimer.290 High levels of NIK induce activation of the noncanonical NF-κB pathway to regulate the self-renewal and metastasis of breast CSCs.291 Moreover, stromal cell-derived factor-1 (SDF-1) also has the same effect by regulating the translocation of p65 from the cytoplasm to the nucleus.292 The inflammatory mediator prostaglandin E2 (PGE2) contributes to tumor formation, maintenance, and metastasis by activating NF-κB via EP4-PI3K (phosphoinositide 3-kinase) and EP4-MAPK pathways in colorectal CSCs.293 Chemokines, low-molecular-weight proinflammatory cytokines, are important mediators of cell proliferation, metastasis, and apoptosis.294 C-C chemokine receptor 7 interacts with its ligand chemokine ligand 21 to inhibit apoptosis and induce survival and migration in CD133+ pancreatic cancer stem-like cells by increasing the expression of extracellular signal-regulated kinase 1/2 (Erk1/2) and p65.295 Furthermore, B cell-specific Moloney murine leukemia virus integration site 1 (Bmi-1) also enhances the p65 protein in gastric CSCs.296 MicroRNAs also play an important role in promoting the proliferation of CSCs. Mir-221/222 promotes self-renewal, migration, and invasion in breast CSCs by inhibiting the expression of PTEN and then inducing the phosphorylation of AKT, resulting in elevated p65, p-p65, and COX2.297

NF-κB signaling pathway in cancer stem cells NF-κB proteins are involved in the dimerization of transcription factors, regulate gene expression, and affect various CSC biological processes, including inflammation, stress responses, growth, and development of CSCs. The main physiological function of NF-κB is the p50-p65 dimer. The active p50-p65 dimer is further activated by post-translational modification (phosphorylation, acetylation, or glycosylation) and transported into the nucleus, which induces the expression of target genes in combination with other transcription factors. Some proteins (CD44, CD146, TNFRSF19, Bmi-1, FOXP3, and SDF-1) and microRNAs (miR-221 and miR-222) directly regulate the NF-κB pathway. In addition, some proteins (PGE2, GIT-1 (G protein-coupled receptor kinase-interacting protein 1), C-C chemokine receptor 7 (CCR7), and TGF-β) and miR-491 indirectly affect the NF-κB pathway via the ERK and MAPK pathways in CSCs
In addition, other transcription factors also inhibit self-renewal and metastasis in CSCs by the NF-κB pathway. Increased expression of FOXP3 has been identified in different cancers.298 FOXP3 interacts with NF-κB, inhibits the expression of COX2 located downstream of NF-κB, and affects self-renewal and metastasis in colorectal CSCs.299 Overexpression of miR-491 blocks the activation of NF-κB in liver CSCs by targeting G protein-coupled receptor kinase-interacting protein 1, which inhibits ERKs.300 Moreover, some drugs inhibit cell proliferation and metastasis of CSCs by the NF-κB pathway. Disulfiram, an anti-alcoholism drug, inhibits tumor growth factor-β (TGF-β)-induced metastasis via the ERK/NF-κB/Snail pathway in breast CSCs.301 Sulforaphane preferentially inhibits self-renewal in triple-negative breast CSCs by inhibiting NF-κB p65 subunit translocation and downregulating p52 and its transcriptional activity.302 Curcumin regulates the proliferation, metastasis, and apoptosis of HCC stem cells by inhibiting the NF-κB pathway.303 These data demonstrate that amplified NF-κB signaling is important for regulating apoptosis, proliferation, and metastasis of CSCs.
JAK-STAT signaling pathway
The Janus kinase/signal transducers and activators of transcription (JAK-STAT) signaling pathway is a signal transduction pathway that is stimulated by cytokines. This pathway is involved in many important biological processes, such as cell proliferation, differentiation, apoptosis, and immune regulation. Compared with the complexity of other signaling pathways, this signaling pathway is relatively simple. There are three components: the tyrosine kinase-related receptor, the tyrosine kinase JAK, and the transcription factor STAT.304 Many cytokines and growth factors transmit signals through the JAK-STAT signaling pathway, including interleukin-2-7, granulocyte/macrophage colony-stimulating factor, growth hormone, EGF, PDGF, and interferon.305 These cytokines and growth factors have corresponding receptors on the cell membrane. The common characteristic of these receptors is that the receptor itself does not have kinase activity, but there is a binding site for the tyrosine kinase JAK in the cells. After binding with ligands, tyrosine residues of various target proteins are phosphorylated through JAK activation to achieve signal transduction from the extracellular to intracellular space. The JAK protein family consists of four members: JAK1, JAK2, JAK3, and Tyk2.306 JAK proteins have seven JAK homology (JH) domains in their structures. The JH1 domain is the kinase domain, the JH2 domain is the "pseudo" kinase domain, and JH6 and JH7 are the receptor binding domains.307 STAT is called "signal transducer and activator of transcription". As the name implies, STAT plays a key role in signal transduction and transcriptional activation. At present, seven members of the STAT family (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, STAT6) have been identified. The structure of STAT protein can be divided into the following functional regions: N-terminal conserved sequence, DNA-binding region, SH3 domain, SH2 domain, and C-terminal transcriptional activation region.308 Generally, many cytokines and growth factors integrate with tyrosine kinase-related receptors. After receiving the signal from the upstream receptor molecule, JAK is quickly recruited to and activates the receptor, resulting in JAK activation to catalyze tyrosine phosphorylation of the receptor. The phosphorylated tyrosine on the receptor molecule, which is a signaling molecule, can bind with the SH2 site of STAT.309 When STAT binds to the receptor, tyrosine phosphorylation of STAT also occurs, which forms a dimer and enters the nucleus.310 As an active transcription factor, the STAT dimer directly affects the expression of related genes and then changes the proliferation or differentiation of target cells.311
Constitutive activation of JAKs and STATs was first recognized as being associated with malignancy in the 1990s.312 Based on current studies, JAK2 mutation and abnormal activation of STAT3 are prone to occur in many tumors.313 Mutations in JAK2 have been reported in the majority of patients with myeloproliferative neoplasms,314 such as polycythemia vera, myelofibrosis, and thrombocythemia.315,316 These disorders are caused by the overexpansion of hematopoietic precursors, which are often clonal and can result in leukemia.314 Several lines of evidence show that constitutive activation of JAK2 and STAT3 in the absence of any stimulating ligand occurs in polycythemia vera.317,318 Moreover, studies have also found aberrant activation of STATs in human cancers, such as head and neck cancer,319 endometrial cancer,320 breast cancer, diffuse large B cell lymphoma,321 HCC,322 colorectal cancer, glioma,323 and colon cancer.324 Furthermore, aberrant STAT5 signaling has been found in the pathogenesis of hematologic and solid organ malignancies.325,326
The JAK/STAT pathway is evolutionarily conserved. This pathway promotes the survival, self-renewal, hematopoiesis, and neurogenesis of ESCs.327 This pathway is also activated in CSCs. The persistent activation of STAT3 significantly promotes cell survival and the maintenance of stemness in breast CSCs.328 IL-10 induces cell self-renewal, migration, and invasion in non-small-cell lung CSCs.329 IL-6 activates the JAK1/STAT3 pathway in ALDHhigh CD126+ endometrial CSCs.320 Furthermore, IL-6 also induces the conversion of nonstem cancer cells into cancer stem-like cells in breast cancer by the activating downstream Oct4 gene.330 Oct4 also activates the JAK1/STAT6 pathway in ovarian CSCs.331 In CD44+CD24− breast and colorectal CSCs, erythropoietin, and IL-6 activate the JAK2/STAT3 pathway.332,333,334 Retinol-binding protein 4 activates JAK2/STAT3 signaling by its STRA6 receptor in colon CSCs.319 HIF-1α enhances the self-renewal of glioma stem-like cells by the JAK1/STAT3 pathway.335 AJUBA is a scaffold protein that participates in the regulation of cell adhesion, differentiation, proliferation, and migration and promotes the survival and proliferation of colorectal CSCs via the JAK1/STAT1 pathway.336
Moreover, microRNAs are also involved in activating JAK/STAT signaling by inhibiting negative regulatory factors of JAK2/STAT3. For example, miR-500a-3p targets multiple negative regulators of the JAK2/STAT3 signaling pathway, such as SOCS2, SOCS4, and PTPN, in HCC stem cells, leading to constitutive activation of STAT3 signaling.322 MiR-30 targets SOCS3 in glioma stem cells.337 Mir-93 downregulates the expression of JAK1 and STAT3 to induce the differentiation of breast CSCs. Mir-218 negatively regulates the IL-6 receptor and JAK3 gene expression in lung CSCs.338 In addition, some endogenous or exogenous genes inhibit JAK/STAT signaling in CSCs. Von Hippel–Lindau suppresses the tumorigenicity and self-renewal ability of glioma stem cells by inhibiting JAK2/STAT3.323 Although there are few studies on JAK in CSCs, there is a role for JAK/STAT signaling in the survival, self-renewal, and metastasis of CSCs.
TGF/SMAD signaling pathway in CSCs
The TGF-β signaling pathway is involved in many cellular processes associated with organism and embryo development, including cell proliferation, differentiation, apoptosis, and homeostasis. Although the TGF-β signaling pathway regulates a wide range of cellular processes, its structure is relatively simple. TGF-β superfamily ligands bind to a type II receptor, which recruits a type I receptor and phosphorylates it. This type I receptor phosphorylates receptor-regulated Smads (R-Smads), which bind to common pathway Smad (co-Smad). The R-Smad/co-Smad complex acts as a transcription factor and accumulates in the nucleus to regulate the expression of target genes. TGF-β superfamily ligands include BMPs, growth and differentiation factors (GDFs), anti-Mullerian hormone (AMH), activin Nodal, and TGF-β.339 These ligands can be divided into two groups, TGF-β/activin and BMP/GDF. The TGF-β/activin group includes TGF-β, activin, and Nodal, and the BMP/GDF group includes BMP, GDF, and AMH ligands.340 Based on Smad structure and functions, Smad proteins can be divided into three subfamilies: receptor-activated or pathway-restricted Smad (R-Smads), Co-Smad, and inhibitory Smad (I-Smads), which includes at least nine Smad proteins.341,342 R-Smads are activated by type I receptors and form transient complexes with these receptors. There are two types of Smad complexes: AR-Smads are activated by activin TGF-β, including Smad2 and Smad3, and BR-Smads are activated by BMP, including Smad1, Smad5, Smad8, and Smad9. Co-Smad, including Smad4, is a common medium in various TGF-β signal transduction processes. I-Smads, including Smad6 and Smad7, bind to activated type I receptors and inhibit or regulate signal transduction of the TGF-β family.343
Many studies have shown that activation of TGF/Smad signaling also occurs in human cancers. Dkk-3, a secreted protein, inhibits TGF-β-induced expression of matrix metallopeptidase 9 (MMP9) and MMP13 to prevent migration and invasion of prostate cancer.344 Cancer upregulated gene 2 promotes cellular transformation and stemness, which is mediated by nuclear NPM1 protein and TGF-β signaling in lung cancer.345 TGF/Smad also plays an important role in the cell proliferation of CSCs. Cyclin D1 interacts with and activates Smad2/3 and Smad4, promoting cyclin D1-Smad2/3-Smad4 signaling to regulate self-renewal of liver CSCs.346 CD51 binds to TGF-β receptors to upregulate TGF-β/Smad signaling in colorectal CSCs.341 Upregulation of TGF-β1 induces the expression of smad4, p-Smad2/3, and CD133 in liver CSCs.347 TGF-β1 also upregulates the expression of PFKFB3 through activation of the p38 MAPK and PI3K/Akt signaling pathways to regulate glycolysis in glioma stem cells.348 Furthermore, silencing ShcA expression also induces activation of STAT4 in breast CSCs.349 Moreover, miR-148a inhibits the TGF-β/Smad2 signaling pathway in HCC stem cells.350 Smad7, a newly discovered target gene of miR-106b, is an inhibitor of TGF-β/Smad signaling, which inhibits sphere formation of gastric cancer stem-like cells.351 Although there are few studies on the TGF/Smad signaling pathway in CSCs, this pathway still plays a very important role.
PI3K/AKT/mTOR signaling pathway in CSCs
Phosphatidylinositol-3-kinase (PI3K) is an intracellular phosphatidylinositol kinase.352 It consists of the regulatory subunit p85 and catalytic subunit p110, which have serine/threonine (Ser/Thr) kinase and phosphatidylinositol kinase activities.353 AKT is a serine/threonine kinase that is expressed as three isoforms: AKT1, AKT2, and AKT3.354 AKT proteins are crucial effectors of PI3K and are directly activated in response to PI3K. One of the key downstream target genes of AKT is the mammalian target of rapamycin (mTOR) complex, which is a conserved serine/threonine kinase. It forms two distinct multiprotein complexes: mTORC1 and mTORC2.355 mTORC1 consists of mTOR, raptor, mLST8, and two negative regulators, PRAS40 and DEPTOR.356,357 mTORC2 phosphorylates AKT at serine residue 473, which leads to full AKT activation.358
Studies show that mutations in PTEN lead to the inhibition of PI3K/mTOR signaling in glioblastoma multiforme. However, deletion of PTEN in neural stem cells leads to a neoplastic phenotype that includes cell growth promotion, resistance to cell apoptosis, and increased migratory and invasive properties in vivo.359 Inactivation of PTEN and activation of protein kinase B have been found in other solid tumors, such as myeloproliferative neoplasia and leukemia.360 Therefore, the PI3K/mTOR signaling pathway is vital for cell proliferation and survival. Abnormal activation of PI3K/mTOR signaling is found in some cancers, such as non-small-cell lung cancer,361 breast cancer,362 prostate cancer,363 Burkitt lymphoma,364 esophageal adenocarcinoma,365 and colorectal cancer.366
Although PI3K/AKT/mTOR has been extensively studied in cancers, there are few studies in CSCs.358 PI3K/Akt/mTOR signaling is involved in ovarian cancer cell proliferation and the epithelial–mesenchymal transition.367 This signaling activation also enhances the migration and invasion of prostate and pancreatic CSCs.368,369 Downregulation of PTEN induces PI3K activation to promote survival, maintenance of stemness, and tumorigenicity of CD133+/CD44+ prostate cancer stem-like cell populations.370 PI3K activation promotes cell proliferation, migration, and invasion in ALDH+CD44high head and neck squamous CSCs.371 Activation of mTOR promotes the survival and proliferation of breast CSCs and nasopharyngeal carcinoma stem cells.328,372 mTORC1 activation also increases aldehyde dehydrogenase 1 (ALDH1) activity in colorectal CSCs.373 Activation of mTORC2 upregulates the expression of the hepatic CSC marker EpCAM (epithelial cellular adhesion molecule) and tumorigenicity in hepatocellular CSCs.374 Nucleotide-binding domain and leucine-rich repeats (NLRs) belong to a large family of cytoplasmic sensors. NLRC3 (also known as CLR16.2 or NOD3) is associated with PI3Ks and blocks activation of PI3K-dependent kinase AKT in colorectal CSCs.375
In addition, some studies have shown that the mTOR signaling pathway is closely related to the metabolism of CSCs. For example, low folate (LF) stress reprograms metabolic signals through the activated mTOR signaling pathway, promoting the metastasis and tumorigenicity of lung cancer stem-like cells.376 However, matcha green tea (MGT), an inhibitor of mTOR, inhibits the proliferation of breast CSCs by targeting mitochondrial metabolism, glycolysis, and multiple cell signaling pathways.377 A link between the PI3K/Akt/mTOR pathway and CSCs is clearly evident.
PPAR signaling pathways in CSCs
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear transcription factors that were first cloned from mouse liver by Isseman and Green.378 PPARs are also members of the ligand-activated transcription factor superfamily of nuclear hormone receptors that are associated with retinoic acid, steroids and thyroid hormone receptors. PPARs act as fat sensors to regulate the transcription of lipid metabolic enzymes.379 At present, three subtypes, PPARα, PPARβ, and PPARγ (encoded by the PPARA, PPARD, and PPARG genes, respectively), have been found.380 PPARα is highly expressed in hepatocytes, cardiac myocytes, intestinal cells, and renal proximal convoluted tubule cells. PPARγ is abundantly expressed in adipose tissue, vascular parietal cells (such as monocytes/macrophages, ECs, and smooth muscle cells), and myocardial cells.381 PPARβ is expressed in almost all tissues of the body, and its expression level is higher than that of PPARα or PPARγ.382 In recent years, studies have found that PPARs are closely related to energy (lipid and sugar) metabolism, cell differentiation, proliferation, apoptosis, and inflammatory reactions.383 PPARs can exert positive or negative effects to regulate target gene expression by binding to a specific peroxisome located at each gene regulatory site and a proliferative response element.378 Their natural ligands are unsaturated fatty acids, eicosane acids, oxidized low-density lipoprotein, very low-density lipoprotein, and linoleic acid derivatives.384
To date, there have been many reports about the role of PPARs in cancer cells, including prostate cancer, breast cancer, glioblastoma, neuroblastoma, pancreatic cancer, hepatic cancer, leukemia, and bladder cancer and thyroid tumors.385 However, the function of PPARs in CSCs is not well understood, except for some reports on PPARγ. As a tumor suppressor, PPARγ binds and activates a canonical response element in the miR-15a gene in breast CSCs to reduce the CD49high/CD24+ mesenchymal stem cell (MSC) population and inhibit angiogenesis.386 PPARγ activation also prevents cell spheroid formation and stem cell-like properties in bladder CSCs and induces adipocyte differentiation and β-catenin degradation in adipose tissues.387 Furthermore, expression of PPARγ restrains YAP transcriptional activity to induce differentiation in osteosarcoma stem cells388 and melanoma cells.389 The PPARγ/NF-κB pathway promotes M2 polarization of macrophages to prevent cell death in ovarian CSCs4.390 PPARγ activation promotes expression of its target gene PTEN to inhibit PI3K/Akt/mTOR signaling, which stunts self-renewal, tumorigenicity, and metastasis in cervical CSCs, glioblastoma stem cells, and liver CSCs.391,392 However, combined expression of Dnmt3a and Dnmt3b inhibits PPARγ expression by direct methylation of its promoter in squamous carcinomas.393 PPARs are also closely related to the metabolism of CSCs. PPARα and PPARβ/δ regulate metabolic reprogramming in glioblastoma stem cells, lung CSCs, and mouse mammary gland cancer.394 The transcription coactivator peroxisome proliferator-activated receptor gamma coactivator 1α (PPARGC1A, also known as PGC-1α) promotes CSC proliferation and invasion by enhancing oxidative phosphorylation, mitochondrial biogenesis, and the oxygen consumption rate of breast CSCs.395 In addition, the AMPK signaling pathway (adenosine 5′-monophosphate (AMP)-activated protein kinase) is an AMP-dependent protein kinase that is a key molecule in the regulation of bioenergy metabolism and is the core of the study of diabetes and other metabolic-related diseases. AMPK is expressed in various CSCs related to metabolism. Some studies have shown that AMPK is necessary for prostate CSCs to maintain glucose balance.396 Metformin, an antidiabetic drug that fights cancer, targets AMPK signaling to inhibit cell proliferation and metabolism in colorectal CSCs397 and HCC stem cells.398 Therefore, metformin may be a potential therapeutic regent by regulating the energy metabolism of CSCs. These studies suggest that PPARs play an important role in the growth of CSCs.
Interactions between signaling pathways in CSCs
As mentioned previously, these complex signal transduction pathways are not linear. In some cases, crosstalk between and among various pathways occurs to regulate CSCs.399 Wnt/β-catenin and NF-κB signaling work together to promote cell survival and proliferation of CSCs. TNFRSF19, a member of the TNF receptor superfamily, is regulated in a β-catenin-dependent manner, but its receptor molecules activate NF-κB signaling to regulate the development of colorectal cancer.400 Knockdown of CD146 results in inhibition of NF-κB/p65-initiated GSK3β expression, which promotes nuclear translocation and activation of β-catenin.401 In addition, there is negative regulation between Wnt/β-catenin and NF-κB signaling. Studies have revealed a negative effect of β-catenin on NF-κB activity in liver, breast, and colon cancer cells.402,403 Leucine zipper tumor suppressor 2 (LZTS2) is a putative tumor suppressor, and NF-κB activation inhibits β-catenin/TCF activity through upregulation of LZTS2 in liver, colon, and breast cancer cells.404,405,406 Wnt/β-catenin and Hh signaling have important functions in embryogenesis, stem cell maintenance, and tumorigenesis. Wnt/β-catenin signaling induces the expression of CRD-BP, an RNA-binding protein, which results in the binding and stabilization of Gli1 mRNA, leading to an increase in Gli1 expression and transcriptional activity, which promotes the survival and proliferation of colorectal CSCs.407 However, a report showed that noncanonical Hh signaling is a positive regulator of Wnt signaling in colon CSCs.408
In addition, crosstalk between pathways promotes cell growth and metastasis through maintenance of the CSC population. Downregulation of Notch1 and IKKα enhances NF-κB activation to promote the CD133+ cell population in melanoma CSCs.409 IL-6/JAK/STAT3 and TGF-β/Smad signaling induce the proliferation and metastasis of lung CSCs.410 IL-17E binding to IL-17RB activates the NF-κB and JAK/STAT3 pathways to promote proliferation and sustain self-renewal of CSCs in HCC.411 TGF-β1 silencing decreases the expression of Smad2/3, β-catenin, and cleaved-Notch1 to inhibit the activation of Wnt and Notch signaling in liver CSCs.346 Activation of TGF-β1 induces lncRNA NKILA expression to block NF-κB signaling, which inhibits metastasis of breast CSCs.412 TGF-β also directly regulates the expression of Wnt5a in breast CSCs to limit the stem cell population.413 Furthermore, Notch, IKK/NF-κB, and other pathways together regulate the proliferation and metastasis of CD133+ cutaneous SCC stem cells.409 PI3K/mTOR signaling upregulates the expression of STAT3 to promote the survival and proliferation of breast CSCs.328 Inhibition of TORC1/2 increases FGF1 and Notch1 expression. The PI3K/AKT/mTOR and Sonic Hh pathways cooperate to inhibit the growth of pancreatic CSCs.414 Increasing evidence shows that crosstalk regulates the survival, self-renewal, and metastasis of CSCs.
The microenvironment of CSCs
CSCs interact with the microenvironment through adhesion molecules and paracrine factors. The microenvironment provides a suitable space for the self-renewal and differentiation of CSCs, protects CSCs from genotoxicity, and increases their chemical and radiological tolerance. The TME mainly consists of the tumor stroma, adjacent tissue cells, microvessels, immune cells, and immune molecules.415 CSCs not only adapt to changes in the TME but also affect the TME. Concurrently, the microenvironment also promotes the self-renewal of CSCs, induces angiogenesis, recruits immune and stromal cells, and promotes tumor invasion and metastasis (Fig. 4).

The microenvironment of cancer stem cells. Proliferation, self-renewal, differentiation, metastasis, and tumorigenesis of CSCs in the CSC microenvironment. The CSC microenvironment is mainly composed of vascular niches, hypoxia, tumor-associated macrophages, cancer-associated fibroblasts, cancer-associated mesenchymal stem cells, and extracellular matrix. These cells in response to hypoxic stress and matrix induce growth factors and cytokines (such as IL-6 and VEGF) to regulate the growth of CSCs via Wnt, Notch, and other signaling pathways
Vascular niche microenvironments and CSCs
The normal vasculature is composed of ECs, basement membranes, and parietal cells. ECs are the basis for the formation of the inner surface of blood vessels.416 Studies reported that glioblastoma stem cells are located around the blood vessels, and the concept of the cancer microvascular environment was first proposed. Calabrese et al.417 demonstrated that direct contact between ECs and CSCs occurs in brain tumors. CSCs are also found near ECs in other cancers, such as papilloma and colorectal cancer.418,419 A study also showed that CD133+/CD144− glioma stem cell-like cells differentiate into cancer cells and endothelial progenitor cells and finally into mature ECs.420 CSCs differentiate into cancer vascular stem cells/progenitor cells and are directly involved in angiogenesis or form vasculogenic mimicry that is directly involved in the microcirculation of tumors.421,422 ECs also promote CSC-like transformation and cell growth through Shh activation of Hh signaling.423 Moreover, secreted microvesicles of CSCs promote the proliferation of human umbilical vein ECs and form a tube-like structure in vitro and in vivo in mice.424,425,426 This CSC plasticity has also been demonstrated in other tumors, including neuroblastoma, renal, breast, and ovarian cancer.427,428,429,430
The vascular microenvironment maintains the initial undifferentiated dormancy of stem cells, supports self-renewal, invasion and metastasis of CSCs, and protects CSCs from any injury.431 The role of the EC signaling system has been proven in maintaining the survival and self-renewal of head and neck SC stem cells.432 Pasquier and colleagues433 showed that treatment with EC microparticles in breast and ovarian cancer models increased the number of CSCs and promoted sphere formation of CSCs. The interaction between CSCs and blood vessels promotes the self-renewal of CSCs through the VEGF-Nrp1 loop.418 CSCs promote cancer angiogenesis by inducing secretion of the cytokines VEGF and hepatocyte growth factor (HGF) from ECs.434 VEGF receptor 2 plays a key role in vasculogenic mimicry formation, neovascularization, and tumor initiation of glioma stem-like cells.435 As a result, the secretion of VEGF in stem cell-like glioma cells is higher than that in normal cancer cells424 and regulates the proliferation of glioma stem cells through the mTOR signaling pathway.436 Subsequent studies have further shown that multiple signals, such as integrin, Notch, and growth factor receptors, are linked to each other on the cell surface to maintain the stemness of CSCs.437,438
The hypoxia microenvironment and CSCs
Hypoxia is a key component for CSC formation and maintenance.439 The hypoxic microenvironment maintains the undifferentiated state of cancer cells, enhances their cloning rate, and induces the expression of CD133 as a specific biomarker of CSCs.440 HIFs are important transcription factors that regulate cellular hypoxia responsiveness441 and inhibit cell apoptosis.442 As a heterodimer, HIF is composed of HIFα and HIFβ.443 HIF-1α regulates the proliferation and fate of CSCs in medulloblastoma and glioblastoma multiforme444 and activates the NF-κB pathway to promote CSC survival and tumorigenesis.445 HIF-2α maintains the survival and phenotype of CSCs.446 HIFα also regulates the expression of the target genes GLUT1, GLUT3, LDHA, and PDK1. Thus, CSCs can adapt to a new method of cell energy metabolism and avoid apoptosis caused by hypoxia.447
HIFs also regulate the stemness of CSCs. Previous studies have shown that CSCs need to activate HIF-1α and HIF-2α to maintain their self-sustainability under hypoxic conditions448 and obtain pluripotency by upregulating the Sox2 and Oct4 genes.440 More importantly, activation of C-MYC by HIF-2α is necessary to ensure undifferentiated CSCs.449 The Wnt and Notch signaling pathways regulated by hypoxia and can induce the EMT, which promotes the stemness of CSCs and increases the invasiveness and resistance to radiotherapy and chemotherapy.450 HIF-1α binds the Notch ICD and enhances its transcriptional activity. In the hypoxic microenvironment of glioma, both HIF-1α and HIF-2α require the Notch signaling pathway to ensure the self-renewal and undifferentiated status of CSCs.451
Tumor-associated macrophages and CSCs
Macrophages are an important component of the innate immune response and are a group of cells with plasticity and heterogeneity.452 Infiltrating and inflammatory macrophages originate from the precursors of bone marrow mononuclear cells.453 These precursor cells infiltrate various tissues from blood vessels and differentiate into different subtypes in different microenvironments. There are two subtypes of macrophages: the M1 and M2 phenotypes. The M1 phenotype has anti-inflammatory and anti-tumor effects and secretes proinflammatory factors such as interleukin-1 (IL-1), IL-12, IL-23, TNF-α, chemokine (C-X-C motif) ligand 5 (CXCL5), CXCL9, and CXCL10. M2 macrophages are generally considered to be the phenotype of tumor-associated macrophages (TAMs),454,455,456 have immunosuppressive and angiogenesis-promoting effects, and are considered to be a tumor-promoting cell type.456,457 M2 macrophages secrete CCL17 (C-C chemokine ligand 17), CCL22, and CCL24 and have low expression of IL-12 and high expression of IL-10. Cytokines secreted by macrophages affect the proliferation, tumorigenic transformation, or apoptosis of CSCs through various signaling pathways.458
TAMs are closely related to CSCs or stem cell transformation. Renal epithelial cells cocultured with macrophages induce the EMT to transform renal cancer cells into CSCs expressing CD117, Nanog, and CD133.459 Another study also showed that mucin-1 secreted by M2 macrophages induces the transdifferentiation of non-small-cell lung cancer cells into CSCs that express CD133 and Sox2.460 Jinushi and colleagues461 also reported that TAMs secrete MFG-E8, which maintains the self-renewal ability of colon and breast CSCs, and knockout of MFG-E8 significantly inhibits the tumorigenic ability in SCID mice.461 TAMs are closely related to glioma stem cell growth.462 TAMs are mainly distributed near CD133+ glioma stem cells and accumulate in pericapillary and hypoxic areas.463 Glioma stem cells recruit and maintain macrophages by secreting a potent chemokine membrane protein.464 The ablation of TAMs inhibits the tumorigenesis of glioma stem cells.465 Recent studies have shown that the interaction between the TME and CSCs is regulated by a variety of signaling pathways.466 Macrophages enhance the invasion of glioma stem-like cells through the TGF-β1 signaling pathway.467 TAMs activate the STAT3/Sox2 signaling pathway in mouse breast CSCs by secreting EGF, which promotes the self-renewal ability of CSCs.468 IL-8 secreted by TAMs also induces the EMT in hepatocellular cancer cells by activating the JAK2/STAT3/Snail pathway.469
Cancer-associated fibroblasts and CSCs
CAFs are one of the most important components of the TME and are critical in tumor development and metastasis.470 The origin of these cells in the stroma is not entirely clear. Current studies hypothesize that there are five possible sources: (1) transference of fibroblasts in the host stroma;471 (2) EMT;472 (3) transdifferentiation of perivascular cells;473 (4) EMT;474 and (5) differentiation of MSCs derived from bone marrow.475 In addition, CAFs are also derived from other cell types, such as smooth muscle cells, pericytes, adipocytes, and immune cells.476 It is not clear whether there are differences in the functions of CAFs from different sources. CAFs affect cancer cell growth through cell–cell interactions and the secretion of various invasive molecules, such as cytokines, chemokines, and inflammatory mediators.477,478,479
CAFs in the TME play an indispensable role in the generation and maintenance of CSCs.480 CAFs transform cancer cells into CSCs.481 Studies have shown that CAFs promote the EMT and enhance the expression of prostate CSC markers482 by secreting IL-6 and IL-1β in breast cancer.483,484 CAFs also secrete TGF-β and activate related pathways to increase ZEB1 transcription, which stimulate lung cancer cells to undergo EMT and CSC transformation.485 CAFs secrete matrix metalloproteinases, which induce the EMT and promote the growth of stem cell-specific components in tumors.482 Paracrine interaction between CAFs and CSCs is critical for maintaining the CSC niche of lung CSCs.486 Fibroblast-derived CCL-2 regulates CSCs through gap activation, thus promoting the progression of tumors.487 CAFs and adipocytes also secrete leptin, which increases the globulation rate of breast CSCs in vitro.488
CAFs also regulate the proliferation of CSCs by other signaling pathways. For example, CAFs increase the secretion of CCL-2 to activate the Notch1/STAT3 pathway, which increases the expression of stem cell markers and upregulates the globulation rate in breast cancer.489 CAFs regulate TIC plasticity in HCC through c-Met/FRA1/HEY1 signaling.490 CAFs secrete high levels of IL-6 to activate Notch signaling through STAT3 Tyr705 phosphorylation, thus promoting the stem cell-like characteristics of HCC cells.491 Similar studies have shown that CAF-derived exons enhance colon stem cell resistance to 5-fluorouracil by activating the Wnt signaling pathway.492
Cancer-associated MSCs and CSCs
MSCs have high self-renewal ability and multidirectional differentiation potential.493 MSCs also specifically migrate to the injured site and tumor tissue and are easy to isolate and expand in vitro.494,495 MSCs are considered to be an ideal vector for gene therapy because of their characteristics of homing to and secreting cytokines in tumors.496 However, these tumorigenic characteristics of MSCs still need to be studied. MSCs not only promote tumor development497,498 but also inhibit cancer cell growth.499 Bone marrow MSCs promote tumor growth by promoting angiogenesis, metastasis, and the survival of CSCs.500 MSCs in the TME are conducive to the proliferation, carcinogenesis, and metastasis of breast CSCs through ionic purinergic signal transduction.501 MSCs can differentiate into CAFs, and CAFs further regulate CSCs and promote the occurrence and metastasis of cancers.502 The possible mechanism is related to the spontaneous fusion between cancer cells and MSCs.503 The fusion of MSCs with breast cancer, ovarian cancer, gastric cancer, and lung cancer cells in vitro and in vivo has been confirmed.504,505 MSCs regulate the TME by secreting IL-6 to maintain the undifferentiated state of osteosarcoma cells.506,507 IL-1 stimulates the secretion of PGE2 via autocrine signaling, which ultimately activates β-catenin signaling in cancer cells in a paracrine manner and transforms cancer cells into CSCs.508 In the ECM, bone mesenchymal stem cells activate the NF-κB pathway and induce a CSC phenotype by secreting a variety of cytokines and chemokines, such as CXCL12, CXCL7, and IL-6/IL-8.509 The interaction between MDSCs and CSCs via IL-6/STAT3 and Notch signaling is critical to the progression of breast cancer.510
Extracellular matrix and CSCs
The ECM is an insoluble structural component of the matrix in mesenchymal and epithelial vessels. The ECM includes collagen, elastin, aminoglycan, proteoglycan, and noncollagen glycoprotein.511,512 At present, increasing evidence shows that the ECM is an integral part of stem cell niches that regulates the balance of stem cells in three different biological states: static, self-renewal, and differentiation.513 Experiments in vitro and in vivo have shown that ECM receptors can be used to aggregate CSCs514 and induce drug resistance.513,515 Fibronectin, vimentin, collagen, and proteoglycan in the ECM bind to cytokines such as FGF, HGF, VGF, BMP, and TGF-β in the TME and regulate their activities.516 In HCC, an increased matrix promotes cell proliferation and chemotherapeutic resistance and increases the expression of CSC-related markers, including CD44, CD133, c-kit, cxcr4, Oct4, and Nanog. Hyaluronic acid in the ECM is a ligand for the CD44 receptor and can regulate the acquisition and maintenance of CSC stemness during mutual contact.517 The ECM also binds the Wnt ligand Wnt5b via molecular MMP3 and leads to the expansion and proliferation of mammary epithelial stem cells.518 In addition, tenascin C in the ECM maintains the stability of breast CSCs by increasing the activity of the Wnt and Notch signaling pathways.519
Exosomes in the TME and CSCs
Exosomes are nanovesicles secreted by various types of living cells (30–100 nm in diameter)520 and are widely distributed in peripheral blood, saliva, urine, ascites, pleural effusion, breast milk, and other body fluids.521 Exosomes contain a large number of functional proteins, RNA, microRNAs, DNA fragments, and other bioactive substances.522,523,524,525 These bioactive substances mediate material transport and information exchange between cells, thus affecting the physiological function of cells.526,527 The exosomes secreted by cancer cells promote angiogenesis,528 induce differentiation of tumor-related fibroblasts,529 participate in immune regulation of the TME,530 and regulate the microenvironment before metastasis.531 Clinical analysis has revealed that exosomes are released at higher levels in cancer cells.532
Recent studies have shown that endocytosis of lipid rafts in MSCs is associated with increased secretion of exosomes.533 Exosome signaling mediates the interaction of CSCs and normal stem cells, thereby regulating oncogenesis and tumor development.534 Exosomes also regulate CSC growth by targeting specific signaling pathways, such as Wnt, Notch, Hippo, Hh, and NF-κB.535,536,537 Extracellular vesicles released by glioblastoma stem cells promote neurosphere formation, endothelial tube formation, and the invasion of glioblastoma.538 CSCs promote cell proliferation and self-renewal through crosstalk between exosome signal transduction and the surrounding microenvironment.539 The exosomes released from CSCs affect signal transduction in nearby breast cancer cells540 and increase the stemness of breast cancer cells.540 Similarly, fibroblast-derived exosomes contribute to chemoresistance by promoting colorectal CSC growth.491 Exosomes in the TME promote the transformation of non-CSCs into CSCs. CAF-derived exosomes significantly increase the ability to form mammary globules and promote the stemness of breast cancer cells.541 Similarly, CAF-derived exosomes also promote sphere formation of colorectal cancer cells by activating Wnt signaling and ultimately increase the percentage of CSCs.491 Exosomes from glioma-associated MSCs increase the tumorigenicity of glioma stem-like cells by transferring miR-1587.542 In addition, exosomes regenerate stem cell phenotypes by mediating the EMT or regulating stem cell-related signaling pathways, such as the Wnt pathway, Notch pathway, Hh pathway and other pathways, which convert cells into CSCs.543 Exosomes have many advantages, such as low immunogenicity, biocompatibility, easy production, cytotoxicity, easy storage, high drug loading capacity, and long life and have become ideal drug carriers for cancer therapy.544,545,546,547,548
Therapeutic targeting of CSCs
Agents targeting CSC-associated surface biomarkers in clinical trials
Monoclonal antibodies (mAbs) that target CSC-specific surface biomarkers have become an emerging technology for cancer therapy. Rituximab, a CD20 mAb, is an active agent for the treatment of follicular lymphoma and mantle-cell lymphoma, but some serious adverse reactions still occur.549 Subsequently, to improve the availability and affordability of radioimmunotherapy for refractory or recurrent non-Hodgkin’s lymphoma (NHL), a phase II clinical trial for a radioiodine replacement of rituximab was carried out, which showed a response rate of 71% and a complete remission rate of 54% in 35 patients, with only two cases of grade IV hematologic toxicity observed.550 Encouragingly, alemtuzumab, a humanized CD52 antibody, has been approved for the treatment of chronic lymphocytic leukemia (CLL) in patients who failed to respond to alkylating agents and purine. Furthermore, the combination of the CD20 and CD52 antibodies in the treatment of refractory CLL was safe, nontoxic, feasible, and positive.551 Another antibody drug, relabeled bivatuzumab, is an anti-CD44v6 mAb,71 which was found to be safe when it was used for the treatment of head and neck SCC.552 These results have been obtained in subsequent clinical research553 and safety/efficacy studies.554 Unfortunately, in a stage I dose escalation study with the CD44v6 antibody, one patient with head and neck SCC of the esophagus suffered deadly skin toxicity.555
Several CD123 antibodies have been developed, XmAb14045 and MGD006, and were designed with biospecific effects against CD3 and CD123. Talacotuzumab is also effective against CD16 and CD123. CSL360, another CD123 antibody, was used to treat relapsed, refractory, or high-risk acute myeloid leukemia (AML) and displayed no anti-leukemic activity in most cases.556 SL-401, another CD123 antibody, was used to treat blastic plasmacytoid dendritic cell neoplasm patients. The results showed major positive responses in seven out of nine patients, including five complete responses and two partial responses.557 An ongoing phase II study of SL-401 in 29 patients with blastic plasmacytoid dendritic cell neoplasms demonstrated robust single-agent activity with an 86% overall response rate.558 The latest antibodies against CSC surface markers, such as XmAb14045 (NCT02730312), flotetuzumab (NCT02152956), and talacotuzumab (NCT02472145), are also in clinical study. Furthermore, several other markers that can distinguish LSCs from other cells are under clinical development, such as IL-1 receptor accessory protein, CD27/70, CD33, CD38, CD138, CD93, CD99, C-type lectin-like molecule-1, and T cell immunoglobulin mucin-3.
EpCAM, a common CSC biomarker, has also received attention in clinical trials.559 Adecatumumab, an EpCAM antibody, was used in patients with hormone-resistant prostate cancer, and the results showed that the EpCAM-specific antibody has great clinical potential.560 Catumaxomab, a multifunctional mAb against EpCAM, binds and recognizes EpCAM and the T cell antigen CD3 (anti-EpCAM × anti-CD3).561 Intraperitoneal injection of catumaxomab to treat EpCAM-positive ovarian cancer and malignant ascites has shown high efficacy in killing cancer cells and reducing the formation of ascites.562 Catumaxomab has been used in non-small-cell lung cancer and also had a good survival rate.561 However, other types of EpCAM antibodies, such as edrecolomab563 and adecatumumab,564 have minimal efficacy in colorectal and breast cancers. Combining EpCAM antibodies with chimeric antigen receptor T cell (CAR-T) technology has also been used in various types of cancers in phase I trials, such as NCT02915445, NCT03563326, NCT02729493, and NCT02725125. With a deeper understanding of CSC surface biomarkers, there has been significant progress in developing antibodies targeting CSCs (Table 2). However, CSC surface phenotypes can vary in different patients or different cancers, and different CSC populations with different phenotypes might coexist. CSCs also diverge or evolve into different cancer cells, acquiring distinct phenotypes upon relapse. Therefore, the strategies used in clinical trials should be determined according to the phenotypes of the different cancers. At the same time, combining different surface antibodies with relevant chemotherapy drugs can achieve an ideal therapeutic effect.
Agents targeting CSC-associated signaling pathways in clinical trials
The signaling pathways that regulate the maintenance and survival of CSCs have become targets for cancer treatment. At present, the main signaling pathways are the Wnt, Notch, and Hh signaling pathways, as well as the TGF-β, JAK-STAT, PI3K, and NF-κB signaling pathways. These pathways often interact with each other during tumor development and in CSCs. Considerable progress has been made in early clinical trials for Notch and Hh pathway inhibitors, while targeting the Wnt pathway has proven to be difficult.10
The Notch signaling pathway plays an important role in the maintenance of CSCs565,566 and can induce CSC differentiation. Abnormal activity of the Notch signaling pathway has been observed in many cancers, such as leukemia,567 glioblastoma,568,569 breast cancer,570 lung cancer,571 ovarian cancer,572 pancreatic cancer,573 and colon cancer.574 At present, there are three major clinical methods used to inhibit Notch signaling, secretase inhibition (γ-secretase inhibitor (GSI)), Notch receptor or ligand antibodies, and combination therapy with other pathways. For example, GSIs have been tested in clinical trials. Among them, MK-0752 (NCT00100152) was the first GSI used to treat T cell acute lymphoblastic leukemia in children in a phase I trial. However, the study was terminated because of poor results.575 MK-0752 also had no clinical activity in extracranial solid tumors in subsequent phase II trials. Only one complete response with interdegenerative astrocytoma and SD extension out of 10 patients with different types of glioma was observed.576 MK-0752 is well tolerated and shows targeted inhibition in recurrent pediatric central nervous system tumors.577 In addition, combining MK-0752 with cisplatin treatment for ovarian cancer,578,579 docetaxel treatment for locally advanced or metastatic breast cancer,569 and gemcitabine treatment for ductal adenocarcinoma of the pancreas580 has shown good efficacy. However, the clinical effect was minimal in patients with advanced solid tumors,576,581 including metastatic pancreatic cancer.582
In addition, RO4929097, another selective GSI, showed good anti-tumor activity in preclinical and early trials,583,584 but was not good for metastatic colorectal cancer,585 metastatic pancreatic adenocarcinoma,586 or recurrent platinum-resistant ovarian cancer.587 Combinations of RO4929097 with gemcitabine,588 temsirolimus,587 cediranib,589 or capecitabine590 in advanced solid tumors, as well as with bevacizumab in recurrent high-grade glioma, are well tolerated and have modest clinical benefits. However, NCT01154452, the combination of RO4929097 with vismodegib and vismodegib alone for patients with advanced osteosarcoma, showed no significant difference in a phase Ib trial. The third oral GSI, PF-03084014, had good efficacy in desmoid tumors either in phase I or subsequent phase II studies.591 Preliminary evidence of its clinical efficacy was demonstrated in patients with solid tumors,592 as well as in patients with recurrent acute T cell lymphoblastic leukemia.593 Other selective GSIs, such as BMS-906024 (NCT01292655), BMS-986115 (NCT01986218), CB-103 (NCT03422679), LY3039478 (NCT02836600), and LY900009 (NCT01158404), have also entered the clinical trial stage, and the results still need to be verified.
DLL4 plays a vital role in regulating tumor angiogenesis.594 Therefore, targeting DLL4 is another strategy to block Notch signaling, and this is being tested in the clinic. Demcizumab (OMP-21M18), a humanized IgG2 mAb that targets DLL4 and blocks its interactions with Notch receptors, was tested in a phase I dose escalation study with 55 patients with previously treated solid tumors.595 The results have shown that demcizumab had good efficacy against solid tumors, but was not good for metastatic pancreatic cancer treatment when combined with gemcitabine and Abraxane (NCT02289898). NCT02259582, a combination of demcizumab with carboplatin and pemetrexed to treat lung cancers (DENALI study), is ongoing in another phase II study.595 Enoticumab, another fully human IgG1 antibody against DLL4, has promising activity in phase I clinical trials for advanced solid malignancies.
Activation of Hh signaling has been implicated in a variety of cancers.596,597,598 Activation of Hh signaling in CSCs contributes to CSC stemness, chemoresistance, and metastatic dissemination. The Hh signaling pathway mainly regulates target gene expression via smoothened (SMO)-mediated nuclear transfer of transcription factors. Three oral SMO antagonists, vismodegib (GDC-0449), sonidegib (LDE225), and glasdegib (PF-04449913), have been approved by the Food and Drug Administration (FDA) and show significant activity in locally advanced and metastatic basal cell carcinoma, as well as in AML.599,600,601 Vismodegib was the first proposed Hh pathway inhibitor in cancer research602 and is approved by the FDA603 for local or advanced metastatic basal cell carcinoma treatment.599 Subsequently, phase I and phase II trials targeting recurrent medulloblastoma have shown that the progression-free survival (PFS) of Shh-mb patients treated with vismodegib is longer and more effective than that of non-Shh-mb patients. Vismodegib even has better activity in patients with recurrent Shh-mb but not in patients with recurrent non-Shh-mb.604,605 Vismodegib has also been tested in metastatic colorectal cancer,606 pancreatic cancer,607 chondrosarcoma,608 relapsed/refractory NHL, CLL,609 and ovarian cancer.610 Disappointingly, these treatments with vismodegib have not resulted in better survival.
Sonidegib was the second SMO antagonist approved for the treatment of locally advanced basal cell carcinoma that recurred after surgery or radiotherapy and is not suitable for surgery or radiation therapy.611 In addition, the results of a multicenter, randomized, double-blind phase II trial have shown that 200 mg sonidegib for patients with advanced basal cell carcinoma is the most clinically appropriate dose.600
In a phase I study of a 3 + 3 dose escalation to treat small-cell lung cancer patients, sonidegib combined with cisplatin and etoposide sustained PFS in patients with Sox2 amplification.224 These combinations in a phase II trial for patients with recurrent medulloblastoma resulted in a complete or partial response in 50% of patients612 and have been used for other cancer treatments in phase I/II clinical trials, such as NCT02111187 for prostate cancer, NCT02027376 for breast cancer, and NCT02195973 for recurrent ovarian cancer.
Glasdegib was the first Hh pathway inhibitor approved for the treatment of AML in patients older than 75 years or those unable to use intensive induction chemotherapy601 and showed good safety and tolerability in a phase I trial for patients with partial hematologic malignancies in Japan.613 In a phase II trial, glasdegib combined with cytarabine/daunorubicin had a significant efficacy in patients with AML, chronic myeloid leukemia (CML) or high-risk myelodysplastic syndromes.614 Glasdegib combined with low-dose cytarabine (LDAC) is a potential option for AML patients who are not suitable for intensive chemotherapy.615 Other selective SMO inhibitors, including taladegib (LY2940680) and saridegib (IPI-926), have also entered clinical trials for other cancers. As single-target agents, these SMO inhibitors have drug resistance problems. To reduce this problem, some novel inhibitors of terminal components of Hh signaling pathway are being developed, such as arsenic trioxide (ATO)616 and GANT-61.617
The Wnt signaling pathway is associated with tumor development in breast cancer,618 ovarian cancer,619 esophageal squamous cell cancer,620 colon cancer,621 prostate cancer,622 and lung cancer.623 Until now, several drugs aimed at the Wnt signaling pathway have been in clinical trials, while the majority of Wnt inhibitors are in preclinical testing. Clinical data from initial trials have shown that ipafricept (OMP-54f28/FZD8-Fc) is a first-in-class recombinant fusion protein that antagonizes Wnt signaling.624 However, its role in patients with desmoid cancers and germ cell cancers is negligible.625 NCT02050178, ipafricept combined with ab-paclitaxel and gemcitabine in patients with untreated stage IV pancreatic cancer, NCT02092363, ipafricept combined with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer, and NCT02069145, ipafricept combined with sorafenib in patients with HCC, are currently being investigated. PRI-724, a β-catenin inhibitor, inhibits the interaction between β-catenin and its transcriptional coactivators. Safety and efficacy testing of PRI-724 for patients with advanced myeloid malignancies (NCT01606579) and advanced or metastatic pancreatic cancer (NCT01764477) have been completed in phase I studies. CWP232291, another inhibitor of β-catenin activity, has also been shown to be effective for AML (NCT03055286) in a phase I clinical study and for recurrent or refractory myeloma (NCT02426723) in a phase I/II clinical study.626 Other Wnt signaling inhibitors have also been under clinical trial, including LGK974 (NCT02278133), ETC-159 (NCT02521844), and OMP-18R5 (NCT01973309, NCT01957007, and NCT02005315).
In addition, the mitochondrial glycolysis pathway also plays a key role in regulating the proliferation and apoptosis of CSCs. Venetoclax, a BCL-2 inhibitor, was initially approved by the FDA recently and shows good tolerance and activity for AML patients with adverse reactions.627 Two arachidonate 5-lipoxygenase inhibitors, VIA-2291 and GSK2190915, might be potent agents for targeting LSCs in CML,628 as shown in Table 3.
Other abnormal signaling pathways have also been found in CSCs, such as TGF-β, JAK-STAT, PI3K, and NF-κB. These signaling pathways are not independent of each other but rather form a complex signaling network. Agents targeting CSC-associated signaling pathways in ongoing clinical trials are listed in Table 3.
Targeting the CSC microenvironment
The CSC microenvironment contributes to the self-renewal and differentiation of CSCs and regulates CSC fate by providing cues in the form of secreted factors and cell contact. CXCR4 has been found in most cancers, especially in CSCs. The most well-characterized drug-targeting CXCR4 is plerixafor (AMD3100), and this drug is an effective hematopoietic stem cell mobilizer for patients with multiple myeloma and NHL.629 AMD3100 treatment for relapsed or refractory AML resulted in 46% of patients with complete remission with or without white count recovery in a phase I/II study.630 In addition, plerixafor with high-dose cytarabine and etoposide treatment for children with relapsed or refractory acute leukemia or myelodysplasia syndrome resulted in two patients with complete remission and one patient with incomplete hematologic recovery out of 18 patients in a phase I study.631 LY2510924, a small cyclic peptide, is a potent and selective antagonist of CXCR4 and is well tolerated with no serious adverse events in a phase I trial.632 However, the combination of LY2510924 with sunitinib for patients with metastatic renal cell carcinoma has no better effect than sunitinib alone in a phase II trial.633 The combination of LY2510924 with carboplatin/etoposide for patients with extensive small-cell lung cancer also had no significant effect compared with that of carboplatin/etoposide alone in a phase II study.634 The combination of LY2510924 with other drugs for gliomas (NCT03746080, NCT01977677, and NCT01288573) and multiple myeloma (NCT00103662, NCT01220375, and NCT00903968) is also under clinical trial.
The microenvironment plays an important role in CSC growth, and it is also a promising target for treatment. Agents targeting the microenvironment in ongoing clinical trials are listed in Table 3.
CSC-directed immunotherapy
In the early twentieth century, Paul Ehrlich first proposed the idea that an intact immune system suppresses tumor development (advancing cancer therapy with present and Emerging Immuno-Oncology Approaches). Based on the understanding of cellular immune regulation, new methods for cancer treatment have emerged. In addition to the antibodies against the CSC molecules mentioned above, some novel anti-CSC immunotherapeutic approaches, such as immunologic checkpoint blocking or CAR-T cell therapies, have been developed. Some drugs that target the immune checkpoint receptors CTLA-4,635 PD-1 (nivolumab,636 pembrolizumab,637 and cemiplimab,638) and PD-L1 (avelumab,639 durvalumab,640 and atezolizumab641) have also been in clinical trials. I ipilimumab, a CTLA-4 antibody, is approved by the FDA, and initial clinical results showed good effectiveness in patients with metastatic melanoma.642 For CAR-T cell therapy, as shown in Table 4, CD19, CD20, CD22, CD123, EpCAM, and ALDH have been used for CSC-directed immunotherapy, and most of them are recruited.
Conclusions and perspectives
We can conclude that CSCs are a small population of cancer cells that have self-renewal capacity and differentiation potential, thereby conferring tumor relapse, metastasis,643 heterogeneity,644 multidrug resistance,645,646 and radiation resistance.647 Several pluripotent transcription factors, including Oct4, Sox2, Nanog, KLF4, and MYC and some intracellular signaling pathways, including Wnt, NF-κB, Notch, Hh, JAK-STAT, PI3K/AKT/mTOR, TGF/Smad, and PPAR, as well as extracellular factors, including vascular niches, hypoxia, TAM, CAF, cancer-associated MSCs, the ECM, and exosomes, are essential regulators of CSCs. Drugs, vaccines, antibodies, and CAR-T cells targeting these pathways have also been developed to target CSCs.648 Importantly, many clinical trials on CSCs have also been performed and show a promising future for cancer therapy.
However, there are also multiple hurdles that need to be solved to effectively eliminate CSCs. First, the characteristics of many CSCs in specific types of tumors are not well identified.649 Second, since most studies on CSCs are performed in immune-deficient mice in the absence of an adaptive immune system, these models do not recapitulate the biological complexity of tumors in the clinic.650 Third, CSCs exist in a specific niche that sustains their survival. However, isolated CSCs are used in most current studies that lacks a microenvironment.651 Fourth, the environmental factors in CSC niches are not well understood, and the relationship between TAMs/CAFs and CSCs has not been well studied.645 Fifth, since CSCs also share some signaling pathways with normal stem cells, not all the regulatory factors that contribute to CSCs are appropriate for use as therapeutic targets in cancer treatment. Sixth, whether CSCs should be activated or arrested is an open question in cancer therapy.652 Seventh, novel signaling and more regulatory levels, such as RNA editing,653 epigenetics,654 and cellular metabolism,655 should be considered in cancer therapy because they also contribute to the stemness of CSCs. Eighth, some inhibitors that target CSC signaling are not very specific, and so new inhibitors need to be designed.656 Ninth, natural products that target CSCs should also be studied in the future.657 Finally, novel ways of targeting the microenvironment of CSCs are also promising and need to be explored.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics. CA Cancer J. Clin. 69, 7–34 (2019).
Sun, Y. Translational horizons in the tumor microenvironment: harnessing breakthroughs and targeting cures. Med. Res. Rev. 35, 408–436 (2015).
Batlle, E. & Clevers, H. Cancer stem cells revisited. Nat. Med. 23, 1124–1134 (2017).
Reya, T., Morrison, S. J., Clarke, M. F. & Weissman, I. L. Jn Stem cells, cancer, and cancer stem cells. Nature 414, 105 (2001).
Chen, W., Dong, J., Haiech, J., Kilhoffer, M. C. & Zeniou, M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016, 1740936 (2016).
Lapidot, T. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648 (1994).
Bonnet, D. & Dick, J. E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737 (1997).
Shimokawa, M. et al. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 545, 187–192 (2017).
Shibata, M. & Hoque, M. O. Targeting cancer stem cells: a strategy for effective eradication of cancer. Cancers 11, 732 (2019).
Visvader, J. E. & Lindeman, G. J. Cancer stem cells: current status and evolving complexities. Cell Stem Cell 10, 717–728 (2012).
Ajani, J. A., Song, S., Hochster, H. S. & Steinberg, I. B. Cancer stem cells: the promise and the potential. Semin. Oncol. 42(Suppl. 1), S3–S17 (2015).
Bjerkvig, R., Tysnes, B. B., Aboody, K. S., Najbauer, J. & Terzis, A. J. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat. Rev. Cancer 5, 899–904 (2005).
Matsui, W. et al. Characterization of clonogenic multiple myeloma cells. Blood 103, 2332–2336 (2004).
Hill, R. P. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res. 66, 1891–1895 (2006). discussion 1890.
Huntly, B. J. & Gilliland, D. G. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Rev. Cancer 5, 311–321 (2005).
Kanwar, S. S., Yu, Y., Nautiyal, J., Patel, B. B. & Majumdar, A. P. The Wnt/beta-catenin pathway regulates growth and maintenance of colonospheres. Mol. Cancer 9, 212 (2010).
Li, C. et al. Identification of pancreatic cancer stem cells. Cancer Res. 67, 1030–1037 (2007).
Wang, J. et al. Notch promotes radioresistance of glioma stem cells. Stem Cells 28, 17–28 (2010).
Pardal, R., Clarke, M. F. & Morrison, S. J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 3, 895–902 (2003).
Hemmati, H. D. et al. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl Acad. Sci. USA 100, 15178–15183 (2003).
Jin, X., Jin, X. & Kim, H. Cancer stem cells and differentiation therapy. Tumour Biol. 39, 1010428317729933 (2017).
Huang, Z., Wu, T., Liu, A. Y. & Ouyang, G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget 6, 39550–39563 (2015).
Bussolati, B., Bruno, S., Grange, C., Ferrando, U. & Camussi, G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 22, 3696–3705 (2008).
Ricci-Vitiani, L. et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468, 824–828 (2010).
Xiong, Y. Q. et al. Human hepatocellular carcinoma tumor-derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin. Cancer Res. 15, 4838–4846 (2009).
Chaffer, C. L. & Weinberg, R. A. A perspective on cancer cell metastasis. Science 331, 1559–1564 (2011).
Le, N. H., Franken, P. & Fodde, R. Tumour–stroma interactions in colorectal cancer: converging on beta-catenin activation and cancer stemness. Br. J. Cancer 98, 1886–1893 (2008).
Mani, S. A. et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J. & Clarke, M. F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl Acad. Sci. USA 100, 3983–3988 (2003).
Hermann, P. C. et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1, 313–323 (2007).
Collins, A. T., Berry, P. A., Hyde, C., Stower, M. J. & Maitland, N. J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 65, 10946–10951 (2005).
Garcia-Mayea, Y., Mir, C., Masson, F., Paciucci, R. & ME, L. L. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. S1044-579X (2019).
Gottesman, M. M., Fojo, T. & Bates, S. E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58 (2002).
Ginestier, C. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 (2007).
Singh, S. et al. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic. Biol. Med. 56, 89–101 (2013).
Diehn, M. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–783 (2009).
Cojoc, M., Mabert, K., Muders, M. H. & Dubrovska, A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin. Cancer Biol. 31, 16–27 (2015).
Tang, D. G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 22, 457–472 (2012).
Hirschmann-Jax, C. et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl Acad. Sci. USA 101, 14228–14233 (2004).
Quintana, E. et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 18, 510–523 (2010).
Singh, S. K. et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004).
van den Hoogen, C. et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 70, 5163–5173 (2010).
Zhang, W. C. et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 148, 259–272 (2012).
Miltenyi, S., Muller, W., Weichel, W. & Radbruch, A. High gradient magnetic cell separation with MACS. Cytometry 11, 231–238 (1990).
de Wynter, E. A. et al. Comparison of purity and enrichment of CD34+ cells from bone marrow, umbilical cord and peripheral blood (primed for apheresis) using five separation systems. Stem Cells 13, 524–532 (1995).
Moghbeli, M., Moghbeli, F., Forghanifard, M. M. & Abbaszadegan, M. R. Cancer stem cell detection and isolation. Med. Oncol. 31, 69 (2014).
Goodell, M. A., Brose, K., Paradis, G., Conner, A. S. & Mulligan, R. C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 183, 1797–1806 (1996).
Pattabiraman, D. R. & Weinberg, R. A. Tackling the cancer stem cells—what challenges do they pose? Nat. Rev. Drug Discov. 13, 497–512 (2014).
Zhou, S. et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 7, 1028–1034 (2001).
Moserle, L., Ghisi, M., Amadori, A. & Indraccolo, S. Side population and cancer stem cells: therapeutic implications. Cancer Lett. 288, 1–9 (2010).
Haraguchi, N. et al. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells 24, 506–513 (2006).
Szotek, P. P. et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl Acad. Sci. USA 103, 11154–11159 (2006).
Feuring-Buske, M. & Hogge, D. E. Hoechst 33342 efflux identifies a subpopulation of cytogenetically normal CD34(+)CD38(−) progmetabolism, cell growth, and tumorigenesisenitor cells from patients with acute myeloid leukemia. Blood 97, 3882–3889 (2001).
Wang, M., Wang, Y. & Zhong, J. Side population cells and drug resistance in breast cancer. Mol. Med. Rep. 11, 4297–4302 (2015).
Ho, M. M., Ng, A. V., Lam, S. & Hung, J. Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 67, 4827–4833 (2007).
Wang, J., Guo, L. P., Chen, L. Z., Zeng, Y. X. & Lu, S. H. Identification of cancer stem cell-like side population cells in human nasopharyngeal carcinoma cell line. Cancer Res. 67, 3716–3724 (2007).
Chiba, T. et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 44, 240–251 (2006).
Montanaro, F. et al. Demystifying SP cell purification: viability, yield, and phenotype are defined by isolation parameters. Exp. Cell Res. 298, 144–154 (2004).
Fang, D. et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 65, 9328–9337 (2005).
Lobo, N. A., Shimono, Y., Qian, D. & Clarke, M. F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 23, 675–699 (2007).
Taylor, M. D. et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8, 323–335 (2005).
O'Brien, C. A., Kreso, A. & Jamieson, C. H. Cancer stem cells and self-renewal. Clin. Cancer Res. 16, 3113–3120 (2010).
van Stijn, A. et al. Differences between the CD34+ and CD34− blast compartments in apoptosis resistance in acute myeloid leukemia. Haematologica 88, 497–508 (2003).
Chiodi, I., Belgiovine, C., Dona, F., Scovassi, A. I. & Mondello, C. Drug treatment of cancer cell lines: a way to select for cancer stem cells? Cancers 3, 1111–1128 (2011).
Bhaskara, V. K., Mohanam, I., Rao, J. S. & Mohanam, S. Intermittent hypoxia regulates stem-like characteristics and differentiation of neuroblastoma cells. PLoS ONE 7, e30905 (2012).
Liu, W. H. et al. Efficient enrichment of hepatic cancer stem-like cells from a primary rat HCC model via a density gradient centrifugation-centered method. PLoS ONE 7, e35720 (2012).
Rahimi, K., Fuchtbauer, A. C., Fathi, F., Mowla, S. J. & Fuchtbauer, E. M. Isolation of cancer stem cells by selection for miR-302 expressing cells. PeerJ 7, e6635 (2019).
Eun, K., Ham, S. W. & Kim, H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 50, 117–125 (2017).
Zhang, H. & Wang, Z. Z. Mechanisms that mediate stem cell self-renewal and differentiation. J. Cell. Biochem. 103, 709–718 (2008).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Maherali, N. et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 1, 55–70 (2007).
Okita, K., Ichisaka, T. & Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 448, 313–317 (2007).
Nichols, J. et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 95, 379–391 (1998).
Jerabek, S., Merino, F., Scholer, H. R. & Cojocaru, V. OCT4: dynamic DNA binding pioneers stem cell pluripotency. Biochim. Biophys. Acta 1839, 138–154 (2014).
Du, Z. et al. Oct4 is expressed in human gliomas and promotes colony formation in glioma cells. Glia 57, 724–733 (2009).
Murakami, S. et al. SRY and OCT4 are required for the acquisition of cancer stem cell-like properties and are potential differentiation therapy targets. Stem Cells 33, 2652–2663 (2015).
Ponti, D. et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 65, 5506–5511 (2005).
Chen, Y. C. et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE 3, e2637 (2008).
Gwak, J. M., Kim, M., Kim, H. J., Jang, M. H. & Park, S. Y. Expression of embryonal stem cell transcription factors in breast cancer: Oct4 as an indicator for poor clinical outcome and tamoxifen resistance. Oncotarget 8, 36305–36318 (2017).
Song, B. et al. OCT4 directly regulates stemness and extracellular matrix-related genes in human germ cell tumours. Biochem. Biophys. Res. Commun. 503, 1980–1986 (2018).
Rodriguez-Pinilla, S. M. et al. Sox2: a possible driver of the basal-like phenotype in sporadic breast cancer. Mod. Pathol. 20, 474–481 (2007).
Hagerstrand, D. et al. Identification of a SOX2-dependent subset of tumor- and sphere-forming glioblastoma cells with a distinct tyrosine kinase inhibitor sensitivity profile. Neuro-Oncology 13, 1178–1191 (2011).
Gangemi, R. M. et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 27, 40–48 (2009).
Basu-Roy, U. et al. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene 31, 2270–2282 (2012).
Boumahdi, S. et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511, 246–250 (2014).
Chambers, I. et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113, 643–655 (2003).
Nagata, T. et al. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer 21, 96–101 (2014).
Lin, T., Ding, Y. Q. & Li, J. M. Overexpression of Nanog protein is associated with poor prognosis in gastric adenocarcinoma. Med. Oncol. 29, 878–885 (2012).
Yu, C. C. et al. MicroRNA let-7a represses chemoresistance and tumourigenicity in head and neck cancer via stem-like properties ablation. Oral Oncol. 47, 202–210 (2011).
Chiou, S. H. et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial–mesenchymal transdifferentiation. Cancer Res. 70, 10433–10444 (2010).
Meng, H. M. et al. Over-expression of Nanog predicts tumor progression and poor prognosis in colorectal cancer. Cancer Biol. Ther. 9, 295–302 (2010).
Ibrahim, E. E. et al. Embryonic NANOG activity defines colorectal cancer stem cells and modulates through AP1- and TCF-dependent mechanisms. Stem Cells 30, 2076–2087 (2012).
Wang, X. Q. et al. Epigenetic regulation of pluripotent genes mediates stem cell features in human hepatocellular carcinoma and cancer cell lines. PLoS ONE 8, e72435 (2013).
Jeter, C. R. et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells 27, 993–1005 (2009).
Lu, Y. et al. Knockdown of Oct4 and Nanog expression inhibits the stemness of pancreatic cancer cells. Cancer Lett. 340, 113–123 (2013).
Flandez, M., Guilmeau, S., Blache, P. & Augenlicht, L. H. KLF4 regulation in intestinal epithelial cell maturation. Exp. Cell Res. 314, 3712–3723 (2008).
Cho, Y. G. et al. Genetic and epigenetic analysis of the KLF4 gene in gastric cancer. APMIS 115, 802–808 (2007).
Bianchi, F. et al. Lung cancers detected by screening with spiral computed tomography have a malignant phenotype when analyzed by cDNA microarray. Clin. Cancer Res. 10, 6023–6028 (2004).
Li, Q. et al. Dysregulated Kruppel-like factor 4 and vitamin D receptor signaling contribute to progression of hepatocellular carcinoma. Gastroenterology 143, 799–810 (2012). e792.
Yasunaga, J. et al. Identification of aberrantly methylated genes in association with adult T-cell leukemia. Cancer Res. 64, 6002–6009 (2004).
Tang, H. et al. KLF4 is a tumor suppressor in anaplastic meningioma stem-like cells and human meningiomas. J. Mol. Cell. Biol. 9, 315–324 (2017).
Ohnishi, S. et al. Downregulation and growth inhibitory effect of epithelial-type Kruppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem. Biophys. Res. Commun. 308, 251–256 (2003).
Luo, A. et al. Discovery of Ca2+-relevant and differentiation-associated genes downregulated in esophageal squamous cell carcinoma using cDNA microarray. Oncogene 23, 1291–1299 (2004).
Piestun, D. et al. Nanog transforms NIH3T3 cells and targets cell-type restricted genes. Biochem. Biophys. Res. Commun. 343, 279–285 (2006).
Foster, K. W. et al. Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ. 10, 423–434 (1999).
Riverso, M., Montagnani, V. & Stecca, B. KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene 36, 3322–3333 (2017).
Tien, Y. T. et al. Downregulation of the KLF4 transcription factor inhibits the proliferation and migration of canine mammary tumor cells. Vet. J. 205, 244–253 (2015).
Bretones, G., Delgado, M. D. & Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 1849, 506–516 (2015).
Dang, C. V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harbor Perspect. Med. 3, a014217 (2013).
Conzen, S. D. et al. Induction of cell cycle progression and acceleration of apoptosis are two separable functions of c-Myc: transrepression correlates with acceleration of apoptosis. Mol. Cell. Biol. 20, 6008–6018 (2000).
Galardi, S. et al. Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 17, 1872–1889 (2016).
Beer, S. et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol. 2, e332 (2004).
Jacobs, J. J. et al. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 13, 2678–2690 (1999).
Cartwright, P. et al. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132, 885–896 (2005).
Liu, Z. et al. SOD2 is a C-myc target gene that promotes the migration and invasion of tongue squamous cell carcinoma involving cancer stem-like cells. Int. J. Biochem. Cell Biol. 60, 139–146 (2015).
Pfister, S. et al. Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J. Clin. Oncol. 27, 1627–1636 (2009).
Rickman, D. S., Schulte, J. H. & Eilers, M. The expanding world of N-MYC-driven tumors. Cancer Discov. 8, 150–163 (2018).
Hirvonen, H., Hukkanen, V., Salmi, T. T., Pelliniemi, T. T. & Alitalo, R. L-myc and N-myc in hematopoietic malignancies. Leuk. Lymphoma 11, 197–205 (1993).
Shachaf, C. M. et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 431, 1112–1117 (2004).
Ellwood-Yen, K. et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 4, 223–238 (2003).
Logan, C. Y. & Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 (2004).
Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 (2014).
Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 51, 1357–1369 (2017).
Latres, E., Chiaur, D. S. & Pagano, M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene 18, 849–854 (1999).
Metcalfe, C., Mendoza-Topaz, C., Mieszczanek, J. & Bienz, M. Stability elements in the LRP6 cytoplasmic tail confer efficient signalling upon DIX-dependent polymerization. J. Cell Sci. 123, 1588–1599 (2010).
Tree, D. R. et al. Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell 109, 371–381 (2002).
Habas, R., Kato, Y. & He, X. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell 107, 843–854 (2001).
Kikuchi, A., Yamamoto, H., Sato, A. & Matsumoto, S. New insights into the mechanism of Wnt signaling pathway activation. Int. Rev. Cell. Mol. Biol. 291, 21–71 (2011).
Gao, C. & Chen, Y. G. Dishevelled: the hub of Wnt signaling. Cell. Signal. 22, 717–727 (2010).
Thompson, J. J. & Williams, C. S. Protein phosphatase 2A in the regulation of Wnt signaling stem cells, and cancer. Genes 9, 121 (2018).
Abd El-Rehim, D. & Ali, M. M. Aberrant expression of beta-catenin in invasive ductal breast carcinomas. J. Egypt. Natl Cancer Inst. 21, 185–195 (2009).
Adachi, S. et al. Role of a BCL9-related beta-catenin-binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res. 64, 8496–8501 (2004).
Ishigaki, K. et al. Aberrant localization of beta-catenin correlates with overexpression of its target gene in human papillary thyroid cancer. J. Clin. Endocrinol. Metab. 87, 3433–3440 (2002).
Kudo, J. et al. Aberrant nuclear localization of beta-catenin without genetic alterations in beta-catenin or Axin genes in esophageal cancer. World J. Surg. Oncol. 5, 21 (2007).
Pan, T., Xu, J. & Zhu, Y. Self-renewal molecular mechanisms of colorectal cancer stem cells. Int. J. Mol. Med. 39, 9–20 (2017).
Mazzoni, S. M. & Fearon, E. R. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 355, 1–8 (2014).
Morin, P. J. Beta-catenin signaling and cancer. BioEssays 21, 1021–1030 (1999).
Laurent-Puig, P. et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 120, 1763–1773 (2001).
Clements, W. M. et al. Beta-catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res. 62, 3503–3506 (2002).
Liang, J. et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 27, 329–351 (2017).
Zhang, K. et al. WNT/beta-catenin directs self-renewal symmetric cell division of hTERT(high) prostate cancer stem cells. Cancer Res. 77, 2534–2547 (2017).
Hwang, W. L. et al. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat. Cell Biol. 16, 268–280 (2014).
Fang, L. et al. Aberrantly expressed miR-582-3p maintains lung cancer stem cell-like traits by activating Wnt/β-catenin signalling. Nat. Commun. 6, 8640 (2015).
Clevers, H. & Nusse, R. Wnt/β-catenin signaling and disease. Cell 149 (2012).
Weinberg, R. A. The many faces of tumor dormancy. APMIS 116, 548–551 (2008).
Reya, T. λ et al. Stem cells, cancer, and cancer stem cells. Nature 414, 105 (2001).
Giancotti, F. G. Mechanisms governing metastatic dormancy and reactivation. Cell 155, 750–764 (2013).
O"Connell, J. T. et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl Acad. Sci. USA 108, 16002–16007 (2011).
Zhao, Z. et al. PKM2 promotes stemness of breast cancer cell by through Wnt/beta-catenin pathway. Tumour Biol. 37, 4223–4234 (2016).
Chen, J. F. et al. EZH2 promotes colorectal cancer stem-like cell expansion by activating p21cip1-Wnt/β-catenin signaling. Oncotarget 7, 41540–41558 (2016).
Ji, C. et al. Capillary morphogenesis gene 2 maintains gastric cancer stem-like cell phenotype by activating a Wnt/beta-catenin pathway. Oncogene 37, 3953–3966 (2018).
Wang, T. et al. SMYD3 controls a Wnt-responsive epigenetic switch for ASCL2 activation and cancer stem cell maintenance. Cancer Lett. 430, 11–24 (2018).
Wang, Y. et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 16, 413–425 (2015).
Zhu, P. et al. lnc-beta-Catm elicits EZH2-dependent beta-catenin stabilization and sustains liver CSC self-renewal. Nat. Struct. Mol. Biol. 23, 631–639 (2016).
Chen, Z., Yao, L., Liu, Y. & Zhu, P. LncTIC1 interacts with beta-catenin to drive liver TIC self-renewal and liver tumorigenesis. Cancer Lett. 430, 88–96 (2018).
Chai, S. et al. Octamer 4/microRNA-1246 signaling axis drives Wnt/beta-catenin activation in liver cancer stem cells. Hepatology 64, 2062–2076 (2016).
Mo, X. M., Li, H. H., Liu, M. & Li, Y. T. Downregulation of GSK3beta by miR-544a to maintain self-renewal ability of lung caner stem cells. Oncol. Lett. 8, 1731–1734 (2014).
Wu, K., Ma, L. & Zhu, J. miR4835p promotes growth, invasion and selfrenewal of gastric cancer stem cells by Wnt/betacatenin signaling. Mol. Med. Rep. 14, 3421–3428 (2016).
Ordonez-Moran, P., Dafflon, C., Imajo, M., Nishida, E. & Huelsken, J. HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 28, 815–829 (2015).
Cai, W. et al. PMP22 regulates self-renewal and chemoresistance of gastric cancer cells. Mol. Cancer Ther. 16, 1187–1198 (2017).
Lettini, G. et al. TRAP1 regulates stemness through Wnt/beta-catenin pathway in human colorectal carcinoma. Cell Death Differ. 23, 1792–1803 (2016).
Lv, Z. et al. Expression and functional regulation of stemness gene Lgr5 in esophageal squamous cell carcinoma. Oncotarget 8, 26492–26504 (2017).
Mu, J. et al. Dickkopf-related protein 2 induces G0/G1 arrest and apoptosis through suppressing Wnt/β-catenin signaling and is frequently methylated in breast cancer. Oncotarget 8, 39443–39459 (2017).
Yin, X. et al. DACT1, an antagonist to Wnt/beta-catenin signaling, suppresses tumor cell growth and is frequently silenced in breast cancer. Breast Cancer Res. 15, R23 (2013).
Li, L. et al. The human cadherin 11 is a pro-apoptotic tumor suppressor modulating cell stemness through Wnt/beta-catenin signaling and silenced in common carcinomas. Oncogene 31, 3901–3912 (2012).
Zhu, J. et al. Wnt/beta-catenin pathway mediates (−)-Epigallocatechin-3-gallate (EGCG) inhibition of lung cancer stem cells. Biochem. Biophys. Res. Commun. 482, 15–21 (2017).
Kim, J. Y. et al. CWP232228 targets liver cancer stem cells through Wnt/beta-catenin signaling: a novel therapeutic approach for liver cancer treatment. Oncotarget 7, 20395–20409 (2016).
Shi, L., Fei, X., Wang, Z. & You, Y. PI3K inhibitor combined with miR-125b inhibitor sensitize TMZ-induced anti-glioma stem cancer effects through inactivation of Wnt/beta-catenin signaling pathway. Vitr. Cell Dev. Biol. Anim. 51, 1047–1055 (2015).
DiMeo, T. A. et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 69, 5364–5373 (2009).
Li, Q. et al. FZD8, a target of p53, promotes bone metastasis in prostate cancer by activating canonical Wnt/beta-catenin signaling. Cancer Lett. 402, 166–176 (2017).
de Lau, W. et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293–297 (2011).
Carmon, K. S., Gong, X., Lin, Q., Thomas, A. & Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl Acad. Sci. USA 108, 11452–11457 (2011).
Todaro, M. et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 14, 342–356 (2014).
Matzke-Ogi, A. et al. Inhibition of tumor growth and metastasis in pancreatic cancer models by interference with CD44v6 signaling. Gastroenterology 150, 513–525 (2016). e510.
Su, J., Wu, S., Wu, H., Li, L. & Guo, T. CD44 is functionally crucial for driving lung cancer stem cells metastasis through Wnt/beta-catenin-FoxM1-Twist signaling. Mol. Carcinogen. 55, 1962–1973 (2016).
Imajo, M., Miyatake, K., Iimura, A., Miyamoto, A. & Nishida, E. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/beta-catenin signalling. EMBO J. 31, 1109–1122 (2012).
Diamantopoulou, Z. et al. TIAM1 antagonizes TAZ/YAP both in the destruction complex in the cytoplasm and in the nucleus to inhibit invasion of intestinal epithelial cells. Cancer Cell 31, 621–634 (2017). e626.
Bray, S. J. Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell. Biol. 7, 678–689 (2006).
Schroeter, E. H., Kisslinger, J. A. & Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386 (1998).
Kovall, R. A. More complicated than it looks: assembly of Notch pathway transcription complexes. Oncogene 27, 5099–5109 (2008).
Ranganathan, P., Weaver, K. L. & Capobianco, A. J. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat. Rev. Cancer 11, 338–351 (2011).
Zhou, W. et al. The AKT1/NF-kappaB/Notch1/PTEN axis has an important role in chemoresistance of gastric cancer cells. Cell Death Dis. 4, e847 (2013).
Wu, F., Stutzman, A. & Mo, Y. Y. Notch signaling and its role in breast cancer. Front. Biosci. 12, 4370–4383 (2007).
Zhang, Y., Li, B., Ji, Z. Z. & Zheng, P. S. Notch1 regulates the growth of human colon cancers. Cancer 116, 5207–5218 (2010).
Gupta, A. et al. Neuroendocrine differentiation in the 12T-10 transgenic prostate mouse model mimics endocrine differentiation of pancreatic beta cells. Prostate 68, 50–60 (2008).
Lefort, K. et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 21, 562–577 (2007).
Konishi, J. et al. Notch3 cooperates with the EGFR pathway to modulate apoptosis through the induction of bim. Oncogene 29, 589–596 (2010).
Viatour, P. et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J. Exp. Med. 208, 1963–1976 (2011).
Parr, C., Watkins, G. & Jiang, W. G. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int. J. Mol. Med. 14, 779–786 (2004).
Li, L. et al. Notch signaling pathway networks in cancer metastasis: a new target for cancer therapy. Med. Oncol. 34, 180 (2017).
Espinoza, I. & Miele, L. Notch inhibitors for cancer treatment. Pharmacol. Ther. 139, 95–110 (2013).
Stylianou, S., Clarke, R. B. & Brennan, K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 66, 1517–1525 (2006).
Harrison, H. et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 70, 709–718 (2010).
Kang, L. et al. MicroRNA-34a suppresses the breast cancer stem cell-like characteristics by downregulating Notch1 pathway. Cancer Sci. 106, 700–708 (2015).
Miao, Z. F. et al. DLL4 overexpression increases gastric cancer stem/progenitor cell self-renewal ability and correlates with poor clinical outcome via Notch-1 signaling pathway activation. Cancer Med. 6, 245–257 (2017).
Zhang, C. et al. Actin cytoskeleton regulator Arp2/3 complex is required for DLL1 activating Notch1 signaling to maintain the stem cell phenotype of glioma initiating cells. Oncotarget 8, 33353–33364 (2017).
Garcia-Heredia, J. M., Lucena-Cacace, A., Verdugo-Sivianes, E. M., Perez, M. & Carnero, A. The Cargo Protein MAP17 (PDZK1IP1) regulates the cancer stem cell pool activating the notch pathway by abducting NUMB. Clin. Cancer Res. 23, 3871–3883 (2017).
Wang, R. et al. iNOS promotes CD24(+)CD133(+) liver cancer stem cell phenotype through a TACE/ADAM17-dependent Notch signaling pathway. Proc. Natl Acad. Sci. USA 115, E10127–E10136 (2018).
Lee, S. H. et al. TNFα enhances cancer stem cell-like phenotype via Notch-Hes1 activation in oral squamous cell carcinoma cells. Biochem. Biophys. Res. Commun. 424, 58–64 (2012).
Zhang, F. et al. Overexpression of PER3 inhibits self-renewal capability and chemoresistance of colorectal cancer stem-like cells via inhibition of notch and beta-catenin signaling. Oncol. Res. 25, 709–719 (2017).
Yu, F. et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene 30, 2161–2172 (2011).
Choi, S. et al. BMP-4 enhances epithelial–mesenchymal transition and cancer stem cell properties of breast cancer cells via Notch signaling. Sci. Rep. 9, 11724 (2019).
Buckley, N. E. et al. BRCA1 is a key regulator of breast differentiation through activation of Notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic Acids Res. 41, 8601–8614 (2013).
Rodrigues, M. et al. GLI3 knockdown decreases stemness, cell proliferation and invasion in oral squamous cell carcinoma. Int. J. Oncol. 53, 2458–2472 (2018).
Kao, S. H., Wu, K. J. & Lee, W. H. Hypoxia, epithelial–mesenchymal transition, and TET-mediated epigenetic changes. J. Clin. Med. 5, (2016).
Sahlgren, C., Gustafsson, M. V., Jin, S., Poellinger, L. & Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl Acad. Sci. USA 105, 6392–6397 (2008).
Xing, F. et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 30, 4075–4086 (2011).
Xie, M. et al. Activation of Notch-1 enhances epithelial–mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 113, 1501–1513 (2012).
Qiang, L. et al. HIF-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 19, 284–294 (2012).
Nwaeburu, C. C., Abukiwan, A., Zhao, Z. & Herr, I. Quercetin-induced miR-200b-3p regulates the mode of self-renewing divisions in pancreatic cancer. Mol. Cancer 16, 23 (2017).
Lu, J. et al. MiR-26a inhibits stem cell-like phenotype and tumor growth of osteosarcoma by targeting Jagged1. Oncogene 36, 231–241 (2017).
Merchant, A. A. & Matsui, W. Targeting Hedgehog—a cancer stem cell pathway. Clin. Cancer Res. 16, 3130–3140 (2010).
Binder, M. et al. Functionally distinctive Ptch receptors establish multimodal hedgehog signaling in the tooth epithelial stem cell niche. Stem Cells 37, 1238–1248 (2019).
Hui, C. C. & Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 27, 513–537 (2011).
Wang, B., Fallon, J. F. & Beachy, P. A. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434 (2000).
Sasaki, H., Nishizaki, Y., Hui, C., Nakafuku, M. & Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 126, 3915–3924 (1999).
Li, S. H., Fu, J., Watkins, D. N., Srivastava, R. K. & Shankar, S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog-GLI pathway. Mol. Cell. Biochem. 373, 217–227 (2013).
Petrova, R. & Joyner, A. L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 141, 3445–3457 (2014).
Chaudhry, P., Singh, M., Triche, T. J., Guzman, M. & Merchant, A. A. GLI3 repressor determines Hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood 129, 3465–3475 (2017).
Zhou, Q. & Kalderon, D. Hedgehog activates fused through phosphorylation to elicit a full spectrum of pathway responses. Dev. Cell 20, 802–814 (2011).
Robbins, D. J., Fei, D. L. & Riobo, N. A. The Hedgehog signal transduction network. Sci. Signal. 5, re6 (2012).
McMillan, R. & Matsui, W. Molecular pathways: the hedgehog signaling pathway in cancer. Clin. Cancer Res. 18, 4883–4888 (2012).
Villegas, V. E. et al. Tamoxifen treatment of breast cancer cells: impact on hedgehog/GLI1 signaling. Int. J. Mol. Sci. 17, 308 (2016).
Pietanza, M. C. et al. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer 99, 23–30 (2016).
Amantini, C. et al. Capsaicin triggers autophagic cell survival which drives epithelial mesenchymal transition and chemoresistance in bladder cancer cells in an Hedgehog-dependent manner. Oncotarget 7, 50180–50194 (2016).
Xu, Y., An, Y., Wang, X., Zha, W. & Li, X. Inhibition of the Hedgehog pathway induces autophagy in pancreatic ductal adenocarcinoma cells. Oncol. Rep. 31, 707–712 (2014).
Campbell, V. T. et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor IPI-926 directly inhibits sarcoma cell growth. Mol. Cancer Ther. 13, 1259–1269 (2014).
Zibat, A. et al. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene 29, 6323–6330 (2010).
Louis, C. U. & Shohet, J. M. Neuroblastoma: molecular pathogenesis and therapy. Annu. Rev. Med. 66, 49–63 (2015).
Buonamici, S. et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2, 51ra70 (2010).
Yoon, C. et al. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin. Cancer Res. 20, 3974–3988 (2014).
Hahn, H. et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85, 841–851 (1996).
Pietsch, T. et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 57, 2085–2088 (1997).
Taylor, M. D. et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 31, 306–310 (2002).
Wong, A. J. et al. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl Acad. Sci. USA 84, 6899–6903 (1987).
Zeng, C. et al. SPOP suppresses tumorigenesis by regulating Hedgehog/Gli2 signaling pathway in gastric cancer. J. Exp. Clin. Cancer Res. 33, 75 (2014).
Yang, L., Xie, G., Fan, Q. & Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 29, 469–481 (2010).
Dierks, C. et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 14, 238–249 (2008).
Zhao, C. et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458, 776–779 (2009).
Po, A. et al. Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene 36, 4641–4652 (2017).
Clement, V., Sanchez, P., de Tribolet, N., Radovanovic, I. & Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 17, 165–172 (2007).
Zhou, A. et al. Gli1-induced deubiquitinase USP48 aids glioblastoma tumorigenesis by stabilizing Gli1. EMBO Rep. 18, 1318–1330 (2017).
Zhang, S. et al. Inhibition of CK2alpha down-regulates Hedgehog/Gli signaling leading to a reduction of a stem-like side population in human lung cancer cells. PLoS ONE 7, e38996 (2012).
Wen, J. et al. WIP1 modulates responsiveness to Sonic Hedgehog signaling in neuronal precursor cells and medulloblastoma. Oncogene 35, 5552–5564 (2016).
Pandolfi, S. et al. WIP1 phosphatase modulates the Hedgehog signaling by enhancing GLI1 function. Oncogene 32, 4737–4747 (2013).
Raducu, M. et al. SCF (Fbxl17) ubiquitylation of Sufu regulates Hedgehog signaling and medulloblastoma development. EMBO J. 35, 1400–1416 (2016).
Yang, Y. et al. RARalpha2 expression confers myeloma stem cell features. Blood 122, 1437–1447 (2013).
Song, K. B., Liu, W. J. & Jia, S. S. miR-219 inhibits the growth and metastasis of TSCC cells by targeting PRKCI. Int. J. Clin. Exp. Med. 7, 2957–2965 (2014).
Justilien, V. et al. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 25, 139–151 (2014).
Airoldi, I. et al. Interleukin-27 re-educates intratumoral myeloid cells and down-regulates stemness genes in non-small cell lung cancer. Oncotarget 6, 3694–3708 (2015).
Li, C. et al. GALNT1-mediated glycosylation and activation of sonic hedgehog signaling maintains the self-renewal and tumor-initiating capacity of bladder cancer stem cells. Cancer Res. 76, 1273–1283 (2016).
Memmi, E. M. et al. p63 sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc. Natl Acad. Sci. USA 112, 3499–3504 (2015).
Han, B. et al. FOXC1 activates smoothened-independent hedgehog signaling in basal-like breast cancer. Cell Rep. 13, 1046–1058 (2015).
Wu, J. et al. The long non-coding RNA LncHDAC2 drives the self-renewal of liver cancer stem cells via activation of Hedgehog signaling. J. Hepatol. 70, 918–929 (2019).
Valenti, G. et al. Cancer stem cells regulate cancer-associated fibroblasts via activation of hedgehog signaling in mammary gland tumors. Cancer Res. 77, 2134–2147 (2017).
Gu, D. et al. Combining hedgehog signaling inhibition with focal irradiation on reduction of pancreatic cancer metastasis. Mol. Cancer Ther. 12, 1038–1048 (2013).
Joost, S. et al. GLI1 inhibition promotes epithelial-to-mesenchymal transition in pancreatic cancer cells. Cancer Res. 72, 88–99 (2012).
Liu, S. et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 66, 6063–6071 (2006).
Song, Z. et al. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS ONE 6, e17687 (2011).
Takahashi, T. et al. Cyclopamine induces eosinophilic differentiation and upregulates CD44 expression in myeloid leukemia cells. Leuk. Res. 35, 638–645 (2011).
Takebe, N., Harris, P. J., Warren, R. Q. & Ivy, S. P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 8, 97–106 (2011).
Tanaka, H. et al. The Hedgehog signaling pathway plays an essential role in maintaining the CD44+CD24−/low subpopulation and the side population of breast cancer cells. Anticancer Res. 29, 2147–2157 (2009).
Kong, Y. et al. Twist1 and Snail link Hedgehog signaling to tumor-initiating cell-like properties and acquired chemoresistance independently of ABC transporters. Stem Cells 33, 1063–1074 (2015).
Zhu, R. et al. TSPAN8 promotes cancer cell stemness via activation of sonic Hedgehog signaling. Nat. Commun. 10, 2863 (2019).
Guo, E., Liu, H. & Liu, X. Overexpression of SCUBE2 inhibits proliferation, migration, and invasion in glioma cells. Oncol. Res. 25, 437–444 (2017).
Tiberi, L. et al. A BCL6/BCOR/SIRT1 complex triggers neurogenesis and suppresses medulloblastoma by repressing Sonic Hedgehog signaling. Cancer Cell 26, 797–812 (2014).
Kim, B. R. et al. RUNX3 suppresses metastasis and stemness by inhibiting Hedgehog signaling in colorectal cancer. Cell Death Differ. 27, 676-694 (2019).
Wang, F. et al. Hedgehog signaling regulates epithelial–mesenchymal transition in pancreatic cancer stem-like cells. J. Cancer 7, 408–417 (2016).
Zinke, J. et al. Beta-catenin-Gli1 interaction regulates proliferation and tumor growth in medulloblastoma. Mol. Cancer 14, 17 (2015).
Tang, B. et al. MicroRNA-324-5p regulates stemness, pathogenesis and sensitivity to bortezomib in multiple myeloma cells by targeting hedgehog signaling. Int. J. Cancer 142, 109–120 (2018).
Miele, E. et al. β-Arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer 17, 488 (2017).
Du, W. et al. Targeting the SMO oncogene by miR-326 inhibits glioma biological behaviors and stemness. Neuro-Oncology 17, 243–253 (2015).
Chandimali, N. et al. MicroRNA-122 negatively associates with peroxiredoxin-II expression in human gefitinib-resistant lung cancer stem cells. Cancer Gene Ther. 26(9–10):292–304(2018).
Zhang, Q., Lenardo, M. J. & Baltimore, D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 (2017).
Hoesel, B. & Schmid, J. A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 12, 86 (2013).
Hayden, M. S. & Ghosh, S. Shared principles in NF-kappaB signaling. Cell 132, 344–362 (2008).
May, M. J. & Ghosh, S. Rel/NF-kappa B and I kappa B proteins: an overview. Semin. Cancer Biol. 8, 63–73 (1997).
Novack, D. V. Role of NF-κB in the skeleton. Cell Res. 21, 169–182 (2011).
Perkins, N. D. & Gilmore, T. D. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ. 13, 759–772 (2006).
Sun, S. C. Non-canonical NF-kappaB signaling pathway. Cell Res. 21, 71–85 (2011).
Xiao, G., Harhaj, E. W. & Sun, S. C. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 7, 401–409 (2001).
Greten, F. R. et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118, 285–296 (2004).
Taniguchi, K. & Karin, M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol. 18, 309–324 (2018).
Denz, U., Haas, P. S., Wasch, R., Einsele, H. & Engelhardt, M. State of the art therapy in multiple myeloma and future perspectives. Eur. J. Cancer 42, 1591–1600 (2006).
Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 1, a000141 (2009).
Prasad, S., Ravindran, J. & Aggarwal, B. B. NF-kappaB and cancer: how intimate is this relationship. Mol. Cell. Biochem. 336, 25–37 (2010).
Brown, M., Cohen, J., Arun, P., Chen, Z. & Van Waes, C. NF-kappaB in carcinoma therapy and prevention. Expert Opin. Ther. Targets 12, 1109–1122 (2008).
Hinz, M. et al. Nuclear factor kappaB-dependent gene expression profiling of Hodgkin’s disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J. Exp. Med. 196, 605–617 (2002).
Richmond, A. Nf-kappa B chemokine gene transcription and tumour growth. Nat. Rev. Immunol. 2, 664–674 (2002).
Gonzalez-Torres, C. et al. NF-kappaB participates in the stem cell phenotype of ovarian cancer cells. Arch. Med. Res. 48, 343–351 (2017).
Vazquez-Santillan, K. et al. NF-kappaBeta-inducing kinase regulates stem cell phenotype in breast cancer. Sci. Rep. 6, 37340 (2016).
Kong, L. et al. Overexpression of SDF-1 activates the NF-kappaB pathway to induce epithelial to mesenchymal transition and cancer stem cell-like phenotypes of breast cancer cells. Int. J. Oncol. 48, 1085–1094 (2016).
Wang, D., Fu, L., Sun, H., Guo, L. & DuBois, R. N. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology 149, 1884–1895 (2015). e1884.
Smith, H. A. & Kang, Y. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. 91, 411–429 (2013).
Zhang, L. et al. CCL21/CCR7 axis contributed to CD133+ pancreatic cancer stem-like cell metastasis via EMT and Erk/NF-κB pathway. PLoS ONE 11, e0158529 (2016).
Wang, X. et al. Bmi-1 regulates stem cell-like properties of gastric cancer cells via modulating miRNAs. J. Hematol. Oncol. 9, 90 (2016).
Li, B. et al. miR-221/222 promote cancer stem-like cell properties and tumor growth of breast cancer via targeting PTEN and sustained Akt/NF-kappaB/COX-2 activation. Chem. Biol. Interact. 277, 33–42 (2017).
Karanikas, V. et al. Foxp3 expression in human cancer cells. J. Transl. Med. 6, 19 (2008).
Liu, S. et al. FOXP3 inhibits cancer stem cell self-renewal via transcriptional repression of COX2 in colorectal cancer cells. Oncotarget 8, 44694–44704 (2017).
Yang, X. et al. MiR-491 attenuates cancer stem cells-like properties of hepatocellular carcinoma by inhibition of GIT-1/NF-kappaB-mediated EMT. Tumour Biol. 37, 201–209 (2016).
Han, D. et al. Disulfiram inhibits TGF-β-induced epithelial–mesenchymal transition and stem-like features in breast cancer via ERK/NF-κB/Snail pathway. Oncotarget 6, 40907–40919 (2015).
Burnett, J. P. et al. Sulforaphane enhances the anticancer activity of taxanes against triple negative breast cancer by killing cancer stem cells. Cancer Lett. 394, 52–64 (2017).
Marquardt, J. U. et al. Curcumin effectively inhibits oncogenic NF-kappaB signaling and restrains stemness features in liver cancer. J. Hepatol. 63, 661–669 (2015).
Levy, D. E. & Darnell, J. E. Jr. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell. Biol. 3, 651–662 (2002).
O’Shea, J. J., Gadina, M. & Schreiber, R. D. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell 109(Suppl.), S121–S131 (2002).
Ihle, J. N. & Kerr, I. M. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 11, 69–74 (1995).
Haan, C., Kreis, S., Margue, C. & Behrmann, I. Jaks and cytokine receptors—an intimate relationship. Biochem. Pharmacol. 72, 1538–1546 (2006).
Yoshimura, A. et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 14, 2816–2826 (1995).
Quintás-Cardama, A. & Verstovsek, S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin. Cancer Res. 19, 1933–1940 (2013).
Stroud, R. M. & Wells, J. A. Mechanistic diversity of cytokine receptor signaling across cell membranes. Sci. STKE 2004, re7 (2004).
Chikuma, S., Kanamori, M., Mise-Omata, S. & Yoshimura, A. Suppressors of cytokine signaling: potential immune checkpoint molecules for cancer immunotherapy. Cancer Sci. 108, 574–580 (2017).
Leonard, W. J. & O'Shea, J. J. Jaks and STATs: biological implications. Annu. Rev. Immunol. 16, 293–322 (1998).
Constantinescu, S. N., Girardot, M. & Pecquet, C. Mining for JAK-STAT mutations in cancer. Trends Biochem. Sci. 33, 122–131 (2008).
Kralovics, R. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 (2005).
Kiladjian, J. J. The spectrum of JAK2-positive myeloproliferative neoplasms. Hematol. Am. Soc. Hematol. Educ. Program 2012, 561–566 (2012).
Levine, R. L. et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7, 387–397 (2005).
Roder, S., Steimle, C., Meinhardt, G. & Pahl, H. L. STAT3 is constitutively active in some patients with Polycythemia rubra vera. Exp. Hematol. 29, 694–702 (2001).
James, C. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 (2005).
Song, J. I. & Grandis, J. R. STAT signaling in head and neck cancer. Oncogene 19, 2489–2495 (2000).
van der Zee, M. et al. IL6/JAK1/STAT3 signaling blockade in endometrial cancer affects the ALDHhi/CD126+ stem-like component and reduces tumor burden. Cancer Res. 75, 3608–3622 (2015).
Lam, L. T. et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 111, 3701–3713 (2008).
Jiang, C. et al. miR-500a-3p promotes cancer stem cells properties via STAT3 pathway in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 36, 99 (2017).
Kanno, H., Sato, H., Yokoyama, T. A., Yoshizumi, T. & Yamada, S. The VHL tumor suppressor protein regulates tumorigenicity of U87-derived glioma stem-like cells by inhibiting the JAK/STAT signaling pathway. Int. J. Oncol. 42, 881–886 (2013).
Karunanithi, S. et al. RBP4-STRA6 pathway drives cancer stem cell maintenance and mediates high-fat diet-induced colon carcinogenesis. Stem Cell Rep. 9, 438–450 (2017).
Yu, H., Pardoll, D. & Jove, R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 (2009).
Rajala, H. L. et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 121, 4541–4550 (2013).
Chambers, I. The molecular basis of pluripotency in mouse embryonic stem cells. Cloning Stem Cells 6, 386–391 (2004).
Zhou, J. et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl Acad. Sci. USA 104, 16158–16163 (2007).
Yang, L. et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-kappaB/Notch1 pathway in non-small cell lung cancer. Int. J. cancer 145, 1099–1110 (2019).
Kim, S. Y. et al. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell. Signal. 25, 961–969 (2013).
Ruan, Z., Yang, X. & Cheng, W. OCT4 accelerates tumorigenesis through activating JAK/STAT signaling in ovarian cancer side population cells. Cancer Manag. Res. 11, 389–399 (2019).
Marotta, L. L. et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Invest. 121, 2723–2735 (2011).
Zhang, X. et al. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 9, 25 (2018).
Zhou, B. et al. Erythropoietin promotes breast tumorigenesis through tumor-initiating cell self-renewal. J. Clin. Invest. 124, 553–563 (2014).
Almiron Bonnin, D. A. et al. Secretion-mediated STAT3 activation promotes self-renewal of glioma stem-like cells during hypoxia. Oncogene 37, 1107–1118 (2018).
Jia, H. et al. The LIM protein AJUBA promotes colorectal cancer cell survival through suppression of JAK1/STAT1/IFIT2 network. Oncogene 36, 2655–2666 (2017).
Che, S. et al. miR-30 overexpression promotes glioma stem cells by regulating Jak/STAT3 signaling pathway. Tumour Biol. 36, 6805–6811 (2015).
Yang, Y. et al. MicroRNA-218 functions as a tumor suppressor in lung cancer by targeting IL-6/STAT3 and negatively correlates with poor prognosis. Mol. Cancer 16, 141 (2017).
Frolik, C. A., Dart, L. L., Meyers, C. A., Smith, D. M. & Sporn, M. B. Purification and initial characterization of a type beta transforming growth factor from human placenta. Proc. Natl Acad. Sci. USA 80, 3676–3680 (1983).
Weiss, A. & Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2, 47–63 (2013).
Brown, J. A. et al. TGF-beta-induced quiescence mediates chemoresistance of tumor-propagating cells in squamous cell carcinoma. Cell Stem Cell 21, 650–664 (2017). e658.
Moustakas, A., Souchelnytskyi, S. & Heldin, C. H. Smad regulation in TGF-beta signal transduction. J. Cell Sci. 114, 4359–4369 (2001).
Massague, J., Seoane, J. & Wotton, D. Smad transcription factors. Genes Dev. 19, 2783–2810 (2005).
Romero, D. et al. Dickkopf-3 regulates prostate epithelial cell acinar morphogenesis and prostate cancer cell invasion by limiting TGF-beta-dependent activation of matrix metalloproteases. Carcinogenesis 37, 18–29 (2016).
Kaowinn, S. et al. Cancer upregulated gene 2 (CUG2), a novel oncogene, promotes stemness-like properties via the NPM1-TGF-beta signaling axis. Biochem. Biophys. Res. Commun. 514, 1278–1284 (2019).
Xia, W. et al. Smad inhibitor induces CSC differentiation for effective chemosensitization in cyclin D1- and TGF-beta/Smad-regulated liver cancer stem cell-like cells. Oncotarget 8, 38811–38824 (2017).
Wang, X. H. et al. TGF-beta1 pathway affects the protein expression of many signaling pathways, markers of liver cancer stem cells, cytokeratins, and TERT in liver cancer HepG2 cells. Tumour Biol. 37, 3675–3681 (2016).
Rodriguez-Garcia, A. et al. TGF-beta1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 284, 3437–3454 (2017).
Muthusamy, B. P. et al. ShcA protects against epithelial–mesenchymal transition through compartmentalized inhibition of TGF-beta-induced Smad activation. PLoS Biol. 13, e1002325 (2015).
Jiang, F. et al. The repressive effect of miR-148a on TGF beta-SMADs signal pathway is involved in the glabridin-induced inhibition of the cancer stem cells-like properties in hepatocellular carcinoma cells. PLoS ONE 9, e96698 (2014).
Yu, D., Shin, H. S., Lee, Y. S. & Lee, Y. C. miR-106b modulates cancer stem cell characteristics through TGF-beta/Smad signaling in CD44-positive gastric cancer cells. Lab. Invest. 94, 1370–1381 (2014).
Tasian, S. K., Teachey, D. T. & Rheingold, S. R. Targeting the PI3K/mTOR pathway in pediatric hematologic malignancies. Front. Oncol. 4, 108 (2014).
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M. & Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell. Biol. 11, 329–341 (2010).
Wang, Q., Chen, X. & Hay, N. Akt as a target for cancer therapy: more is not always better (lessons from studies in mice). Br. J. Cancer 117, 159–163 (2017).
Loewith, R. et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468 (2002).
Kim, D. H. et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 (2002).
Sancak, Y. et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 (2007).
Knowles, M. A., Platt, F. M., Ross, R. L. & Hurst, C. D. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer metastasis Rev. 28, 305–316 (2009).
Duan, S. et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat. Commun. 6, 10068 (2015).
Yuzugullu, H. et al. A PI3K p110beta-Rac signalling loop mediates Pten-loss-induced perturbation of haematopoiesis and leukaemogenesis. Nat. Commun. 6, 8501 (2015).
Fumarola, C., Bonelli, M. A., Petronini, P. G. & Alfieri, R. R. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem. Pharmacol. 90, 197–207 (2014).
Dey, N., De, P. & Leyland-Jones, B. PI3K-AKT-mTOR inhibitors in breast cancers: from tumor cell signaling to clinical trials. Pharmacol. Ther.175, 91–106 (2017).
Offermann, A. et al. MED15 overexpression in prostate cancer arises during androgen deprivation therapy via PI3K/mTOR signaling. Oncotarget 8, 7964–7976 (2017).
Giulino-Roth, L. et al. Inhibition of Hsp90 suppresses PI3K/AKT/mTOR signaling and has antitumor activity in Burkitt lymphoma. Mol. cancer therapeutics 16, 1779–1790 (2017).
Zaidi, A. H. et al. PI3K/mTOR dual inhibitor, LY3023414, demonstrates potent antitumor efficacy against esophageal adenocarcinoma in a rat model. Ann. Surg. 266, 91–98 (2017).
Karki, R., Malireddi, R. K. S., Zhu, Q. & Kanneganti, T. D. NLRC3 regulates cellular proliferation and apoptosis to attenuate the development of colorectal cancer. Cell Cycle 16, 1243–1251 (2017).
Deng, J. et al. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 19, 618 (2019).
Chang, L. et al. Acquisition of epithelial–mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell death Dis. 4, e875 (2013).
Fitzgerald, T. L. et al. Roles of EGFR and KRAS and their downstream signaling pathways in pancreatic cancer and pancreatic cancer stem cells. Adv. Biol. Regul. 59, 65–81 (2015).
Dubrovska, A. et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl Acad. Sci. USA 106, 268–273 (2009).
Keysar, S. B. et al. Regulation of head and neck squamous cancer stem cells by PI3K and SOX2. J. Natl Cancer Inst. 109 (2017).
Yang, C. et al. Downregulation of cancer stem cell properties via mTOR signaling pathway inhibition by rapamycin in nasopharyngeal carcinoma. Int. J. Oncol. 47, 909–917 (2015).
Huang, E. H. et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 69, 3382–3389 (2009).
Nishitani, S., Horie, M., Ishizaki, S. & Yano, H. Branched chain amino acid suppresses hepatocellular cancer stem cells through the activation of mammalian target of rapamycin. PLoS ONE 8, e82346 (2013).
Karki, R. et al. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature 540, 583–587 (2016).
Chen, W. J. & Huang, R. F. S. Low folate stress reprograms cancer stem cell-like potentials and bioenergetics metabolism through activation of mTOR signaling pathway to promote in vitro invasion and in vivo tumorigenicity of lung cancers. J. Nutr. Biochem. S0955286316306994 (2017).
Bonuccelli, G., Sotgia, F. & Lisanti, M. P. Matcha green tea (MGT) inhibits the propagation of cancer stem cells (CSCs), by targeting mitochondrial metabolism, glycolysis and multiple cell signalling pathways. Aging (Albany, NY) 10:1867–1883 (2018).
Issemann, I. & Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347, 645–650 (1990).
Peters, J. M., Shah, Y. M. & Gonzalez, F. J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 12, 181–195 (2012).
Kuenzli, S. & Saurat, J. H. Peroxisome proliferator-activated receptors in cutaneous biology. Br. J. Dermatol. 149, 229–236 (2003).
Elbrecht, A. et al. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem. Biophys. Res. Commun. 224, 431–437 (1996).
Tyagi, S., Gupta, P., Saini, A. S., Kaushal, C. & Sharma, S. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2, 236–240 (2011).
Sertznig, P. & Reichrath, J. Peroxisome proliferator-activated receptors (PPARs) in dermatology: challenge and promise. Dermatol. Endocrinol. 3, 130–135 (2011).
Houseknecht, K. L., Cole, B. M. & Steele, P. J. Peroxisome proliferator-activated receptor gamma (PPARgamma) and its ligands: a review. Domest. Anim. Endocrinol. 22, 1–23 (2002).
Yousefnia, S., Momenzadeh, S., Seyed Forootan, F., Ghaedi, K. & Nasr Esfahani, M. H. The influence of peroxisome proliferator-activated receptor gamma (PPARgamma) ligands on cancer cell tumorigenicity. Gene 649, 14–22 (2018).
Kramer, K., Wu, J. & Crowe, D. L. Tumor suppressor control of the cancer stem cell niche. Oncogene 35, 4165–4178 (2016).
Wang, Y. et al. The combinatory effects of PPAR-gamma agonist and survivin inhibition on the cancer stem-like phenotype and cell proliferation in bladder cancer cells. Int. J. Mol. Med. 34, 262–268 (2014).
Basu-Roy, U. et al. PPARgamma agonists promote differentiation of cancer stem cells by restraining YAP transcriptional activity. Oncotarget 7,60954–60970 (2016).
Giampietri, C. et al. Lipid storage and autophagy in melanoma cancer cells. Int. J. Mol. Sci. 18, (2017).
Deng, X. et al. Ovarian cancer stem cells induce the M2 polarization of macrophages through the PPARgamma and NF-kappaB pathways. Int. J. Mol. Med. 36, 449–454 (2015).
Bigoni-Ordonez, G. D. et al. Molecular iodine inhibits the expression of stemness markers on cancer stem-like cells of established cell lines derived from cervical cancer. BMC Cancer 18, 928 (2018).
Liu, L. et al. Inhibition of oxidative stress-elicited AKT activation facilitates PPARgamma agonist-mediated inhibition of stem cell character and tumor growth of liver cancer cells. PLoS ONE 8, e73038 (2013).
Rinaldi, L. et al. Loss of Dnmt3a and Dnmt3b does not affect epidermal homeostasis but promotes squamous transformation through PPAR-gamma. eLife 6, pii: e21697 (2017).
Wang, H., Zheng, S., Tu, Y. & Zhang, Y. Screening and identification of novel drug-resistant genes in CD133+ and CD133− lung adenosarcoma cells using cDNA microarray. Chin. J. Lung Cancer 17, 437–443 (2014).
Lebleu, V. S. et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 16, 992–1003 (2014).
Wang, X. et al. AMPK promotes SPOP-mediated NANOG degradation to regulate prostate cancer cell stemness. Dev. Cell. 48, 345–360 (2019). e7.
Kim, J. H. et al. Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci. Rep. 8, 409 (2018).
Maehara, O. et al. Metformin regulates the expression of CD133 through the AMPK-CEBPβ pathway in hepatocellular carcinoma cell lines. Neoplasia 21, 545–556 (2019).
Katoh, M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 3, 30–38 (2007).
Schon, S. et al. Beta-catenin regulates NF-kappaB activity via TNFRSF19 in colorectal cancer cells. Int. J. Cancer 135, 1800–1811 (2014).
Liu, D. et al. Reduced CD146 expression promotes tumorigenesis and cancer stemness in colorectal cancer through activating Wnt/beta-catenin signaling. Oncotarget 7, 40704–40718 (2016).
Du, Q. et al. Wnt/beta-catenin signaling regulates cytokine-induced human inducible nitric oxide synthase expression by inhibiting nuclear factor-kappaB activation in cancer cells. Cancer Res. 69, 3764–3771 (2009).
Moreau, M., Mourah, S. & Dosquet, C. Beta-catenin and NF-kappaB cooperate to regulate the uPA/uPAR system in cancer cells. Int. J. Cancer 128, 1280–1292 (2011).
Thyssen, G. et al. LZTS2 is a novel beta-catenin-interacting protein and regulates the nuclear export of beta-catenin. Mol. Cell. Biol. 26, 8857–8867 (2006).
Gwak, J. et al. Polysiphonia japonica extract suppresses the Wnt/beta-catenin pathway in colon cancer cells by activation of NF-kappaB. Int. J. Mol. Med. 17, 1005–1010 (2006).
Albanese, C. et al. IKKalpha regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Mol. Biol. Cell 14, 585–599 (2003).
Noubissi, F. K. et al. Wnt signaling stimulates transcriptional outcome of the Hedgehog pathway by stabilizing GLI1 mRNA. Cancer Res. 69, 8572–8578 (2009).
Regan, J. L. et al. Non-canonical hedgehog signaling is a positive regulator of the WNT pathway and is required for the survival of colon cancer stem cells. Cell Rep. 21, 2813–2828 (2017).
Quan, X. X. et al. Targeting Notch1 and IKKα enhanced NF-κB activation in CD133(+) skin cancer stem cells. Mol. Cancer Ther. 17, 2034–2048 (2018).
Liu, R. Y. et al. JAK/STAT3 signaling is required for TGF-beta-induced epithelial-mesenchymal transition in lung cancer cells. Int. J. Oncol. 44, 1643–1651 (2014).
Luo, Y. et al. Non-CSCs nourish CSCs through interleukin-17E-mediated activation of NF-kappaB and JAK/STAT3 signaling in human hepatocellular carcinoma. Cancer Lett. 375, 390–399 (2016).
Wu, W. et al. LncRNA NKILA suppresses TGF-beta-induced epithelial-mesenchymal transition by blocking NF-kappaB signaling in breast cancer. Int. J. Cancer 143, 2213–2224 (2018).
Serra, R., Easter, S. L., Jiang, W. & Baxley, S. E. Wnt5a as an effector of TGFbeta in mammary development and cancer. J. Mammary Gland Biol. Neoplasia 16, 157–167 (2011).
Zuo, M. et al. Novel therapeutic strategy targeting the Hedgehog signalling and mTOR pathways in biliary tract cancer. Br. J. Cancer 112, 1042–1051 (2015).
Barker, A. et al. Validation of a non-targeted LC-MS approach for identifying ancient proteins: method development on bone to improve artifact residue analysis. Ethnobiol. Lett. 6, 162–174 (2015).
Sturtzel, C. Endothelial cells. Adv. Exp. Med. Biol. 1003, 71–91 (2017).
Calabrese, C. et al. A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82 (2007).
Beck, B. et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 478, 399–403 (2011).
Lu, J. et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 23, 171–185 (2013).
Rong, W. et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468, 829–833 (2010).
Scully, S. et al. Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J. Neurosci. 32, 12950–12960 (2012).
Liu, T. J. et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 32, 544–553 (2013).
Yan, G. N. et al. Endothelial cells promote stem‐like phenotype of glioma cells through activating the Hedgehog pathway. J. Pathol. 234, 11–22 (2015).
Bao, S. et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 66, 7843–7848 (2006).
Kaidi, A., Williams, A. C. & Paraskeva, C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 9, 210–217 (2007).
Grange, C. et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 71, 5346 (2011).
Bussolati, B., Grange, C., Sapino, A. & Camussi, G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J. Cell Mol. Med. 13, 309–319 (2009).
Alvero, A. B. et al. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells 27, 2405–2413 (2009).
Pezzolo, A. et al. Oct-4+/Tenascin C+ neuroblastoma cells serve as progenitors of tumor-derived endothelial cells. Cell Res. 21, 1470–1486 (2011).
Zhao, Y. et al. Endothelial cell transdifferentiation of human glioma stem progenitor cells in vitro. Brain Res. Bull. 82, 308–312 (2010).
Ping, Y. F., Zhang, X. & Bian, X. W. Cancer stem cells and their vascular niche: Do they benefit from each other? Cancer Lett. 380, 561–567 (2016).
Krishnamurthy, S. et al. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res. 70, 9969–9978 (2010).
Jennifer, P. et al. Microparticles mediated cross-talk between tumoral and endothelial cells promote the constitution of a pro-metastatic vascular niche through Arf6 up regulation. Cancer Microenviron. 7, 41–59 (2014).
Lyden, D. et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 7, 1194–1201 (2001).
Yao, X. et al. Vascular endothelial growth factor receptor 2 (VEGFR-2) plays a key role in vasculogenic mimicry formation, neovascularization and tumor initiation by glioma stem-like cells. PLoS ONE 8, e57188- (2013).
Galan-Moya, E. M. et al. Endothelial secreted factors suppress mitogen deprivation-induced autophagy and apoptosis in glioblastoma stem-like cells. PLoS ONE 9, e93505 (2014).
Gilbertson, R. J. & Rich, J. N. Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 7, 733–736 (2007).
Bao, S. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006).
Li, Z. et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513 (2009).
Kim, R. J. et al. High aldehyde dehydrogenase activity enhances stem cell features in breast cancer cells by activating hypoxia-inducible factor-2alpha. Cancer Lett. 333, 18–31 (2013).
Keith, B. & Simon, M. C. Hypoxia-inducible factors, stem cells cancer. Cell 129, 465–472 (2007).
Hur, H. et al. Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int. J. Oncol. 42, 44–54 (2013).
Samanta, D., Gilkes, D. M., Chaturvedi, P., Xiang, L. & Semenza, G. L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl Acad. Sci. USA 111, E5429–E5438 (2014).
Heddleston, J. M., Li, Z., McLendon, R. E., Hjelmeland, A. B. & Rich, J. N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8, 3274–3284 (2009).
Mendez, O. et al. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 9, 133 (2010).
Seidel, S. et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 133, 983–995 (2010).
Ye, X. Q. et al. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 129, 820–831 (2011).
Civenni, G. et al. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer Res. 73, 6816–6827 (2013).
Takebe, N., Warren, R. Q. & Ivy, S. P. Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 13, 211 (2011).
Bhat, K. M. Notch signaling acts before cell division to promote asymmetric cleavage and cell fate of neural precursor cells. Sci. Signal. 7, ra101 (2014).
Lavin, Y. et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326 (2014).
Davies, L. C., Jenkins, S. J., Allen, J. E. & Taylor, P. R. Tissue-resident macrophages. Nat. Immunol. 14, 986–995 (2013).
Sica, A. & Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest 122, 787–795 (2012).
Hao, N. B. et al. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 948098 (2012).
Medrek, C., Ponten, F., Jirstrom, K. & Leandersson, K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 12, 306 (2012).
Yamaguchi, T. et al. Tumor-associated macrophages of the M2 phenotype contribute to progression in gastric cancer with peritoneal dissemination. Gastric Cancer 19, 1052–1065 (2016).
Zhou, P. et al. The epithelial to mesenchymal transition (EMT) and cancer stem cells: implication for treatment resistance in pancreatic cancer. Mol. Cancer 16, 52 (2017).
Yang, Z., Xie, H., He, D. & Li, L. Infiltrating macrophages increase RCC epithelial mesenchymal transition (EMT) and stem cell-like populations via AKT and mTOR signaling. Oncotarget 7, 44478–44491 (2016).
Huang, W. C., Chan, M. L., Chen, M. J., Tsai, T. H. & Chen, Y. J. Modulation of macrophage polarization and lung cancer cell stemness by MUC1 and development of a related small-molecule inhibitor pterostilbene. Oncotarget 7, 39363–39375 (2016).
Masahisa, J. et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl Acad. Sci. USA 108, 12425–12430 (2011).
Wu, A. et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 12, 1113–1125 (2010).
Yi, L. et al. Glioma-initiating cells: a predominant role in microglia/macrophages tropism to glioma. J. Neuroimmunol. 232, 75–82 (2011).
Yamashina, T. et al. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res. 74, 2698–2709 (2014).
Leslie, C. S. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264 (2013).
Shi, Y. et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 8, 15080 (2017).
Xie, K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 12, 375–391 (2001).
Yang, J. et al. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 31, 248–258 (2013).
Fu, X. T. et al. Macrophage-secreted IL-8 induces epithelial–mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int. J. Oncol. 46, 587–596 (2015).
Wang, H. et al. HepG2 cells acquire stem cell-like characteristics after immune cell stimulation. Cell Oncol. (Dordr.) 39, 35–45 (2016).
Kalluri, R. & Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401 (2006).
Potenta, S., Zeisberg, E. & Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 99, 1375–1379 (2008).
Crisan, M. et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3, 301–313 (2008).
Kalluri, R. & Weinberg, R. A. The basics of epithelial–mesenchymal transition. J. Clin. Invest 119, 1420–1428 (2009).
Buchsbaum, R. J. & Young, O. S. Breast cancer-associated fibroblasts: where we are and where we need to go. Cancers 8, 19 (2016).
Jotzu, C. et al. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Cell. Oncol. 34, 55–67 (2011).
Wikstrom, P., Marusic, J., Stattin, P. & Bergh, A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 69, 799–809 (2009).
Goicoechea, S. M. et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene 33, 1265–1273 (2014).
Xia, L. et al. A CCL2/ROS autoregulation loop is critical for cancer-associated fibroblasts-enhanced tumor growth of oral squamous cell carcinoma. Carcinogenesis 35, 1362–1370 (2014).
Nair, N. et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci. Rep. 7, 6838 (2017).
Scheel, C. et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145, 926–940 (2011).
Giannoni, E. et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial–mesenchymal transition and cancer stemness. Cancer Res. 70, 6945–6956 (2010).
Liao, C. P., Adisetiyo, H., Liang, M. & Roy-Burman, P. Cancer-associated fibroblasts enhance the gland-forming capability of prostate cancer stem cells. Cancer Res. 70, 7294–7303 (2010).
Wolfson, B., Eades, G. & Zhou, Q. Adipocyte activation of cancer stem cell signaling in breast cancer. World J. Biol. Chem. 6, 39–47 (2015).
Chaffer, C. L. et al. Poised chromatin at the ZEB1 promoter enables cell plasticity and enhances tumorigenicity. Cell 154, 61–74 (2013).
Chen, W. J. et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 5, 3472 (2014).
Tsuyada, A. et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 72, 2768 (2012).
Giordano, C. et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 7, 1262–1275 (2016).
Mccuaig, R., Wu, F., Dunn, J., Rao, S. & Dahlstrom, J. E. The biological and clinical significance of stromal-epithelial interactions in breast cancer. Pathology 49, 133–140 (2016).
Lau, E. Y. et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 15, 1175–1189 (2016).
Xiong, S. et al. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am. J. Cancer Res. 8, 302 (2018).
Yibing, H. et al. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE 10, e0125625 (2015).
Bagley, R. G. et al. Human mesenchymal stem cells from bone marrow express tumor endothelial and stromal markers. Int. J. Oncol. 34, 619–627 (2009).
Cavarretta, I. T. et al. Adipose tissue-derived mesenchymal stem cells expressing prodrug-converting enzyme inhibit human prostate tumor growth. Mol. Ther. 18, 223–231 (2010).
Kidd, S. et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 27, 2614–2623 (2009).
Martin, F. T. et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res. Treat. 124, 317–326 (2010).
Xue, J. et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer 15, 793 (2015).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Brunel, M., Herr, F. & Durrbach, A. Immunosuppressive properties of mesenchymal stem cells. Curr. Transplant. Rep. 3, 348–357 (2016).
Abdel, A. M. T. et al. Efficacy of mesenchymal stem cells in suppression of hepatocarcinorigenesis in rats: possible role of wnt signaling. J. Exp. Clin. Cancer Res. 30, 49–49 (2011).
Maffey, A. et al. Mesenchymal stem cells from tumor microenvironment favour breast cancer stem cell proliferation, cancerogenic and metastatic potential, via ionotropic purinergic signalling. Sci. Rep. 7, 13162 (2017).
Tu, B., L, D., Fan, Q. M., Tang, Z. & Tang, T. T. STAT3 activation by IL-6 from mesenchymal stem cells promotes the proliferation and metastasis of osteosarcoma. Cancer Lett. 325, 80–88 (2012).
Spaeth, E. L. et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 8, e4992 (2013).
Reagan, M. R. & Kaplan, D. L. Concise review: mesenchymal stem cell tumor-homing: detection methods in disease model systems. Stem Cells 29, 920–927 (2011).
Yang, Y., Otte, A. & Hass, R. Human mesenchymal stroma/stem cells exchange membrane proteins and alter functionality during interaction with different tumor cell lines. Stem Cells Dev. 24, 1205–1222 (2014).
Xu, M. H. et al. EMT and acquisition of stem cell-like properties are involved in spontaneous formation of tumorigenic hybrids between lung cancer and bone marrow-derived mesenchymal stem cells. PLoS ONE 9, e87893 (2014).
Yang, X. et al. Increased invasiveness of osteosarcoma mesenchymal stem cells induced by bone-morphogenetic protein-2. Vitr. Cell Dev. Biol. Anim. 49, 270–278 (2013).
Ma, Y. C. et al. The tyrosine kinase c-Src directly mediates growth factor-induced Notch-1 and Furin interaction and Notch-1 activation in pancreatic cancer cells. PLoS ONE 7, e33414 (2012).
Carnero, A. et al. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat. Rev. 49, 25–36 (2016).
Peng, D. et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res. 76, 3156 (2016).
Hynes, R. O. & Naba, A. Overview of the matrisome—an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 4, a004903 (2012).
Mouw, J. K., Ou, G. & Weaver, V. M. Extracellular matrix assembly: a multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 15, 771–785 (2014).
Pengfei, L., Ken, T., Weaver, V. M. & Zena, W. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harbor Perspectives in. Cold Spring Harbor Perspect. Biol. 3, 1750–1754 (2011).
Pengfei, L., Weaver, V. M. & Zena, W. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395 (2012).
Li, L., John, C. & Margolin, D. A. Cancer stem cell and stromal microenvironment. Ochsner J. 13, 109–118 (2013).
Schrader, J. et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 53, 1192–1205 (2011).
Casazza, A. et al. Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene 33, 1743–1754 (2014).
Murai, T. Lipid raft-mediated regulation of hyaluronan–CD44 interactions in inflammation and cancer. Front. Immunol. 6, 420 (2015).
Kai, K. et al. A role for matrix metalloproteinases in regulating mammary stem cell function via the Wnt signaling pathway. Cell Stem Cell 13, 300–313 (2013).
Thordur, O. et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 17, 867–874 (2011).
Yuan, A. et al. Transfer of microRNAs by embryonic stem cell microvesicles. PLoS ONE 4, e4722 (2009).
Vlassov, A. V., Susan, M., Robert, S. & Rick, C. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 1820, 940–948 (2012).
Kawikova, I. & Askenase, P. W. Diagnostic and therapeutic potentials of exosomes in CNS diseases. Brain Res. 1617, 63–71 (2015).
Pete, H. et al. A family of thermostable fungal cellulases created by structure-guided recombination. Proc. Natl Acad. Sci. USA 106, 5610–5615 (2009).
Alonso, M. A. & Millán, J. The role of lipid rafts in signalling and membrane trafficking in T lymphocytes. J. Cell Sci. 114, 3957 (2001).
Chow, A. et al. Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-κB. Sci. Rep. 4, 5750 (2014).
Lopatina, T., Gai, C., Deregibus, M. C., Kholia, S. & Camussi, G. Cross talk between cancer and mesenchymal stem cells through extracellular vesicles carrying nucleic acids. Front. Oncol. 6, 125 (2016).
Abels, E. R. & Breakefield, X. O. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell. Mol. Neurobiol. 36, 301–312 (2016).
Gajos-Michniewicz, A., Duechler, M. & Czyz, M. MiRNA in melanoma-derived exosomes. Cancer Lett. 347, 29–37 (2014).
Webber, J. P. et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 34, 290 (2015).
Mrizak, D. et al. 19 Effect of nasopharyngeal carcinoma-derived exosomes on human regulatory T cells. J. Natl Cancer Inst. 51, e33–e33 (2015).
Rana, S., Malinowska, K. & Zöller, M. Exosomal tumor microRNA modulates premetastatic organ cells 1 2. Neoplasia 15, 281 (2013). IN214–IN295, IN231.
Melo, S. et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 26, 707–721 (2014).
Andrew, R., Elaine, C., Eva, B. & Simon, P. Regulation of exosome release from mammary epithelial and breast cancer cells—a new regulatory pathway. Eur. J. Cancer 50, 1025–1034 (2014).
Nakano, I., Garnier, D., Minata, M. & Rak, J. Extracellular vesicles in the biology of brain tumour stem cells—implications for inter-cellular communication, therapy and biomarker development. Semin. Cell Dev. Biol. 40, 17–26 (2015).
Rinkenbaugh, A. L. & Baldwin, A. S. The NF-κB pathway and cancer stem cells. Cells 5, 16 (2016).
Dandawate, P. R., Subramaniam, D., Jensen, R. A. & Anant, S. Targeting cancer stem cells and signaling pathways by phytochemicals: novel approach for breast cancer therapy. Semin. Cancer Biol. 40–41, 192–208 (2016).
Huang, R. & Rofstad, E. K. Cancer stem cells (CSCs), cervical CSCs and targeted therapies. Oncotarget 8, 35351–35367 (2017).
Bronisz, A. et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 74, 738–750 (2014).
Ye, J., Wu, D., Wu, P., Chen, Z. & Huang, J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumor Biol. 35, 3945–3951 (2014).
Shimoda, M. et al. Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat. Cell Biol. 16, 889–901 (2014).
Donnarumma, E. et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 8, 19592–19608 (2017).
Figueroa, J. et al. Exosomes from glioma-associated mesenchymal stem cells increase the tumorigenicity of glioma stem-like cells via transfer of miR-1587. Cancer Res. 77, 5808 (2017).
Xu, J., Liao, K. & Zhou, W. Exosomes regulate the transformation of cancer cells in cancer stem cell homeostasis. Stem Cells Int. 2018, 4837370 (2018).
Munagala, R., Aqil, F., Jeyabalan, J. & Gupta, R. C. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 371, 48–61 (2016).
Srivastava, A. et al. Exploitation of exosomes as nanocarriers for gene-, chemo-, and immune-therapy of cancer. J. Biomed. Nanotechnol. 12, 1159 (2016).
Pitt, J. M. et al. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Invest. 126, 1224 (2016).
Tian, X., Zhu, M. & Nie, G. How can nanotechnology help membrane vesicle-based cancer immunotherapy development? Hum. Vaccin. Immunother. 9, 222–225 (2013).
Wang, J. et al. Extracellular vesicle cross-talk in the bone marrow microenvironment: implications in multiple myeloma. Oncotarget 7, 38927–38945 (2016).
Ghielmini, M. et al. The effect of Rituximab on patients with follicular and mantle-cell lymphoma. Swiss Group for Clinical Cancer Research (SAKK). Ann. Oncol. 11(Suppl. 1), 123–126 (2000).
Turner, J. H., Martindale, A. A., Boucek, J., Claringbold, P. G. & Leahy, M. F. 131I-anti CD20 radioimmunotherapy of relapsed or refractory non-Hodgkins lymphoma: a phase II clinical trial of a nonmyeloablative dose regimen of chimeric rituximab radiolabeled in a hospital. Cancer Biother. Radiopharm. 18, 513–524 (2003).
Nabhan, C. et al. A pilot trial of rituximab and alemtuzumab combination therapy in patients with relapsed and/or refractory chronic lymphocytic leukemia (CLL). Leuk. Lymphoma 45, 2269–2273 (2004).
Börjesson, P. K. et al. Phase I therapy study with (186)Re-labeled humanized monoclonal antibody BIWA 4 (bivatuzumab) in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 9, 3961s–3972s (2003).
Postema, E. J. et al. Dosimetric analysis of radioimmunotherapy with 186Re-labeled bivatuzumab in patients with head and neck cancer. J. Nucl. Med. 44, 1690–1699 (2003).
Colnot, D. R. et al. Safety, biodistribution, pharmacokinetics, and immunogenicity of 99mTc-labeled humanized monoclonal antibody BIWA 4 (bivatuzumab) in patients with squamous cell carcinoma of the head and neck. Cancer Immunol. Immunother. 52, 576–582 (2003).
Riechelmann, H. et al. Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral. Oncol. 44, 823–829 (2008).
He, S. Z. et al. A phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk. Lymphoma 56, 1406–1415 (2015).
Frankel, A. E. et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood 124, 385–392 (2014).
Pemmaraju, N. et al. Results from phase 2 trial ongoing expansion stage of SL-401 in patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood 128, 342–342 (2016).
Osta, W. A. et al. EpCAM is overexpressed in breast cancer and is a potential target for breast cancer gene therapy. Cancer Res. 64, 5818–5824 (2004).
Oberneder, R. et al. A phase I study with adecatumumab, a human antibody directed against epithelial cell adhesion molecule, in hormone refractory prostate cancer patients. Eur. J. Cancer 42, 2530–2538 (2006).
Sebastian, M. et al. Treatment of non-small cell lung cancer patients with the trifunctional monoclonal antibody catumaxomab (anti-EpCAM × anti-CD3): a phase I study. Cancer Immunol. Immunother. 56, 1637–1644 (2007).
Jaeger, M. et al. Immunotherapy with the trifunctional antibody removab leads to significant elimination of tumor cells from malignant ascites in ovarian cancer: results of a phase I/II study. J. Clin. Oncol. 22, 2504–2504 (2004).
Niedzwiecki, D. et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J. Clin. Oncol. 29, 3146–3152 (2011).
Schmidt, M. et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann. Oncol. 21, 275–282 (2010).
Quail, D. F., Taylor, M. J. & Postovit, L. M. Microenvironmental regulation of cancer stem cell phenotypes. Curr. Stem Cell Res Ther. 7, 197–216 (2012).
Meurette, O. & Mehlen, P. Notch signaling in the tumor microenvironment. Cancer Cell 34, 536–548 (2018).
Nowell, C. S. & Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 17, 145–159 (2017).
Rajakulendran, N. et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 33, 498–510 (2019).
van Groningen, T. et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 10, 1530 (2019).
Zanotti, S. & Canalis, E. Notch signaling and the skeleton. Endocr. Rev. 37, 223–253 (2016).
Gazdar, A. F., Bunn, P. A. & Minna, J. D. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat. Rev. Cancer 17, 725–737 (2017).
Dai, W., Peterson, A., Kenney, T., Burrous, H. & Montell, D. J. Quantitative microscopy of the Drosophila ovary shows multiple niche signals specify progenitor cell fate. Nat. Commun. 8, 1244 (2017).
Mamidi, A. et al. Mechanosignalling via integrins directs fate decisions of pancreatic progenitors. Nature 564, 114–118 (2018).
Hayakawa, Y. et al. BHLHA15-positive secretory precursor cells can give rise to tumors in intestine and colon in mice. Gastroenterology 156, 1066–1081 (2019). e1016.
Fouladi, M. et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J. Clin. Oncol. 29, 3529–3534 (2011).
Krop, I. et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 30, 2307–2313 (2012).
Hoffman, L. M. et al. Phase I trial of weekly MK-0752 in children with refractory central nervous system malignancies: a pediatric brain tumor consortium study. Childs Nerv. Syst. 31, 1283–1289 (2015).
Chen, X. et al. Sequential combination therapy of ovarian cancer with cisplatin and γ-secretase inhibitor MK-0752. Gynecol. Oncol. 140, 537–544 (2016).
Schott, A. F. et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin. Cancer Res. 19, 1512–1524 (2013).
Cook, N. et al. A phase I trial of the γ-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br. J. Cancer 118, 793–801 (2018).
Brana, I. et al. A parallel-arm phase I trial of the humanised anti-IGF-1R antibody dalotuzumab in combination with the AKT inhibitor MK-2206, the mTOR inhibitor ridaforolimus, or the NOTCH inhibitor MK-0752, in patients with advanced solid tumours. Br. J. Cancer 111, 1932–1944 (2014).
Zhang, S., Chung, W. C., Miele, L. & Xu, K. Targeting Met and Notch in the Lfng-deficient, Met-amplified triple-negative breast cancer. Cancer Biol. Ther. 15, 633–642 (2014).
Luistro, L. et al. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res. 69, 7672–7680 (2009).
Tolcher, A. W. et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J. Clin. Oncol. 30, 2348–2353 (2012).
Strosberg, J. R. et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur. J. Cancer 48, 997–1003 (2012).
De Jesus-Acosta, A. et al. A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest. N. Drugs 32, 739–745 (2014).
Diaz-Padilla, I. et al. A phase II study of single-agent RO4929097, a gamma-secretase inhibitor of Notch signaling, in patients with recurrent platinum-resistant epithelial ovarian cancer: a study of the Princess Margaret, Chicago and California phase II consortia. Gynecol. Oncol. 137, 216–222 (2015).
Richter, S. et al. A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL-078/CTEP 8575). Invest N. Drugs 32, 243–249 (2014).
Sahebjam, S. et al. A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC-004/NCI 8503). Br. J. Cancer 109, 943–949 (2013).
LoConte, N. K. et al. A multicenter phase 1 study of γ -secretase inhibitor RO4929097 in combination with capecitabine in refractory solid tumors. Invest. N. Drugs 33, 169–176 (2015).
Kummar, S. et al. Clinical activity of the γ-secretase inhibitor PF-03084014 in adults with desmoid tumors (aggressive fibromatosis). J. Clin. Oncol. 35, 1561–1569 (2017).
Messersmith, W. A. et al. A phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin. Cancer Res. 21, 60–67 (2015).
Papayannidis, C. et al. A phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 5, e350 (2015).
Yang, J. et al. Role of Jagged1/STAT3 signalling in platinum-resistant ovarian cancer. J. Cell. Mol. Med. 23, 4005–4018 (2019).
Smith, D. C. et al. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin. Cancer Res. 20, 6295–6303 (2014).
Mukherjee, S. et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 5, 674–683 (2006).
Yuan, Z. et al. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene 26, 1046–1055 (2007).
Thayer, S. P. et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425, 851–856 (2003).
Sekulic, A. et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 366, 2171–2179 (2012).
Dummer, R. et al. The 12-month analysis from basal cell carcinoma outcomes with LDE225 treatment (BOLT): a phase II, randomized, double-blind study of sonidegib in patients with advanced basal cell carcinoma. J. Am. Acad. Dermatol 75, 113–125 (2016). e115.
Norsworthy, K. J. et al. FDA approval summary: glasdegib for newly diagnosed acute myeloid leukemia. Clin. Cancer Res. 25, 6021–6025 (2019).
Scales, S. J. & de Sauvage, F. J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 30, 303–312 (2009).
Zito, P. M. & Scharf, R. in StatPearls (StatPearls Publishing StatPearls Publishing LLC., Treasure Island, FL, 2019).
Robinson, G. W. et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J. Clin. Oncol. 33, 2646–2654 (2015).
Gajjar, A. et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin. Cancer Res. 19, 6305–6312 (2013).
Berlin, J. et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res. 19, 258–267 (2013).
Catenacci, D. V. et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 33, 4284–4292 (2015).
Italiano, A. et al. GDC-0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 24, 2922–2926 (2013).
Houot, R. et al. Inhibition of Hedgehog signaling for the treatment of lymphoma and CLL: a phase II study from the LYSA. Ann. Oncol. 27, 1349–1350 (2016).
Kaye, S. B. et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res. 18, 6509–6518 (2012).
Pan, S. et al. Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med. Chem. Lett. 1, 130–134 (2010).
Kieran, M. W. et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 19, 1542–1552 (2017).
Magnani, L. et al. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc. Natl Acad. Sci. USA 110, E1490–E1499 (2013).
Cortes, J. E. et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: phase 2 study results. Am. J. Hematol. 93, 1301–1310 (2018).
Cortes, J. E. et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 33, 379–389 (2019).
List, A. et al. Opportunities for Trisenox (arsenic trioxide) in the treatment of myelodysplastic syndromes. Leukemia 17, 1499–1507 (2003).
Lauth, M., Bergström, A., Shimokawa, T. & Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl Acad. Sci. USA 104, 8455–8460 (2007).
Pohl, S. G. et al. Wnt signaling in triple-negative breast cancer. Oncogenesis 6, e310 (2017).
Kraggerud, S. M. et al. Molecular characteristics of malignant ovarian germ cell tumors and comparison with testicular counterparts: implications for pathogenesis. Endocr. Rev. 34, 339–376 (2013).
Fu, L. et al. Wnt2 secreted by tumour fibroblasts promotes tumour progression in oesophageal cancer by activation of the Wnt/β-catenin signalling pathway. Gut 60, 1635–1643 (2011).
Hua, F. et al. TRIB3 interacts with β-catenin and TCF4 to increase stem cell features of colorectal cancer stem cells and tumorigenesis. Gastroenterology 156, 708–721 (2019). e715.
Pal, S. K., Swami, U. & Agarwal, N. Characterizing the Wnt pathway in advanced prostate cancer: when, why, and how. Eur. Urol. (2019).
Lin, W. et al. Mesenchymal stem cells and cancer: clinical challenges and opportunities. Biomed. Res. Int. 2019, 2820853 (2019).
Le, P. N., McDermott, J. D. & Jimeno, A. Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP-54F28. Pharmacol. Ther. 146, 1–11 (2015).
Jimeno, A. et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. 19, 2766–2774 (2013).
Cortes, J. E. et al. Phase 1 study of CWP232291 in relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). J. Clin. Oncol. 33, 7044–7044 (2015).
Konopleva, M. et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 6, 1106–1117 (2016).
Saygin, C., Matei, D., Majeti, R., Reizes, O. & Lathia, J. D. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell 24, 25–40 (2019).
Cashen, A. et al. A phase II study of plerixafor (AMD3100) plus G-CSF for autologous hematopoietic progenitor cell mobilization in patients with Hodgkin lymphoma. Biol. Blood Marrow Transplant. 14, 1253–1261 (2008).
Uy, G. L. et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 119, 3917–3924 (2012).
Cooper, T. M. et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: a Pediatric Oncology Experimental Therapeutics Investigators' Consortium study (POE 10-03). Pediatr. Blood Cancer 64, (2017).
Galsky, M. D. et al. A phase I trial of LY2510924, a CXCR4 peptide antagonist, in patients with advanced cancer. Clin. Cancer Res. 20, 3581–3588 (2014).
Hainsworth, J. D. et al. A randomized, open-label phase 2 study of the CXCR4 inhibitor LY2510924 in combination with sunitinib versus sunitinib alone in patients with metastatic renal cell carcinoma (RCC). Target Oncol. 11, 643–653 (2016).
Salgia, R. et al. A randomized phase II study of LY2510924 and carboplatin/etoposide versus carboplatin/etoposide in extensive-disease small cell lung cancer. Lung Cancer 105, 7–13 (2017).
Sakamuri, D. et al. Phase I dose-escalation study of anti-CTLA-4 antibody ipilimumab and lenalidomide in patients with advanced cancers. Mol. Cancer Ther. 17, 671–676 (2018).
Meindl-Beinker, N. M. et al. A multicenter open-label phase II trial to evaluate nivolumab and ipilimumab for 2nd line therapy in elderly patients with advanced esophageal squamous cell cancer (RAMONA). BMC Cancer 19, 231 (2019).
Cortese, I. et al. Pembrolizumab treatment for progressive multifocal leukoencephalopathy. N. Engl. J. Med. 380, 1597–1605 (2019).
Migden, M. R. et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N. Engl. J. Med. 379, 341–351 (2018).
Motzer, R. J. et al. Avelumab plus Axitinib versus Sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 380, 1103–1115 (2019).
Fujiwara, Y. et al. Tolerability and efficacy of durvalumab in Japanese patients with advanced solid tumors. Cancer Sci. 110, 1715–1723 (2019).
Sullivan, R. J. et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat. Med. 25, 929–935 (2019).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Nandy, S. B. & Lakshmanaswamy, R. Cancer stem cells and metastasis. Prog. Mol. Biol. Transl. Sci. 151, 137–176 (2017).
Rich, J. N. Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine 95, S2–S7 (2016).
Prieto-Vila, M., Takahashi, R. U., Usuba, W., Kohama, I. & Ochiya, T. Drug resistance driven by cancer stem cells and their niche. Int. J. Mol. Sci. 18, 2574 (2017).
Zhao, J. Cancer stem cells and chemoresistance: the smartest survives the raid. Pharmacol. Ther. 160, 145–158 (2016).
Chang, L. et al. Cancer stem cells and signaling pathways in radioresistance. Oncotarget 7, 11002–11017 (2016).
Liu, B., Yan, L. & Zhou, M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am. J. Cancer Res. 9, 228–241 (2019).
Bao, B. et al. Overview of Cancer Stem Cells (CSCs) and Mechanisms of Their Regulation: Implications for Cancer Therapy. Current protocols in pharmacology Chapter 14:Unit14.25 (2013).
Shultz, L. D., Brehm, M. A., Garcia-Martinez, J. V. & Greiner, D. L. Humanized mice for immune system investigation: progress, promise and challenges. Nat. Rev. Immunol. 12, 786–798 (2012).
Plaks, V., Kong, N. & Werb, Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16, 225–238 (2015).
Takeishi, S. & Nakayama, K. I. To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci. 107, 875–881 (2016).
Jiang, Q., Crews, L. A., Holm, F. & Jamieson, C. H. M. RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat. Rev. Cancer 17, 381–392 (2017).
Wainwright, E. N. & Scaffidi, P. Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer 3, 372–386 (2017).
Sancho, P., Barneda, D. & Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 114, 1305–1312 (2016).
Du, F.-Y., Zhou, Q.-F., Sun, W.-J. & Chen, G.-L. Targeting cancer stem cells in drug discovery: Current state and future perspectives. World J. Stem Cells 11, 398–420 (2019).
Moselhy, J., Srinivasan, S., Ankem, M. K. & Damodaran, C. Natural products that target cancer stem cells. Anticancer Res. 35, 5773–5788 (2015).
Shackleton, M. et al. Generation of a functional mammary gland from a single stem cell. Nature 439, 84–88 (2006).
Pece, S. et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 140, 62–73 (2010).
Lu, H. H. et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 16, 1105 (2014).
Liu, T. J. et al. CD133(+) cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 32, 544–553 (2013).
Ricardo, S. et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 64, 937–946 (2011).
Fillmore, C. M. & Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 10, R25 (2008).
Wright, M. H. et al. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 10, R10 (2008).
Moreb, J., Schweder, M., Suresh, A. & Zucali, J. R. Overexpression of the human aldehyde dehydrogenase class I results in increased resistance to 4-hydroperoxycyclophosphamide. Cancer Gene Ther. 3, 24–30 (1996).
Azevedo, R. et al. CD44 glycoprotein in cancer: a molecular conundrum hampering clinical applications. Clin. Proteom. 15, 22 (2018).
Kumar, A., Bhanja, A., Bhattacharyya, J. & Jaganathan, B. G. Multiple roles of CD90 in cancer. Tumour Biol. 37, 11611–11622 (2016).
Yin, A. H. et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 90, 5002–5012 (1997).
Baumann, P. et al. CD24 expression causes the acquisition of multiple cellular properties associated with tumor growth and metastasis. Cancer Res. 65, 10783–10793 (2005).
Vassilopoulos, A., Chisholm, C., Lahusen, T., Zheng, H. & Deng, C. X. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene 33, 5477–5482 (2014).
Deng, Z. et al. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC immunology. 16, 1 (2015).
Kerr, B. A. et al. CD117(+) cells in the circulation are predictive of advanced prostate cancer. Oncotarget 6, 1889–1897 (2015).
Patrawala, L. et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 25, 1696–1708 (2006).
Ugolkov, A. V., Eisengart, L. J., Luan, C. Y. & Yang, X. M. J. Expression analysis of putative stem cell markers in human benign and malignant prostate. Prostate 71, 18–25 (2011).
Darash-Yahana, M. et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 18, 1240-+ (2004).
Bae, K. M., Parker, N. N., Dai, Y., Vieweg, J. & Siemann, D. W. E-cadherin plasticity in prostate cancer stem cell invasion. Am. J. Cancer Res. 1, 71–84 (2011).
Richardson, G. D. et al. CD133, a novel marker for human prostatic epithelial stem cells. J. Cell Sci. 117, 3539–3545 (2004).
Fukamachi, H. et al. CD49f(high) cells retain sphere-forming and tumor-initiating activities in human gastric tumors. PLoS ONE 8, e72438 (2013).
He, J. T. et al. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol. Cell. Proteom. 11, M111.010744 (2012).
Jackson, M., Hassiotou, F. & Nowak, A. Glioblastoma stem-like cells: at the root of tumor recurrence and a therapeutic target. Carcinogenesis 36, 177–185 (2015).
Hale, J. S. et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 32, 1746–1758 (2014).
Mazzoleni, S. et al. Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res. 70, 7500–7513 (2010).
Chiocca, E. A., Liau, L. M., Lim, D. A., Berger, M. S. & Piepmeier, J. M. Identification of A2B5(+)CD133-tumor-initiating cells in adult human gliomas—Comments. Neurosurgery 62, 514–515 (2008).
Bao, S. D. et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 68, 6043–6048 (2008).
Cheng, J. X., Liu, B. L. & Zhang, X. How powerful is CD133 as a cancer stem cell marker in brain tumors? Cancer Treat. Rev. 35, 403–408 (2009).
Wang, J. C. & Li, Y. S. CD36 tango in cancer: signaling pathways and functions. Theranostics 9, 4893–4908 (2019).
Drago, J., Reid, K. L. & Bartlett, P. F. Induction of the ganglioside marker-A2b5 on cultured cerebellar neural cells by growth factors. Neurosci. Lett. 107, 245–250 (1989).
Nishikawa, S. et al. Aldehyde dehydrogenase high gastric cancer stem cells are resistant to chemotherapy. Int. J. Oncol. 42, 1437–1442 (2013).
Takaishi, S. et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 27, 1006–1020 (2009).
Lau, W. M. et al. CD44v8-10 is a cancer-specific marker for gastric cancer stem cells. Cancer Res. 74, 2630–2641 (2014).
Zhu, Y. L. et al. Overexpression of CD133 enhances chemoresistance to 5-fluorouracil by activating the PI3K/Akt/p70S6K pathway in gastric cancer cells. Oncol. Rep. 32, 2437–2444 (2014).
Fujikuni, N. et al. Hypoxia-mediated CD24 expression is correlated with gastric cancer aggressiveness by promoting cell migration and invasion. Cancer Sci. 105, 1411–1420 (2014).
Chen, T. et al. Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell Res. 22, 248–258 (2012).
Xue, Z. F. et al. Identification of cancer stem cells in vincristine preconditioned SGC7901 gastric cancer cell line. J. Cell. Biochem. 113, 302–312 (2012).
Ohkuma, M. et al. Absence of CD71 transferrin receptor characterizes human gastric adenosquamous carcinoma stem cells. Ann. Surg. Oncol. 19, 1357–1364 (2012).
Du, W. Q. et al. EpCAM is overexpressed in gastric cancer and its downregulation suppresses proliferation of gastric cancer. J. Cancer Res. Clin. 135, 1277–1285 (2009).
Zhang, S. S., Huang, Z. W., Li, L. X., Fu, J. J. & Xiao, B. Identification of CD200+ colorectal cancer stem cells and their gene expression profile. Oncol. Rep. 36, 2252–2260 (2016).
Tseng, J. Y. et al. Circulating CD133(+)/ESA(+) cells in colorectal cancer patients. J. Surg. Res. 199, 362–370 (2015).
Ren, F., Sheng, W. Q. & Du, X. CD133: a cancer stem cells marker, is used in colorectal cancers. World J. Gastroenterol. 19, 2603–2611 (2013).
Fan, W. et al. Identification of CD206 as a potential biomarker of cancer stem-like cells and therapeutic agent in liver cancer. Oncol. Lett. 18, 3218–3226 (2019).
Dalerba, P. et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl Acad. Sci. USA 104, 10158–10163 (2007).
Lugli, A. et al. Prognostic impact of the expression of putative cancer stem cell markers CD133, CD166, CD44s, EpCAM, and ALDH1 in colorectal cancer. Br. J. Cancer 103, 382–390 (2010).
Ko, Y. C. et al. Endothelial CD200 is heterogeneously distributed, regulated and involved in immune cell-endothelium interactions. J. Anat. 214, 183–195 (2009).
Jiao, J. et al. Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PLoS ONE 7, e42564 (2012).
Huang, L. et al. Functions of EpCAM in physiological processes and diseases (Review). Int. J. Mol. Med. 42, 1771–1785 (2018).
Lee, T. K. et al. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 9, 50–63 (2011).
Suetsugu, A. et al. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem. Biophys. Res. Commun. 351, 820–824 (2006).
Sun, J. H., Luo, Q., Liu, L. L. & Song, G. B. Liver cancer stem cell markers: progression and therapeutic implications. World J. Gastroenterol. 22, 3547–3557 (2016).
Zhu, Z. et al. Cancer stem/progenitor cells are highly enriched in CD133(+)CD44(+) population in hepatocellular carcinoma. Int. J. Cancer 126, 2067–2078 (2010).
Yang, Z. F. et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 13, 153–166 (2008).
Nio, K. et al. Defeating EpCAM(+) liver cancer stem cells by targeting chromatin remodeling enzyme CHD4 in human hepatocellular carcinoma. J. Hepatol. 63, 1164–1172 (2015).
Pasqualini, R. et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 60, 722–727 (2000).
Sidney, L. E., Branch, M. J., Dunphy, S. E., Dua, H. S. & Hopkinson, A. Concise review: evidence for CD34 as a common marker for diverse progenitors. Stem Cells 32, 1380–1389 (2014).
Orciani, M., Trubiani, O., Guarnieri, S., Ferrero, E. & Di Primio, R. CD38 is constitutively expressed in the nucleus of human hematopoietic cells. J. Cell. Biochem. 105, 905–912 (2008).
Allden, S. J. et al. The Transferrin receptor CD71 delineates functionally distinct airway macrophage subsets during idiopathic pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 200, 209–219 (2019).
Li, X. et al. CD19, from bench to bedside. Immunol. Lett. 183, 86–95 (2017).
Okroj, M., Osterborg, A. & Blom, A. M. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat. Rev. 39, 632–639 (2013).
Maguer-Satta, V., Besancon, R. & Bachelard-Cascales, E. Concise review: neutral endopeptidase (CD10): a multifaceted environment actor in stem cells, physiological mechanisms, and cancer. Stem Cells 29, 389–396 (2011).
Henson, S. M., Riddell, N. E. & Akbar, A. N. Properties of end-stage human T cells defined by CD45RA re-expression. Curr. Opin. Immunol. 24, 476–481 (2012).
Liu, K. et al. CD123 and its potential clinical application in leukemias. Life Sci. 122, 59–64 (2015).
Lang, D., Mascarenhas, J. B. & Shea, C. R. Melanocytes, melanocyte stem cells, and melanoma stem cells. Clin. Dermatol. 31, 166–178 (2013).
Boiko, A. D. et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 466, 133–137 (2010).
Luo, Y. C. et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells 30, 2100–2113 (2012).
Quintana, E. et al. Efficient tumour formation by single human melanoma cells. Nature 456, 593–598 (2008).
Alvarez-Viejo, M., Menendez-Menendez, Y. & Otero-Hernandez, J. CD271 as a marker to identify mesenchymal stem cells from diverse sources before culture. World J. Stem Cells 7, 470–476 (2015).
van der Horst, G., Bos, L. & van der Pluijm, G. Epithelial plasticity, cancer stem cells, and the tumor-supportive stroma in bladder carcinoma. Mol. Cancer Res. 10, 995–1009 (2012).
Verma, A., Kapoor, R. & Mittal, R. D. Cluster of differentiation 44 (CD44) gene variants: a putative cancer stem cell marker in risk prediction of bladder cancer in North Indian population. Indian J. Clin. Biochem. 32, 74–83 (2017).
Su, Y. et al. Aldehyde dehydrogenase 1 A1-positive cell population is enriched in tumor-initiating cells and associated with progression of bladder cancer. Cancer Epidemiol. Biomark. Prev. 19, 327–337 (2010).
Klatte, T. et al. Absent CD44v6 expression is an independent predictor of poor urothelial bladder cancer outcome. J. Urol. 183, 2403–2408 (2010).
Gao, M. Q., Choi, Y. P., Kang, S., Youn, J. H. & Cho, N. H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 29, 2672–2680 (2010).
Silva, I. A. et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 71, 3991–4001 (2011).
Zhang, S. et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 68, 4311–4320 (2008).
Wei, X. et al. Mullerian inhibiting substance preferentially inhibits stem/progenitors in human ovarian cancer cell lines compared with chemotherapeutics. Proc. Natl Acad. Sci. USA 107, 18874–18879 (2010).
Kryczek, I. et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 130, 29–39 (2012).
Miettinen, M. & Lasota, J. KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl. Immunohistochem. Mol. Morphol. 13, 205–220 (2005).
Zhan, H. X., Xu, J. W., Wu, D., Zhang, T. P. & Hu, S. Y. Pancreatic cancer stem cells: new insight into a stubborn disease. Cancer Lett. 357, 429–437 (2015).
Ishiwata, T. et al. Pancreatic cancer stem cells: features and detection methods. Pathol. Oncol. Res 24, 797–805 (2018).
Marechal, R. et al. High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br. J. Cancer 100, 1444–1451 (2009).
Krishnamurthy, S. & Nor, J. E. Head and neck cancer stem cells. J. Dent. Res. 91, 334–340 (2012).
Baumann, M. & Krause, M. CD44: a cancer stem cell-related biomarker with predictive potential for radiotherapy. Clin. Cancer Res. 16, 5091–5093 (2010).
Yan, M. et al. Plasma membrane proteomics of tumor spheres identify CD166 as a novel marker for cancer stem-like cells in head and neck squamous cell carcinoma. Mol. Cell. Proteom. 12, 3271–3284 (2013).
Shi, C. et al. CD44+ CD133+ population exhibits cancer stem cell-like characteristics in human gallbladder carcinoma. Cancer Biol. Ther. 10, 1182–1190 (2010).
Corro, C. & Moch, H. Biomarker discovery for renal cancer stem cells. J. Pathol. Clin. Res. 4, 3–18 (2018).
Zhang, Y. H. et al. Clinical significances and prognostic value of cancer stem-like cells markers and vasculogenic mimicry in renal cell carcinoma. J. Surg. Oncol. 108, 414–419 (2013).
Duff, S. E., Li, C., Garland, J. M. & Kumar, S. CD105 is important for angiogenesis: evidence and potential applications. FASEB J. 17, 984–992 (2003).
Tachezy, M. et al. Activated leukocyte cell adhesion molecule (CD166): an “inert” cancer stem cell marker for non-small cell lung cancer? Stem Cells 32, 1429–1436 (2014).
Yan, X. P. et al. Identification of CD90 as a marker for lung cancer stem cells in A549 and H446 cell lines. Oncol. Rep. 30, 2733–2740 (2013).
Gutova, M. et al. Identification of uPAR-positive chemoresistant cells in small cell lung cancer. PLos ONE 2, e243 (2007).
Jiang, F. et al. Aldehyde dehydrogenase 1 Is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 7, 330–338 (2009).
Leung, E. L. et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 5, e14062 (2010).
Janikova, M. et al. Identification of CD133+/nestin+ putative cancer stem cells in non-small cell lung cancer. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. 154, 321–326 (2010).
Ghani, F. I. et al. Identification of cancer stem cell markers in human malignant mesothelioma cells. Biochem. Biophys. Res. Commun. 404, 735–742 (2011).
Powner, D., Kopp, P. M., Monkley, S. J., Critchley, D. R. & Berditchevski, F. Tetraspanin CD9 in cell migration. Biochem. Soc. Trans. 39, 563–567 (2011).
Ohnuma, K., Dang, N. H. & Morimoto, C. Revisiting an old acquaintance: CD26 and its molecular mechanisms in T cell function. Trends Immunol. 29, 295–301 (2008).
Ghuwalewala, S. et al. CD44(high)CD24(low) molecular signature determines the cancer stem cell and EMT phenotype in oral squamous cell carcinoma. Stem Cell Res. 16, 405–417 (2016).
Ming, X. Y. et al. Integrin alpha7 is a functional cancer stem cell surface marker in oesophageal squamous cell carcinoma. Nat. Commun. 7, 13568 (2016).
Burkin, D. J. & Kaufman, S. J. The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res. 296, 183–190 (1999).
Goldie, S. J., Chincarini, G. & Darido, C. Targeted therapy against the cell of origin in cutaneous squamous cell carcinoma. Int. J. Mol. Sci. 20 (2019).
Xu, R. et al. The expression status and prognostic value of cancer stem cell biomarker CD133 in cutaneous squamous cell carcinoma. JAMA Dermatol. 152, 305–311 (2016).
Ghosh, N. & Matsui, W. Cancer stem cells in multiple myeloma. Cancer Lett. 277, 1–7 (2009).
Matsui, W. et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 68, 190–197 (2008).
O'Connell, F. P., Pinkus, J. L. & Pinkus, G. S. CD138 (syndecan-1), a plasma cell marker immunohistochemical profile in hematopoietic and nonhematopoietic neoplasms. Am. J. Clin. Pathol. 121, 254–263 (2004).
Borst, J., Hendriks, J. & Xiao, Y. CD27 and CD70 in T cell and B cell activation. Curr. Opin. Immunol. 17, 275–281 (2005).
Huang, R. X. & Rofstad, E. K. Cancer stem cells (CSCs), cervical CSCs and targeted therapies. Oncotarget 8, 35351–35367 (2017).
Liu, S. Y. & Zheng, P. S. High aldehyde dehydrogenase activity identifies cancer stem cells in human cervical cancer. Oncotarget 4, 2462–2475 (2013).
Su, J. et al. Identification of cancer stem-like CD44(+) cells in human nasopharyngeal carcinoma cell line. Arch. Med. Res. 42, 15–21 (2011).
Zhuang, H. W. et al. Biological characteristics of CD133(+) cells in nasopharyngeal carcinoma. Oncol. Rep. 30, 57–63 (2013).
Wu, A. B. et al. Aldehyde dehydrogenase 1, a functional marker for identifying cancer stem cells in human nasopharyngeal carcinoma. Cancer Lett. 330, 181–189 (2013).
Yang, C. H. et al. Identification of CD24 as a cancer stem cell marker in human nasopharyngeal carcinoma. PLoS ONE 9, e99412 (2014).
Greco, A. et al. Cancer stem cells in laryngeal cancer: what we know. Eur. Arch. Oto Rhino Laryngol. 273, 3487–3495 (2016).
Zhou, L., Wei, X., Cheng, L., Tian, J. & Jiang, J. J. CD133, one of the markers of cancer stem cells in Hep-2 cell line. Laryngoscope 117, 455–460 (2007).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (nos. 2016YFC1302204 and 2017YFC1308600), the National Science Foundation of China (nos. 81672502, 81872071, and 81902664) and the Natural Science Foundation of Chongqing (no. cstc2019jcyj-zdxmX0033).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, L., Shi, P., Zhao, G. et al. Targeting cancer stem cell pathways for cancer therapy. Sig Transduct Target Ther 5, 8 (2020). https://doi.org/10.1038/s41392-020-0110-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-0110-5
This article is cited by
-
Crosstalk between colorectal CSCs and immune cells in tumorigenesis, and strategies for targeting colorectal CSCs
Experimental Hematology & Oncology (2024)
-
TSP50 facilitates breast cancer stem cell-like properties maintenance and epithelial-mesenchymal transition via PI3K p110α mediated activation of AKT signaling pathway
Journal of Experimental & Clinical Cancer Research (2024)
-
Transcriptional regulation of cancer stem cell: regulatory factors elucidation and cancer treatment strategies
Journal of Experimental & Clinical Cancer Research (2024)
-
An exosomal strategy for targeting cancer-associated fibroblasts mediated tumors desmoplastic microenvironments
Journal of Nanobiotechnology (2024)
-
Regulation of cancer stem cells by CXCL1, a chemokine whose secretion is controlled by MCM2
BMC Cancer (2024)
