
Abstract
Ubiquitination, an important type of protein posttranslational modification (PTM), plays a crucial role in controlling substrate degradation and subsequently mediates the “quantity” and “quality” of various proteins, serving to ensure cell homeostasis and guarantee life activities. The regulation of ubiquitination is multifaceted and works not only at the transcriptional and posttranslational levels (phosphorylation, acetylation, methylation, etc.) but also at the protein level (activators or repressors). When regulatory mechanisms are aberrant, the altered biological processes may subsequently induce serious human diseases, especially various types of cancer. In tumorigenesis, the altered biological processes involve tumor metabolism, the immunological tumor microenvironment (TME), cancer stem cell (CSC) stemness and so on. With regard to tumor metabolism, the ubiquitination of some key proteins such as RagA, mTOR, PTEN, AKT, c-Myc and P53 significantly regulates the activity of the mTORC1, AMPK and PTEN-AKT signaling pathways. In addition, ubiquitination in the TLR, RLR and STING-dependent signaling pathways also modulates the TME. Moreover, the ubiquitination of core stem cell regulator triplets (Nanog, Oct4 and Sox2) and members of the Wnt and Hippo-YAP signaling pathways participates in the maintenance of CSC stemness. Based on the altered components, including the proteasome, E3 ligases, E1, E2 and deubiquitinases (DUBs), many molecular targeted drugs have been developed to combat cancer. Among them, small molecule inhibitors targeting the proteasome, such as bortezomib, carfilzomib, oprozomib and ixazomib, have achieved tangible success. In addition, MLN7243 and MLN4924 (targeting the E1 enzyme), Leucettamol A and CC0651 (targeting the E2 enzyme), nutlin and MI‐219 (targeting the E3 enzyme), and compounds G5 and F6 (targeting DUB activity) have also shown potential in preclinical cancer treatment. In this review, we summarize the latest progress in understanding the substrates for ubiquitination and their special functions in tumor metabolism regulation, TME modulation and CSC stemness maintenance. Moreover, potential therapeutic targets for cancer are reviewed, as are the therapeutic effects of targeted drugs.
Similar content being viewed by others

Introduction
Ubiquitin (Ub), a highly conserved regulatory protein containing 76 amino acids, can be covalently tagged to target proteins via a cascade of enzymatic reactions, including Ub-activating (E1), Ub-conjugating (E2) and Ub-ligating (E3) enzymes. Subsequently, mono- or polyubiquitination regulates the function of a large number of proteins in various physiological and/or pathological conditions.1,2 Polyubiquitin with different chain topologies and lengths linked to specific lysine residues on substrates is associated with different functional consequences.3 Moreover, the function of Ub ligases can also be reversed by deubiquitinases (DUBs), which are also critical for almost all cellular signaling pathways, such as the cell cycle, apoptosis, receptor downregulation and gene transcription, by removing Ub from substrate proteins.4,5
Proteins are the fundamental units in regulating cellular functions, and ubiquitination is the second most common posttranslational modification (PTM) for proteins, behind only phosphorylation.6 Thus, aberrant ubiquitination may lead to disease development and progression, especially cancer.7 Mounting evidence suggests that alterations in the activity of many E3 ligases are significantly associated with the etiology of human malignancies.8 Mutations of E3 ligases may result in the rapid degradation of tumor suppressors or, conversely, the lack of ubiquitination of oncogenic proteins.9 The pathological processes not only involve tumor metabolism regulation but also contribute to immunological tumor microenvironment (TME) modulation and cancer stem cell (CSC) stemness maintenance.10 Moreover, due to their high substrate specificity, E3 ligases and DUBs are promising potential therapeutic targets for cancer treatment. Currently, anti-cancer drugs targeting the proteasome, E3 and DUBs have been actively developed, and their therapeutic effects have been suggested by animal experiments and clinical trials.11,12 Here, we specifically summarize the mechanisms of the different components of the ubiquitin proteasome system (UPS), including E1, E2, E3, the proteasome and deubiquitinating enzymes, in mediating substrate ubiquitination/deubiquitination, highlight the unique functions of ubiquitination in tumorigenesis, including tumor metabolism regulation, immunological TME modulation and CSC stemness maintenance, and review potential therapeutic targets and the therapeutic effects of targeted drugs.
The components and processes of the UPS
Ub
Ub, named for its wide distribution in various types of cells among eukaryotes, was first identified by Gideon Goldstein et al. in 1975 and further confirmed over the next several decades.13,14 In the human genome, Ub is encoded by four genes, namely, UBB, UBC, UBA52 and RPS27A. The UBA52 and RPS27A genes encode single copy Ub, which is fused to the N-terminus of the ribosomal protein subunits L40 and S27a, respectively; the UBB and UBC genes encode polyubiquitin molecules that repeat the tandem 3 and 9 times, respectively. In cells, DUBs specifically cleave these fusion proteins to produce active Ub molecules. Occasionally, the monomeric Ub unit cannot be directly utilized by E1, E2 or E3. For example, PTEN-induced putative kinase 1 (PINK1)-mediated phosphorylation of Ser at position 65 of Ub is necessary for the ubiquitination of mitochondrial membrane proteins. Therefore, phosphorylation at Ser65 of Ub plays an important role in mitophagy.15,16,17,18 In addition to Ser65, Ub can also be phosphorylated at Thr7, Thr12, Thr14, Ser20, Ser57, Tyr59 and Thr66, and phosphorylated monoubiquitin and polyubiquitin chains may alter their recognition by E3 ligases or Ub-binding proteins.19,20,21,22 Additionally, the Ub molecule can also be modified by other PTMs. For instance, the acetylation of Ub at K6 and K48 inhibits the formation and elongation of Ub chains.23,24 These characteristics further complicate the Ub codes, including the length of the Ub chain, the degree of mixing and the state of the branch.
Ubiquitination
In 1977, Goldknopf et al. discovered that intracellular histones could be modified by ubiquitination, and ubiquitination emerged as a new protein PTM. In 2004, the Royal Swedish Academy of Sciences awarded the Nobel Prize in Chemistry to three scientists, Aaron Ciechanover, Avram Hershko and Irwin Rose, for their significant contributions in the field of ubiquitination.
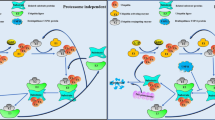
Ubiquitination is carried out in a highly specific manner that labels substrate proteins with Ub. The attachment of Ub to the substrate requires an enzymatic cascade consisting of E1, E2 and E3.13 Specifically, these processes include a three-step enzymatic reaction. Initially, Ub is activated by E1 in an adenosine triphosphate-dependent manner and then is transferred to E2. This process involves the formation of a thioester bond between the active site Cys residue of E1 and the C-terminal carboxyl group of Ub (E1~Ub). The human genome encodes only two kinds of E1, namely, UBa1 and UBa6 (Fig. 1a).25 In the second step, E1 delivers the activated Ub to E2 and assists the specific E3s in transferring the activated Ub to the substrate. Generally, humans have 35 distinct Ub-binding enzymes. Although all E2s contain a very conserved Ub-binding catalytic domain, members of this family exhibit significant specificity in their interaction with E3s (Fig. 1a).26,27 Finally, E3 ligases catalyze the transfer of Ub from E2~Ub to a specific substrate protein. When this process is completed, an isopeptide bond is formed between the lysine ε-amino group of the substrate and the C-terminal carboxyl group of Ub (Fig. 1a). The E3 ligase is the largest and most complex component of the UPS.26,28 To date, more than 600 E3 Ub ligases have been identified in the human genome (Fig. 1a). Although some E2s can directly transfer Ub to substrate proteins, in most ubiquitination processes, substrate selection and Ub linkage are achieved by E3.28,29

The components and processes of the UPS. a The components of the UPS and different classes of E3 ligases. b The ubiquitination linkage
Ubiquitination linkage
According to the structural characteristics, three main types of ubiquitination linkages have been identified: monoubiquitination, polyubiquitination and branched ubiquitination (Fig. 1b). Monoubiquitination refers to the attachment of a single Ub to a specific lysine residue of the substrate under the enzymatic cascade of E1, E2 and E3.5 Accumulating evidence has revealed that monoubiquitination is involved in the regulation of DNA damage repair. For example, the E3 ligase Rad18 regulates proliferating cell nuclear antigen (PCNA) monoubiquitination in response to DNA damage repair via the recruitment of DNA polymerases.30 In addition, the monoubiquitination of H2AX driven by TNF receptor-associated factor 6 (TRAF6) is a prerequisite for recruiting ataxia telangiectasia mutated (ATM).31 In other cases, monoubiquitination does not regulate DNA damage repair but mediates other cellular processes, such as autophagy and chromatin remodeling. For instance, monoubiquitination of membrane proteins can modulate their interaction with the autophagy adapter protein p62, thereby promoting mitochondrial autophagy and peroxisome autophagy.32 In addition, a well-defined case is the lysine-specific monoubiquitination of histone, whose modification takes part in chromatin remodeling.33 Moreover, Ras can also undergo multiple ubiquitination events at multiple sites and then regulate various signaling pathways.34
Polyubiquitination refers to the attachment of more than two Ub molecules to the same lysine residue of the substrate. Compared to monoubiquitination, there are many types of polyubiquitination, which can be linked by any lysine residues in Ub (K6, K11, K27, K29, K33, K48 and K63) or through its N-terminal Met.35,36 Initially, the ubiquitination of the K48 type was considered to be the only polyubiquitination. It serves as a degradation signal for transferring proteins to the 26S proteasome.37 With the deepening of research, scientists have realized that K48-type polyubiquitination is only the tip of the ubiquitination iceberg.23,38,39 To date, eight types of polyubiquitin linkages have been identified (K6, K11, K27, K29, K33, K48, K63 and Met1) with specific functions. Unlike K48 polyubiquitination, K6 polyubiquitination takes part in the process of DNA damage repair;40,41 K11 polyubiquitination plays an important role in the cell cycle and trafficking events;42,43 K27 polyubiquitination regulates mitochondrial autophagy;44,45 K29 polyubiquitination modulates ubiquitin-fusion degradation (UFD)-mediated protein degradation;46,47 and K33 polyubiquitination participates in Toll receptor-mediated signaling pathways.48,49 K63 polyubiquitination typically takes part in protein–protein interactions, protein activity and trafficking, thereby regulating various biological processes.50,51,52 Moreover, Met1 is usually involved in the coupling of the C-terminus of Ub to the methionine (M1) residue on the substrate to form a peptide bond. This type of ubiquitination modification is catalyzed by a specific E3 ligase and usually controls the TNFα signaling pathway.53,54,55
A Ub chain with a single linkage is called a homologous chain, and a branched polyubiquitin chain contains a variety of linkages. To date, in addition to the polyubiquitination of K6, K11, K27, K29, K33, K48, K63 and Met1, branched polyubiquitination also plays an important role in regulating various cellular processes.51 For example, the mixed K11 and K63 linkages participate in the Epsin1-mediated endocytosis of major histocompatibility complex I (MHCI).51,56
E3 ligases
E3 ligases are extraordinarily important in determining the specific type of ubiquitinated substrate. According to the catalytic structure, the E3 ligases are historically grouped into three types: the RING (really interesting new gene) family, the HECT (homologous to the E6-AP carboxyl terminus) family and the RBR (ring between ring fingers) family (Fig. 1a).57
RING type of E3 ligases
RING E3 is characterized by its RING or U-box folding catalytic domain that facilitates direct Ub transfer from E2 to the substrate (Fig. 1a). There are more than 600 E3 ligases in the human genome, and the RING family, encoded by ~270 human genes, is the largest family of E3 ligases. The RING finger protein generally contains the following amino acid sequence: Cys-X2-Cys-X9–39-Cys-X1–3-His-X2–3-Cys/His-X2-Cys-X4–48-Cys-X2-Cys, wherein X represents any amino acid. An E3 can bind directly to the substrate without the assistance of other proteins in catalyzing the ubiquitination of the substrate.58,59 For example, Mdm2 (murine double minute2)/Hdm2 (HDM2 being the human enzyme) and RNF152 (ring finger protein 152) belong to this class of E3s. The former promotes p53 degradation,60 and the latter mediates the polyubiquitination of RagA.61 Instead, some E3 catalytic domains and substrate recruitment modules are composed of multiple proteins, including SCF (Skp1-cullin1-F-box) and APC/C (anaphase promoting complex/cyclosome).62,63 SCF is a multisubunit complex consisting of four proteins: invariant Rbx1 (recruit the E2 enzyme), Cul1 (scaffold protein), Skp1 (bridge F-box proteins (FBPs)) and a different FBP (harbor catalytic activity). Approximately 70 FBPs have been identified in humans. Generally, the FBP takes effect in substrate recognition in the complex and selectively regulates many downstream biological processes.64 F-box/WD repeat-containing protein 7 (FBXW7) and S-phase kinase-associated protein 2 (SKP2) are well-studied FBPs. As an important tumor suppressor, FBXW7 participates in the degradation of many oncogenes, such as Myc, c-Jun, cyclin E, mTOR, Notch-1 and Mcl-1. Its mutation and deletion are often associated with tumorigenesis.65 SKP2, an important oncogene, regulates a number of CDK inhibitors (such as p27) and cell cycle proteins (such as p21, p57, cyclin A, cyclin E and cyclin D1).66 Another representative example is APC/C, which is the most sophisticated RING E3 ligase. APC/C contains a cullin-related scaffolding protein, APC2, to catalyze the ubiquitination reaction and further precisely controls cell cycle progression by alternately engaging with the substrate binding module, CDC20 (recruiting cell division cycle 20) or CDC20-like protein 1 (CDH1). APC/C-CDC20 promotes the cell cycle transition from metaphase to anaphase, while APC/C-CDH1 mediates mitotic exit and early G1 entry.67,68
HECT-type of E3 ligases
The second category of E3s is the HECT Ub ligase, which can be further divided into three subfamilies: Nedd4/Nedd4-like E3s containing a WW domain, HERC E3s containing an RLD domain and other E3s without a WW or RLD domain. Compared with RING E3s, there are fewer HECT E3s, with only 28 coding genes in the human genome.59,69 The most obvious feature of HECT E3s is the HECT domain, which forms a transiently covalent bound to Ub through a conserved Cys. Unlike RING E3s, HECT E3s bind to Ub in E2-Ub and form a thioester-linked intermediate before being ligated to the lysine residue of the substrate. That is, Ub is transferred from E2 to E3, and then E2 activates HECT, thereby linking Ub to the HECT E3s via a thioester bond and transferring Ub to the substrate (Fig. 1a).70
The polyubiquitination linkage promoted by HECT E3s is determined by the C-terminal region of E3s rather than E2s.71 For instance, E6-related protein (E6AP), the first identified HECT E3, promotes K48-linkage polyubiquitination and substrate degradation; Rsp5, which belongs to the Nedd4 family of HECT, adds K63 linkage polyubiquitination to the substrate and regulates cellular endocytosis (receptors, ion channels, etc.).72,73 It is surprising that replacing the 62 amino acid sequence in the C-terminal amino acid of Rsp5 with the corresponding sequence of E6AP makes Rsp5 form a specific K48 polyubiquitin chain.74 In addition to E6AP and Nedd4, HECT E3s bind directly to the PY motifs or variant regions of the substrate through the WW domain. This interaction is critical in regulating signaling pathways, especially in the Hippo and TGFβ signaling pathways.75,76,77,78
RBR-type of E3 ligases
The RBR family is a special type of E3 ligase with an activation mechanism that is different from those of the RING and HECT types. The human genome encodes more than a dozen RBR E3s, and the family members are all multidomain proteins consisting of really interesting new gene 1 (RING1), in-between RING (IBR) and really interesting new gene 2 (RING2) (Fig. 1a).79 Among them, RING1 binds to E2 and has the characteristics of RING-type E3s. RING2, which contains a catalytic Cys nucleophile, has a similar activity as HECT E3. It forms a thioester bond intermediate with Ub and transfers Ub to the substrate (Fig. 1a).80,81,82 The most striking E3 of the RBR family is the linear ubiquitin chain assembly complex (LUBAC) complex, consisting of HOIP, HOIL-1L and Sharpin. It is specifically responsible for regulating the linear ubiquitination of substrates, which plays a very important role in various biological processes, such as innate immunity and inflammation.55,83,84,85
All RBR E3s have a special regulation of self-inhibition due to their special structure. Mechanically, in the RBR E3 ligase, the domain outside the RING1, IBR and RING2 domains separates the RING2 domain from the RING1–IBR domain and structurally masks the active site Cys. The spatial distance between the active site of RING2 and E2 inhibits the thiol-transfer reaction and decreases the activity of RBR.80,86,87 Thus, the E3 ligase of the RBR family needs to undergo a conformational change to expose the Cys of RING2 and activate the E3 ligase.86
The activity of the RBR E3 family needs to be regulated in an orderly manner, and aberrant activity may lead to a number of diseases, including cancer and Parkinson’s disease (PD). For example, although its mutation is a major cause of familial PD,88,89 Parkin can function as a tumor suppressor to downregulate some substrates, such as cyclin D and cyclin E, and subsequently control cell cycle progression.90 Additionally, Parkin can promote the degradation of TRAF2 and TRAF6, thereby inhibiting the nuclear factor-kappa-B (NF-κB) signaling pathway and inducing tumor apoptosis.91
Nonclassical ubiquitination
Although the UPS is exclusive to eukaryotes, a recent report revealed that the SidE (siderophore E) effector family could perform atypical ubiquitination on a variety of host proteins.92 It is derived from Legionella pneumophila and works as an E3 ligase independent of E1 and E2 enzymes. The diverse strategies adopted by SidE are divided into two steps. First, the mART domain of SidE catalyzes the attachment of ADP-ribose to Arg42 of Ub and forms ADP-ribosylated Ub (ADPr-Ub). Second, the PDE domain of SidE further cleaves the phosphodiester bond in ADPr-Ub to form phospho-ribosylated Ub (Pr-Ub), which is covalently attached to the Ser of the substrate through the PDE domain of SidE. Further mechanistic studies successfully revealed a high-resolution crystal structure of the pre-reaction (SidE protein alone), the first step reaction complex (mART-Ub-NAD) and the second step reaction complex (PDE-Ub-ADP ribose). Combined with a large number of biochemical experiments and mutant analysis, the interaction between the novel E3 ligase SidE with Ub and ligand is completely presented.93,94 These exciting findings not only open a new chapter in the ubiquitination field but also provide a theoretical basis for developing targeted drugs.
Ub-like proteins
In addition to Ub, the Ub superfamily also contains Ub-like (UBL) proteins, which includes NEDD8, SUMO, FAT10, ISG15, ATG8, ATG1, HUB1 and FUB1. These UBL proteins not only have sequence homology and structural similarity to Ub but also use a similar enzymatic cascade to modify their substrate proteins.95,96 Due to space limitations, we will mainly discuss neddylation and SUMOylation below in this review.
NEDD8
In the UBL superfamily, NEDD8 has the highest homology with Ub and is indispensable in various biological processes. The specific attachment of NEDD8 to the substrate protein is called neddylation, which is a dynamic and reversible process. To date, there are many kinds of NEDD8-specific E3 ligases that determine the specificity of substrates, along with one E1 (NEDD8 activating enzyme, NAE) and two NEDD8-specific E2 ligases. The NEDD8 modification can be reversed by the COP9 signalosome (CSN), which deconjugates NEDD8 from the cullin protein.96,97,98
Unlike ubiquitination, neddylation does not degrade the substrate. However, as a PTM, neddylation also regulates the activation of substrates and subsequently controls a variety of cellular biological functions, such as cell cycle regulation and signal transduction. For example, neddylation mediates the biological function of the cullin-RING ubiquitin ligase (CRL) family and regulates the activity of the E3 complex by cullins, the key subunit of CRLs. Blocking the neddylation of cullins leads to substrate accumulation.99,100
Small ubiquitin-related modifier
Small ubiquitin-related modifier (SUMO), a widely expressed UBL protein in eukaryotes, is named for its similar structure and enzymatic cascade with Ub.101 SUMOylation is the process in which SUMO links to a substrate by forming an isopeptide bond between its terminal glycine and the lysine of the substrate.96,102 Currently, more than 500 substrates have been reported to undergo SUMOylation and take part in regulating the localization, stability and activity of many proteins.103,104 For instance, the SUMOylation of RPA1 (RPA subunit) regulates the affinity between RPA and RAD51 and promotes homologous recombination (HR)-mediated DNA double-strand break (DSB) repair.105 RNF4, a SUMO-targeted E3 ligase, has been identified as the link between ubiquitination and SUMOylation. SUMOylation of PML recruits RNF4 and triggers its degradation in a ubiquitination-proteasome-dependent way.106,107,108 Therefore, SUMOylation takes part in a variety of cellular physiological activities, such as gene stability maintenance and transcriptional regulation, and aberrant SUMOylation is closely related to the development and progression of certain diseases, including cancer.
Deubiquitinating enzymes
Ubiquitination, a dynamic and reversible process, is regulated by DUBs and E3 ligases.109 DUBs belong to the family of Cys proteases and cleave the isopeptide bond (the attachment of Ub to lysine) or the peptide bond (the connection of Ub to the N-terminal methionine of the protein) with high specificity.70,109
Currently, the human genome encodes no less than 100 DUBs (Fig. 1a). According to their sequence and structural similarities, they can be divided into six families: ubiquitin-specific proteases (USPs), ubiquitin carboxyl-terminal hydrolases (UCHs), otubain proteases (OTUs), Machado-Joseph disease protein domain proteases (MJDs), JAMM/MPN domain-associated metallopeptidases (JAMMs) and monocyte chemotactic protein-induced proteins (MCPIPs). Among them, all DUBs are Cys proteases except the JAMM family of metalloproteinases. These enzymes are capable of directly binding to different types, topologies or lengths of Ub chains and removing Ub chains from the substrate.110 Engineered deubiquitination synthesis reveals that the OTU specifically removes the K29 linkage Ub chain from the substrate,111 and the JAMM, such as AMSH, AMSH-LP, BRCC36 and POH1, are often specific for the Ub chain for K63 linkage ubiquitination.112,113 CYLD is more likely to act on linear ubiquitination and the K63 linkage Ub chain.114 Similarly, OTU domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) specifically acts on K48-linked ubiquitination,115,116 with Cezanne specifically removing the K11 linkage Ub chain,117,118 and TRABID specifically recognizing the K29-linked or K33-linked Ub chain.119 These specific Ub-type deubiquitinating enzymes cannot remove the last molecule of Ub-modified on the substrate, which may generate a monoubiquitinated substrate protein.
To date, many DUBs have been found to be associated with p53 regulation in tumorigenesis. For example, USP7 regulates the stability of both p53 and Mdm2 and maintains p53 ubiquitination levels;120 USP2 mediates the stability of Mdm2;121 USP10 modulates p53 localization and stability;122 OTUB1 abrogates p53 ubiquitination and activates p53.123 Interestingly, USP10 can stabilize both mutated and wild-type p53, with a dual role in tumorigenesis. USP11 participates in the regulation of DNA DSB repair. USP11 is often overexpressed in cancer and induces resistance to poly(ADP-ribose) polymerase 1 (PARP1) inhibitors.124
Ubiquitination in tumor metabolism regulation
Ubiquitination in the mTORC1 signaling pathway
As an important nutrient and key environmental stimulus, amino acids play a critical role in the mechanistic target of rapamycin complex 1 (mTORC1) signaling pathway. The mechanism of the amino acid-induced mTORC1 signaling pathway is still under continuous research. One well-demonstrated model has proposed that the activation of mTORC1 is induced by amino acid sensing cascades, including Rag GTPase, Ragulator and vacuolar H+-ATPase (v-ATPase), at lysosomes. During this process, amino acids can promote RagA/B binding to GTP, which is essential for mTORC1 lysosome localization.125,126,127 Moreover, many regulators of RagA/B have been identified, and these include the SLC38A9 functioning as the guanosine exchange factor (GEF) of RagA/B,128 Sestrin2 identified as the guanine nucleotide dissociation inhibitor (GDI) of RagA/B,129 and the GATOR1 complex acting as a GTPase-activating protein (GAP) to RagA/B.130 However, the role of ubiquitination in the RagA-mTORC1 pathway in response to amino acids is still poorly understood.
Ubiquitination of RagA
Recently, RagA and mTORC1 were found to be inactivated upon acute amino acid withdrawal. In this study, RagA was modified by polyubiquitination in an amino acid-sensitive manner. By screening a series of E3 ligases, RNF152, a lysosomal E3 ligase, was identified to mediate K63-linked polyubiquitination of RagA. In addition, ubiquitination of RagA recruited GATOR1, led to the inactivation of RagA and caused mTORC1 release from the lysosomal surface, thereby blocking the inactivation of the mTORC1 signaling pathway.61 Moreover, SKP2, another E3 ligase, could mediate RagA polyubiquitination on lysine 15.131 Thus, the polyubiquitination of RagA plays an important role in regulating the mTORC1 signaling pathway (Fig. 2a).

Ubiquitination in tumor metabolism regulation. a Ubiquitination in the mTORC1 signaling pathway. b Ubiquitination in the PTEN-AKT signaling pathway. c Ubiquitination of key transcription factors in cell metabolism regulation
Ubiquitination of mTOR
Undoubtedly, mTOR occupies a decisive position in the amino acid-induced mTORC1 signaling pathway. As mentioned above, being located on lysosomes via RagA deubiquitination is the premise of mTORC1 activation.130,132,133,134 In addition to RNF152/SKP2, TRAF6, an E3 ligase, is also reported to regulate mTOR translocation to the lysosome in response to amino acid stimulation by catalyzing the K63 ubiquitination of mTOR in the form of the p62-TRAF6 heterodimer complex. Thus, TRAF6 regulates autophagy and cancer cell proliferation by activating mTORC1.135 In addition to K63 ubiquitination, other types of polyubiquitin linkages have also been identified on mTOR. K48 ubiquitination is reported to be involved in the stability of mTOR. In this process, FBXW7 directly binds to mTOR and mediates its degradation by the proteasome (Fig. 2a).136 These results highlight the dominant role of ubiquitination in the mTORC1 pathway and reveal that different types of ubiquitination linkages lead to different functions.
Ubiquitination of DEPDC5
Amino acid stimulation can abolish the interaction between Sestrin2/CASTOR1/2 and the GATOR2 complex, which is essential for the activation of Rag GTPase. GATOR2 dissociates from Sestrin2/CASTOR1/2 and activates RagA/B by inhibiting the activity of GATOR1, which consists of DEPDC5, NPRL3 and NPRL2, and displays GAP activity to RagA/B.129,130,132,137 In addition, ubiquitination is also involved in the regulation of GATOR1 activity, in which Cullin3-KLHL22 E3 ligase promotes K48 linkage polyubiquitination of DEPDC5 and mediates its degradation by proteasomes under amino acid-stimulated conditions (Fig. 2a). KLHL22 plays a conserved role in the mTORC1-mediated autophagy, cell size and regulation of the nematode lifespan through DEPDC5. Moreover, the expression of KLHL22 is significantly negatively correlated with DEPDC5 in patients with breast cancer. Therefore, pharmacological interventions targeting KLHL22 may have therapeutic potential for relevant diseases, such as breast cancer and age-related diseases.138
Ubiquitination of mLST8
mTOR predominantly exists in two multicomponent kinase complexes, mTORC1 and mTORC2, which are structurally related but functionally distinct. The mTORC1 and mTORC2 signaling pathways are not independent.127 The activation of mTORC1 is inseparable from AKT activated by mTORC2, and the feedback inhibition of mTORC2 activation requires mTORC1-mediated Sin1 phosphorylation.139 mTORC2 contains six components, of which mTOR, DEPTOR and mLST8 are identical to mTORC1. Therefore, the dynamic assembly of mammalian lethality with SEC13 protein 8 (mLST8) in the two complexes is important for both complexes. Previous studies have shown that the K63 linkage polyubiquitination of mLST8, promoted by TRAF2, determines the homeostasis of mTORC1 formation and activation. Specifically, the K63 linkage polyubiquitination of mLST8 disrupts its interaction with the mTORC2 component Sin1 to favor mTORC1 formation. In addition, the deubiquitinating enzyme OTUD7B was reported to facilitate the formation of mTORC2 by removing the polyubiquitin chain on mLST8 and then promoting the interaction between mLST8 and Sin1. Collectively, the dynamic assembly and activation of mTORC1 and mTORC2 are dependent on the ubiquitination of mLST8, further demonstrating the importance of ubiquitination in the mTOR signaling pathway (Fig. 2a).140
Ubiquitination of DEPTOR
DEP domain-containing mTOR-interacting protein (DEPTOR) is an important component and negative regulator of both mTORC1 and mTORC2.141 Its stability is governed in a Ub-proteasome pattern by the E3 ligase beta-transducin repeat containing protein 1 (β-TrCP1), simultaneously proven by three different teams.142,143,144 In these studies, DEPTOR was recognized by β-TrCP1 via its degron sequence and subsequently ubiquitinated and degraded. Moreover, DEPTOR accumulation upon β-TrCP1 knockdown or the degron mutation could promote autophagy by inactivating mTORC1 (Fig. 2a).
The regulatory mechanisms of DEPTOR stability have also been explored. OTUB1 specifically interacts with DEPTOR via its N-terminal domain, removes the Ub chain on DEPTOR and stabilizes DEPTOR via DUB activity in an Asp88-dependent but not Cys91-dependent manner (Fig. 2a). Thus, β-TrCP1 and OTUB1 can balance cell survival and autophagy by activating mTORC1 through regulating DEPTOR ubiquitination, which also illuminates the importance of ubiquitination in the mTORC1 signaling pathway.145
Ubiquitination of TSC-Rheb
As a major regulator of Ras homolog enriched in brain (Rheb), the TSC complex is the central node for many growth and stress signals, ranging from growth factors, glucose, oxygen and energy to oncogenes and tumor suppressors. TSC2, a short-lived protein, is regulated by PTM in response to upstream signals.146 ERK- and AKT-mediated phosphorylation of TSC2 can result in the activation of Rheb,147,148 while ubiquitination can regulate the stability of TSC2. For example, TSC2 can bind to FBW5, a compound of the FBW5-DDB1-CUL4-ROC1 E3 ligase. The overexpression of FBW5 or CUL4A promotes TSC2 ubiquitination and degradation. Thus, FBW5 is a specific E3 ligase targeting TSC2 for its degradation and promoting TSC complex turnover (Fig. 2a).149
mTORC1 is recruited to lysosomes, where it is activated by its interaction with GTP-bound Rheb.133 The ubiquitination of Rheb regulates its activity. It has been reported that the ubiquitination of Rheb governs its nucleotide-bound status and controls the transformation between Rheb-GDP and Rheb-GTP. The lysosomal E3 ligase RNF152 can induce Rheb ubiquitination and promote its binding to the TSC complex in an epidermal growth factor (EGF)-sensitive manner. Upon growth factor stimulation, USP4 removes the Ub chain from Rheb in an AKT-dependent manner, which leads to the release of Rheb from the TSC complex, resulting in the subsequent activation of both Rheb and mTORC1. Therefore, the ubiquitination of Rheb, determined by RNF152 and USP4, also plays an important role in mTORC1 activation and consequent tumorigenesis (Fig. 2a).150
Ubiquitination in the adenylate-activated protein kinase signaling pathway
Adenylate-activated protein kinase (AMPK), the master sensor of energy in cells and organisms, is the core in regulating intracellular metabolic homeostasis, and its mutation is associated with tumorigenesis.151,152 When the cellular level of ATP decreases and AMP/ATP increases, the activation of AMPK increases. For instance, in response to a high AMP/ATP ratio in the cytosol, AMPK enhances glucose uptake and utilization by regulating key proteins in the cellular metabolic pathway, as well as fatty acid oxidation, to produce more energy.151,153 In addition to being phosphorylated, AMPK can also undergo ubiquitination. Specifically, the E3 ligase MAGE-A3/6-TRIM28 can ubiquitinate AMPK and promote its degradation. Furthermore, two homologs, MAGE-A3 and MAGE-A6, originally expressed only in the male germline, are reactivated in tumors. In mice, overexpressing MAGE-A3/A6 in cell lines promotes tumor growth and metastasis. In this process, MAGE interacts with the E3 ligase TRIM28, which controls the stability of AMPKα by mediating the K48 linkage polyubiquitination of AMPKα (Fig. 2a).154
In addition to K48 linkage polyubiquitination, the activation of AMPKα is also regulated by K63-specific polyubiquitination, which may mask its structure to block the access of liver kinase B1 (LKB1) and inhibit the activation of AMPKα. The deubiquitinating enzyme USP10 can remove the Ub chain from AMPKα and promote AMPKα activation by facilitating LKB1-mediated AMPKα phosphorylation, thereby participating in glucose and lipid metabolism in cells.155 In addition, ubiquitination also acts on LKB1. Generally, LKB1 functions as an oncoprotein and is activated by a complex with STRAD and MO25.156,157,158 SKP2 promotes the K63 polyubiquitination of LKB1 and plays an important role in LKB1 activation by maintaining the intact LKB1-STRAD-MO25 complex (Fig. 2a). Additionally, in a hepatocellular carcinoma model, SKP2-mediated LKB1 polyubiquitination is required for its activation and cell survival.159
In addition, ubiquitination is also involved in the regulation of the calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2)–AMPK signaling pathway. For example, the stability of CaMKK2 is controlled by the E3 ligase Fbxl12, which facilitates the degradation of CaMKK2 by promoting its ubiquitination (Fig. 2a).160 Thus, to maintain intracellular metabolic homeostasis, ubiquitination should not be ignored in the regulation of AMPK.
Ubiquitination in the PTEN-AKT signaling pathway
Unlike amino acid stimulation, growth factors are sensed by PTEN-AKT. It has been validated that both the PTEN-AKT and mTOR signaling pathways are important for the growth factor response. However, the two pathways were not unified until the identification of two key proteins: the small GTPase Rheb and its negative regulator TSC complex (Fig. 2a).148,161,162,163
Ubiquitination of PTEN
PTEN, a tumor suppressor and lipid phosphatase, plays an important role in tumorigenesis by inhibiting the PI3K signaling pathway. Generally, PTEN can be ubiquitinated and deubiquitinated. Nedd4-1, WW domain-containing ubiquitin E3 ligase 2 (WWP2), X-linked inhibitor of apoptosis protein (XIAP) and C-terminus of HSC70-interacting protein (CHIP) have been identified as the specific E3 ligases for PTEN, and the ubiquitination of PTEN mediated by each of them has different functions.164,165,166,167 For example, Nedd4-1, an E3 ligase of the HECT family, can promote both monoubiquitination and polyubiquitination of PTEN at K13 and K289, leading to the cytoplasmic localization and subsequent degradation of PTEN. PTEN is usually stable and not polyubiquitinated in the nucleus. The monoubiquitination of PTEN induces its translocation from the nucleus to the cytoplasm, and further polyubiquitination functions as a proteolytic signal to degrade PTEN via the proteasome.168 Moreover, WWP2 is also found to ubiquitinate PTEN and regulate cell apoptosis by mediating PTEN degradation.165 Additionally, the E3 ligase WWP1-induced PTEN ubiquitination inhibits PTEN dimerization, membrane recruitment and function. Inhibiting the activity of WWP1 leads to PTEN reactivation and blocks MYC-driven tumorigenesis.169 Moreover, the E3 ligases XIAP and CHIP can also target PTEN for ubiquitination and degradation and further activate the AKT signaling pathway (Fig. 2b).166,167
Due to the reversibility of ubiquitination, the deubiquitination of PTEN has also attracted the attention of researchers. USP7, a highly expressed DUB in prostate cancer and progressive multifocal leukoencephalopathy (PML), plays a direct role in PTEN deubiquitination and regulates its localization rather than protein stability.170 In addition, USP13 and OTUD3 can interact with PTEN and remove its polyubiquitin chain. Subsequently, blocking the degradation of PTEN inhibits the activity of the AKT signaling pathway and tumor growth (Fig. 2b).171,172
Ubiquitination of AKT
As a critical upstream target of the mTORC1 signaling pathway, AKT kinase transfers growth factor signals from the extracellular environment to the intercellular space. Its activation, which depends on the localization to the plasma membrane and is associated with K63-linked ubiquitination, is essential for cell growth, proliferation and metabolism.173,174 TRAF6, SKP2, tetratricopeptide repeat domain 3 (TTC3), CHIP, Nedd4 and MULAN have been identified as E3 ligases for AKT and participate in AKT kinase activation. TRAF6 is a direct E3 ligase for AKT in response to IGF-1 stimulation, and K63 polyubiquitination by TRAF6 is necessary for AKT membrane recruitment, phosphorylation and activation. The cancer-associated AKT mutation displays an increasing trend in AKT ubiquitination.175 In addition, SKP2 is also the E3 ligase for ErbB-receptor-mediated AKT ubiquitination. In a breast cancer metastasis model, SKP2 deficiency decreases the activation of the AKT kinase.176 Moreover, K48 linkage ubiquitination was also identified to regulate the stability of AKT instead of its activation. In addition, many studies have been identified that CHIP, MULAN and TTC3, an E3 Ub ligase, can ubiquitinate AKT and mediate its degradation (Fig. 2b).177,178,179,180
Corresponding to ubiquitination, “eraser” DUBs can also regulate protein degradation, localization, activation and protein–protein interactions of AKT. The cylindromatosis (CYLD), a well-known tumor suppressor, can interact directly with AKT and deubiquitinate its K63-linked ubiquitination in response to the stimulation of growth factors, which results in K48 linkage polyubiquitination via BRCA1 or TTC3 (Fig. 2b).181 The loss of CYLD accelerates tumorigenesis and triggers cisplatin resistance in melanoma and oral squamous cell carcinoma.182,183 Thus, CYLD is considered a molecular switch for the ubiquitination of AKT and determines the localization and activation of AKT during cancer progression.
To date, it has been well documented that the AKT kinase can be modified by phosphorylation, ubiquitination, acetylation, methylation and hydroxylation.184,185 Moreover, SUMOylation can also be responsible for AKT activation (Fig. 2b). Lysine 276, located in the SUMOylation consensus motif, is essential for AKT activation, while the mutation of K276R can reduce the SUMOylation of AKT, and AKT E17K can mediate cell proliferation, migration and tumorigenesis.186
Ubiquitination of key transcription factors in cell metabolism regulation
Transcription factors also play crucial roles in regulating cellular metabolism. When cells are in a state of limited energy intake or starvation, transcription factors can activate the related genes in glycolysis and the tricarboxylic acid cycle, increase hepatic glucose production, reduce insulin secretion and provide a substrate for gluconeogenesis. Among them, hypoxia-inducible factor-1α (HIF-1α), Myc and p53 are closely related to cell metabolism.151,187,188
Ubiquitination regulates HIF-1α
HIF1, a transcription factor widely expressed under hypoxic conditions, is the key regulator of oxygen homeostasis in cells.189 It can induce the expression of many glycolytic genes, such as glucose transporter member 1, hexokinase 1 and hexokinase 2, lactate dehydrogenase A, monocarboxylate transporters 4 and PDK1, which are indispensable in glucose uptake. HIFs include three subtypes: HIF1, HIF2 and HIF3. They are composed of α and β subunits, wherein the α subtype, which is sensitive to oxygen, is easily degraded via the proteasome pathway; and in contrast, the β subunit is more stable.190,191
Due to the important role of HIF in cells, many studies have been performed to investigate the regulatory mechanism of HIF.192,193 Among them, E3 ligases and DUBs have been found to regulate the stability of HIF. Under normal conditions, HIF-1α is extremely unstable. The tumor suppressor E3 ligase von Hippel-Lindau (VHL), which is widely involved in tumor vascularization, interacts with HIF-1α in a proline hydroxylation-dependent manner and mediates its degradation, thereby inhibiting tumor growth (Fig. 2c).194
It has been found that glycogen synthase kinase 3 (GSK3β) phosphorylates HIF-1α and promotes K48 polyubiquitination by FBW7, thereby mediating the degradation of HIF-1α and inhibiting angiogenesis, cell migration and tumor growth.195 On the other hand, the FBW7-mediated proteolytic signal can be removed by the deubiquitinating enzyme USP28.196 In addition to VHL, the tumor suppressors p53, Tap73 and PTEN also recruit the E3 ligase Mdm2 to HIF-1α, leading to the ubiquitination and degradation of HIF-1α by the proteasome.197 Unlike K48 linkage polyubiquitination, TRAF6 can mediate the K63 linkage polyubiquitination of HIF-1α and block its degradation (Fig. 2c).198 Moreover, the E3 ligase FBXO11 can reduce the mRNA level of HIF-1α but has no effect on its protein stability.199
Similarly, HIF-1α can also be deubiquitinated. For example, OTU deubiquitinase 7B (OTUD7B) can deubiquitinate HIF-1α and inhibit its degradation by the lysosome.200 By screening an siRNA library, the deubiquitinating enzyme USP8 interacts with HIF-1α, removes the Ub chain from HIF-1α, and maintains its expression and transcriptional activity under normal oxygen.201 Moreover, HIF-1α can also undergo SUMOylation. Overexpressing the SUMO molecule and SUMO ligase in lymphatic endothelial cells can induce the SUMOylation of HIF-1α and maintain the stability and transcriptional activity of HIF-1α (Fig. 2c).202
Ubiquitination regulates c-Myc
The transcription factor c-Myc can regulate cell proliferation, metabolism and metastasis by mediating a variety of cellular metabolism pathways, such as glucose metabolism, fatty acid and nucleotide biosynthesis. c-Myc is involved in glucose uptake and glycolysis, and its activation upregulates the expression of glucose transporters and hexokinases.203,204 As a very unstable protein with a very short half-life, c-Myc can be degraded in a proteasome-dependent manner, and many studies have identified the E3 ligase and DUB of c-Myc. For example, in the G1 to S phases of the cell cycle, the E3 ligase SKP2 can interact with c-Myc and mediate its degradation by ubiquitination, thereby blocking the cell cycle and inhibiting tumorigenesis.205,206 Additionally, the phosphorylation of c-Myc on Thr58 by GSK3 promotes its interaction with Fbw7 and facilitates K48 linkage polyubiquitination. The subsequent degradation inhibits cell proliferation and tumor growth. Missense mutations of Fbw7 are found in many malignancies; for example, the mutation R465C fails to degrade c-Myc in T-cell acute lymphoblastic leukemia.207 In addition to Fbw7, other E3 ligases, such as β-TrCP1, CHIP and FBXO32, can also ubiquitinate c-Myc, mediate its subsequent degradation and inhibit tumorigenesis.208,209,210 Moreover, the USP37 and USP36 can promote tumorigenesis by stabilizing c-Myc.211,212 By mass spectrometry, SUMO ligase protein inhibitor of activated STAT (PIAS) and Sentrin-specific protease 7 (SENP7) were also found to control the SUMOylation of c-Myc at K326 and regulate its ubiquitination and degradation (Fig. 2c).213
Ubiquitination regulates p53
p53, one of the most important tumor suppressors, works in multiple cellular processes, such as cell cycle regulation, DNA repair and apoptosis. In addition, p53 also plays an important role in cell metabolism by inhibiting glycolysis and promoting oxidative phosphorylation in response to nutrient stimulation.214,215
Under the stimulation of low carcinogenicity and genotoxicity, p53 is persistently expressed, but its protein level is often maintained at low levels. Under the stimulation of the external environment, the degradation of p53 is inhibited, resulting in the improvement of its stabilization and transcriptional activity.214 To date, more than 15 E3 ligases of p53 have been identified, and they are divided into the RING family (Mdm2, Pirh2, Trim24, Cul1/Skp2, Cul4a/DDB1/Roc, Cul5, Cul7, Synoviolin, Cop1, CARP1/2, CHIP, UBE4B) and HECT family (ARF-BP1, Msl2/WP1) ligases. Moreover, both K48- and K63-linked ubiquitination have been found on p53. The former can promote the degradation of p53 via the proteasome, while the latter is required for the translocation of p53 to the cytoplasm. More specifically, the E3 ligases Mdm2, Pirh2, Trim24, Cul1/Skp2, Cul4a/DDB1/Roc, Cul5, Synoviolin, Cop1, CARP1/2, ARF-BP1, Msl2/WP1, CHIP and UBE4B mediate K48 linkage polyubiquitination, which degrades p53 via the proteasome, while Cul7-mediated polyubiquitination regulates the localization and activity of p53 (Fig. 2c).9,216,217
As mentioned above, deubiquitination is also a key regulatory step in cell metabolism. The deubiquitination enzymes HAUSP and USP10 can remove the proteolytic signal of p53 and abolish its degradation. HAUSP, which is localized in the nucleus, can prevent the degradation of p53 by deubiquitinating even under the circumstance of highly expressed Mdm2.120,218 Unlike HAUSP, USP10 is generally located in the cytoplasm. In the case of DNA damage, the phosphorylation of USP10 at Thr42 and Ser337 mediated by ATM is essential for its translocation to the nucleus, where USP10 induces the deubiquitination of p53 and makes p53 work as a tumor suppressor (Fig. 2c).122
In addition to ubiquitination, p53 can also undergo other ubiquitination-like modifications, such as neddylation and SUMOylation. For example, Mdm2 can inhibit p53-mediated transcriptional activity via neddylation on K370, K372 and K373 of p53.219 SUMOylation of p53 can regulate the transcriptional activity under genotoxic stress (Fig. 2c).220
Ubiquitination in immunological TME modulation
The innate immune system can recognize invading pathogenic microorganisms by inducing the expression of proinflammatory and anti-infective genes. During the process of tumorigenesis, premalignant lesions, regarded as invaders, can lead to inflammation and activate local innate immune surveillance to the malignant cells in the early stages.221 Then, the inflammatory, immunological and metabolic processes of the tumor and the tumor-draining lymph nodes (TDLNs), constituting the immunological TME, are also reprogrammed.222 According to Dvorak’s 1986 comment, malignancies are regarded as “wounds that do not heal”.223,224 As an important risk factor for malignancy, chronic immune activation and inflammation persistently promote TME formation by providing inflammatory mediators such as TNF-α, IL-1β, IL-6 and TGF-β and ultimately lead to angiogenesis and antitumor immunity.225,226
Ubiquitination, a ubiquitous PTM in cells, appears to be a critical mediator of the host cell defense and immunological TME modulation by regulating cell signal transduction pathways. On the one hand, as a multifunctional signal regulator, ubiquitination can precisely regulate the process of the immune response in a time and space manner.227 On the other hand, it can effectively induce antitumor immunity by mediating the degradation of key signal transduction molecules to stabilize and maintain the balance between tumor suppressors and oncoproteins.228,229 The Toll-like receptor (TLR), RIG-like receptor (RLR) and DNA recognition receptor signaling pathways are very important in the immune system; thus, we introduce the functions of ubiquitination in TLR, RLR and DNA recognition receptor signaling pathways, and related molecular regulatory mechanisms are relatively highly studied.
Ubiquitination in the TLR signaling pathway
As innate immune receptors, TLRs are involved in the recognition of microorganisms by the immune system. Generally, TLRs recognize a conserved component of the pathogen and then activate the signaling pathway.230 TLR signaling in immune and inflammatory cells of the TME also induces the production of proinflammatory cytokines and leads to the polarization of tumor-associated macrophages (TAMs), activation of protumorigenic functions of immature myeloid cells and transformation from fibroblasts into cancer-associated fibroblasts (CAFs).231 TLR, a family of receptors, has 13 members. Among them, TLR4/7/8/9 activates the MyD88-dependent signaling pathway and subsequently elevates the activity of the downstream TRAF6. In the TLR4 signaling pathway, the K63 polyubiquitin chain catalyzed by TRAF6 recruits the TGF-β-activated kinase 1 (TAK1) complex and IκB kinase (IKK) complex and then increases the expression of inflammatory factors downstream of NF-κB. The K63 polyubiquitin chain also recruits TRAF3, IKKα and IRF7 and ultimately increases the expression of type I interferon in the TLR7/8/9 signaling pathway.232,233 In addition, the K63 polyubiquitination of receptor-interacting serine/threonine-protein kinase 1 (RIPK1), catalyzed by the E3 ubiquitin ligase Peli1, plays an important role in the TIR-domain-containing adapter-inducing interferon-β (TRIF)-dependent TLR signaling pathway, which significantly enhances the activation of NF-κB by transferring NF-κB or IRF3 to the nucleus to regulate the transcription of target genes.234 MARCH5, an E3 ligase located on mitochondria, catalyzes K63-linked polyubiquitination of TRAF family member-associated NF-κB activator (TANK) and then enhances the activation of the TLR signaling pathway (Fig. 3a).235

Ubiquitination in immunological tumor microenvironment (TME) modulation. a Ubiquitination in the TLR signaling pathway. b Ubiquitination in the RLR signaling pathway. c Ubiquitination in the STING-dependent signaling pathway
NF-κB signaling pathway inhibitors are degraded by the ubiquitination-proteasome pathway. For example, the transcription factor NF-κB is retained in the cytoplasm due to its interaction with the inhibitor IκBα in the remaining cells, while lipopolysaccharide (LPS) stimulation induces the phosphorylation of IκBα by IKKβ and degradation by the E3 ligase β-TrCP1.236 In some cases, polyubiquitination at K48 and K63 can also synergistically promote the activation of signaling pathways. For example, MyD88 is crucial for gathering TRAF6, TRAF3 and cIAP1/2 in the TLR4-MyD88 signaling pathway. More specifically, TRAF6, recruited by MyD88, activates cIAP by catalyzing the K63-linked polyubiquitination of cIAP, while activated cIAP induces the K48 polyubiquitination of TRAF3, leading to the degradation of TRAF3 by the proteasome.237 TRAF6 can also result in the ubiquitination of ECSIT and increase mitochondrial and cellular TLR-induced ROS generation.238 Recently, USP4 has been identified as a new DUB of TRAF6 and can negatively regulate the NF-κB signaling pathway.239
The ubiquitination process is also a safeguard to prevent tumorigenesis by inhibiting the overactivation of NF-κB at multiple sites. A negative regulator A20 can cooperate with RNF11, ITCH and TAX1BP1 to remove the K63-linked polyubiquitin chain catalyzed by TRAF6 from cIAP.240 Additionally, Nrdp1, Trim38, WWP2 and PDLIM2 trigger the K48-linked polyubiquitination of MyD88, TRAF6, TRIF and p65, respectively, and promote the degradation of target proteins by proteasomes (Fig. 3a).241,242,243,244
Ubiquitination in the RLR signaling pathway
The RLR family comprises three members: retinoic acid-inducible gene 1 (RIG-I), melanoma differentiation-associated protein 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2). The RIG-I/MDA5 receptor recognizes and binds to viral RNA and regulates the expression of antiviral genes through the MAVS-TBK1-IRF3 signaling pathway.245 Many E3 ligases are involved in regulating the downstream signaling of MAVS. For example, the E3 ligase Trim25 catalyzes the K63 polyubiquitination of RIG-I. Subsequently, MAVS is recruited, and the activation signal is transferred to the MAVS signal complex.246 Another is LUBAC, which decreases the activation of RIG-I by inhibiting the binding of Trim25 and RIG-I or mediating the polyubiquitination and degradation of Trim25.247 Similar to Trim25, RNF135 (also known as Riplet) also catalyzes the K63 polyubiquitination of RIG-I and activates the RIG-I signaling pathway.248 Additionally, the E3 ligase MIB1/2 regulates the activation of TANK-binding kinase 1 (TBK1) by catalyzing TBK1 K63-linked polyubiquitination. The K27-linked polyubiquitination of NF-κB essential modulator (NEMO), which is mediated by Trim23 and Shigella effector IpaH9.8, can also promote the activation of the TBK1 and IKK complexes (Fig. 3b).249,250 Thus, E3 ligases have a key regulatory function in the RLR signaling pathway, and the regulation of ubiquitination on the immune response is complex and precise.
To avoid tumorigenesis caused by excessive activation of the RLR signaling pathway, host cells inhibit the overproduction of downstream inflammatory factors and interferons by ubiquitinating and degrading key proteins in the RLR signaling pathway. The E3 ligase RNF125 catalyzes the K48 linkage ubiquitination of RIG-I/MDA5 and promotes the degradation of RIG-I/MDA5 through the proteasome.251 More importantly, upon stimulation with an RNA virus, the lectin family member Siglec-G recruits the E3 ligase c-Cbl, catalyzes the K48-linked ubiquitination of RIG-I, and promotes the degradation of RIG-I.252 As a pivotal protein of the RLR signaling pathway, mitochondrial antiviral signaling (MAVS) is also regulated by ubiquitination. The poly(rC)-binding protein PCBP2 recruits the E3 ligase ITCH, which catalyzes the K48 ubiquitination of MAVS, regulates its degradation, and inhibits the activation of the RLR signaling pathway mediated by MAVS.253 Many E3s have been identified to regulate the stability of MAVS downstream signaling components. For example, NACHT, LRR and PYD domains-containing protein 4 (NLRP4) recruits the E3 ligase DTX4 and promotes the ubiquitination and degradation of TBK1.254 Triad3A catalyzes the K48 polyubiquitination of TRAF3.255 The E3 ligase RNF5 promotes the K48-linked ubiquitination and degradation of MAVS (Fig. 3b).256
Ubiquitination in the STING-dependent signaling pathway
STING, an adapter transmembrane protein residing in the endoplasmic reticulum (ER), is an important innate immune sensor for tumor detection.257,258,259 The STING pathway is activated by antigen-presenting cells (APCs) and produces type I IFNs. Subsequently, adequately activated APCs in the TME induce CD8+ T cell priming and lead to adaptive anticancer immune responses.260 Recently, many DNA-binding proteins have been reported in the cytoplasm and include cGAS, Mre11, IFI16 (p204), DDX41 and DNA-PKcs. They recognize DNA in the cytoplasm and strongly initiate the type I interferon gene through the STING-TBK1-IRF3 signaling axis. In response to the stimulation of cytoplasmic DNA, STING on the ER can rapidly dimerize and transfer from the ER to the nuclear peripheral bodies. Interestingly, TBK1 also aggregates into the nuclear peripheral bodies and forms the STING-TBK1 complex, which is essential for the activation of TBK1 (Fig. 3c).261,262
Currently, various polyubiquitinations of STING have been identified, including polyubiquitination of K63, K48, K11 and K27, all of which play important roles in the innate immune response against RNA and DNA infections. The different connections between these polyubiquitin chains not only broaden the functional spectrum of STING but also determine its strength and duration in regulating the expression of type I interferon genes. Under the stimulation of exogenous DNA, Trim56 induces K63 linkage ubiquitination of STING and promotes STING dimerization and recruitment to TBK1.263 In addition to Trim56, the E3 ligase Trim32 promotes the interaction between STING and TBK1 by catalyzing the K63-linked polyubiquitination of STING and finally increases the expression of STING-mediated interferon-β.264 To control cancer cells, HER2 also connects with STING and recruits AKT1 to directly phosphorylate TBK1, which prevents TBK1 K63-linked ubiquitination.265 Additionally, K48 polyubiquitination also inhibits signal transduction by promoting the degradation of STING. In detail, under the stimulation of DNA or RNA, RNF5 catalyzes the K48 polyubiquitination of STING at K150 and K48. This modification serves as a proteolytic signal by targeting STING for degradation via the 26S proteasome.266 The E3 ligase RNF26 localized on the ER catalyzes the ubiquitination of the K11 linkage of STING. In the early stage of viral infection, the K11 polyubiquitin chain catalyzed by RNF26 competes with the K48 ubiquitination of STING, prevents the RNF5-mediated degradation of STING, and increases the expression of type I interferon, whereas in the late stage of a viral infection, RNF26 inhibits the expression of type I interferon by promoting lysosomal degradation of IRF3.267 In addition, K27-linked polyubiquitination of STING induced by autocrine motility factor receptor (AMFR) works as a molecular platform to recruit TBK1 and promotes the translocation of TBK1 to the nuclear peripheral bodies (Fig. 3c).268
Modification of the K27- and K63-linked ubiquitination chains of STING activates anti-DNA viral effects in cells. USP21 can interact directly with STING and remove the K27 and K63 Ub chains on STING, thereby inhibiting the production of type I interferons. In the late stage of herpes simplex virus 1 (HSV-1) infection, protein kinase p38 phosphorylates USP21 and recruits it to bind to STING. Inhibiting the activity of p38 in mice blocks the binding of USP21 to STING, which in turn protects mice from an HSV-1 infection by inhibiting the production of type I interferons. Additionally, in USP21 knockout mice, resistance to DNA viruses was enhanced (Fig. 3c).269
Ubiquitination in CSC stemness maintenance
The “stemness” state of stem cells is the key ability to self-renew and differentiate into the germline. Stem cells can be found in adult and embryonic tissues and play an extremely important role in cell regeneration, growth and embryonic development. CSCs are a subpopulation of tumor masses with pluripotent tumorigenesis, metastasis dissemination, drug resistance and cancer recurrence.270 In CSCs, a fine-tuning circuit consisting of a core set of transcription factors regulates stemness-specific gene expression profiles, including the core stem cell regulator triplet, Oct4, Sox2 and Nanog.271,272,273 In addition, some signaling pathways, including the Hippo and Wnt signaling pathways, also participate in CSC stemness maintenance. Ubiquitination plays an important role in CSC characteristics, such as self-renewal, maintenance, differentiation and tumorigenesis. By comparing the protein expression and ubiquitination levels between pluripotent and differentiated stem cells via quantitative proteomics, Iannis surprisingly found the ubiquitination of core transcription factors, which included Nanog, Oct4 and Sox2, indicating the crucial roles of the ubiquitination-mediated transcriptional regulatory network in maintaining the stemness and pluripotency of stem cells.274 This section focuses on the recent progress in the ubiquitination-mediated transcriptional regulatory network and signaling pathways in maintaining the stemness and pluripotency of stem cells.
Nanog ubiquitination
As the key transcription factor for maintaining stem cell pluripotency and promoting somatic cell reprogramming, Nanog is mainly regulated by its allele, transcription factors and PTM in stem cells. Nanog contains a degradation determinant PEST sequence with a very short half-life. However, the regulatory mechanism of Nanog stability was unclear until 2014. Researchers identified that ERK1 phosphorylated the Ser52 of Nanog and promoted its interaction with Fbxw8, which played an important role in Nanog's proteasome pathway degradation and the differentiation of stem cells (Fig. 4a).275

Ubiquitination in cancer stem cell (CSC) stemness maintenance. a The ubiquitination-mediated regulation of the transcriptional regulatory network in maintaining the stemness of stem cells. b Ubiquitination in the Wnt and HIPPO signaling pathways
In addition to Fbxw8, another investigation proposed that SPOP, the Cullin3-dependent E3 ligase, could also mediate Nanog degradation. SPOP contains a BTB domain linked to Cullin3 and a MATH domain that specifically recognizes and binds to substrates. A variety of biological processes, including cell proliferation, apoptosis and cell senescence, are regulated by SPOP by degrading a variety of substrates, such as AR, DEK, ERG, SRC3, DAXX and SENP7.276,277,278,279,280,281 Two independent studies revealed that the Pin1 or AMPK-BRAF signaling pathway phosphorylated Nanog-Ser68; then, the modified Nanog was recognized and polyubiquitinated by SPOP and finally degraded via the proteasome (Fig. 4a).282,283
As ubiquitination is a reversible process, Nanog may also be regulated by deubiquitinating enzymes during stem cell or somatic cell reprogramming. To screen the DUB of Nanog, the deubiquitinating enzyme USP21 was identified by the efficient dual-luciferase reporter assay system. USP21 could significantly enhance the stability of Nanog and then maintain the self-renewal of stem cells. The interaction between USP21 and Nanog could be blocked by ERK-mediated phosphorylation at position 539 of USP21, which subsequently promoted the differentiation of stem cells by downregulating the stability of Nanog.284,285 Additionally, USP21 was also reported to maintain the self-renewal of embryonic stem cells by stabilizing Nanog (Fig. 4a).285,286
Oct4 ubiquitination
As a member of the POU transcription factor family, Oct4 plays a key role in maintaining the stemness and pluripotency of stem cells.287 In stem cells, the protein level of Oct4 is accurately regulated, and the abnormal expression of Oct4 is the main cause of somatic cell cloning failure. In terms of the regulation of Oct4, the UPS plays an important role. WWP2, an E3 ligase of the HECT family, interacts directly with Oct4 and mediates its proteasome degradation by promoting its ubiquitination.288,289 Additionally, the E3 ligase ITCH can also interact with Oct4, promote its ubiquitination and then regulate Oct4 transcription activation; however, ITCH cannot mediate the degradation of Oct4 (Fig. 4a).288 The different functions of the two E3 ligases for Oct4 in stem cells suggest that the same substrate can be regulated by different E3 ligases. In addition, ERK1 can phosphorylate Ser111 of Oct4, induce its ubiquitination and promote its degradation and cytoplasm location.289
Sox2 ubiquitination
Similar to Oct4 and Nanog, the protein level of the core transcription factor Sox2 is also regulated by the UPS in stem cells.290 In 2014, the methylation enzyme Set7 was found to induce the monomethylation of Sox2. It not only inhibited the expression of Sox2 but also promoted the ubiquitination of Sox2 by facilitating the interaction between WWP2 and Sox2. Thus, Set7 can mediate the degradation of Sox2 and stem cell differentiation. In contrast, AKT phosphorylates Sox2 and inhibits the Set7-mediated methylation of Sox2, thereby inhibiting the ubiquitination of Sox2 and maintaining the self-renewal of stem cells, suggesting that Sox2 is precisely regulated by PTM to maintain stem cell pluripotency and direct differentiation.291 As an important regulator of stem cell self-renewal, Sox2 can also interact with APC and Ube2s directly, which mediates the degradation of Sox2 by promoting K11 linkage ubiquitination at the Lys123 of Sox2 (Fig. 4a).292,293,294,295
In addition, USP7 is able to maintain stem cell self-renewal by inhibiting the E3 ubiquitin ligase β-TrCP1-mediated ubiquitination of REST, a stemness transcription factor that plays an important role in neural differentiation (Fig. 4a).296 In addition to USP7, many deubiquitinating enzymes have been identified in transcriptional regulation in stem cells based on genome-wide localization analysis. For example, USP10, USP16, USP3, USP37 and USP44 are reported to bind to the promoter of Nanog; USP44 is capable of binding to the promoter of Oct4; and USP22, USP25, USP44 and USP49 are proven to bind to the promoter of Sox2.273
Ubiquitination in the Wnt signaling pathway
The Wnt signaling pathway is an evolutionarily conserved signaling pathway that is critical in regulating CSCs.297 In the absence of Wnt on the cell surface, the “destructive complex” of β-catenin, a multisubunit complex consisting of four proteins, GSK3β, Axin, casein kinase 1α (CK1α) and adenomatous polyposis coli (APC), can bind to β-catenin and promote its phosphorylation at the N-terminus via CK1α and GSK3β. β-TrCP1 can recognize phosphorylated β-catenin and promote its ubiquitination and degradation, thereby negatively regulating the Wnt signaling pathway.298,299,300 As a ligand, Wnt bridges the Frizzled-LRP5/6 protein and phosphorylates LRP5/6 through CK1a. Subsequently, it recruits the “destructive complex” to the cell membrane, inhibits the phosphorylation and ubiquitination of β-catenin and promotes its accumulation in the nucleus to regulate CSC stemness maintenance.301,302
In addition to the degradation of β-catenin, the internalization and lysosomal degradation of Frizzled-LRP5/6 can be regulated by the E3 ligases ZNRF3- and RNF43-mediated ubiquitination. It is also an important way to negatively regulate Wnt signaling.303,304,305,306 As a feedback regulator of Wnt signaling, inactivation of RNF43 and ZNRF3 leads to a significant expansion of the crypt proliferation region and promotes tumorigenesis. Moreover, RNF43 inactivating mutations can be found in various cancers.307,308
Axin is a key scaffold protein in the “destructive complex”, and its regulation is associated with the Wnt signaling pathway. For example, the E3 ligase RNF146 promotes ubiquitination-mediated proteasomal degradation of Axin on the basis of the PARsylation of Axin,309,310 while SIAH binds to Axin and mediates its degradation, which amplifies the feedback regulation of the Wnt signaling pathway.311 Unlike the degradation signal, SMURF1 inhibits its interaction with LRP5/6 by mediating the K29-linked polyubiquitination of Axin and negatively regulates the Wnt signaling pathway.312
Notably, UPS also regulates the ubiquitination of different components in the Wnt signaling pathway. For example, Nedd4L, ITCH and KLHL12 are able to negatively regulate the Wnt signaling pathway by targeting Disheveled (Dvl) degradation.313,314,315 In addition, the deubiquitinating enzymes USP34/USP7, CYLD/USP9X and USP4 can bind to Axin, Dvl and β-catenin, respectively, thereby promoting the nuclear localization of β-catenin and the Wnt signaling pathway by inhibiting their ubiquitination.316,317,318,319,320
Ubiquitination in the Hippo-YAP signaling pathway
In mammals, the Hippo signaling pathway can also maintain CSC stemness, regulate cell growth, control the size of organs and take part in tumorigenesis.321,322 Ubiquitination also plays an important role in regulating the Hippo signaling pathway, with a variety of E3 ligases being identified. For example, CRL4DCAF1 negatively regulates the Hippo signaling pathway by ubiquitinating and degrading Lats1 while promoting the monoubiquitination of Lats2 and inhibiting its activity.323 Similarly, the stability of Last2 is also regulated by the E3 ligase SIAH2. SIAH2, an important regulator of the HIF signaling pathway, degrades PHD3/1 by ubiquitination in a hypoxic environment. In turn, the activation of the Hippo signaling pathway also controls the stability of HIF1α and the HIF1α signaling pathway (Fig. 4b).194,324 This part of the work highlights the important roles of the hypoxic environment in regulating the ubiquitination of the Hippo signaling pathway.
YAP/TAZ is the key component in the Hippo signaling pathway. The stability of YAP/TAZ is also controlled by PTM. For example, phosphorylated YAP is recognized and ubiquitinated by the E3 ligase β-TrCP1.325 The CK1ε-mediated phosphorylation of TAZ is recognized by β-TrCP1 and promotes the Κ48-linkage ubiquitination of TAZ. The ubiquitination of TAZ mediates its entrance into the proteasome for degradation (Fig. 4b).326 Unlike K48 linkage ubiquitination, a recent report indicated that the E3 ligase SKP2 induces the nonproteolytic K63 linkage ubiquitination of YAP and leads to its nuclear localization and interaction with the nuclear binding partner TEAD. In this process, OTUD1 could remove the K63 linkage ubiquitination of YAP and negatively regulate transcriptional activity and cell growth (Fig. 4b).327
The Wnt signaling pathway, associated with CSC stemness maintenance, also plays an important role in regulating the stability and degradation of TAZ. Phosphorylated β-catenin can serve as a platform for TAZ and β-TrCP1 and promotes TAZ degradation by β-TrCP1.328 Moreover, YAP/TAZ is essential for β-TrCP1 recruitment to the APC complex and β-catenin inactivation. Under Wnt OFF conditions, YAP/TAZ is sequestered in the APC complex by binding to Axin1 and then recruiting β-TrCP1 to degrade β-catenin. Under Wnt ON conditions, YAP/TAZ is dissociated from Axin1 and accumulates in the nucleus to regulate the Wnt/β-catenin signaling pathway.329 In addition to the Ub ligases reported above, the K48 linkage polyubiquitination mediated by Nedd4 also functions as a proteolytic signal and degrades WW45 and Last1/2 via the proteasome (Fig. 4b).330 Taken together, these clues indicate that ubiquitination can regulate the Hippo and Wnt signaling pathways by controlling the stability of different substrates.
Cancer therapeutic strategy via targeting the UPS
As mentioned above, the UPS plays an essential role in protein degradation and fundamental cellular process regulation.331,332 Genetic alterations, abnormal expression or dysfunction of the UPS often lead to human pathogenesis, especially cancer. Thus, these components can serve as potential drug targets for therapeutic strategies against cancer.8 Currently, many small molecule inhibitors have been developed that target different components of the UPS, which include the proteasome, E3 ligases, E1 enzymes, E2 enzymes and DUBs, and their therapeutic effects are gradually being tested.333
Targeting the proteasome activity
Among all UPS components, only the proteasome has been successfully exploited as a therapeutic target for the clinical treatment of cancer. Tangible success has been achieved using proteasome inhibitors (PIs), such as bortezomib, carfilzomib, oprozomib and ixazomib (Fig. 4b).334,335 Under normal physiological conditions, selective tagging of proteins with Ub is targeted to the proteasome and results in proteasome-mediated proteolysis.336 The proteasome exhibits three distinct activities, namely, chymotrypsin-like, trypsin-like and caspase-like activities. Its alterations are found in various human diseases. In tumorigenesis, proteasome abnormalities are not observed; thus, the function of the proteasome in tumor cells may be on the basis of their own needs.11
The boronic acid derivative bortezomib (Velcade, Millennium Pharmaceuticals), a unique first-in-class compound, can slowly and reversibly block chymotrypsin-like and decrease trypsin-like and caspase-like activities of the 20S proteasome.337 Previous studies have proven that bortezomib can inhibit proliferation and induce cell apoptosis by blocking the NF-κB pathway, activating the c-Jun/AP-1 pathway and increasing cyclin-CDK inhibitors (p21 and p27) in various tumor cell lines, such as squamous cell carcinoma, multiple myeloma (MM), mantle cell lymphoma (MCL), hepatocellular carcinoma and non-small-cell lung cancer (NSCLC).338,339,340,341,342,343 In the clinic, it is the first approved PI by the U.S. Food and Drug Administration (FDA) for relapsed MM344 and MCL.345 Later, it was expanded for use in patients with NSCLC and pancreatic cancer.346 Despite its promising results, some off-target and adverse effects, such as fatigue, asthenia, thrombocytopenia, peripheral neuropathy and gastrointestinal symptoms, also limit its application.347,348 The off-target effects may lead to dose-limiting toxicity and subsequently result in permanent nerve damage to the extremities, called bortezomib-induced peripheral neuropathy (BIPN).349 Moreover, bortezomib resistance may occur within an average of 1 year, especially for solid tumors.350,351,352 The resistance mechanism includes an enhanced aggresome-autophagy pathway, increased expression of proinflammatory macrophages, alterations in apoptotic signaling and decreased ER stress response.353,354
Another approved PI for relapsed or refractory MM is carfilzomib (PR-171; Kyprolis; Onyx Pharmaceutical), a second-in-class PI (Fig. 5).355 Similar to bortezomib, carfilzomib also inhibits the chymotrypsin-like activity of the 20S proteasome. However, unlike bortezomib, the activity of carfilzomib is irreversible. In addition, carfilzomib is more effective than bortezomib in acute myeloid leukemia (AML) cells by inducing apoptosis and inhibiting proliferative activity.356 It also shows improved safety in terms of peripheral neurotoxicity and maintains its cytotoxic potential in bortezomib-resistant cell lines.357 Due to the good tolerance and promising efficacy for MM in phase I and II clinical trials, carfilzomib was approved by the FDA for the treatment of relapsed MM patients who experience disease progression within 60 days after the treatment of bortezomib and immunomodulatory drugs.358 Carfilzomib treatment can also cause adverse effects, such as cardiovascular complications (hypertension, heart failure), hematologic complications (thrombocytopenia, anemia), gastrointestinal complications (diarrhea, nausea/vomiting) and systemic symptoms (fever, fatigue). Therefore, its treatment should also be monitored carefully359,360

Cancer therapeutic strategy by targeting the UPS
As a new generation of PIs, oprozomib (ONX0912; PR-047) is designed as a tripeptide analog of carfilzomib (Table 1).361 In contrast to intravenously administered bortezomib and carfilzomib, oprozomib has better oral bioavailability and is suitable for oral administration. It also has a similar antitumor activity, potency and selectivity as carfilzomib in MM and can be used to treat bortezomib-, dexamethasone- or lenalidomide-resistant MM.362 In the treatment of solid tumors, it induces cell apoptosis by upregulating proapoptotic Bik and Mcl-1.363 However, due to oral administration, oprozomib has a high rate of gastrointestinal toxicities and unstable pharmacokinetics.364
Ixazomib citrate (MLN9708) and its biologically active form ixazomib (MLN2238) are the first orally administered PIs with improved pharmacokinetic and pharmacodynamic profiles (Table 1).365 Like bortezomib, Ixazomib citrate is a boron-containing peptidic agent that reversibly targets the β5 proteasome subunit and inhibits chymotrypsin-like proteasome activity. In the clinic, ixazomib citrate is also recommended for the treatment of relapsed and refractory MM with a positive safety profile.357,366,367 Moreover, in a phase 3 trial (NCT02181413), ixazomib maintenance can prolong progression-free survival (PFS) and is regarded as an additional option for posttransplant maintenance therapy in patients with newly diagnosed MM.368 Delanzomib (CEP-18770) is also a reversible and orally bioavailable structural analog of bortezomib (Fig. 5). Although it overcomes the resistance of bortezomib to peripheral neuropathy, severe skin toxicity limits its clinical application.369
Marizomib (NPI-0052), a novel PI, can affect chymotrypsin-like, trypsin-like and caspase-like activities of the 20S proteasome (Table 1). It can overcome bortezomib resistance and exhibit broader anticancer activities, with a better therapeutic ratio.370 Additionally, marizomib exhibits synergistic effects with bortezomib and lenalidomide or pomalidomide and low-dose dexamethasone in patients with refractory and relapsed MM.362,371 In addition, marizomib has the ability to penetrate the blood–brain barrier and induce apoptosis in glioma cells with low toxicity on normal cells.372 Thus, it has been applied in newly diagnosed glioblastoma (NCT03345095). The side effects of marizomib are limited to gastrointestinal symptoms without neuropathy or other significant systemic toxicities.373
As a hierarchical biological system, the UPS displays multilevel regulation of protein ubiquitination (Table 2). The PI targets only the last step of the ubiquitination process and thus leads to the accumulation of upstream ubiquitinated proteins, which may lead to other side effects. For instance, BIPN occurrence is associated with the aggregation of Ub-laden proteins in the dorsal root ganglia, which may result in bortezomib dose reduction or even discontinuation.352 Thus, other agents targeting other aspects of the UPS should also be developed.
Targeting E3 enzymes
As the most important components of the Ub conjugation machinery, E3 ligases mediate degradation with high substrate specificity.374 Thus, targeting the active site of E3 enzymes or their interactions with substrates offers promising options for developing drugs with fewer side effects.
Mdm2/Mdmx/p53
As noted above, the high-profile target is Mdm2/Hdm2, which binds to the amino terminus of p53 and mediates the degradation of p53 by Ub-dependent mechanisms.375 Due to the critical role of p53, many efforts have been made to find an antagonist of the E3 ligase Mdm2/Hdm2 (Fig. 5).
The crystal structure reveals that the N-terminal domain of Mdm2 complexes with a small peptide of p53, including three critical amino acids: Phe19, Trp23 and Leu26.376 Moreover, the peptide has a far higher affinity for Mdm2 and can serve as the “druggable” pocket.377 The first nonpeptidic molecule demonstrated to interrupt the p53–Mdm2 interface is 4,5-dihydroimidazoline (nutlin; Roche).378 Nutlin and its derivatives (for example, nutlin-3a, also known as RG7112, and RG7388) are the most successful chemical compounds identified by high-throughput screening (Table 1). Structurally, they are mimics of p53 and specifically bind to the pocket on Mdm2, thereby disrupting the protein–protein interface.379 Nutlins have been shown to inhibit the growth of hematological malignancies, glioblastomas and AML cells by activating the p53-dependent apoptotic pathway.378,380,381 In addition, they do not induce apoptosis in normal cells.382 Although the primary results of nutlins are promising, their shortcomings have recently been uncovered. Nutlins can only be used in tumor cells with wild-type p53, whereas they are insensitive to p53-deleted or p53-mutated cells. Moreover, it has been reported that nutlins are not specific for p53 and may compete with other proteins for Mdm2 binding. The same shortcomings are also found in RITA (reactivation of p53 and induction of tumor cell apoptosis), a small molecule inhibitor preventing the interaction between p53 and Mdm2 both in vitro and in vivo.383 Guided by the interactions among the p53 peptide, nutlin and Mdm2, a new class of inhibitors was developed and includes MI-219,384 SyI-155,385 MI-63,386 PRIMA1387 and RG7112 (Table 1).375 For example, MI-219 is an orally available compound with a subnanomolar affinity for Mdm2 and can increase the expression of p53 and p53-targeted genes.384
In addition, other small molecules, which include HLI98,388 HLI373,389 MEL24 (ref. 390) and the natural product sempervirine (Table 1),391 can also target the E3 ligase activity of Mdm2 directly and prevent the degradation of p53. HLI98 has been shown to activate p53 signaling and inhibit tumor cell growth in a p53-dependent manner. HLI373 is the highly water-soluble derivative of HLI98, and it has a greater potential to be a cancer therapeutic agent due to this characteristic.389
Although Mdmx (murine/human double minute X) has no ubiquitination activity, it can either bind to the N-terminus of p53 and inactivate it directly or ubiquitinate p53 by heterodimerizing with Mdm2.375 Nutlin-3 exhibits poor inhibition of Mdmx–p53 interactions, and there is a need for dual inhibitors of both Mdm2 and Mdmx due to the high expression of Mdmx in some cancers.392,393 ATSP-7041, a dual inhibitor, can induce p53-dependent tumor growth suppression.394 NSC207895, targeting Mdmx specifically, can act additively with nutlin-3a to activate p53 and induce apoptosis (Fig. 5).395
SCF E3 ligases
As mentioned before, SCF E3 ligases are also a well-known family of E3 ligases and includes FBXW7, SKP2 and β-TrCP1.396 Since FBPs are responsible for the specificity of SCFs, many small molecules have been designed to target them and may provide attractive therapeutic agents.63
Mutations in FBW7 and its targets often block the degradation of these oncogenic substrates (c-Myc, c-Jun and Notch) and subsequently promote tumorigenesis.397 Some efforts have been made to develop agonists for the FBW7 E3 ligase complex. The natural compound oridonin promotes FBW7-mediated proteasomal degradation of c-Myc, thereby inducing apoptosis in leukemia and lymphoma cells.398 By small molecule screening, SCF-12 allosterically inhibits the recognition of the substrate FBP Cdc4 (a homolog of FBW7 in yeast) but not its human ortholog FBXW7 (Table 1).399
For SKP2, compound ZL25 can inhibit SKP2 directly and subsequently result in cancer cell senescence in a p53-independent way.400 Compound A, another SKP2 inhibitor, was also found to induce cell cycle arrest and cell death in a p27-dependent manner. The inhibition blocks SCF–SKP2 complex formation (Fig. 5).401
β-TrCP1 can ubiquitinate the phosphorylated inhibitor of nuclear factor-κB (IκB) and lead to its degradation by the proteasomal pathway.402,403 A small phosphopeptide agonist has been designed to target the IκB Ub ligase, and this peptide can inhibit IκB degradation in TNF-stimulated HeLa cells.404 Erioflorin, an inhibitor of β-TrCP1, has been shown to suppress the activity of NF-κB and decelerate cell proliferation in various cancer cells by stabilizing the tumor suppressor PDCD4 (Fig. 5).405 GS143, another inhibitor of β-TrCP1, can also inhibit the NF-κB signaling pathway by decreasing IκB ubiquitination (Fig. 5).406
Generally, the FBP MET30, a member of the SCF E3-ligase family, regulates various cellular processes, including cell proliferation, transcription and immune response.407 By a yeast-based screen, small-molecule enhancers of rapamycin 3 (SMER3) were found to directly bind to MET30 and block its Ub ligase (Fig. 5). In addition, APC/C initiates the metaphase–anaphase transition and mitotic exit by targeting proteins such as securin and cyclin B1 for Ub-dependent destruction by the proteasome. CDC20 binds to the APC/C complex and induces its activation in mitosis. N-4-Tosyl-l-arginine methyl ester (TAME) binds to the APC E3 ligase and inhibits its activation by targeting both CDH1 and CDC20 and inducing mitotic arrest.408 Another small molecule called apcin (APC inhibitor) binds directly to CDC20, competitively inhibits the ubiquitination of D-box-containing substrates, blocks mitotic exit, and then induces tumor cell death (Table 1).409
VHL E3 Ub Ligase
Mutations in the VHL E3 ligase often result in tumors, especially renal cell carcinoma.410 Its mutations can block the degradation of HIF and eventually lead to high vascularity and promote tumor growth even under normal oxygen conditions. A small molecule was generated by Buckley et al. to target the VHL E3 ligase protein and mimic the binding mode of HIF-1α, which might provide a novel therapeutic strategy for anemia and ischemia.411
TRAFs
TRAF6, another member of the RING-domain family, is mentioned above to induce the K63-linked polyubiquitination and activation of IκappaB kinase (IκK), which in turn promotes the activation of downstream NF-κB signaling. Benzoxadiazole derivatives inhibiting TRAF6 can block the proliferation of lung and prostate cancer cells.412 BC-1215, an inhibitor of the FBP Fbxo3, degrades TRAF adapter proteins by inhibiting the degradation of Fbxl2 and then blocks inflammation and tumorigenesis (Fig. 5).413,414,415,416
Targeting the E1 enzyme
The E1 enzyme is responsible for activating Ub molecules in the protein degradation process and plays an important role in tumorigenesis. Currently, many efforts have been made to explore compounds targeting the E1 enzyme. The adenosine sulfamate analogs, MLN7243 and MLN4924, have been reported as UBA1 and NAE inhibitors, respectively, and both are currently used in Phase I/II and Phase I clinical trials (Table 1).417,418,419 The latter can form an irreversible covalent adduct with NEDD8 and block the formation of thioester bonds between NAE and NEDD8. This process inhibits the neddylation of cullins and leads to the accumulation of cullin-mediated degradation of proteins such as p21, p27 and IκBα.420 Moreover, experimental inhibitors of E1 have also been developed.421 PYR-41 (4[4-3,5-dioxo-pyrazolidin-1-yl]-benzoic acid ethyl ester), an irreversible Ub E1, can inhibit E1 and block the initiation of ubiquitination (Fig. 5). It inhibits the degradation of p53 and promotes apoptotic cell death in a p53-dependent manner. In addition, it also controls inflammation by inactivating NF-κB and inhibiting the expression of cytokines, chemokines and inflammatory mediators.422
Targeting E2 enzymes
E2 enzymes mainly mediate the conjugation of Ub to substrates. Currently, some efforts have been made to identify inhibitors that prevent the interaction between E1s and E2s or E2s and E3s. For instance, Leucettamol A, isolated from a marine sponge, Leucetta aff. microrhaphis, can inhibit the Ubc13–Uev1A interaction and block the formation of their complex (Fig. 5).423 With the same target of Ubc13–Uev1A, manadosterols A and B, isolated from the marine sponge Lissodendoryx fibrosa manadosterols, are also identified. They are regarded as the second and third natural compounds against the Ubc13–Uev1A interaction. Additionally, they are more potent than the abovementioned Leucettamol A (Fig. 5).424
CC0651, a small-molecule selective allosteric site inhibitor of the E2 enzyme hCdc34, can block the ubiquitination and degradation of p27 and then inhibit tumor cell proliferation (Fig. 5).425 In addition, small-molecule microarray-based screening has also been successfully used to identify the inhibitor of the SUMO E2 enzyme Ubc9, and a few small-molecule inhibitors have been found.426
Targeting DUB activity
As mentioned, ubiquitination is a dynamic and reversible process, and DUBs mediate the removal and processing of Ub or polyubiquitin chains from ubiquitinated proteins.427 Many DUBs have been found to participate in various events during the cell cycle progression, genomic instability regulation and tumorigenesis processes.428 As such, a number of DUB inhibitors have been developed ranging from broad-spectrum inhibitors to specific inhibitors and identified as potential anticancer agents.7,429,430,431
As broad-spectrum inhibitors, compounds G5 and F6 were identified by a cell-level drug screening.432 They are chalcone DUB inhibitors and are reported to induce Bcl-2-independent apoptosis.432,433 By activity-based chemical proteomics, compound PR-619 was identified as a broad-range DUB inhibitor (Fig. 5).434 Another identified broad-spectrum DUB inhibitor is NSC632839. It can target USP2 and USP7 and trigger apoptotic cell death in cancer cell lines (Fig. 5).432 Pimozide, a specific USP1 inhibitor, can block glioma stem cell maintenance and radioresistance (Table 1).435 WP1130, a small-molecule compound, also inhibits the activity of several DUBs, including USP9x, USP5, USP14, UCHL5 and UCH37. Furthermore, it downregulates the anti-apoptotic protein MCL-1 and upregulates the proapoptotic protein p53, leading to anti-tumor activity.436 As a natural product, betulinic acid can be isolated from a variety of plants, including the Betula/birch tree. Recently, it was reported to be a nonselective DUB inhibitor and induce the loss of transmembrane potential and cancer cell apoptosis.437,438 However, as unspecific DUB inhibitors, these broad-spectrum inhibitors may amplify their biological effects and unspecific toxicity, including (1) accumulation of polyubiquitinated proteins or unanchored polyubiquitin chains; (2) accumulation of misfolded proteins; (3) reduction in the individual DUB activities; and (4) aberrant biological activities of DUB-regulated oncoproteins.439 Thus, specific DUB inhibitors are recommended for clinical application.
Due to the important roles of USP7 in controlling p53 stability, it is the famous USP target for drug development. Many small-molecule antagonists of USP7 have been developed. For example, compounds p5091, p220077 and p50429 have been reported to enhance the ubiquitination and degradation of Mdm2 and induce apoptosis of bortezomib-resistant MM cells434,440,441; in addition, compounds FT671 and FT827 were identified by a cocrystal structure and proved to target a dynamic pocket near the catalytic center of the autoinhibited apo form of USP7 (Table 1).442 Along with FT671, XL188 designed by the DUB costructure can destabilize USP7 substrates, increase the expression of p53 and p53 downstream target genes, including the tumor suppressor p21, and subsequently inhibit tumor growth.442,443 A structural class of small molecules represented by HBX 19,818 and HBX 28,258, as well as P22077 and P50429, have been identified by biochemical assays and activity-based protein profiling in living systems to specifically inhibit USP7.441,444
Additionally, the proteasome-associated DUB USP14 is also well known for suppressing substrate degradation by separating proteasome-bound polyubiquitin chains.445 USP14 works as an oncogene and is overexpressed in several cancers, which may be associated with WNT/β-catenin signaling.446,447 Moreover, USP14 is also positively correlated with tumor recurrence and poor prognosis.448,449 After chemical library screening, the inhibitor IU1 was identified to bind specifically to the activated form of USP14 and abrogate its enzymatic activity (Table 1). Further high-resolution cocrystal structure analysis revealed that IU1 and its analogs can bind to a previously unknown steric binding site in USP14 (ref. 450) and enhance proteasome function by blocking the access of the C-terminus of Ub to the active site of USP14.451
Currently, many new screening methods have been developed and used to select small-molecule inhibitors and compounds for DUB. For example, high-throughput screening is used to identify small-molecule inhibitors selectively targeting Ub C-terminal hydrolase (UCH-L1). A class of isatin O-acyl oximes (LDN-57444) were found and shown to induce apoptotic cell death in lung tumor cell lines (Fig. 5).452 Due to the limited aqueous solubility of LDN-57444, its soluble form, LDN-Pox, was then developed and proved to have the potential to treat invasive carcinomas, including EBV-positive malignancies.453 In addition, a cell-based screening was also used to select compounds inducing cathepsin-dependent apoptosis. Intriguingly, b-AP15 was identified to induce the accumulation of high-molecular-mass Ub complexes in cells (Fig. 5).454 It is a 19S regulatory particle inhibitor that selectively inhibits the deubiquitinating activity of USP14 and UCHL5 without inhibiting the proteasome activity.455 Additionally, it can also block the degradation of a proteasome-degraded reporter protein, resulting in the accumulation of polyubiquitin and inducing strong proteotoxic stress and mitochondrial damage.456,457 In many solid tumors and MM, b-AP15 can induce tumor cell apoptosis, which may be associated with c-Myc-Noxa-mediated apoptosis.455,456,458
Multitarget combination treatment
Drug adverse effects and resistance are major obstacles in preclinical and clinical cancer treatments, and UPS inhibitors are no exception.459,460,461 For drug adverse effects, the balance between effective dose and dose limiting toxicity is the principal contradiction.462 The molecular mechanisms of anti-cancer drug resistance are also associated with tumor metabolism, the TME and CSCs, such as increasing drug metabolism and degradation of drug target proteins, enhance the tolerability of stressful TME conditions, and enhance the DNA damage response and anti-apoptotic mechanisms of CSCs.461,463,464,465,466 Thus, to improve therapeutic effects, a multitargeted combination treatment is proposed for UPS inhibitors.
Due to the good curative effects of PIs in MM, a monotherapy of PIs has become the standard of care for patients with MM.467 In regard to relapsed MM, recommended therapies usually involve PIs and immunomodulatory drugs (IMiDs; e.g., lenalidomide, pomalidomide) in doublet or triplet combinations with corticosteroids or other systemic therapies including the anti-CD38 monoclonal antibody daratumumab and the immunoglobulin G1 (IgG1) monoclonal antibody isatuximab for the CD38 receptor.468,469,470 For patients with MCL and diffuse large B-cell lymphoma, combination treatment of PIs and chemotherapies or histone deacetylase inhibitors also yields benefits,471,472 which may also overcome the impact of gain-of-function p53 mutations in solid tumors.473 In the clinic, with regard to bortezomib-resistant tumors, the combination treatments of bortezomib with chemotherapy drugs such as doxorubicin, plerixafor and daratumumab have shown improved clinical outcomes, suggesting that conventional chemotherapy could increase the sensitivity of bortezomib to malignancies.474,475,476 Moreover, combined treatment is also very common for carfilzomib. Combined with lenalidomide and dexamethasone, carfilzomib achieves a near complete clinical response in naive MM patients.477 In patients with relapsed/refractory MM, carfilzomib and dexamethasone (Kd56) demonstrate a longer PFS than that of bortezomib and dexamethasone (Vd).478 Panobinostat, an HDAC inhibitor, is also used with carfilzomib in MM patients and achieves a good response rate (ClinicalTrial.gov: NCT01549431).359
In addition to combination treatment targeting UPS and other signaling pathways, combined inhibitors within the UPS also work in cancer treatment. In PI-resistant MM, inhibiting upstream components of UPS is a promising interest. For example, high expression of USP7 is found in bortezomib-resistant MM and is associated with a short overall survival and poor outcome. As a preclinical practice, the usage of USP7 inhibitors combined with bortezomib triggers synergistic antitumor activity.479 Additionally, in PI-resistant MM, the E1 Ub-activating enzyme inhibitor TAK-243 can also block myeloma cell proliferation and induce apoptosis.480
Conclusions and future perspectives
The UPS, a network of enzymes, has been researched for nearly 40 years since its first discovery in 1975. As an important PTM, ubiquitination can regulate a large number of signaling pathways and take part in many biological processes. E3 ligases, regarded as the “brain” in the UPS, can select a specific E2 and substrate and directly transfer Ub to the substrate.481,482 However, the results obtained from current studies have not explained how E3 recognizes a specific E2 and selects a substrate, along with the selectivity mechanism of specific lysines in the substrate. It will be necessary to explain the mechanisms underlying the regulation of E3s via structure–function studies.
In ubiquitination, an E3 ligase can mediate divergent functions of substrates by regulating different types of ubiquitination.6 SKP2 can not only regulate the stability of c-Myc by promoting its K48 linkage polyubiquitination but also control the activity of AKT/RagA/LKB instead of its stability by mediating K63 linkage polyubiquitination.131,159,176 A similar situation occurs with Mdm2, which can promote both the K48 linkage ubiquitination and the neddylation of p53.219,483 To understand the dynamics and complexity of such events, it will be necessary to place special emphasis on dissecting the diverse mechanisms of Ub chain assembly by E3s.
In addition to classical ubiquitination, nonclassical ubiquitination, including ubiquitination mediated by SidE that does not depend on E1 and E2,93,94 and nonclassical ubiquitination sites (Ser/Thr/Cys) via the formation of thio- or hydroxy-bond esters, are also important parts of ubiquitination.484,485,486,487,488,489,490,491,492 Although many efforts have been made, there are still some unresolved problems in the UPS. For example, although K48- and K11-type ubiquitination can serve as a degradation signal for transferring the target to the 26S proteasome,37,493 it is still unclear how the proteasome recognizes K11-type ubiquitination and distinguishes different types of ubiquitination. In addition, the length of the Ub chain is also an open question. There is an urgent need to identify the length of the Ub chain in cells and its befitting length in regulating the function of the substrate. Moreover, it has been reported that the K48-type Ub chain consisting of four Ubs can be degraded by the proteasome.494,495 Why four Ubs? These lengths of the Ub chain are also required for other linkage Ub chains. The existence of these problems is closely related to the technical deficiency in detecting the length of the polyubiquitin chain; thus, useful technology to address the looming question is the key to unlocking new areas of UPS.
Because of the importance of the UPS in normal biological processes, its alterations often contribute to the etiology of many diseases, particularly cancer.3 The roles of the UPS in tumorigenesis are not only associated with tumor metabolism regulation (mTORC1/AMPK/AKT) but also related to immunological TME modulation (TLR/RLR/STING) and CSC stemness maintenance (Nanog/Oct4/Sox2/Hippo/Wnt).496,497,498,499 As targeting the TME is a hotspot in the field of cancer treatment, many studies have reported the roles of ubiquitination in tumor immunology. For example, the stability of PD-L1 is regulated by SPOP via proteasome-mediated degradation in cancer cells.500 FBXO38, an E3 ligase of PD-1, mediates K48-linked polyubiquitination and subsequent proteasome degradation in activated T cells.501 Additionally, COP9 signalosome 5 (CSN5) is required for PD-L1 stabilization by inhibiting its ubiquitination and degradation in cancer cells.502 However, the specific role of ubiquitination in T cells, macrophages and DC cells is still unclear, especially for immunotherapies targeting ubiquitination, and need further development.
Based on aberrant UPS activity frequently observed in human cancers, potential therapeutic targets have been identified, and corresponding inhibitors have been developed.11,65,503 Currently, the proteasome is a successful target in the clinic, and good therapeutic results are achieved for some FDA-approved PIs, such as bortezomib, carfilzomib, oprozomib and ixazomib. However, as the last step of the ubiquitination process, these PIs can result in some side effects due to the accumulation of upstream ubiquitinated proteins, which limits their widespread application. Thus, attempts have been made to explore targeted inhibitors for E2, E3 ligases, the UBL system and the process of UBD binding to Ub.504,505 However, most of these inhibitors work well in cell culture studies and not well enough in animal models and clinical trials.164,168,506 One reason for this unsatisfactory situation is the incomplete understanding of the structural analysis of target proteins, pharmaceutical chemistry and combinatorial chemistry, which needs the advances in technology. In addition, high-throughput screening may also help to identify the most feasible inhibitors. Moreover, aberrant activity of the UPS along with other oncogenic signaling pathways may occur simultaneously during the process of tumorigenesis, which makes targeted therapies more complicated. Thus, multitarget combination treatment is recommended as a future direction. Moreover, genomics and proteomics studies based on a large number of patient tumor tissue samples should be adopted to better understand the dynamic process of tumorigenesis. In the end, in-depth exploration of the functions of the UPS and conducting more clinical studies are needed to elucidate the roles of the UPS in tumorigenesis and to develop novel strategies for the treatment and prevention of human cancers.
References
Shmueli, A. & Oren, M. Life, death, and ubiquitin: taming the mule. Cell 121, 963–965 (2005).
Lopez-Otin, C. & Hunter, T. The regulatory crosstalk between kinases and proteases in cancer. Nat. Rev. Cancer 10, 278–292 (2010).
Suryadinata, R., Roesley, S. N., Yang, G. & Sarcevic, B. Mechanisms of generating polyubiquitin chains of different topology. Cells 3, 674–689 (2014).
Ikeda, F. & Dikic, I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 9, 536–542 (2008).
Rajalingam, K. & Dikic, I. SnapShot: expanding the ubiquitin code. Cell 164, 1074–1074.e1071 (2016).
Gross, S. et al. Targeting cancer with kinase inhibitors. J. Clin. Invest. 125, 1780–1789 (2015).
Morrow, J. K., Lin, H. K., Sun, S. C. & Zhang, S. Targeting ubiquitination for cancer therapies. Future Med. Chem. 7, 2333–2350 (2015).
Hoeller, D., Hecker, C. M. & Dikic, I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer 6, 776–788 (2006).
Mansour, M. A. Ubiquitination: friend and foe in cancer. Int. J. Biochem. Cell Biol. 101, 80–93 (2018).
Telerman, A. & Amson, R. The molecular programme of tumour reversion: the steps beyond malignant transformation. Nat. Rev. Cancer 9, 206–216 (2009).
Hoeller, D. & Dikic, I. Targeting the ubiquitin system in cancer therapy. Nature 458, 438–444 (2009).
Senft, D., Qi, J. & Ronai, Z. A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 18, 69–88 (2018).
Komander, D. & Rape, M. The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (2012).
Husnjak, K. & Dikic, I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 81, 291–322 (2012).
Koyano, F. et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166 (2014).
Kane, L. A. et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153 (2014).
Kazlauskaite, A. et al. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139 (2014).
Wauer, T. et al. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 34, 307–325 (2015).
Swaney, D. L. et al. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 10, 676–682 (2013).
Hunter, T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 (2007).
Lundby, A. et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2, 419–431 (2012).
Webb, K. & Bennett, E. J. Eavesdropping on PTM cross-talk through serial enrichment. Nat. Methods 10, 620–621 (2013).
Peng, J. et al. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926 (2003).
Zheng, N. & Shabek, N. Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem. 86, 129–157 (2017).
Ciechanover, A., Heller, H., Katz-Etzion, R. & Hershko, A. Activation of the heat-stable polypeptide of the ATP-dependent proteolytic system. Proc. Natl Acad. Sci. USA 78, 761–765 (1981).
Ye, Y. & Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10, 755–764 (2009).
Stewart, M. D., Ritterhoff, T., Klevit, R. E. & Brzovic, P. S. E2 enzymes: more than just middle men. Cell Res. 26, 423–440 (2016).
Varshavsky, A. The ubiquitin system, an immense realm. Annu. Rev. Biochem. 81, 167–176 (2012).
Hershko, A. Ubiquitin-mediated protein degradation. J. Biol. Chem. 263, 15237–15240 (1988).
Geng, L., Huntoon, C. J. & Karnitz, L. M. RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J. Cell Biol. 191, 249–257 (2010).
Rezaeian, A. H. et al. A hypoxia-responsive TRAF6-ATM-H2AX signalling axis promotes HIF1alpha activation, tumorigenesis and metastasis. Nat. Cell Biol. 19, 38–51 (2017).
Kwon, Y. T. & Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 42, 873–886 (2017).
Bonnet, J., Devys, D. & Tora, L. Histone H2B ubiquitination: signaling not scrapping. Drug Discov. Today Technol. 12, e19–e27 (2014).
Pfleger, C. M. Ubiquitin on ras: warden or partner in crime? Sci. Signal. 4, pe12 (2011).
Yau, R. & Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586 (2016).
Swatek, K. N. & Komander, D. Ubiquitin modifications. Cell Res. 26, 399–422 (2016).
Hochstrasser, M. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30, 405–439 (1996).
Chen, Z. J. & Sun, L. J. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 33, 275–286 (2009).
Xu, P. et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 (2009).
Elia, A. E. et al. Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol. Cell 59, 867–881 (2015).
Michel, M. A., Swatek, K. N., Hospenthal, M. K. & Komander, D. Ubiquitin linkage-specific affimers reveal insights into K6-linked ubiquitin signaling. Mol. Cell 68, 233–246.e235 (2017).
Bremm, A. & Komander, D. Emerging roles for Lys11-linked polyubiquitin in cellular regulation. Trends Biochem. Sci. 36, 355–363 (2011).
Matsumoto, M. L. et al. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol. Cell 39, 477–484 (2010).
Geisler, S. et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 (2010).
Glauser, L., Sonnay, S., Stafa, K. & Moore, D. J. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 118, 636–645 (2011).
Metzger, M. B. & Weissman, A. M. Working on a chain: E3s ganging up for ubiquitylation. Nat. Cell Biol. 12, 1124–1126 (2010).
Hwang, C. S., Shemorry, A., Auerbach, D. & Varshavsky, A. The N-end rule pathway is mediated by a complex of the RING-type Ubr1 and HECT-type Ufd4 ubiquitin ligases. Nat. Cell Biol. 12, 1177–1185 (2010).
Ye, W. et al. TRIM8 negatively regulates TLR3/4-mediated innate immune response by blocking TRIF-TBK1 interaction. J. Immunol. 199, 1856–1864 (2017).
Miao, Y., Wu, J. & Abraham, S. N. Ubiquitination of innate immune regulator TRAF3 orchestrates expulsion of intracellular bacteria by exocyst complex. Immunity 45, 94–105 (2016).
Datta, A. B., Hura, G. L. & Wolberger, C. The structure and conformation of Lys63-linked tetraubiquitin. J. Mol. Biol. 392, 1117–1124 (2009).
Kulathu, Y. & Komander, D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 13, 508–523 (2012).
Takahashi, T. S. et al. Structural insights into two distinct binding modules for Lys63-linked polyubiquitin chains in RNF168. Nat. Commun. 9, 170 (2018).
Heger, K. et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 559, 120–124 (2018).
Iwai, K., Fujita, H. & Sasaki, Y. Linear ubiquitin chains: NF-kappaB signalling, cell death and beyond. Nat. Rev. Mol. Cell Biol. 15, 503–508 (2014).
Ikeda, F. et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 471, 637–641 (2011).
Boname, J. M. et al. Efficient internalization of MHC I requires lysine-11 and lysine-63 mixed linkage polyubiquitin chains. Traffic 11, 210–220 (2010).
Morreale, F. E. & Walden, H. Types of ubiquitin ligases. Cell 165, 248–248.e241 (2016).
Deshaies, R. J. & Joazeiro, C. A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 (2009).
Metzger, M. B., Hristova, V. A. & Weissman, A. M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 125, 531–537 (2012).
Wade, M., Li, Y. C. & Wahl, G. M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 13, 83–96 (2013).
Deng, L. et al. The ubiquitination of rag A GTPase by RNF152 negatively regulates mTORC1 activation. Mol. Cell 58, 804–818 (2015).
Chang, L. F. et al. Molecular architecture and mechanism of the anaphase-promoting complex. Nature 513, 388–393 (2014).
Skaar, J. R., Pagan, J. K. & Pagano, M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 13, 889–903 (2014).
Wang, Z., Liu, P., Inuzuka, H. & Wei, W. Roles of F-box proteins in cancer. Nat. Rev. Cancer 14, 233–247 (2014).
Wang, D. et al. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 36, 683–702 (2017).
Ostrowska, H. The ubiquitin-proteasome system: a novel target for anticancer and anti-inflammatory drug research. Cell. Mol. Biol. Lett. 13, 353–365 (2008).
Wu, W. J. et al. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J. Transl. Med. 11, 142 (2013).
Fujita, T. et al. Dissection of the APCCdh1-Skp2 cascade in breast cancer. Clin. Cancer Res. 14, 1966–1975 (2008).
Rotin, D. & Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409 (2009).
Pickart, C. M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 (2001).
Huang, L. et al. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science 286, 1321–1326 (1999).
French, M. E., Kretzmann, B. R. & Hicke, L. Regulation of the RSP5 ubiquitin ligase by an intrinsic ubiquitin-binding site. J. Biol. Chem. 284, 12071–12079 (2009).
Kim, H. C. & Huibregtse, J. M. Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol. Cell Biol. 29, 3307–3318 (2009).
Kim, H. C. et al. Structure and function of a HECT domain ubiquitin-binding site. EMBO Rep. 12, 334–341 (2011).
Bae, S. J. et al. NEDD4 controls intestinal stem cell homeostasis by regulating the Hippo signalling pathway. Nat. Commun. 6, 6314 (2015).
Ho, K. C. et al. Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability [corrected]. Proc. Natl Acad. Sci. USA 108, 4870–4875 (2011).
Moren, A. et al. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J. Biol. Chem. 280, 22115–22123 (2005).
Imamura, T., Oshima, Y. & Hikita, A. Regulation of TGF-beta family signalling by ubiquitination and deubiquitination. J. Biochem. 154, 481–489 (2013).
Dove, K. K. & Klevit, R. E. RING-between-RING E3 ligases: emerging themes amid the variations. J. Mol. Biol. 429, 3363–3375 (2017).
Wenzel, D. M., Lissounov, A., Brzovic, P. S. & Klevit, R. E. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474, 105–108 (2011).
Huang, A. et al. E2-c-Cbl recognition is necessary but not sufficient for ubiquitination activity. J. Mol. Biol. 385, 507–519 (2009).
Shimura, H. et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305 (2000).
Stieglitz, B. et al. Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature 503, 422–426 (2013).
Tokunaga, F. & Iwai, K. LUBAC, a novel ubiquitin ligase for linear ubiquitination, is crucial for inflammation and immune responses. Microbes Infect. 14, 563–572 (2012).
Tokunaga, F. et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature 471, 633–636 (2011).
Wenzel, D. M. & Klevit, R. E. Following Ariadne's thread: a new perspective on RBR ubiquitin ligases. BMC Biol. 10, 24 (2012).
Duda, D. M. et al. Structure of HHARI, a RING-IBR-RING ubiquitin ligase: autoinhibition of an Ariadne-family E3 and insights into ligation mechanism. Structure 21, 1030–1041 (2013).
Kitada, T. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998).
Nussbaum, R. L. Putting the parkin into Parkinson's. Nature 392, 544–545 (1998).
Gong, Y. et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat. Genet. 46, 588–594 (2014).
Zhang, X. et al. Parkin facilitates proteasome inhibitor-induced apoptosis via suppression of NF-kappaB activity in hepatocellular carcinoma. Cell Death Dis. 10, 719 (2019).
Qiu, J. et al. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 533, 120–124 (2016).
Bhogaraju, S. & Dikic, I. Cell biology: ubiquitination without E1 and E2 enzymes. Nature 533, 43–44 (2016).
Wang, Y. et al. Structural insights into non-canonical ubiquitination catalyzed by SidE. Cell 173, 1231–1243.e1216 (2018).
Bedford, L. et al. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat. Rev. Drug Discov. 10, 29–46 (2011).
Cappadocia, L. & Lima, C. D. Ubiquitin-like protein conjugation: structures, chemistry, and mechanism. Chem. Rev. 118, 889–918 (2018).
Schwechheimer, C. NEDD8-its role in the regulation of Cullin-RING ligases. Curr. Opin. Plant Biol. 45, 112–119 (2018).
Wei, N. & Deng, X. W. The COP9 signalosome. Annu. Rev. Cell Dev. Biol. 19, 261–286 (2003).
Enchev, R. I., Schulman, B. A. & Peter, M. Protein neddylation: beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 16, 30–44 (2015).
Kerscher, O., Felberbaum, R. & Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22, 159–180 (2006).
Seeler, J. S. & Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 17, 184–197 (2017).
Gareau, J. R. & Lima, C. D. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 (2010).
Geiss-Friedlander, R. & Melchior, F. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956 (2007).
Lamoliatte, F. et al. Large-scale analysis of lysine SUMOylation by SUMO remnant immunoaffinity profiling. Nat. Commun. 5, 5409 (2014).
Bartek, J. & Hodny, Z. SUMO boosts the DNA damage response barrier against cancer. Cancer Cell 17, 9–11 (2010).
de The, H., Pandolfi, P. P. & Chen, Z. Acute promyelocytic leukemia: a paradigm for oncoprotein-targeted cure. Cancer Cell. 32, 552–560 (2017).
Gartner, A. & Muller, S. PML, SUMO, and RNF4: guardians of nuclear protein quality. Mol. Cell 55, 1–3 (2014).
Dassouki, Z. et al. ATL response to arsenic/interferon therapy is triggered by SUMO/PML/RNF4-dependent Tax degradation. Blood 125, 474–482 (2015).
Komander, D., Clague, M. J. & Urbe, S. Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 (2009).
David, R. DUBs' key to selectivity. Nat. Rev. Mol. Cell Biol. 13, 64 (2011).
Virdee, S. et al. Engineered diubiquitin synthesis reveals Lys29-isopeptide specificity of an OTU deubiquitinase. Nat. Chem. Biol. 6, 750–757 (2010).
Cooper, E. M. et al. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 28, 621–631 (2009).
Komander, D. et al. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 10, 466–473 (2009).
Sato, Y. et al. Structures of CYLD USP with Met1- or Lys63-linked diubiquitin reveal mechanisms for dual specificity. Nat. Struct. Mol. Biol. 22, 222–229 (2015).
Wiener, R., Zhang, X., Wang, T. & Wolberger, C. The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 483, 618–622 (2012).
Juang, Y. C. et al. OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Mol. Cell 45, 384–397 (2012).
Bonacci, T. & Emanuele, M. J. Impressionist portraits of mitotic exit: APC/C, K11-linked ubiquitin chains and Cezanne. Cell Cycle 18, 652–660 (2019).
Bonacci, T. et al. Cezanne/OTUD7B is a cell cycle-regulated deubiquitinase that antagonizes the degradation of APC/C substrates. EMBO J. 37, e98701 (2018).
Licchesi, J. D. et al. An ankyrin-repeat ubiquitin-binding domain determines TRABID's specificity for atypical ubiquitin chains. Nat. Struct. Mol. Biol. 19, 62–71 (2011).
Cummins, J. M. et al. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 428, 1–2 (2004).1 p following 486.
Stevenson, L. F. et al. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 26, 976–986 (2007).
Yuan, J. et al. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 140, 384–396 (2010).
Sun, X. X., Challagundla, K. B. & Dai, M. S. Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO J. 31, 576–592 (2012).
Wiltshire, T. D. et al. Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J. Biol. Chem. 285, 14565–14571 (2010).
Jewell, J. L., Russell, R. C. & Guan, K. L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 14, 133–139 (2013).
Efeyan, A., Zoncu, R. & Sabatini, D. M. Amino acids and mTORC1: from lysosomes to disease. Trends Mol. Med. 18, 524–533 (2012).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 (2017).
Shen, K. & Sabatini, D. M. Ragulator and SLC38A9 activate the Rag GTPases through noncanonical GEF mechanisms. Proc. Natl Acad. Sci. USA 115, 9545–9550 (2018).
Peng, M., Yin, N. & Li, M. O. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 159, 122–133 (2014).
Bar-Peled, L. et al. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106 (2013).
Jin, G. et al. Skp2-mediated RagA ubiquitination elicits a negative feedback to prevent amino-acid-dependent mTORC1 hyperactivation by recruiting GATOR1. Mol. Cell 58, 989–1000 (2015).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Sancak, Y. et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008).
Kim, J. & Guan, K. L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 21, 63–71 (2019).
Linares, J. F. et al. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 51, 283–296 (2013).
Mao, J. H. et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321, 1499–1502 (2008).
Bar-Peled, L., Schweitzer, L. D., Zoncu, R. & Sabatini, D. M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 (2012).
Chen, J. et al. KLHL22 activates amino-acid-dependent mTORC1 signalling to promote tumorigenesis and ageing. Nature 557, 585–589 (2018).
Xie, J. & Proud, C. G. Signaling crosstalk between the mTOR complexes. Translation (Austin) 2, e28174 (2014).
Wang, B. et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 545, 365–369 (2017).
Peterson, T. R. et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886 (2009).
Duan, S. et al. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol. Cell 44, 317–324 (2011).
Zhao, Y., Xiong, X. & Sun, Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell 44, 304–316 (2011).
Gao, D. et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell 44, 290–303 (2011).
Zhao, L. et al. OTUB1 protein suppresses mTOR complex 1 (mTORC1) activity by deubiquitinating the mTORC1 inhibitor DEPTOR. J. Biol. Chem. 293, 4883–4892 (2018).
Xie, J., Wang, X. & Proud, C. G. mTOR inhibitors in cancer therapy. F1000Res. 5, (F1000 Faculty Rev):2078 (2016).
Sengupta, S., Peterson, T. R. & Sabatini, D. M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 40, 310–322 (2010).
Tee, A. R. et al. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268 (2003).
Hu, J. et al. WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1-CUL4-ROC1 ligase. Genes Dev. 22, 866–871 (2008).
Deng, L. et al. Ubiquitination of Rheb governs growth factor-induced mTORC1 activation. Cell Res. 29, 136–150 (2019).
Steinberg, G. R. & Carling, D. AMP-activated protein kinase: the current landscape for drug development. Nat. Rev. Drug Discov. 18, 527–551 (2019).
Han, F. et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat. Commun. 9, 4728 (2018).
Green, D. R., Galluzzi, L. & Kroemer, G. Cell biology. Metabolic control of cell death. Science 345, 1250256 (2014).
Pineda, C. T. et al. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160, 715–728 (2015).
Deng, M. et al. Deubiquitination and Activation of AMPK by USP10. Mol. Cell 61, 614–624 (2016).
Sanchez-Cespedes, M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene 26, 7825–7832 (2007).
Hemminki, A. et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391, 184–187 (1998).
Jeon, S. M., Chandel, N. S. & Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (2012).
Lee, S. W. et al. Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol. Cell 57, 1022–1033 (2015).
Mallampalli, R. K. et al. Fbxl12 triggers G1 arrest by mediating degradation of calmodulin kinase I. Cell. Signal. 25, 2047–2059 (2013).
Dibble, C. C. & Cantley, L. C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555 (2015).
Inoki, K., Li, Y., Xu, T. & Guan, K. L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 (2003).
Mossmann, D., Park, S. & Hall, M. N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757 (2018).
Wang, X. et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128, 129–139 (2007).
Maddika, S. et al. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 13, 728–733 (2011).
Van Themsche, C., Leblanc, V., Parent, S. & Asselin, E. X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J. Biol. Chem. 284, 20462–20466 (2009).
Xu, T. et al. The E3 ubiquitin ligase CHIP/miR-92b/PTEN regulatory network contributes to tumorigenesis of glioblastoma. Am. J. Cancer Res. 7, 289–300 (2017).
Trotman, L. C. et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156 (2007).
Lee, Y. R. et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science. 364, eaau0159 (2019).
Song, M. S. et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455, 813–817 (2008).
Zhang, J. et al. Deubiquitylation and stabilization of PTEN by USP13. Nat. Cell Biol. 15, 1486–1494 (2013).
Yuan, L. et al. Deubiquitylase OTUD3 regulates PTEN stability and suppresses tumorigenesis. Nat. Cell Biol. 17, 1169–1181 (2015).
Liu, P., Cheng, H., Roberts, T. M. & Zhao, J. J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8, 627–644 (2009).
Manning, B. D. & Cantley, L. C. AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 (2007).
Yang, W. L. et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 (2009).
Chan, C. H. et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 149, 1098–1111 (2012).
Suizu, F. et al. The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Dev. Cell 17, 800–810 (2009).
Su, C. H. et al. Akt phosphorylation at Thr308 and Ser473 is required for CHIP-mediated ubiquitination of the kinase. Cell. Signal. 23, 1824–1830 (2011).
Fan, C. D. et al. Ubiquitin-dependent regulation of phospho-AKT dynamics by the ubiquitin E3 ligase, NEDD4-1, in the insulin-like growth factor-1 response. J. Biol. Chem. 288, 1674–1684 (2013).
Bae, S. et al. Akt is negatively regulated by the MULAN E3 ligase. Cell Res. 22, 873–885 (2012).
Lim, J. H. et al. CYLD negatively regulates transforming growth factor-beta-signalling via deubiquitinating Akt. Nat. Commun. 3, 771 (2012).
de Jel, M. M. et al. Loss of CYLD accelerates melanoma development and progression in the Tg(Grm1) melanoma mouse model. Oncogenesis 8, 56 (2019).
Suenaga, N. et al. Loss of tumor suppressor CYLD expression triggers cisplatin resistance in oral squamous cell carcinoma. Int. J. Mol. Sci. 20, 5194 (2019).
Guo, J. et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 353, 929–932 (2016).
Guo, J. et al. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat. Cell Biol. 21, 226–237 (2019).
Li, R. et al. Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer Res. 73, 5742–5753 (2013).
Prabhakar, N. R. & Semenza, G. L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 92, 967–1003 (2012).
Stine, Z. E. et al. MYC, metabolism, and cancer. Cancer Discov. 5, 1024–1039 (2015).
Hsieh, J. J. et al. Chromosome 3p loss-orchestrated VHL, HIF, and epigenetic deregulation in clear cell renal cell carcinoma. J. Clin. Oncol. 36, 3533–3539 (2018).
Denko, N. C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705–713 (2008).
Parks, S. K., Chiche, J. & Pouyssegur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 13, 611–623 (2013).
Rankin, E. B. & Giaccia, A. J. Hypoxic control of metastasis. Science 352, 175–180 (2016).
Pouyssegur, J., Dayan, F. & Mazure, N. M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441, 437–443 (2006).
Nakayama, K. et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell 117, 941–952 (2004).
Cassavaugh, J. M. et al. Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J. Cell Biochem. 112, 3882–3890 (2011).
Flugel, D., Gorlach, A. & Kietzmann, T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood 119, 1292–1301 (2012).
Kojima, K. et al. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1alpha-mediated down-regulation of CXCL12. Blood 118, 4431–4439 (2011).
Sun, H. et al. TRAF6 upregulates expression of HIF-1alpha and promotes tumor angiogenesis. Cancer Res. 73, 4950–4959 (2013).
Ju, U. I. et al. FBXO11 represses cellular response to hypoxia by destabilizing hypoxia-inducible factor-1alpha mRNA. Biochem. Biophys. Res. Commun. 464, 1008–1015 (2015).
Bremm, A. et al. Cezanne (OTUD7B) regulates HIF-1alpha homeostasis in a proteasome-independent manner. EMBO Rep. 15, 1268–1277 (2014).
Zhang, Y. et al. Nrdp1 increases ischemia induced primary rat cerebral cortical neurons and pheochromocytoma cells apoptosis via downregulation of HIF-1alpha protein. Front. Cell Neurosci. 11, 293 (2017).
Carbia-Nagashima, A. et al. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell 131, 309–323 (2007).
Kress, T. R., Sabo, A. & Amati, B. MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593–607 (2015).
Dang, C. V., Reddy, E. P., Shokat, K. M. & Soucek, L. Drugging the ‘undruggable' cancer targets. Nat. Rev. Cancer 17, 502–508 (2017).
Hydbring, P., Castell, A. & Larsson, L. G. MYC modulation around the CDK2/p27/SKP2 axis. Genes (Basel). 8, e174 (2017).
Jin, J. & Harper, J. W. A license to kill: transcriptional activation and enhanced turnover of Myc by the SCF(kp2) ubiquitin ligase. Cancer Cell 3, 517–518 (2003).
Wei, W. et al. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 8, 25–33 (2005).
Popov, N., Schulein, C., Jaenicke, L. A. & Eilers, M. Ubiquitylation of the amino terminus of Myc by SCF(beta-TrCP) antagonizes SCF(Fbw7)-mediated turnover. Nat. Cell Biol. 12, 973–981 (2010).
Mei, Z. et al. FBXO32 targets c-Myc for proteasomal degradation and inhibits c-Myc activity. J. Biol. Chem. 290, 16202–16214 (2015).
Paul, I. et al. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene 32, 1284–1295 (2013).
Pan, J. et al. USP37 directly deubiquitinates and stabilizes c-Myc in lung cancer. Oncogene 34, 3957–3967 (2015).
Sun, X. X. et al. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl Acad. Sci. USA 112, 3734–3739 (2015).
Kessler, J. D. et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353 (2012).
Hafner, A., Bulyk, M. L., Jambhekar, A. & Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 20, 199–210 (2019).
Bykov, V. J. N., Eriksson, S. E., Bianchi, J. & Wiman, K. G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 18, 89–102 (2018).
Vucic, D., Dixit, V. M. & Wertz, I. E. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 12, 439–452 (2011).
Brooks, C. L. & Gu, W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 21, 307–315 (2006).
Li, M. et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416, 648–653 (2002).
Harper, J. W. Neddylating the guardian; Mdm2 catalyzed conjugation of Nedd8 to p53. Cell 118, 2–4 (2004).
Park, J. H. et al. Modification of DBC1 by SUMO2/3 is crucial for p53-mediated apoptosis in response to DNA damage. Nat. Commun. 5, 5483 (2014).
Vesely, M. D. & Schreiber, R. D. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann. N. Y. Acad. Sci. 1284, 1–5 (2013).
Kaewkangsadan, V. et al. Tumour-draining axillary lymph nodes in patients with large and locally advanced breast cancers undergoing neoadjuvant chemotherapy (NAC): the crucial contribution of immune cells (effector, regulatory) and cytokines (Th1, Th2) to immune-mediated tumour cell death induced by NAC. BMC Cancer 18, 123 (2018).
Dvorak, H. F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659 (1986).
Federico, A. et al. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 121, 2381–2386 (2007).
Landskron, G. et al. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 149185 (2014).
Torisu, H. et al. Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: possible involvement of TNFalpha and IL-1alpha. Int. J. Cancer 85, 182–188 (2000).
Chang, S. C. & Ding, J. L. Ubiquitination and SUMOylation in the chronic inflammatory tumor microenvironment. Biochim. Biophys. Acta Rev. Cancer 1870, 165–175 (2018).
Tognon, C. E. et al. Insulin-like growth factor 1 receptor stabilizes the ETV6-NTRK3 chimeric oncoprotein by blocking its KPC1/Rnf123-mediated proteasomal degradation. J. Biol. Chem. 293, 12502–12515 (2018).
Rabl, J. et al. Structural basis of BRCC36 function in DNA repair and immune regulation. Mol. Cell 75, 483–497.e489 (2019).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010).
Mai, C. W., Kang, Y. B. & Pichika, M. R. Should a Toll-like receptor 4 (TLR-4) agonist or antagonist be designed to treat cancer? TLR-4: its expression and effects in the ten most common cancers. OncoTargets Ther. 6, 1573–1587 (2013).
Wang, C. et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 (2001).
Deng, L. et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361 (2000).
Meylan, E. et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 5, 503–507 (2004).
Shi, H. X. et al. Mitochondrial ubiquitin ligase MARCH5 promotes TLR7 signaling by attenuating TANK action. PLoS Pathog. 7, e1002057 (2011).
Liu, S. & Chen, Z. J. Expanding role of ubiquitination in NF-kappaB signaling. Cell Res. 21, 6–21 (2011).
Tseng, P. H. et al. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 11, 70–75 (2010).
West, A. P. et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480 (2011).
Xiao, N. et al. Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFalpha-induced cancer cell migration. Biochem J. 441, 979–986 (2012).
Shembade, N., Parvatiyar, K., Harhaj, N. S. & Harhaj, E. W. The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-kappaB signalling. EMBO J. 28, 513–522 (2009).
Wang, C. et al. The E3 ubiquitin ligase Nrdp1 ‘preferentially' promotes TLR-mediated production of type I interferon. Nat. Immunol. 10, 744–752 (2009).
Zhao, W. et al. E3 ubiquitin ligase tripartite motif 38 negatively regulates TLR-mediated immune responses by proteasomal degradation of TNF receptor-associated factor 6 in macrophages. J. Immunol. 188, 2567–2574 (2012).
Tanaka, T., Grusby, M. J. & Kaisho, T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 8, 584–591 (2007).
Yang, Y. et al. E3 ligase WWP2 negatively regulates TLR3-mediated innate immune response by targeting TRIF for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 110, 5115–5120 (2013).
Yoneyama, M. et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004).
Gack, M. U. et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920 (2007).
Inn, K. S. et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 41, 354–365 (2011).
Oshiumi, H., Matsumoto, M., Hatakeyama, S. & Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 284, 807–817 (2009).
Arimoto, K. et al. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl Acad. Sci. USA 107, 15856–15861 (2010).
Li, S. et al. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35, 426–440 (2011).
Arimoto, K. et al. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl Acad. Sci. USA 104, 7500–7505 (2007).
Chen, W. et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152, 467–478 (2013).
You, F. et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 10, 1300–1308 (2009).
Cui, J. et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat. Immunol. 13, 387–395 (2012).
Nakhaei, P. et al. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 5, e1000650 (2009).
Zhong, B. et al. The E3 ubiquitin ligase RNF5 targets virus-induced signaling adaptor for ubiquitination and degradation. J. Immunol. 184, 6249–6255 (2010).
Yang, H. et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Investig. 130, 4350–4364 (2019).
Woo, S. R. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014).
Barber, G. N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 35, 88–93 (2014).
Corrales, L. & Gajewski, T. F. Molecular pathways: targeting the stimulator of interferon genes (STING) in the immunotherapy of cancer. Clin. Cancer Res. 21, 4774–4779 (2015).
Sun, L. et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Motwani, M., Pesiridis, S. & Fitzgerald, K. A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 20, 657–674 (2019).
Tsuchida, T. et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776 (2010).
Zhang, J., Hu, M. M., Wang, Y. Y. & Shu, H. B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287, 28646–28655 (2012).
Wu, S. et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat. Cell Biol. 21, 1027–1040 (2019).
Zhong, B. et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30, 397–407 (2009).
Qin, Y. et al. RNF26 temporally regulates virus-triggered type I interferon induction by two distinct mechanisms. PLoS Pathog. 10, e1004358 (2014).
Wang, Q. et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933 (2014).
Chen, Y. et al. p38 inhibition provides anti-DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J. Exp. Med. 214, 991–1010 (2017).
Lytle, N. K., Barber, A. G. & Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 18, 669–680 (2018).
Silva, J. & Smith, A. Capturing pluripotency. Cell 132, 532–536 (2008).
Young, R. A. Control of the embryonic stem cell state. Cell 144, 940–954 (2011).
Boyer, L. A. et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956 (2005).
Buckley, S. M. et al. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell. Stem Cell. 11, 783–798 (2012).
Kim, S. H. et al. ERK1 phosphorylates Nanog to regulate protein stability and stem cell self-renewal. Stem Cell Res. 13, 1–11 (2014).
Kwon, J. E. et al. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J. Biol. Chem. 281, 12664–12672 (2006).
Geng, C. et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc. Natl Acad. Sci. USA 110, 6997–7002 (2013).
An, J. et al. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 6, 657–669 (2014).
Theurillat, J. P. et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 346, 85–89 (2014).
Li, C. et al. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene 30, 4350–4364 (2011).
Zhu, H. et al. SPOP E3 ubiquitin ligase adaptor promotes cellular senescence by degrading the SENP7 deSUMOylase. Cell Rep. 13, 1183–1193 (2015).
Wang, X. et al. AMPK promotes SPOP-mediated NANOG degradation to regulate prostate cancer cell stemness. Dev. Cell. 48, 345–360.e347 (2019).
Zhang, J. et al. SPOP promotes nanog destruction to suppress stem cell traits and prostate cancer progression. Dev. Cell. 48, 329–344.e325 (2019).
Jin, J. et al. The deubiquitinase USP21 maintains the stemness of mouse embryonic stem cells via stabilization of Nanog. Nat. Commun. 7, 13594 (2016).
Liu, X. et al. USP21 deubiquitylates Nanog to regulate protein stability and stem cell pluripotency. Signal Transduct. Target Ther. 1, 16024 (2016).
Liu, X. et al. Erratum: USP21 deubiquitylates Nanog to regulate protein stability and stem cell pluripotency. Signal Transduct. Target Ther. 2, 16046 (2017).
Brehm, A., Ohbo, K. & Scholer, H. The carboxy-terminal transactivation domain of Oct-4 acquires cell specificity through the POU domain. Mol. Cell Biol. 17, 154–162 (1997).
Liao, B. et al. Itch, an E3 ligase of Oct4, is required for embryonic stem cell self-renewal and pluripotency induction. J. Cell Physiol. 228, 1443–1451 (2013).
Spelat, R., Ferro, F. & Curcio, F. Serine 111 phosphorylation regulates OCT4A protein subcellular distribution and degradation. J. Biol. Chem. 287, 38279–38288 (2012).
Wang, Z. et al. Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell 10, 440–454 (2012).
Fang, L. et al. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol. Cell 55, 537–551 (2014).
Garnett, M. J. et al. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat. Cell Biol. 11, 1363–1369 (2009).
Wu, T. et al. UBE2S drives elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. Proc. Natl Acad. Sci. USA 107, 1355–1360 (2010).
Meyer, H. J. & Rape, M. Enhanced protein degradation by branched ubiquitin chains. Cell 157, 910–921 (2014).
Wang, J. et al. Ube2s regulates Sox2 stability and mouse ES cell maintenance. Cell Death Differ. 23, 393–404 (2016).
Huang, Z. et al. Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat. Cell Biol. 13, 142–152 (2011).
Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480 (2006).
Marikawa, Y. & Elinson, R. P. beta-TrCP is a negative regulator of Wnt/beta-catenin signaling pathway and dorsal axis formation in Xenopus embryos. Mech. Dev. 77, 75–80 (1998).
Fearon, E. R. & Spence, J. R. Cancer biology: a new RING to Wnt signaling. Curr. Biol. 22, R849–R851 (2012).
Ikeda, S. et al. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 17, 1371–1384 (1998).
Gammons, M. & Bienz, M. Multiprotein complexes governing Wnt signal transduction. Curr. Opin. Cell Biol. 51, 42–49 (2018).
Ling, L., Nurcombe, V. & Cool, S. M. Wnt signaling controls the fate of mesenchymal stem cells. Gene 433, 1–7 (2009).
Jiang, X. et al. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol. Cell 58, 522–533 (2015).
Szenker-Ravi, E. et al. RSPO2 inhibition of RNF43 and ZNRF3 governs limb development independently of LGR4/5/6. Nature 557, 564–569 (2018).
Koo, B. K. et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488, 665–669 (2012).
Hao, H. X. et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 485, 195–200 (2012).
Parry, W. H., Martorano, F. & Cotton, E. K. Management of life-threatening asthma with intravenous isoproterenol infusions. Am. J. Dis. Child. 130, 39–42 (1976).
Stamm, O., Latscha, U., Janecek, P. & Campana, A. Development of a special electrode for continuous subcutaneous pH measurement in the infant scalp. Am. J. Obstet. Gynecol. 124, 193–195 (1976).
Huang, S. M. et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620 (2009).
Zhang, Y. et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 13, 623–629 (2011).
Ji, L. et al. The SIAH E3 ubiquitin ligases promote Wnt/beta-catenin signaling through mediating Wnt-induced Axin degradation. Genes Dev. 31, 904–915 (2017).
Fei, C. et al. Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/beta-catenin signaling. Mol. Cell Biol. 33, 4095–4105 (2013).
Ding, Y. et al. HECT domain-containing E3 ubiquitin ligase NEDD4L negatively regulates Wnt signaling by targeting dishevelled for proteasomal degradation. J. Biol. Chem. 288, 8289–8298 (2013).
Wei, W. et al. The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol. Cell Biol. 32, 3903–3912 (2012).
Angers, S. et al. The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation. Nat. Cell Biol. 8, 348–357 (2006).
Lui, T. T. et al. The ubiquitin-specific protease USP34 regulates axin stability and Wnt/beta-catenin signaling. Mol. Cell Biol. 31, 2053–2065 (2011).
Tauriello, D. V. et al. Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol. Cell 37, 607–619 (2010).
Yun, S. I. et al. Ubiquitin specific protease 4 positively regulates the WNT/beta-catenin signaling in colorectal cancer. Mol. Oncol. 9, 1834–1851 (2015).
Nielsen, C. P. et al. USP9X deubiquitylates DVL2 to regulate WNT pathway specification. Cell Rep. 28, 1074–1089.e1075 (2019).
Ji, L. et al. USP7 inhibits Wnt/beta-catenin signaling through promoting stabilization of Axin. Nat. Commun. 10, 4184 (2019).
Moya, I. M. & Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 20, 211–226 (2019).
Fang, L. et al. SET1A-mediated mono-methylation at K342 regulates YAP activation by blocking its nuclear export and promotes tumorigenesis. Cancer Cell. 34, 103–118.e109 (2018).
Li, W., Cooper, J., Karajannis, M. A. & Giancotti, F. G. Merlin: a tumour suppressor with functions at the cell cortex and in the nucleus. EMBO Rep. 13, 204–215 (2012).
Ma, B. et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 17, 95–103 (2015).
Zhao, B. et al. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 24, 72–85 (2010).
Liu, C. Y. et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J. Biol. Chem. 285, 37159–37169 (2010).
Yao, F. et al. SKP2- and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nat. Commun. 9, 2269 (2018).
Azzolin, L. et al. Role of TAZ as mediator of Wnt signaling. Cell 151, 1443–1456 (2012).
Azzolin, L. et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170 (2014).
Salah, Z., Cohen, S., Itzhaki, E. & Aqeilan, R. I. NEDD4 E3 ligase inhibits the activity of the Hippo pathway by targeting LATS1 for degradation. Cell Cycle 12, 3817–3823 (2013).
Cadena, C. et al. Ubiquitin-dependent and -independent roles of E3 ligase RIPLET in innate immunity. Cell 177, 1187–1200.e1116 (2019).
Koren, I. et al. The Eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C-terminal degrons. Cell 173, 1622–1635.e1614 (2018).
Appel, A. Drugs: more shots on target. Nature 480, S40–S42 (2011).
Richardson, P. G., Hideshima, T. & Anderson, K. C. Bortezomib (PS-341): a novel, first-in-class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control 10, 361–369 (2003).
Kouroukis, T. C. et al. Bortezomib in multiple myeloma: systematic review and clinical considerations. Curr. Oncol. 21, e573–e603 (2014).
Mofers, A., Pellegrini, P., Linder, S. & D'Arcy, P. Proteasome-associated deubiquitinases and cancer. Cancer Metastasis Rev. 36, 635–653 (2017).
Robak, T. et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N. Engl. J. Med. 372, 944–953 (2015).
Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5, 417–421 (2004).
Sunwoo, J. B. et al. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. 7, 1419–1428 (2001).
Hideshima, T. et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 61, 3071–3076 (2001).
Baiz, D. et al. Bortezomib arrests the proliferation of hepatocellular carcinoma cells HepG2 and JHH6 by differentially affecting E2F1, p21 and p27 levels. Biochimie 91, 373–382 (2009).
Kontopodis, E. et al. A phase II, open-label trial of bortezomib (VELCADE((R))) in combination with gemcitabine and cisplatin in patients with locally advanced or metastatic non-small cell lung cancer. Cancer Chemother. Pharmacol. 77, 949–956 (2016).
Dispenzieri, A. Bortezomib for myeloma–much ado about something. N. Engl. J. Med. 352, 2546–2548 (2005).
Kane, R. C., Bross, P. F., Farrell, A. T. & Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 8, 508–513 (2003).
Fisher, R. I. et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 24, 4867–4874 (2006).
Frankland-Searby, S. & Bhaumik, S. R. The 26S proteasome complex: an attractive target for cancer therapy. Biochim. Biophys. Acta 1825, 64–76 (2012).
Richardson, P. G. et al. Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. J. Clin. Oncol. 24, 3113–3120 (2006).
Gelman, J. S. et al. Alterations of the intracellular peptidome in response to the proteasome inhibitor bortezomib. PLoS ONE 8, e53263 (2013).
Cavaletti, G. & Jakubowiak, A. J. Peripheral neuropathy during bortezomib treatment of multiple myeloma: a review of recent studies. Leuk. Lymphoma 51, 1178–1187 (2010).
Chanukuppa, V. et al. XPO1 is a critical player for bortezomib resistance in multiple myeloma: a quantitative proteomic approach. J. Proteom. 209, 103504 (2019).
Richardson, P. G. et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 348, 2609–2617 (2003).
Suzuki, E. et al. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS ONE 6, e27996 (2011).
Beyar-Katz, O. et al. Proinflammatory macrophages promote multiple myeloma resistance to bortezomib therapy. Mol. Cancer Res. 17, 2331–2340 (2019).
Zhang, H. et al. ClC5 decreases the sensitivity of multiple myeloma cells to bortezomib via promoting prosurvival autophagy. Oncol. Res. 26, 421–429 (2018).
Piva, R. et al. CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood 111, 2765–2775 (2008).
Stapnes, C. et al. The proteasome inhibitors bortezomib and PR-171 have antiproliferative and proapoptotic effects on primary human acute myeloid leukaemia cells. Br. J. Haematol. 136, 814–828 (2007).
Park, J. E. et al. Next-generation proteasome inhibitors for cancer therapy. Transl. Res. 198, 1–16 (2018).
Herndon, T. M. et al. U.S. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 19, 4559–4563 (2013).
Kaufman, J. L. et al. Combining carfilzomib and panobinostat to treat relapsed/refractory multiple myeloma: results of a Multiple Myeloma Research Consortium Phase I Study. Blood Cancer J. 9, 3 (2019).
Berenson, J. R. et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood 127, 3360–3368 (2016).
Zhou, H. J. et al. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J. Med. Chem. 52, 3028–3038 (2009).
Chauhan, D. et al. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 116, 4906–4915 (2010).
Zang, Y. et al. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin. Cancer Res. 18, 5639–5649 (2012).
Hari, P. et al. Oprozomib in patients with newly diagnosed multiple myeloma. Blood Cancer J. 9, 66 (2019).
Baljevic, M. & Orlowski, R. Z. Pharmacodynamics and pharmacokinetics of proteasome inhibitors for the treatment of multiple myeloma. Expert Opin. Drug Metab. Toxicol. 15, 459–473 (2019).
Dhakal, B. et al. Phase I/II trial of bendamustine, ixazomib, and dexamethasone in relapsed/refractory multiple myeloma. Blood Cancer J. 9, 56 (2019).
Dimopoulos, M. A. et al. Ixazomib maintenance therapy in newly diagnosed multiple myeloma: an integrated analysis of four phase I/II studies. Eur. J. Haematol. 102, 494–503 (2019).
Dimopoulos, M. A. et al. Oral ixazomib maintenance following autologous stem cell transplantation (TOURMALINE-MM3): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet 393, 253–264 (2019).
Gallerani, E. et al. A first in human phase I study of the proteasome inhibitor CEP-18770 in patients with advanced solid tumours and multiple myeloma. Eur. J. Cancer 49, 290–296 (2013).
Potts, B. C. et al. Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 11, 254–284 (2011).
Spencer, A. et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): final study results. Br. J. Haematol. 180, 41–51 (2018).
Palumbo, A. et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N. Engl. J. Med. 375, 754–766 (2016).
Di, K. et al. Marizomib activity as a single agent in malignant gliomas: ability to cross the blood-brain barrier. Neuro-Oncol. 18, 840–848 (2016).
Bernassola, F., Karin, M., Ciechanover, A. & Melino, G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 14, 10–21 (2008).
Khoo, K. H., Verma, C. S. & Lane, D. P. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 13, 217–236 (2014).
Chi, S. W. et al. Structural details on mdm2-p53 interaction. J. Biol. Chem. 280, 38795–38802 (2005).
Chene, P. et al. A small synthetic peptide, which inhibits the p53-hdm2 interaction, stimulates the p53 pathway in tumour cell lines. J. Mol. Biol. 299, 245–253 (2000).
Vassilev, L. T. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004).
Vu, B. et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4, 466–469 (2013).
Tisato, V. et al. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 10, 133 (2017).
Sarisozen, C. et al. MDM2 antagonist-loaded targeted micelles in combination with doxorubicin: effective synergism against human glioblastoma via p53 re-activation. J. Drug Target. 27, 624–633 (2019).
Kojima, K. et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood 106, 3150–3159 (2005).
Issaeva, N. et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 10, 1321–1328 (2004).
Shangary, S. et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl Acad. Sci. USA 105, 3933–3938 (2008).
Li, W. D. et al. Cytotoxic effect of a non-peptidic small molecular inhibitor of the p53-HDM2 interaction on tumor cells. World J. Gastroenterol. 11, 2927–2931 (2005).
Ding, K. et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 49, 3432–3435 (2006).
Bykov, V. J. et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 8, 282–288 (2002).
Yang, Y. et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 7, 547–559 (2005).
Kitagaki, J. et al. Targeting tumor cells expressing p53 with a water-soluble inhibitor of Hdm2. Mol. Cancer Ther. 7, 2445–2454 (2008).
Herman, A. G. et al. Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov. 1, 312–325 (2011).
Sasiela, C. A. et al. Identification of inhibitors for MDM2 ubiquitin ligase activity from natural product extracts by a novel high-throughput electrochemiluminescent screen. J. Biomol. Screen. 13, 229–237 (2008).
Joseph, T. L. et al. Differential binding of p53 and nutlin to MDM2 and MDMX: computational studies. Cell Cycle 9, 1167–1181 (2010).
Wade, M. & Wahl, G. M. Targeting Mdm2 and Mdmx in cancer therapy: better living through medicinal chemistry? Mol. Cancer Res. 7, 1–11 (2009).
Chang, Y. S. et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl Acad. Sci. USA 110, E3445–3454 (2013).
Wang, H. et al. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol. Cancer Ther. 10, 69–79 (2011).
Nakayama, K. I. & Nakayama, K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 (2006).
Yang, Y. et al. Targeting the ubiquitin-proteasome system for cancer therapy. Cancer Sci. 100, 24–28 (2009).
Huang, H. L. et al. Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Mol. Cancer Ther. 11, 1155–1165 (2012).
Orlicky, S. et al. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat. Biotechnol. 28, 733–737 (2010).
Chan, C. H. et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 154, 556–568 (2013).
Chen, Q. et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 111, 4690–4699 (2008).
Benary, U. & Wolf, J. Controlling nuclear NF-kappaB dynamics by beta-TrCP-insights from a computational model. Biomedicines 7, 40 (2019).
Nalepa, G., Rolfe, M. & Harper, J. W. Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 5, 596–613 (2006).
Yaron, A. et al. Inhibition of NF-kappa-B cellular function via specific targeting of the I-kappa-B-ubiquitin ligase. EMBO J. 16, 6486–6494 (1997).
Blees, J. S. et al. Erioflorin stabilizes the tumor suppressor Pdcd4 by inhibiting its interaction with the E3-ligase β-TrCP1. PLoS ONE 7, e46567 (2012).
Nakajima, H. et al. A novel small-molecule inhibitor of NF-kappaB signaling. Biochem. Biophys. Res. Commun. 368, 1007–1013 (2008).
Aghajan, M. et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat. Biotechnol. 28, 738–742 (2010).
Zeng, X. et al. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 18, 382–395 (2010).
Sackton, K. L. et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 514, 646–649 (2014).
Zhang, J. et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 361, 290–295 (2018).
Buckley, D. L. et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J. Am. Chem. Soc. 134, 4465–4468 (2012).
Silva, V. A. O. et al. Rapid diminution in the level and activity of DNA-dependent protein kinase in cancer cells by a reactive nitro-benzoxadiazole compound. Int. J. Mol. Sci. 17, 703 (2016).
Chen, B. B. et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat. Immunol. 14, 470–479 (2013).
Mallampalli, R. K. et al. Targeting F box protein Fbxo3 to control cytokine-driven inflammation. J. Immunol. 191, 5247–5255 (2013).
Chen, B. B., Glasser, J. R., Coon, T. A. & Mallampalli, R. K. F-box protein FBXL2 exerts human lung tumor suppressor-like activity by ubiquitin-mediated degradation of cyclin D3 resulting in cell cycle arrest. Oncogene 31, 2566–2579 (2012).
Chen, B. B., Glasser, J. R., Coon, T. A. & Mallampalli, R. K. Skp-cullin-F box E3 ligase component FBXL2 ubiquitinates Aurora B to inhibit tumorigenesis. Cell Death Dis. 4, e759 (2013).
Hyer, M. L. et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 24, 186–193 (2018).
Nawrocki, S. T., Griffin, P., Kelly, K. R. & Carew, J. S. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin. Investig. Drugs 21, 1563–1573 (2012).
Soucy, T. A. et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009).
Watson, I. R., Irwin, M. S. & Ohh, M. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell 19, 168–176 (2011).
Yang, Y. et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 67, 9472–9481 (2007).
Matsuo, S., Sharma, A., Wang, P. & Yang, W. L. PYR-41, A. Ubiquitin-activating enzyme E1 inhibitor, attenuates lung injury in sepsis. Shock 49, 442–450 (2018).
Tsukamoto, S. et al. Leucettamol A: a new inhibitor of Ubc13-Uev1A interaction isolated from a marine sponge, Leucetta aff. microrhaphis. Bioorg. Med. Chem. Lett. 18, 6319–6320 (2008).
Ushiyama, S. et al. Manadosterols A and B, sulfonated sterol dimers inhibiting the Ubc13-Uev1A interaction, isolated from the marine sponge Lissodendryx fibrosa. J. Nat. Prod. 75, 1495–1499 (2012).
Ceccarelli, D. F. et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 145, 1075–1087 (2011).
Zlotkowski, K. et al. A small-molecule microarray approach for the identification of E2 enzyme inhibitors in ubiquitin-like conjugation pathways. SLAS Discov. 22, 760–766 (2017).
Nijman, S. M. et al. A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786 (2005).
Fang, Y., Fu, D. & Shen, X. Z. The potential role of ubiquitin c-terminal hydrolases in oncogenesis. Biochim. Biophys. Acta 1806, 1–6 (2010).
Liu, J. et al. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta 1855, 50–60 (2015).
Wei, R. et al. Deubiquitinases in cancer. Oncotarget 6, 12872–12889 (2015).
Harrigan, J. A., Jacq, X., Martin, N. M. & Jackson, S. P. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat. Rev. Drug Discov. 17, 57–78 (2018).
Aleo, E. et al. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer Res. 66, 9235–9244 (2006).
Nicholson, B. et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 17, 1035–1043 (2008).
Altun, M. et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 18, 1401–1412 (2011).
Lee, J. K. et al. USP1 targeting impedes GBM growth by inhibiting stem cell maintenance and radioresistance. Neuro-Oncol. 18, 37–47 (2016).
Kapuria, V. et al. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 70, 9265–9276 (2010).
Fulda, S. et al. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem. 273, 33942–33948 (1998).
Reiner, T. et al. Betulinic acid selectively increases protein degradation and enhances prostate cancer-specific apoptosis: possible role for inhibition of deubiquitinase activity. PLoS ONE 8, e56234 (2013).
D'Arcy, P., Wang, X. & Linder, S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 147, 32–54 (2015).
Chauhan, D. et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 22, 345–358 (2012).
Pozhidaeva, A. et al. USP7-specific inhibitors target and modify the enzyme's active site via distinct chemical mechanisms. Cell Chem. Biol. 24, 1501–1512.e1505 (2017).
Turnbull, A. P. et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 550, 481–486 (2017).
Lamberto, I. et al. Structure-guided development of a potent and selective non-covalent active-site inhibitor of USP7. Cell Chem. Biol. 24, 1490–1500.e1411 (2017).
Reverdy, C. et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 19, 467–477 (2012).
Lee, M. J. et al. Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol. Cell. Proteom. 10, R110.003871 (2011).
Shinji, S. et al. Ubiquitin-specific protease 14 expression in colorectal cancer is associated with liver and lymph node metastases. Oncol. Rep. 15, 539–543 (2006).
Wu, N. et al. Over-expression of deubiquitinating enzyme USP14 in lung adenocarcinoma promotes proliferation through the accumulation of beta-catenin. Int. J. Mol. Sci. 14, 10749–10760 (2013).
Zhu, Y. et al. Function of deubiquitinating enzyme USP14 as oncogene in different types of cancer. Cell. Physiol. Biochem. 38, 993–1002 (2016).
Zhang, B. et al. Overexpression of ubiquitin specific peptidase 14 predicts unfavorable prognosis in esophageal squamous cell carcinoma. Thorac. Cancer 8, 344–349 (2017).
Wang, Y. et al. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. 28, 1186–1194 (2018).
Lee, B. H. et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184 (2010).
Liu, Y. et al. Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem. Biol. 10, 837–846 (2003).
Kobayashi, E. et al. Inhibition of UCH-L1 deubiquitinating activity with two forms of LDN-57444 has anti-invasive effects in metastatic carcinoma cells. Int. J. Mol. Sci. 20, 3733 (2019).
Berndtsson, M. et al. Induction of the lysosomal apoptosis pathway by inhibitors of the ubiquitin-proteasome system. Int. J. Cancer 124, 1463–1469 (2009).
Tian, Z. et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 123, 706–716 (2014).
D'Arcy, P. et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 17, 1636–1640 (2011).
Zhang, X. et al. The deubiquitinase inhibitor b-AP15 induces strong proteotoxic stress and mitochondrial damage. Biochem. Pharmacol. 156, 291–301 (2018).
Sha, B. et al. Deubiquitylatinase inhibitor b-AP15 induces c-Myc-Noxa-mediated apoptosis in esophageal squamous cell carcinoma. Apoptosis 24, 826–836 (2019).
Kikuchi, H., Yuan, B., Hu, X. & Okazaki, M. Chemopreventive and anticancer activity of flavonoids and its possibility for clinical use by combining with conventional chemotherapeutic agents. Am. J. Cancer Res. 9, 1517–1535 (2019).
Selvaraju, K. et al. Inhibition of proteasome deubiquitinase activity: a strategy to overcome resistance to conventional proteasome inhibitors? Drug Resist. Updat. 21-22, 20–29 (2015).
Qiu, G. Z. et al. The bad seed gardener: deubiquitinases in the cancer stem-cell signaling network and therapeutic resistance. Pharmacol. Ther. 172, 127–138 (2017).
Cengiz Seval, G. & Beksac, M. The safety of bortezomib for the treatment of multiple myeloma. Expert Opin. Drug Saf. 17, 953–962 (2018).
Munakata, K. et al. Cancer stem-like properties in colorectal cancer cells with low proteasome activity. Clin. Cancer Res. 22, 5277–5286 (2016).
Tanaka, N. et al. Acquired platinum resistance involves epithelial to mesenchymal transition through ubiquitin ligase FBXO32 dysregulation. JCI Insight 1, e83654 (2016).
Jing, X. et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 18, 157 (2019).
Majzner, R. G. & Mackall, C. L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 25, 1341–1355 (2019).
Strifler, S. & Knop, S. The role of carfilzomib in treatment of newly diagnosed multiple myeloma. Future Oncol. 14, 3123–3134 (2018).
Kumar, S. K. et al. NCCN guidelines insights: multiple myeloma, version 3.2018. J. Natl Compr. Canc. Netw. 16, 11–20 (2018).
Mikhael, J. et al. A phase 1b study of isatuximab plus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma. Blood 134, 123–133 (2019).
Jakubowiak, A. J. et al. Phase 1 study of selinexor plus carfilzomib and dexamethasone for the treatment of relapsed/refractory multiple myeloma. Br. J. Haematol. 186, 549–560 (2019).
Li, S. J., Hao, J., Mao, Y. & Si, Y. L. Effects of the proteasome inhibitor bortezomib in combination with chemotherapy for the treatment of mantle cell lymphoma: a meta-analysis. Turk. J. Haematol. https://doi.org/10.4274/tjh.galenos.2019.2019.0128 (2019).
Yazbeck, V. et al. A phase II trial of bortezomib and vorinostat in mantle cell lymphoma and diffuse large B-cell lymphoma. Clin. Lymphoma Myeloma Leuk. 18, 569–575.e561 (2018).
Meng, X. et al. Combination of proteasome and histone deacetylase inhibitors overcomes the impact of gain-of-function p53 mutations. Dis. Markers 2018, 3810108 (2018).
Ghobrial, I. M. et al. Phase I/II trial of the CXCR4 inhibitor plerixafor in combination with bortezomib as a chemosensitization strategy in relapsed/refractory multiple myeloma. Am. J. Hematol. 94, 1244–1253 (2019).
Moreau, P. et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet 394, 29–38 (2019).
Tomlinson, B. K. et al. A phase II study of bortezomib in combination with pegylated liposomal doxorubicin for acute myeloid leukemia. Am. J. Hematol. 94, E291–E294 (2019).
Kuhn, D. J. et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 110, 3281–3290 (2007).
Dimopoulos, M. et al. Carfilzomib vs bortezomib in patients with multiple myeloma and renal failure: a subgroup analysis of ENDEAVOR. Blood 133, 147–155 (2019).
Yao, Y. et al. Blockade of deubiquitinase USP7 overcomes bortezomib resistance by suppressing NF-kappaB signaling pathway in multiple myeloma. J. Leukoc. Biol. 104, 1105–1115 (2018).
Zhuang, J. et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 133, 1572–1584 (2019).
Erpapazoglou, Z., Walker, O. & Haguenauer-Tsapis, R. Versatile roles of k63-linked ubiquitin chains in trafficking. Cells 3, 1027–1088 (2014).
Haglund, K., Di Fiore, P. P. & Dikic, I. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem. Sci. 28, 598–603 (2003).
Wang, W. et al. Targeting MDM2 for novel molecular therapy: beyond oncology. Med. Res. Rev. doi: https://doi.org/10.1002/med.21637 (2019).
Wang, X., Herr, R. A. & Hansen, T. H. Ubiquitination of substrates by esterification. Traffic 13, 19–24 (2012).
Cadwell, K. & Coscoy, L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science 309, 127–130 (2005).
Carvalho, A. F. et al. Ubiquitination of mammalian Pex5p, the peroxisomal import receptor. J. Biol. Chem. 282, 31267–31272 (2007).
Williams, C., van den Berg, M., Sprenger, R. R. & Distel, B. A conserved cysteine is essential for Pex4p-dependent ubiquitination of the peroxisomal import receptor Pex5p. J. Biol. Chem. 282, 22534–22543 (2007).
Leon, S. & Subramani, S. A conserved cysteine residue of Pichia pastoris Pex20p is essential for its recycling from the peroxisome to the cytosol. J. Biol. Chem. 282, 7424–7430 (2007).
Hensel, A. et al. Cysteine-dependent ubiquitination of Pex18p is linked to cargo translocation across the peroxisomal membrane. J. Biol. Chem. 286, 43495–43505 (2011).
Liu, X. & Subramani, S. Unique requirements for mono- and polyubiquitination of the peroxisomal targeting signal co-receptor, Pex20. J. Biol. Chem. 288, 7230–7240 (2013).
Wang, X. et al. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell Biol. 177, 613–624 (2007).
Pao, K. C. et al. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 556, 381–385 (2018).
Rape, M. Assembly of k11-linked ubiquitin chains by the anaphase-promoting complex. Subcell. Biochem. 54, 107–115 (2010).
Thrower, J. S., Hoffman, L., Rechsteiner, M. & Pickart, C. M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102 (2000).
Lasker, K. et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl Acad. Sci. USA 109, 1380–1387 (2012).
Patel, C. H., Leone, R. D., Horton, M. R. & Powell, J. D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 18, 669–688 (2019).
Zhu, J. & Thompson, C. B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 20, 436–450 (2019).
Liu, L., Michowski, W., Kolodziejczyk, A. & Sicinski, P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 21, 1060–1067 (2019).
McDowell, S. A. C. & Quail, D. F. Immunological regulation of vascular inflammation during cancer metastasis. Front. Immunol. 10, 1984 (2019).
Zhang, J. et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95 (2018).
Meng, X. et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature 564, 130–135 (2018).
Lim, S. O. et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939 (2016).
Veggiani, G., Gerpe, M. C. R., Sidhu, S. S. & Zhang, W. Emerging drug development technologies targeting ubiquitination for cancer therapeutics. Pharmacol. Ther. 199, 139–154 (2019).
Khaminets, A., Behl, C. & Dikic, I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 26, 6–16 (2016).
Duncan, K. et al. Targeting neddylation in cancer therapy. Future Oncol. 8, 1461–1470 (2012).
Fouladkou, F. et al. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc. Natl Acad. Sci. USA 105, 8585–8590 (2008).
Hideshima, T., Richardson, P. G. & Anderson, K. C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 10, 2034–2042 (2011).
Hajek, R. et al. Design and rationale of FOCUS (PX-171-011): a randomized, open-label, phase 3 study of carfilzomib versus best supportive care regimen in patients with relapsed and refractory multiple myeloma (R/R MM). BMC Cancer 12, 415 (2012).
Lee, E. C. et al. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin. Cancer Res. 17, 7313–7323 (2011).
Bartlett, J. B., Dredge, K. & Dalgleish, A. G. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat. Rev. Cancer 4, 314–322 (2004).
Ito, T. et al. Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010).
Zhu, Y. X. et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 118, 4771–4779 (2011).
Lopez-Girona, A. et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 26, 2326–2335 (2012).
Syed, Y. Y. Lenalidomide: a review in newly diagnosed multiple myeloma as maintenance therapy after ASCT. Drugs 77, 1473–1480 (2017).
Lehmann, S. et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 30, 3633–3639 (2012).
Bykov, V. J. N. et al. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 292, 19607 (2017).
Chargari, C. et al. Preclinical assessment of JNJ-26854165 (Serdemetan), a novel tryptamine compound with radiosensitizing activity in vitro and in tumor xenografts. Cancer Lett. 312, 209–218 (2011).
Tabernero, J. et al. A phase I first-in-human pharmacokinetic and pharmacodynamic study of serdemetan in patients with advanced solid tumors. Clin. Cancer Res. 17, 6313–6321 (2011).
Andreeff, M. et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 22, 868–876 (2016).
Ding, Q. et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 56, 5979–5983 (2013).
Phelps, D. et al. Inhibition of MDM2 by RG7388 confers hypersensitivity to X-radiation in xenograft models of childhood sarcoma. Pediatr. Blood Cancer 62, 1345–1352 (2015).
Voltan, R. et al. Nanoparticles engineered with rituximab and loaded with Nutlin-3 show promising therapeutic activity in B-leukemic xenografts. Clin. Cancer Res. 19, 3871–3880 (2013).
Bista, M. et al. On the mechanism of action of SJ-172550 in inhibiting the interaction of MDM4 and p53. PLoS ONE 7, e37518 (2012).
Ling, X. et al. FL118 induces p53-dependent senescence in colorectal cancer cells by promoting degradation of MdmX. Cancer Res. 74, 7487–7497 (2014).
Kategaya, L. et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 550, 534–538 (2017).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos. 31801177, 81702659, 31830053, 31920103007 and 81625019), the Science Technology Commission of Shanghai Municipality (No. 18410722000), the Shanghai Sailing Program (No. 18YF1419500), and the Fundamental Research Funds for the Central Universities (No. 22120180043).
Author information
Authors and Affiliations
Contributions
L.D., T.M., L.C. and P.W. designed the study. L.D., T.M. and L.C. contributed to the literature search, drew the figures and cowrote the paper. W.W. and P.W. contributed their experiences and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, L., Meng, T., Chen, L. et al. The role of ubiquitination in tumorigenesis and targeted drug discovery. Sig Transduct Target Ther 5, 11 (2020). https://doi.org/10.1038/s41392-020-0107-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-0107-0
This article is cited by
-
Incorporating genetic similarity of auxiliary samples into eGene identification under the transfer learning framework
Journal of Translational Medicine (2024)
-
Ubiquitination and deubiquitination in cancer: from mechanisms to novel therapeutic approaches
Molecular Cancer (2024)
-
Transcriptional regulation of cancer stem cell: regulatory factors elucidation and cancer treatment strategies
Journal of Experimental & Clinical Cancer Research (2024)
-
Rolapitant treats lung cancer by targeting deubiquitinase OTUD3
Cell Communication and Signaling (2024)
-
Deubiquitinase PSMD7 facilitates pancreatic cancer progression through activating Nocth1 pathway via modifying SOX2 degradation
Cell & Bioscience (2024)
