
Abstract
Gallbladder cancer (GBC) is rare, but is the most malignant type of biliary tract tumor. Unfortunately, only a small population of cancer patients is acceptable for the surgical resection, the current effective regimen; thus, the high mortality rate has been static for decades. To substantially circumvent the stagnant scenario, a number of therapeutic approaches owing to the creation of advanced technologic measures (e.g., next-generation sequencing, transcriptomics, proteomics) have been intensively innovated, which include targeted therapy, immunotherapy, and nanoparticle-based delivery systems. In the current review, we primarily focus on the targeted therapy capable of specifically inhibiting individual key molecules that govern aberrant signaling cascades in GBC. Global clinical trials of targeted therapy in GBC are updated and may offer great value for novel pathologic and therapeutic insights of this deadly disease, ultimately improving the efficacy of treatment.
Similar content being viewed by others


Introduction
Gallbladder cancer (GBC) is the most common cancer of the biliary tract system and ranked as the top six in general gastrointestinal tract neoplasms worldwide.1,2,3 While the incidence rate of GBC varies widely, it has a unique distribution pattern in some regions, where Chile, India, some other Asian countries, Eastern European, and Latin American countries have reported more cases than the rest of the world every year.4,5,6 This geographical distribution signature probably ascribed to the difference of genetic susceptibility contributes to an important risk factor of GBC. The other factors, which associated with chronic inflammation and disease pathogenesis, such as hepatobiliary stones, liver flukes, and Clostridium frequently observed in these areas, also constitute the other high-risk factors of bile tract cancer (BTC) including GBC.7 In addition to these regionally limited factors, plenty of ubiquitous risk factors have been documented globally and taken into account seriously, which include gallstone, gender, age, obesity, reproductive factors, race, primary sclerosis cholangitis, gallbladder polyps, congenital biliary cysts, typhoid, Helicobacter pylori infection, alcohol intake, smoking, fatty liver disease, unhealthy diet, and environmental exposure to specific chemicals.8,9,10 As a result, the early protection from carcinogenesis has been enforced in clinical practice and the occurrence of this disease only accounts for 1.2% of all cancers diagnosed in the world.
Currently, radical resection is the most effective strategy to potentially cure GBC. Unfortunately, the population falling into this operational course is largely limited, as a large number of patients (>70–90%) can only accept non-surgical treatment. Such unfavorable outcome is because of the atypical clinical symptoms at earlier stages, contrary to the noticeable symptoms that emerge in most cases with advanced disease.11,12 The non-surgical therapies engaged in patients were primarily composed of chemotherapy and radiotherapy. However, over the past decades, additional therapeutic strategies have been continuously renovated, given rapid discoveries of the advanced technology, including next-generation sequencing (NGS), whole-exome sequencing (WES), RNA-sequencing (RNAseq), and single-cell isolation, as well as characterization that have fundamentally opened a novel view enabled to globally identify genetic and epigenetic features and key molecules as potential therapeutic targets. In particular, specific target treatment, immune therapy, vaccine therapy, biotherapy, and nanoparticles have been intensively developed in the preclinical and clinical trials. In the present review, we will focus on the targeted regimen and immune therapy as these treatments have recently received considerable attention with the hope of improving quality of life and overall survival (OS) of GBC patients in the clinic.
Chemotherapy and radiotherapy of GBC
Chemotherapy involves agents used for non-specific inhibition of tumor cell proliferation usually via blockade of DNA synthesis, which has been extensively engaged in the treatment of a variety of cancers. National Comprehensive Cancer Network has provided two options for GBC treatment: single-agent therapy, which is fluoropyrimidine or gemcitabine-based treatment, and multiagents regimen, which includes oxaliplatin, cisplatin, and capecitabine.13,14,15 Although there are limited data to define a standard regimen or definitive benefit, the combined therapy regimens of FOLFOX (5-fluorouracil and oxaliplatin),16 CAPOX (capecitabine and oxaliplatin),17,18 GC (gemcitabine and cisplatin),19 and Gemox (gemcitabine and oxaliplatin)20 still remain the mainstream chemotherapy programs in clinical trials. It is noticeable that some clinical trials have shown that combination chemotherapy with fluorouracil, leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) yield promising results in patients with BTC.21,22 However, none of the single program has been widely offered, since unexpected responses were deliberately taken into account, such as systemic toxicity, insufficient drug responses, and drug resistance.23 Therefore, there are currently a large number of preclinical and clinical researches that are adding significant efforts in order to define the overall benefit of drug treatment even in the presence of adversary responses, which can be otherwise governed at a minimal level. For example, transplantation of freshly resected patient tissue of GBC in mice as a mini patient-derived xenograft (mini-PDX) model to examine individual drug sensitivity and select more effective drugs from combined agent trials for the guidance of clinic drug-selective treatment in the patient. They tested gemcitabine, oxaliplatin, 5‐fluorouracil, nanoparticle albumin‐bound nab‐paclitaxel, and irinotecan after surgery, and found that patients in the PDX-guided chemotherapy group had significantly longer median OS (mOS; 18.6 months; 95% confidence interval (CI) 15.9–21.3 months) and disease-free survival (DFS 17.6 months; 95% CI 14.5–20.6 months) than patients in the conventional random drug-selective chemotherapy group with mOS (13.9 months; 95% CI 11.7–16.2 months) (P = 0.030; hazard ratio (HR) 3.18; 95% CI 1.47–6.91) and DFS (12.0 months; 95% CI 9.7–14.4 months) (P = 0.014; HR 3.37; 95% CI 1.67–6.79). Thus, mini-PDX may devise the regimes of chemotherapy to improve the outcomes; in particular, providing an optimal opportunity for personalized medicine.24,25
In addition to intervention of most chemotherapeutic agents on DNA synthesis, over the past decade, there has been growing research work focusing on RNA molecules that controls oncogene or tumor suppressor gene expression. It is emerging that microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) act as indispensable factors to coordinately manipulate multiple gene expression underlying carcinogenesis, in sharp contrast to the traditional paradigm that non-coding RNAs are noisy and non-functional in the regulation of gene expression. Multiple laboratories including ours have reported that miRNAs and lncRNAs mediate the proliferation, invasion, and chemotherapy resistance of GBC, and serve as new therapeutic targets for the treatment of advanced GBC, including miR-125b-5p,26 miR-122,27 miR-223,28 miR-31,29,30 miR-30a-5p,31 lncRNA-HGBC,32 lncRNA-PAGBC,33 lncRNA PVT1,34 and lncRNA GBCDRlnc1.35 Concomitantly, advanced nanotechnology employing various modified materials that offer optimal cargos to efficiently deliver RNA molecules has also given rise to promising benefit in the chemotherapy of GBC. Cai and co-workers36,37 found that nanomaterial-induced photothermal therapy in combination with chemotherapy and chloroquine inhibited GBC cell proliferation. It is worthwhile monitoring the efficacy of this novel delivery system with RNA molecules as an alternative therapy for GBC in future.
Albeit the benefit of adjuvant radiotherapy (ART) is evident in many other types of cancers, it remains unclear in GBC, since there is a lack of strong evidence of improved endpoints. Up to date, no standard regimens are available for the selection of ART; thus, combination therapy of ART with chemotherapy is usually engaged in the clinic. Some of the clinical settings have yielded encouraging results.38 For instance, a phase II study led by the Southwest Oncology Group (SWOG) investigated the clinical outcomes of extrahepatic cholangiocarcinoma and GBC treated with adjuvant chemotherapy GC followed by chemoradiation (combined capecitabine and radiotherapy) in a single-arm study. The 2-year OS was 65% for all patients (67% for R0 and 60% for R1), the mOS time was 35 months, and only 14 patients had local recurrence, demonstrating the feasibility and benefit of adjuvant chemotherapy followed by chemoradiation therapy for GBC.39 Thus, the combination of radiotherapy is well tolerated, and has a considerable effect; however, it still needs to be confirmed by large-scale, multicenter randomized case–control phase III clinical trials.
Targeted therapy of GBC
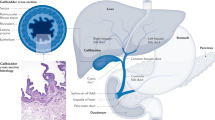
Targeted therapy for cancers was initially developed in 1988 based on the concept of specific chemicals that was able to eliminate some microorganisms in the early 1900.40 Since then the efficacy of targeted therapy has been extensively investigated in multiple cancers to specifically block a number of molecular targets that are keenly associated with tumor cell proliferation, differentiation, migration, cancer stemness, vascular angiogenesis, and antitumor immune responses.41,42 A great volume of drugs for targeted therapy have been created, which mainly consist of small molecules and immunized antibodies. The small molecules with molecular weight <900 Da can be readily transported into cells to inactivate specific proteins or enzymes, thus inhibiting tumor cell growth,43 while therapeutic antibodies specifically bind to cell membrane receptors or their ligands to regulate cell proliferation or apoptosis.44 Some drugs were developed to target extracellular molecules that mediate angiogenesis or immune reaction in the tumor microenvironment, resulting in inhibition of tumor growth, angiogenesis, and metastasis.45,46 With regards to recently intense clinical research on varied specific agents that intervene intracellular signaling pathways dysfunctional in GBC (Table 1), here we updated these targeted therapies on individual signaling pathways, including human epidermal growth factor receptor 2 (HER2), growth factor receptor tyrosine kinases (RTKs) (EGFR, vascular endothelial growth factor receptor (VEGFR)), programmed death receptor 1 (PD-1)/programmed death ligand 1 (PD-L1), TP53, KIT, CDKN2A/B, phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR), and RAS/BRAF/MEK/MAK (Fig. 1).

Overview of GBC targeted sites and agents. Boxes highlight drugs undergoing clinical investigation as reviewed, with arrows indicating pathway/target activation and blocked lines indicating pathway/target inhibition. c-MET mesenchymal–epithelial transition factor, VEGF vascular endothelial growth factor, VEGFR vascular endothelial growth factor receptor, EGFR epidermal growth factor receptor, ERBB2 human epidermal growth factor 2, PD-1 programmed death-1, PD-L1 programmed death ligand 1, PI3K phosphoinositide 3-kinase, AKT protein kinase B, also known as PKB, mTOR mammalian target of rapamycin, MEK mitogen-activated protein kinase, ERK extracellular signal-regulated kinase
HER2
HER2, also known as ErbB2, is a member of HER family, which includes HER1, HER2, HER3, and HER4, also called ErbB1 (EGFR), ErbB2, ErbB3, and ErbB4, respectively.47,48 HERs are cell-surface receptors that harbor a transmembrane tyrosine kinase domain capable of activating multiple downstream signal pathways upon binding with epidermal growth factor (EGF).49 HER2 has been well appreciated to play a crucial role in cancer biology and is an essential functional partner for the other family member receptor binding orchestrating a heterodimer, while HER2 itself forms a homodimer as cognate ligand binding.50 HER2/HER3 interaction leads to PI3K/Akt phosphorylation and downstream signaling activation that mediates cell polarity, cell adhesion, and cell cycles.51,52 HER2 can also trigger mitogen-activated protein kinase (MAPK) signaling.53 Many elegant mechanistic studies on these signaling pathways reported previously were not elucidated here.
Abnormality of HER2 with gene overexpression and/or activated mutations has been reported in multiple cancers, such as breast cancer, colon cancer, lung cancer, stomach cancer, and BTC.50,54,55
Kiguchi et al.56 first reported that overexpression of HER2 in the basal layer of biliary tract epithelium in transgenic mice led to the development of gallbladder adenocarcinoma at the age of 3 months. In human GBC, HER overexpression was found between 9.8 and 12.8%.54,57 Since 2014, we have added notable effort to gain GBC WES and identify potential mutations of ERBB family. In Fig. 2b, we summarized the detailed mutation information of HER2 discovered in our previous two studies. Frequency of mutations of HER2 and HER3 was found to be 9.8% and 11.8%, respectively, which accounted for ErbB signaling pathway activations in GBC.58 Supporting the evidence, these activated mutations of HER2/3 in GBC cell lines resulted in a significant increase in cell proliferation and tumor development in animals,59 underscoring the essential role of HER2/3 mutations in the development of GBC. In concert with our findings, Sebastian and co-workers60 also recently demonstrated the high alteration frequency (12.6%) of HER2 in GBC and the S310 of HER2 as a hot-spot mutation.

Summary of HER2 mutations in the COSMIC database (a) and mutation information of HER2 in our previous two studies (b)
There was growing research evidence reported from preclinical and clinical therapeutic trials in GBC. Ah-Rong and co-workers61 found that HER2+ SNU-2670 and SNU-2773 GBC cell lines were more sensitive to trastuzumab, dacomitinib, and afatinib than HER2− BTC cell lines. In addition, in the mouse xenograft model of SNU-2670, trastuzumab as a monotherapy and in combination with gemcitabine demonstrated a stronger antitumor effect and greater cell apoptosis than gemcitabine treatment. Iyer et al.62 discovered that treatment of GBC cells isolated from patients with ErbB2-specific short hairpin RNA (shRNA), EGFR-specific shRNA, or afatinib can inhibit the invasiveness of GBC cells. Interestingly, Wang et al.63 showed that gemcitabine/5-fluorouracil increased the expressions of total and phosphorylated forms of HER2 in GBC cells, thus enhancing the cytotoxicity of trastuzumab, suggesting that sequential therapy with gemcitabine/5-fluorouracil followed by trastuzumab perhaps devises a novel and promising therapeutic strategy aiming at HER2− GBCs that are currently short of targeted drugs.
Inagaki et al.64 reported that a GBC case harboring HER2 mutation on the primary and metastatic site underwent HER2-targeted treatment with lapatinib and capecitabin. After two cycles of treatment, contrast computed tomography imaging showed a decrease in the size of tumor emboli and hepatic lesions. Likewise, Prieto et al.65 demonstrated favorable benefit of 5-year survival without recurrence after treatment with chemotherapy and trastuzumab on a stage IV GBC patient who presented progression during first-line chemotherapy treatment following radical savage surgery. It is emerging that the insufficiency of GBC cohorts enrolled in the studies is the primary obstacle for the clinical trial practice. In 2009, Ramanathan et al.66 reported a phase II study with lapatinib on patients with advanced biliary tree and hepatocellular cancer previously treated with no more than one line of chemotherapy. Of 17 BTC patients, 5 patients with GBC did not achieve any improvement with the treatment. In 2019, Harding et al.67 operated another phase II SUMMIT “basket” trial, a multi-histology, open-label, phase II “basket” study for patients who harbored somatic HER2 mutations and received neratinib (ClinicalTrial.gov NCT01953926; EudraCT 2013-002872-42). A total of 19 BTC patients containing 8 GBCs were recruited for a single-arm trial with neratinib monotherapy at 240 mg oral daily. Calculating all BTC patients as a whole, the confirmed objective response rate (ORR) was 10.5% (95% CI 1.3–33.1) and clinical benefit rate was 31.6% (95% CI 12.6–56.6), including two confirmed partial response (PR) and four patients achieving stable disease. mPFS was 1.8 months (95% CI 1.0–3.7). These data indicate that HER2-directed targeted therapy represents a novel and tolerable treatment approach for advanced BTCs that express somatic HER2 mutations.
We have recently begun a clinical trial named “A Multicenter, Open-label, Randomized, Controlled Study of Target Therapy Based on Tumor Molecular Profiling With GEMOX in Resectable GBC Patients Monitored by ctDNA.” (ClinicalTrial.gov NCT04183712). A total of 54 GBC participants will be enrolled from multiple hospitals and the feasibility, efficacy, and safety of target therapy with afatinib will be evaluated and the outcomes of this study will be reported in 2 years.
VEGF/VEGFR
VEGF/VEGFR axis plays a key role in both physiological and pathological vascularization in diseases such as tumor angiogenesis.68,69,70,71 There are five distinct VEGF family members in a mammal: VEGF-A (also referred to as VEGF), VEGF-B, VEGF-C, VEGF-D, and placenta growth factor (PLGF). VEGFR family consists of RTK member, including VEGFR1, VEGFR2, and VEGFR3, as well as the non-tyrosine kinase co-receptors neuropilin-1 (NP-1) and NP-2. The tyrosine kinase receptors are composed of an extracellular ligand-binding region with distinct binding affinities for individual VEGF ligands, seven immunoglobulin-like loop domains, and a cytoplasmic catalytic domain.72,73 VEGF-A, VEGF-B, and PLGF are mainly involved in angiogenesis, while VEGF-C and VEGF-D regulate lymphangiogenesis. VEGF-A and VEGF-B display the rigorous ability to interact with VEGFR1 and VEGFR2 expressed on vascular endothelial cells and vascular smooth muscle cells.74 VEGF-C and VEGF-D have high affinity to bind to VEGFR3 expressed on lymphoendothelium, stimulating lymphangiogenesis.75
VEGFR1 was expressed on many types of cells, including endothelial cells, epithelial cells, inflammatory cells, and cancer cells. Interestingly, VEGFR1 does not seem to regulate endothelial cell migration or proliferation;76,77 instead, VEGFR1 regulates epithelial cell differentiation and migration.78 In endothelial cells, VEGFR1 is recognized as a decoy to regulate free VEGF-A as it binds to and activates VEGFR2.79 The binding of VEGF to VEGFR2 leads to phosphorylation of VEGFR2 at Tyr951 and Tyr1175, in which phosphorylated Tyr951 regulates vascular permeability mediated by SRC tyrosine kinase activation,80 whereas phosphorylated Tyr1175 of VEGFR2 recruits phospholipase Cγ (PLC-γ) and activates downstream of both MAPK cascade and PI3K/AKT pathway, stimulating endothelial cell proliferation and survival.72,81 Activated VEGFR3 induces the RAS/MAPK/ERK pathway and the PI3K-AKT/PKB pathway, leading to increased differentiation, migration, proliferation, and survival of lymphatic endothelial cells.82,83 Intriguingly, VEGFR3 was also documented to fenestrate VEGF-A/VEGFR2 signaling, participating in angiogenesis.84
In addition to vast research evidence revealing the regulatory role of the VEGF–VEGFR axis, the cooperative or independent impacts of the axis-associated molecules in GBC have also been concomitantly explored. For example, HIF‑1α was found to promote tumor cell migration via upregulation of VEGF-A in GBC; this effect was inhibited by metformin.85 Tumor necrosis factor-α (TNF-α)-induced ERK1/2-AP-1 pathway-dependent transcriptional activation of VEGF-D, leading to lymphangiogenesis and lymphatic metastasis of GBC.86 Likewise, receptor-interacting protein 1, a multifunctional protein in the TNF-α signaling pathway, was highly expressed in GBC and promoted lymphangiogenesis and lymph node metastasis via nuclear factor-κB-mediated transcriptional activation of VEGF-C.87 Dual-specificity MAP kinase phosphatase1 (DUSP1/MKP1) suppressed VEGF expression, abrogating angiogenesis in GBC mouse model.88 In addition, overexpression of miR-1 in GBC cells inhibited VEGF-A mRNA expression.89
It is well appreciated that microvessel density correlated with cancer progression, metastasis, and prognosis in GBC90 and VEGF-A was overexpressed to serve as an independent prognostic factor of survival in GBC.91 Recently, Xu et al.92 found that VEGF was notably elevated in the serum of patients with GBC and VEGF promoted angiogenesis, cell proliferation, and invasion, but inhibited apoptosis in GBC cells. Inconsistent with the data, Zhang et al.93 reported that the expression of estrogen receptor 1 (ER1) or VEGF-A alone was not correlated with OS of GBC patients. However, combined high expression of VEGF-A with ER1 predicted poor prognosis for GBC patients, suggesting that VEGF-A combined with hormone receptor ER may provide a biomarker for GBC prognosis. Resembling tumor angiogenesis, lymphoangiogenesis was known to play a central role in GBC metastasis. In a case–control study with 50 patients of GBC, 10 samples of normal gallbladder tissues and 19 samples of chronic cholecystitis, VEGF-C and -D were overexpressed in GBC tissues relative to normal or inflammation tissues.94 Strongly supporting this evidence, a number of independent studies have found that serum and tumor VEGF-C levels were increased in patients with GBC compared with healthy donors. In addition, the elevated serum VEGF-C was positively correlated with decreased OS and increased lymph node metastasis.95,96 In a large cohort study with 195 GBC patients and 300 healthy serum samples, polymorphisms of c.*237C > T and g.43737830A > G of VEGF gene were associated with the disease development, indicating that VEGF polymorphisms may offer a valuable marker to predict the susceptibility of carcinogenesis.97 In line with clinic trials, blocking VEGF-C by short interfering RNA or a neutralizing antibody in cultured GBC cells inhibited tumor cell proliferation and invasion. Indeed, using orthotopic xenograft models, Lin et al.98 found that inhibition of VEGF-D led to the suppression of lymphangiogenesis and lymphatic metastasis.
As a variety of anti-angiogenic inhibitors, including antibodies and small molecules, were frequently engaged in multiple cancer patients, these inhibitors have also been increasingly employed in the clinical practice for GBC patients. A multicentric phase II study of VEGF antibody bevacizumab (NCT00361231) in combination with gemcitabine and oxaliplatin in advanced BTC with a single-arm trial demonstrated that response rate was 40% and mPFS was 7 months, and OS was 12.7 months.99 A similar single-arm phase II trial (NCT00356889) of bevacizumab in combination with erlotinib but no traditional cytotoxic drugs in patients with unresectable BTC demonstrated a response rate of 18.4%, mOS of 9.9 months, and time to progression (TTP) of 4.4 months.100 In addition, atrial of multicenter phase II study (NCT01007552) of bevacizumab in combination with GC in advanced BTCs reported a 24% PR, 8.1 months of median PFS (mPFS), and 10.2 months of mOS.101A phase II study (NCT02053376) suggested promising efficacy of regorafenib (inhibitor of VEGFR1-3) in chemotherapy-refractory advanced/metastatic BTC, which demonstrated that mPFS was 15.6 weeks, mOS was 31.8 weeks, PR was 11%, and stable disease was 44% with a disease control rate of 56%. During the courses of these clinical trials, adverse reactions included hypertension (23%), hyperbilirubinemia (26%), hypophosphatemia (40%), and hand–foot skin reaction (7%).102 Some of trial failure events were also documented. Sorafenib, a multi-kinase inhibitor of VEGFR2/3, B-Raf, PDGFR-β, and C-Raf, showed a minimal level of drug efficacy in advanced BTC in a non-randomized phase II clinical trial with an ORR of 2%, the rate of stable disease at 12 weeks of 32.6%, PFS of 2.3 months (range: 0–12 months), and a mOS of 4.4 months (range: 0–22 months).103 Agreeing with this study, a multicenter, multinational phase II study (NCT01082809) revealed that sunitinib (inhibitor of multiple RTKs including VEGFR) monotherapy showed marginal efficacy in metastatic BTC patients. The median TTP was 1.7 months, the ORR was 8.9%, and the disease control rate was 50.0%.104 VEGFR2 antagonist vandetanib monotherapy or chemo combinations did not yield noticeable benefits in PFS in advanced BTC in a phase II trial (NCT00753675).105 Furthermore, a phase I trial (NCT02443324) of ramucirumab, a fully humanized monoclonal VEGFR2-targeted IgG antibody, reported that OR rate was 4%, and mPFS and mOS were 1.6 months and 6.4 months, respectively, in advanced BTC.106 A similar new phase II study of ramucirumab in patients with advanced BTC is presently ongoing (NCT02520141). Although the discrepancy from these individual clinical studies on angiogenic blockade remains to be mechanistically deciphered, a number of potential factors may be taken into account. First, monoclonal antibodies (e.g., bevacizumab) usually have higher specificity to bind to single proteins compared to small-molecule inhibitors that display a broader binding ability to block multiple proteins/kinases, likely eliciting off-target reactions. Second, some of the individual drug resistance cannot be neglected, as acquired drug resistance may rapidly develop in some patients who were not timely evaluated for the dynamic alterations of targeted receptors/molecules expression during the therapy. In addition, the divergent expression levels of VEGF/VEGFR and/or polymorphisms in GBC should be evaluated with respect to susceptibility to specific blockers. Finally, substantially longitudinal, case–control studies with large cohorts may be essential to establish the benefits of anti-angiogenic blockers. Nevertheless, at present, the combination regimen with angiogenic-targeted antibodies and other chemotherapeutic agents offers the optimal means to treat GBC.
EGFR
EGFR, also commonly known as ErbB1 or HER1, was discovered to be associated with cancer development when v-ErbB oncogene of the avian erythroblastosis virus was observed in transformed chicken cells.107 Like HER2, EGFR activation triggers multiple intracellular downstream signaling cascades, including ERK/MAPK, PI3K-AKT, SRC, PLC-γ1-PKC, JNK, and JAK-STAT pathways,108,109 thus mediating cancer proliferation, angiogenesis, cell motility, adhesion, and metastasis.110,111 Elevated expression levels of EGFR were identified in non-small cell lung cancer (NSCLC) and malignant gliomas.112,113 In addition, constitutively activated mutations of EGFR existed in multiple cancers,114,115 thereby EGFR serves as a diagnostic and prognostic cancer biomarker, and also a potential target for cancer treatment.116,117,118
Identical to the earlier description regarding VEGF-targeted drugs, drugs that were developed to target EGFR mainly involve humanized monoclonal antibodies against the EGFR extracellular domain and small molecules of tyrosine kinase inhibitors (TKIs). Typical antibodies are cetuximab and panitumumab that are able to prevent EGFR from activated dimerization, thus inhibiting the downstream signaling.119,120 TKIs primarily include gefitinib, erlotinib, and afatinib, which have the ability to bind to the ATP-binding pockets on the intracellular catalytic kinase domain of RTKs, committing to the disruption of downstream signaling.121 To date, three generations of EGFR-TKI drugs have been evolutionarily devised to fight against mutation activity of EGFR.122,123,124 Erlotinib and gefitinib, representing the first generation of these TKIs, have the ability to compete reversibly with ATP binding at the tyrosine kinase domain of EGFR. As point mutation of EGFR T790M developed during the treatment of NSCLC patients with the first-generation EGFR TKIs,125,126,127 drug resistance emerged. To improve the efficacy, the second-generation EGFR TKIs such as afatinib and dacomitinib were created, which irreversibly inhibit ATP binding at the tyrosine kinase domain.128 In some of the clinical trials with afatinib, the drug failed to reach a level by which it effectively abolishes activity of T790M mutant EGFR.129,130 Subsequently, the third-generation TKIs osimertinib and olmutinib were formulated.124 Both drugs exhibited robust inhibition on the mutation activities of EGFR, as a favorable responses to osimertinib and olmutinib were achieved in 50–60% of patients with the T790M mutations.131,132 Thus, these two drugs have been approved as the second-line treatment drugs of patients who resist to the first-generation EGFR TKIs.
The overexpression population of EGFR in GBC was observed between 44 and 77% of patients in different independent studies.133,134,135 Elevated expression of EGFR in GBC tissues was positively correlated with poor prognosis of the patients.136 We also found that somatic mutations of EGFR were quite low ranging from 2.5 to 3.9% in GBC patients.58,59
A variety of therapeutic trials targeting EGFR in GBC patients have been completed, but endpoints were varied. Mody et al.137 reported a metastatic GBC case who received a combination treatment with gemcitabine (1000 mg/m2) on days 1 and 8 every 21 days and daily erlotinib (100 mg). The disease remained for 18 months with no progression after 12 cycles of combination therapy followed by maintenance with only erlotinib for 6 months. This result indicated the potential effective responses of EGFR-TKI on GBC therapy. Philip et al.138 reported a phase II study of erlotinib in 42 patients with advanced biliary cancer, in which 16 cases were GBCs. The overall confirmed response rate was 8% (3 patients; 95% CI 2–20) and the median TTP was 2.6 months (95% CI 2–4 months), while EGFR level was not associated significantly with clinical outcome. Lubner et al.100 published results of a multicenter phase II trial testing a combination of bevacizumab with erlotinib in 53 patients with unresectable biliary cancer (ClinicalTrial.gov NCT00356889). Twelve percent of patients (95% CI 6–27) had a confirmed PR. mOS was 9.9 months and TTP was 4.4 months. El-Khoueiry et al.139 reported ineffective outcomes in the phase II SWOG study on sorafenib and erlotinib in patients with 14 advanced GBCs and 20 cholangiocarcinomas. The combination of the two drugs resulted in two unconfirmed PRs (6%, 95% CI 1–20) with a mPFS of 2 months (95% CI 2–3), and a mOS of 6 months (95% CI 3–8 months) in the single-arm study. Cai et al.140 employed a meta-analysis to evaluate a combination therapy of EGFR-targeted drugs (erlotinib, cetuximab, or panitumumab) with GEMOX (gemcitabine and oxaliplatin) in 612 BTCs. The combination of GEMOX and EGFR-targeted therapy demonstrated improved PFS (HR 0.80, 95% CI 0.66–0.94, P = 0.03) compared with GEMOX alone, although OS was not significantly different. They also found that cholangiocarcinoma exhibited significantly greater benefits with 44% reduction from targeted therapy than non-cholangiocarcinoma including GBC and ampulla of Vater carcinomas that showed only 6% reduction in cholangiocarcinoma risk. Nonetheless, more clinical settings with combined regimens targeting EGFR are essential to offer a powerful tool for clinical practice.
MAPK (RAS/RAF /MEK/ERK) pathway
MAPK signaling pathway is a crucial intracellular signal transduction that regulates varied cellular activities and is frequently dysfunctional in cancer.141 RAS/RAF/MEK/ERK is the most common pathway in MAPK signaling by which a variety of cancerous cells promote cell proliferation, death, differential, cell cycle progression, apoptosis, survival, metastasis, metabolism, and angiogenesis.142,143,144 A number of membrane receptors and intracellular proteins are directly or indirectly able to activate RAS, which includes K‐Ras, H‐Ras, and N‐Ras family members.145 Once the RAS protein is activated, it recruits RAF kinase family members such as Araf, Braf, or Craf to the plasma membrane and elicits downstream MEK.146,147 MEK has dual-specific serine/threonine and tyrosine kinase activity and share 80% sequence homology in MEK1 and MEK2.148 Activated MEK1/2 by RAFs induce phosphorylation of ERK1 or ERK2; then, pERK1 or pERK2 dimerizes and translocates to the nucleus, where it activates transcription factors to regulate gene expression.149,150 Approximately 40% of all human cancers involve altered MAPK pathway, including mutations of BRAF (~10%) and RAS (~30%).151 In GBC, KRAS point mutations were detected between 0 and 41% and BRAF gene amplifications existed in 5% patients.152,153,154,155,156,157
There was ample convincing evidence demonstrating that KRAS mutations mediate carcinogenesis of BTC by multiple research groups.156,158,159,160 Gln25His polymorphism of KRAS gene was identified to connect with GBC pathogenesis.161 Consistent with this result, KRAS rs61764370 polymorphism was intimately associated with risk and prognosis of cancers in 307 healthy controls and 541 GBCs in Indians.162 With regard to BRAF mutation frequency in GBC, it is still inconsistent, as no BRAF mutations were reported in the United States and Chile patients, but 33% of patients with BRAF mutations were present in Europe.154,158 However, the role of MEK/ERK in GBC has been relatively confirmed. MEK1/2 inhibitor trametinib inhibited GBC cells’ proliferation, migration, and invasion in a dose- and time-dependent manner, and induced GBC cell apoptosis in vivo and in vitro.163 Likewise, Horiuchi et al.164 demonstrated that MEK inhibitor U0126 abolished tumor liver invasion and increased survival of nude mice bearing human GBC cells.16 In addition, some traditional Chinese medicines, such as bufalin, pachymic acid, and artemisinin, had the ability to block GBC cell proliferation and invasion via interrupting MEK/ERK signaling.165,166 MiR-663a impaired MAPK/ERK pathway via altered regulation of EMP3 to suppress GBC progression.167 In contrast, lncRNA MALAT1, SLC25A22, and miR-101 augmented GBC cell proliferation through activating the MAPK/ERK pathway, leading to metastasis.168,169,170
A number of divergent therapeutic approaches in treatment with MAPK pathway blockers have been employed to unveil the potential targets for GBC patients. Giannini et al.171 reported that two cases yielded better therapeutic efficacy with PFS6months after receiving BRAF/MEK inhibitors treated for secondary GBC. Consistent with the report, Yu et al.172 also published an advanced secondary GBC case with a successful combination therapy with BRAF and MEK inhibitors after surgical excision, as there was no evidence of metastasis with PFS for 14 months and OS for 26 months after 8 months of treatment. Interestingly, a phase II study of GEMOX in combination with EGFR inhibitor cetuximab declared that KRAS mutations did not affect the difference in ORR and PFS between GEMOX and combination with EGFR inhibitor (NCT01308840, NCT01389414) in GBC,173,174 suggesting that the addition of cetuximab to gemcitabine and oxaliplatin did not seem to enhance the activity of chemotherapy in patients with GBC. In a phase II study of trametinib (GSK1120212, JTP-74057), the first generation of MEK1/2 inhibitor approved by Food and Drug Administration,175 trametinib showed safer and more effective drug responses than single gemcitabine treatment. Trametinib led to 10.6 months of PFS (95% CI 4.6–12.1), 20.0% of 1-year OS, 65% of stable disease, and 35% of PD in 20 Japanese patients with advanced BTC refractory to gemcitabine‐based therapy (NCT01943864).176 Similar results were obtained in SWOG S1310 study that recruited 44 patients (32% GBC patients) for trametinib treatment. The ORR of trametinib therapy was 10% (95% CI 0–23) vs. 8% (95% CI 0–19) seen in fluoropyrimidine therapy and the mPFS in trametinib therapy was 3.3 months in contrast to 1.4 months in fluoropyrimidine therapy.177 Some drug candidates with notable on-target adverse and toxicities events were limited to extensive clinical trials, including CI-1040 and PD0325901.178,179 Selumetinib (AZD6244, ARRY-142886), a second generation of MEK1/2 drug, was developed for the selective and uncompetitive small-molecule inhibitor of MEK1/2.180 A recent multi-institutional phase II study (NCT00553332) of selumetinib demonstrated that the drug response is of acceptable tolerability in patients with metastatic BTC, and that mPFS and mOS in selumetinib-treated cases were 3.7 months (95% CI 3.5–4.9 months) and 9.8 months (95% CI 5.97–not available), respectively.181
PI3K/AKT/mTOR pathway
PI3K/AKT/mTOR signaling pathway is well known to participate in various biological and physiological cellular processes, including cell growth, mobility, differentiation, metabolic activity, and apoptosis.182,183,184 A wealth of oncogenic research evidence has established the notion that PI3K/AKT/mTOR pathway is one of the most key signaling pathways remarkably upregulated in a broad spectrum of cancers that involve breast cancer,185 NSCLC,186 gastric cancer,187 hepatocellular carcinoma,188 colorectal cancer,189 pancreatic cancer,190 cholangiocarcinoma,191 and GBC.192 Transmembrane growth factor receptors VEGFR, EGFR, insulin growth factor receptor 1, G protein-coupled receptors, and RAS proteins are capable of activating PI3K that phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 subsequently binds to phosphoinositide-dependent kinase 1 (PDK1), thus phosphorylating and activating the serine/threonine kinase AKT.193,194 PTEN (phosphatase and tensin homolog), a tumor suppressor, is a phosphatase that dephosphorylates PIP3 into inactive PIP2, thereby dampening AKT and PDK1.195,196 Activated AKT can phosphorylate and activate mTOR or indirectly promote mTOR activity by phosphorylation and inactivation of tuberous sclerosis complex 1/2, a mTOR inhibitor. mTOR exists in two distinct complexes: mTOR complex 1 (mTORC1), which is composed of mTOR, Raptor, mLST8, and PRAS40; mTOR complex 2 (mTORC2) that consist of mTOR, Rictor, Sin1, and mLST8. mTORC1 activates S6 kinase 1 (also known as p70SK6) and promotes dissociation of eukaryotic initiation factor 4E binding protein 1 from eIF4E, stimulating cell growth and protein synthesis.197,198 As relatively limited understanding is available of mTORC1, the mTORC2 complex, insensitive to rapamycin, regulates actin cytoskeleton activity and controls AKT phosphorylation at Ser 473.199,200
Like other types of cancers that revealed important pathological impacts of the PI3K/AKT/mTOR pathway in cancer growth and metastases, mechanistic insights of GBC pathogenesis have advanced our knowledge that this pathway predominantly contributes to the initiation and progression of GBC. Lunardi et al.201 found that 90% of Pten+/− mice with a high level of phosphorylated AKT developed GBC, highlighting an active role of PI3K/AKT signaling in the transformation of gallbladder epithelial cells. Leal et al.202 demonstrated that phospho-mTOR was positive in 64.1% of GBC patients and in 24% of chronic cholecystitis cases and that a high phospho-mTOR level in immunohistochemical analyses predicted poorer prognosis in patients with advanced GBC. PI3KCA mutations in GBC were differently reported in individual studies in the world. While no PIK3CA mutations were found in Brazil patients, 12.5%, 16.9%, and 21.4% of GBC with PIK3CA mutations were identified in USA, Japan, and Chile, respectively.154,203,204,205 We previously found that PIK3CA mutations E545K occurred in ~5.9% of GBC, and that these patients exhibited a worse prognosis.58,206 Epigenetic alteration of PTEN also contributes to the development of GBC as 30% GBCs exhibited PTEN promoter hypermethylated.207
Accumulating research evidence, not limited to PI3K, AKT, and mTOR, has also unveiled tumor-promoting function of important molecules that manipulate activation of the PI3K/AKT/mTOR pathway. For instance, M2 macrophages secrete CCL18 to promote GBC cell migration and invasion via activating PI3K/AKT pathway.208 EIF3D stabilizes GRK2 kinase that activates PI3K/AKT signaling pathway, rendering GBC cells invasive.209 Exploiting the same pathway, Nectin-4 and STYK1 promote GBC cell proliferation, metastasis, and tumor growth.210,211 In addition, ubiquitin protein ligase E3 component N-recognin 5 decreases the degradation of PTEN/PI3K/AKT signal pathway, facilitating tumor growth.212 Given the growing evidence that lncRNAs emerge to regulate gene expression, a number of laboratories have paid considerable attention in the identification of novel lncRNAs that prompt GBC progression. We recently discovered that lncRNA-HGBC stabilized by HuR promotes GBC cell proliferation, migration, and invasion by regulating miR-502-3p/SET/AKT axis.32 Analogous to our findings, PABPC1-stabilized lncRNA-PAGBC acted as a miRNA sponge to activate the PI3K/AKT/mTOR pathway, committing to tumor growth and metastasis.33 Specificity protein 1-induced lncRNA LINC00152 up-regulates PI3K/AKT pathway and contributes to GBC cell growth and tumor metastasis.213 Jin et al.214 demonstrated that miR-143-3p targets ITGA6 to inhibit PI3K/AKT pathway, thus suppressing GBC growth and angiogenesis. Overall, the identification of a great number of key factors has pointed to novel targets potential for therapy in GBC. Several inhibitors targeting the PI3K/AKT/mTOR pathway, including A66, Wortmannin, and LY294002, have been demonstrated to inhibit GBC cell proliferation, migration, and invasion both in vitro and in vivo.215,216,217 In addition, rapamycin, RAD001, and AZD8055 reported by Leal et al.218 were able to block mTOR and inhibit the growth and migration of GBC cells in vitro. In a transgenic mouse model, rapamycin can also inhibit the incidence of GBC.219 OSI-027 blocked mTOR, enhancing the sensitivity of GBC cells to 5-fluorouracil.220 Some of the traditional Chinese medicines, such as bufalin, liensinine, and dioscin, also received striking attention capable of inhibiting GBC cell proliferation and inducing cell apoptosis via targeting the PI3K/AKT pathway.166,221,222
In the clinic, a phase I trial (NCT00949949) was performed to determine the maximum tolerated dose (MTD) of mTOR inhibitor everolimus (5 mg) combined with either gemcitabine (800 mg/m2, Cohort I) or gemcitabine plus cisplatin (12.5 mg/m2, Cohort II) in cancers. In this clinical setting, 10 patients with cholangiocarcinoma or GBCs were abstracted for the Cohort III trial treated with three drugs. The results showed that six patients remained stable, whereas four developed a progressive stage, suggesting that the three-drug combination course may offer favorable benefit to GBC.223 A multi-institutional phase II study of MK-2206 (NCT01425879) demonstrated that MK-2206, a single-agent targeting AKT, exhibited acceptable tolerability in eight patients with advanced, refractory BC.224 Regarding the multiple drug candidates available from the preclinical trials described early, it is worthwhile interrogating if some of these agents function to block the PI3K/AKT/mTOR pathway in GBC patient, thereof offering additional opportunities for patient treatment.225
PD-1/PD-L1
In the recent few years, intense research evidence focusing on antitumor immunity has established the proof of concept that blockade of the interaction between tumor-derived checkpoint ligands and their corresponding binding receptors expressed by T cells elicits T cell immunity against tumors.106,226 PD-L1, also known as B7-H1 or CD274, is a membrane-associated protein and specifically binds to PD-1 expressed on T cells.227 Interaction of these immune checkpoint proteins leads to disruption of major histocompatibility complex coupled with T cell receptors, the identical interaction of membrane proteins by which cytotoxic T cells recognize and eliminate tumor cells (Fig. 3). While the aberrant expression of PD-L1 in multiple cancers enables tumor cells to escape the host immune surveillance and drive tumor metastasis, little is known regarding mechanistic regulation of PD-L1 and PD-1 underlying GBC development. We are the first group, to our knowledge, to report that ectopic expression of ErbB2/ErbB3 mutants in GBC cells upregulated PD-L1 expression, which suppressed T cell-mediated cytotoxicity and drove tumor growth and metastasis.228 Mechanistically, ErbB2/ErbB3-mediated expression of PD-L1 was dependent on activation of the PI3K/Akt signaling pathway. The results demonstrated that acquired ERBB2/ERBB3 mutations by tumor cells are essential to induce checkpoint PD-L1, rendering tumor cells evasive from cytotoxic T cell immunity against tumor. Agreeing with our findings, Gong et al.229 found that PLAC8 increased PD-L1 expression and conferred tumor resistance to gemcitabine and oxaliplatin, providing an alternative opportunity with PD-1/PD-L1 blockers to treat chemotherapy-refractory patients with GBC.

Immunoregulation of PD-L1/PD-1 in GBC. a Activated ERBB2/ERBB3 mutations upregulate PD-L1 expression through activation of the PI3K/AKT signaling and RAS/RAF/MEK/ERK pathway to induce immune evasion of GBC cells. b Yellow boxes highlight that drug targeted ERBB2, PD-1, or PD-L1 could dampen immune evasion of GBC cells
There were accumulating literature reports analyzing the relationship between expression levels of PD-L1 and/or PD-1 and clinical outcomes. Elevated PD-L1/PD-1 levels were correlated with poor survival of cancer patients with NSCLC,230 melanoma,231 gastric cancer,232 adrenocortical carcinoma,233 breast cancer,234 hepatic cancer,235 and pancreatic cancer.236,237 Specifically, PD‐L1 expression in GBC (23%) was comparable to breast cancer (23%), urothelial cancer (20%), and pulmonary squamous cell carcinoma (27%).238,239 Mody et al.240 found that 12% patients with tumor cell-expressing PD-L1 and 55% patients with tumor-infiltrating lymphocyte (TIL)-expressing PD-1 in a total of 203 GBC patients were associated with corresponding genetic aberrations and tumor mutational burden (TMB) status. Likewise, Lin et al.241 showed that 18% of 66 GBC patients were positive for PD-L1 expressed by tumor cells. Ha et al.242 measured the serum level of the soluble form of PD-L1 (sPD-L1) in 158 advanced BTC patients, and found that patients with high sPD-L1 (≥0.94 ng/mL) showed decreased OS than patients with low sPD-L1 (7.93 vs. 14.10 months, p < 0.001). Kim et al.243 observed that the mOS of low and high PD-L1 expression of 101 primary GBC cases was 50.13 ± 3.14 and 27.88 ± 6.69 months (P value = 0.049), respectively, and the mPFS was 49.48 ± 3.29 and 23.33 ± 7.47 months (P value = 0.028), respectively. These individual studies underscore that PD-L1 serves as an independent marker for the prognosis of GBC. Not surprisingly, there was a limited observation that did not fully support these earlier findings. Neyaz et al.239 recruited 174 cases of GBC and found that mOS in PD‐L1‐negative (PD-L1−) and PD‐L1-positive (PD-L1+) cases were 12.0 and 14.0 months, respectively (P = 0.546). Another study showed that PD-1+ in TILs and PD-L1+ in tumor cells did not correlate with OS or PFS, but a high density of CD8+ TILs in PD-L1− tumors was positively correlated with OS (P = 0.002) and PFS (P = 0.014), indicating that CD8+ TILs is an additional marker for disease prognosis, except PD-L1 and PD-1. Although we do not have sufficient knowledge to explain the inconsistency, the research focusing on tumor immune checkpoint proteins has opened a new era to explore their potential roles in mediating tumor ability to escape immune surveillance.
Treatment of GBC patients with PD-1/PD-L1 inhibitors has emerged as a promising strategy for targeted therapies (Table 2).244,245 TMB-H and MSI-H are potentially useful for assessing neoantigen presentation and viability of immune checkpoint inhibition.246,247 Weinberg et al.248 reported that PD-L1 overexpression was seen on GBC tumor cells from 19 of 237 (8.0%) tumors using IHC. Increased MSI-H and TMB-H were, respectively, seen in 1 out of 104 tumors (1.0%) and 6 out of 104 tumors (5.8%) using NGS. All of these results suggested that PD-L1 in GBCs is a therapeutic marker for immune checkpoint blockade (12%). In 2019, Kong et al.249 reported a successful immune therapy case that expressed strong PD-L1 expression (≥50%) and gave rise to substantial responses to nivolumab immunotherapy after the failure of multiple lines of therapies. Indeed, multiple clinical studies have demonstrated the efficacy of PD-L1/PD-1 inhibitors in the treatment of GBC. In a multicenter, case-controlled phase I trial named MakotoUeno (ID: JapicCTI-153098), 30 unresectable and recurrent BTC patients were enrolled into two cohort trials: patients received nivolumab only (240 mg every 2 weeks) as the monotherapy; patients received nivolumab (240 mg every 2 weeks) and cisplatin (25 mg/m2) plus gemcitabine (1000 mg/m2) as combined regimens.250 The results showed that the mOS was 5.2 months (90% CI 4.5–8.7) vs. 15.4 months (90% CI 11.8–not estimable), and mPFS was 1.4 months (90% CI 1.4–1.4) vs. 4.2 months (90% CI 2.8–5.6), respectively, for the first and second cohort trials. In a single-arm phase II study of nivolumab in BTC patients (NCT02829918), the mPFS was 3.68 months (95% CI: 2.33–5.98) and the mOS was 14.24 months (95% CI 6.64–NA).251 In addition, 6- and 12-month OS was obtained in 71.4% and 52.3% populations and 6- and 12-month PFS was seen in 35.2% and 24.1%, respectively. These results indicated that nivolumab is a manageable and effective agent for BTC patients. In the KEYNOTE-028 (NCT02054806; phase I) and KEYNOTE-158 (NCT02628067; phase II) study, BTC patients received pembrolizumab treatment.252,253 In KEYNOTE-028 study, objective response rate (ORR) was 13.0% (3/23, all PR; 95% CI 2.8‒33.6) and median duration of response (DOR) was not reached (NR; range 21.5–29.4+ month). mOS and mPFS were 6.2 months (95% CI 3.8‒10.3) and 1.8 months (95% CI 1.4‒3.7), respectively. Furthermore, ORR of PD‐L1+ patients (n = 61) and PD‐L1− patients (n = 34) was 6.6% (4/61) and 2.9% (1/34), respectively. In KEYNOTE-158, ORR was 5.8% (6/104, all PR; 95% CI 2.1–12.1) and median DOR was NR (range 6.2–23.2+ months). mOS and mPFS were 7.4 months (95% CI 5.5–9.6) and 2.0 months (95% CI 1.9–2.1), respectively. The results showed that pembrolizumab provides durable antitumor activity, regardless of PD-L1 expression. Unlike KEYNOTE‐028 study, the trail (NCT02443324) recruited both PD-L1+ and PD‐L1− patients, in which 46.2% of PD-L1 patients underwent tumor recurrence or metastasis. ORR was 4%. mOS and mPFS were 6.4 and 1.6 months, respectively. PD‐L1+ patients had improved OS compared with PD‐L1− cases (11.3 vs. 6.1 months), but there is no difference in mPFS (1.5 vs. 1.6 months).106 All the results suggest that PD-1/PD-L1 inhibitors are safe and effective in treating GBC.
Combined regimens of anti-PD-L1/PD-1 checkpoint agents with other therapies have also been increasingly practiced in clinic, including additional immunotherapies, chemotherapy, and targeted therapies.254,255,256,257,258,259,260 In a phase I study led by Xie’s group (NCT01853618) showed that PFS and OS were 3.4 months (95% CI 2.5–5.2) and 6.0 months (95% CI 3.8–8.8),261 which declared that tremelimumab (anti-CTLA-4 monoclonal antibody (mAb)) is a potential treatment strategy for patients with advanced BTC. A phase I study (NCT01938612) evaluated durvalumab (anti-PD-L1 mAb) with/without tremelimumab in Asian GBC patients. Median DOR was 9.7 and 8.5 months for single durvalumab and durvalumab + tremelimumab treatment, respectively. mOS was 8.1 (95% CI 5.6–10.1) months and 10.1 (95% CI 6.2–11.4) months for the single and dual treatment, respectively, suggesting that conjunction of anti-PD-L1/PD-1 with anti-CTLA-4 therapies may hold promising efficacy for patients with GBC. Thus, comprehensive trials utilizing anti-CTLA-4 antibody combined with anti-PD-L1/PD-1 antibodies in GBC are currently underway, as this approach yielded noticeable benefits to other types of cancer patients. Fujiwara et al.262 employed a new therapeutic tool engineering a bifunctional fusion protein that targets PD-L1 and transforming growth factor-β (M7824) to treat Asian patients with advanced solid tumors (including 40% GBC patients) (NCT02699515). In this trial, three patients discontinued M7824 treatment due to treatment-related adverse events (the cases of bullous pemphigoid, colitis, and gastroparesis). This endogenous interruption targeting both molecules needs to be substantially evaluated in future. While multiple clinic settings with immune checkpoint therapies combined with other therapies wait for outcomes at present, we are fully confident with this novel approach able to yield greater benefits to GBC patients, ultimately improving life quality.
DNA damage repair (DDR) pathway
DDR can execute full repairing or elimination of damaged cells to protect host organisms against possible carcinogenesis.263 There are four major DDR pathways identified in the cells, for example, base excision repair, nucleotide excision repair (NER), double-strand break repair and mismatch repair (MMR).264 As the intracellular events participate in the pathogenesis of GBC, DDR pathways act an active role to contribute to the development of this disease. Fang et al.265 reported that a dual-specific phosphatase DUSP1 enhances the chemoresistance of GBC via the modulation of the p38 pathway and DNA damage/repair system. Suppression of cholesterol biosynthesis by lovastatin could inhibit GBC cell proliferation, possibly through attenuating the DDR process.266
Abdel-Wahab et al.267 have clustered 20 “direct” DDR genes (ATM, ATR, BRCA1, BRCA2, FANCA, FANCD2, MLH1, MSH2, MSH6, PALB2, POLD1, POLE, PRKDC, RAD50, SLX4) and “caretaker” genes that regulate DDR process (BAP1, CDK12, MLL3, TP53, BLM) in BTC (270 ICC, 60 ECC, and 92 GBC specimens) by hybrid capture-based NGS. BRCA-associated BTC is uncommon as BRCA1/2 mutations were detected in 4.0% of 353 GBC samples by Spizzo. However, they found a correlation of BRCA-mutant BTC with MSI-H/dMMR, which represented an additional predictive marker for response to checkpoint inhibition.268 Consistent with these reports, Javle et al.269 identified 7.8% BRCA2 or ATM mutations in 623 advanced GBC patients.
The alterations of DDR genes increase the sensitivity of anti-cancer chemotherapy and radiation treatments. The recent researches suggest that specific DDR gene mutation or expression may have an impact on response to platinum-based chemotherapy in patients diagnosed with BTC.270 Hwang et al.271 evaluated the effect of ERCC1 on treatment outcomes in advanced BTC patients treated with platinum-based chemotherapy, which is the 5′ endonuclease of the NER complex to prevent damage to DNA by NER. They found that mPFS and mOS were significantly longer in ERCC1− group than in ERCC1+ group of cisplatin-treated group (4.6 vs. 1.9 months, P = 0.014; 9.1 vs. 7.9 months, P = 0.017). Baek-Yeol Ryoo et al.272 revealed that DDR gene mutations were found in 62.5% of BTC patients (including 20.2% GBC patients), and that DDR gene mutations associated with longer mPFS (6.9 vs. 5.7 months; P = 0.013) and mOS (21.0 vs. 13.3 months; P = 0.009) in patients with BTC treated with first-line platinum-based chemotherapy for unresectable or metastatic disease. These results indicate that mutations in DDR genes may serve as predictive biomarkers for the response to platinum-based chemotherapy in patients with BTC.
To date, clinical trials clarifying the efficacy of DDR inhibitors in GBC patients have not been reported, but poly (ADP-ribose) polymerase (PARP) inhibitors have been developed in monotherapy that is engaged for patients with HR deficiency (i.e., BRCA mutation) and also in BRCA-like tumors. At present, a clinical trial (NCT03878095) targeting PARP is waiting for final results.
C-mesenchymal–epithelial transition factor (MET)
MET is an oncogene encoding tyrosine kinase receptor of the hepatocyte growth factor (HGF). Once HGF binds to MET, the receptor undergoes dimerization and induces downstream signaling pathways, such as PI3K/AKT, RAS/RAF/MEK/ERK, and Wnt/β-catenin signaling,273,274,275 which regulate cell proliferation, metastasis, and drug resistance. Elevated MET was associated with poor prognosis in liver cancer, breast cancer, pancreatic cancer, gastric cancer, NSCLC, cervical cancer, and colorectal cancer.79,276,277,278,279,280,281,282,283,284,285 In GBC, MET overexpression ranged from 5 to 74% of patients, and was also associated with clinical poor outcome.286,287,288,289,290 The conclusion was further supported by different groups that found a similar correlation.288 In addition, NK4, an HGF inhibitor, inhibited tumor growth and invasion of GBC in animal models.291,292,293,294 Inconsistent with these findings, Kim et al.295 reported dissimilar results that no such correlation between MET expression and poor prognosis was significantly obtained. Up to date, there are three categories of MET inhibitors available in the clinic: small molecules targeting MET receptors (e.g., crizotinib, tivantinib, savolitinib, tepotinib, cabozantinib, and foretinib), MET receptor mAbs (e.g., onartuzumab), and antibodies against its ligand HGF (e.g., ficlatuzumab and rilotumumab).296,297,298,299 A clinical trial (NCT03027284) is currently settled to evaluate the feasibility and efficacy of MET inhibitors in the treatment of GBC.
TP53
TP53 is an important tumor suppressor gene and its mutations are commonly detected in 50% of almost all human cancers, such as colon cancer, pancreatic cancer, breast cancer, and hepatobiliary cancer.TP53 is known to participate in the cellular DNA damage response, and induction of cell cycle arrest and apoptosis.300,301 Magnolol, an organic compound derived from Chinese traditional medicine, was found to upregulate P53, resulting in interrupting cell cycle progression at theG0/G1 phase and inducing mitochondrial-mediated apoptosis. However, this cell death effect was prevented by pretreatment with a p53 inhibitor pifithrin-a.302 Tian et al.303 also demonstrated that apoptosis-stimulating of p53 protein 2 activated p53 and recruited macrophages via PKC-ɩ/GLI1 pathway, committing to inhibition of metastasis.
It is of note that the expression of p53 in GBC is ethnically related to different race populations. The TP53 mutations in Greek GBC patients were lower than those in Japan and Chile GBC patients. One-third of the north Indian patients with GBC have mutations in exons 5–8 of p53 gene.304 Moreover, different types of TP53 mutations in GBC were defined in Japan, Chile, and Hungary.159,305,306 It is quite interesting to mechanistically understand the genetic variations associated with geographic difference.
Overexpression and high mutation frequency of p53 protein, which has tumor-promoting signature rather than cell apoptotic activity, were correlated with a poor survival of a broad type of cancers, including GBC,307,308,309 thus serving as a cancer biomarker. We previously analyzed GBC mutagenesis and found that TP53 was ranked as the top one in a large spectrum of mutated genes in GBC.58,228 In addition, Singh et al.310 demonstrated that p53 is an independent prognostic factor for the poor prognosis of GBC (P = 0.03; HR: 5.63; 95% CI 1.21–26.26). Supporting this notion, a variety of studies also pointed to the mutated p53 as a prognostic marker of GBC.300,311 P53 expression together with other factors was engaged to predict poorer prognosis of GBC, such as cyclin D1, Ki-67, p16, and MSH2.300,307,312 Interestingly, some divergent evidence was also documented in the literature. One hundred and three (44.8%) of 230 GBC cases expressed mutant p53 protein that was not correlated with clinical parameters such as tumor growth.313 Likewise, Hidalgo Grau et al.314 reported p53 protein nuclear overexpression in 41 GBCs was not associated with poor histological differentiation, gallbladder wall invasion, or patient survival. Although these distinct conclusions are still controversial, mechanistic insights should be in parallel taken into account, as a variety of p53 mutation forms together with multiple phosphorylation sites coexist, implicating dissimilar roles played by p53 in tumor development, such as tumor-promoting effects and loss of tumor suppressor activity.
Although TP53 has the highest mutation rate in GBC, very limited clinical trials targeting mutated p53 were reported to evaluate potential therapeutic benefit. Makower et al.315 led to a phase II clinical trial of oncolytic adenovirus ONYX-015(dl1520, CI-1042), which intervenes the p53 pathway in 19 patients with hepatobiliary tumors, in which 15 cases expressed p53 mutations, and 5 patients had GBCs.316 In this study, 16 patients responded to the viral product in intralesional treatment, while serious toxicities (>grade 2) were rarely observed. The comprehensive analyses of this approach for therapeutic efficacy and safety remain to be clarified.
CDKN2A/B
CDKN2A/B, a cyclin-dependent kinase inhibitor 2A/B, inhibits CDK4 and CDK6, and also prevents pRB phosphorylation, thus leading to cell cycle arrest at the G1/S phase.317,318,319 In an analysis of the comprehensive genomic profiling with NGS, CDKN2A/B gene is one of the most frequently mutated genes in 108 Chinese and 107 US GBC patients. The altered rate with 26% in Chinese patients was identical to 25% seen in US populations.320 More interestingly, the coincidence of ERBB2 genetic mutations with CDKN2A/B variations in US patients was stronger (odds ratio 10.8, P = 0.0001) than those in Chinese cohort (odds ratio 5.4, P = 0.0014), which suggests that CDKN2A/B alterations were significantly associated with distant metastases.321 Our previous study showed that CDKN2A/B mutation rate was ~5.9% in GBC.58,228 There were other reports supporting the notion that CDKN2A/B mutations mediate the pathogenesis of GBC.248,322 Collectively, all the data indicate that CDKN2A/B is a potential target for GBC therapy. It was also noted that Leiting et al.323 found that CDKN2A was not associated with the survival of BTC. Nonetheless, to date, no potential drug candidates have been explored to target CDKN2A/B in cancer treatment.
KIT
KIT is a type III transmembrane RTK expressed in a variety of human cells.324 The downstream signaling of KIT mainly involves MAPK, PI3K/AKT, and JAK/STAT pathways, thus participating in the regulation of cell proliferation, differentiation, growth, survival, and migration.325,326,327 Interestingly, the expression of KIT of patients with gallstones was lower in gallbladder tissue than that in healthy subjects.328 However, in GBC, the expression of KIT was elevated.329 At present, at least 16 KIT inhibitors are available for the blockade of its activity elevated in various types of cancers, including leukemia, prostate cancer, gastrointestinal stromal tumor, and renal carcinoma.330,331,332,333 Typical therapeutic blocking agents employed to target KIT include cabozantinib, dovitinib, masitinib, and pazopanib.334,335,336 At present, two additional clinical trials (NCT01153750, NCT02115542) wait for final results.
Challenges and future perspective
Given that GBC, a high-grade malignant tumor, is mostly of contraindication to surgical removal, great effort has been added to devise the therapeutic strategies and improve the efficacy of non-surgical treatment, including developing new specific agents and therapeutic means, such as targeted drugs, vaccines, and nanoparticles. We are particularly encouraged by the tremendous achievement in a number of recent clinical trials focusing on the efficacy of targeted drugs as described earlier in the text, since the favorable endpoints of these trials are at least, in part, attributed to the rapid development of the state-of-art measures, including NGS, human WES, transcriptomic (RNA array, RNAseq), proteomic, epigenetic, and metabolomic profiling that assist evaluation of genetic signature, drug action mechanisms, and therapeutic responses.337 Therefore, these clinical attempts are anticipated to be of great benefits for the treatment devises. It is concomitantly worthwhile to note that a number of unexpected obstacles coexist and even persist throughout the entire clinical trials, resulting in the elimination of a majority of drug candidates, terminating trials, or leading to testing failure. These typical unfavorable events involve apparent drug side effects or off-target toxicities particularly in the multiple chemotherapeutic drug applications, and decreased responses or no responses due to rapid development of resistance. In addition, other scenarios cannot be ignored, including individual genetic difference, tumor gene mutations, and tumor heterogeneity that otherwise coordinately bypass the targeted therapy. Hence, the therapeutic efficacy of promising drug candidates together with potentially adversary impacts should be warily taken into account. To achieve overall benefits, it is highly recommended to exploit combination targeted therapies aiming at different key pathways underpinning cancer metastasis, which expectedly yield synergistic efficacy with minimal toxicities. Of note, the newly created tumor models transplanted directly with patient-derived tumors such as PDX/PDTX and patient-derived organoids have offered a great opportunity for drug sensitivity and/or resistance screening, as it is of paramount importance for devising sensitive treatment in personalized medicine. In addition, it is also emerging that the tumor immune therapy has paved an exciting novel therapeutic avenue to improve the efficacy of GBC treatment. In context with recent findings of the key roles played by PD-L1 immune checkpoint and other signaling molecules in GBC, we have begun a multicenter clinical phase II trial (NCT03768375) employing an umbrella design, which aims to evaluate the efficacy of different courses of treatment based on genomic and proteomic profiling combined with FORFIRINOX in advanced or recurrent GBC patients (Fig. 4). We expect that these multiple combination targeted settings yield distinct levels of benefits to participants, thus offering great value for future clinical practice, particularly in personalized medicine. To this end, it is urgent to establish a fruitful platform to collaborate globally with divergent research institutions, laboratories, and hospitals, ultimately giving rise to an optimal course to fight against this fatal disease.

Schematic of the clinical trial in our center (NCT03768375). Based on genomic and proteomic profiling of participants, patients received personalized targeted drugs with FOLFRINOX treatment or just FOLFRINOX treatment
References
Acharya, M. R., Patkar, S., Parray, A. & Goel, M. Management of gallbladder cancer in India. Chin. Clin. Oncol. 8, 35 (2019).
Krell, R. W. & Wei, A. C. Gallbladder cancer: surgical management. Chin. Clin. Oncol. 8, 36 (2019).
Thomas, T. S. et al. Advancing age and the risk of bleomycin pulmonary toxicity in a largely older cohort of patients with newly diagnosed Hodgkin Lymphoma. J. Geriatr. Oncol. 11, 69–74 (2020).
Noel, R., Arnelo, U., Lundell, L. & Sandblom, G. Does the frequency of cholecystectomy affect the ensuing incidence of gallbladder cancer in Sweden? A population-based study with a 16-year coverage. World J. Surg. 40, 66–72 (2016).
Hundal, R. & Shaffer, E. A. Gallbladder cancer: epidemiology and outcome. Clin. Epidemiol. 6, 99–109 (2014).
Goetze, T. O. Gallbladder carcinoma: Prognostic factors and therapeutic options. World J. Gastroenterol. 21, 12211–12217 (2015).
Shin, H.-R. et al. Epidemiology of cholangiocarcinoma: an update focusing on risk factors. Cancer Sci. 101, 579–585 (2010).
Larsson, S. C., Håkansson, N. & Wolk, A. Healthy dietary patterns and incidence of biliary tract and gallbladder cancer in a prospective study of women and men. Eur. J. Cancer 70, 42–47 (2017).
Rawla, P., Sunkara, T., Thandra, K. C. & Barsouk, A. Epidemiology of gallbladder cancer. Clin. Exp. Hepatol. 5, 93–102 (2019).
Makiuchi, T. et al. Reproductive factors and gallbladder/bile duct cancer: a population-based cohort study in Japan. Eur. J. Cancer Prev. 26, 292–300 (2017).
Baiu, I. & Visser, B. Gallbladder cancer. JAMA 320, 1294 (2018).
Hickman, L. & Contreras, C. Gallbladder cancer: diagnosis, surgical management, and adjuvant therapies. Surg. Clin. N. Am. 99, 337–355 (2019).
Primrose, J. N. et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): a randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 20, 663–673 (2019).
Ben-Josef, E. et al. SWOG S0809: a Phase II Intergroup Trial of adjuvant capecitabine and gemcitabine followed by radiotherapy and concurrent capecitabine in extrahepatic cholangiocarcinoma and gallbladder carcinoma. J. Clin. Oncol. 33, 2617–2622 (2015).
Valle, J. et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 362, 1273–1281 (2010).
Sinn, M., Wege, H. & Stein, A. Biliary tract cancer: on the way to a personalized therapy. Dtsch. Med. Wochenschr. 145, 442–446 (2020).
Graham, J. S. et al. A phase II study of capecitabine and oxaliplatin combination chemotherapy in patients with inoperable adenocarcinoma of the gall bladder or biliary tract. BMC Res. Notes 9, 161 (2016).
Gunnlaugsson, A., Anderson, H., Lind, P., Glimelius, B. & Johnsson, A. Multicentre phase I–II trial of capecitabine and oxaliplatin in combination with radiotherapy for unresectable pancreatic and biliary tract cancer: The CORGI-U study. Radiother. Oncol. 95, 292–297 (2010).
Sharma, A. et al. Modified gemcitabine and oxaliplatin or gemcitabine + cisplatin in unresectable gallbladder cancer: results of a phase III randomised controlled trial. Eur. J. Cancer 123, 162–170 (2019).
Qin, T.-J. et al. Efficacy and safety of gemcitabine-oxaliplatin combined with huachansu in patients with advanced gallbladder carcinoma. World J. Gastroenterol. 14, 5210–5216 (2008).
Phelip, J.-M. et al. Modified FOLFIRINOX versus CisGem first-line chemotherapy for locally advanced non resectable or metastatic biliary tract cancer (AMEBICA)-PRODIGE 38: Study protocol for a randomized controlled multicenter phase II/III study. Dig. Liver Dis. 51, 318–320 (2019).
Belkouz, A. et al. Efficacy and safety of FOLFIRINOX as salvage treatment in advanced biliary tract cancer: an open-label, single arm, phase 2 trial. Br. J. Cancer 122, 634–639 (2020).
Yoo, C. et al. Multicenter phase II study of oxaliplatin, irinotecan, and S-1 as first-line treatment for patients with recurrent or metastatic biliary tract cancer. Cancer Res. Treat. 50, 1324–1330 (2018).
Hernandez, M. C. et al. Patient-derived xenografts can be reliably generated from patient clinical biopsy specimens. J. Gastrointest. Surg. 23, 818–824 (2019).
Owen, D. J. & Evans, P. R. A structural explanation for the recognition of tyrosine-based endocytotic signals. Science 282, 1327–1332 (1998).
Yang, D. et al. miR-125b-5p enhances chemotherapy sensitivity to cisplatin by down-regulating Bcl2 in gallbladder cancer. Sci. Rep. 7, 43109 (2017).
Lu, W. et al. miR-122 inhibits cancer cell malignancy by targeting PKM2 in gallbladder carcinoma. Tumour Biol. 37, 15615–15625 (2015).
Lu, W. et al. miR-223 increases gallbladder cancer cell sensitivity to docetaxel by downregulating STMN1. Oncotarget 7, 62364–62376 (2016).
Li, M. et al. MiR-31 regulates the cisplatin resistance by targeting Src in gallbladder cancer. Oncotarget 7, 83060–83070 (2016).
Ishigami, K. et al. MicroRNA-31 reflects IL-6 expression in cancer tissue and is related with poor prognosis in bile duct cancer. Carcinogenesis 39, 1127–1134 (2018).
Ye, Y.-Y. et al. MicroRNA-30a-5p inhibits gallbladder cancer cell proliferation, migration and metastasis by targeting E2F7. Cell Death Dis. 9, 410 (2018).
Hu, Y.-P. et al. LncRNA-HGBC stabilized by HuR promotes gallbladder cancer progression by regulating miR-502-3p/SET/AKT axis. Mol. Cancer 18, 167 (2019).
Wu, X.-S. et al. LncRNA-PAGBC acts as a microRNA sponge and promotes gallbladder tumorigenesis. EMBO Rep. 18, 1837–1853 (2017).
Chen, J. et al. Long non-coding RNA PVT1 promotes tumor progression by regulating the miR-143/HK2 axis in gallbladder cancer. Mol. Cancer 18, 33 (2019).
Cai, Q. et al. Long non-coding RNA GBCDRlnc1 induces chemoresistance of gallbladder cancer cells by activating autophagy. Mol. Cancer 18, 82 (2019).
Zhan, M. et al. Guided chemotherapy based on patient-derived mini-xenograft models improves survival of gallbladder carcinoma patients. Cancer Commun. 38, 48 (2018).
Cai, Q. et al. Gallbladder cancer progression is reversed by nanomaterial-induced photothermal therapy in combination with chemotherapy and autophagy inhibition. Int. J. Nanomed. 15, 253–262 (2020).
Ren, B. et al. A meta-analysis of the efficacy of postoperative adjuvant radiotherapy versus no radiotherapy for extrahepatic cholangiocarcinoma and gallbladder carcinoma. Radiat. Oncol. 15, 15 (2020).
Bridgewater, J. A., Goodman, K. A., Kalyan, A. & Mulcahy, M. F. Biliary tract cancer: epidemiology, radiotherapy, and molecular profiling. Am. Soc. Clin. Oncol. Educ. Book 35, e194–e203 (2016).
Brodsky, F. M. Monoclonal antibodies as magic bullets. Pharm. Res. 5, 1–9 (1988).
Lee, Y. T., Tan, Y. J. & Oon, C. E. Molecular targeted therapy: treating cancer with specificity. Eur. J. Pharmacol. 834, 188–196 (2018).
Chung, C. Current targeted therapies in lymphomas. Am. J. Health Syst. Pharm. 76, 1825–1834 (2019).
Oh, D.-Y. & Bang, Y.-J. HER2-targeted therapies—a role beyond breast cancer. Nat. Rev. Clin. Oncol. 17, 33–48 (2020).
Gasser, M. & Waaga-Gasser, A. M. Therapeutic antibodies in cancer therapy. Adv. Exp. Med. Biol. 917, 95–120 (2016).
Wen, Y.-M. & Shi, Y. Immune complex vaccination. Curr. Top. Microbiol. Immunol. 423, 95–118 (2019).
Seebacher, N. A., Stacy, A. E., Porter, G. M. & Merlot, A. M. Clinical development of targeted and immune based anti-cancer therapies. J. Exp. Clin. Cancer Res. 38, 156 (2019).
Riese, D. J. 2nd & Stern, D. F. Specificity within the EGF family/ErbB receptor family signaling network. BioEssays 20, 41–48 (1998).
Vermeulen, Z., Segers, V. F. & De Keulenaer, G. W. ErbB2 signaling at the crossing between heart failure and cancer. Basic Res. Cardiol. 111, 60 (2016).
Tebbutt, N., Pedersen, M. W. & Johns, T. G. Targeting the ERBB family in cancer: couples therapy. Nat. Rev. Cancer 13, 663–673 (2013).
Iqbal, N. & Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol. Biol. Int. 2014, 852748 (2014).
Mishra, R., Patel, H., Alanazi, S., Yuan, L. & Garrett, J. T. HER3 signaling and targeted therapy in cancer. Oncol. Rev. 12, 355 (2018).
Ortega-Cava, C. F. et al. Continuous requirement of ErbB2 kinase activity for loss of cell polarity and lumen formation in a novel ErbB2/Neu-driven murine cell line model of metastatic breast cancer. J. Carcinog. 10, 29 (2011).
Lenferink, A. E., Busse, D., Flanagan, W. M., Yakes, F. M. & Arteaga, C. L. ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res. 61, 6583–6591 (2001).
Yan, M. et al. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 34, 157–164 (2015).
Nagaraja, V. & Eslick, G. D. HER2 expression in gastric and oesophageal cancer: a meta-analytic review. J. Gastrointest. Oncol. 6, 143–154 (2015).
Kiguchi, K. et al. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 61, 6971–6976 (2001).
Roa, I. et al. Overexpression of the HER2/neu gene: a new therapeutic possibility for patients with advanced gallbladder cancer. Gastrointest. Cancer Res. 7, 42–48 (2014).
Li, M. et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 46, 872–876 (2014).
Li, M. et al. Genomic ERBB2/ERBB3 mutations promote PD-L1-mediated immune escape in gallbladder cancer: a whole-exome sequencing analysis. Gut 68, 1024–1033 (2019).
Mondaca, S. et al. Genomic characterization of ERBB2-driven biliary cancer and a case of response to ado-trastuzumab emtansine. JCO Precision Oncol. 3, 1–9 (2019).
Nam, A. R. et al. Therapeutic implication of HER2 in advanced biliary tract cancer. Oncotarget 7, 58007–58021 (2016).
Iyer, P. et al. ERBB2 and KRAS alterations mediate response to EGFR inhibitors in early stage gallbladder cancer. Int. J. Cancer 144, 2008–2019 (2019).
Wang, W. et al. Pretreatment with gemcitabine/5-fluorouracil enhances the cytotoxicity of trastuzumab to HER2-negative human gallbladder cancer cells in vitro and in vivo. Biomed. Res. Int. 2019, 9205851 (2019).
Inagaki, C. et al. Gallbladder cancer harboring ERBB2 mutation on the primary and metastatic site: a case report. World J. Gastrointest. Oncol. 11, 761–767 (2019).
Prieto, M. et al. Long term recurrence free survival in a stage IV gallbladder cancer treated with chemotherapy plus trastuzumab and salvage liver resection. Ann. Hepatobiliary Pancreat. Surg. 23, 403–407 (2019).
Ramanathan, R. K. et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother. Pharmacol. 64, 777–783 (2009).
Harding, J. et al. Treating HER2-mutant advanced biliary tract cancer with neratinib: benefits of HER2-directed targeted therapy in the phase 2 SUMMIT ‘basket’ trial. Ann. Oncol. 30, iv127 (2019).
Costache, M. I. et al. VEGF expression in pancreatic cancer and other malignancies: a review of the literature. Rom. J. Intern. Med. 53, 199–208 (2015).
Yang, J., Yan, J. & Liu, B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front. Immunol. 9, 978 (2018).
Stevens, M. & Oltean, S. Modulation of receptor tyrosine kinase activity through alternative splicing of ligands and receptors in the VEGF-A/VEGFR axis. Cells 8, 288 (2019).
Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 69, 4–10 (2005).
Takahashi, T., Yamaguchi, S., Chida, K. & Shibuya, M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 20, 2768–2778 (2001).
Ivy, S. P., Wick, J. Y. & Kaufman, B. M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 6, 569–579 (2009).
Lee, Y. J. et al. Differential effects of VEGFR-1 and VEGFR-2 inhibition on tumor metastases based on host organ environment. Cancer Res. 70, 8357–8367 (2010).
Terman, B. I. et al. Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene 6, 1677–1683 (1991).
Gille, H. et al. A repressor sequence in the juxtamembrane domain of Flt-1 (VEGFR-1) constitutively inhibits vascular endothelial growth factor-dependent phosphatidylinositol 3'-kinase activation and endothelial cell migration. EMBO J. 19, 4064–4073 (2000).
Ito, N., Wernstedt, C., Engstrom, U. & Claesson-Welsh, L. Identification of vascular endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of SH2 domain-containing molecules. J. Biol. Chem. 273, 23410–23418 (1998).
Takahashi, H. & Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 109, 227–241 (2005).
Xie, Y. H., Chen, Y. X. & Fang, J. Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 5, 22 (2020).
Li, X. et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat. Commun. 7, 11017 (2016).
Ferrara, N., Gerber, H. P. & LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 9, 669–676 (2003).
Cebe-Suarez, S., Zehnder-Fjallman, A. & Ballmer-Hofer, K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell. Mol. Life Sci. 63, 601–615 (2006).
Kiselyov, A., Balakin, K. V. & Tkachenko, S. E. VEGF/VEGFR signalling as a target for inhibiting angiogenesis. Expert Opin. Investig. Drugs 16, 83–107 (2007).
Heinolainen, K. et al. VEGFR3 modulates vascular permeability by controlling VEGF/VEGFR2 signaling. Circ. Res. 120, 1414–1425 (2017).
Ye, J. et al. Metformin suppresses hypoxiainduced migration via the HIF1alpha/VEGF pathway in gallbladder cancer in vitro and in vivo. Oncol. Rep. 40, 3501–3510 (2018).
Hong, H. et al. TNF-alpha promotes lymphangiogenesis and lymphatic metastasis of gallbladder cancer through the ERK1/2/AP-1/VEGF-D pathway. BMC Cancer 16, 240 (2016).
Li, C. Z. et al. RIP1 regulates TNF-alpha-mediated lymphangiogenesis and lymphatic metastasis in gallbladder cancer by modulating the NF-kappaB-VEGF-C pathway. OncoTargets Ther. 11, 2875–2890 (2018).
Shen, J. et al. DUSP1 inhibits cell proliferation, metastasis and invasion and angiogenesis in gallbladder cancer. Oncotarget 8, 12133–12144 (2017).
Letelier, P. et al. miR-1 and miR-145 act as tumor suppressor microRNAs in gallbladder cancer. Int. J. Clin. Exp. Pathol. 7, 1849–1867 (2014).
Chen, Y., Chen, Y., Yu, G. & Ding, H. Lymphangiogenic and angiogentic microvessel density in gallbladder carcinoma. Hepato-Gastroenterology 58, 20–25 (2011).
Sun, X. N. et al. Prognostic impact of vascular endothelial growth factor-A expression in resected gallbladder carcinoma. Tumour Biol. 32, 1183–1190 (2011).
Xu, D., Li, J., Jiang, F., Cai, K. & Ren, G. The effect and mechanism of vascular endothelial growth factor (VEGF) on tumor angiogenesis in gallbladder carcinoma. Iran. J. Public health 48, 713–721 (2019).
Zhang, L. Q. et al. Prognostic implications of estrogen receptor 1 and vascular endothelial growth factor A expression in primary gallbladder carcinoma. World J. Gastroenterol. 21, 1243–1250 (2015).
Liu, M. C. et al. Serum vascular endothelial growth factors C and D as forecast tools for patients with gallbladder carcinoma. Tumour Biol. 36, 6305–6312 (2015).
Jiang, L. et al. Serum vascular endothelial growth factor-C levels predict lymph node metastasis and prognosis of patients with gallbladder cancer. Oncol. Lett. 16, 6065–6070 (2018).
Nakashima, T. et al. Vascular endothelial growth factor-C expression in human gallbladder cancer and its relationship to lymph node metastasis. Int. J. Mol. Med. 11, 33–39 (2003).
Mishra, K. et al. Vascular endothelial growth factor single-nucleotide polymorphism in gallbladder cancer. J. Gastroenterol. Hepatol. 28, 1678–1685 (2013).
Lin, W. et al. Vascular endothelial growth factor-D promotes growth, lymphangiogenesis and lymphatic metastasis in gallbladder cancer. Cancer Lett. 314, 127–136 (2012).
Zhu, A. X. et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: a phase 2 study. Lancet Oncol. 11, 48–54 (2010).
Lubner, S. J. et al. Report of a multicenter phase II trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: a phase II Consortium study. J. Clin. Oncol. 28, 3491–3497 (2010).
Iyer, R. V. et al. A Multicenter Phase II Study of Gemcitabine, Capecitabine, and Bevacizumab for Locally Advanced or Metastatic Biliary Tract Cancer. Am. J. Clin. Oncol. 41, 649–655 (2018).
Sun, W. et al. A phase 2 trial of regorafenib as a single agent in patients with chemotherapy-refractory, advanced, and metastatic biliary tract adenocarcinoma. Cancer 125, 902–909 (2019).
Bengala, C. et al. Sorafenib in patients with advanced biliary tract carcinoma: a phase II trial. Br. J. Cancer 102, 68–72 (2010).
Yi, J. H. et al. A phase II study of sunitinib as a second-line treatment in advanced biliary tract carcinoma: a multicentre, multinational study. Eur. J. Cancer 48, 196–201 (2012).
Santoro, A. et al. A randomized, multicenter, phase II study of vandetanib monotherapy versus vandetanib in combination with gemcitabine versus gemcitabine plus placebo in subjects with advanced biliary tract cancer: the VanGogh study. Ann. Oncol. 26, 542–547 (2015).
Arkenau, H. T. et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced or metastatic biliary tract cancer: Nonrandomized, Open-Label, Phase I Trial (JVDF). Oncologist 23, 1407–e1136 (2018).
Ullrich, A. et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 309, 418–425 (1984).
Wee, P. & Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 9, 52 (2017).
Tomas, A., Futter, C. E. & Eden, E. R. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 24, 26–34 (2014).
Gazzeri, S. Nuclear EGFR: a new mode of oncogenic signalling in cancer. Biol. Aujourdhui 212, 27–33 (2018).
Rajaram, P. et al. Epidermal growth factor receptor: role in human cancer. Indian J. Dent. Res. 28, 687–694 (2017).
Veale, D., Ashcroft, T., Marsh, C., Gibson, G. J. & Harris, A. L. Epidermal growth factor receptors in non-small cell lung cancer. Br. J. Cancer 55, 513–516 (1987).
Wong, A. J. et al. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl Acad. Sci. USA 84, 6899–6903 (1987).
Castellanos, E., Feld, E. & Horn, L. Driven by mutations: the predictive value of mutation subtype in EGFR-mutated non-small cell lung cancer. J. Thorac. Oncol. 12, 612–623 (2017).
Zhang, Y. L. et al. The prevalence of EGFR mutation in patients with non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget 7, 78985–78993 (2016).
Yarden, Y. & Pines, G. The ERBB network: at last, cancer therapy meets systems biology. Nat. Rev. Cancer 12, 553–563 (2012).
Roskoski, R. Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharm. Res. 139, 395–411 (2019).
Singh, D., Attri, B. K., Gill, R. K. & Bariwal, J. Review on EGFR inhibitors: critical updates. Mini Rev. Med. Chem. 16, 1134–1166 (2016).
Burgess, A. W. et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 12, 541–552 (2003).
Capdevila, J. et al. Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment. Cancer Treat. Rev. 35, 354–363 (2009).
Xu, M. J., Johnson, D. E. & Grandis, J. R. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 36, 463–473 (2017).
Yamaoka, T., Ohba, M. & Ohmori, T. Molecular-targeted therapies for epidermal growth factor receptor and its resistance mechanisms. Int. J. Mol. Sci. 18, 2420 (2017).
Karachaliou, N., Fernandez-Bruno, M., Paulina Bracht, J. W. & Rosell, R. EGFR first- and second-generation TKIs-there is still place for them in EGFR-mutant NSCLC patients.Transl. Cancer Res. 8, S23–S47 (2018).
Tan, C. S. et al. Third generation EGFR TKIs: current data and future directions. Mol. Cancer 17, 29 (2018).
Kobayashi, S. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 (2005).
Yun, C. H. et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl Acad. Sci. USA 105, 2070–2075 (2008).
Yu, H. A. et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 19, 2240–2247 (2013).
Kosaka, T., Yamaki, E., Mogi, A. & Kuwano, H. Mechanisms of resistance to EGFR TKIs and development of a new generation of drugs in non-small-cell lung cancer. J. Biomed. Biotechnol. 2011, 165214 (2011).
Sequist, L. V. et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 31, 3327–3334 (2013).
Wu, Y. L. et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 15, 213–222 (2014).
Park, K. et al. 190TiP: ELUXA 1: phase II study of BI 1482694 (HM61713) in patients (pts) with T790M-positive non-small cell lung cancer (NSCLC) after treatment with an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR TKI). J. Thorac. Oncol. 11, S139 (2016).
Janne, P. A. et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 372, 1689–1699 (2015).
Barreto, S. G., Dutt, A. & Chaudhary, A. A genetic model for gallbladder carcinogenesis and its dissemination. Ann. Oncol. 25, 1086–1097 (2014).
Gomes, R. V., Vidigal, P., Damasceno, K., Rodrigues, M. & Resende, V. Epidermal growth factor receptor (EGFR) in biliary tract cancer. HPB 18, e466 (2016).
Hadi, R. et al. EGFR and HER-2/neu expression in gallbladder carcinoma: an institutional experience. Gulf J. Oncol. 1, 12–19 (2016).
Zhang, M. et al. Arctigenin induced gallbladder cancer senescence through modulating epidermal growth factor receptor pathway. Tumour Biol. 39, 1010428317698359 (2017).
Mody, K., Strauss, E., Lincer, R. & Frank, R. C. Complete response in gallbladder cancer to erlotinib plus gemcitabine does not require mutation of the epidermal growth factor receptor gene: a case report. BMC Cancer 10, 570 (2010).
Philip, P. A. et al. Phase II study of erlotinib in patients with advanced biliary cancer. J. Clin. Oncol. 24, 3069–3074 (2006).
El-Khoueiry, A. B. et al. S0941: a phase 2 SWOG study of sorafenib and erlotinib in patients with advanced gallbladder carcinoma or cholangiocarcinoma. Br. J. Cancer 110, 882–887 (2014).
Cai, W. et al. EGFR target therapy combined with gemox for advanced biliary tract cancers: a meta-analysis based on RCTs. J. Cancer 9, 1476–1485 (2018).
Lee, S., Rauch, J. & Kolch, W. Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 21, 1102 (2020).
Peluso, I., Yarla, N. S., Ambra, R., Pastore, G. & Perry, G. MAPK signalling pathway in cancers: olive products as cancer preventive and therapeutic agents. Semin. Cancer Biol. 56, 185–195 (2019).
Sun, M. et al. Circular RNA CEP128 promotes bladder cancer progression by regulating Mir-145-5p/Myd88 via MAPK signaling pathway. Int. J. Cancer 145, 2170–2181 (2019).
Wu, Y. et al. Amplification of USP13 drives non-small cell lung cancer progression mediated by AKT/MAPK signaling. Biomed. Pharmacother. 114, 108831 (2019).
You, Z., Liu, S.-P., Du, J., Wu, Y.-H. & Zhang, S.-Z. Advancements in MAPK signaling pathways and MAPK-targeted therapies for ameloblastoma: a review. J. Oral Pathol. Med. 48, 201–205 (2019).
Degen, M., Natarajan, E., Barron, P., Widlund, H. R. & Rheinwald, J. G. MAPK/ERK-dependent translation factor hyperactivation and dysregulated laminin γ2 expression in oral dysplasia and squamous cell carcinoma. Am. J. Pathol. 180, 2462–2478 (2012).
Mandal, R., Becker, S. & Strebhardt, K. Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: an emerging threat to anticancer therapy. Oncogene 35, 2547–2561 (2016).
Crews, C. M., Alessandrini, A. & Erikson, R. L. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science 258, 478–480 (1992).
Raoul, C. et al. Motoneuron death triggered by a specific pathway downstream of Fas. Potentiation by ALS-linked SOD1 mutations. Neuron 35, 1067–1083 (2002).
Khokhlatchev, A. V. et al. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 93, 605–615 (1998).
Santarpia, L., Lippman, S. M. & El-Naggar, A. K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 16, 103–119 (2012).
Huang, W.-C., Tsai, C.-C. & Chan, C.-C. Mutation analysis and copy number changes of KRAS and BRAF genes in Taiwanese cases of biliary tract cholangiocarcinoma. J. Formos. Med. Assoc. 116, 464–468 (2017).
Kazmi, H. R. et al. Prognostic significance of K-ras codon 12 mutation in patients with resected gallbladder cancer. Dig. Surg. 30, 233–239 (2013).
Deshpande, V. et al. Mutational profiling reveals PIK3CA mutations in gallbladder carcinoma. BMC Cancer 11, 60 (2011).
Kim, Y. T. et al. Genetic alterations in gallbladder adenoma, dysplasia and carcinoma. Cancer Lett. 169, 59–68 (2001).
Vidaurre, T. et al. Tumor protein p53 and K-ras gene mutations in Peruvian patients with gallbladder cancer. Asian Pac. J. Cancer Prev. 20, 289–294 (2019).
Saetta, A. A. et al. Mutational analysis of BRAF in gallbladder carcinomas in association with K-ras and p53 mutations and microsatellite instability. Virchows Arch. 445, 179–182 (2004).
Goldenberg, D. et al. The V599E BRAF mutation is uncommon in biliary tract cancers. Mod. Pathol. 17, 1386–1391 (2004).
Nagahashi, M. et al. Genetic changes of p53, K-ras, and microsatellite instability in gallbladder carcinoma in high-incidence areas of Japan and Hungary. World J. Gastroenterol. 14, 70–75 (2008).
Asai, T. et al. High frequency of TP53 but not K-ras gene mutations in Bolivian patients with gallbladder cancer. Asian Pac. J. Cancer Prev. 15, 5449–5454 (2014).
Pramanik, V. et al. A novel polymorphism in codon 25 of the KRAS gene associated with gallbladder carcinoma patients of the eastern part of India. Genet. Test. Mol. Biomark. 15, 431–434 (2011).
Kazmi, H. R. et al. A let-7 microRNA binding site polymorphism in the KRAS 3'UTR is associated with increased risk and reduced survival for gallbladder cancer in North Indian population. J. Cancer Res. Clin. Oncol. 142, 2577–2583 (2016).
Li, M. et al. Identification of MAP kinase pathways as therapeutic targets in gallbladder carcinoma using targeted parallel sequencing. Oncotarget 8, 36319–36330 (2017).
Horiuchi, H. et al. A MEK inhibitor (U0126) markedly inhibits direct liver invasion of orthotopically inoculated human gallbladder cancer cells in nude mice. J. Exp. Clin. Cancer Res. 23, 599–606 (2004).
Cao, Y. et al. Prohibitin overexpression predicts poor prognosis and promotes cell proliferation and invasion through ERK pathway activation in gallbladder cancer. J. Exp. Clin. Cancer Res. 35, 68 (2016).
Qian, L. et al. Anti-tumor activity of bufalin by inhibiting c-MET mediated MEK/ERK and PI3K/AKT signaling pathways in gallbladder cancer. J. Cancer 11, 3114–3123 (2020).
Ma, Q. et al. EMP3, which is regulated by miR-663a, suppresses gallbladder cancer progression via interference with the MAPK/ERK pathway. Cancer Lett. 430, 97–108 (2018).
Wu, X.-S. et al. MALAT1 promotes the proliferation and metastasis of gallbladder cancer cells by activating the ERK/MAPK pathway. Cancer Biol. Ther. 15, 806–814 (2014).
Du, P. et al. SLC25A22 promotes proliferation and metastasis by activating MAPK/ERK pathway in gallbladder cancer. Cancer Cell Int. 19, 33 (2019).
Bao, R.-F. et al. miR-101 targeting ZFX suppresses tumor proliferation and metastasis by regulating the MAPK/Erk and Smad pathways in gallbladder carcinoma. Oncotarget 7, 22339–22354 (2016).
Giannini, I., Cutrignelli, D. A., Resta, L., Gentile, A. & Vincenti, L. Metastatic melanoma of the gallbladder: report of two cases and a review of the literature. Clin. Exp. Med. 16, 295–300 (2016).
Yu, Z., Quiroz, E., Shen, Y. & Jaiyesimi, I. A. Pathological complete response induced by neoadjuvant treatment using BRAF and MEK inhibitors in a patient with unresectable BRAF V600E-mutant malignant melanoma of the gallbladder. OncoTargets Ther. 11, 8723–8728 (2018).
Hezel, A. F. et al. Phase II study of gemcitabine, oxaliplatin in combination with panitumumab in KRAS wild-type unresectable or metastatic biliary tract and gallbladder cancer. Br. J. Cancer 111, 430–436 (2014).
Chen, J. S. et al. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann. Oncol. 26, 943–949 (2015).
Gilmartin, A. G. et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res. 17, 989–1000 (2011).
Ikeda, M. et al. Efficacy and safety of trametinib in Japanese patients with advanced biliary tract cancers refractory to gemcitabine. Cancer Sci. 109, 215–224 (2018).
Kim, R. D. et al. Randomised phase II trial (SWOG S1310) of single agent MEK inhibitor trametinib versus 5-fluorouracil or capecitabine in refractory advanced biliary cancer. Eur. J. Cancer 130, 219–227 (2020).
Rinehart, J. et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 22, 4456–4462 (2004).
Huang, W. et al. PD0325901, a mitogen-activated protein kinase kinase inhibitor, produces ocular toxicity in a rabbit animal model of retinal vein occlusion. J. Ocul. Pharmcol. Ther. 25, 519–530 (2009).
Davies, B. R. et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action <em>in vivo</em>, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol. Cancer Ther. 6, 2209–2219 (2007).
Bekaii-Saab, T. et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J. Clin. Oncol. 29, 2357–2363 (2011).
Hay, N. & Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 (2004).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371 (2017).
Lien, E. C., Dibble, C. C. & Toker, A. PI3K signaling in cancer: beyond AKT. Curr. Opin. Cell Biol. 45, 62–71 (2017).
Ippen, F. M. et al. Targeting the PI3K/Akt/mTOR pathway with the pan-Akt inhibitor GDC-0068 in PIK3CA-mutant breast cancer brain metastases. Neuro-Oncology 21, 1401–1411 (2019).
Umemura, S. et al. Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. J. Thorac. Oncol. 9, 1324–1331 (2014).
Hu, M., Zhu, S., Xiong, S., Xue, X. & Zhou, X. MicroRNAs and the PTEN/PI3K/Akt pathway in gastric cancer (Review). Oncol. Rep. 41, 1439–1454 (2019).
Yang, Y. F. et al. SPAG5 interacts with CEP55 and exerts oncogenic activities via PI3K/AKT pathway in hepatocellular carcinoma. Mol. Cancer 17, 117 (2018).
Ma, J. et al. Fibroblast-derived CXCL12 regulates PTEN expression and is associated with the proliferation and invasion of colon cancer cells via PI3k/Akt signaling. Cell Commun. Signal. 17, 119 (2019).
Xu, X., Yu, Y., Zong, K., Lv, P. & Gu, Y. Up-regulation of IGF2BP2 by multiple mechanisms in pancreatic cancer promotes cancer proliferation by activating the PI3K/Akt signaling pathway. J. Exp. Clin. Cancer Res. 38, 497 (2019).
Tiemin, P. et al. MUC13 promotes intrahepatic cholangiocarcinoma progression via EGFR/PI3K/AKT pathways. J. Hepatol. 72, 761–773 (2020).
Shu, Y. J. et al. SPOCK1 as a potential cancer prognostic marker promotes the proliferation and metastasis of gallbladder cancer cells by activating the PI3K/AKT pathway. Mol. Cancer 14, 12 (2015).
LoPiccolo, J., Blumenthal, G. M., Bernstein, W. B. & Dennis, P. A. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist. Updat.11, 32–50 (2008).
Weichhart, T. & Saemann, M. D. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann. Rheum. Dis. 67, iii70–iii74 (2008).
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M. & Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341 (2010).
Courtney, K. D., Corcoran, R. B. & Engelman, J. A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 28, 1075–1083 (2010).
Memmott, R. M. & Dennis, P. A. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell. Signal. 21, 656–664 (2009).
Sciarretta, S., Forte, M., Frati, G. & Sadoshima, J. New insights into the role of mTOR signaling in the cardiovascular system. Circ. Res. 122, 489–505 (2018).
Li, X. & Gao, T. mTORC2 phosphorylates protein kinase Czeta to regulate its stability and activity. EMBO Rep. 15, 191–198 (2014).
Sarbassov, D. D., Guertin, D. A., Ali, S. M. & Sabatini, D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005).
Lunardi, A. et al. Role of aberrant PI3K pathway activation in gallbladder tumorigenesis. Oncotarget 5, 894–900 (2014).
Leal, P. et al. Immunohistochemical expression of phospho-mTOR is associated with poor prognosis in patients with gallbladder adenocarcinoma. Arch. Pathol. Lab. Med. 137, 552–557 (2013).
Sabato, C. et al. Genetic analysis of brazilian patients with gallbladder cancer. Pathol. Oncol. Res. 25, 811–814 (2019).
Noguchi, R. et al. Genetic alterations in Japanese extrahepatic biliary tract cancer. Oncol. Lett. 14, 877–884 (2017).
Roa, I. et al. Somatic mutations of PI3K in early and advanced gallbladder cancer: additional options for an orphan cancer. J. Mol. Diagn. 18, 388–394 (2016).
Zhao, S. et al. The E545K mutation of PIK3CA promotes gallbladder carcinoma progression through enhanced binding to EGFR. J. Exp. Clin. Cancer Res. 35, 97 (2016).
Tekcham, D. S., Gupta, S., Shrivastav, B. R. & Tiwari, P. K. Epigenetic downregulation of PTEN in gallbladder cancer. J. Gastrointest. Cancer 48, 110–116 (2017).
Zhou, Z. et al. CCL18 secreted from M2 macrophages promotes migration and invasion via the PI3K/Akt pathway in gallbladder cancer. Cell. Oncol. 42, 81–92 (2019).
Zhang, F. et al. EIF3D promotes gallbladder cancer development by stabilizing GRK2 kinase and activating PI3K-AKT signaling pathway. Cell Death Dis. 8, e2868 (2017).
Hu, Y. P. et al. STYK1 promotes cancer cell proliferation and malignant transformation by activating PI3K-AKT pathway in gallbladder carcinoma. Int. J. Biochem. Cell Biol. 97, 16–27 (2018).
Zhang, Y. et al. A novel PI3K/AKT signaling axis mediates Nectin-4-induced gallbladder cancer cell proliferation, metastasis and tumor growth. Cancer Lett. 375, 179–189 (2016).
Zhang, Z. et al. Overexpression of UBR5 promotes tumor growth in gallbladder cancer via PTEN/PI3K/Akt signal pathway. J. Cell. Biochem. 120, 11517–11524 (2019).
Cai, Q. et al. Upregulation of long non-coding RNA LINC00152 by SP1 contributes to gallbladder cancer cell growth and tumor metastasis via PI3K/AKT pathway. Am. J. Transl. Res. 8, 4068–4081 (2016).
Jin, Y. P. et al. miR-143-3p targeting of ITGA6 suppresses tumour growth and angiogenesis by downregulating PLGF expression via the PI3K/AKT pathway in gallbladder carcinoma. Cell Death Dis. 9, 182 (2018).
Zhang, Y. et al. TASP1 promotes gallbladder cancer cell proliferation and metastasis by up-regulating FAM49B via PI3K/AKT pathway. Int. J. Biol. Sci. 16, 739–751 (2020).
Li, C., Wang, C., Xing, Y., Zhen, J. & Ai, Z. CD133 promotes gallbladder carcinoma cell migration through activating Akt phosphorylation. Oncotarget 7, 17751–17759 (2016).
Li, Z. et al. LASP-1 induces proliferation, metastasis and cell cycle arrest at the G2/M phase in gallbladder cancer by down-regulating S100P via the PI3K/AKT pathway. Cancer Lett. 372, 239–250 (2016).
Leal, P. et al. AKT/mTOR substrate P70S6K is frequently phosphorylated in gallbladder cancer tissue and cell lines. OncoTargets Ther. 6, 1373–1384 (2013).
Wu, Q. et al. Therapeutic effect of rapamycin on gallbladder cancer in a transgenic mouse model. Cancer Res. 67, 3794–3800 (2007).
Li, Q. et al. Inhibition of mTOR suppresses human gallbladder carcinoma cell proliferation and enhances the cytotoxicity of 5-fluorouracil by downregulating MDR1 expression. Eur. Rev. Med. Pharmacol. Sci. 20, 1699–1706 (2016).
Shen, Y. et al. Liensinine induces gallbladder cancer apoptosis and G2/M arrest by inhibiting ZFX-induced PI3K/AKT pathway. Acta Biochim. Biophys. Sin. 51, 607–614 (2019).
Song, X. et al. Dioscin induces gallbladder cancer apoptosis by inhibiting ROS-mediated PI3K/AKT signalling. Int. J. Biol. Sci. 13, 782–793 (2017).
Costello, B. A. et al. Phase I trial of everolimus, gemcitabine and cisplatin in patients with solid tumors. Investig. N. Drugs 32, 710–716 (2014).
Ahn, D. H. et al. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in patients with advanced biliary cancer. Sci. Rep. 5, 12122 (2015).
Mohri, D. et al. A potent therapeutics for gallbladder cancer by combinatorial inhibition of the MAPK and mTOR signaling networks. J. Gastroenterol. 51, 711–721 (2016).
Duan, J., Wang, Y. & Jiao, S. Checkpoint blockade-based immunotherapy in the context of tumor microenvironment: opportunities and challenges. Cancer Med. 7, 4517–4529 (2018).
Boussiotis, V. A. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N. Engl. J. Med. 375, 1767–1778 (2016).
Li, M. et al. Genomic/mutations promote PD-L1-mediated immune escape in gallbladder cancer: a whole-exome sequencing analysis. Gut 68, 1024–1033 (2019).
Gong, K. et al. PLAC8 overexpression correlates with PD-L1 upregulation and acquired resistance to chemotherapies in gallbladder carcinoma. Biochem. Biophys. Res. Commun. 516, 983–990 (2019).
Millares, L. et al. Tumor-associated metabolic and inflammatory responses in early stage non-small cell lung cancer: local patterns and prognostic significance. Lung Cancer 122, 124–130 (2018).
Buchbinder, E. I. et al. Therapy with high-dose Interleukin-2 (HD IL-2) in metastatic melanoma and renal cell carcinoma following PD1 or PDL1 inhibition. J. Immunother. Cancer 7, 49 (2019).
Li, H. et al. The immune checkpoint regulator PDL1 is an independent prognostic biomarker for biochemical recurrence in prostate cancer patients following adjuvant hormonal therapy. J. Cancer 10, 3102–3111 (2019).
Billon, E. et al. expression is associated with longer postoperative, survival in adrenocortical carcinoma. Oncoimmunology 8, e1655362 (2019).
Ren, X. et al. PD1 protein expression in tumor infiltrated lymphocytes rather than PDL1 in tumor cells predicts survival in triple-negative breast cancer. Cancer Biol. Ther. 19, 373–380 (2018).
Mocan, T., Sparchez, Z., Craciun, R., Bora, C. N. & Leucuta, D. C. Programmed cell death protein-1 (PD-1)/programmed death-ligand-1 (PD-L1) axis in hepatocellular carcinoma: prognostic and therapeutic perspectives. Clin. Transl. Oncol. 21, 702–712 (2019).
Birnbaum, D. J. et al. Prognostic value of PDL1 expression in pancreatic cancer. Oncotarget 7, 71198–71210 (2016).
Wang, X. et al. PD-L1 is a direct target of cancer-FOXP3 in pancreatic ductal adenocarcinoma (PDAC), and combined immunotherapy with antibodies against PD-L1 and CCL5 is effective in the treatment of PDAC. Signal Transduct. Target. Ther. 5, 38 (2020).
Xylinas, E. et al. Association of T-cell co-regulatory protein expression with clinical outcomes following radical cystectomy for urothelial carcinoma of the bladder. Eur. J. Surg. Oncol. 40, 121–127 (2014).
Neyaz, A. et al. Clinical relevance of PD-L1 expression in gallbladder cancer: a potential target for therapy. Histopathology 73, 622–633 (2018).
Mody, K. et al. Patterns and genomic correlates of PD-L1 expression in patients with biliary tract cancers. J. Gastrointest. Oncol. 10, 1099–1109 (2019).
Lin, J. et al. Classification of gallbladder cancer by assessment of CD8 TIL and PD-L1 expression. BMC Cancer 18, 766 (2018).
Ha, H. et al. Soluble programmed death-ligand 1 (sPDL1) and neutrophil-to-lymphocyte ratio (NLR) predicts survival in advanced biliary tract cancer patients treated with palliative chemotherapy. Oncotarget 7, 76604–76612 (2016).
Kim, J. H. et al. Programmed death-ligand 1 expression and its correlation with clinicopathological parameters in gallbladder cancer. J. Pathol. Transl. Med. 54, 154–164 (2020).
Jakubowski, C. D. & Azad, N. S. Immune checkpoint inhibitor therapy in biliary tract cancer (cholangiocarcinoma). Chin. Clin. Oncol. 9, 2 (2020).
Kang, J. et al. Efficacy and safety of pembrolizumab in patients with refractory advanced biliary tract cancer: tumor proportion score as a potential biomarker for response. Cancer Res. Treat. 52, 594–603 (2020).
Fashoyin-Aje, L. et al. FDA approval summary: pembrolizumab for recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma expressing PD-L1. Oncologist 24, 103–109 (2019).
Salem, M. E. et al. Landscape of tumor mutation load, mismatch repair deficiency, and PD-L1 expression in a large patient cohort of gastrointestinal cancers. Mol. Cancer Res. 16, 805–812 (2018).
Weinberg, B. A. et al. Molecular profiling of biliary cancers reveals distinct molecular alterations and potential therapeutic targets. J. Gastrointest. Oncol. 10, 652–662 (2019).
Kong, W. et al. Significant benefit of nivolumab combining radiotherapy in metastatic gallbladder cancer patient with strong PD-L1 expression: a case report. Onco Targets Ther. 12, 5389–5393 (2019).
Ueno, M. et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: a non-randomised, multicentre, open-label, phase 1 study. Lancet Gastroenterol. Hepatol. 4, 611–621 (2019).
Kim, R. D. et al. A phase II study of nivolumab in patients with advanced refractory biliary tract cancers (BTC). J. Clin. Oncol. 37, 4097–4097 (2019).
Bang, Y.-J. et al. Pembrolizumab (pembro) for advanced biliary adenocarcinoma: Results from the KEYNOTE-028 (KN028) and KEYNOTE-158 (KN158) basket studies. J. Clin. Oncol. 37, 4079–4079 (2019).
Piha-Paul, S. A. et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: results from the KEYNOTE-158 and KEYNOTE-028 studies. Int. J. Cancer 147, 2190–2198 (2020).
Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
Borghaei, H. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
Carbone, D. P. et al. First-line Nivolumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med. 376, 2415–2426 (2017).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Ribas, A. & Hu-Lieskovan, S. What does PD-L1 positive or negative mean? J. Exp. Med. 213, 2835–2840 (2016).
Zitvogel, L., Galluzzi, L., Smyth, M. J. & Kroemer, G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity 39, 74–88 (2013).
Bezu, L. et al. Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 6, 187 (2015).
Xie, C. et al. Tremelimumab in combination with microwave ablation in patients with refractory biliary tract cancer. Hepatology 69, 2048–2060 (2019).
Fujiwara, Y., Koyama, T., Helwig, C., Watanabe, M. & Doi, T. M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGF-β, in Asian patients with advanced solid tumors. J. Clin. Oncol. 36, 762–762 (2018).
Banimohamad-Shotorbani, B. et al. DNA damage repair response in mesenchymal stromal cells: from cellular senescence and aging to apoptosis and differentiation ability. Ageing Res. Rev. 62, 101125 (2020).
Reuvers, T. G. A., Kanaar, R. & Nonnekens, J. DNA damage-inducing anticancer therapies: from global to precision damage. Cancers 12, 2098 (2020).
Fang, J. et al. DUSP1 enhances the chemoresistance of gallbladder cancer via the modulation of the p38 pathway and DNA damage/repair system. Oncol. Lett. 16, 1869–1875 (2018).
Zhang, Y. et al. Cholesterol depletion sensitizes gallbladder cancer to cisplatin by impairing DNA damage response. Cell Cycle 18, 3337–3350 (2019).
Abdel-Wahab, R. et al. Variations in DNA repair genomic alterations and tumor mutation burden in biliary tract cancer (BTC) subtypes. J. Clin. Oncol. 36, 263–263 (2018).
Spizzo, G. et al. Molecular profile of BRCA-mutated biliary tract cancers. ESMO Open 5, e000682 (2020).
Javle, M. M. et al. Precision medicine for gallbladder cancer using somatic copy number amplifications (SCNA) and DNA repair pathway gene alterations. J. Clin. Oncol. 35, 4076–4076 (2017).
Ahn, D. H. et al. Next-generation sequencing survey of biliary tract cancer reveals the association between tumor somatic variants and chemotherapy resistance. Cancer 122, 3657–3666 (2016).
Hwang, I. G. et al. Different relation between ERCC1 overexpression and treatment outcomes of two platinum agents in advanced biliary tract adenocarcinoma patients. Cancer Chemother. Pharmacol. 68, 935–944 (2011).
Chae, H. et al. Therapeutic relevance of targeted sequencing in management of patients with advanced biliary tract cancer: DNA damage repair gene mutations as a predictive biomarker. Eur. J. Cancer 120, 31–39 (2019).
Lee, Y. Y. et al. Phosphoproteomic analysis identifies activated MET-axis PI3K/AKT and MAPK/ERK in lapatinib-resistant cancer cell line. Exp. Mol. Med. 45, e64 (2013).
Scagliotti, G. V., Novello, S. & von Pawel, J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat. Rev. 39, 793–801 (2013).
Organ, S. L. & Tsao, M.-S. An overview of the c-MET signaling pathway.Ther. Adv. Med. Oncol. 3, S7–S19 (2011).
Liang, H. & Wang, M. MET oncogene in non-small cell lung cancer: mechanism of MET dysregulation and agents targeting the HGF/c-Met axis. Onco Targets Ther. 13, 2491–2510 (2020).
Finocchiaro, G., Toschi, L., Gianoncelli, L., Baretti, M. & Santoro, A. Prognostic and predictive value of MET deregulation in non-small cell lung cancer. Ann. Transl. Med. 3, 83 (2015).
Giordano, S. & Columbano, A. Met as a therapeutic target in HCC: facts and hopes. J. Hepatol. 60, 442–452 (2014).
Qiao, Z. et al. Cancer cell derived small extracellular vesicles contribute to recipient cell metastasis through promoting HGF/c-Met pathway. Mol. Cell. Proteom. 18, 1619–1629 (2019).
You, W.-K. & McDonald, D. M. The hepatocyte growth factor/c-Met signaling pathway as a therapeutic target to inhibit angiogenesis. BMB Rep. 41, 833–839 (2008).
Kim, K.-H. & Kim, H. Progress of antibody-based inhibitors of the HGF-cMET axis in cancer therapy. Exp. Mol. Med. 49, e307 (2017).
Fu, R., Jiang, S., Li, J., Chen, H. & Zhang, X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med. Oncol. 37, 24 (2020).
Blumenschein, G. R., Mills, G. B. & Gonzalez-Angulo, A. M. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J. Clin. Oncol. 30, 3287–3296 (2012).
Bouattour, M. et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 67, 1132–1149 (2018).
Xing, F. et al. Activation of the c-Met pathway mobilizes an inflammatory network in the brain microenvironment to promote brain metastasis of breast cancer. Cancer Res. 76, 4970–4980 (2016).
Heo, M. H. et al. The clinical impact of c-MET over-expression in advanced biliary tract cancer (BTC). J. Cancer 8, 1395–1399 (2017).
Nakazawa, K. et al. Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J. Pathol. 206, 356–365 (2005).
Yang, L., Guo, T., Jiang, S. & Yang, Z. Expression of ezrin, HGF and c-met and its clinicopathological significance in the benign and malignant lesions of the gallbladder. Hepatogastroenterology 59, 1769–1775 (2012).
Sanada, Y. et al. Critical role of c-Met and Ki67 in progress of biliary carcinoma. Am. Surg. 76, 372–379 (2010).
Moon, W. S. et al. Co-expression of cox-2, C-met and beta-catenin in cells forming invasive front of gallbladder cancer. Cancer Res. Treat. 37, 171–176 (2005).
Date, K. et al. Inhibition of tumor growth and invasion by a four-kringle antagonist (HGF/NK4) for hepatocyte growth factor. Oncogene 17, 3045–3054 (1998).
Tanaka, T. et al. Gallbladder cancer treatment using adenovirus expressing the HGF/NK4 gene in a peritoneal implantation model. Cancer Gene Ther. 11, 431–440 (2004).
Matsumoto, K., Date, K., Shimura, H. & Nakamura, T. Acquisition of invasive phenotype in gallbladder cancer cells via mutual interaction of stromal fibroblasts and cancer cells as mediated by hepatocyte growth factor. Jpn J. Cancer Res. 87, 702–710 (1996).
Li, H. et al. Hepatocyte growth factor stimulates the invasion of gallbladder carcinoma cell lines in vitro. Clin. Exp. Metastasis 16, 74–82 (1998).
Kim, Y. et al. Prevalence and clinicopathological significance of MET overexpression and gene amplification in patients with gallbladder carcinoma. Cancer Res. Treat. 52, 481–491 (2020).
Wang, Q., Yang, S., Wang, K. & Sun, S.-Y. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J. Hematol. Oncol. 12, 63 (2019).
Nandagopal, L., Sonpavde, G. P. & Agarwal, N. Investigational MET inhibitors to treat renal cell carcinoma. Expert Opin. Investig. Drugs 28, 851–860 (2019).
Hardy-Werbin, M., Del Rey-Vergara, R., Galindo-Campos, M. A., Moliner, L. & Arriola, E. MET inhibitors in small cell lung cancer: from the bench to the bedside. Cancers 11, 1404 (2019).
Pasquini, G. & Giaccone, G. C-MET inhibitors for advanced non-small cell lung cancer. Expert Opin. Investig. Drugs 27, 363–375 (2018).
Ghosh, M., Sakhuja, P., Singh, S. & Agarwal, A. K. p53 and beta-catenin expression in gallbladder tissues and correlation with tumor progression in gallbladder cancer. Saudi J. Gastroenterol. 19, 34–39 (2013).
Coggi, G. et al. p53 protein accumulation and p53 gene mutation in esophageal carcinoma. A molecular and immunohistochemical study with clinicopathologic correlations. Cancer 79, 425–432 (1997).
Li, M. et al. Magnolol inhibits growth of gallbladder cancer cells through the p53 pathway. Cancer Sci. 106, 1341–1350 (2015).
Tian, L. et al. Downregulation of ASPP2 promotes gallbladder cancer metastasis and macrophage recruitment via aPKC-ι/GLI1 pathway. Cell Death Dis. 9, 1115–1115 (2018).
Nigam, P., Misra, U., Negi, T. S., Mittal, B. & Choudhuri, G. Alterations of p53 gene in gallbladder cancer patients of North India. Trop. Gastroenterol. 31, 96–100 (2010).
Yokoyama, N. et al. Mutations of p53 in gallbladder carcinomas in high-incidence areas of Japan and Chile. Cancer Epidemiol. Biomark. Prev. 7, 297–301 (1998).
Rai, R., Tewari, M., Kumar, M., Singh, A. K. & Shukla, H. S. p53: its alteration and gallbladder cancer. Eur. J. Cancer Prev. 20, 77–85 (2011).
Chaube, A., Tewari, M., Garbyal, R. S., Singh, U. & Shukla, H. S. Preliminary study of p53 and c-erbB-2 expression in gallbladder cancer in Indian patients manuscript id: 8962091628764582. BMC Cancer 6, 126 (2006).
Yadav, S. et al. Targeted gene sequencing of gallbladder carcinoma identifies high-impact somatic and rare germline mutations. Cancer Genomics Proteom. 14, 495–506 (2017).
Shukla, S. K., Singh, G., Shahi, K. S., Bhuvan & Pant, P. Genetic changes of P and Kras in gallbladder carcinoma in Kumaon region of Uttarakhand. J. Gastrointest. Cancer 51, 552–559 (2019).
Singh, A. et al. Prognostic significance of HER-2 and p53 expression in gallbladder carcinoma in North Indian patients. Oncology 91, 354–360 (2016).
Shu, G.-s, Lv, F., Yang, Z.-l & Miao, X.-y Immunohistochemical study of PUMA, c-Myb and p53 expression in the benign and malignant lesions of gallbladder and their clinicopathological significances. Int. J. Clin. Oncol. 18, 641–650 (2013).
Legan, M., Luzar, B., Marolt, V. F. & Cor, A. Expression of cyclooxygenase-2 is associated with p53 accumulation in premalignant and malignant gallbladder lesions. World J. Gastroenterol. 12, 3425–3429 (2006).
Neyaz, A. et al. Investigation of targetable predictive and prognostic markers in gallbladder carcinoma. J. Gastrointest. Oncol. 9, 111–125 (2018).
Hidalgo Grau, L. A. et al. Gallbladder carcinoma: the role of p53 protein overexpression and Ki-67 antigen expression as prognostic markers. HPB 6, 174–180 (2004).
Makower, D. et al. Phase II clinical trial of intralesional administration of the oncolytic adenovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin. Cancer Res. 9, 693–702 (2003).
Bischoff, J. R. et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274, 373–376 (1996).
Li, W.-Q. et al. Genetic polymorphisms in the 9p21 region associated with risk of multiple cancers. Carcinogenesis 35, 2698–2705 (2014).
Zhang, L. et al. Genomic analysis of primary and recurrent gliomas reveals clinical outcome related molecular features. Sci. Rep. 9, 16058 (2019).
Tramontano, A. et al. Methylation of the Suppressor gene: mechanism and consequences. Biomolecules 10, 446 (2020).
Hannou, S. A., Wouters, K., Paumelle, R. & Staels, B. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: what have we learned from GWASs? Trends Endocrinol. Metab. 26, 176–184 (2015).
Yang, P. et al. Somatic genetic aberrations in gallbladder cancer: comparison between Chinese and US patients. Hepatobiliary Surg. Nutr. 8, 604–614 (2019).
Lin, J. et al. Precision oncology for gallbladder cancer: insights from genetic alterations and clinical practice. Ann. Transl. Med. 7, 467 (2019).
Leiting, J. L. et al. Biliary tract cancer patient-derived xenografts: surgeon impact on individualized medicine. JHEP Rep. 2, 100068 (2020).
Meng, D. & Carvajal, R. D. KIT as an oncogenic driver in melanoma: an update on clinical development. Am. J. Clin. Dermatol. 20, 315–323 (2019).
Joensuu, H. & DeMatteo, R. P. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu. Rev. Med. 63, 247–258 (2012).
Lv, M. et al. MicroRNA-664 suppresses the growth of cervical cancer cells via targeting c-Kit. Drug Des. Dev. Ther. 13, 2371–2379 (2019).
Ou, W.-B. et al. Cyclin D1 is a mediator of gastrointestinal stromal tumor KIT-independence. Oncogene 38, 6615–6629 (2019).
Tan, Y. Y. et al. Decreased SCF/C-kit signaling pathway contributes to loss of interstitial cells of cajal in gallstone disease. Int. J. Clin. Exp. Med. 7, 4099–4106 (2014).
Mochizuki, K., Hata, H., Naitou, K., Motosugi, U. & Kondo, T. Carcinosarcoma (adenocarcinoma, neuroendocrine carcinoma, undifferentiated carcinoma and chondrosarcoma) of the gallbladder. Clin. J. Gastroenterol. 13, 110–115 (2020).
Martinez-Anton, A., Gras, D., Bourdin, A., Dubreuil, P. & Chanez, P. KIT as a therapeutic target for non-oncological diseases. Pharm. Ther. 197, 11–37 (2019).
Grüllich, C. in Small Molecules in Oncology (ed. Uwe, M. M.) 207–214 (Springer, Berlin, Heidelberg, 2014).
Dubreuil, P. et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 4, e7258 (2009).
Renhowe, P. A. et al. Design, structure−activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: a novel class of receptor tyrosine kinase inhibitors. J. Med. Chem. 52, 278–292 (2009).
Ergün, S. et al. Tr-KIT/c-KIT ratio in renal cell carcinoma. Mol. Biol. Rep. 46, 5287–5294 (2019).
Hsueh, Y.-S. et al. Nuclear KIT induces a NFKBIB-RELA-KIT autoregulatory loop in imatinib-resistant gastrointestinal stromal tumors. Oncogene 38, 6550–6565 (2019).
Guan, Y., Wang, Y., Bhandari, A., Xia, E. & Wang, O. IGSF1: a novel oncogene regulates the thyroid cancer progression. Cell Biochem. Funct. 37, 516–524 (2019).
Xuan, J., Yu, Y., Qing, T., Guo, L. & Shi, L. Next-generation sequencing in the clinic: promises and challenges. Cancer Lett. 340, 284–295 (2013).
Acknowledgements
This study was supported by Shanghai Key Laboratory of Biliary Tract Disease Research Foundation (17DZ2260200), the National Natural Science Foundation of China (Nos. 81572819, 91940305, 81773043, 91440203, 81902361, 81702315), National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (2019ZX09301158), Shanghai joint research projects on emerging frontier technologies (SHDC12018107), Clinical research program of Xinhua Hospital (19XHCR3D), multicenter clinical research project of Shanghai Jiao Tong University School of Medicine (DLY201507), the Peak Plateau Discipline Construction Project of Shanghai Jiao Tong University School of medicine (No. 20181808), the Program of Shanghai Academic Research Leader (No. 19XD1422700), the Precision Medicine Research Program of Shanghai Jiao Tong University School of Medicine (Nos. 15ZH4003, DKY201507), Shanghai Sailing Program (19YF1432900, 19YF1433000), and the Shanghai Artificial Intelligence Innovation and Development Project (2019-RGZN-01096).
Author information
Authors and Affiliations
Contributions
Y. Liu conceived and designed the study. X.S., Y.H., Y. Li, and F.L. searched and reviewed the literature and drafted the illustrations and initial manuscript. R.S. critically reviewed and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, X., Hu, Y., Li, Y. et al. Overview of current targeted therapy in gallbladder cancer. Sig Transduct Target Ther 5, 230 (2020). https://doi.org/10.1038/s41392-020-00324-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-00324-2
This article is cited by
-
The relationship and clinical significance of lactylation modification in digestive system tumors
Cancer Cell International (2024)
-
MST1R-targeted therapy in the battle against gallbladder cancer
Cell & Bioscience (2024)
-
Acylcarnitines promote gallbladder cancer metastasis through lncBCL2L11-THOC5-JNK axis
Journal of Translational Medicine (2024)
-
TGF-β1 facilitates gallbladder carcinoma metastasis by regulating FOXA1 translation efficiency through m6A modification
Cell Death & Disease (2024)
-
Establishment and characterization of a novel human gallbladder cancer cell line, GBC-X1
Scientific Reports (2024)
