
Abstract
The MYC family oncogene is deregulated in >50% of human cancers, and this deregulation is frequently associated with poor prognosis and unfavorable patient survival. Myc has a central role in almost every aspect of the oncogenic process, orchestrating proliferation, apoptosis, differentiation, and metabolism. Although Myc inhibition would be a powerful approach for the treatment of many types of cancers, direct targeting of Myc has been a challenge for decades owing to its “undruggable” protein structure. Hence, alternatives to Myc blockade have been widely explored to achieve desirable anti-tumor effects, including Myc/Max complex disruption, MYC transcription and/or translation inhibition, and Myc destabilization as well as the synthetic lethality associated with Myc overexpression. In this review, we summarize the latest advances in targeting oncogenic Myc, particularly for cancer therapeutic purposes.
Similar content being viewed by others


Introduction
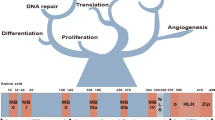
The MYC oncogene family consists of three members, C-MYC, MYCN, and MYCL, which encode c-Myc, N-Myc, and L-Myc, respectively.1,2,3 The Myc oncoproteins belong to a family of so-called “super-transcription factors” that potentially regulate the transcription of at least 15% of the entire genome.4 The major downstream effectors of Myc include those involved in ribosome biogenesis, protein translation, cell-cycle progression and metabolism, orchestrating a broad range of biological functions, such as cell proliferation, differentiation, survival, and immune surveillance (Fig. 1).4, 5

Myc regulates a spectrum of cellular functions. Myc regulates a large number of protein-coding or non-coding genes that are involved in distinct cellular functions, including cell cycle, protein biogenesis, cell adhesion, metabolism, signal transduction, transcription, and translation, among others
Myc family members exhibit high-structural homology, including the basic-region /helix-loop-helix/leucine-zipper (BR/HLH/LZ) motif at the C terminus and three highly conserved elements, known as Myc boxes 1–3 at the N terminus (Fig. 2a).1, 2, 5 As a master transcription factor, Myc binds to Max through the common BR/HLH/LZ motif, which is required for DNA–protein interactions.1, 2, 5 The Myc/Max heterodimer recruits a chromatin-modifying complex (TRRAP, GCN5, TIP60, and TIP48) and activates transcription by binding to the conserved E-box DNA sequence (CACGTG) located in the transcriptional regulatory region of target genes (Fig. 2b).1, 2, 5 Recent studies have shown that, in addition to recognizing specific E-box sequences, c-Myc also accumulates in the promoter regions of active genes, leading to transcriptional amplification (Fig. 2c).6, 7

Transcriptional activation of target genes by Myc family members. a protein structure of Myc family members. The N terminus of Myc comprises a transactivation domain (TAD) and three highly conserved elements, known as Myc boxes 1–3. Myc box 1 (MB1) possesses a phosphodegron, which is targeted by the ubiquitin E3 ligase FBW7. MB2 is required for all the known functions of Myc and recruits a histone acetyltransferase (HAT) complex, MB3 regulates Myc protein stability and transcriptional activities. The C-terminal domain contains a basic-region /helix-loop-helix/leucine-zipper (BR/HLH/LZ) motif that is necessary for DNA–protein interactions. Max, the partner of Myc, binds with Myc through the C-terminal BR/HLH/LZ motif. b Myc functions as a transcription factor. Upon binding to CACGTG (E-box), the Myc–Max dimeric complex recruits chromatin-modifying complexes, including GCN5, TIP60, TIP48, and TRRAP, leading to transcriptional activation. GCN5 and TIP60 are histone acetyltransferases; TIP48 is an ATP-binding protein, TRRAP transactivation/transformation-associated protein. c Myc functions as a transcriptional signal amplifier. In this model, Myc binding is not E-box dependent. Myc accumulates in the promoter and enhancer region of all active genes and causes transcriptional signal amplification
The expression of Myc family members is tightly controlled under normal circumstances.1, 5 Yet, Myc is frequently deregulated in human cancers. Excess Myc expression can be induced upon retroviral promoter insertion, chromosomal translocation/amplification, activation of super-enhancers within the MYC gene, and/or mutation of upstream signaling pathways that enhance Myc stability.5 Studies in transgenic mouse models have demonstrated that even transient inactivation of Myc elicits tumor regression, suggesting that regulation of oncogenic Myc could be harnessed to treat cancer patients.8,9,10 Yet, drug development aimed at directly targeting Myc has proved challenging. First, as a transcription factor, Myc lacks a specific active site for small molecules, making it difficult to functionally inhibit its activities using strategies similar to those used for kinases. Second, Myc is predominantly located in the nucleus, thus, targeting nuclear Myc with specific monoclonal antibodies is technically impractical. To overcome these obstacles, alternative approaches to indirectly abrogate Myc oncogenic functions have been extensively investigated.
Indirect targeting of Myc
Because strategies to directly target Myc have not been achieved thus far, essential targets involved in Myc deregulation have been exploited as new approaches to treat Myc-driven cancers. Targeting MYC transcription by interfering with chromatin-dependent signal transduction to RNA polymerase, a process in which BRD4 has been implicated, has shown great promise.11, 12 Myc stability is tightly controlled by the ubiquitin-proteasome system, thus, a potential strategy to target Myc is to selectively inhibit the kinases and/or deubiquitinases that stabilize Myc.13, 14 Myc strictly depends on its partner Max to regulate gene transcription, so interrupting the Myc–Max complex is therefore an additional approach to inhibit Myc signaling.15 Here, we provide a concise overview of the key factors involved in the transcription, translation, stability, and activation of Myc, which could be targeted for the treatment of Myc-addicted cancers (Fig. 3).

Various strategies to target Myc. Inhibitors of BRD4, CDK7, and CDK9 inhibit MYC expression at the transcriptional level. Inhibition of the PI3K/AKT/mTOR pathway blocks MYC translation, whereas USP7, AURKA, and PLK1 inhibitors destabilize Myc at the posttranslational level. 10058-F4 and Omomyc function to interrupt the Myc–Max dimeric complex. BRD4 bromodomain-containing 4, CDK7 cyclin-dependent kinase 7, CDK9 cyclin-dependent kinase 9, PLK1 polo-like kinases 1, PI3K/AKT/mTOR phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin
Targeting MYC transcription
Bromodomain-containing 4 (BRD4)
BRD4 is a member of the mammalian bromodomain and extraterminal (BET) family.16 BRD4 regulates transcription through recruitment of the positive transcription elongation factor b (P-TEFb), which phosphorylates the carboxy-terminal domain of RNA polymerase II (pol II), to the site of hyperacetylated chromatin.17 These changes lead to the release of RNA pol II from pausing in the promoter-proximal region, ultimately resulting in transcriptional elongation.17, 18 MYC transcription is under BRD4 regulation. JQ1, a powerful inhibitor of BRD4, competes with BRD4 for binding to acetylated lysines and displaces BRD4 from the super-enhancers within the MYC oncogene.11, 12 As such, inhibition of the BET bromodomain with JQ1 showed potent anti-cancer effects both in vitro and in vivo in multiple hematopoietic cancers and pancreatic ductal adenocarcinoma (PDAC) exhibiting C-MYC overexpression.19,20,21,22 Neuroblastomas and other MYCN-driven cancers are also sensitive to BET inhibitors.23 GSK525762, a specific BET inhibitor, is currently in early-phase clinical trials for treating these hematopoietic malignancies and solid tumors (ClinicalTrials.gov: NCT01943851, NCT03266159).
Cyclin-dependent kinase 7 and 9 (CDK7 and CDK9)
In contrast to the classical cell-cycle CDKs which are largely responsible for cell-cycle transition, CDK7 and CDK9 are CDKs that have critical roles in transcription initiation and elongation.18, 24 CDK7 is a catalytic subunit of the transcription factor IIH complex (TFIIH), and CDK9 is a kinase subunit of P-TEFb.25, 26 These two transcriptional kinases phosphorylate specific serine residues within the carboxy-terminal domain of Pol II, facilitating efficient transcriptional initiation, pause release and elongation.27 Numerous studies demonstrate that inhibition of transcriptional CDKs primarily affects the accumulation of transcripts critical for the control of cell identity, growth, and proliferation.28,29,30
A general feature of MYC deregulation is its transcriptional regulation by Super-Enhancers (SEs), clusters of enhancers that are densely occupied by transcription factors and chromatin regulators, including CDK7 and CDK9, rendering this group of kinases ideal candidates for blocking Myc-dependent transcriptional amplification.30, 31 Indeed, inhibition of CDK7 and/or CDK9 substantially reduces MYC expression, attendant to widespread transcriptional downregulation of Myc target genes.30, 32, 33 Administration of specific inhibitors against CDK7 (THZ1) and/or CDK9 (PC585) induced potent anti-tumor effects in MYC-overexpressing T-cell acute lymphoblastic leukemia, mixed-lineage leukemia, neuroblastomas, and small cell lung cancers, validating these newly developed transcriptional CDK inhibitors as a potential treatment strategy that targets global transcriptional amplification in Myc-driven cancers.30, 32, 33
Targeting MYC mRNA translation
Mammalian target of rapamycin (mTOR)
The phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR pathway is frequently altered in various cancers.34 mTOR is a serine/threonine kinase that functions as the catalytic subunit of two distinct complexes called mTOR complexes 1 and 2 (mTORC1 and mTORC2).35 The central role of mTOR in protein synthesis is largely attributed to mTORC1.35, 36 mTORC1-dependent phosphorylation of eukaryotic translation initiation factor 4E (eIF4E) binding protein 1 (4EBP1) blocks its ability to negatively regulate the translation initiation factor eIF4E, thus promoting the translation of mRNAs containing long 5′-untranslated regions (5′-UTRs) with complex RNA secondary structures, such as MYC.35, 36 As such, pharmacological inhibition of the PI3K/AKT/mTOR pathway markedly decreased Myc level and exhibited remarkable therapeutic efficacy in Myc-driven cancers, including neuroblastoma, small-cell lung carcinoma, breast cancer, and multiple hematopoietic cancers.34, 37,38,39
Cytoplasmic polyadenylation element-binding protein (CPEB)
The CPEB-family proteins are sequence-specific RNA-binding proteins which control the elongation of the poly(A) tail and polyadenylation-induced translation.40 CPEB binds the cytoplasmic polyadenylation element (CPE) containing the conserved UUUUAU or UUUUAAU sequence within the 3′-UTRs of responding mRNAs.40 A recent study revealed that the C-MYC mRNA contains CPEs that can be recognized by CPEB.41 Mechanistically, CPEB recruits Caf1 deadenylase through an interaction with Tob, an antiproliferative protein, and inhibits c-Myc expression by accelerating the deadenylation and decay of its mRNA.42 Expression of CPEB-family proteins are frequently downregulated in human cancers.40 Therefore, pharmacological approaches aimed at reactivating CPEB expression would lead to Myc inhibition in Myc-driven cancers.
Targeting MYC stability
USP28, USP36, and USP7
Myc stability is tightly controlled by the ubiquitin-proteasome system.1 Upon phosphorylation at Thr58, Myc is polyubiquitinated by the E3 ligase FBW7 and degraded by the proteasome.43 The human FBW7 locus encodes three protein isoforms, FBW7α, FBW7β, and FBW7ɣ, that differ in their N-terminal sequences and in their subcellular localization.44 Both FBW7α and FBW7ɣ are responsible for the selective degradation of endogenous Myc in human cells.44 Several deubiquitinating enzymes are involved in Myc stabilization. USP28 was shown to bind c-Myc through an interaction with FBW7α and antagonize its E3 ligase activities in the nucleus, leading to Myc stabilization and tumor cell proliferation.45 USP36 deubiquitinates and stabilizes c-Myc through interactions with FBW7ɣ in the nucleolus.46 USP7 directly binds to and stabilizes N-Myc through deubiquitination in neuroblastomas cells, and a small-molecule inhibitor of USP7, P22077, markedly suppressed growth of MYCN-amplified neuroblastoma in a xenograft model.13 In principle, targeting these deubiquitinases could cause Myc destabilization and tumor suppression.
AURKA
The Aurora family includes AURKA, AURKB, and AURKC, which are key regulators of mitosis.47 AURKA contributes to tumorigenesis through interactions with P53 and Myc.48,49,50 Recently, Otto et al.14 showed that Aurora A and N-Myc acted as oncogenic partners in neuroblastomas. AURKA forms a complex with N-Myc, which protects N-Myc from FBW7-mediated proteasomal degradation.14 Two AURKA inhibitors, MLN8054 and MLN8237, disrupt the Myc–AURKA complex, resulting in N-Myc degradation and tumor regression in MYCN-amplified neuroblastomas.14, 51 MLN8237 also induced c-Myc degradation in P53-mutant human hepatocellular carcinoma cells.52 These data suggest that AURKA inhibitors may be potential therapeutics for the treatment of Myc-dependent cancers.
Polo-like kinase 1 (PLK1)
The Polo-like kinases (PLKs) comprise a family of five serine/threonine protein kinases that control many crucial biological processes.53 The best characterized PLK family member is PLK1. Using MYCN-amplified neuroblastomas and small cell lung carcinomas as model systems, we recently demonstrated that PLK1 and Myc created a positive, feedforward activation loop that was essential for sustaining mutual high expression, leading to Myc-dependent transcriptional amplification and aggressive tumor progression. Inhibitors of PLK1, such as BI 6727 or BI2356, preferentially induce potent apoptosis of Myc-overexpressing tumor cells and synergistically potentiate the therapeutic efficacies of BCL-2 antagonists. These findings reveal a PLK1-FBW7-Myc signaling circuit that underlies tumorigenesis and validate PLK1 inhibitors, alone or with BCL-2 antagonists, as potential effective therapeutics for Myc-overexpressing cancers.54
Targeting the MYC–MAX complex
The Myc–Max complex is required for the binding of Myc to DNA and its subsequent activation of target gene transcription.1 The Myc–Max dimer interface is a parallel, left-handed, four-helix bundle, with each monomer comprising two R-helices separated by a loop.55 Although studies have shown that this structure has no apparent sites for positioning a small-molecule inhibitor, several labs have screened small molecules that block this interaction. The peptide mimetic IIA6B17 was first reported as a small-molecule inhibitor of Myc–Max dimerization.15 A compound called 10058-F4 was capable of disrupting the Myc–Max complex in HL60 cells.56 Another widely known inhibitor, Omomyc, a mutant basic helix-loop-helix peptide that sequesters Myc in a transcriptionally incompetent complex, prevents Myc-induced tumorigenesis in multiple mouse tumor models.57,58,59
Synthetic lethal interaction with MYC
Two genes (“A” and “B”) are said to be “synthetic lethal” if mutation of either gene alone is compatible with viability but simultaneous mutation of both genes causes death.60 Synthetic lethal interactions are most commonly described for loss-of-function alleles but can also involve gain-of-function alleles.60 For example, gene A might become essential for survival when a particular gene B is overexpressed. This situation describes Myc-mediated synthetic lethality. MYC overexpression is found in many cancers, and MYC overexpression sensitizes cells to apoptosis, enabling targeting a gene that is synthetic lethal to a cancer-relevant MYC overexpression should kill only cancer cells but spare normal counterparts. Here, we describe key factors that exhibit synthetic lethal interactions with Myc deregulation (Fig. 4).

Synthetic lethal interactions with Myc deregulation. Myc-mediated synthetic lethality has been observed with various targets, including CDK1, CHK1, and GLS. CDK1 cyclin-dependent kinase 1, CHK1 checkpoint kinase 1, GLS glutaminase
Cyclin-dependent kinase 1 (CDK1)
CDK1 is a catalytic subunit of the highly conserved protein kinase complex known as the M-phase-promoting factor, which is essential for the G1/S and G2/M-phase transitions of the eukaryotic cell cycle.61 RNAi screens for synthetic lethality in MYC overexpressing cells highlight the promise of targeting this cell-cycle kinase for Myc-dependent cancers.62 Indeed, inhibiting CDK1 function using the small molecule purvalanol A selectively induced apoptosis in cells with MYC overexpression and significantly decreased tumor growth in Myc-dependent lymphoma and hepatoblastoma mouse models.63 It appears that the selective induction of apoptosis upon CDK1 inhibition is associated with upregulation of the pro-apoptotic molecule BIM and/or downregulation of the anti-apoptotic molecule survivin.62, 63 It should be noted that CDK1 inhibition could also selectively kill transformed cells by targeting E2F-1 and/or enhancer of zeste homolog 2 (EZH2).64, 65 Most likely, multiple mechanisms contributed to the CDK1 inhibition-induced tumor regression. Nevertheless, these results suggest the potential value of targeting CDK1 in Myc-driven cancers.
Checkpoint kinase 1 (CHK1)
CHK1 has a key role in cell-cycle progression and DNA damage checkpoint control.66, 67 The use of CHK1 inhibitors to treat cancer was derived from observations that tumor cells without DNA damage checkpoints during tumorigenesis or therapy are highly sensitive to additional genomic instability.68 Myc deregulation is sufficient to induce genome instability.69 Myc induces replication stresses and DNA damages through excessive replication-fork firing, making Myc-overexpressing tumors substantially more sensitive to CHK1 inhibition.69 As such, CHK1 inhibition leads to massive cell death in Myc-overexpressing lymphomas, neuroblastomas, breast and lung cancers.70,71,72
Glutaminase (GLS)
Many tumor cells rely on glutamine metabolism to fuel their unabated growth and proliferation.73, 74 Oncogenic Myc increases the surface expression of glutamine transporters and alters mitochondrial metabolism, making the cell dependent on exogenous glutamine for survival.75,76,77,78,79 Glutamine is converted to glutamate by GLS, an enzyme that is highly expressed in tumor cells. Accordingly, inhibition of glutamine metabolism by GLS inhibitors selectively induces apoptosis in Myc-overexpressing tumor cells.75,76,77,78,79,80 Of note, CB-839, a potent and selective GLS inhibitor, is currently in phase I clinical studies for treating leukemias and other hematological tumors with Myc deregulation.81
Conclusion and perspectives
Here, we have described multiple pharmacological approaches to indirectly target Myc at different levels (Table 1). These approaches should be translated as a strategy to move forward in future patient care, as patients with Myc deregulation are likely to respond. Although direct targeting of Myc has not yet been achieved, promise remains in developing innovative approaches to effectively and specifically target this cancer super-controller. As a matter of fact, BCL-2 was also considered undruggable until a decade of fragment-based nuclear magnetic resonance (NMR) screening altered and broadened the view of this potential inhibitory molecule.82 Whether through direct or indirect targeting of Myc, better therapeutics to target Myc-dependent cancers will be required in the future.
References
Adhikary, S. & Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 6, 635–645 (2005).
Pelengaris, S., Khan, M. & Evan, G. c-MYC: more than just a matter of life and death. Nat. Rev. Cancer 2, 764–776 (2002).
Dang, C. V. MYC on the path to cancer. Cell 149, 22–35 (2012).
Dang, C. V. et al. The c-Myc target gene network. Semin. Cancer Biol. 16, 253–264 (2006).
Meyer, N. & Penn, L. Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990 (2008).
Nie, Z. et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79 (2012).
Lin, C. Y. et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67 (2012).
Shachaf, C. M. & Felsher, D. W. Tumor dormancy and MYC inactivation: pushing cancer to the brink of normalcy. Cancer Res. 65, 4471–4474 (2005).
Soucek, L. et al. Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683 (2008).
Arvanitis, C. & Felsher, D. W. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin. Cancer Biol. 16, 313–317 (2006).
Delmore, J. E. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Tavana, O. et al. HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nat. Med. 22, 1180–1186 (2016).
Otto, T. et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 15, 67–78 (2009).
Berg, T. et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl Acad. Sci. USA 99, 3830–3835 (2002).
Haynes, S. R. et al. The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 20, 2603 (1992).
Yang, Z. et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19, 535–545 (2005).
Peterlin, B. M. & Price, D. H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23, 297–305 (2006).
Dawson, M. A. et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533 (2011).
Zuber, J. et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528 (2011).
Mertz, J. A. et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl Acad. Sci. USA 108, 16669–16674 (2011).
Mazur, P. K. et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 21, 1163–1171 (2015).
Puissant, A. et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 3, 308–323 (2013).
Fisher, R. P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell. Sci. 118, 5171–5180 (2005).
Shiekhattar, R. et al. CDK-activating kinase complex is a component of human transcription factor TFIIH. Nature 374, 283–287 (1995).
Peng, J., Marshall, N. F. & Price, D. H. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J. Biol. Chem. 273, 13855–13860 (1998).
Heidemann, M., Hintermair, C., Voß, K. & Eick, D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta 1829, 55–62 (2013).
Chen, R., Keating, M. J., Gandhi, V. & Plunkett, W. Transcription inhibition by flavopiridol: mechanism of chronic lymphocytic leukemia cell death. Blood 106, 2513–2519 (2005).
Rahl, P. B. et al. c-Myc regulates transcriptional pause release. Cell 141, 432–445 (2010).
Chipumuro, E. et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159, 1126–1139 (2014).
Chapuy, B. et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790 (2013).
Kwiatkowski, N. et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620 (2014).
Garcia-Cuellar, M. P. et al. Efficacy of cyclin-dependent-kinase 9 inhibitors in a murine model of mixed-lineage leukemia. Leukemia 28, 1427–1435 (2014).
Bjornsti, M. A. & Houghton, P. J. The TOR pathway: a target for cancer therapy. Nat. Rev. Cancer 4, 335–348 (2004).
Wullschleger, S., Loewith, R. & Hall, M. N. TOR signaling in growth and metabolism. Cell 124, 471–484 (2006).
Kim, D. H. et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 (2002).
Frost, P. et al. In vivo antitumor effects of the mTOR inhibitor CCI-779 against human multiple myeloma cells in a xenograft model. Blood 104, 4181–4187 (2004).
Chapuis, N. et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin. Cancer Res. 16, 5424–5435 (2010).
Yu, K. et al. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr. Relat. Cancer 8, 249–258 (2001).
Fernández-Miranda, G. & Méndez, R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res. Rev. 11, 460–472 (2012).
Groisman, I. et al. Control of cellular senescence by CPEB. Genes Dev. 20, 2701–2712 (2006).
Ogami, K., Hosoda, N., Funakoshi, Y. & Hoshino, S. Antiproliferative protein Tob directly regulates c-myc proto-oncogene expression through cytoplasmic polyadenylation element-binding protein CPEB. Oncogene 33, 55–64 (2014).
Yada, M. et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23, 2116–2125 (2004).
Welcker, M. et al. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr. Biol. 14, 1852–1857 (2004).
Popov, N. et al. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 9, 765–774 (2007).
Sun, X. X. et al. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl Acad. Sci. USA 112, 3734–3739 (2015).
Bischoff, J. R. & Plowman, G. D. The Aurora/Ipl1p kinase family: regulators of chromosome segregation and cytokinesis. Trends Cell Biol. 9, 454–459 (1999).
Sasai, K., Treekitkarnmongkol, W., Kai, K., Katayama, H. & Sen, S. Functional significance of Aurora kinases-p53 protein family interactions in cancer. Front Oncol. 6, 247 (2016).
Takahashi, Y. et al. The AURKA/TPX2 axis drives colon tumorigenesis cooperatively with MYC. Ann. Oncol. 26, 935–942 (2015).
Yang, S. et al. Suppression of Aurora-A oncogenic potential by c-Myc downregulation. Exp. Mol. Med. 42, 759–767 (2010).
Brockmann, M. et al. Small molecule inhibitors of Aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 24, 75–89 (2013).
Dauch, D. et al. A MYC-Aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 22, 744–753 (2016).
Golsteyn, R. M., Lane, H. A., Mundt, K. E., Arnaud, L. & Nigg, E. A. The family of polo-like kinases. Prog. Cell Cycle Res. 2, 107–114 (1996).
Xiao, D. et al. Polo-like kinase-1 regulates Myc stabilization and activates a feedforward circuit promoting tumor cell survival. Mol. Cell 64, 493–506 (2016).
Nair, S. K. & Burley, S. K. X-ray structures of Myc–Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 112, 193–205 (2003).
Wang, H. et al. Improved low molecular weight Myc–Max inhibitors. Mol. Cancer Ther. 6, 2399–2408 (2007).
Soucek, L. et al. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 17, 2463–2472 (1998).
Soucek, L., Nasi, S. & Evan, G. I. Omomyc expression in skin prevents Myc-induced papillomatosis. Cell Death Differ. 11, 1038–1045 (2004).
Annibali, D. et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 5, 4632 (2014).
Kaelin, W. G. Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 5, 689–698 (2005).
Malumbres, M. & Barbacid, M. Cell cycle, cdks and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 (2009).
Kang, J., Sergio, C. M., Sutherland, R. L. & Musgrove, E. A. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer 14, 32 (2014).
Goga, A., Yang, D., Tward, A. D., Morgan, D. O. & Bishop, J. M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 13, 820–827 (2007).
Peeper, D. S. et al. Phosphorylation of a specific cdk site in E2F-1 affects its electrophoretic mobility and promotes pRB-binding in vitro. Oncogene 10, 39–48 (1995).
Göllner, S. et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 23, 69–78 (2017).
Zhao, H. & Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 21, 4129–4139 (2001).
Furnari, B., Rhind, N. & Russell, P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science 277, 1495–1497 (1997).
Höglund, A. et al. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin. Cancer Res. 17, 7067–7079 (2011).
Dominguez-Sola, D. et al. Non-transcriptional control of DNA replication by c-Myc. Nature 448, 445–451 (2007).
Cole, K. A. et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl Acad. Sci. USA 108, 3336–3341 (2011).
Murga, M. et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 18, 1331–1335 (2011).
Ferrao, P. T., Bukczynska, E. P., Johnstone, R. W. & McArthur, G. A. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 31, 1661–1672 (2012).
DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G. & Thompson, C. B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 (2008).
Dang, C. V. A time for MYC: metabolism and therapy. Cold Spring Harb. Symp. Quant. Biol. 81, 79–83 (2016).
Yuneva, M., Zamboni, N., Oefner, P., Sachidanandam, R. & Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 178, 93–105 (2007).
Gao, P. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 (2009).
Mullen, A. R. et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388 (2011).
Metallo, C. M. et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2011).
Qing, G. et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 22, 631–644 (2012).
Xiao, D. et al. Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget 6, 40655–40666 (2015).
Jacque, N. et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 126, 1346–1356 (2015).
Superti-Furga, G. et al. Where is the future of drug discovery for cancer? Cell 168, 564–565 (2017).
Fong, C. Y. et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–542 (2015).
Matkar, S. et al. An epigenetic pathway regulates sensitivity of breast cancer cells to HER2 inhibition via FOXO/c-Myc axis. Cancer Cell 28, 472–485 (2015).
Cicenas, J. et al. Roscovitine in cancer and other diseases. Ann. Transl. Med 3, 135 (2015).
Awan, F. T. et al. A phase 1 clinical trial of flavopiridol consolidation in chronic lymphocytic leukemia patients following chemoimmunotherapy. Ann. Hematol. 95, 1137–1143 (2016).
Vignot, S., Faivre, S., Aguirre, D. & Raymond, E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann. Oncol. 16, 525–537 (2005).
Xu, C. X. et al. The combination of RAD001 and NVP-BEZ235 exerts synergistic anticancer activity against non-small cell lung cancer in vitro and in vivo. PLoS ONE 6, e20899 (2011).
Pal, S. K., Reckamp, K., Yu, H. & Figlin, R. A. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin. Investig. Drugs 19, 1355–1366 (2010).
Blackburn, J. S. et al. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell 25, 366–378 (2014).
Gjertsen, B. T. & Schoffski, P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 29, 11–19 (2015).
Schoffski, P. et al. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur. J. Cancer 48, 179–186 (2012).
Sebastian, M. et al. The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase II clinical trial. J. Thorac. Oncol. 5, 1060–1067 (2010).
Awad, M. M. et al. An open-label, phase II study of the polo-like kinase-1 (Plk-1) inhibitor, BI 2536, in patients with relapsed small cell lung cancer (SCLC). Lung Cancer 104, 126–130 (2017).
Cassaday, R. D. et al. A phase II, single-arm, open-label, multicenter study to evaluate the efficacy and safety of P276-00, a cyclin-dependent kinase inhibitor, in patients with relapsed or refractory mantle cell lymphoma. Clin. Lymphoma Myeloma Leuk. 15, 392–397 (2015).
Sen, T. et al. CHK1 inhibition in small-cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res. 77, 3870–3884 (2017).
Hong, D. et al. Phase I study of LY2606368, a checkpoint kinase 1 inhibitor, in patients with advanced cancer. J. Clin. Oncol. 34, 1764–1771 (2016).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81470332 to H.L. and 81372205 to G.Q.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, H., Liu, H. & Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Sig Transduct Target Ther 3, 5 (2018). https://doi.org/10.1038/s41392-018-0008-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-018-0008-7
This article is cited by
-
C-Myc protein expression indicates unfavorable clinical outcome in surgically resected small cell lung cancer
World Journal of Surgical Oncology (2024)
-
Enhancing protein production and growth in chinese hamster ovary cells through miR-107 overexpression
AMB Express (2024)
-
Marinopyrrole derivative MP1 as a novel anti-cancer agent in group 3 MYC-amplified Medulloblastoma
Journal of Experimental & Clinical Cancer Research (2024)
-
Exploration of tumor growth regression of quinoa and chia oil nanocapsules via the control of PIK3CA and MYC expression, anti-inflammation and cell proliferation inhibition, and their hepatorenal safety in rat breast cancer model
Bulletin of the National Research Centre (2024)
-
CLK3 promotes tumor proliferation by activating MYC signaling
Cell Division (2024)
