
Abstract
Prostate cancer (PrCa) is one of the most common cancers in men, but little is known about factors affecting its clinical outcomes. Genome-wide association studies have identified more than 170 germline susceptibility loci, but most of them are not associated with aggressive disease. We performed a genome-wide analysis of 185,478 SNPs in Finnish samples (2738 cases, 2400 controls) from the international Collaborative Oncological Gene-Environment Study (iCOGS) to find underlying PrCa risk variants. We identified a total of 21 common, low-penetrance susceptibility loci, including 10 novel variants independently associated with PrCa risk. Novel risk loci were located in the 8q24 (CASC8 rs16902147, OR 1.86, padj = 3.53 × 10−8 and rs58809953, OR 1.71, padj = 4.00 × 10−6; intergenic rs79012498, OR 1.81, padj = 4.26 × 10−8), 17q21 (SP6 rs2074187, OR 1.66, padj = 3.75 × 10−5), 11q13 (rs12795301, OR 1.42, padj = 2.89 × 10−5) and 8p21 (rs995432, OR 1.38, padj = 3.00 × 10−11) regions. Here, we describe SP6, a transcription factor gene, as a new, potentially high-risk gene for PrCa. The intronic variant rs2074187 in SP6 was associated not only with overall susceptibility to PrCa (OR 1.66) but also with a higher odds ratio for aggressive PrCa (OR 1.89) and lower odds for non-aggressive PrCa (OR 1.43). Furthermore, the new intergenic variant rs79012498 at 8q24 conferred risk for aggressive PrCa. Our findings highlighted the power of a population-stratified approach to identify novel, clinically actionable germline PrCa risk loci and strongly suggested SP6 as a new PrCa candidate gene that may be involved in the pathogenesis of PrCa.
Similar content being viewed by others

Introduction
Prostate cancer (PrCa) is the second most common cancer in males and the fifth leading cause of cancer death in men worldwide (2018) [1, 2]. In Finland, it accounts for 29.6% of all newly diagnosed cancer cases and for 13.6% of all cancer deaths based on the latest NORDCAN (Cancer statistics for the Nordic countries) data (2012–2016) [3].
PrCa has a major heritable component with genetic factors accounting for 57% (95% CI 51–63%) of the variation in risk in the Nordic Twin Study of Cancer [4]. To date, genome-wide association studies (GWAS) have identified over 170 low-penetrance PrCa susceptibility loci, predominantly in populations of mixed European ancestry [5,6,7,8,9]. However, only a few of the identified susceptibility variants are associated with clinically relevant aggressive or advanced disease.
Previous studies have established that genetic loci, effect allele frequencies (EAF) and the strength of association (odds ratio, OR) are highly variable across geographic regions [8, 10]. In bottlenecked and isolated populations such as the Finns, many functional variants are present at relatively high frequencies because of increased drift and reduced selective pressure [11], whereas in larger outbred populations, deleterious alleles occur at low frequencies due to selection [12]. Hence, recent isolates like the Finns provide an ideal opportunity to discover disease-associated genes as underlying and initially rare variants can be encountered at higher frequencies.
In an effort to discover novel, potentially clinically actionable germline PrCa biomarkers, we conducted a genome-wide association analysis of the Finnish samples in the international Collaborative Oncological Gene-Environment Study (iCOGS).
Materials and methods
Study design
This study is a Finnish population-specific analysis of the single nucleotide polymorphisms (SNPs) in the iCOGS genome-wide custom genotyping array [5] for overall PrCa risk and subsequent testing for aggressive disease. The iCOGS study was designed by the international consortium to detect genetic variants related to prostate, breast and ovarian cancers [13]. The study protocol was approved as described in the iCOGS study [5].
Study participants
After application of quality control criteria, the final analysis was based on 2738 PrCa cases and 2400 controls without a known diagnosis of PrCa. Of the cases, 2283 were clinically diagnosed patients from the Pirkanmaa Hospital District confirmed from medical records. Another set of patients consisted of 455 cases of the Finnish Randomized Study of Screening for Prostate Cancer (FinRSPC) [14, 15]. The FinRSPC trial population and the study protocol have been described in detail elsewhere [16]. Cancer free control subjects (n = 2.400) were identified through the FinRSPC trial [14]. All of the samples were collected with written and signed informed consent. The cancer diagnoses were confirmed using medical records and the Finnish Cancer Registry. The study was approved by the research Ethics committee at Pirkanmaa Hospital District (tracking numbers R10167, 90577, R03203) and by the National Supervisory Authority for Welfare and Health (VALVIRA).
Clinical characteristics of the genotyped PrCa patients are summarised in Table 1. PSA at diagnosis was classified as ≤20 versus >20 ng/mL. Gleason score was divided into ≤6, 7 and ≥8. Stage was divided into organ-confined (T1-2, N0/x, M0/x) versus advanced disease (T3–4, or N1 or M1). PrCa death was defined based on the underlying cause recorded as the official cause of death by Statistics Finland. Aggressive PrCa was defined as having PSA at diagnosis >20 ng/mL, or Gleason Score ≥8, or T3/T4, or N1, or M1, or PrCa-specific mortality (PCM). Comprehensive definition of non-aggressive PrCa was the following: PSA at diagnosis ≤20 ng/mL, and Gleason Score ≤6, and not T3/T4, and not N1, and not M1, and not PCM.
Genotype quality control
Altogether, n = 211,155 SNPs were genotyped in iCOGS for Finnish subjects. Systematic quality control (QC) steps were conducted on the raw iCOGS genotyping data. Females, individuals with low call rate, individuals with extreme heterozygosity, known or cryptic duplicates, individuals not matching previous genotyping and ethnic outliers have been left out. Subsequently, first-degree relatives, duplicate subjects, and cases missing clinical data have been removed as well. The exclusion criteria for SNPs were a genotyping call rate less than 95%, failing the missingness test (GENO > 1, default ---geno value of 0.0 was used), minor allele frequency (MAF < 1 × 10−6 or > 0.499) and genotype frequency that deviated from expected Hardy–Weinberg equilibrium among control samples (P ≤ 0.05). After frequency and genotype pruning, 185,478 SNPs were retained for analysis [5].
Statistical analyses
Standard procedures for case-control GWAS were executed [17, 18]. The association between each SNP and PrCa was estimated by per-allele OR and 95% CI using unconditional logistic regression implemented in PLINK (v1.07) [19] assuming an additive genetic model. We used a p value threshold of 5.0 × 10−8 to determine genome-wide significance. False discovery rate (FDR)-adjusted significance was set to padj < 4 × 10−5 using the Benjamini–Hochberg method. The EAF was set to >5%. The Hardy–Weinberg equilibrium equation was used to determine whether the proportion of each genotype obtained was in agreement with the expected values as calculated from the allele frequencies.
Identified PrCa susceptibility variants were pruned by pairwise threshold, removing loci with a high level of linkage disequilibrium (LD) (r2 > 0.5), resulting in independent signals for PrCa risk (PLINK v1.07) [19]. The variants were then tested in a case-control setting for the risk of aggressive PrCa defined by a Gleason Score ≥8 and for the risk of non-aggressive PrCa defined by a Gleason Score ≤6. In addition, we assessed the association of the genetic variants with the comprehensively defined entity of aggressive PrCa and with the comprehensively defined non-aggressive PrCa (for definition see above) (IBM® SPSS® Statistics Version26 for Mac SPSS Inc., Chicago, IL, USA).
Annotation
Ensembl was used for gene annotation [20] indicating HGNC gene symbols from the HUGO Gene Nomenclature Committee [21]. Variant annotation and functional effect prediction was performed with the Variant Effect Predictor (VEP) [22] and SnpEff [23].
Results
Prostate cancer susceptibility
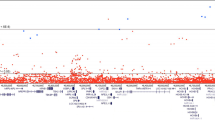
Altogether, we identified 160 PrCa susceptibility loci at GWAS significance (p < 5 × 10−8, padj < 4 × 10−5, Supplementary Table 1 and Supplementary Fig. 1). After genotype pruning, 21 common, low-penetrance susceptibility loci were independently associated with PrCa risk with per-allele ORs ranging between 1.86 and 0.74 (Table 2.). Association of the 10 novel variants with malignant neoplasm of prostate has been validated using the FinnGen and UKBB biobank data (Supplementary Table 2, http://r3.finngen.fi)
In this study, the EAFs of common PrCa susceptibility variants ranged between 0.06 and 0.53. The identified PrCa risk loci spanned nine different gene regions altogether with five of the associated loci being intergenic. Most of the PrCa susceptibility variants were detected in the CASC8 gene (n = 8), whereas SP6, CASC17, JAZF1, HNF1B, KLK2, KLK3, AC011523.2, and LINC02086 possessed a single variant each.
Based on functional annotation of the identified PrCa-associated variants, intronic variants were most frequent (10 SNPs, 48%), followed by intergenic variants (5 SNPs, 24%), and there were equal numbers of upstream and downstream intronic gene variants (both 3 SNPs, 14%). These findings highlight the possible importance of transcriptional regulation in PrCa.
The identified 13 risk signals were condensed at chromosomal regions 8q24, 17q21, 11q13, 8p21, and 17q12 (OR 1.86–1.26), whereas the eight protective variants were situated at 19q13, 8q24, 7p15, and 17q24 (OR 0.72–0.80). Chromosomes 11 and 17 appeared to be exclusively risk-conferring, whereas chromosomes 7 and 19 possessed solely protective variants. Exclusively risk genes identified in this study were predominantly transcription factors (SP6, HNF1B, LINC02086). On the other hand, SNPs in CASC17, KLK2, KLK3, JAZF1, and AC011523.2 were solely protective.
The strongest risk effect was found for the novel intronic variant rs16902147 in the CASC8 (cancer susceptibility candidate 8) gene at 8q24 with an OR of 1.86 (95% CI 1.56–2.23; padj = 3.53 × 10−8) and EAF of 0.07. The statistically most significant signal originated from the intergenic variant (RP11-583M2.2-NKX3-1) rs995432 at 8p21 (padj = 3.00 × 10−11). This finding confirmed the previous GWAS findings at these genomic locations [24, 25] and strengthened these observations with the new variants.
Out of the 21 identified PrCa susceptibility hits, 10 (48%) were novel variants not reported earlier in association with PrCa susceptibility. Novel loci with high effect sizes were located in 8q24 (CASC8 rs16902147, OR 1.86, padj = 3.53 × 10−8 and rs58809953, OR 1.71, padj = 4.00 × 10−6; intergenic rs79012498, OR 1.81, padj = 4.26 × 10−8) and 17q21 (SP6 rs2074187, OR 1.66, padj = 3.75 × 10−5) regions and had low EAF (≤0.08). Additionally, two novel intergenic variants, rs12795301 at 11q13 (OR 1.42, padj = 2.89 × 10−5) and rs995432 at 8p21 (OR 1.38, padj = 3.00 × 10−11), showed risk for overall PrCa. Novel protective variants were located in CASC8 (rs4871790 and rs587948, for both OR 0.80), KLK2 (rs2739459, OR 0.79) and JAZF1 (rs757138, OR 0.78) genes. Interestingly, they showed relatively high EAFs of 0.26–0.45. The most important finding was a possible new PrCa risk gene, SP6, that had not yet been implicated as a potential causal gene for PrCa.
Aggressive prostate cancer susceptibility
To explore whether the identified PrCa susceptibility loci were associated with aggressive disease, we analysed their association with a high Gleason Score ≥8 and a low Gleason Score ≤6 and with comprehensively defined aggressive PrCa and non-aggressive PrCa (see Methods). Findings are summarised in Table 3. The intronic variant rs2074187 in SP6 was associated with higher OR for high Gleason score disease (OR 2.09, p = 0.000005) than for low Gleason score disease (OR 1.50, p = 0.0004) or overall PrCa (OR 1.66, p = 3.752 × 10−5). Similarly, it was associated with a higher effect size for comprehensively defined aggressive PrCa (OR 1.89, p = 4.738 × 10−8) than non-aggressive PrCa (OR 1.43, p = 0.008) or overall PrCa (OR 1.66, p = 3.752 × 10−5). Furthermore, we revealed an association between the new intergenic variant rs79012498 at 8q24 (PRNCR1-CASC19) and aggressive PrCa. The ORs for high Gleason score and aggressive PrCa (OR 2.14 and OR 2.10, respectively) were higher than for low Gleason score and non-aggressive PrCa (OR 1.76 and OR 1.57, respectively), or for overall PrCa (OR 1.81).
The EAF for both the SP6 rs2074187 and the intergenic rs79012498 variant was clearly higher in aggressive PrCa compared to non-aggressive PrCa (p ≤ 0.05) or in controls (p < 0.00001).
Discussion and conclusions
This population-specific GWAS addressed the major challenge of the basis of inheritance of PrCa by discovering germline biomarkers for aggressive disease in the Finnish population. We identified 21 independent PrCa susceptibility loci demonstrating statistically significant association after FDR correction, including 10 novel germline variants. In addition, we not only proposed SP6 as a new PrCa risk gene that had not yet been implicated as a potential causal gene for PrCa, but we also linked the SP6 rs2074187 intronic variant to aggressive disease outcomes. Furthermore, we showed a new intergenic variant (rs79012498) at 8q24 PRNCR1-CASC19 conferred risk of aggressive PrCa.
The vast majority of the 21 identified PrCa susceptibility variants were intronic in this study. Non-coding variants were reported to play a role in distinguishing PrCa, metastatic PrCa, and castration-resistant metastatic PrCa [26] and could pave the way for identifying novel treatment paradigms [27]. Mechanistic explanations for the effect of some non-coding variants do exist. For example, the rs11672691 SNP at 19q13 was associated with aggressive PrCa and creates a transcription factor binding site that in turn promotes oncogenesis by impacting expression of nearby genes [28].
Previous studies have demonstrated the utility of bottleneck populations to enable the discovery of rare but high-impact, disease-associated variants due to their enrichment in these populations [29,30,31]. Our study suggests a similar phenomenon with the 10 newly identified PrCa susceptibility loci. Interestingly, the EAF of the new risk variants was rather low (EAF 0.07–0.013), which might be the result of genetic drift [11]. Except for the rs995432 SNP at 8p21 (EAF 0.53). In contrast, the EAF of the new protective variants are condensed at high levels (EAF 0.26–0.45), and the EAFs of earlier reported PrCa risk alleles are uniformly distributed [6, 7].
The direction and strength of the associations of the PrCa-related variants often differ across populations. The per-allele OR of the new PrCa risk variants found in this study was in the higher range (OR 1.86–1.38) of previously identified, common, low-penetrance PrCa susceptibility loci as reviewed [5,6,7, 32], where each variant individually modestly modified the risk of PrCa. Similarly, the protective variants described here (OR 0.78–0.80) were more protective than the earlier established SNPs [5,6,7, 32].
To date, one of the strongest PrCa risk factors is the newly identified rs16902147 and rs58809953 in CASC8. CASC8 is a long non-coding RNA (lncRNA) gene located in the gene desert region of 8q24 near the MYC gene [33]. CASC8 gene itself has been implicated in PrCa risk [34] as its variants could potentially affect transcription factor binding [33]. The 8q24 region is a known PrCa susceptibility hot-spot, harbouring multiple risk variants where lncRNAs have been implicated [35]. Our findings support earlier observations that lncRNAs at 8q24 play a key role in PrCa aetiology [36,37,38].
The three newly identified intergenic risk variants were located in 11q13, 8p21 and 8q24. The 11q13 region has been previously linked to PrCa risk, where a rare intronic variant (IVS6-43A > G) in the EMSY gene has been associated with aggressive unselected PrCa cases [39]. Nurminen et al. found two more independent regions at 11q13 associated with PrCa risk (rs10899221 in EMSY, rs12277366 intergenic) [40]. Previous research has pointed to the 8p21 region [41] where frequent alteration in the prostate oncogenome has been associated with loss of androgen-regulated prostate-specific NKX3.1 homeobox transcription factor gene [42].
The rs79012498 novel intergenic variant at 8q24 was associated with aggressive PrCa in this study. It lies at the hypothetical locus LOC105375752 of a lncRNA gene between PRNCR1 and CASC19. The LOC105375752 locus itself has been reported to be a PrCa GWAS locus [43, 44] but has not been associated with aggressive PrCa. PRNCR1 (PCAT8) is similarly a lncRNA and reported PrCa risk locus [44]. PRNCR1 is highly overexpressed in aggressive PrCa [45]. PRNCR1, together with PCGEM1, bind to an androgen receptor (AR) and strongly enhance androgen-receptor-mediated gene activation programmes and proliferation in PrCa cells, thereby circumventing androgen-deprivation therapy [45]. PRNCR1 is upregulated in PrCa and prostatic intraepithelial neoplasia cells and attenuates cell viability and activity of the AR when knocked down [46]. The other nearest gene to the rs79012498 variant is CASC19 (cancer susceptibility 19), which is likewise a tumour risk lncRNA gene [44]. A rare segregating haplotype, including PRNCR1 and CASC19 gene variants in the region of 8q24, has been identified in familial PrCa samples as a cancer predisposition locus [37].
The newly identified SP6 candidate gene for PrCa is a transcription factor gene [47]. Transcription factors are cellular proteins, and by regulating the transcription of genes they offer promising therapeutic targets for RNA interference therapy in PrCa [48]. The SP6 gene, also known as EPFN or KLF14 or EPIPROFIN, encodes an intracellular transcription factor protein. It belongs to a family of transcription factors that contain 3 classical zinc finger DNA-binding domains consisting of a zinc atom tetrahedrally coordinated by 2 cysteines and 2 histidines (C2H2 motif). These transcription factors bind to GC-rich sequences and related GT and CACCC boxes [49]. Interestingly, SP6 RNA expression is enhanced in ductus deferens, seminal vesicles and placenta, but not in prostate [50]. Predicted localisation is intracellular and, mainly in the nucleoplasm. SP6 has two transcripts and different splice variants. Variant rs2074187 in SP6 was associated with aggressive PrCa risk and suggestively shows potential as a novel germline genetic marker. This SNP encodes transcript variant 1, which represents the longer transcript of the gene [51]. The higher effect size of rs2074187, differentiating aggressive PrCa (OR 1.89) from non-aggressive disease (OR 1.43), is remarkable compared to previously identified aggressive loci (OR 1.12–2.3) [52,53,54], and Supplementary Table 3. The EAF of 0.07 in Finnish cases in our discovery cohort is comparable with EAFs of earlier identified aggressive PrCa risk loci [53,54,55].
Interestingly, the SP6 transcription factor gene is located in 17q21, which is close to HOXB13. The G84E mutation of HOXB13 has been linked to significantly increased PrCa risk [56, 57], especially in Finns. Previously, we showed a synergistic effect between HOXB13 (G84E) and CIP2A (R229Q) strongly predisposing patients to aggressive PrCa [55]. However, the HOXB13 G84E risk variant only partially explained the linkage signal to 17q21 observed in Finns earlier [58]. Our finding of SP6 as a new, potential PrCa risk gene may explain the remaining part of this linkage, which warrants follow-up.
SP6 was previously associated with β-catenin-mediated prostate tumourigenesis [59]. The confounding role of androgen signalling in β-catenin-mediated oncogenic transformation in prostate tumourigenesis has been shown through upregulation of the SP6 gene among others in microarray analyses of transcriptional profiles in mice [59].
SP6 has also been implicated in breast cancer therapy resistance and linked to the regulation of the Wnt-BMP signalling pathway [60]. An important paralog of the SP6 protein coding gene is SP8, which was previously identified as a candidate gene (rs12155172, p = 4.95 × 10−13) associated with PrCa susceptibility in European ancestry samples [5].
Like the SP6 gene, many of the previously identified PrCa genes are transcription factors (e.g., HOXB13, AR, HNF1B, FOXA1, NKX3.1), and their binding is often affected by sequence variations [61]. DNA transcription-related genes have been justified as the largest molecular functional group in gene set enrichment analyses [62]. This finding may point to the possible implications of RNA interference therapy in the future [48].
In summary, we report a new PrCa risk gene, SP6, that is also associated with aggressive disease outcomes. Findings in this study demonstrate the utility of population-specific approach and the power of homogenous populations to discover disease-specific SNPs that have not been revealed in mixed European studies.
At the same time, homogeneous population material provided a resource to validate previous findings from mixed European populations shown by finding a number of previously identified, important PrCa susceptibility genes (CASC8, HNF1B, JAZF1, CASC17, KLK2, KLK3).
This population-specific approach is further strengthened and justified by the FinnGen study identifying top hits for malignant neoplasma of prostate, e.g. POU5F1B, HOXB13, HOXB7, SKAP, NPEPPS, GNGT2 (http://r3.finngen.fi/top_hits).
Consequently, this study reports a novel gene and candidate variants for investigation of the pathogenesis of PrCa. Variants presented in this study are optimal candidates for functional studies to further investigate the molecular mechanisms and biological effects underlying this association and the role of the 17q21 and 8q24 regions in PrCa development.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2018;68:394–42.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. Cancer J Clin. 2019;69:7–34.
Engholm G, Ferlay J, Christensen N, Bray F, Gjerstorff ML, Klint A, et al. NORDCAN — A Nordic tool for cancer information, planning, quality control and research. Acta Oncol. 2010;49:725-36.
Mucci LA, Hjelmborg JB, Harris JR, Czene K, Havelick DJ, Scheike T, et al. Nordic Twin Study of Cancer (NorTwinCan) Collaboration. Familial risk and heritability of cancer among twins in Nordic countries. JAMA 2016;315:68–76.
Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, et al. UK ProtecT (Prostate testing for cancer and Treatment) Study Collaborators, PRACTICAL (Prostate Cancer Association Group to Investigate Cancer-Associated Alterations in the Genome) Consortium, Kote-Jarai Z, Easton DF. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet. 2013;45:385–91.
AAl Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet. 2014;46:1103–9.
Eeles R, Goh C, Castro E, Bancroft E, Guy M, Al Olama AA, et al. The genetic epidemiology of prostate cancer and its clinical implications. Nat Rev Urol. 2014;11:18.
Virlogeux V, Graff RE, Hoffmann TJ, Witte JS. Replication and heritability of prostate cancer risk variants: impact of population-specific factors. Cancer Epidemiol Prev Biomark. 2015;24:938–43.
Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, Saunders EJ, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet. 2018;50:928–36.
Kote-Jarai Z, Mikropoulos C, Leongamornlert DA, Dadaev T, Tymrakiewicz M, Saunders EJ, et al. Prevalence of theHOXB13 G84E germline mutation in British men and correlation with prostate cancer risk, tumour characteristics and clinical outcomes. Ann Oncol. 2015;26:756–61.
Chheda H, Palta P, Pirinen M, McCarthy S, Walter K, Koskinen S, et al. Whole-genome view of the consequences of a population bottleneck using 2926 genome sequences from Finland and United Kingdom. Eur J Hum Genet. 2017;25:477–84.
Goldstein DB, Allen A, Keebler J, Margulies EH, Petrou S, Petrovski S, et al. Sequencing studies in human genetics: design and interpretation. Nat Rev Genet. 2013;14:460–70.
Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium. http://practical.icr.ac.uk
Schröder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, Nelen V, et al. Screening and prostate-cancer mortality in a randomized European study. N. Engl J Med. 2009;360:1320–8.
Schröder FH, Hugosson J, Roobol MJ, Tammela TL, Zappa M, Nelen V, et al. Screening and prostate cancer mortality: results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow-up. Lancet. 2014;384:2027–35.
Finne P, Stenman UH, Määttänen L, Mäkinen T, Tammela TL, Martikainen P, et al. The Finnish trial of prostate cancer screening: where are we now? BJU Int. 2003;92:22–6.
Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT. Data quality control in genetic case-control association studies. Nat Protoc. 2010;5:1564–73.
Bush WS, Moore JH. Genome-wide association studies. PLoS Comput Biol. 2012;8:e1002822.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, et al. Ensembl 2017. Nucleic Acids Res. 2017;45:D635–42.
Yates B, Braschi B, Gray KA, Seal RL, Tweedie S, Bruford EA. Genenames. org: the HGNC and VGNC resources in 2017. Nucleic Acids Res. 2016. https://doi.org/10.1093/nar/gkw1033.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–70.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92.
Eeles RA, Kote-Jarai Z, Al Olama AA, Giles GG, Guy M, Severi G, et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet. 2009;41:1116–21.
Takata R, Akamatsu S, Kubo M, Takahashi A, Hosono N, Kawaguchi T, et al. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat Genet. 2010;42:751–4.
Alanazi IO, Al Shehri ZS, Ebrahimie E, Giahi H, Mohammadi-Dehcheshmeh M. Non-coding and coding genomic variants distinguish prostate cancer, castration-resistant prostate cancer, familial prostate cancer, and metastatic castration-resistant prostate cancer from each other. Mol Carcinogen. 2019;58:862–74.
Baumgart SJ, Nevedomskaya E, Haendler B. Dysregulated transcriptional control in prostate cancer. Int J Mol Sci. 2019;20:2883.
Gao P, Xia JH, Sipeky C, Dong XM, Zhang Q, Yang Y, et al. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell. 2018;174:576–89.
Stoll G, Pietiläinen OP, Linder B, Suvisaari J, Brosi C, Hennah W, et al. Deletion of TOP3β, a component of FMRP-containing mRNPs, contributes to neurodevelopmental disorders. Nat Neurosci. 2013;16:1228–37.
Lim ET, Würtz P, Havulinna AS, Palta P, Tukiainen T, Rehnström K, et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10:e1004494.
Sidore C, Busonero F, Maschio A, Porcu E, Naitza S, Zoledziewska M, et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet. 2015;47:1272.
Eeles RA, Kote-Jarai Z, Giles GG, Al Olama AA, Guy M, Jugurnauth SK, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316.
Loo LW, Fong AY, Cheng I, Le Marchand L. In silico functional pathway annotation of 86 established prostate cancer risk variants. PloS ONE. 2015;10:e0117873.
Hao Q, Wei D, Zhang Y, Chen X, Yang F, Yang Z, et al. Systematic meta-analyses of gene-specific genetic association studies in prostate cancer. Oncotarget. 2016;7:22271.
Han Y, Rand KA, Hazelett DJ, Ingles SA, Kittles RA, Strom SS, et al. Prostate cancer susceptibility in men of African ancestry at 8q24. J Natl Cancer Inst. 2016;108:djv431.
Li C, Yang L, Lin C. Long noncoding RNAs in prostate cancer: mechanisms and applications. Mol Cell Oncol. 2014;1:e963469.
Teerlink CC, Leongamornlert D, Dadaev T, Thomas A, Farnham J, Stephenson RA, et al. Genome-wide association of familial prostate cancer cases identifies evidence for a rare segregating haplotype at 8q24. 21. Hum Genet. 2016;135:923–38.
Xu T, Lin CM, Cheng SQ, Min J, Li L, Meng XM, et al. Pathological bases and clinical impact of long noncoding RNAs in prostate cancer: a new budding star. Mol Cancer. 2018;17:1–7.
Nurminen R, Wahlfors T, Tammela TL, Schleutker J. Identification of an aggressive prostate cancer predisposing variant at 11q13. Int J Cancer. 2011;129:599–606.
Nurminen R, Lehtonen R, Auvinen A, Tammela TL, Wahlfors T, Schleutker J. Fine mapping of 11q13. 5 identifies regions associated with prostate cancer and prostate cancer death. Eur J Cancer. 2013;49:3335–43.
Zeegers MP, Nekeman D, Khan HS, Van Dijk BA, Goldbohm RA, Schalken J, et al. Prostate cancer susceptibility genes on 8p21–23 in a Dutch population. Prostate Cancer Prostat Dis. 2013;16:248–53.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22.
Rebbeck TR. Prostate cancer genetics: variation by race, ethnicity, and geography. Semin Radiat Oncol. 2017;27:3–10.
GWAS Catalog. 2020. https://www.ebi.ac.uk/gwas/.
Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013;500:598–602.
Chung S, Nakagawa H, Uemura M, Piao L, Ashikawa K, Hosono N, et al. Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 2011;102:245–52.
Gene Database SP6 gene. 2020. https://www.ncbi.nlm.nih.gov/gene/80320.
Fitzgerald KA, Evans JC, McCarthy J, Guo J, Prencipe M, Kearney M, et al. The role of transcription factors in prostate cancer and potential for future RNA interference therapy. Expert Opin Therap Targets. 2014;18:633–49.
Scohy S, Gabant P, Van Reeth T, Hertveldt V, Drèze PL, Van Vooren P, et al. Identification of KLF13 and KLF14 (SP6), novel members of the SP/XKLF transcription factor family. Genomics. 2000;70:93–101.
Human Protein Atlas. https://www.proteinatlas.org/ENSG00000189120-SP6/tissue. 2020. https://www.proteinatlas.org/ENSG00000189120-SP6/tissue.
dbSNP rs2074187. 2020. https://www.ncbi.nlm.nih.gov/snp/rs2074187.
Duggan D, Zheng SL, Knowlton M, Benitez D, Dimitrov L, Wiklund F, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst. 2007;99:1836–44.
FitzGerald LM, Kwon EM, Conomos MP, Kolb S, Holt SK, Levine D, et al. Genome-wide association study identifies a genetic variant associated with risk for more aggressive prostate cancer. Cancer Epidemiol Prev Biomark. 2011;20:1196–203.
Amin Al Olama A, Kote-Jarai Z, Schumacher FR, Wiklund F, Berndt SI, Benlloch S, et al. PRACTICAL Consortium A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum Mol Genet. 2013;22:408–15.
Sipeky C, Gao P, Zhang Q, Wang L, Ettala O, Talala KM, et al. Synergistic interaction of hoxb13 and cip2a predisposes to aggressive prostate cancer. Clin Cancer Res. 2018;24:6265–76.
Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, Wiley KE, Isaacs SD, Johng D, et al. Germline mutations in HOXB13 and prostate-cancer risk. N. Engl J Med. 2012;366:141–9.
Laitinen VH, Wahlfors T, Saaristo L, Rantapero T, Pelttari LM, Kilpivaara O, et al. HOXB13 G84E mutation in Finland: population-based analysis of prostate, breast, and colorectal cancer risk. Cancer Epidemiol Prev Biomark 2013;22:452–6.
Laitinen VH, Rantapero T, Fischer D, Vuorinen EM, Tammela TL, Practical Consortium. et al. Fine-mapping the 2q37 and 17q11. 2-q22 loci for novel genes and sequence variants associated with a genetic predisposition to prostate cancer. Int J Cancer. 2015;136:2316–27.
Lee SH, Luong R, Johnson DT, Cunha GR, Rivina L, Gonzalgo ML, et al. Androgen signaling is a confounding factor for β-catenin-mediated prostate tumorigenesis. Oncogene. 2016;35:702–14.
Ibarretxe G, Aurrekoetxea M, Crende O, Badiola I, Jimenez-Rojo L, Nakamura T, et al. Epiprofin/Sp6 regulates Wnt-BMP signaling and the establishment of cellular junctions during the bell stage of tooth development. Cell Tissue Res. 2012;350:95–107.
Hazelett DJ, Rhie SK, Gaddis M, Yan C, Lakeland DL, Coetzee SG, et al. Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet. 2014;10:e1004102.
Dai JY, Wang X, Wang B, Sun W, Jordahl KM, Kolb S, et al. DNA methylation and cis-regulation of gene expression by prostate cancer risk SNPs. PLoS Genet. 2020;16:e1008667.
Acknowledgements
The authors particularly thank the patients, who participated in this study. FinRSPC Study Group members are acknowledged for the FinRSPC cohort: Ulf-Håkan Stenman, Faculty of Medicine, University of Helsinki, Finland and Department of Clinical Chemistry, Helsinki University Hospital, Helsinki, Finland; Paula Kujala, Department of Pathology, Fimlab Laboratories, Tampere, Finland; Kirsi Talala Finnish Cancer Registry, Mass Screening Registry, Helsinki, Finland; Kimmo Taari Department of Urology, University of Helsinki and Helsinki University Hospital, Helsinki, Finland. We thank Dr Samuel Heron for language check. This work was financially supported by the Academy of Finland (#310105 to JS, #123054 and #260931 to AA), the Sigrid Juselius Foundation (to JS), Cancer Foundation Finland sr (to JS), Cancer Foundation Finland sr (Movember grant for prostate cancer research to AA) and State Research Funding (VTR) Turku University Hospital (#M3002 to JS) and Tampere University Hospital (grants # 9E089, 9F100, 9G096, 9H099, 9L085, 9N064 and 9R002 to TLJT). We thank the members from the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium who are provided in the Supplement/footnotes. Information of the consortium can be found at http://practical.icr.ac.uk/. This study would not have been possible without the contributions of the following: Per Hall (COGS); Douglas F. Easton, Paul Pharoah, Kyriaki Michailidou, Manjeet K. Bolla, Qin Wang (BCAC), Andrew Berchuck (OCAC), Rosalind A. Eeles, Douglas F. Easton, Ali Amin Al Olama, Zsofia Kote-Jarai, Sara Benlloch (PRACTICAL), Georgia Chenevix-Trench, Antonis Antoniou, Lesley McGuffog, Fergus Couch and Ken Offit (CIMBA), Joe Dennis, Alison M. Dunning, Andrew Lee, and Ed Dicks, Craig Luccarini and the staff of the Centre for Genetic Epidemiology Laboratory, Javier Benitez, Anna Gonzalez-Neira and the staff of the CNIO genotyping unit, Jacques Simard and Daniel C. Tessier, Francois Bacot, Daniel Vincent, Sylvie LaBoissière and Frederic Robidoux and the staff of the McGill University and Génome Québec Innovation Centre, Stig E. Bojesen, Sune F. Nielsen, Borge G. Nordestgaard, and the staff of the Copenhagen DNA laboratory, and Julie M. Cunningham, Sharon A. Windebank, Christopher A. Hilker, Jeffrey Meyer and the staff of Mayo Clinic Genotyping Core Facility. Funding for the iCOGS infrastructure came from: the European Community’s Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A 10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112—the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. Authors declare no conflicts of interest, including relevant financial interests, activities, relationships, and affiliations.
Funding
Open access funding provided by University of Turku (UTU) including Turku University Central Hospital.
Author information
Authors and Affiliations
Contributions
CS: concept, study plan, data acquisition, statistical analyses, tables, figure and explanation text preparation, literature assembly, manuscript writing, final approval of the manuscript. TLJT: patient material collection, clinical input to the study plan, final approval of manuscript. AA: patient material collection, statistical input, approval of the analyses, interpretation of results, manuscript writing, final approval of the manuscript. JS: study plan preparation and adjustment, interpretation of results, manuscript writing and correcting, final approval of manuscript, correspondence.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sipeky, C., Tammela, T.L.J., Auvinen, A. et al. Novel prostate cancer susceptibility gene SP6 predisposes patients to aggressive disease. Prostate Cancer Prostatic Dis 24, 1158–1166 (2021). https://doi.org/10.1038/s41391-021-00378-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41391-021-00378-5
