
Abstract
Background
Childhood cancer has a poorly known etiology, and investigating the underlying genetic background may provide novel insights. A recognized association exists between non-chromosomal birth defects and childhood cancer susceptibility.
Methods
We performed whole-exome sequencing and chromosomal microarray analysis in a cohort of childhood cancer (22 individuals, 50% with congenital anomalies) to unravel deleterious germline variants.
Results
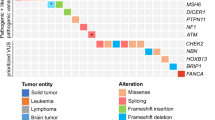
A diagnostic yield of 14% was found, encompassing heterozygous variants in bona fide dominant Cancer Predisposition Genes (CPGs). Considering candidate and recessive CPGs harboring monoallelic variants, which were also deemed to play a role in the phenotype, the yield escalated to 45%. Most of the deleterious variants were mapped in genes not conventionally linked to the patient’s tumor type. Relevant findings were detected in 55% of the syndromic individuals, mostly variants potentially underlying both phenotypes.
Conclusion
We uncovered a remarkable prevalence of germline deleterious CPG variants, highlighting the significance of a comprehensive genetic analysis in pediatric cancer, especially when coupled with additional clinical signs. Moreover, our findings emphasized the potential for oligogenic inheritance, wherein multiple genes synergistically increase cancer risk. Lastly, our investigation unveiled potentially novel genotype-phenotype associations, such as SETD5 in neuroblastoma, KAT6A in gliomas, JAG1 in hepatoblastomas, and TNFRSF13B in Langerhans cell histiocytosis.
Impact
-
Novel gene-phenotype associations and candidate genes for pediatric cancer were unraveled, such as KAT6A in gliomas, SETD5 in neuroblastoma, JAG1 in hepatoblastomas, and TNFRSF13B in Langerhans cell histiocytosis.
-
Our analysis revealed a high frequency of deleterious germline variants, particularly in cases accompanied by additional clinical signs, highlighting the importance of a comprehensive genetic evaluation in childhood cancer.
-
Our findings also underscored the potential for oligogenic inheritance in pediatric cancer risk.
-
Understanding the cancer etiology is crucial for genetic counseling, often influencing therapeutic decisions and offering valuable insights into molecular targets for the development of oncological therapies.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 14 print issues and online access
$259.00 per year
only $18.50 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Sweet-Cordero, E. A. & Biegel, J. A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 363, 1170–1175 (2019).
Spector, L. G., Pankratz, N. & Marcotte, E. L. Genetic and nongenetic risk factors for childhood cancer. Pediatr. Clin. North Am. 62, 11–25 (2015).
Schraw, J. M. et al. Cancer diagnostic profile in children with structural birth defects: An assessment in 15,000 childhood cancer cases. Cancer 126, 3483–3492 (2020).
Plon, S. E. & Lupo, P. J. Genetic predisposition to childhood cancer in the genomic era. Annu. Rev. Genom. Hum. Genet. 20, 241–263 (2019).
Lupo, P. J. et al. Association between birth defects and cancer risk among children and adolescents in a population-based assessment of 10 million live births. JAMA Oncol. 5, 1150–1158 (2019).
Rahman, N. Realizing the promise of cancer predisposition genes. Nature 505, 302–308 (2014).
Sylvester, D. E. et al. Rare germline variants in childhood cancer patients suspected of genetic predisposition to cancer. Genes Chromosom. Cancer 61, 81–93 (2022).
Bertelsen, B. et al. High frequency of pathogenic germline variants within homologous recombination repair in patients with advanced cancer. NPJ Genom. Med. 4, 13 (2019).
Capellini, A., Williams, M., Onel, K. & Huang, K. L. The functional hallmarks of cancer predisposition genes. Cancer Manag. Res. 13, 4351–4357 (2021).
Filbin, M. & Monje, M. Developmental origins and emerging therapeutic opportunities for childhood cancer. Nat. Med. 25, 367–376 (2019).
Zhang, J. et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 373, 2336–2346 (2015).
Gröbner, S. N. et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018).
Newman, S. et al. Genomes for kids: The scope of pathogenic mutations in pediatric cancer revealed by comprehensive dna and rna sequencing. Cancer Discov. 11, 3008–3027 (2021).
Krepischi, A. C. V. et al. Large germline copy number variations as predisposing factor in childhood neoplasms. Futur. Oncol. 10, 1627–1633 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genome 00, 1–3 (2013).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Challis, D. et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinform. 13, 8 (2012).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Naslavsky, M. S. et al. Whole-genome sequencing of 1171 elderly admixed individuals from Brazil. Nat. Commun. 13, 1004 (2022).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Stelzer, G. et al. VarElect: The phenotype-based variation prioritizer of the GeneCards Suite. BMC Genom. 17, 444 (2016).
Köhler, S. et al. The human phenotype ontology in 2021. Nucleic Acids Res. 49, D1207–D1217 (2021).
Franklin by Genoox. https://franklin.genoox.com.
Miller, D. T. et al. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 24, 1407–1414 (2022).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Biesecker, L. G. & Harrison, S. M. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet. Med. 20, 1687–1688 (2018).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro. Oncol. 23, 1231–1251 (2021).
Aguiar, T. F. et al. Atypical presentation of a germline APC mutation in a child with supratentorial primitive neuroectodermal tumor. Pediatr. Blood Cancer 66, 1–2 (2019).
Pires, S. F. et al. Expanding the role of SETD5 haploinsufficiency in neurodevelopment and neuroblastoma. Pediatr. Blood Cancer 67, 3–5 (2020).
Dangoni, G. D. et al. A rare case of hepatoblastoma in a syndromic child with a de novo germline JAG1 mutation. Pediatr. Blood Cancer 70, e30311 (2023).
Fisher, P. G. et al. Cancer in children with nonchromosomal birth defects. J. Pediatr. 160, 978–983 (2012).
Gillentine, M. A. & Schaaf, C. P. The human clinical phenotypes of altered CHRNA7 copy number. Biochem. Pharmacol. 97, 352–362 (2015).
Tham, E. et al. Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am. J. Hum. Genet. 96, 507–513 (2015).
Huang, F., Abmayr, S. M. & Workman, J. L. Regulation of KAT6 acetyltransferases and their roles in cell cycle progression, stem cell maintenance, and human disease. Mol. Cell. Biol. 36, 1900–1907 (2016).
Wiesel-Motiuk, N. & Assaraf, Y. G. The key roles of the lysine acetyltransferases KAT6A and KAT6B in physiology and pathology. Drug Resist. Updat. 53, 100729 (2020).
Lv, D. et al. Histone acetyltransferase KAT6A upregulates PI3K/AKT signaling through TRIM24 binding. Cancer Res. 77, 6190–6201 (2017).
Pilarski, R. et al. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J. Natl Cancer Inst. 105, 1607–1616 (2013).
Hollander, M. C., Blumenthal, G. M. & Dennis, P. A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 11, 289–301 (2011).
Yehia, L., Keel, E. & Eng, C. The clinical spectrum of PTEN mutations. Annu. Rev. Med. 71, 103–116 (2020).
Morotti, A. et al. The role of PTEN in myeloid malignancies. Hematol. Rep. 7, 84–87 (2015).
Yilmaz, Ö. H. et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482 (2006).
Aggerholm, A., Grønbaek, K., Guldberg, P. & Hokland, P. Mutational analysis of the tumour suppressor gene MMAC1/PTEN in malignant myeloid disorders. Eur. J. Haematol. 65, 109–113 (2000).
Yehia, L. et al. Longitudinal analysis of cancer risk in children and adults with germline PTEN variants. JAMA Netw. Open 6, e239705 (2023).
Deliu, E. et al. Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat. Neurosci. 21, 1717–1727 (2018).
Johnsen, J. I., Dyberg, C. & Wickström, M. Neuroblastoma—A Neural Crest Derived Embryonal Malignancy. Front. Mol. Neurosci. 12, 9 (2019).
Wang, L. L. et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. JNCI J. Natl Cancer Inst. 95, 669–674 (2003).
Cao, F. et al. Generalized metabolic bone disease and fracture risk in Rothmund-Thomson syndrome. Hum. Mol. Genet. 26, 3046–3055 (2017).
Carlo, M. I. et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol. 4, 1228–1235 (2018).
Truong, H. et al. Germline variants identified in patients with early-onset renal cell carcinoma referred for germline genetic testing. J. Urol. 207, 1151–1152 (2022).
Orgueira, A. M. et al. Detection of rare germline variants in the genomes of patients with b-cell neoplasms. Cancers (Basel). 13, 1–18 (2021).
Muskens, I. S. et al. Germline cancer predisposition variants and pediatric glioma: a population-based study in California. Neuro. Oncol. 22, 864–874 (2020).
Maciaszek, J. L. et al. Enrichment of heterozygous germline RECQL4 loss-of-function variants in pediatric osteosarcoma. Cold Spring Harb. Mol. Case Stud. 5, 1–15 (2019).
Martin-Giacalone, B. A., Rideau, T.-T., Scheurer, M. E., Lupo, P. J. & Wang, L. L. Cancer risk among RECQL4 heterozygotes. Cancer Genet. 262–263, 107–110 (2022).
Smetsers, S. et al. Heterozygote FANCD2 mutations associated with childhood T Cell ALL and testicular seminoma. Fam. Cancer 11, 661–665 (2012).
Reid, S. et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 39, 162–164 (2007).
van Os, N. J. H. et al. Health risks for ataxia-telangiectasia mutated heterozygotes: a systematic review, meta-analysis and evidence-based guideline. Clin. Genet. 90, 105–117 (2016).
Gruber, S. B. et al. BLM heterozygosity and the risk of colorectal. Cancer Sci. 297, 2013 (2002).
Allen, C. E., Merad, M. & McClain, K. L. Langerhans-cell histiocytosis. N. Engl. J. Med. 379, 856–868 (2018).
Abla, O., Rollins, B. & Ladisch, S. Langerhans cell histiocytosis: progress and controversies. Br. J. Haematol. 187, 559–562 (2019).
Bogaert, D. J. A. et al. Genes associated with common variable immunodeficiency: One diagnosis to rule them all? J. Med. Genet. 53, 575–590 (2016).
Resnick, E. S., Moshier, E. L., Godbold, J. H. & Cunningham-Rundles, C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 119, 1650–1657 (2012).
Ardeniz, Ö. & Cunningham-Rundles, C. Granulomatous disease in common variable immunodeficiency. Clin. Immunol. 133, 198–207 (2009).
Waller, R. G. et al. Sequencing at lymphoid neoplasm susceptibility loci maps six myeloma risk genes. Hum. Mol. Genet. 30, 1142–1153 (2021).
Lee, J. J. et al. The C104R mutant impairs the function of transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) through haploinsufficiency. J. Allergy Clin. Immunol. 126, 1234–1241.e2 (2010).
Mather, M. W. et al. Mutation of TNFRSF13B in a child with 22q11 deletion syndrome associated with granulomatous lymphoproliferation. J. Allergy Clin. Immunol. 135, 559–561 (2015).
Thaventhiran, J. E. D. et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature 583, 90–95 (2020).
Gross, J. A. et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease: Impaired B cell maturation in mice lacking BLyS. Immunity 15, 289–302 (2001).
Celik, S., Tangi, F. & Oktenli, C. Increased frequency of Mediterranean fever gene variants in multiple myeloma. Oncol. Lett. 8, 1735–1738 (2014).
Stankovic, K. & Grateau, G. Auto inflammatory syndromes: diagnosis and treatment. Jt. Bone Spine 74, 544–550 (2007).
Celik, S. et al. The rate of MEFV gene mutations in hematolymphoid neoplasms. Int. J. Immunogenet. 37, 387–391 (2010).
Oktenli, C. & Celik, S. High frequency of inherited variants in the MEFV gene in patients with hematologic neoplasms: a genetic susceptibility? Int. J. Hematol. 95, 380–385 (2012).
Sayan, O. et al. High frequency of inherited variants in the MEFV gene in acute lymphocytic leukemia. Indian J. Hematol. Blood Transfus. 27, 164–168 (2011).
Tariq, H. et al. Refractory hemophagocytic lymphohistiocytosis in an adult patient with occult ALK-Positive anaplastic large cell lymphoma and a heterozygous MEFV mutation. Leuk. Lymphoma 63, 495–498 (2022).
Rossi-Semerano, L., Hermeziu, B., Fabre, M. & Koné-Paut, I. Macrophage activation syndrome revealing familial mediterranean fever. Arthritis Care Res. 63, 780–783 (2011).
Crasto, S., My, I. & Di Pasquale, E. The broad spectrum of LMNA cardiac diseases: from molecular mechanisms to clinical phenotype. Front. Physiol. 11, 1–11 (2020).
Shin, J.-Y. & Worman, H. J. Molecular pathology of laminopathies. Annu. Rev. Pathol. Mech. Dis. 17, 159–180 (2022).
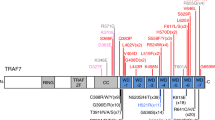
Ko, A. et al. LZTR1 mutation mediates oncogenesis through stabilization of EGFR and AXL. Cancer Discov. 13, 702–723 (2023).
Piotrowski, A. et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 46, 182–187 (2014).
Akhavanfard, S., Padmanabhan, R., Yehia, L., Cheng, F. & Eng, C. Comprehensive germline genomic profiles of children, adolescents and young adults with solid tumors. Nat. Commun. 11, 2206 (2020).
Foss-Skiftesvik, J. et al. Redefining germline predisposition in children with molecularly characterized ependymoma: a population-based 20-year cohort. Acta Neuropathol. Commun. 10, 1–12 (2022).
Bosse, D. van den. The role of germline heterozygous LZTR1 variants in pediatric cancer predisposition. (Utrecht University, 2022).
Abe, T. et al. LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ. 27, 1023–1035 (2020).
Schindler, E. A. et al. Alagille syndrome and risk for hepatocellular carcinoma: need for increased surveillance in adults with mild liver phenotypes. Am. J. Med. Genet. Part A 185, 719–731 (2021).
Torrezan, G. T. et al. Mutational spectrum of the APC and MUTYH genes and genotype-phenotype correlations in Brazilian FAP, AFAP, and MAP patients. Orphanet J. Rare Dis. 8, 1–12 (2013).
Attard, T. M., Giglio, P., Koppula, S., Snyder, C. & Lynch, H. T. Brain tumors in individuals with Familial Adenomatous Polyposis: A cancer registry experience and pooled case report analysis. Cancer 109, 761–766 (2007).
Mendoza, P. R. & Grossniklaus, H. E. The Biology of Retinoblastoma. Prog. Mol. Biol. Transl. Sci. 134, 503–516 (2015).
Aretz, S. et al. MUTYH-associated polyposis (MAP): Evidence for the origin of the common European mutations p.Tyr179Cys and p.Gly396Asp by founder events. Eur. J. Hum. Genet. 22, 923–929 (2014).
Frans, G. et al. Conventional and single-molecule targeted sequencing method for specific variant detection in IKBKG while bypassing the IKBKGP1 pseudogene. J. Mol. Diagn. 20, 195–202 (2018).
Beetz, C. & Bauer, P. Dual genetic diagnoses - underappreciated ‘double trouble’. J. Biochem. Clin. Genet. 3, 52–53 (2020).
McBride, K. A. et al. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 11, 260–271 (2014).
Pietragalla, A., Arcieri, M., Marchetti, C., Scambia, G. & Fagotti, A. Ovarian cancer predisposition beyond BRCA1 and BRCA2 genes. Int. J. Gynecol. Cancer 30, 1803–1810 (2020).
Palmero, E. I. et al. Detection of R337H, a germline TP53 mutation predisposing to multiple cancers, in asymptomatic women participating in a breast cancer screening program in Southern Brazil. Cancer Lett. 261, 21–25 (2008).
Custódio, G. et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J. Clin. Oncol. 31, 2619–2626 (2013).
Pereira Caminha, I. Prevalência da mutação germinativa TP53 p.R337H na região metropolitana de Campinas e cidades circunvizinhas. (Universidade Estadual de Campinas, https://doi.org/10.47749/T/UNICAMP.2015.949595 (2015).
Ribeiro, R. C. et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc. Natl Acad. Sci. USA. 98, 9330–9335 (2001).
Seidinger, A. L. et al. Association of the highly prevalent TP53 R337H mutation with pediatric choroid plexus carcinoma and osteosarcoma in Southeast Brazil. Cancer 117, 2228–2235 (2011).
Custodio, G. et al. Increased incidence of choroid plexus carcinoma due to the germline TP53 R337H mutation in southern Brazil. PLoS One 6, e18015 (2011).
Mastellaro, M. J. et al. Contribution of the TP53 R337H mutation to the cancer burden in southern Brazil: Insights from the study of 55 families of children with adrenocortical tumors. Cancer 123, 3150–3158 (2017).
Pinto, E. M. & Zambetti, G. P. What 20 years of research has taught us about the TP53 p.R337H mutation. Cancer 126, 4678–4686 (2020).
Schayek, H. et al. The rate of recurrent BRCA1, BRCA2, and TP53 mutations in the general population, and unselected ovarian cancer cases, in Belo Horizonte, Brazil. Cancer Genet. 209, 50–52 (2016).
Mathias, C. et al. Frequency of the TP53 R337H variant in sporadic breast cancer and its impact on genomic instability. Sci. Rep. 10, 1–12 (2020).
Achatz, M. I. & Zambetti, G. P. The Inherited p53 Mutation in the Brazilian Population. Cold Spring Harb. Perspect. Med. 6, a026195 (2016).
Jara, L. et al. Mutations in BRCA1, BRCA2 and other breast and ovarian cancer susceptibility genes in Central and South American populations. Biol. Res. 50, 35 (2017).
Diets, I. J. et al. High yield of pathogenic germline mutations causative or likely causative of the cancer phenotype in selected children with cancer. Clin. Cancer Res. 24, 1594–1603 (2018).
Acknowledgements
We would like to thank the patients and their families for participating in this study.
Funding
This research was carried out with financial support from CAPES (88887.606266/2021-00); FAPESP (2013/08028-1, 2018/21047-9, 2018/05961-2, 2022/03980-5); CNPq (grant number 305806/2019-0; 305101/2022-6). We are grateful for the confidence of these institutions in investing public funds in our research.
Author information
Authors and Affiliations
Contributions
A.C.V.K: study design. G.D.D., L.N.S., E.M.N., C.S.C.V., S.M.M.S., V.O.F. participant enrollment and data collection. G.D.D., A.C.B.T., S.S.C., A.C.V.K.: data analysis and interpretation. G.D.D., A.C.V.K.: manuscript writing. G.D.D., A.C.B.T., L.M.L.C., M.C.M., A.C.V.K. manuscript revision. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guidelines on human experimentation (Resolution 466/12) and with the Helsinki Declaration of 1975, as revised in 2008. The project was approved by the Research Ethics Committee of the Clinical Hospital - Faculty of Medicine of the University of São Paulo (CAAE 47277115.0.0000.0068), and Institute of Biosciences (University of São Paulo, São Paulo, Brazil) (CAAE 09163818.4.0000.5464). Informed consent was obtained from all study participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementry Information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dangoni, G.D., Teixeira, A.C.B., da Costa, S.S. et al. Germline mutations in cancer predisposition genes among pediatric patients with cancer and congenital anomalies. Pediatr Res 95, 1346–1355 (2024). https://doi.org/10.1038/s41390-023-03000-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-023-03000-7
