
Abstract
Neonatal encephalopathy (NE) results from impaired cerebral blood flow and oxygen delivery to the brain. The pathophysiology of NE is complex and our understanding of its underlying pathways continues to evolve. There is considerable evidence that cholesterol dysregulation is involved in several adult diseases, including traumatic brain injury, stroke, Huntington’s disease, and Parkinson’s disease. Although the research is less robust in pediatrics, there is emerging evidence that aberrations in cholesterol metabolism may also be involved in the pathophysiology of neonatal NE. This narrative review provides an overview of cholesterol metabolism in the brain along with several examples from the adult literature where pathologic alterations in cholesterol metabolism have been associated with inflammatory and ischemic brain injury. Using those data as a background, the review then discusses the current preclinical data supporting the involvement of cholesterol in the pathogenesis of NE as well as how brain-specific cholesterol metabolites may serve as serum biomarkers for brain injury. Lastly, we review the potential for using the cholesterol metabolic pathways as therapeutic targets. Further investigation of the shifts in cholesterol synthesis and metabolism after hypoxia–ischemia may prove vital in understanding NE pathophysiology as well as providing opportunities for rapid diagnosis and therapeutic interventions.
Impact
-
This review summarizes emerging evidence that aberrations in cholesterol metabolism may be involved in the pathophysiology of NE.
-
Using data from NE as well as analogous adult disease states, this article reviews the potential for using cholesterol pathways as targets for developing novel therapeutic interventions and using cholesterol metabolites as biomarkers for injury.
-
When possible, gaps in the current literature were identified to aid in the development of future studies to further investigate the interactions between cholesterol pathways and NE.
Similar content being viewed by others


Introduction
Neonatal encephalopathy (NE) is the result of impaired cerebral blood flow and oxygen delivery to the brain. It affects up to 1.5 per 1000 live births each year in the United States and is one of the most serious complications affecting full-term infants.1 There are many diverse etiologies underlying the impaired blood flow and lack of oxygen that can come from maternal, fetal, or placental pathologies. Unfortunately, the rate of NE has not significantly decreased despite advancements in perinatal care aimed at preventing this devastating disorder. Postnatally, the current standard of care for infants suffering from NE is hypothermia; however, many infants still die or sustain significant morbidity despite treatment. The pathophysiology of NE is complex, and our understanding of the various underlying pathways continues to evolve.
There are three predominant stages of NE. The first stage involves primary cellular energy failure secondary to the initial injury and resulting reduction of cerebral blood flow. The extent of the cerebral blood flow reduction contributes to further secondary energy failure 6–48 h after the injury.2 The third stage of NE begins ~72 h after the initial injury and involves the subacute effects of NE, including remodeling and gliosis.3 Excitotoxicity and inflammation appear to be involved throughout the first two phases, with the first phase also resulting in cellular necrosis and the secondary phase primarily resulting in apoptotic cell death.2 Several brain cell populations are affected by NE, with the most sensitive being oligodendrocytes, hippocampal neurons, and microglia.2,4 Further investigation and identification of key pathways involved in NE is vital for the development of more effective and targeted therapies.
In addition to the need for novel effective therapies, the care of neonates with HIE is currently suffering due to a lack of sensitive and specific biomarkers. Several risk factors, including maternal and placental abnormalities, increase suspicion for NE but have poor specificity.2 Currently, low Apgar scores, severe metabolic acidosis, and clinical neurological features of encephalopathy serve as the primary diagnostic triggers to initiate hypothermia, although these also have relatively low sensitivity and specificity and both the Apgar scores and neurological examination are prone to subjectivity. Electroencephalography (EEG) or amplitude-integrated EEG (aEEG) have also been used to both evaluate for seizure activity and to assess background cerebral function as a surrogate for encephalopathy severity.5 In addition, the background pattern can act as a prognostic marker, as a persistently abnormal aEEG background pattern 48 h after injury has been associated with poor neurodevelopmental outcomes.6 Although both sensitive and specific starting ~48 h after injury, aEEG lacks specificity early in the course of NE (0.61 at 6 h after injury).7 Magnetic resonance imaging (MRI) has demonstrated some potential as a biomarker, particularly in neonates that are more significantly affected. Unfortunately, patients are often not stable enough for MRI, especially in the first 6 h, which is when the decision to initiate hypothermia must be made.3 In addition, many MRI sequences do not begin to demonstrate the effects of injury until ~3 days after injury. Due to these challenges in the rapid and accurate diagnosis of NE, significant advances in the treatment of NE will likely require the identification of sensitive and specific biomarkers.
Altered levels of cholesterol and its metabolites have been demonstrated in the brain and serum after injury in several adult diseases, including traumatic brain injury (TBI), stroke, and multiple sclerosis.8,9 In addition, aberrations in cholesterol metabolites have been associated with brain abnormalities and significant developmental delay in genetic disorders, such as cerebrotendinous xanthomatosis (CTX) and Smith–Lemli–Opitz syndrome (SLOS).10,11 A few trials have demonstrated effects of NE on brain cholesterol synthesis and the regulation of cholesterol metabolism, suggesting not only that cholesterol pathways may provide a therapeutic target in NE, but also that its metabolites (specifically those released from the brain into the serum) may potentially act as biomarkers to aid in the identification or severity stratification of hypoxic–ischemic brain injury.4,12,13 Despite these early study results, investigators are still far from having a thorough understanding of the interactions between NE and cholesterol. Further understanding of the alterations in cholesterol synthesis and metabolism after hypoxia–ischemia may prove essential in understanding NE pathophysiology as well as providing opportunities for rapid diagnosis and therapeutic interventions. This article will review the importance of cholesterol and cholesterol metabolism in the brain during normal fetal and neonatal development, as well as after acute injury. In addition, we will describe the interactions between brain injury and cholesterol seen in the adult literature and correlate those findings to the studies that have been performed to date assessing the role of cholesterol in NE.
Cholesterol metabolism in the brain
Cholesterol synthesis is necessary for brain growth and development and costly with respect to energy expenditure. Excess cholesterol can be toxic and result in cell death. Lipids in the brain are composed of cholesterol, sphingolipids, and glycerophospholipids.14 The majority of cholesterol in the central nervous system is concentrated in myelin (~80%) as well as in the membranes of neurons and glia.12 Cholesterol homeostasis is dependent on balanced synthesis, absorption, and conversion to bile acids.15 The concentration of sterols in the central nervous system is higher than other parts of the body16 and peripheral cholesterol is unable to cross the blood–brain barrier (BBB), so cholesterol synthesis in the brain is almost completely de novo.17 Cholesterol biosynthesis occurs from a series of reactions in the Kandutsch–Russell and Bloch pathways (Fig. 1).18 Neurons and astroglia use the Bloch pathway preferentially.18 Maintenance of adequate brain cholesterol levels is vital to neurodevelopment, and interruptions of any of the several steps of the cholesterol biosynthesis pathway may result in abnormal cholesterol levels, potentially affecting neurodevelopment.4

Cholesterol is unable to cross an intact blood–brain barrier (BBB), but may be able to cross an impaired BBB. 24S-hydroxycholesterol is produced in the brain and is able to cross the BBB. Hypoxic–ischemic injury increases cholesterol 24S-hydroxylase (CYP46) and impairs the BBB, and hypoxia inhibits cholesterol synthesis due to the need for oxygen in the metabolic pathways. Statins, including simvastatin, may induce overproduction of DHCR24 and/or inhibit 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). DHCR7 7-dehydrocholesterol reductase, HMGCS hydroxymethylglutaryl-CoA synthase.
Desmosterol is one of the key metabolites in the cholesterol biosynthesis pathway. In the Bloch pathway, desmosterol is the immediate precursor to cholesterol, as it is directly converted to cholesterol via 24-dehydrocholesterol reductase (DHCR24, a.k.a. 3β-hydroxysteroid delta 24 reductase).19 An accumulation of desmosterol signals sterol overload in the cell, which in astrocytes stimulates liver X receptor (LXR) signaling to increase sterol secretion from the cell.19,20 DHCR24 is highly localized to areas that are steroidogenic and cholesterogenic and, as such, is expressed at high levels in the spinal cord and medulla, as well as in the liver and adrenal glands.19 DHCR24 may also play a role in modulating oxidative stress.19 A deficiency in DHCR24 leads to decreased cholesterol levels in plasma membranes and, because desmosterol is not a good surrogate for cholesterol in lipid rafts, the replacement of cholesterol with desmosterol in the plasma membrane increases caveolar openings and decreases the stability of caveolin oligomers. This impaired formation/stability of lipid rafts can further alter downstream cellular signaling.21
Lanosterol is another key metabolite in the cholesterol biosynthesis pathway. The conversion of lanosterol to cholesterol requires the removal of three methyl groups. Hypoxia inhibits the demethylation of lanosterol in vitro, thus leading to intracellular accumulation of lanosterol. The accumulation of lanosterol may accelerate the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), which controls one of the rate-limiting steps in cholesterol synthesis.22 Studies of the brain myelin fraction in vivo have not shown an increase in lanosterol after hypoxia, however. In fact, no or trace amounts of lanosterol were detected in the brains of rats exposed to moderate or severe hypoxia,23 and in a model of global ischemia, there was actually a significant decrease in lanosterol levels after injury.24
Several other metabolites and transcription factors have been identified as critical for cholesterol metabolism in the brain as well. Brain-specific cytochrome P450 46A1 (CYP46 or CYP46A1, a.k.a. cholesterol 24S-hydroxylase) is an endoplasmic reticulum enzyme that is expressed in neurons and microglia.25,26 CYP46 is mainly localized in cortical layers V and VI in large pyramidal neurons, but is also found in the thalamus and hippocampus.27 CYP46 converts cholesterol into 24S-hydroxycholesterol (24S-HC) via hydroxylation in the brain and upregulates cholesterol efflux through nuclear transcription factor X activation. CYP46 knockout studies indicate that it is responsible for a majority of cholesterol efflux in the brain.26 Since elevated intracellular levels of cholesterol can be toxic, CYP46-mediated conversion of cholesterol to 24S-HC may be protective to the injured brain, and alterations in its activity have been seen in the glia during pathologic states such as TBI and Alzheimer’s disease.26
24S-HC is specific to the brain, is able to cross the BBB, and is cleared by the liver.28 It is a positive N-methyl-d-aspartate receptor (NMDAR) modulator, and as such, administration of a 24S-HC analog at the time of hypoxia has been shown to increase neuronal injury in a hippocampal cell culture.29 Excess levels of 24S-HC can contribute to cell death and, since it passes through membranes such as the BBB easier than cholesterol, it may serve as a serum biomarker for brain injury.27 Increases in serum 24S-HC levels have been demonstrated in the early injury phase of multiple disease processes including multiple sclerosis and Parkinson’s disease (PD), as well as in developmental alterations such as autism spectrum disorders, while decreased levels may suggest a loss of metabolically active neurons and brain atrophy in Alzheimer’s disease and multiple sclerosis.30,31,32 Recently, high serum levels of 24S-HC have also been seen acutely after NE.27
24S-HC further impacts cholesterol levels in the brain via reduced sterol regulatory elementary binding proteins 1 and 2 (SREBP-1 and -2). SREBPs are transcription factors involved in lipid homeostasis. Proteolytic release of SREBP-2 from the membrane and movement to the nucleus prompts transcription of genes for the synthesis, uptake, and use of lipids,33 and SREBP-2 is induced when sterols are depleted.34 SREBP-2 is important to cholesterol synthesis, and the absence of SREBP-2 is lethal.34,35 In addition, SREBP-2 plays significant roles in the development and function of astrocytes,36 limb patterning,35 and neurite growth.36 Mice that have lost SREBP-2 demonstrate impairments in social behavior, memory, and motor coordination.36
Cholesterol metabolism during brain development
In early development, the fetal brain derives the majority of its cholesterol from the mother, with maternal–fetal exchange occurring at the yolk sac and placenta.37 In the placenta, cholesterol is transported from the maternal trophoblasts to fetal trophoblasts via receptor-dependent and -independent channels.38 As such, alterations of maternal serum cholesterol levels early in pregnancy may affect fetal brain sterol concentrations.39 The majority of brain cholesterol continues to be derived from the mother until the BBB develops at around E10–11 in the mouse (corresponding to 4–6 weeks of gestation in humans).40 Once the BBB develops in the fetus, the brain must generate the majority of its cholesterol de novo, receiving only ~10% of its cholesterol from the peripheral circulation by the time of birth.41,42
Although developing neurons are able to synthesize their own cholesterol,13 fully developed neurons are predominantly dependent on astrocytes for cholesterol synthesis43 (Fig. 2). Neurons require cholesterol for synapse development44. Separating neurons from astrocytes in vitro results in the significant decrease of the number and efficacy of neuronal synapses.44,45 The roles of other glial cells such as the lipid-rich oligodendrocytes in the production of cholesterol remain unclear.13

Astroglia and neurons preferentially use the Bloch pathway for cholesterol synthesis. Cholesterol, bound to ApoE to form ApoE-lipoprotein, effluxes through the ATP-binding cassette transporter 1 (ABCA1) receptor and can enter the neuron by endocytosis facilitated by the low-density lipoprotein receptor (LDLR). Cholesterol in the neuron is metabolized to 24-hydroxycholesterol via cholesterol 24S-hydroxylase (CYP46A1) and is able to efflux to the cerebrospinal fluid (CSF), through the blood–brain barrier (BBB), as well as to astrocytes. LXR liver X receptor, HMGCR 3-hydroxy-3-methylglutaryl-CoA reductase, SRE sterol regulatory element, SREBP SRE-binding protein.
As mentioned in the section above, desmosterol accumulation generally signals sterol overload in the cell. Even in normal fetal brain development, however, desmosterol can accumulate to levels up to 30% of that of the total sterols in the brain; this is significantly elevated compared to other tissues where cholesterol precursors rarely make up >1% of the total sterols.20 Desmosterol accumulation increases during fetal development of the brain and peaks in the first postnatal week. This period corresponds to active growth of neuronal cell processes and is followed by synaptogenesis.20
During this developmental accumulation of desmosterol, DHCR24 activity appropriately increases, so the accumulation does not appear to be due to DHCR24 deficiency.20,46 Rather, progesterone has been implicated as one cause of rising desmosterol levels as it can cause post-transcriptional inhibition of DHCR24. It has been postulated that fetal and early neonatal desmosterol accumulation is evolutionarily conserved because desmosterol cannot be esterified or metabolized to 24S-HC, thus increasing the pool of brain sterols during the period of rapid brain growth. In addition, the increased desmosterol levels may stimulate LXR-mediated sterol secretion from astrocytes.20 Since it is not possible to accurately and non-invasively measure human fetal brain cholesterol levels, however, much of our current understanding of the effects of altered cholesterol in the human brain during development comes from genetic disorders of altered cholesterol metabolism such as CTX and SLOS.
Cerebrotendinous xanthomatosis
CTX is a rare autosomal recessive disorder caused by a mutation in the 27-hydroxylase gene (CYP27A1) that normally helps to break down cholesterol into the bile acid chenodeoxycholic acid.10 Without CYP27A1, the body uses alternate metabolic pathways forming cholestanol, which, along with cholesterol, builds up in tissues to form xanthomas.47 CTX can have broad manifestations, but is often associated with progressive neurological dysfunction, including epilepsy, neuropathy, and movement disorders, as well as juvenile cataracts, premature atherosclerosis, and osteoporosis.47,48 MRI studies of patients with CTX indicate cerebellar atrophy, hyperintensity in the dentate nuclei, and white matter changes.47 CTX is managed with supplementation of the bile acid chenodeoxycholic acid, which can normalize cholestanol levels and can improve both neurological and non-neurological symptoms.47
Smith–Lemli–Opitz syndrome
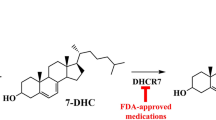
SLOS is an autosomal recessive inborn error of metabolism resulting from missense, nonsense, or splice site mutations in 7-dehydrocholesterol reductase (DHCR7).11 Much like DHCR24, DHCR7 acts in both the Bloch and Kandutsch–Russell pathways. DHCR7 dysfunction results in reduced cholesterol levels with elevations in 7-dehydrocholesterol compared to controls.49 Patients with the lowest serum cholesterol levels tend to have increased mortality.50,51
There is a wide clinical spectrum with a variable phenotypic expression for patients with SLOS.52 Manifestations associated with SLOS include craniofacial or other skeletal abnormalities, endocardial cushion defects, and central nervous system abnormalities, such as holoprosencephaly or partial agenesis of the corpus callosum.52,53 A DHCR7 knockout mouse model of SLOS has been developed, which mimics the sterol compositions in human patients with SLOS. The knockout animals also develop some of the clinical findings of SLOS, such as cleft palates, incomplete lung development, and bladder distension.54 Homozygote mice have labored breathing at delivery, lack of movement, and tend to die within 18 h of delivery.54
In addition to their roles in brain development, many of the steps in cholesterol metabolism can be altered in traumatic, neurodegenerative, and ischemic disease states, demonstrating significant interactions between cholesterol and brain pathology.55
Pathological alterations in cholesterol metabolism
Although there have been very few studies directly assessing the interaction between neonatal NE and cholesterol metabolism, there is much that can be learned by the research that has been performed to date assessing cholesterol in other inflammatory and ischemic brain injuries.
Stroke
It has long been known that patients with elevated levels of serum cholesterol are at increased risk for vascular disease and stroke. Interestingly, although, there appears to be a U-shaped association between serum cholesterol and the incidence of intracranial pathology, with increased risk seen in patients with both high and low levels of total serum cholesterol. Low levels are associated with intracerebral hemorrhage, while high levels are associated with ischemic stroke.8 In addition to increasing the risk of stroke, excess cholesterol accumulation in peripheral vasculature induces an inflammatory response and a positive feedback loop that subsequently induces further cholesterol deposition.56 An increase in cholesterol has also been associated with an increase in leukocyte recruitment as well as the release of chemokines and proinflammatory cytokines.56
In extreme cases of cholesterol deficiency, such as that seen in mice deficient in DHCR24, DHCR24-deficient mice demonstrate larger areas of infarct than wild-type animals in a stroke model.57 Furthermore, the knockout model for DHCR24 demonstrates increased proinflammatory markers after induced stroke. Conversely, in DHCR24-sufficient animals, DHCR24 expression increases after stroke and those increases in DHCR24 have been shown to be neuroprotective in acute stress conditions.57 This is likely related to the observation that overexpression of DHCR24 increases the number of dendritic spines and mushroom spines in mature hippocampal neurons.57
Similar to DHCR24, cholesterol levels do not only affect stroke risk, but suffering a stroke can also have effects on cholesterol levels. In a stroke model, researchers demonstrated increased stimulation of glutamate receptors after stroke injury, which was shown to result in the loss of membrane cholesterol.55 There was no significant difference in the overall amounts of phospholipids, cholesterol, and galactolipids in the brain tissue; however, when comparing the ipsilateral and contralateral cerebrum in the injured brain versus controls.58
Huntington’s disease
Huntington’s disease (HD) has also been associated with aberrations of cholesterol metabolism.59 Expression of the mutant huntingtin protein, the protein involved in the development of HD, has been shown to lead to an accumulation of cholesterol and a reduction of cholesterol synthesis in cell culture.60 Adeno-associated virus vector-induced upregulation of CYP46 was shown in a knock-in mouse model of HD to restore cholesterol homeostasis, alleviate dysfunction with an improvement of synaptic activity, and increase of both 24S-HC and desmosterol.59 In addition, this study demonstrated that lanosterol, desmosterol, and CYP46 all reduced mutant huntingtin protein aggregate formation and increased clearance of aggregates via autophagy.59 Lastly, cholesterol metabolism in HD may also be altered by SREBPs, as a reduction in SREBP-2 has been demonstrated in HD cell culture.61
Parkinson’s disease
Loss of dopaminergic neurons in the striatum is fundamental in the development of PD. Cholesterol is reduced in membrane lipid rafts of patients afflicted with Parkinson’s disease and several preclinical and clinical trials have suggested that elevated plasma cholesterol may be linked to the development of PD.62,63,64,65 In addition to increasing the risk of PD development, high-fat diets are associated with increased loss of dopamine neurons in animal models of PD, suggesting that cholesterol may also play a role in PD severity.66,67 In a PD animal model, lanosterol increased autophagy, regulated mitochondrial function, and appeared to serve as a survival factor in dopaminergic neurons. A decrease in levels of lanosterol was seen in the striatum and ventral midbrain of the PD animals.68
Traumatic brain injury
TBI can affect patients of all ages and is a complex injury resulting from a combination of the primary impact and secondary injury mechanisms.69 Secondary effects of TBI result from glial cell activation, leukocyte recruitment, upregulation of inflammatory mediators, and impairment of the BBB.70 TBI is associated with changes in total cholesterol and phospholipid concentrations.9,71 CYP46 activity is upregulated in the ipsilateral cortex and microglia 3 and 7 days after TBI injury, likely reflecting the injured brain’s attempt at normalizing the increased cholesterol levels through lipid phagocytosis and increased efflux from the cells.25,26 Despite the upregulation of CYP46, however, no increase in serum 24S-HC was seen after TBI.72
Cholesterol metabolism after hypoxic–ischemic brain injury
Although the immature brains of neonates react differently to insults as compared to adult brains, there are many similarities between NE and the adult brain injuries described above. Specifically, the inflammatory, excitotoxic, and oxidative injuries seen after NE overlap considerably with those seen after stroke, both in neonates and adults. Many of the same mechanisms are also integral in the secondary response seen after TBI. Despite the presence of significant differences between these different pathologies that restrict the ability to directly apply their results to the neonatal population, the adult literature described above can at least provide a general roadmap for the types of studies that need to be performed in neonates to better define the interactions between NE and cholesterol.
The limited neonatal data that exist (Table 1) have demonstrated that cholesterol levels are decreased in the brain white matter after neonatal hypoxic–ischemic brain injury.4 Since cholesterol is an important component of myelin, decreases in cholesterol levels may result in impaired axon myelination.13,73 Levels of cholesterol, as well as other lipids such as gangliosides and phospholipids, are decreased in the ipsilateral hippocampus of neonatal rats for 7–90 days after hypoxic–ischemic brain injury.13 The decreased lipid levels seen after hypoxic–ischemic injury were significantly greater than those seen with ischemic stroke-like injury alone or with hypoxia alone, suggesting that the combined injury has a greater effect on cholesterol metabolism than either injury alone.13 This is consistent with the adult study described above that found no change in brain cholesterol levels after stroke injury.58
CYP46 is upregulated at 6 and 24 h after NE with an associated increase in 24S-HC in serum and ipsilateral cortex at those time points, suggesting that one mechanism resulting in the decreased cholesterol levels may be cholesterol efflux.12,27 This is in contrast to TBI, where upregulation of CYP46 is also seen after injury, but serum 24S-HC is not elevated. The elevated brain levels of 24S-HC may potentiate the brain injury, as 24S-HC functions to exaggerate oxygen glucose deprivation-induced damage through activation of NMDA receptors.17 25-Hydroxycholesterol (25-HC) is an oxysterol synthesized by macrophages and possibly microglia that may antagonize the NMDA receptor-mediated effects of 24S-HC. This decrease in NMDA receptor activation may provide neuroprotection through a reduction in excitotoxicity.74
The hypoxic–ischemic injury itself may not be the only aspect of the infant’s care, however, that alters cholesterol metabolism. The current standard management for NE is therapeutic hypothermia. In conjunction with hypoxia, mild hypothermia has been shown to alter the body’s metabolic response, resulting in increased triglycerides, total cholesterol, and LDL cholesterol (LDL-C), as well as inhibition of lipid uptake.75 As such, studies of cholesterol metabolism and cholesterol-related therapies should take into account the potential difficulty in translating any animal studies performed at normothermia to human subjects with NE who will be undergoing hypothermia.
Cholesterol metabolites as biomarkers for brain injury
Infants with NE have limited time for diagnosis as hypothermia must be initiated within the first 6 h after injury for full effect, and ideally should be started in the first 3 h.76 Unfortunately, the brain injury from the hypoxic–ischemic injury evolves over the first hours of life, resulting in variable examination findings and each of the current diagnostic criteria individually have relatively low sensitivity and specificity. Because of this, several investigators have attempted to identify novel biomarkers for diagnosis and prognosis of infants after NE. Many biomarkers have been evaluated, although none have yet been validated as providing benefit in clinical practice. A few studies have examined cholesterol metabolites as serum biomarkers for brain injury, and some have shown early promise.
As mentioned above, 24S-HC is brain-specific and is able to cross the BBB much easier than cholesterol, so it is a promising candidate for a serum biomarker of brain injury. 24S-HC has been found to be decreased in the plasma of patients with Parkinson’s disease but increased in Alzheimer’s disease.77,78,79 In an NE model, animals with higher serum 24S-HC at 6 and 24 h corresponded to larger infarct volume and more significant injury at 35–40 days after injury.27 Functionally, the higher serum 24S-HC levels were associated with increased anxiety and poorer novel object recognition in mice after stroke.27
Desmosterol is another cholesterol-related biomarker prospect, as levels of desmosterol have been found to be decreased in the plasma of patients with Alzheimer’s disease as compared to controls.80 Conversely, desmosterol was increased in patients with myotonic dystrophy.81 These changes were not identified in patients with schizophrenia or Parkinson disease;80 however, and to date, no study has assessed serum desmosterol levels after neonatal hypoxic–ischemic brain injury.
Cholesterol modulation as therapy for brain injury
In addition to clarifying pathophysiology and providing potential biomarkers, the identification of aberrations in brain cholesterol metabolism in infants with NE could also provide novel avenues for future therapy.
Dietary cholesterol supplementation
Although cholesterol does not cross the BBB under healthy conditions, brain injury often affects BBB integrity. For example, an experimental model of demyelination via cuprizone demonstrated increased permeability of the BBB after injury, which resulted in the ability of dietary cholesterol to penetrate into the brain.82 Those animals that received cholesterol supplementation had a significant increase in the number of astrocytes, a reduction in the number of activated microglial cells, and attenuated axonal damage.82
In a rat model of TBI, however, exposure to a high-fat diet prior to injury was associated with higher baseline serum cholesterol, but had no impact on neuronal apoptosis or neuroinflammation, suggesting that cholesterol penetration of the BBB was not increased despite injury.83 In addition, patients with Smith–Lemli–Opitz did not show improvement in developmental quotients or behavior when administered dietary cholesterol.52,84 The therapeutic effects of dietary cholesterol supplementation in NE has not been evaluated.
HMGCR inhibitors
HMGCR inhibitors (a.k.a. statins) have been used for many years to treat disorders of cholesterol metabolism; however, recently there has been increasing interest in their use after brain injuries such as TBI and NE. Statins are a class of drug that inhibit HMGCR, thereby decreasing the levels of LDL-C, apolipoprotein B, and triglycerides, as well as inhibiting the biosynthesis of cholesterol in the liver.56,85 Statins have also been shown to have anti-inflammatory effects that appear to be unrelated to cholesterol homeostasis, such as reducing C-reactive protein levels and decreasing other proinflammatory markers, including tumor necrosis factor-α (TNF-α) and interleukin-6.56,85
Statins appear to impact TBI via a combination of cholesterol-dependent and -independent effects.83 They were shown to cause a decrease in the formation of isoprenoids and inflammatory mediators with the improvement of BBB integrity and cerebral edema.86 Furthermore, in rats that experienced TBI and then were given simvastatin, there was decreased TNF-α expression in astrocytes and microglia.83 Functionally, administration of statins after TBI was associated with a reduction in neurologic deficits and improvement in motor function.83,87
Statins also may improve endothelial function and increase cerebral blood flow. They have been shown to decrease intravascular thrombosis and promote angiogenesis, as well as reducing infarct volume and glial cell activation in stroke.86,87 In addition, lovastatin limited levels of 24S-HC, cholesterol, and 7-ketocholesterol in an animal model of excitotoxic injury.88
When considering the use of statins in neonates such as those affected by NE, data regarding safety are limited, although statins have been used with some success in some inherited disorders of cholesterol metabolism, including CTX and Smith–Lemli–Opitz.89,90,91 A few small studies have assessed the use of statins in animal models of NE. Prophylactic simvastatin had a neuroprotective effect in animal models of NE, potentiating autophagy and resulting in smaller infarcts compared to the ischemic group. In addition, prophylactic simvastatin improved memory as demonstrated by improved times in a circular water maze that was similar to uninjured controls.92 Expression of sirtuin 1, a gene involved in the regulation of metabolism and autophagy, was preserved in simvastatin-treated animals, suggesting that it may play a role in the neuroprotective findings.93 In another study, there was a reduction in pyknotic cells, microglial activation, and neuronal loss after NE with the administration of simvastatin. Hypomyelination in the external and internal capsule was also reduced in the treatment group at 3 and 7 days after injury.94 These effects may be due, in part, to simvastatin inducing the overproduction of DHCR24, thus exerting its previously characterized neuroprotective effects.72
Current research gaps
Dysregulation of cholesterol metabolism is found in neurodegenerative, traumatic, and ischemic disease states. Early studies have similarly suggested that the underlying pathophysiology of NE may also be linked to cholesterol metabolism. Future studies could help to answer several questions that remain in the neonatal literature regarding the interaction between cholesterol and neonatal hypoxic–ischemic brain injury. For instance, is it possible to use serum levels of cholesterol metabolites such as 24S-HC or desmosterol as biomarkers for effective diagnosis and/or prognosis in infants after NE? How are the levels of cholesterol and its precursors affected by neonatal hypoxic–ischemic brain injury and can those effects on cholesterol metabolism be ameliorated by targeted therapies? Lastly, can systemic therapies such as dietary cholesterol supplementation or administration of statins after injury improve the outcomes of neonates suffering from NE? Continued investigation of cholesterol dysregulation in NE may provide an avenue for novel interventions and improvements in therapy could decrease the significant neurodevelopmental morbidities seen in infants after NE.
References
Kurinczuk, J. J., White-Koning, M. & Badawi, N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum. Dev. 86, 329–338 (2010).
Allen, K. A. & Brandon, D. H. Hypoxic ischemic encephalopathy: pathophysiology and experimental treatments. Newborn Infant Nurs. Rev. 11, 125–133 (2011).
Douglas-Escobar, M. & Weiss, M. D. Hypoxic-ischemic encephalopathy: a review for the clinician. JAMA Pediatr. 169, 397–403 (2015).
Yu, Z. et al. Hypoxia-ischemia brain damage disrupts brain cholesterol homeostasis in neonatal rats. Neuropediatrics 40, 179–185 (2009).
Bruns, N. et al. Application of an Amplitude-integrated EEG Monitor (Cerebral Function Monitor) to neonates. J. Vis. Exp. 127, (2017).
Chandrasekaran, M., Chaban, B., Montaldo, P. & Thayyil, S. Predictive value of amplitude-integrated EEG (aEEG) after rescue hypothermic neuroprotection for hypoxic ischemic encephalopathy: a meta-analysis. J. Perinatol. 37, 684–689 (2017).
Del Río, R. et al. Amplitude integrated electroencephalogram as a prognostic tool in neonates with hypoxic-ischemic encephalopathy: a systematic review. PLoS ONE 11, e0165744 (2016).
Hackam, D. G. & Hegele, R. A. Cholesterol lowering and prevention of stroke. Stroke 50, 537–541 (2019).
Kay, A. D. et al. Remodeling of cerebrospinal fluid lipoprotein particles after human traumatic brain injury. J. Neurotrauma 20, 717–723 (2003).
Björkhem, I. & Hansson, M. Cerebrotendinous xanthomatosis: an inborn error in bile acid synthesis with defined mutations but still a challenge. Biochem. Biophys. Res. Commun. 396, 46–49 (2010).
Fitzky, B. U. et al. Mutations in the Delta7-sterol reductase gene in patients with the Smith–Lemli–Opitz syndrome. Proc. Natl Acad. Sci. USA 95, 8181–8186 (1998).
Lu, F. et al. Upregulation of cholesterol 24-hydroxylase following hypoxia-ischemia in neonatal mouse brain. Pediatr. Res. 83, 1218–1227 (2018).
Ramirez, M. R. et al. Neonatal hypoxia-ischemia reduces ganglioside, phospholipid and cholesterol contents in the rat hippocampus. Neurosci. Res. 46, 339–347 (2003).
Zhang, J. & Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 6, 254–264 (2015).
Alphonse, P. A. S. & Jones, P. J. H. Revisiting human cholesterol synthesis and absorption: the reciprocity paradigm and its key regulators. Lipids 51, 519–536 (2016).
Dietschy, J. M. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 390, 287–293 (2009).
Sun, M.-Y. et al. 24(S)-Hydroxycholesterol as a modulator of neuronal signaling and survival. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 22, 132–144 (2016).
Genaro-Mattos, T. C. et al. Cholesterol biosynthesis and uptake in developing neurons. ACS Chem. Neurosci. 10, 3671–3681 (2019).
Zerenturk, E. J., Sharpe, L. J., Ikonen, E. & Brown, A. J. Desmosterol and DHCR24: unexpected new directions for a terminal step in cholesterol synthesis. Prog. Lipid Res. 52, 666–680 (2013).
Jansen, M. et al. What dictates the accumulation of desmosterol in the developing brain? FASEB J. 27, 865–870 (2013).
Hernández-Jiménez, M. et al. Seladin-1/DHCR24 is neuroprotective by associating EAAT2 glutamate transporter to lipid rafts in experimental stroke. Stroke 47, 206–213 (2016).
Nguyen, A. D., McDonald, J. G., Bruick, R. K. & DeBose-Boyd, R. A. Hypoxia stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase through accumulation of lanosterol and hypoxia-inducible factor-mediated induction of insigs. J. Biol. Chem. 282, 27436–27446 (2007).
Dorszewska, J. & Adamczewska-Goncerzewicz, Z. Patterns of free and esterified sterol fractions of the cerebral white matter in severe and moderate experimental hypoxia. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 6, 227–231 (2000).
Wender, M., Adamczewska-Goncerzewicz, Z., Doroszewska, J. & Szczech, J. Free sterols in the rat white matter following experimental global ischemia. Exp. Toxicol. Pathol. 49, 57–59 (1997).
Cartagena, C. M. et al. Cortical injury increases cholesterol 24S hydroxylase (Cyp46) levels in the rat brain. J. Neurotrauma 25, 1087–1098 (2008).
Cartagena, C. M., Burns, M. P. & Rebeck, G. W. 24S-hydroxycholesterol effects on lipid metabolism genes are modeled in traumatic brain injury. Brain Res. 1319, 1–12 (2010).
Lu, F. et al. Serum 24S-hydroxycholesterol predicts long-term brain structural and functional outcomes after hypoxia-ischemia in neonatal mice. J. Cereb. Blood Flow Metab. 271678X20911910 (2020).
Björkhem, I. et al. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39, 1594–1600 (1998).
Emnett, C. M. et al. Interaction between positive allosteric modulators and trapping blockers of the NMDA receptor channel. Br. J. Pharm. 172, 1333–1347 (2015).
Grayaa, S. et al. Plasma oxysterol profiling in children reveals 24-hydroxycholesterol as a potential marker for Autism Spectrum Disorders. Biochimie 153, 80–85 (2018).
Björkhem, I. et al. Oxysterols and Parkinson’s disease: evidence that levels of 24S-hydroxycholesterol in cerebrospinal fluid correlates with the duration of the disease. Neurosci. Lett. 555, 102–105 (2013).
Leoni, V. et al. Changes in human plasma levels of the brain specific oxysterol 24S-hydroxycholesterol during progression of multiple sclerosis. Neurosci. Lett. 331, 163–166 (2002).
Ye, J. & DeBose-Boyd, R. A. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb. Perspect. Biol. 3, 7. https://cshperspectives.cshlp.org/content/3/7/a004754.long (2011).
Eberlé, D. et al. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86, 839–848 (2004).
Vergnes, L. et al. SREBP-2-deficient and hypomorphic mice reveal roles for SREBP-2 in embryonic development and SREBP-1c expression. J. Lipid Rs. 57, 410–421 (2016).
Ferris, H. A. et al. Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc. Natl Acad. Sci. USA 114, 1189–1194 (2017).
Woollett, L. A. Review: Transport of maternal cholesterol to the fetal circulation. Placenta 32, S218–S221 (2011).
Woollett, L. A. Maternal cholesterol in fetal development: transport of cholesterol from the maternal to the fetal circulation. Am. J. Clin. Nutr. 82, 1155–1161 (2005).
McConihay, J. A., Horn, P. S. & Woollett, L. A. Effect of maternal hypercholesterolemia on fetal sterol metabolism in the Golden Syrian hamster. J. Lipid Res. 42, 1111–1119 (2001).
Tint, G. S. et al. The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J. Lipid Res. 47, 1535–1541 (2006).
Tint, G. S. et al. Desmosterol in brain is elevated because DHCR24 needs REST for robust expression but REST is poorly expressed. Dev. Neurosci. 36, 132–142 (2014).
Jurevics, H. A., Kidwai, F. Z. & Morell, P. Sources of cholesterol during development of the rat fetus and fetal organs. J. Lipid Res. 38, 723–733 (1997).
Pfrieger, F. W. & Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 50, 357–371 (2011).
Nägler, K., Mauch, D. H. & Pfrieger, F. W. Glia-derived signals induce synapse formation in neurones of the rat central nervous system. J. Physiol. 533, 665–679 (2001).
Ullian, E. M., Sapperstein, S. K., Christopherson, K. S. & Barres, B. A. Control of synapse number by glia. Science 291, 657–661 (2001).
Hinse, C. H. & Shah, S. N. The desmosterol reductase activity of rat brain during development. J. Neurochem. 18, 1989–1998 (1971).
Nie, S., Chen, G., Cao, X. & Zhang, Y. Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 9, 179 (2014).
Ben Hamida, M. et al. Peripheral neuropathy in a sporadic case of cerebrotendinous xanthomatosis. Rev. Neurol. 147, 385–388 (1991).
Tint, G. S. et al. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 330, 107–113 (1994).
Tint, G. S. et al. Correlation of severity and outcome with plasma sterol levels in variants of the Smith-Lemli-Opitz syndrome. J. Pediatr. 127, 82–87 (1995).
Nowaczyk, M. J. M. & Irons, M. B. Smith-Lemli-Opitz syndrome: phenotype, natural history, and epidemiology. Am. J. Med. Genet. C 160C, 250–262 (2012).
Tierney, E., Conley, S. K., Goodwin, H. & Porter, F. D. Analysis of short-term behavioral effects of dietary cholesterol supplementation in Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. A 152A, 91–95 (2010).
Porter, F. D. Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 16, 535–541 (2008).
Fitzky, B. U. et al. 7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome. J. Clin. Invest. 108, 905–915 (2001).
Sodero, A. O. et al. Cholesterol loss during glutamate-mediated excitotoxicity. EMBO J. 31, 1764–1773 (2012).
Catapano, A. L., Pirillo, A. & Norata, G. D. Vascular inflammation and low-density lipoproteins: is cholesterol the link? A lesson from the clinical trials. Br. J. Pharm. 174, 3973–3985 (2017).
Martiskainen, H. et al. DHCR24 exerts neuroprotection upon inflammation-induced neuronal death. J. Neuroinflamm. 14, 215 (2017).
Yatsu, F. M. & Moss, S. A. Brain lipid changes following hypoxia. Stroke 2, 587–593 (1971).
Kacher, R. et al. CYP46A1 gene therapy deciphers the role of brain cholesterol metabolism in Huntington’s disease. Brain J. Neurol. 142, 2432–2450 (2019).
del Toro, D. et al. Altered cholesterol homeostasis contributes to enhanced excitotoxicity in Huntington’s disease. J. Neurochem. 115, 153–167 (2010).
Valenza, M. et al. Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J. Neurosci. J. Soc. Neurosci. 25, 9932–9939 (2005).
Hu, G. et al. Total cholesterol and the risk of Parkinson disease. Neurology 70, 1972–1979 (2008).
Hu, G. et al. Body mass index and the risk of Parkinson disease. Neurology 67, 1955–1959 (2006).
Doria, M. et al. Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free Radic. Biol. Med. 101, 393–400 (2016).
Lee, C.-Y. J. et al. Different patterns of oxidized lipid products in plasma and urine of dengue fever, stroke, and Parkinson’s disease patients: cautions in the use of biomarkers of oxidative stress. Antioxid. Redox Signal. 11, 407–420 (2009).
Choi, J.-Y., Jang, E.-H., Park, C.-S. & Kang, J.-H. Enhanced susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in high-fat diet-induced obesity. Free Radic. Biol. Med. 38, 806–816 (2005).
Bousquet, M. et al. High-fat diet exacerbates MPTP-induced dopaminergic degeneration in mice. Neurobiol. Dis. 45, 529–538 (2012).
Lim, L. et al. Lanosterol induces mitochondrial uncoupling and protects dopaminergic neurons from cell death in a model for Parkinson’s disease. Cell Death Differ. 19, 416–427 (2012).
Xiong, Y., Zhang, Y., Mahmood, A. & Chopp, M. Investigational agents for treatment of traumatic brain injury. Expert Opin. Investig. Drugs 24, 743–760 (2015).
Morganti-Kossmann, M. C., Satgunaseelan, L., Bye, N. & Kossmann, T. Modulation of immune response by head injury. Injury 38, 1392–1400 (2007).
Wang, J. et al. Traumatic brain injury leads to accelerated atherosclerosis in apolipoprotein E deficient mice. Sci. Rep. 8, 5639 (2018).
Weiner, M. F. et al. Plasma 24S-hydroxycholesterol and other oxysterols in acute closed head injury. Brain Inj. 22, 611–615 (2008).
Saher, G. & Stumpf, S. K. Cholesterol in myelin biogenesis and hypomyelinating disorders. Biochim. Biophys. Acta 1851, 1083–1094 (2015).
Linsenbardt, A. J. et al. Different oxysterols have opposing actions at N-methyl-D-aspartate receptors. Neuropharmacology 85, 232–242 (2014).
Jun, J. C. et al. Thermoneutrality modifies the impact of hypoxia on lipid metabolism. Am. J. Physiol. 304, E424–E435 (2013).
Denihan, N. M., Boylan, G. B. & Murray, D. M. Metabolomic profiling in perinatal asphyxia: a promising new field. BioMed. Res. Int. 2015, 254076 (2015).
Fabelo, N. et al. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 17, 1107–1118 (2011).
Schönknecht, P. et al. Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer’s disease compared to healthy controls. Neurosci. Lett. 324, 83–85 (2002).
Papassotiropoulos, A. et al. Plasma 24S-hydroxycholesterol: a peripheral indicator of neuronal degeneration and potential state marker for Alzheimer’s disease. NeuroReport 11, 1959–1962 (2000).
Sato, Y. et al. Identification of a new plasma biomarker of Alzheimer’s disease using metabolomics technology. J. Lipid Res. 53, 567–576 (2012).
Wakamatsu, H. et al. Serum desmosterol and other lipids in myotonic dystrophy-a possible pathogenesis of myotonic dystrophy. Keio J. Med. 19, 145–149 (1970).
Berghoff, S. A. et al. Dietary cholesterol promotes repair of demyelinated lesions in the adult brain. Nat. Commun. 8, 14241 (2017).
Chong, A. J. et al. The neuroprotective effects of simvastatin on high cholesterol following traumatic brain injury in rats. World Neurosurg. 132, e99–e108 (2019).
Sikora, D. M. et al. Cholesterol supplementation does not improve developmental progress in Smith-Lemli-Opitz syndrome. J. Pediatr. 144, 783–791 (2004).
Ridker, P. M. et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N. Engl. J. Med. 344, 1959–1965 (2001).
Wible, E. F. & Laskowitz, D. T. Statins in traumatic brain injury. Neurother. J. Am. Soc. Exp. Neurother. 7, 62–73 (2010).
Xu, X. et al. Anti-inflammatory and immunomodulatory mechanisms of atorvastatin in a murine model of traumatic brain injury. J. Neuroinflamm. 14, 167 (2017).
He, X. et al. Lovastatin modulates increased cholesterol and oxysterol levels and has a neuroprotective effect on rat hippocampal neurons after kainate injury. J. Neuropathol. Exp. Neurol. 65, 652–663 (2006).
Wassif, C. A. et al. A placebo-controlled trial of simvastatin therapy in Smith-Lemli-Opitz syndrome. Genet. Med. 19, 297–305 (2017).
Jira, P. E. et al. Simvastatin. A new therapeutic approach for Smith-Lemli-Opitz syndrome. J. Lipid Res. 41, 1339–1346 (2000).
Verrips, A. et al. Effect of simvastatin in addition to chenodeoxycholic acid in patients with cerebrotendinous xanthomatosis. Metabolism 48, 233–238 (1999).
Balduini, W., De Angelis, V., Mazzoni, E. & Cimino, M. Simvastatin protects against long-lasting behavioral and morphological consequences of neonatal hypoxic/ischemic brain injury. Stroke 32, 2185–2191 (2001).
Carloni, S. & Balduini, W. Simvastatin preconditioning confers neuroprotection against hypoxia-ischemia induced brain damage in neonatal rats via autophagy and silent information regulator 1 (SIRT1) activation. Exp. Neurol. 324, 113117 (2020).
Li, A. et al. Simvastatin attenuates hypomyelination induced by hypoxia-ischemia in neonatal rats. Neurol. Res. 32, 945–952 (2010).
Rufini, S. et al. Cholesterol depletion inhibits electrophysiological changes induced by anoxia in CA1 region of rat hippocampal slices. Brain Res. 1298, 178–185 (2009).
Balduini, W. et al. Prophylactic but not delayed administration of simvastatin protects against long-lasting cognitive and morphological consequences of neonatal hypoxic-ischemic brain injury, reduces interleukin-1beta and tumor necrosis factor-alpha mRNA induction, and does not affect endothelial nitric oxide synthase expression. Stroke 34, 2007–2012 (2003).
Acknowledgements
We thank Dr. Zeljka Korade for her support and advice during the process of writing this review.
Author information
Authors and Affiliations
Contributions
A.M.D and E.S.P. both contributed substantially to the conception, design, drafting, and critical revisions of the article. Both authors reviewed and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dave, A.M., Peeples, E.S. Cholesterol metabolism and brain injury in neonatal encephalopathy. Pediatr Res 90, 37–44 (2021). https://doi.org/10.1038/s41390-020-01218-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-01218-3
This article is cited by
-
Proteomic analysis discovers potential biomarkers of early traumatic axonal injury in the brainstem
International Journal of Legal Medicine (2024)
