
Abstract
Thyroid cancer is the fifth most common cancer diagnosed in women worldwide. Notwithstanding advancements in the prognosis and treatment of thyroid cancer, 10–20% of thyroid cancer patients develops chemotherapeutic resistance and experience relapse. According to previous reports and TCGA database, MUC15 (MUCIN 15) upregulation is highly correlated with thyroid cancer progression. However, the role of MUC15 in tumor progression and metastasis is unclear. This study aimed to investigate factors mediating cancer stemness in thyroid cancer. MUC15 plays an important role in sphere formation, as an evident from the expression of stemness markers including SOX2, KLF4, ALDH1A3, and IL6. Furthermore, ectopic expression of MUC15 activated extracellular signal-regulated kinase (ERK) signaling via G-protein–coupled receptor (GPCR)/cyclic AMP (cAMP) and integrin/focal adhesion kinase pathways. Interestingly, ectopic expression of MUC15 did not affect RAF/mitogen-activated protein kinase kinase (MEK)-mediated ERK activation. The present findings may provide novel insights into the development of diagnostic, prognostic, and therapeutic applications of MUC15 in thyroid cancer.
Similar content being viewed by others


Introduction
According to reports of National Cancer Institutes (NCI), thyroid cancer has shown a significant increase over the last 30 years1. Although thyroid cancer has a good prognosis and is considered easily curable via surgical resection and radio-iodine based therapies, 10–20% of thyroid cancer cases involve aggressive behavior including local invasion, distant metastasis, drug resistance, recurrence, and mortality2,3,4,5. Since thyroid carcinoma is at a high risk of invasion, recurrence, and metastasis, it is important to study the underlying molecular mechanism.
A crucial cancer stem cell (CSC) population causes these malignant phenotypes in various cancers6,7,8,9,10, especially thyroid cancer11,12. Indeed CSCs can be verified through clonogenic assays of cells to assess proliferative capacity including sphere formation in vitro13,14 or via in vivo tumorigenesis experiments15. Current radiotherapy and chemotherapy often eliminate the bulk of cancer cells but not CSCs, which are protected via specific resistance mechanisms16,17. These unsolved issues may be explained on the basis of CSC-like properties of many tumor types6,11,18,19. We found that one isoform of mucins, MUC15 play a critical role of mediating cancer stemness in thyroid cancers.
Mucins are high-molecular-weight membrane glycoproteins (>200 kDa) in various types of epithelial cells20,21,22. Secreted form of mucins have a protective functional epithelial barrier to protect against from bacteria and virus infections21 while membrane-associated-mucin proteins are intracellular receptors involved in signal transduction, leading to coordinated cellular responses including proliferation, differentiation, apoptosis, and secretion of specialized cellular products23,24.
MUC15 upregulation is significantly correlated with various types of cancers such as colon cancer, hepatocellular carcinoma, and especially thyroid cancer25,26,27. High cancer scores of MUC15 expression are significantly correlated with age, distant metastasis, and the presence of multifocality26. In addition, ectopic MUC1 expression upregulates CSC markers in breast cancer and lung cancer such as OCT4, SOX2, ALDH1A1, IL6, and CXCR417,28,29. Moreover, MUC1 upregulation has been considered to significantly contribute to the aggressiveness of the papillary thyroid cancer (PTC)30. Similarly, MUC4 is known to be upregulated in 20% of PTC cells compared to normal cells26,31 and is correlated with the development of chemoresistance in pancreatic CSC32. These results imply that mucins play an important role in mediating tumor development and progression and are related to cancer stemness properties in thyroid cancer and various types of cancer17,29,33,34. However, its physiological role and its underlying molecular mechanisms in thyroid cancer progression and metastasis are unclear.
The RAS-ERK pathway mediates cancer progression, metastasis, and cancer stemness in various types of cancer35,36,37. Furthermore, G-protein-coupled receptor (GPCR) and integrin-focal adhesion kinase (FAK) signaling are upstream targets of activated extracellular signal-regulated kinase (ERK)38,39, thereby increasing the stemness of thyroid cancer cells.
This study aimed to investigate the correlation between CSC-like properties and thyroid cancer progression and metastasis because CSCs are crucial for cancer recurrence, metastasis, and drug resistance, which are yet unclear. Our results may provide novel insights into help understand physiological and cellular mechanisms of recurrence and metastasis in thyroid cancer and the characteristics of thyroid CSC, and in developing novel therapeutic targets for thyroid cancer patients.
Results
MUC15 is upregulated in later stages of thyroid cancer
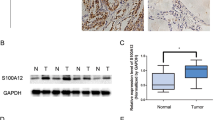
Thyroid tumors display greater MUC15 expression than neighboring normal thyroid epithelial cells26. These findings are concurrent with those of our clinical studies. Mostly thyroid tumors in patients have greater MUC15 expression than normal tissue, especially in higher-grade tumors (Fig. 1a, b). To clarify the role of MUC15 in thyroid cancer progression, we first examined MUC15 expression in thyroid cancer cells (FTC-238 and TPC-1), relative to that of normal thyroid cells (Nthy-ori-3-1). Metastatic thyroid cancer cells (FTC-238) displayed upregulated MUC15 expression compared to normal thyroid epithelial cells; in particular, FTC-238 cells displayed significantly upregulated MUC15 expression (greater than 20 folds) than Nthy-ori-3.1 cells (p = 0.0014); however, TPC-1 (non-metastatic cancer cells) did not display a significant difference (Fig. 1c and Supplementary Figure S1D). Consistent with reverse transcription PCR (RT-PCR) results, western blot as well as and immuno-fluorescence analyses confirmed increased MUC15 expression in thyroid cancer cells (Fig. 1e). These findings are confirmed via TCGA database using GEPIA website based on a published dataset at 2014 (http://gepia.cancer-pku.cn/) (Fig. 1d and Supplementary Figure S1A)40,41. In addition, thyroid cancer patient with high level of MUC15 show worse prognosis analyzed by cBioPortal (http://www.cbioportal.org/) and GEPIA (http://gepia.cancer-pku.cn/) websites respectively (Supplementary Figure S1B, C)41,42.

a Relative expression level of MUC15 between tumor (25 cases) and their neighboring normal thyroid tissue (27 cases) in thyroid cancer patients. b MUC15 Immunohistochemical (IHC) positivity in the patients was scored from 0 to 3 according to the percentage positivity (grade 0, 0%; grade 1, 1–33%; grade 2, 34–66%; grade 3, 67–100%). c Relative expression level of MUC15 normalized by GAPDH expression level among Nthy-ori-3-1, TPC-1, and FTC-238 cells. Western blots showing the expression level of the MUC15 protein. d Thyroid tumors have enhanced expression of MUC15 in patients according to TCGA database. e Immunostaining of MUC15 in Nthy-ori-3-1, TPC1 and FTC-238 cells, respectively
Ectopic MUC15 expression promotes CSC-like properties
Metastasis and chemotherapeutic resistance are associated with properties of CSCs in various types of cancers18,43,44,45. Sphere formation is a key characteristic of stem cells and CSCs; hence, this characteristic is harnessed in analyzing properties of cancer stemness34,46,47,48. While investigating the physiological role of MUC15 in thyroid cancer, we found that FTC-238 cancer cells display greater sphere formation abilities; however, Nthy-ori-3-1 and TPC-1 cells did not adequately generate spheres (Fig. 2a, b).

a, b Representative images showing sphere formation among Nthy-ori-3-1 cells, TPC-1 and FTC-238 cells and quantitative values measured size of sphere from the results. c, d Representative images show sphere formation ability among transfected with Empty vector and indicated MUC15 shRNA respectively and quantitative values measured size of sphere from the results. e–g Representative images showing sphere formation between Nthy-ori-3-1 and MUC15-OV (MUC15 overexpression) cells and quantitative values measured size of sphere from the results. h RT-qPCR data show that MUC15 over expression cell increased the expression of stem cell markers SOX2, OCT4, KLF4 and cancer stem cell markers ALDH1A3 and IL6
To confirm the function of MUC15 in sphere formation, we knocked down MUC15 in FTC-238 with lentiviral MUC15 shRNA (Supplementary Figure S2A and C). Consequently, sphere formation abilities of MUC15 shRNA-transduced cells were lower than those of control cells (cells transfected with the vector control) (Fig. 2c, d). These data together suggest MUC15 expression is strongly associated with sphere formation among thyroid cancer cells.
To further verify whether MUC15 regulates sphere formation, we transduced MUC15 expressing lentivirus to Nthy-ori-3-1 cells to generate cells stably overexpressing MUC15, referred to as MUC15 overexpress cells (MUC15-OV cell) (Supplementary Figure S2B, D, E and F). Interestingly, these cells displayed enhanced sphere formation ability, implying CSC-like properties (Fig. 2e, f). We also quantified the number and size of spheres between Nthy-ori-3-1 and MUC15-OV cells. Although both Nthy-ori-3-1 and MUC15-OV cells could form spheres of 50–100 and 100–150 µm, MUC15-OV cells exhibited numerous colonies. In particular, spheres of greater than 250 µm were only found in MUC15-OV cells (Fig. 2g).
The expression level of stem cell markers SOX2, OCT4, and KLF4 in MUC15-OV cells were 2.38, 1.45, and 40.1 folds higher than those in normal thyroid epithelial cells, NThy-ori-3.1, respectively (Fig. 2h). Additionally, the expression level of ALDH1A3, a CSC marker among various types of cancer cells49, and IL6 known as a downstream functional target to induce sphere formation in many types of cancer cells50,51,52,53, were 22.9 and 3.09 folds higher in MUC15-OV cells than in Nthy-ori-3.1 cells (Fig. 2h).
In addition, ectopic MUC15 expression increases cellular survival and proliferation even in adhered cells. The data show that the number of MUC15-OV cells was higher than that of Nthy-ori-3.1 cells during suspension culture (Supplementary Figure S3A). Moreover, MUC15-OV cells showed a low population of apoptotic cells in suspension culture, especially 3 and 5d after incubation, as determined via FACS (Supplementary Figure S3B, C). Since MUC15 could inhibit apoptosis, thereby increasing the number of survived cells in suspension culture, this property is correlated with metastasis and CSC-like properties25.
MUC15 activates the MEK-ERK pathway independent of BRAF
MUC15 induces the activation of oncogenic ERK activation, thereby mediating metastasis in colon cancer25. MUC1 could activate ERK-C/EBP beta signaling in breast cancer cells28. MUC4 triggers ERK activation dependent on confluence of mammary epithelial and tumor cells54. These findings together suggest that MUC15 may also activate ERK signaling in thyroid epithelial cells. Thus, we investigated whether over-expression of MUC15 increases ERK phosphorylation. Indeed, MUC15-OV cells showed increased MEK and ERK phosphorylation (Fig. 3a). However, we could not observe differences in BRAF activation on adding hEGF (100 ng/ml) between Nthy-ori-3.1 and MUC15-OV cells, although the initial state of MEK and ERK phosphorylation was enhanced in MUC15-OV cells (Fig. 3b). Previously, activation of ERK is associated with CSC-like properties and sphere formation34. ERK suppression with pharmacological inhibitor UO126 decreased sphere formation in MUC15-OV cells (Fig. 3c, d). These observations suggest that MUC15 plays an important role in developing CSCs via the MEK-ERK pathway.

a The level of phosphorylation ERK1/2 and total ERK1/2 in MUC15 over expression cell are higher than Nthy-ori cell. b Nthy-ori and MUC15 over expression cells were treated with either serum or hEGF for 10, 30, 60 min and RAF/MEK/ERK protein expression were determined by immunoblotting. c, d Blocking ERK activity by UO126 (10 µM) were determined by immunoblotting and inhibition of ERK decreased mammosphere formation ability of MUC15 over expression cells
MUC15 induces expression of genes related to the GPCR-cAMP pathway, thereby increasing ERK phosphorylation
Sphere formation is regulated by various signaling pathways that promote CSCs. To determine signaling pathway involves MUC15, we performed next-generation sequencing (NGS) for total mRNA and compared the expression of numerous genes between Nthy-ori-3-1 and MUC15-OV cells, using a log2 scale heat-map via MeV program (Fig. 4a)55. Differentially expressed target genes were analyzed in silico to further analyze enriched signaling pathways through the Carcinogenic Potency Database (CPDB) molgen website (http://cpdb.molgen.mpg.de/) and GO-term analysis using PANTHER (http://pantherdb.org/)56,57. These two different tools revealed that the molecular signaling pathway related to MUC15 is the GPCR pathway (Supplementary Figure S4A, B, Table S1 and Table S2). We confirmed the expression of GPCR-related genes in response to MUC15 via RT-qPCR and western blot analysis (Fig. 4b–d). As shown in Fig. 4b; CCR7, C3 expression level in MUC15-OV cell was greater than eight-folds and two-folds than those in Nthy-ori-3.1 cells, respectively.

MUC15 drives activation of ERK via GPCR-cAMP signaling pathway independent to RAF/MEK, associated to FAK signaling. a GPCR signaling was determined as a downstream target of MUC15 by using RNA-SEQ analysis. b–d RT-PCR and RT-qPCR and western blot data represented the gene upregulated of GPCR signaling pathway in MUC15 overexpression cell and Nthy-ori cell. e, f Nthy-ori and MUC15 overexpression cell were treated with PTX, an inhibitor of GPCR and Caffeine, an inhibitor of cAMP. The protein expression was determined by immunoblotting. g Nthy-ori and MUC15 over expression cells were treated PF-573228 as 10 μM, as an inhibitor of FAK, and protein levels of MEK/ERK were determined by immunoblotting
Together, RNA-SEQ analysis and western blot analysis displaying that over-expression of MUC15 mediates activation of ERK, we hypothesized that MUC15 may induce activation of ERK via a GPCR pathway rather than RAS-RAF pathway, in accordance with a previous study58. Previously, cells were treated with pertusis toxin (PTX) (200 ng/ml) to inhibit the GPCR signaling pathway59. PTX is known to catalyze the ADP-ribosylation of the αi subunits of the heterotrimeric G protein60. Interestingly, phosphorylation level of ERK of MUC15-OV cells was decreased with PTX treatment after 4 h (Fig. 4e). This inhibitor can block the activation of ERK, although phosphorylation levels of BRAF and MEK were not altered mediated by MUC15 over-expression (Fig. 4e).
These results demonstrate that MUC15 activate GPCR-ERK signaling; however, the involvement of other pathways cannot be eliminated. We were curious about which downstream signaling pathway mediates GPCR-ERK signaling independent on MEK activation. cAMP is known as the second messenger to activate ERK in endocrine cells mediated by GPCR signaling pathway independent of MEK activation61. Therefore, we wanted to investigate whether inhibition of cAMP production by caffeine treatment (1 mM) decreases the activation of ERK without affecting MEK phsophorylation62. Surprisingly, treatment with caffeine drastically decreased the activation of ERK, but not MEK in both Nthy-ori-3-1 and MUC15-OV cell lines (Fig. 4f).
These findings raise the question why MUC15 may drive ERK activation via GPCR/CAMP pathway but stimulate MEK/ERK activation through another pathway. GPCR crosstalk with FAK signaling is critical to activate ERK activation63. MUC5AC mediates metastasis of cancer cells by interacting with integrin β4–FAK signaling in lung cancer cells64. We hence set to investigate whether FAK signaling may be associated with ERK activation triggered by MUC15 over-expression. Interestingly, FAK inhibitor (PF-00562271) can suppress activation of MEK and ERK in MUC15-OV cells (Fig. 4g). Previous findings and these results together indicate that over-expression of MUC15 could activate MEK-ERK signaling by the integrin-FAK signaling pathway.
The GPCR-cAMP pathway is critical to sustain sphere formation and motile behavior of thyroid cancer mediated by MUC15 over-expression
MUC15 enhanced sphere formation and the GPCR-cAMP axis is important for the activation of ERK signaling mediated by MUC15 (Fig. 4). Therefore, we investigated whether the GPCR-cAMP axis is involved in sphere formation in thyroid cancer. Interestingly, PTX or caffeine treatment reduces sphere formation ability of MUC15-OV cells and FTC-238 cells respectively (Fig. 5a, b). In addition, these drugs also decrease known stemness markers such as SOX2, OCT4, KLF4 CD44, and ALDH1A3 (Fig. 5c). Together, we concluded that exogenously expressed MUC15 induces the expression of genes related to the GPCR signaling pathway and triggers activation of the GPCR-cAMP-ERK pathway independent of the RAF/MEK signaling pathway.

a, b Sphere formation ability of cell lines express MUC15, MUC15 overexpression cell and FTC-238, are reduced after treated with PTX (200 ng/ml) and Caffeine (1 mM). c The expression level of stem cell and cancer stem cell in MUC15 overexpression cell were determined by RT-qPCR. d, e The migration of Nthy-ori cell and MUC15 overexpression cell after treated with PTX (200 ng/ml) and caffeine (1 mM)
However, the functional role of MUC15 on cell migration is controversial. In hepatocellular carcinoma and trophoblast-like cells, MUC15 decrease migration capability27,65. Whereas, in case of colon cancer, MUC15 drives invasive migration through boyden chambers, thereby enhancing metastasis25. In our study, there was no major difference between Nthy-ori-3-1 and MUC15-OV cells for wound healing migration capability (Fig. 5d). However, inhibition of GPCR or cAMP signaling by PTX and caffeine, respectively, decreased wound healing migration in MUC15-OV cells (Fig. 5d, e). However, caffeine treatment selectively suppresses migration ability of MUC15-OV cells, but not Nthy-ori-3-1 cells (Fig. 5d, e). These observations imply that MUC15 somehow has an inhibitory role of migration such as strong adhesion; however, activation of GPCR-cAMP signaling pathway compensates the ability of migration.
MUC15 enhanced tumorigenesis in NOD/SCID mice
To confirm cancer stem-like capacity driven by MUC15 in vivo, we additionally performed a tumorigenesis experiment using a xenograft mouse model. First, we generated lentiviral-mediated control or MUC15 shRNA knockdown cells in FTC-238, wherein MUC15 was overexpressed, compared to Nthy-ori-3-1 and TPC-1 cells. These lentiviral vectors containing tGFP thereby transfected cells, which could then be detected by the IVIS imaging system. These cells were injected subcutaneously into the belly in NOD/SCID mice and the mice were monitored for up to 30 days. Interestingly, the signal of tGFP from FTC-238 control cell has significantly higher intensity than that of MUC15 knockdown cells in all of monitoring (Fig. 6a–c and Supplementary Figure S5B). In addition, FTC-238 cells bearing control shRNA generated larger-sized tumors than those transfected with MUC15 shRNA (Supplementary Figure S5A). These data indicate that MUC15 is closely associated with tumorigenesis and carcinogenesis.

a, b The GFP signal imaging after 30 days inject FTC-238 control, FTC-238 MUC15 knockdown (FTC-238-SH) respectively. c The radiant efficiency of GFP signals from 1 day to 30 day after injection thyroid cancer cells
Discussion
Regardless of cell type to be initiated, various types of tumors develop in the thyroid gland, such as papillary (the most common), follicular, medullary, and anaplastic (the least common) tumors. Although most types of thyroid tumors are benign, there are still some types of tumors that can be malignant and metastatic, recurrent, and even display chemotherapeutic resistance. Several studies confirmed various vital signaling pathways that modulate embryonic development and stem cell maintenance. However, aberrant regulation of these intrinsic biological processes may have harmful effects on neoplastic transformation. Nowadays, it is reported that normal stem cells can transform to CSC, thereby promoting malignancy66.
Cancer stemness is considered a critical feature to mediate malignancy of cancer such as metastasis, recurrence, drug resistance in many types of cancer as well as thyroid cancer11,16. It led us to investigate what factors mediate cancer stemness in thyroid cancer. We identified MUC15 plays a key role in the progression of tumorgenesis by enhancing cancer stemness in thyroid cancer. Either gain or loss of function study demonstrates that MUC15 is necessary and sufficient factor to induce feature of thyroid CSC. The MUC15 expression is higher in follicular thyroid cancer cells, especially FTC-238 cell line, while TPC-1 cells derived from primary thyroid cancer does not show enhanced expression of MUC15. In consistent with previous studies, our results show that MUC15 play a positive role of developing thyroid cancer progression67.
Our data indicate that MUC15 expression enhanced sphere formation, the renewal characteristic of stem-like properties. In addition, MUC15 expression increases several vital genes to mediate CSC properties, SOX2, KLF4 and IL-6 in normal thyroid epithelial cells (Fig. 2h)51,53. Consistent with the previous reports, MUC15 indeed drives the invasive behavior of colon cancer and metastasis25. Collectively, our results suggest that MUC15 play an important role in developing CSC thereby mediate metastasis and recurrence of thyroid cancer.
CSC are developed by hijacking intrinsic signaling pathway which mediates progression of normal embryonic development in stem cells. This pathway is also known as key player to modulate normal embryonic development as well68,69. The GPCR pathway is thought to be related to the stem cell function by the fact that the pattern of expression of GPCR varies greatly at each step during embryonic development or differentiation70. This ability allows tumor to be regenerated after anti-cancer therapy by chemotherapy or radiotherapy.
GPCR signaling pathway closely associated with cancer stemness in thyroid cancer according to previous studies71. Consistent with this, we found MUC15 induces expression of several class of GPCR signaling related genes and confer properties of cancer stemness by the GPCR pathway. These genes that mediate GPCR signaling pathway are highly up-regulated in MUC15-OV cells such as CCR7, C3, CCL20, and C5AR1 (Fig. 4b–d). CCR7, one of chemokine receptor involved in GPCR families, it is identified as tumor progression marker in thyroid cancer patients72. In the previous report, CCR7 mediates activation of ERK thereby suppresses apoptosis in lung cancer73. CCR7 also promotes mammary tumorigenesis through amplication of stem-like cell74. CCL20 is identified as inflammatory factor to recruit dendritic cells75. Production of CCL20 by lung cancer could induce cell migration and proliferation via PI3K signaling pathway as well76. Recent study suggests that inflammatory CCL20 is also required to maintain CSC in breast cancer77. In addition, we found some up-regulated genes, ADRA2C, C3, and C5AR1, which were not previously reported in cancer stemness. These findings may provide novel candidate markers to identify CSC in thyroid as well as other types of cancer.
Important downstream target of GPCR signaling pathway is cAMP-PKA pathway. Subunit of GPCR, the Gαs and Gαi/o are able to trigger activation of adenylate cyclase to modulate production of cyclic AMP (cAMP) and cascadic activation of protein kinase A (PKA)78,79. Treatment of caffeine as inhibitor of adenylate cyclase suppresses the proliferation and migration of the cell expressing high level of MUC15 (Fig. 5). This result indicates that cAMP plays a critical role to promote sphere formation and ERK activation mediated by MUC15. It is reported that caffeine consuming such as coffee uptake reduce the risk of several types cancers including breast, colon80. These clinical studies may associate with our finding that caffeine effectively decreases CSC properties and migration ability (Fig. 5).
Ectopic expression of MUC15 increase activation of ERK signaling pathway but BRAF does not seem to be a downstream target of MUC15, although BRAF reported in many case of thyroid cancer81. Furthermore, there are cross-talks between GPCR and integrin signaling in cell proliferation the activation of some GPCR component can activate FAK and stimulate the activation of MEK/ERK signal82,83, it is consistent with the activation of FAK and GPCR association with MUC15 expression (Fig. 4e–g). Taken together, we suggest that MUC15 mediated GPCR-cAMP signaling pathway, which crosstalk with integrin-FAK signaling are critical to driving sphere formation via ERK activation independent to RAS-RAF signaling (Fig. 7).

Over-expression of MUC15 activate MEK-ERK pathway dependent on integrin-FAK and GPCR-cAMP pathway thereby promote cancer stemness and metastasis. Interestingly, cAMP signaling affects ERK activation independent on Raf-MEK signaling pathway driven by MUC15 over-expression
MUC15-mediated signaling pathway contributes a key characteristic of CSC and confers opportunities to development novel therapeutic strategies and diagnostic/prognostic markers for thyroid cancer patients. Further investigation of MUC15-mediated gene expression and downstream signaling pathway will elucidate self-renewal properties of CSC for highly tumorigenic population of CSC in thyroid cancer.
Materials and methods
Cell culture
The cell lines used to recapitulate human thyroid cancer were FTC-238, derived from follicular thyroid carcinoma (FTC) lung metastasis from a 42-year-old man, and TPC-1, derived from a weak metastatic papillary thyroid carcinoma (PTC)70. Human thyroid epithelial cell line Nthy-ori 3-1 constituted the control. Cancer cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech Cellgro, USA) containing 10% fetal bovine serum (FBS) (Gibco, USA), 100 IU/ml penicillin, and 100 µg/ml streptomycin (Invitrogen, USA). Epithelial cells were cultured with Roswell Park Memorial Institute medium (RPMI) (Mediatech Cellgro) containing 10% FBS 100 IU/ml penicillin, and 100 µg/ml streptomycin. Cells were cultured at 37 °C and 5% CO2.
In vivo tumorigenicity assay
All animal work was conducted in accordance with a protocol approved by the Institutional Animal Care and Use Committee at Soonchunhyang University. MUC15 tumorigenicity in vivo was investigated using a xenograft tumor model in the NOD/SCID mice. Eight-week-old male NOD/SCID mice were provided sterilized food and water and equally divided into three groups. Approximately 1 × 106 cells with or without MUC15 shRNA and control shRNA (vector control) were mixed with Matrigel (Corning, USA) and subcutaneously inoculated into the right flank of each NOD/SCID mice. These cells contain GFP-expressing vectors to monitor the GFP fluorescence signal using IVIS image system (Xenogen) every 5 days up to day 30. Once bearing palpable tumors (about 4 weeks after tumor cell inoculation), mice were euthanized and their tumors were isolated, and photographed. Experiments were performed in triplicate.
Human samples
Human tumor samples were collected from patients enrolled on Institutional Review Board approved trials at Yonsei Hospital (IRB 4-2012-0682). Specimens were collected after obtaining written informed consent prior to undergoing any study-specific procedures in accordance with the Declaration of Helsinki. Patient’s identity of pathological specimens remained anonymous in the context of this study.
RNA extraction and cDNA synthesis
In order to perform RT-qPCR and NGS sequencing, total content of cellular RNA was extracted from 80% confluence cells using an RNeasy Mini kit (Qiagen, USA) in accordance with the manufacture’s protocol. For cDNA synthesis, 1 μg of RNA was used as a template to reverse-transcribe the RNA into template DNA in accordance with the instructions of the ReverTra Ace qPCR RT kit (Toyobo, Japan). Polymerase chain reaction (PCR) was carried out to assess differences in mRNA expression of the aforementioned genes. The products were electrophoresed on a 2% agarose gel to visualize the differences in mRNA expression. Similarly, to further validate the results, a more sensitive quantitative PCR (qPCR) was carried out using SYBR green from Biorad.
Transfection
To develop an overexpression vector for MUC15, the pMSCV puro (lentivirus) vector was used. MUC15 gene was isolated from MUC15-expressing colon cancer cells and was used for complementary DNA (cDNA) synthesis. MUC15 cDNA was cloned into pMSCV puro vector and transfected into Nthy-ori 3-1 cells, followed by puromycin treatment for selection.
Lentiviral constructs containing MUC15-specific shRNA conjugated with puromycin resistance genes and tGFP was obtained from Sigma Aldrich (MO, USA). To generate the lentivirus, tGFP-shRNA of Lenti vector and packaging vectors were used to co-transfect with HEK293T cells in accordance with the manufacturer’s instructions. Viral supernatants were harvested and used to transduce FTC-238 cells. Transduction efficiency was analyzed via qPCR and western blotting analyses.
RNA sequencing
Total cellular RNA was extracted from cells grown to 80% confluence, using an RNeasy Mini Kit (Qiagen) in accordance with the manufacturer’s protocol. The total RNA was sent to Macrogen (Korea) for sequencing.
Proliferation assay
Cells were seeded in triplicate wells in 96-well plates at 2000 cells per well and treated with PrestoBlue® Cell Viability Reagent (Invitrogen) for the proliferation assay in accordance with the manufacturer’s protocol. The experiment was performed every day for 7 days.
Apoptosis assay
Cells were seeded in triplicate wells in six-well low-attachment plates at 5 × 104 cells per well. After incubation for 1 days, 3 days, and 5 days, cells were harvested. Apoptosis was assayed using the Annexin-V apoptosis Detection Kit (eBioscience, UK) in accordance with the manufacturer’s instructions. Cells were washed once with 100 µl Binding Buffer and stained for 10 min with Annexin-V at room temperature in dark. Stained cells were washed once with 200 µl Binding Buffer and stained again with 7-Aminoactinomycin D. Stained cells were analyzed using a BD fluorescence-activated cell sorting Canto flow cytometer (BD Biosciences, UK).
Sphere forming assay
Cells with greater stemness or self-renewal ability are likely to form spheres, which are more in number and bigger in diameter, when cultured in special media. This property was exploited in the following assay. Indicated cells were seeded at 5000 cells/well in triplicate in six-well low-attachment plates. Cells were cultured in Phenol-red free DMEM/F12 (Gibco) containing 1 ml of B27 supplement minus vitamin A (50×) (Gibco), 5 µl of rhEGF (100 µg/ml) (R&D System, UK), 100 µl of bFGF (10 µg/ml) (BD), 100 IU/ml penicillin, and 100 µg/ml streptomycin (Invitrogen). The cells were incubated for 5 days at 37 °C and 5% CO2 for sphere formation. UO126 was purchased from cell signaling technology (MA, USA). PTX and caffeine were purchased from Sigma Aldrich (MO, USA).
Western blot analysis
Starvation was induced in the cells for 12 h in serum-free media. Thereafter, growth factor hEGF (100 ng/ml) (BD) was added and cells were incubated for 0, 10, 30, and 60 min. Thereafter, cells were lysed in RIPA lysis buffer containing 1 mL of RIPA buffer (10×) (Millionpore), 100 µl PMSF (Sigma), and 1 table of protease inhibitor. Collected the cell lysate were subject to spin-down by centrifuges at 13,000 × g for 15 min and only supernatant was used for western blot analysis. Protein lysates were quantified and mixed with 4X loading dye (10 ml of NuPAGE LDS Sample Buffer (4×) and 500 µl of beta-mercaptoethanol) at 20 µg/16 µl and heated at 95 °C for 5 min. Sixteen microliters of protein sample was loaded onto a polyacrylamide gel (12%), which was run at 100 V for 2 h and the proteins were then electroblotted onto Immuno-Blot PVDF Membrane (Biorad, USA) for 1 h at 250 mA, on ice. The membrane was then incubated with primary during overnight at 4 °C and secondary antibodies for 1 h at room temperature with agitation. ECL Prime Western blotting reagent (Amersham, UK) was used to develop the membrane. The primary antibodies were as follows: anti-MUC15 (Novus, USA), anti-total BRAF, anti-phospho-BRAF (Ser445), anti-total c-RAF, anti-phospho-c-RAF (Ser338), anti-total MEK, anti-phospho-MEK, anti-total ERK1/2, and anti-phosphor-ERK1/2 (Cell signaling technology, USA). β-actin was used as the control and detected with anti-β-actin rabbit polyclonal antibody (Sigma-Aldrich, USA). Anti-rabbit secondary antibody was purchased from Dako (USA).
Statistical analysis
All quantified experimental data are expressed as mean ± SD values. To test statistical difference, student’s t-test and one-way ANOVA were conducted. GraphPad Prism 7.0 (GraphPad Software Inc., San Diego, CA, USA) was used for statistics analysis and *P < 0.05, **P < 0.01, and ***P < 0.001 values were used as significant values.
References
Nguyen, Q. T. et al. Diagnosis and treatment of patients with thyroid cancer. Am. Health Drug Benefits 8, 30–40 (2015).
Høie, J., Stenwig, A. E., Kullmann, G. & Lindegaard, M. Distant metastases in papillary thyroid cancer. A review of 91 patients. Cancer 61, 1–6 (1988).
Mazzaferri, E. L. & Jhiang, S. M. Long-term impact of initial surgical and medical therapy on papillary and follicular thyroid cancer. Am. J. Med. 97, 418–428 (1994).
Stassi, G. et al. Thyroid cancer resistance to chemotherapeutic drugs via autocrine production of interleukin-4 and interleukin-10. Cancer Res. 63, 6784–6790 (2003).
Xing, M. et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J. Clin. Endocrinol. Metab. 90, 6373–6379 (2005).
Chang, J. C. Cancer stem cells: role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 95, S20–S25 (2016).
Doherty, M. R., Smigiel, J. M., Junk, D. J. & Jackson, M. W. Cancer stem cell plasticity drives therapeutic resistance. Cancers (Basel) 8, 8 (2016).
McCarthy, N. Underground movement. Nat. Rev. Cancer 7, 812–813 (2007).
Sampieri, K. & Fodde, R. Cancer stem cells and metastasis. Semin. Cancer Biol. 22, 187–193 (2012).
Singh, A. & Settleman, J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29, 4741–4751 (2010).
Nagayama, Y., Shimamura, M. & Mitsutake, N. Cancer stem cells in the thyroid. Front Endocrinol. (Lausanne) 7, 20 (2016).
Vicari, L., Colarossi, C., Giuffrida, D., De Maria, R. & Memeo, L. Cancer stem cells as a potential therapeutic target in thyroid carcinoma. Oncol. Lett. 12, 2254–2260 (2016).
Iglesias, J. M. et al. Mammosphere formation in breast carcinoma cell lines depends upon expression of E-cadherin. PLoS ONE 8, e77281 (2013).
Shaker, H. et al. Tissue Factor promotes breast cancer stem cell activity in vitro. Oncotarget 8, 25915–25927 (2017).
Kang, Y. Analysis of cancer stem cell metastasis in xenograft animal models. Cancer Stem Cells 568, 7–19 (2009).
Guo, Z., Hardin, H. & Lloyd, R. V. Cancer stem-like cells and thyroid cancer. Endocr. Relat. Cancer 21, T285–T300 (2014).
Ham, S. Y. et al. Mucin 1-mediated chemo-resistance in lung cancer cells. Oncogenesis 5, e185 (2016).
Shiozawa, Y., Nie, B., Pienta, K. J., Morgan, T. M. & Taichman, R. S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 138, 285–293 (2013).
Vinogradov, S. & Wei, X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine 7, 597–615 (2012).
Allen, A. & Flemstrom, G. Gastroduodenal mucus bicarbonate barrier: protection against acid and pepsin. Am. J. Physiol. Cell. Physiol. 288, C1–C19 (2005).
Corfield, A. P. et al. Mucins and mucosal protection in the gastrointestinal tract: new prospects for mucins in the pathology of gastrointestinal disease. Gut 47, 589–594 (2000).
Corfield, A. P. Mucins: a biologically relevant glycan barrier in mucosal protection. Biochim. Biophys. Acta 1850, 236–252 (2015).
Carraway, K. L., Ramsauer, V. P., Haq, B. & Carothers Carraway, C. A. Cell signaling through membrane mucins. Bioessays 25, 66–71 (2003).
Meerzaman, D., Shapiro, P. S. & Kim, K. C. Involvement of the MAP kinase ERK2 in MUC1 mucin signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L86–L91 (2001).
Huang, J. et al. Overexpression of MUC15 activates extracellular signal-regulated kinase 1/2 and promotes the oncogenic potential of human colon cancer cells. Carcinogenesis 30, 1452–1458 (2009).
Nam, K. H. et al. Expression of the membrane mucins MUC4 and MUC15, potential markers of malignancy and prognosis, in papillary thyroid carcinoma. Thyroid 21, 745–750 (2011).
Wang, R. Y. et al. MUC15 inhibits dimerization of EGFR and PI3K–AKT signaling and is associated with aggressive hepatocellular carcinomas in patients. Gastroenterology 145, 1436–1448 (2013). e1412.
Alam, M., Ahmad, R., Rajabi, H., Kharbanda, A. & Kufe, D. MUC1-C oncoprotein activates ERK-->C/EBPbeta signaling and induction of aldehyde dehydrogenase 1A1 in breast cancer cells. J. Biol. Chem. 288, 30892–30903 (2013).
Engelmann, K., Shen, H. & Finn, O. J. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 68, 2419–2426 (2008).
Patel, K. N. et al. MUC1 plays a role in tumor maintenance in aggressive thyroid carcinomas. Surgery 138, 994–1001 (2005). discussion 1001–1002.
Baek, S. K. et al. Prognostic significance of the MUC1 and MUC4 expressions in thyroid papillary carcinoma. Laryngoscope 117, 911–916 (2007).
Mimeault, M. et al. MUC4 down-regulation reverses chemoresistance of pancreatic cancer stem/progenitor cells and their progenies. Cancer Lett. 295, 69–84 (2010).
Ponnusamy, M. P., Seshacharyulu, P., Vaz, A., Dey, P. & Batra, S. K. MUC4 stabilizes HER2 expression and maintains the cancer stem cell population in ovarian cancer cells. J. Ovarian Res. 4, 7 (2011).
Xu, C. et al. Let-7a regulates mammosphere formation capacity through Ras/NF-κB and Ras/MAPK/ERK pathway in breast cancer stem cells. Cell Cycle 14, 1686–1697 (2015).
Apostolou, P. et al. The MAPK/ERK pathway among colon cancer cells, colon cancer stem cells, embryonic stem cells and normal cells. Onkologie, 36, 1–1.
Ciccarelli, C. et al. Key role of MEK/ERK pathway in sustaining tumorigenicity and in vitro radioresistance of embryonal rhabdomyosarcoma stem-like cell population. Mol. Cancer 15, 16 (2016).
Luo, M. L. et al. The Rab2A GTPase promotes breast cancer stem cells and tumorigenesis via Erk signaling activation. Cell Rep. 11, 111–124 (2015).
Luttrell, L. M., Daaka, Y., Della Rocca, G. J. & Lefkowitz, R. J. G protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in rat 1a fibroblasts Shc phosphorylation and receptor endocytosis correlate with activation of Erk kinases. J. Biol. Chem. 272, 31648–31656 (1997).
Teoh, C. M., Tam, J. K. C. & Tran, T. Integrin and GPCR crosstalk in the regulation of ASM contraction signaling in asthma. J. Allergy 2012, 1–9 (2012).
Agrawal, N. et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 (2014).
Tang, Z. et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102 (2017).
Sloan, M. Kettering Cancer Center. cBioPortal for Cancer Genomics.
Abdullah, L. N. & Chow, E. K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2, 3 (2013).
Diehn, M. & Majeti, R. Metastatic cancer stem cells: an opportunity for improving cancer treatment? Cell. Stem. Cell. 6, 502–503 (2010).
Wang, X. et al. The role of cancer stem cells in cancer metastasis: new perspective and progress. Cancer Epidemiol. 37, 60–63 (2013).
Cioce, M. et al. Mammosphere-forming cells from breast cancer cell lines as a tool for the identification of CSC-like-and early progenitor-targeting drugs. Cell Cycle 9, 2950–2959 (2010).
Wahler, J. et al. Vitamin D compounds reduce mammosphere formation and decrease expression of putative stem cell markers in breast cancer. J. Steroid Biochem. Mol. Biol. 148, 148–155 (2015).
Wolf, J. et al. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 15, R109 (2013).
Sullivan, K. E., Rojas, K., Cerione, R. A., Nakano, I. & Wilson, K. F. The stem cell/cancer stem cell marker ALDH1A3 regulates the expression of the survival factor tissue transglutaminase, in mesenchymal glioma stem cells. Oncotarget 8, 22325–22343 (2017).
Iliopoulos, D., Hirsch, H. A. & Struhl, K. An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706 (2009).
Kim, S.-Y. et al. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell. Signal. 25, 961–969 (2013).
Sansone, P. et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Invest. 117, 3988–4002 (2007).
Xie, G. et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int. J. Oncol. 40, 1171–1179 (2012).
Pino, V., Ramsauer, V. P., Salas, P., Carothers Carraway, C. A. & Carraway, K. L. Membrane mucin Muc4 induces density-dependent changes in ERK activation in mammary epithelial and tumor cells: role in reversal of contact inhibition. J. Biol. Chem. 281, 29411–29420 (2006).
Howe, E. A., Sinha, R., Schlauch, D. & Quackenbush, J. RNA-seq analysis in MeV. Bioinformatics 27, 3209–3210 (2011).
Herwig, R., Hardt, C., Lienhard, M. & Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 11, 1889–1907 (2016).
Thomas, P. D. et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141 (2003).
Eishingdrelo, H. & Kongsamut, S. Minireview: targeting GPCR activated ERK pathways for drug discovery. Curr. Chem. Genom. Transl. Med. 7, 9–15 (2013).
Paudyal, P., Xie, Q., Vaddi, P. K., Henry, M. D. & Chen, S. Inhibiting G protein βγ signaling blocks prostate cancer progression and enhances the efficacy of paclitaxel. Oncotarget 8, 36067 (2017).
Blaukat, A., Barac, A., Cross, M. J., Offermanns, S. & Dikic, I. G protein-coupled receptor-mediated mitogen-activated protein kinase activation through cooperation of Gαq and Gαi signals. Mol. Cell. Biol. 20, 6837–6848 (2000).
Emery, A. C., Eiden, M. V., Mustafa, T. & Eiden, L. E. Rapgef2 connects GPCR-mediated cAMP signals to ERK activation in neuronal and endocrine cells. Sci. Signal. 6, ra51 (2013).
Brenner, M. & Thoms, S. D. Caffeine blocks activation of cyclic AMP synthesis in Dictyostelium discoideum. Dev. Biol. 101, 136–146 (1984).
Li, Y. et al. Stretch activates human myometrium via ERK, caldesmon and focal adhesion signaling. PLoS ONE 4, e7489 (2009).
Lakshmanan, I. et al. MUC5AC interactions with integrin β4 enhances the migration of lung cancer cells through FAK signaling. Oncogene 35, 4112–4121 (2016).
Shyu, M.-K. et al. Mucin 15 is expressed in human placenta and suppresses invasion of trophoblast-like cells in vitro. Hum. Reprod. 22, 2723–2732 (2007).
Kasai, T. et al. Cancer stem cells converted from pluripotent stem cells and the cancerous niche. J. Stem Cells Regen. Med. 10, 2–7 (2014).
Saiselet, M. et al. Thyroid cancer cell lines: an overview. Front Endocrinol. 3, 133 (2012).
Layden, B. T., Newman, M., Chen, F., Fisher, A. & Lowe, W. L. Jr. G protein coupled receptors in embryonic stem cells: a role for Gs-alpha signaling. PLoS ONE 5, e9105 (2010).
Nakamura, K., Salomonis, N., Tomoda, K., Yamanaka, S. & Conklin, B. R. Gi-coupled GPCR signaling controls the formation and organization of human pluripotent colonies. PLoS ONE 4, e7780 (2009).
Shin, S. et al. Long‐term proliferation of human embryonic stem cell–derived neuroepithelial cells using defined adherent culture conditions. Stem Cells 24, 125–138 (2006).
Lynch, J. R. & Wang, J. Y. G protein-coupled receptor signaling in stem cells and cancer. Int. J. Mol. Sci. 17, 707 (2016).
Wagner, P. L. et al. The chemokine receptors CXCR4 and CCR7 are associated with tumor size and pathologic indicators of tumor aggressiveness in papillary thyroid carcinoma. Ann. Surg. Oncol. 15, 2833–2841 (2008).
Xu, Y. et al. CCL21/CCR7 prevents apoptosis via the ERK pathway in human non-small cell lung cancer cells. PLoS ONE 7, e33262 (2012).
Boyle, S. T. et al. The chemokine receptor CCR7 promotes mammary tumorigenesis through amplification of stem-like cells. Oncogene 35, 105–115 (2016).
Schutyser, E., Struyf, S. & Van Damme, J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 14, 409–426 (2003).
Wang, B. et al. Production of CCL20 from lung cancer cells induces the cell migration and proliferation through PI3K pathway. J. Cell. Mol. Med. 20, 920–929 (2016).
Ibrahim, S. A. et al. Syndecan-1 (CD138) modulates triple-negative breast cancer stem cell properties via regulation of LRP-6 and IL-6-mediated STAT3 signaling. PLoS ONE 8, e85737 (2013).
Dupré, D. J., Robitaille, M., Rebois, R. V. & Hébert, T. E. The role of Gβγ subunits in the organization, assembly, and function of GPCR signaling complexes. Annu. Rev. Pharmacol. Toxicol. 49, 31–56 (2009).
Taussig, R., Iñiguez-Lluhi, J. A. & Gilman, A. G. Inhibition of Adenylyl Cyclase by G~ i~. Sci.-NEW Y. THEN Wash. 261, 218–218 (1993).
Wang, A. et al. Coffee and cancer risk: a meta-analysis of prospective observational studies. Sci. Rep. 6, 33711 (2016).
Fagin, J. A. & Wells, S. A. Jr. Biologic and clinical perspectives on thyroid cancer. N. Engl. J. Med. 375, 1054–1067 (2016).
Das, M., Ithychanda, S., Qin, J. & Plow, E. F. Mechanisms of talin-dependent integrin signaling and crosstalk. Biochim. Biophys. Acta 1838, 579–588 (2014).
Ross, R. S. Molecular and mechanical synergy: cross-talk between integrins and growth factor receptors. Cardiovasc. Res. 63, 381–390 (2004).
Acknowledgements
This study was supported by the Ministry of Science and ICT and Business Belt Program (2017K000492), a faculty research grant of Yonsei University College of Medicine for 2013 (6-2013-0108), the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2017R1A2B4010480) and the Research fellowship program of National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2017R1A6A3A11032244).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, C., Thi Thao Tran, N., Van Ngu, T. et al. Promotion of tumor progression and cancer stemness by MUC15 in thyroid cancer via the GPCR/ERK and integrin-FAK signaling pathways. Oncogenesis 7, 85 (2018). https://doi.org/10.1038/s41389-018-0094-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-018-0094-y
This article is cited by
-
LPAR5 confers radioresistance to cancer cells associated with EMT activation via the ERK/Snail pathway
Journal of Translational Medicine (2022)
-
Downregulation of MUC15 by miR-183-5p.1 promotes liver tumor-initiating cells properties and tumorigenesis via regulating c-MET/PI3K/AKT/SOX2 axis
Cell Death & Disease (2022)
-
Establishment of a 5-gene risk model related to regulatory T cells for predicting gastric cancer prognosis
Cancer Cell International (2020)
-
MUC15 inhibits cancer metastasis via PI3K/AKT signaling in renal cell carcinoma
Cell Death & Disease (2020)
-
Nuclear FAM289-Galectin-1 interaction controls FAM289-mediated tumor promotion in malignant glioma
Journal of Experimental & Clinical Cancer Research (2019)
