
Abstract
Sarcomas are malignant tumors derived from mesenchymal tissues and may harbor a subset of cells with cancer stem-like cell (CSC) properties. Platelet-derived growth factor receptors α and β (PDGFR-α/β) play an important role in the maintenance of mesenchymal stem cells. Here we examine the role of PDGFR-α/β in sarcoma CSCs. PDGFR-α/β activity and the effects of PDGFR-α/β inhibition were examined in 3 human sarcoma cell lines using in vitro assays and mouse xenograft models. In all three cell lines, PDGFR-α/β activity was significantly higher in cells grown as spheroids (to enrich for CSCs) and in cells sorted for CD133 expression (a marker of sarcoma CSCs). Self-renewal transcription factors Nanog, Oct4, and Slug and epithelial-to-mesenchymal transition (EMT) proteins Snail, Slug, and Zeb1 were also significantly higher in spheroids cells and CD133(+) cells. Spheroid cells and CD133(+) cells demonstrated 2.9- to 4.2-fold greater migration and invasion and resistance to doxorubicin chemotherapy. Inhibition of PDGFR-α/β in CSCs using shRNA or pharmacologic inhibitors reduced expression of certain self-renewal and EMT proteins, reduced spheroid formation by 74–82%, reduced migration and invasion by 73–80%, and reversed chemotherapy resistance. In mouse xenograft models, combining PDGFR-α/β inhibition (using shRNA or imatinib) with doxorubicin had a more-than-additive effect in blocking tumor growth, with enhanced apoptosis, especially in CD133(+) cells. These results indicate that PDGFR-α/β activity is upregulated in sarcoma CSCs and promote CSC phenotypes including migration, invasion, and chemotherapy resistance. Thus, the PDGFR-α/β pathway represents a new potential therapeutic target to reduce metastatic potential and increase chemosensitivity.
Similar content being viewed by others


Introduction
Mesenchymal tissues are derived from the mesoderm (i.e., middle layer of the embryo) and include the musculoskeletal system, circulatory and lymphatic systems, and connective tissues. Sarcomas are malignant tumors of mesenchymal tissues. These tumors are diagnosed in over 15,000 people in the United States annually, and are fatal for ~40% of patients due to either locoregional disease or distant metastasis1. The treatment of primary tumors typically includes aggressive surgical resection and radiation therapy, but local recurrence remains a significant problem for tumors in difficult locations such as the head and neck, paraspinal region, retroperitoneum, and pelvis. Furthermore, up to half of patients with large, high-grade sarcomas develop distant metastases, most frequently to the lung2. The efficacy of chemotherapy in treating local and distant recurrence is modest at best, and for the minority of patients who do respond, nearly all eventually develop chemotherapy resistance3.
The cancer stem cell (CSC) theory postulates that malignant tumors harbor a subset of cells that share characteristics with normal adult stem cells, namely the capacity for self-renewal and pluripotent differentiation4. Methods to identify CSCs include tumor initiation in immunodeficient mice, spheroid colony formation in vitro, and expression of certain cell surface markers. The most commonly identified cell surface marker for CSCs in sarcomas is CD1335,6. Given there are over 80 distinct histologic subtypes of sarcoma, many have also suggested that sarcomas originate from multipotent cells such as mesenchymal stem cells7. Numerous studies have demonstrated that putative CSCs are more resistant to chemotherapy than non-CSCs and may be a source of distant metastasis8,9. Thus, targeting these CSCs may lead to the development of more effective treatment regimens in advanced sarcomas.
Platelet-derived growth factor (PDGF) ligands and their associated PDGF receptors, PDGFR-α and PDGFR-β, are important regulator proteins for mesenchymal stem cell growth and differentiation10–12. The PDGF ligands exist as homodimers and heterodimers formed by dimerization of A-polypeptide, B-polypeptide, C-polypeptide, and D-polypeptide chains, and transduce the signals intracellularly by binding to PDGF-α and -β tyrosine kinase receptors13. The structures of both PDGF receptors are similar, consisting of five immunoglobulin (Ig)-like domains in the extracellular region, a transmembrane domain, and tyrosine kinase domains located within the intracellular region.
We previously demonstrated in a gene expression microarray analysis of 38 human sarcomas compared to 13 normal tissues that PDGFR-α is one of the top upregulated genes14. In a randomized phase II clinical trial, the combination of olaratumab, a monoclonal antibody targeting PDGFR-α, and doxorubicin chemotherapy improved overall survival in patients with advanced soft-tissue sarcoma over doxorubicin alone15. Given these findings and the known importance of PDGFR signaling in mesenchymal stem cell biology, we chose to examine the role of PDGFR-α and -β in sarcoma CSCs.
Results
CD133 is a marker of sarcoma CSCs
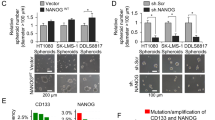
Growth of cancer cells as spheroids in vitro selects for cells with CSC properties9, and the most commonly identified cell surface marker for CSCs in sarcomas is CD133.5,6, Alternative CSC cell surface markers in sarcomas include CD44, CD271, and TNAP16. HT1080 fibrosarcoma cells, SK-LMS-1 leiomyosarcoma cells, and DDLS8817 dedifferentiated liposarcoma cells were examined for expression of these cell surface proteins after being grown as monolayers or as spheroids. CD133 expression was consistently higher in spheroids of all three cell lines as compared to monolayers (Fig. 1a). The other purported CSC markers were variably increased in spheroids compared to monolayer cells. Sarcoma cell lines grown as monolayers and spheroids were then sorted by FACS analysis for CD133 expression, and the proportion of CD133(+) cells was 7–14 times higher in spheroid cells as compared to monolayer cells (Fig. 1b).

a Western blot analysis of cell surface markers for cancer stem-like cells in human sarcoma cell lines HT1080, SK-LMS-1, and DDLS8817. Cells were grown as monolayers or spheroids. β-actin was used as the loading control. b Percentage of CD133(+) cells in human sarcoma cells grown as monolayers or as spheroids as determined by FACS analysis. *p < 0.05 compared to Monolayers. c Western blot analysis of human sarcoma cells grown as monolayers or as spheroids for self-renewal transcription factors. d Western blot analysis of FACS-sorted CD133(+) and CD133(−) spheroid cells for self-renewal transcription factors. Experiments in a–d were performed three times with similar results. e Representative light microscopy images from single cell assays performed in triplicate for human sarcoma spheroid cells FACS-sorted by CD133. Graph displays the mean diameters of the spheroid colonies. Bars represent standard deviation. *p < 0.05 compared to CD133(−)
Western blot analysis of the self-renewal transcription factors Nanog, Oct4, Sox2, and c-Myc was then performed on spheroid and monolayers cells from the three sarcoma cell lines. There were modest increases in the expression of these proteins in spheroid cells compared to monolayer cells (Fig. 1c). When these same factors were examined in spheroid cells after sorting for CD133 expression, there were greater relative increases of these self-renewal proteins in CD133(+) cells compared to CD133(−) cells (Fig. 1d). Immunofluorescence analysis for self-renewal proteins confirmed increased expression of Nanog, Oct4, and Sox2 in CD133(+) cells compared to CD133(−) cells (Supplemental Fig. S1A). Next, the ability to form spheroid colonies from a single cell was assessed in CD133(+) and CD133(−) cells in the three sarcoma cell lines. The average size of spheroids was nearly double in CD133(+) cells as compared to CD133(−) cells (Fig. 1e, Suppl Fig. S1B). Thus, CD133 may be a reliable marker of sarcoma CSCs.
Role of PDGFR-α/β in sarcoma CSCs
Levels of total and phosphorylated PDGFR-α and PDGFR-β were examined by Western blot analysis in the three sarcoma cell lines after growth as monolayers or as spheroids (Fig. 2a). Total PDGFR-α and PDGFR-β expression levels were relatively equal across monolayer and spheroid cells from all three cell lines. However, phosphorylated PDGFR-α and phosphorylated PDGFR-β levels were significantly increased in the spheroid cells compared to monolayer cells. Similar results were seen when comparing CD133(+) cells to CD133(−) cells (Supplemental Fig. S2A).

a Western blot analysis of phosphorylated and total PDGFR-α and -β in human sarcoma cell lines grown as monolayers and as spheroids. β-actin was used as a loading control. b Western blot analysis of PDGFR-α and -β expression and self-renewal transcription factors in human sarcoma cell lines grown as spheroids and treated with imatinib (1 μM) or DMSO control. Experiments in a, b were performed three times with similar results. c Graph displaying the mean diameters of the spheroids for HT1080, SK-LMS-1, and DDLS8817 cells treated with imatinib or DMSO. d Light microscopy images from single cell assays for spheroid cells treated with imatinib or DMSO. e Representative light microscopy images from spheroid formation assays performed in triplicate of human sarcoma cell lines under varying conditions. Cells were grown as spheroids following PDGFR-α and PDGFR-β double knockdown (sh.PDGFR-α/β), transduction with scramble shRNA (sh.Scr), or treatment with imatinib or DMSO. Graphs display the mean numbers of spheroids ≥50 μm per field in each treatment group. Bars represent standard deviation. *p < 0.05 compared to DMSO
Imatinib is an inhibitor of the receptor tyrosine kinases Bcr-Abl, c-Kit, PDGFR-α, and PDGFR-β17. Imatinib effectively blocked phosphorylation of PDGFR-α and PDGFR-β and decreased expression of Nanog in spheroid cells from all three sarcoma cell lines (Fig. 2b). On immunofluorescence analysis, CD133 and Nanog expression were decreased in spheroid cells treated with imatinib (Supplemental Fig. S2B). Spheroid formation under standard conditions or from a single cell was also reduced by 77–82% in the sarcoma spheroid cells treated with imatinib compared to control (Fig. 2c, d). Sarcoma cells were then transduced with PDGFR-α and -β short hairpin RNA (shRNA) (“double knockdown”) or scramble shRNA. Stable double knockdown was confirmed by western blot analysis, and there was only a minor effect on cell growth in vitro over 4 days (Supplemental Fig. S2C, S2D). PDGFR-α and -β shRNA double knockdown cells had a 76–77% decrease in spheroid formation compared to control cells transduced with scramble shRNA (Fig. 2e). Thus, PDGFR-α and -β are upregulated in sarcoma cells grown as spheroids and in CD133(+) cells, and may play an important role in the maintenance of sarcoma CSCs.
PDGFR-α/β promotes sarcoma CSC migration/invasion and anchorage-independent growth
Epithelial-to-mesenchymal transition (EMT) can lead to the acquisition of CSC phenotypes18. Western blot analysis was performed to examine the expression of the EMT-associated cell surface marker N-cadherin and EMT transcription factors Snail, Slug, and Zeb1. Increased expression of N-cadherin, Snail, and Slug was observed in spheroid cells as compared to monolayer cells in all three cell lines (Fig. 3a). Zeb1 had marginally elevated expression in SK-LMS-1 and DDLS8817 spheroid cells as compared to monolayer cells. In migration and invasion in vitro assays, sarcoma cell lines grown under spheroid formation conditions had a 3.7- to 4.2-fold increase in migration and a 2.9- to 3.6-fold increase in invasion as compared to monolayer cells (Fig. 3b). To examine anchorage-independent growth, soft agar colony formation assays were performed. Cells grown under spheroid conditions formed 2.5- to 3.3-fold more colonies as compared to monolayer cells across all three cell lines (Fig. 3c). FACS-sorted CD133(+) cells demonstrated similar increases in anchorage-independent growth when compared to CD133(−) cells (Supplemental Fig. S3A).

a Western blot analysis of cell adhesion protein N-cadherin and EMT transcription factors Snail, Slug, and Zeb1 in human sarcoma cell lines grown as monolayers and as spheroids. β-actin was used as the loading control. b Representative images under light microscopy of migration and invasion assays of human sarcoma cell lines grown as monolayers or as spheroids for 24–48 h. Graphs display the number of migrated or invasive cells per field. c Representative light microscopy images of forrmed colonies in soft agar assay of human sarcoma cell lines grown as monolayers or spheroids for 14–20 days. Graph displays the number colonies per field. d Representative images under light microscopy of migration and invasion assays of human sarcoma cell lines grown as monolayers or as spheroids. Spheroid cells were treated with imatinib or DMSO. e Western blot analysis of N-cadherin and EMT-regulating transcription factors in human sarcoma cell lines grown as spheroids and treated with imatinib or DMSO. Experiments in a and e were performed three times with similar results. Bars represent standard deviation. *p < 0.05 compared to Monolayers
The functional importance of PDGFR signaling in metastasis has been demonstrated in various malignancies19,20. We next examined the effects of PDGFR-α and -β inhibition with imatinib on migration and invasion. Spheroid cells treated with imatinib had 83–87% less migratory capacity and 84–85% less invasive capacity (Fig. 3d, Suppl. Fig. S3B). Similar results were found on migration and invasion when PDGFR-α and –β shRNA were used instead of imatinib (Suppl. Fig. S4A, S4B). On Western blot analysis, spheroid cells treated with imatinib had lower expression of the cell surface marker N-cadherin and the transcription factor Slug (Fig. 3e). Immunofluorescence analysis confirmed the decreased expression of N-cadherin and Slug in all three sarcoma cell lines (Supplemental Fig. S4C). Lastly, anchorage-independent growth in spheroid cells treated with imatinib was also reduced to levels similar to that of monolayer cells (Suppl. Fig. S4D). These data suggest that PDGFR signaling is important for sarcoma CSC EMT, migration, and invasion.
PDGFR-α/β signaling in sarcoma CSCs contributes to chemotherapy resistance
Given CSCs are generally resistant to chemotherapy, we examined the sensitivity of the sarcoma cell lines to doxorubicin, the most commonly used chemotherapy for the treatment of advanced sarcomas. Doxorubicin at 1 μM decreased cell proliferation in monolayer cells by 27–39% and in spheroid cells by 11–15% (Fig. 4a, b). Single isoform shRNA knockdown of PDGFR-α or PDGFR-β was investigated along with imatinib therapy, which inhibits both isoforms. Stable knockdown of PDGFR-α and -β was confirmed by Western blot analysis (Supplemental Fig. S5A, S5B). PDGFR-α shRNA knockdown reduced proliferation in monolayer cells by 2–10% and in spheroid cells by 13–18% (Fig. 4a, b). PDGFR-β shRNA knockdown had a greater effect, with a 10–19% reduction in proliferation of monolayer cells and 16–23% reduction in spheroid cells. Imatinib reduced proliferation of monolayer cells by 19–32% and of spheroid cells by 31–33%. Single isoform knockdown of PDGFR-α or -β or imatinib had marginal to no additive effect over chemotherapy alone in monolayer cells (Fig. 4a). The combination of single isoform shRNA knockdown and chemotherapy had only a modest effect in spheroid cells, reducing proliferation by 20–29% (Fig. 4b). In contrast, there was a more-than-additive effect when imatinib was combined with chemotherapy, decreasing cell proliferation in spheroid cells by 69–77%. To ensure the ability of imatinib to improve the efficacy of chemotherapy in spheroid cells was not due to targets other than PDGFR-α and PDGFR-β, we transduced HT1080 monolayer and spheroid cells with shRNA for both PDGFR-α and PDGFR-β and repeated our proliferation assays. Dual knockdown of PDGFR-α and PDGFR-β demonstrated efficacy similar to imatinib in reducing proliferation in both monolayer and spheroids cells (Fig. 4c, d).

Proliferation assays for human sarcoma cell lines grown as (a) monolayers or (b) spheroids for 48–60 h. Treatment groups included cells with PDGFR-α knockdown (sh.PDGFR-α), PDGFR-β knockdown (sh.PDGFR-β), and cells treated with imatinib; each group was also treated with doxorubicin chemotherapy (Dox) 0.5 μmM or DMSO. Proliferation assays were also performed with both PDGFR-α and -β double knockdown cells (sh.PDGFR-α/β) in human sarcoma cells grown as (c) monolayers or (d) spheroids, following treatment with doxorubicin or DMSO. Experiments in a–d were performed three times with similar results. *p < 0.05 compared to control and monotherapy groups
The effects of PDGFR inhibition and doxorubicin chemotherapy were next examined in an athymic nude mouse xenograft model following subcutaneous flank implantation of HT1080 fibrosarcoma cells. HT1080 cells stably transduced with PDGFR-α shRNA and PDGFR-β shRNA or scrambled control shRNA were injected. Mice were randomized to treatment with DMSO, doxorubicin, and/or imatinib when tumors reached 50–100 mm3. Body weights remained stable throughout the treatment period (Suppl. Fig. S6A). Control tumors treated with DMSO alone grew to over 1000 mm3 by 15 days (Fig. 5a). Tumors in mice treated with doxorubicin grew to an average of 739 mm3, tumors in mice treated with imatinib grew to an average of 641 mm3, and tumors with knockdown of PDGFR-α and PDGFR-β grew on average to 675 mm3. The combination of doxorubicin with either imatinib or PDGFR-α/β knockdown had a more-than-additive effect. Average tumor size was 218–268 mm3, representing a 73–78% decrease compared to control tumors.

a Tumor growth curves for HT1080 xenografts treated with doxorubicin (Dox) 4 mg/kg), imatinib (90 mg/kg), PDGFR-α shRNA and PDGFR-β shRNA (sh.PDGFR-α/β), and/or DMSO. There were five mice per group. b Representative immunofluorescence images of treated HT1080 tumors and stained for PCNA (purple), cleaved caspase-3 (red), and CD133 (green). White scale bars 25 μm; yellow scale bar 50 μm. c,d Graphs displaying the number of cleaved caspase 3(+), CD133(+), and dual cleaved caspase 3(+)/CD133(+) cells on immunofluorescence imaging analysis. Bars represent standard deviation. *p < 0.05 compared to control and monotherapy groups
The collected xenografts were processed and underwent immunofluorescence analysis for the proliferation marker PCNA, apoptosis marker cleaved caspase 3, and the sarcoma CSC marker CD133 (Fig. 5b). Compared to control tumors, the combination of imatinib and doxorubicin demonstrated a modest decrease in cell proliferation (Suppl. Fig. S6B) and more-than-additive increases in overall apoptosis (Fig. 5c) and CD133 expression (Fig. 5d). Double staining with CD133 and cleaved caspase-3 revealed that tumors treated with combination therapy had a significantly larger proportion of CD133(+) cells undergoing apoptosis (Fig. 5d).
PDGFR-α/β signaling in sarcoma CSCs is upregulated by hypoxia
We have previously demonstrated that CD133(+) CSCs in HT1080 fibrosarcoma xenografts are found preferentially in hypoxic regions of tumors but also reside in non-hypoxic regions21. Tumors respond to hypoxic stress through multiple mechanisms, including the stabilization of hypoxia-inducible factor 1α (HIF-1α). HIF-1α levels are significantly higher in CD133(+) cells compared to CD133(−) cells (Suppl. Fig. S7A). We thus examined whether there are differences in HIF-1α levels and PDGFR-α/β activity in sarcoma CSCs under normoxic and hypoxic conditions. HIF-1α levels are higher in sarcoma cell lines grown as spheroids compared to monolayer cells (Fig. 6a). As expected, HIF-1α is upregulated in all three sarcoma cells grown as monolayers or as spheroids in response to hypoxia, and phosphorylation of PDGFR-α and -β is also upregulated in response to hypoxia. The highest levels of HIF-1α and phosphorylated PDGFR-α and PDGFR-β are seen in spheroid cells under hypoxic conditions. For all three sarcoma cell lines, the ability of cells to form spheroids was increased under 1% oxygen compared to 21% oxygen (Fig. 6b). Knockdown of PDGFR-α and -β decreased the ability of sarcoma cells to form spheroids in normoxia by 75–90% and in hypoxia by 82–89% (Fig. 6c). Sarcoma cells also demonstrated increased migration, invasion, and soft agar colony formation under hypoxia, and these phenotypes were similarly reduced with knockdown of PDGFR-α and -β shRNA (Suppl. Fig. S7B, S8). Interestingly, HIF-1α shRNA greatly attenuated PDGFR-α and -β phosphorylation in response to hypoxia (Fig. 6d), suggesting that hypoxia-induced HIF-1α stabilization and transport to the nucleus is upstream of PDGFR-α and -β activation. These data suggest that sarcoma CSCs are able to thrive under hypoxia and that PDGFR-α/β signaling is important in the maintenance of CSCs in hypoxic regions of tumors.

a Western blot analysis of HIF-1α, phosphorylated and total PDGFR-α and -β, and CD133 in human sarcoma cell lines grown as monolayers or as spheroids under normoxia (21% O2) and hypoxia (1% O2) for 18–24 h. b Graph displaying the mean numbers of spheroids >50 μm per field formed by sarcoma spheroid cells under normoxia and hypoxia. c Graphs displaying the mean numbers of spheroids >50 μm per field formed by sarcoma spheroid cells under normoxia and hypoxia following transduction with control shRNA (sh.Scr) or PDGFR-α and -β shRNA (sh.PDGFR-α/β). d Western blot analysis of HIF-1α, phosphorylated and total PDGFR-α and -β in human sarcoma spheroid cells under normoxia (21% O2) and hypoxia (1% O2) following transduction with control shRNA (sh.Scr) or HIF-1α shRNA (sh.HIF-1α). Experiments a and d were performed three times with similar results. Bars represent standard deviation. *p < 0.05 compared to Normoxia
Discussion
The role of PDGFR signaling in mesenchymal stem cell biology has been well described, and PDGFR-α and PDGFR-β have been shown to be expressed on both sarcoma cells and mesenchymal stromal cells in the tumor microenvironment22,23. This study is the first to demonstrate an important role of PDGFR signaling in maintaining sarcoma CSC phenotypes such as self-renewal, migration, invasion, and chemotherapy resistance. Sarcoma cell lines grown as spheroids or sorted for the sarcoma CSC marker CD133 demonstrated higher levels of PDGFR-α and PDGFR-β activity than unselected cells, had greater migratory and invasive capacities, and were more resistant to chemotherapy. PDGFR-α and PDGFR-β knockdown by shRNA or pharmacologic inhibition with imatinib diminished the expression of the self-renewal transcription factor Nanog and blocked spheroid colony formation in vitro. Inhibition of these receptors also blocked expression of the EMT transcription factor Slug and dramatically reduced migration and invasion. Combined inhibition of PDGFR-α and PDGFR-β in HT1080 sarcoma xenografts reversed chemotherapy resistance. Based on these data, targeting the PDGFR-α/β signaling pathway in sarcomas may show promise in combating metastasis and acting as a chemosensitizer.
The existence of CSCs has been validated in multiple human cancers including several sarcoma subtypes, but the clinical relevance and their potential as therapeutic targets remain highly controversial topics. Studies examining CSCs in sarcomas are scarce, but there is evidence that supports mesenchymal stem cells as the origin of both pleomorphic and translocation-associated sarcomas7. Whether this population of cells is responsible for therapeutic resistance and recurrence has not been established. Sarcomas are a heterogenous group of tumors with over 80 subtypes, each with its own tumor biology and response to therapy. However, sarcomas may originate from multipotent stem cells such as mesenchymal stem cells7, and so targeting such cells may have broad application to all sarcoma subtypes. Here we examined three common sarcoma subtypes (fibrosarcoma, leiomyosarcoma, and liposarcoma) and found similar effects when targeting PDGFR-α and PDGFR-β in CSCs from these cell lines.
The EMT program is a naturally occurring transdifferentiation program that regulates changes in cell states between epithelial and mesenchymal24. Sarcomas are mesenchymal neoplasms but may possess varying degrees of the EMT program, particularly between unselected cells and CSCs. The link between EMT and CSCs has been examined in numerous studies, but the recently uncovered link between the passage through EMT and the acquisition of stem-like properties suggests that the EMT program may be a mechanism for generating CSCs18. In this study, we use spheroid formation and expression of CD133 as means of identifying sarcoma CSCs. These spheroid cells or CD133(+) cells have elevated expression of the EMT-associated proteins Slug and N-cadherin and significantly increased migration and invasion. Blockage of PDGFR-α and PDGFR-β activity in sarcoma CSCs downregulates Slug and reduces migration and invasion. Thus, we find in this study that targeting PDGFR-α and PDGFR-β affects both CSCs and EMT-associated phenotypes.
Multidrug chemotherapy with agents such as doxorubicin, ifosfamide, gemcitabine, and docetaxel is standard treatment for patients with advanced or metastatic sarcomas. The addition of targeted agents may improve the efficacy of chemotherapy. The first placebo-controlled phase III clinical trial to examine PDGFR inhibition in non-GIST sarcomas was the PALETTE trial, which tested the angiogenesis inhibitor pazopanib in metastatic non-adipocytic sarcomas25. Pazopanib is a tyrosine kinase inhibitor with a large panel of targets, including vascular endothelial growth factor receptor (VEGFR)-1, -2, and -3 and both PDGFR-α and PDGFR-β. The study showed that pazopanib improved median progression-free survival by 3 months, but there was no significant improvement in overall survival. More recently, a randomized phase II trial demonstrated that the PDGFR-α-specific inhibitor olaratumab, in combination with doxorubicin, improved median overall survival vs. doxorubicin alone in patients with advanced sarcomas15. A larger phase III trial is currently underway. Interestingly, in our present study, single isoform shRNA knockdown failed to reverse chemoresistance in vitro or in the nude mouse xenograft model. Only with inhibition of both PDGFR-α and PDGFR-β was cell proliferation or tumor growth blocked. These findings highlight the need for improving our understanding of the role of PDGFR-α and PDGFR-β in sarcomas.
Prior studies have shown that CSCs may be more reliant on HIF-1α activity and that CSCs may preferentially reside in hypoxic regions of tumors26. We previously found that sarcoma CSCs indeed are found more commonly in hypoxic regions of sarcomas and that multimodal therapy with VEGF-A inhibition, HIF-1α inhibition, and hypoxia-activated chemotherapy effectively blocks sarcoma growth through inhibition of both tumor vasculature and cancer stem-like cells21. This multimodal strategy did not incorporate specific targeting of an intrinsic pathway in sarcoma CSCs. Here we find not only that PDGFR-α and -β are important in sarcoma CSC maintenance but also that these pathways are even more upregulated in CSCs under hypoxic conditions. This PDGFR-α and -β upregulation was found to be downstream of HIF-1α activation. Given PDGFR signaling is also important in tumor endothelial cells and pericytes27, targeting PDGFR signaling may inhibit both sarcoma CSCs and the tumor microenvironment.
In conclusion, this study demonstrates an important role of PDGFR-α and PDGFR-β in sarcomas, specifically in the maintenance and activity of CSC populations. Targeting this signaling pathway may be a potential means of blocking metastasis and overcoming chemoresistance in advanced sarcomas.
Materials and methods
Cell lines and reagents
HT1080 human fibrosarcoma cells and SK-LMS-1 human leiomyosarcoma cells were obtained from the American Type Culture Collection (ATCC). The DDLS8817 dedifferentiated liposarcoma cell line was established a tumor sample from a patient who signed informed consent and was confirmed to harbor 12q amplification by cytogenetic analysis and by DNA copy number array (Agilent 244K). HT1080 and SK-LMS-1 were maintained in Dulbecco’s Modified Eagle’s medium (DMEM), and DDLS8817 was maintained in DMEM-F12. All media were supplemented with 10% fetal bovine serum, penicillin (100 U/mL), streptomycin (100 μg/mL), and l-glutamine (2 mM) (“regular media”). Cancer cell lines were actively passaged for <6 months from the time that they were received, and United Kingdom Coordinating Committee on Cancer Research (UKCCCR) guidelines were followed28. Doxorubicin (RYG02) and imatinib (RS029) were purchased from TSZ Chem/BIOTANG Inc. (Lexington, MA).
Spheroid culture generation
Cells were resuspended in “spheroid media” comprised of DMEM/F12 containing epidermal growth factor (EGF, 20 ng/mL; Sigma-Aldrich), basic fibroblast growth factor (bFGF, 20 ng/mL; Sigma-Aldrich), N-2 supplement (20 ng/mL; ThermoFisher Scientific), and B27 (20 ng/mL; Sigma-Aldrich) and then plated on Ultra-Low Attachment culture dishes (Corning Life Sciences). Spheroids were collected after 5–7 days except when noted otherwise. Protein was extracted for analysis, or cells were dissociated with Accutase (Innovative Cell Technologies) and used for other experiments. To count the number of spheroids, the number of spheroids >50 μm in diameter was counted in five fields after image processing using Imaris 7.6 (Bitplane).
Single cell assay
Spheroid cells stably transduced with GFP-expressing lentivirus were dissociated to single cells and plated onto Ultra-Low Attachment 96-well plates with a density of a single cell per well. After 24 h, cells were visualized with an inverted fluorescence microscope (Leica Microsystems, Buffalo Grove, IL). The clones were grown, and clone formation was monitored at sequential points of 1, 4, 7, and 10 days. The size of clones was measured using MetaMorph software, version 7.8.2 (Molecular Devices).
Western blot analysis
Proteins were extracted by collecting cells in RIPA buffer (Sigma-Aldrich) containing Complete Protease Inhibitor Cocktail (Roche Diagnostics USA, Indianapolis, IN), and protein concentration was determined by the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Western blot analysis was performed using the following antibodies: CD133 (MBS462020; MyBioSource), CD44 (#3578; Cell Signaling Technology), CD271 (LS-C109106; LifeSpan BioSciences), TNAP (sc-23430; Santa Cruz), Nanog (#4893; Cell Signaling), Oct4 (#2788; Cell Signaling), Sox2 (#2748; Cell Signaling), c-Myc (sc-40; Santa Cruz Biotechnology), PDGFR-α (sc-338; Santa Cruz), PDGFR-β (#3169; Cell Signaling), phospho-PDGFR-α (Tyr 754) (sc-12911; Santa Cruz), phospho-PDGFR-β (Tyr 751) (#3166; Cell Signaling), N-cadherin (#14215; Cell Signaling), Snail (#3879; Cell Signaling), Slug (#9585; Cell Signaling), Zeb1 (NBP1-05987; Novus Biologicals), HIF1α (BD610958; BD Biosciences), and β-actin (A5228; Sigma-Aldrich).
Fluorescence activated cell sorting
Cells were dissociated using Accutase and resuspended in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA). The cells were stained with PE-conjugated CD133 (130-098-826; Miltenyi Biotec) or isotype control antibody (BD555742; BD Biosciences) on ice for 30 min. Cells were then washed with PBS and analyzed on a BD FACSCalibur™ (BD Biosciences) using BD CellQuest software (BD Biosciences).
Immunocytochemistry and immunofluorescence
Spheroid cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS. Following cell fixation, cells were incubated with antibodies for CD133 (MBS462020; Miltenyi Biotec), Nanog (#8822; Cell Signaling), Oct4 (#83932; Cell Signaling), Sox2 (#3570; Cell signaling), c-Myc (sc-40; Santa Cruz), N-cadherin (BD610920; BD Biosciences), and/or Slug (#9585; Cell Signaling) in a solution of PBS with 1% BSA and 0.1% Triton X-100 at 4 °C overnight. Cells were then stained with Alexa Fluor 488 (A-11029; ThermoFisher), Alexa Fluor 568 (A-11061; ThermoFisher), Alexa Fluor Plus 555 (A-32732; ThermoFisher, and Alexa Fluor 647 (A-21236; ThermoFisher). Nuclei were counterstained using 4′,6-diamidino-2-phenylindole (DAPI, 28718-90-3; Sigma-Aldrich). Stained cells were visualized with an inverted confocal microscope (Leica Microsystems). Image processing was performed using Imaris, Version 7.6 (Bitplane).
Cell proliferation, migration, and invasion assays
Spheroids were dissociated with accutase, and monolayer cells were collected with trypsin. To assay for proliferation, 2000 cells were plated onto 96-well flat bottom plates and maintained in regular media overnight. A colorimetric MTT assay was used to assess cell number by optical density after the specified number of days, as previously described29. Data reflect the mean of six samples.
To assay for migration and invasion, 2000 cells were suspended in 0.2 mL of serum-free DMEM. The cells were loaded in the upper well of an 8-μm pore Transwell chamber (Corning). For invasion assays, the upper well was pre-coated with 10 mg/mL growth factor-reduced BD Matrigel™ matrix (BD Biosciences). The lower well was filled with 0.8 mL of DMEM with serum. After incubation for 48 h at 37 °C, non-invaded cells on the upper surface of the filter were removed with a cotton swab. The migrated or invaded cells on the lower surface of the filter were fixed and stained with Siemens Diff-Quick Stain Kit, per the manufacturer’s protocol (B4132-1A; Siemens Healthcare Diagnostics). The fixed cells were photographed at ×20 magnification and visualized with an inverted confocal microscope (Leica Microsystems). Image processing was performed using Imaris 7.6 (Bitplane). Cells were counted in five microscopic fields per well, and the extent of migration or invasion was expressed as an average number of cells per microscopic field. Data reflect the mean of six samples.
Soft agar colony formation assay
To examine anchorage-independent growth, a cell suspension of 1 × 104 cells per mL was mixed in 0.4% agarose and seeded in triplicate onto previously set 0.9% soft agar in a 60 mm culture dish. The soft agar suspensions were prepared in either regular or spheroid media, as applicable. Cells were incubated for 3–4 weeks during which growth was observed weekly under an inverted microscope (Leica). Colonies were then photographed and counted in four to five randomly chosen fields and expressed as means of the triplicate cultures.
Lentiviral transduction
Silencing of PDGFR-α and PDGFR-β was achieved via lentiviral transduction of human PDGFR-α shRNA (sc-29443-V; Santa Cruz) and human PDGFR-β shRNA (sc-29442-V; Santa Cruz) per the manufacturer’s protocol. A scramble shRNA control (sc-108080; Santa Cruz) and a GFP-expression vector (sc-108084; Santa Cruz) were also used. Maximal knockdown of PDGFR-α and PDGFR-β occurred 72–96 h after transduction.
Mouse xenograft model
All mouse protocols were approved by the Institutional Animal Care and Use Committee. To generate subcutaneous flank tumors, 1 × 106 HT1080 cells (with shRNA knockdown of PDGFR where indicated) were resuspended in 100 μL of Hank’s balanced salt solution and injected subcutaneously into the right flank of athymic, nude, 6–8 week-old male BALB/c nu/nu mice (Taconic, Hudson, NY) following isoflurane anesthesia. Mice were assigned into treatment groups (5 mice per group) when tumors reached 50–100 mm3 in volume. This time point was designated as “Day 0”. Doxorubicin (4 mg/kg) or control DMSO carrier was administered 2 times per week by intraperitoneal injection. Imatinib (90 mg/kg) was delivered daily by intraperitoneal injection. When mice were treated with combination therapies, imatinib was delivered first. Tumor volume (TV) was calculated by using the following formula: TV = length × (width)2 × 0.52.
Mice were killed and tumors were collected immediately and placed into 10% formalin for 2–3 days at 4 °C. Tumors were then rinsed, stored in saline, and sent to our pathology core facility. At least four sections were analyzed from each tumor. For immunofluorescence analysis, paraffin-embedded sections were deparaffinized and incubated with the primary antibody of PCNA (sc-56; Santa Cruz), cleaved caspase-3 (#9661; Cell Signaling), or CD133 (MBS462020; Miltenyi Biotec), in a solution of PBS with 1% BSA and 0.1% Triton X-100 at 4 °C overnight. Staining was visualized using Alexa Fluor 488 (A-21206; ThermoFisher), Alexa Fluor 568 (A-11011; ThermoFisher), and Alexa Fluor 647 (A-27034; ThermoFisher). Nuclei were counterstained using DAPI. To quantify immunofluorescence, slides were digitally scanned with Panoramic Flash 250 (3DHistech, Budapest, Hungary) using a 20×/0.8 NA objective. Stained tissues were counted in five microscopic fields. Image processing was performed using MetaMorph, version 7.8.2 (Molecular Devices).
Statistical analysis
Statistical analyses were performed using Microsoft Office Excel 2010 software. p-values were calculated using Student’s t-test. For comparisons between more than two groups, treatment groups were compared to the control group using one-way analysis of variance with Bonferroni adjustment for multiple comparisons. p-values <0.05 were considered significant.
Change history
29 May 2024
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41389-024-00520-7
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 67, 7–30 (2017).
Billingsley, K. G. et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann. Surg. 229, 602–610 (1999).
Van Glabbeke, M. et al. Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens--a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J. Clin. Oncol. 17, 150–157 (1999).
Tan, B. T., Park, C. Y., Ailles, L. E. & Weissman, I. L. The cancer stem cell hypothesis: a work in progress. Lab. Invest. 86, 1203–1207 (2006).
Terry, J. & Nielsen, T. Expression of CD133 in synovial sarcoma. Appl. Immunohistochem. Mol. Morphol. 18, 159–165 (2010).
Feng, B. H. et al. CD133+ subpopulation of the HT1080 human fibrosarcoma cell line exhibits cancer stem-like characteristics. Oncol. Rep. 30, 815–823 (2013).
Xiao, W., Mohseny, A. B., Hogendoorn, P. C. & Cleton-Jansen, A. M. Mesenchymal stem cell transformation and sarcoma genesis. Clin. Sarcoma Res. 3, 10 (2013).
Allegra, A. et al. The cancer stem cell hypothesis: a guide to potential molecular targets. Cancer Invest. 32, 470–495 (2014).
Fujii, H. et al. Sphere-forming stem-like cell populations with drug resistance in human sarcoma cell lines. Int. J. Oncol. 34, 1381–1386 (2009).
Chen, C. Y., Liu, S. H., Chen, C. Y., Chen, P. C. & Chen, C. P. Human placenta-derived multipotent mesenchymal stromal cells involved in placental angiogenesis via the PDGF-BB and STAT3 pathways. Biol. Reprod. 93, 103 (2015).
Zhang, J. M. et al. Platelet-derived growth factor-BB protects mesenchymal stem cells (MSCs) derived from immune thrombocytopenia patients against apoptosis and senescence and maintains MSC-mediated immunosuppression. Stem Cells Transl. Med. 5, 1631–1643 (2016).
Ng, F. et al. PDGF, TGF-beta, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 112, 295–307 (2008).
Farooqi, A. A. & Siddik, Z. H. Platelet-derived growth factor (PDGF) signalling in cancer: rapidly emerging signalling landscape. Cell Biochem. Funct. 33, 257–265 (2015).
Yoon, S. S., Segal, N. H., Olshen, A. B., Brennan, M. F., & Singer, S. Circulating angiogenic factor levels correlate with extent of disease and risk of recurrence in patients with soft tissue sarcoma. Ann. Oncol. 15, 1261–1266 (2004).
Tap, W. D. et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 388, 488–497 (2016).
Wirths, S. et al. Shared cell surface marker expression in mesenchymal stem cells and adult sarcomas. Stem Cells Transl. Med 2, 53–60 (2013).
Heldin, C. H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 11, 97 (2013).
Scheel, C. & Weinberg, R. A. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin. Cancer Biol. 22, 396–403 (2012).
Gotzmann, J. et al. A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene 25, 3170–3185 (2006).
Abouantoun, T. J. & MacDonald, T. J. Imatinib blocks migration and invasion of medulloblastoma cells by concurrently inhibiting activation of platelet-derived growth factor receptor and transactivation of epidermal growth factor receptor. Mol. Cancer Ther. 8, 1137–1147 (2009).
Yoon, C. et al. Multimodal targeting of tumor vasculature and cancer stem-like cells in sarcomas with VEGF-A inhibition, HIF-1alpha inhibition, and hypoxia-activated chemotherapy. Oncotarget 7, 42844–42858 (2016).
Ostman, A. & Heldin, C. H. PDGF receptors as targets in tumor treatment. Adv. Cancer Res. 97, 247–274 (2007).
Paulsson, J., Ehnman, M. & Ostman, A. PDGF receptors in tumor biology: prognostic and predictive potential. Future Oncol. 10, 1695–1708 (2014).
Lamouille, S., Xu, J. & Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 (2014).
van der Graaf, W. T. et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379, 1879–1886 (2012).
Peng, G. & Liu, Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol. Sci. 36, 374–383 (2015).
Kaurah, P. et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 297, 2360–2372 (2007).
UKCCCR guidelines for the use of cell lines in cancer research. Br. J. Cancer 82, 1495–1509 (2000).
Yoon, S. S. et al. Mouse endostatin inhibits the formation of lung and liver metastases. Cancer Res 59, 6251–6256 (1999).
Acknowledgements
This study was funded by NIH grants NIH/NCI Grant 1R01 CA158301-01 (S.S.Y.), Cycle for Survival (W.D.T.), and the NIH/NCI Cancer Center Support Grant P30 CA008748 (MSKCC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail:https://doi.org/10.1038/s41389-024-00520-7
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, K.K., Yoon, C., Yi, B.C. et al. RETRACTED ARTICLE: Platelet-derived growth factor receptor-α and -β promote cancer stem cell phenotypes in sarcomas. Oncogenesis 7, 47 (2018). https://doi.org/10.1038/s41389-018-0059-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-018-0059-1
This article is cited by
-
Retraction Note: Lymphatic metastasis-related TBL1XR1 enhances stemness and metastasis in gastric cancer stem-like cells by activating ERK1/2-SOX2 signaling
Oncogene (2024)
-
Retraction Note: ERK1/2-Nanog signaling pathway enhances CD44(+) cancer stem-like cell phenotypes and epithelial-to-mesenchymal transition in head and neck squamous cell carcinomas
Cell Death & Disease (2024)
-
Pediatric cancer—pathology and microenvironment influence: a perspective into osteosarcoma and non-osteogenic mesenchymal malignant neoplasms
Discover Oncology (2024)
-
Integrated analyses reveal two molecularly and clinically distinct subtypes of H3 K27M-mutant diffuse midline gliomas with prognostic significance
Acta Neuropathologica (2024)
-
Atovaquone inhibits colorectal cancer metastasis by regulating PDGFRβ/NF-κB signaling pathway
BMC Cancer (2023)
